Способ получения 3-замещенных 2-амино-5-галогенбензамидов - RU2443679C2
Код документа: RU2443679C2
Описание
Уровень техники
Как указано в патентных публикациях PCT WO 2003/015518, WO 2006/055922 и WO 2006/062978, 3-замещенные 2-амино-5-галогенбензамиды являются полезными исходными соединениями для получения диамидов антраниловой кислоты, проявляющих активность против членистоногих. В WO 2006/062978 раскрыто, что 3-замещенные 2-амино-5-галогенбензамиды могут быть получены галогенированием соответствующих 3-замещенных 2-аминобензамидов. Поскольку аминогруппа в значительной степени активирует электрофильное замещение в бензольном кольце, 3-замещенные 2-аминобензамиды быстро взаимодействуют с электрофильными галогенирующими реагентами по положению 5. Однако поскольку образующиеся продукты являются производными анилина и лишь частично деактивируются в результате моногалогенирования, они могут быть подвергнуты дальнейшему галогенированию. Соответственно, существует потребность в способах получения 3-замещенных 2-амино-5-галогенбензамидов без непосредственного взаимодействия анилина с галогенирующим агентом.
Сущность изобретения
Настоящее изобретение обеспечивает способ получения соединения формулы (1)

где
R1 представляет собой H, C1-C4 алкил, циклопропил, циклопропилметил или метилциклопропил;
R2 представляет собой CH3 или Cl; и
X представляет собой Cl или Br;
включающий взаимодействие соединения формулы 2

с соединением формулы 3
R1-NH2
3
в присутствии карбоновой кислоты.
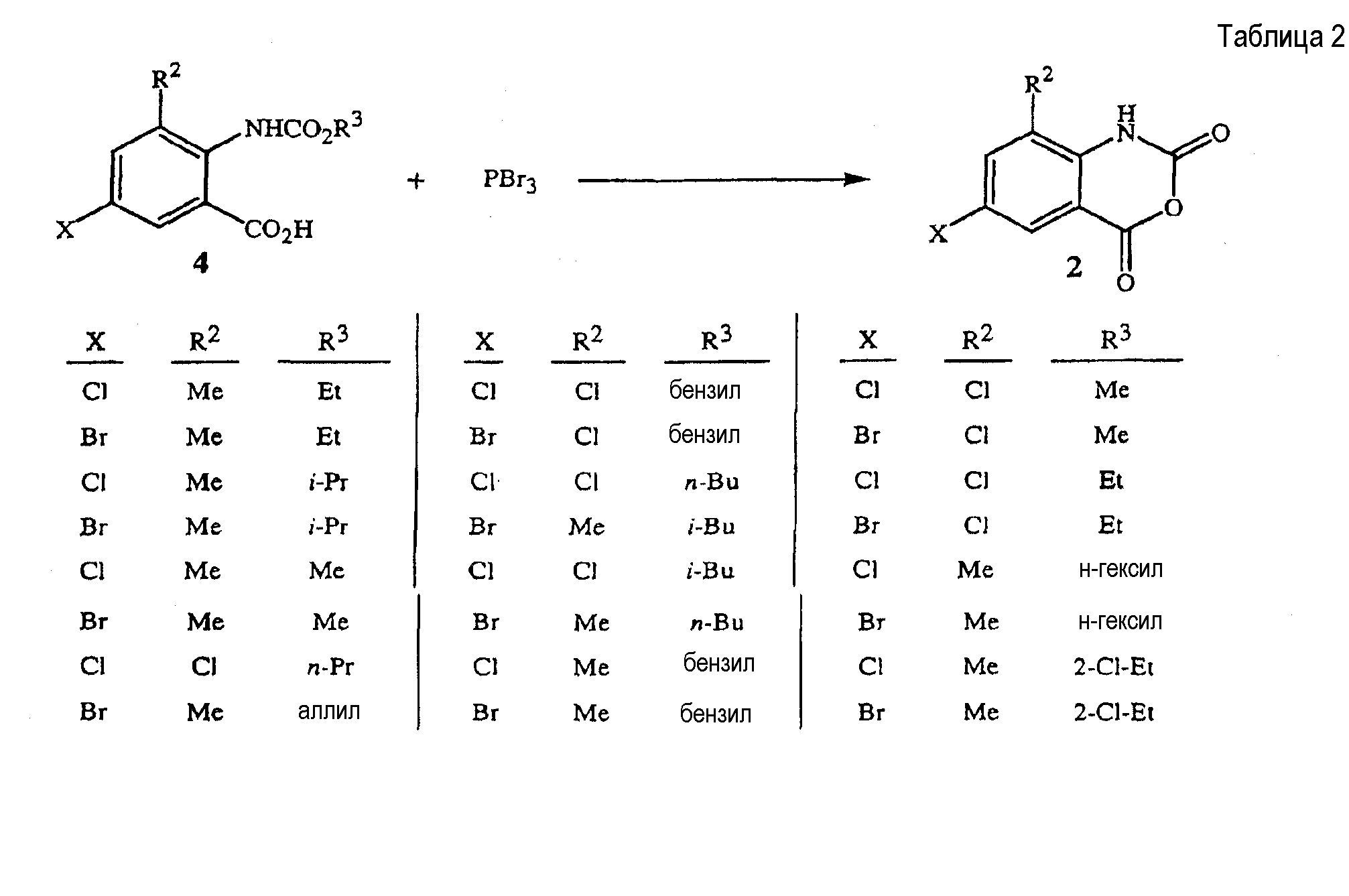
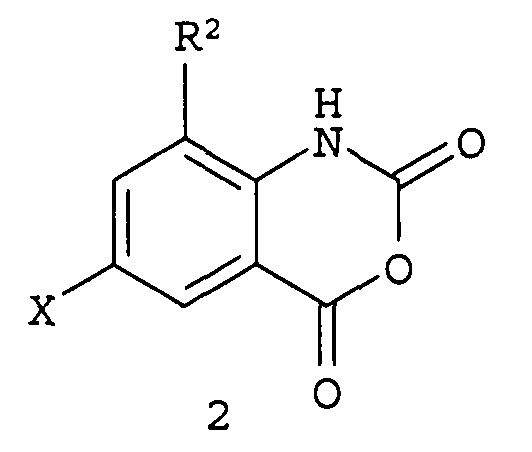
Настоящее изобретение также обеспечивает способ получения соединения формулы 2, где R2 представляет собой CH3 или Cl; и X представляет собой Cl или Br; включающий взаимодействие соединения формулы 4

где R3 представляет собой С1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом;
с трибромидом фосфора.
Настоящее изобретение далее относится к новому соединению формулы 4, где R2 представляет собой CH3 или Cl; R3 представляет собой С1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом; и X представляет собой Cl или Br; при условии что если R2 и X каждый представляет собой Cl, тогда R3 не является CH3; причем указанное соединение представляет собой полезное промежуточное соединение для получения соединений формул 1 и 2 указанными выше способами.
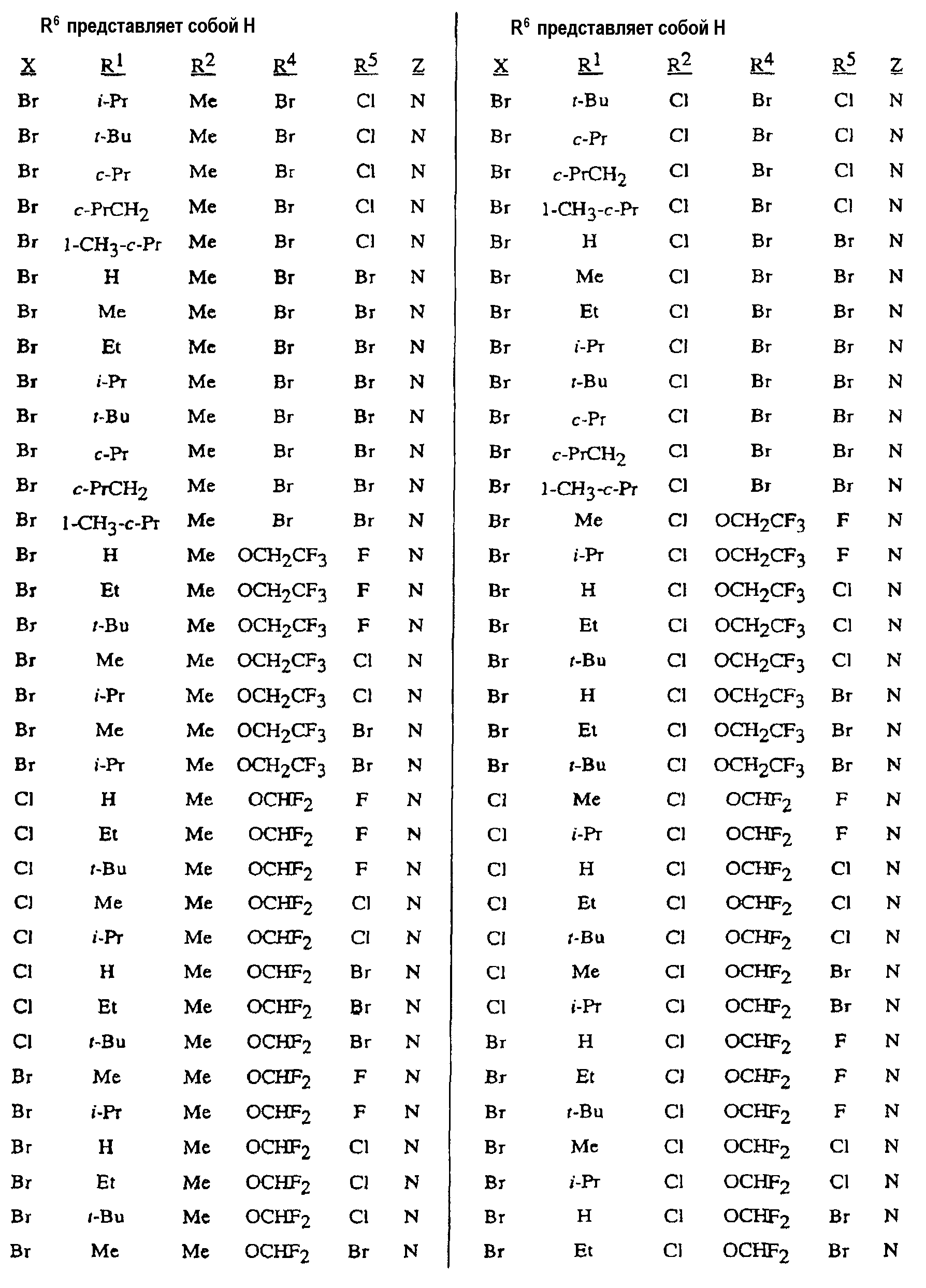
Настоящее изобретение также относится к способу получения соединения формулы 5

где
X представляет собой Cl или Br;
Z представляет собой CR7 или N;
R1 представляет собой H, С1-С4 алкил, циклопропил, циклопропилметил или метилциклопропил;
R2 представляет собой CH3 или Cl;
R4 представляет собой Cl, Br, CF3, OCF2H или OCH2CF3;
R5 представляет собой F, Cl или Br;
R6 представляет собой H, F или Cl; и
R7 представляет собой H, F, Cl или Br;
с использованием соединения формулы 1. Данный способ отличается тем, что соединение формулы 1 получают из соединений формул 2 и 3 способом, показанным выше.
Подробное описание изобретения
Как используется в настоящем описании, термины «содержит», «содержащий», «включает», «включающий», «имеет», «имеющий» или любые другие их варианты относятся к включению, не исключающему других возможностей. Например, композиция, способ, метод, изделие или агрегат, которые включают перечень элементов, необязательно ограничиваются только этими элементами, но могут включать другие элементы, которые не перечислены явно или не присущи такой композиции, способу, методу, изделию или агрегату. Далее, если прямо не указано противоположное, частица «или» имеет включающий, а не исключающий смысл. Например, условие «A или B» удовлетворяется любой из следующих ситуаций: A верно (или присутствует) и B ложно (или не присутствует), A ложно (или не присутствует) и B верно (или присутствует), и A и B верны (или присутствуют).
Кроме того, форма единственного числа, относящаяся к элементу или компоненту изобретения, также включает множественное число, если не очевидно, что число является единственным.
Термин «необязательно замещенный» в определении радикала (например, алкила или алкенила) означает, что данный радикал является незамещенным или замещен одним или несколькими заместителями вплоть до любого установленного предела числа заместителей. Поскольку термин «необязательно замещенный» включает возможность отсутствия замещения, фраза «каждый необязательно замещен 1-3 заместителями» означает, что возможно присутствие 0, 1, 2 или 3 заместителей. Следовательно, фраза «каждый необязательно замещен 1-3 заместителями» является синонимом фразе «каждый необязательно замещен 0-3 заместителями» и фразе «каждый необязательно замещен не более чем 3 заместителями». Родственные фразы, включающие словосочетание «необязательно замещенный», определяются аналогичным образом. В качестве дополнительного примера фраза «каждый необязательно замещен не более чем 3 атомами галогена» является синонимом фразе «каждый необязательно замещен 1-3 атомами галогена» и фраза «каждый необязательно замещен не более чем 1 фенилом» является синонимом «каждый необязательно замещен 0-1 фенилом». Когда галоген приведен в контексте диапазона, включающего 1 или более одного (например, «не более 3 атомов галогена»), форма единственного числа слова «галоген» означает «галогены» или «атомы галогена», когда присутствует более одного атома галогена. Когда присутствует более одного заместителя, каждый заместитель независим от других. Например, если в качестве заместителей присутствуют два атома галогена или более, каждый из атомов галогена может быть одинаковым или различным галогеном.
Соотношения, как правило, приводятся в настоящем описании в виде одного числа, которое соотносится с числом 1; например, соотношение 4 означает 4:1.
В настоящем описании и формуле изобретения термин «карбоновая кислота» означает химическое органическое соединение, включающее по меньшей мере одну карбоксильную функциональную группу (т.е. -C(O)OH). Термин «карбоновая кислота» не включает угольную кислоту (т.е. HOC(O)OH). Карбоновые кислоты включают, например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, хлоруксусную кислоту, бензойную кислоту, малеиновую кислоту и лимонную кислоту. Термин «эффективное значение pKa» относится к pKa функциональной группы карбоновой кислоты или, если соединение имеет более чем одну функциональную группу карбоновой кислоты, термин «эффективное значение pKa» относится к pKa наиболее кислотной функциональной группе карбоновой кислоты. В настоящем описании «эффективное значение pH» неводного вещества или смеси, такой как реакционная смесь, определяют смешиванием аликвоты вещества или смеси примерно с 5-20 объемами воды с последующим измерением pH полученной водной смеси (например, с помощью pH-метра). В настоящем описании термин «по существу безводное» вещество означает, что вещество содержит не более примерно 1% воды по массе. Химическое название «изатоевый ангидрид» является другим наименованием, соответствующим современному наименованию согласно номенклатуре Chemical Abstracts «2H-3,1-бензоксазин-2,4(1H)-дион».
Варианты осуществления настоящего изобретения включают:
Вариант осуществления A1. Способ, описанный в разделе «Сущность изобретения», получения соединения формулы 1, включающий взаимодействие соединения формулы 2 с соединением формулы 3 в присутствии карбоновой кислоты.
Вариант осуществления A2. Способ варианта осуществления A1, где R1 представляет собой C1-C4 алкил, циклопропил, циклопропилметил или метилциклопропил.
Вариант осуществления A3. Способ варианта осуществления A2, где R1 представляет собой C1-C4 алкил или циклопропилметил.
Вариант осуществления A4. Способ варианта осуществления A3, где R1 представляет собой метил.
Вариант осуществления A5. Способ варианта осуществления A1, где мольное соотношение соединения формулы 3 к соединению формулы 2 составляет примерно от 1,1 до примерно 2.
Вариант осуществления A5a. Способ варианта осуществления A5, где мольное соотношение соединения формулы 3 к соединению формулы 2 составляет примерно от 1,1 до примерно 1,5.
Вариант осуществления A5b. Способ варианта осуществления A5a, где мольное соотношение соединения формулы 3 к соединению формулы 2 составляет примерно от 1,1 до примерно 1,3.
Вариант осуществления A5c. Способ варианта осуществления A5b, где мольное соотношение соединения формулы 3 к соединению формулы 2 составляет примерно от 1,2 до примерно 1,3.
Вариант осуществления A6. Способ варианта осуществления A1, где соединение формулы 2 подвергают взаимодействию с соединением формулы 3 в присутствии карбоновой кислоты и в присутствии подходящего органического растворителя.
Вариант осуществления A7. Способ варианта осуществления A1, где соединение формулы 2 подвергают взаимодействию с соединением формулы 3 в присутствии карбоновой кислоты в реакционной среде, включающей подходящий органический растворитель.
Вариант осуществления A8. Способ варианта осуществления A7, где реакционная среда содержит 5 мас.% воды или менее.
Вариант осуществления A9. Способ варианта осуществления A8, где реакционная среда содержит 1 мас.% воды или менее.
Вариант осуществления A10. Способ варианта осуществления A9, где реакционная среда содержит 0,1 мас.% воды или менее.
Вариант осуществления A11. Способ варианта осуществления A7, где реакционная среда является по существу безводной.
Вариант осуществления A12. Способ любого из вариантов осуществления A6 и A7, где в органический растворитель входят один или несколько растворителей, выбранных из сложных эфиров, кетонов, нитрилов, галогеналканов, простых эфиров и галогенированных и негалогенированных ароматических углеводородов.
Вариант осуществления A13. Способ варианта осуществления A12, где органический растворитель включает эфир C2-C3 алкилкарбоновой кислоты и C1-C3 алканола.
Вариант осуществления A14. Способ варианта осуществления A13, где органический растворитель включает этилацетат.
Вариант осуществления A15. Способ варианта осуществления A1, где взаимодействие проводят в реакционной среде, имеющей pH в диапазоне примерно от 3 до примерно 7.
Вариант осуществления A16. Способ варианта осуществления A15, где карбоновая кислота выбрана таким образом, чтобы обеспечить pH в пределах указанного диапазона.
Вариант осуществления A17. Способ варианта осуществления A1, где карбоновая кислота имеет эффективное значение pKa примерно от 2 до примерно 5.
Вариант осуществления A18. Способ варианта осуществления A1, где карбоновая кислота представляет собой C2-C18 алкилкарбоновую кислоту.
Вариант осуществления A19. Способ варианта осуществления A18, где карбоновая кислота представляет собой уксусную кислоту.
Вариант осуществления A20. Способ варианта осуществления A1, где мольное отношение соединения формулы 3 к карбоновой кислоте составляет примерно от 0,6 до примерно 3.
Вариант осуществления A20a. Способ варианта осуществления A20, где мольное отношение соединения формулы 3 к карбоновой кислоте составляет примерно от 0,6 до примерно 1,2.
Вариант осуществления A20b. Способ варианта осуществления A20, где мольное отношение соединения формулы 3 к карбоновой кислоте составляет примерно от 0,8 до примерно 3.
Вариант осуществления A20c. Способ варианта осуществления A20b, где мольное отношение соединения формулы 3 к карбоновой кислоте составляет примерно от 0,8 до примерно 1,2.
Вариант осуществления A21. Способ варианта осуществления A1, где соединение формулы 2 подвергают взаимодействию с соединением формулы 3 и карбоновой кислотой при температуре примерно от 5 до примерно 75°C.
Вариант осуществления A21a. Способ варианта осуществления A21, где температура находится в пределах примерно от 15°C до примерно 70°C.
Вариант осуществления A21b. Способ варианта осуществления A21a, где температура находится в пределах примерно от 35°C до примерно 60°C.
Вариант осуществления A21c. Способ варианта осуществления A21b, где температура находится в пределах примерно от 35°C до примерно 55°C.
Вариант осуществления A21d. Способ варианта осуществления A21b, где температура находится в пределах примерно от 50°C до примерно 60°C.
Вариант осуществления A22. Способ варианта осуществления A21d, где температура находится в пределах примерно от 50°C до примерно 55°C.
Вариант осуществления A23. Способ варианта осуществления A1, где соединение формулы 3 добавляют в смесь соединения формулы 2 и карбоновой кислоты.
Вариант осуществления A24. Способ варианта осуществления A23, где соединение формулы 3 добавляют в безводной форме (т.е. по существу безводной форме).
Вариант осуществления A25. Способ варианта осуществления A1, где соединение формулы 2 получают путем взаимодействия соединения формулы 4 с трибромидом фосфора.
Вариант осуществления A26. Способ варианта осуществления A1, где соединение формулы 3 добавляют в смесь, содержащую соединение формулы 2 и карбоновую кислоту.
Вариант осуществления B1. Способ, описанный в разделе «Сущность изобретения», получения соединения формулы 2, включающий взаимодействие соединения формулы 4 с трибромидом фосфора.
Вариант осуществления B4. Способ варианта осуществления B1, где R3 представляет собой C1-C4 алкил.
Вариант осуществления B5. Способ варианта осуществления B4, где R3 не содержит разветвления у атома углерода, связанного с кислородом.
Вариант осуществления B6. Способ варианта осуществления B5, где R3 представляет собой метил или этил.
Вариант осуществления B7. Способ варианта осуществления B1, где соединение формулы 4 подвергают взаимодействию с трибромидом фосфора в присутствии подходящего органического растворителя.
Вариант осуществления B8. Способ варианта осуществления B1, где органический растворитель включает один или несколько растворителей, выбранных из сложных эфиров, нитрилов, углеводородов и галогенированных углеводородов.
Вариант осуществления B8a. Способ варианта осуществления B8, где органический растворитель включает один или несколько растворителей, выбранных из сложных эфиров, нитрилов, галогеналканов, а также галогенированных и негалогенированных ароматических углеводородов.
Вариант осуществления B9. Способ варианта осуществления B8a, где органический растворитель включает один или несколько растворителей, выбранных из галогеналканов и галогенированных и негалогенированных ароматических углеводородов.
Вариант осуществления B10. Способ варианта осуществления B9, где органический растворитель включает один или несколько растворителей, выбранных из 1,2-дихлорэтана, бензола, толуола, ксилола и хлорбензола.
Вариант осуществления B11. Способ варианта осуществления B10, где органический растворитель включает толуол.
Вариант осуществления B12. Способ варианта осуществления B1, где мольное соотношение трибромида фосфора и соединения формулы 3 составляет примерно от 0,3 до примерно 3.
Вариант осуществления B12a. Способ варианта осуществления B12, где мольное соотношение трибромида фосфора и соединения формулы 3 составляет примерно от 0,3 до примерно 0,5.
Вариант осуществления B13. Способ варианта осуществления B12a, где мольное соотношение трибромида фосфора и соединения формулы 3 составляет примерно от 0,33 до примерно 0,40.
Вариант осуществления B14. Способ варианта осуществления B1, где соединение формулы 3 подвергают взаимодействию с трибромидом фосфора при температуре в диапазоне примерно от 50°C до примерно 90°C.
Вариант осуществления B14a. Способ варианта осуществления B14, где температура находится в диапазоне примерно от 50°C до примерно 80°C.
Вариант осуществления B14b. Способ варианта осуществления B14a, где температура находится в диапазоне примерно от 60°C до примерно 75°C.
Вариант осуществления B15. Способ варианта осуществления B14b, где температура находится в диапазоне примерно от 60°C до примерно 70°C.
Вариант осуществления C1. Способ по любому из вариантов осуществления A1 и B1, где R2 представляет собой метил.
Вариант осуществления C2. Способ по любому из вариантов осуществления A1 и B1, где X представляет собой Cl.
Вариант осуществления C3. Способ по любому из вариантов осуществления A1 и B1, где X представляет собой Br.
Вариант осуществления C4. Способ по любому из вариантов осуществления A1, A4 и B1, где R2 представляет собой CH3 и X представляет собой Cl.
Вариант осуществления D1. Соединение формулы 4, где R2 представляет собой CH3 или Cl; R3 представляет собой C1-C6 алкил или C3-C6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом; и X представляет собой Cl или Br; при условии что когда R2 и X каждый является Cl, R3 отличен от CH3.
Вариант осуществления D2. Соединение по варианту осуществления D1, где R2 представляет собой CH3.
Вариант осуществления D3. Соединение по варианту осуществления D1, где R3 представляет собой C1-C4 алкил.
Вариант осуществления D4. Соединение по варианту осуществления D3, где R3 не содержит разветвления у атома углерода, связанного с кислородом.
Вариант осуществления D5. Соединение по варианту осуществления D4, где R3 представляет собой метил или этил.
Вариант осуществления D6. Соединение по варианту осуществления D1, где X представляет собой Cl.
Вариант осуществления D7. Соединение по варианту осуществления D1, где X представляет собой Br.
Вариант осуществления D8. Соединение по варианту осуществления D1, где R2 представляет собой CH3, и X представляет собой Cl.
Вариант осуществления D9. Соединение по варианту осуществления D1, где R2 представляет собой CH3, и X представляет собой Br.
Вариант осуществления D10. Соединение по любому из вариантов осуществления D1, D8 и D9, где R3 представляет собой C1-C2 алкил.
Вариант осуществления D11. Соединение по варианту осуществления D10, где R2 представляет собой CH3, R3 представляет собой CH3, и X представляет собой Cl.
Вариант осуществления D12. Соединение по варианту осуществления D10, где R2 представляет собой CH3, R3 представляет собой CH3, и X представляет собой Br.
Вариант осуществления D13. Соединение по варианту осуществления D10, где R2 представляет собой CH3, R3 представляет собой CH2CH3, и X представляет собой Cl.
Вариант осуществления D14. Соединение по варианту осуществления D10, где R2 представляет собой CH3, R3 представляет собой CH2CH3, и X представляет собой Br.
Вариант осуществления D15. Соединение по варианту осуществления D1, где если X представляет собой Cl, то R2 отличен от Cl.
Вариант осуществления D16. Соединение по варианту осуществления D1, где R2 представляет собой CH3.
Вариант осуществления E1. Способ, описанный в разделе «Сущность изобретения», получения соединения формулы 5, включающий использование соединения формулы 1, полученного из соединений формул 2 и 3.
Вариант осуществления E2. Способ варианта осуществления E1, где X представляет собой Cl.
Вариант осуществления E3. Способ варианта осуществления E1, где X представляет собой Br.
Вариант осуществления E4. Способ варианта осуществления E1, где Z представляет собой N.
Вариант осуществления E5. Способ варианта осуществления E1, где R1 представляет собой С1-С4 алкил, циклопропил, циклопропилметил или метилциклопропил.
Вариант осуществления E6. Способ варианта осуществления E5, где R1 представляет собой С1-С4 алкил или циклопропилметил.
Вариант осуществления E7. Способ варианта осуществления E6, где R1 представляет собой метил.
Вариант осуществления E8. Способ варианта осуществления E1, где R2 представляет собой CH3.
Вариант осуществления E9. Способ варианта осуществления E1, где R4 представляет собой Br.
Вариант осуществления E10. Способ варианта осуществления E1, где R5 представляет собой Cl.
Вариант осуществления E11. Способ варианта осуществления E1, где R6 представляет собой H.
Вариант осуществления E12. Способ варианта осуществления E1, где R1 представляет собой CH3, R2 представляет собой CH3, R4 представляет собой Br, R5 представляет собой Cl, R6 представляет собой H, X представляет собой Cl, и Z представляет собой N.
Варианты осуществления настоящего изобретения можно комбинировать любым образом.
Способы и промежуточные соединения по настоящему изобретению более подробно описаны ниже. На приведенных ниже схемах определения R1, R2, R3, R4, R5, R6, R7, X и Z являются такими, как определено выше, если не указано иное.
Как показано на схеме 1, в способе по настоящему изобретению замещенный антраниламид формулы 1 получают путем взаимодействия замещенного изатоевого ангидрида формулы 2 с амином формулы 3 в присутствии карбоновой кислоты.
Схема 1

Поскольку амины, такие как соединение формулы 3, являются основаниями, в отсутствие карбоновой кислоты смесь соединений формул 2 и 3 должна являться основной (например, эффективное значение pH>7). В способе по настоящему изобретению карбоновая кислота действует в качестве буфера, уменьшая эффективное значение pH реакционной смеси. В способе по настоящему изобретению могут быть использованы самые различные карбоновые кислоты, поскольку единственным условием является наличие хотя бы одной группы карбоновой кислоты для придания соединению кислотности. В молекуле карбоновой кислоты могут присутствовать другие функциональные группы, а также может присутствовать более чем одна группа карбоновой кислоты. Обычно карбоновая кислота в способе по настоящему изобретению имеет эффективное значение pKa в диапазоне примерно от 2 до примерно 5. Карбоновые кислоты включают, например, муравьиную кислоту, пропионовую кислоту, хлоруксусную кислоту, бензойную кислоту, фталевую кислоту, малеиновую кислоту, винную кислоту и лимонную кислоту. С точки зрения стоимости предпочтительны недорогие карбоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота и бензойная кислота. Особенно предпочтительна уксусная кислота, которая может быть приобретена по невысокой стоимости в безводной форме (известной как «ледяная уксусная кислота»).
Комбинация карбоновой кислоты с основным амином формулы 3 образует соль амина и карбоновой кислоты. Соль амина может быть получена перед добавлением изатоевого ангидрида формулы 2, или соль амина может быть получена in situ путем добавления определенного количества амина формулы 3 в смесь соединения формулы 2 и карбоновой кислоты. При любом способе добавления наилучшее осуществление способа по настоящему изобретению заключается в поддержании эффективного pH смеси во время реакции в диапазоне примерно от 3 до примерно 7.
Поскольку эффективное значение pH смеси является результатом буферного действия карбоновой кислоты в комбинации с амином формулы 3, эффективный pH может быть отрегулирован в соответствии с эффективным значением pKa карбоновой кислоты, за счет регулирования мольного соотношения карбоновой кислоты и амина формулы 3. Как правило, мольные соотношения амина формулы 3 и карбоновой кислоты находятся в пределах примерно от 0,6 до примерно 3, преимущественно примерно от 0,8 до примерно 3. В частности, если способ смешивания реагентов включает добавление определенного количества амина формулы 3 в смесь изатоевого ангидрида формулы 2 и карбоновой кислоты, мольное соотношение амина формулы 3 и карбоновой кислоты предпочтительно составляет примерно от 0,85 до примерно 3. Когда способ смешивания реагентов включает получение соли амина перед добавлением соединения формулы 2, мольное соотношение амина формулы 3 и карбоновой кислоты предпочтительно составляет примерно от 0,8 до примерно 1,05; если применяется почти эквимолярное соотношение (например, приблизительно от 0,95 до приблизительно 1,05) амина формулы 3 и карбоновой кислоты, полученную при этом соль амина используют в мольном соотношении примерно от 1,1 до примерно 5 к соединению формулы 2. Для достижения оптимального превращения молярное отношение амина формулы 3 к изатоевому ангидриду соединения формулы 2 должно составлять не менее 1,0, хотя предпочтительно, чтобы это отношение составляло примерно от 1,1 до примерно 1,5, с точки зрения эффективности и экономии, независимо от способа смешивания компонентов. Количество молей амина формулы 3 по отношению к соединению формулы 2 может значительно превышать 1,5, в частности когда используется почти эквимолярное соотношение амина и кислоты (например, приблизительно от 0,95 до приблизительно 1,05).
Способ, приведенный на схеме 1, обычно дает возможность добиться наиболее высоких выходов и чистоты продукта, когда реакционная среда является по существу безводной. Поэтому в реакционную среду, обычно, добавляют по существу безводные соединения формул 2 и 3 и карбоновую кислоту. Предпочтительно реакционная среда и составляющие ее вещества содержат примерно 5% или менее, более предпочтительно примерно 1% или менее, и наиболее предпочтительно примерно 0,1% или менее воды (по массе). Если карбоновая кислота представляет собой уксусную кислоту, ее предпочтительно используют в виде ледяной уксусной кислоты.
Реакцию по схеме 1, как правило, проводят в жидкой фазе. Во многих случаях реакция может быть осуществлена без специального растворителя, отличного от соединений формул 1, 2, 3 и карбоновой кислоты. Однако предпочтительная методика включает использование растворителя, в котором можно суспендировать или по меньшей мере частично растворить реагенты. В число предпочтительных растворителей входят такие растворители, которые не обладают реакционной способностью в отношении компонентов реакционной смеси и имеют диэлектрическую константу, равную 5 или более, такие как алкилнитрилы, сложные эфиры, простые эфиры или кетоны. Предпочтительно растворитель должен быть по существу безводным, чтобы была возможность получить по существу безводную реакционную среду. Массовое соотношение растворителя к соединению формулы 2 составляет, как правило, примерно от 1 до примерно 20 и предпочтительно примерно 5, по соображениям эффективности и экономичности.
В способе, изображенном на схеме 1, в качестве побочного продукта образуется диоксид углерода. При проведении способа обычно большая часть образовавшегося диоксида углерода выделяется из реакционной смеси в виде газа. Добавление соединения формулы 2 в реакционную среду, содержащую амин формулы 3, или добавление амина формулы 3 в реакционную среду, содержащую соединение формулы 2, предпочтительно проводят с такой скоростью и при такой температуре, которые позволяют контролировать выделение диоксида углерода. Как правило, температура реакционной среды составляет примерно от 5 до 75°C, чаще примерно от 35 до 60°C.
Продукт формулы 1 может быть выделен стандартными методами, известными в данной области, включая регулирование значения pH, экстракцию, выпаривание, кристаллизацию и хроматографию. Например, реакционную смесь можно разбавить примерно 3-15 массовыми частями воды, относительно массы исходного соединения формулы 2, pH необязательно можно изменить до желаемого с помощью кислоты или основания для оптимизации удаления или кислотных, или основных загрязнений, водная фаза может быть необязательно отделена и большая часть органического растворителя может быть удалена отгонкой или выпариванием при пониженном давлении. Поскольку соединения формулы 1 при комнатной температуре являются, как правило, твердыми кристаллическими веществами, их по существу легче всего выделять фильтрованием, необязательно с последующим промыванием водой и затем сушкой. Как правило, регулирование pH необходимо во время обработки реакционной среды, и вода является средой, которую можно использовать для кристаллизации продуктов формулы 1. Следовательно, особенно удобная методика заключается в разбавлении реакционной среды водой, удалении большей части органического растворителя отгонкой при атмосферном давлении и затем охлаждении водной смеси для кристаллизации продукта, который затем может быть отделен фильтрованием. Способ, показанный на схеме 1, проиллюстрирован приведенными ниже примерами 2-5.
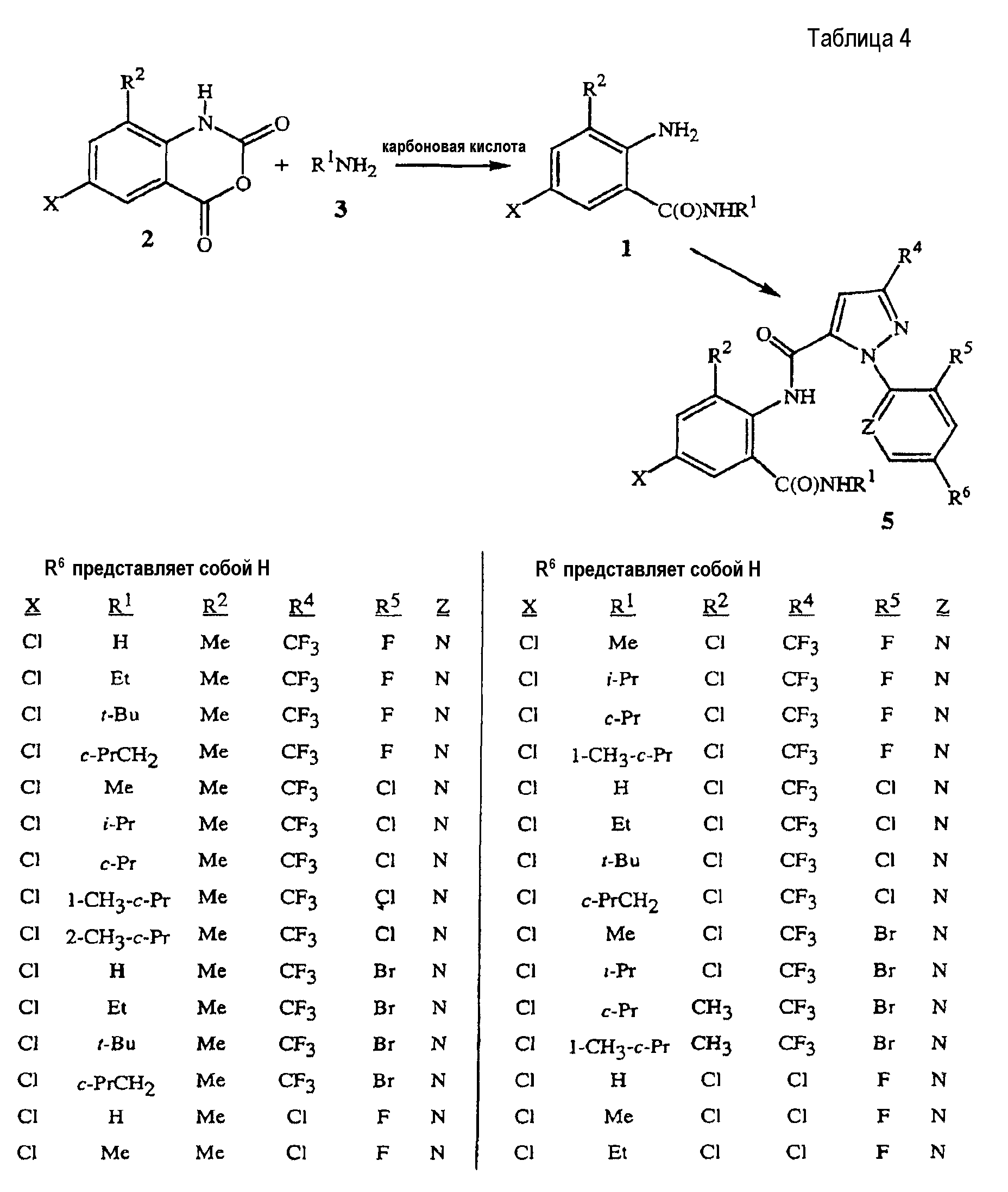
Как показано на схеме 2, в другом аспекте настоящего изобретения замещенный изатоевый ангидрид формулы 2 получают путем взаимодействия соединения формулы 4 с трибромидом фосфора.
Схема 2

Не ограничиваясь какой-либо конкретной теорией, можно предположить, что трибромид фосфора взаимодействует с соединением формулы 4, образуя наряду с бромистым водородом соединение формулы 10, как показано на рис.1, в качестве промежуточного соединения, которое затем взаимодействует с образованием соединения формулы 2 и R3Br, в качестве итогового побочного продукта.
Рисунок 1

В способе по схеме 2 стехиометрическое количество трибромида фосфора, необходимое для достижения полного превращения соединения формулы 4 в соединение формулы 2, составляет одну треть мольного эквивалента. Как правило, количество используемого трибромида фосфора составляет примерно от 0,3 до 3 мольных эквивалентов, причем с точки зрения экономичности предпочтительно количество примерно от 0,33 до примерно 0,4 эквивалентов.
Способ, изображенный на схеме 2, как правило, проводят в жидкой фазе, обычно содержащей растворитель, для по меньшей мере частичного растворения соединения формулы 4. Растворитель должен быть инертен в отношении трибромида фосфора и предпочтительно должен иметь температуру кипения при нормальном давлении выше 50°C, предпочтительно выше 70°C, для обеспечения необходимой температуры реакции. Примерами растворителей, подходящих для данной реакции, являются углеводороды (например, циклогексан, бензол, толуол), галогенированные углеводороды (например, 1-хлорбутан, 1,2-дихлорэтан, хлорбензол, о-дихлорбензол), сложные эфиры (например, н-бутилацетат) или нитрилы (например, ацетонитрил, бензонитрил).
Способ по схеме 2 удобно осуществлять путем разбавления соединения формулы 4 растворителем с последующим добавлением трибромида фосфора. Как правило, трибромид фосфора добавляют в реакционную смесь, содержащую соединение формулы 4, с такой скоростью, чтобы температура реакционной смеси составляла диапазон примерно от 50 до 80°C. Предпочтительно скорость добавления трибромида фосфора выбирают таким образом, чтобы поддерживать температуру реакционной смеси в диапазоне примерно от 60 до 75°C, поскольку это дает возможность управлять экзотермической реакцией и добиться максимальной чистоты продукта.
После завершения реакции продукт формулы 2 может быть выделен с помощью стандартных методик, известных в данной области, включая барботирование газа, регулирование pH, экстракцию, выпаривание, кристаллизацию и хроматографию. Большая часть побочного продукта R3Br и оставшийся в реакционной смеси бромистый водород можно удалить барботированием воздуха или газа, например, азота. Соединения формулы 2, как правило, являются твердыми кристаллическими веществами; при охлаждении реакционной смеси продукт обычно кристаллизуется в виде твердого вещества, которое может быть отделено фильтрованием, промыто водой для удаления остатков фосфорной кислоты и бромистого водорода и высушено. Способ по схеме 2 проиллюстрирован примером 1.
Соединения формулы 4 могут быть получены согласно общим способам, известным в данной области, включая, например, галогенирование соответствующих соединений формулы 11 с хлором или бромом, как показано на схеме 3.
Схема 3

Особенно пригодными для галогенирования по схеме 3 являются хлор или бром в момент выделения, которые образуются при взаимодействии водных растворов хлористоводородной или бромистоводородной кислот с пероксидом водорода, согласно общему способу патентной публикации Германии DE 2750292-A1. Данный способ проиллюстрирован справочным примером 1, в котором X представляет собой хлор. Соответствующие соединения могут быть получены по данной методике при замене хлористоводородной кислоты бромистоводородной кислотой.
Можно надеяться, что специалист в данной области техники сможет использовать настоящее изобретение в самой полной степени без дополнительных исследований. Поэтому следующие далее примеры следует истолковывать только как иллюстративные, а не ограничивающие каким бы то ни было образом раскрытие настоящего изобретения. Процентные соотношения указаны по массе, за исключением смесей растворителей для хроматографии или тех случаев, где это специально оговорено. Части и процентные доли для смесей хроматографических растворителей приведены по объемам, если не указано иное. Чистоту продуктов, содержащих 2-амино-5-хлор-N,3-диметилбензамид, определяли ВЭЖХ с обращенной фазой, используя колонку Ace C4 (Advanced Chromatography Technologies, Aberdeen, Scotland) и градиент смеси ацетонитрил/вода, содержащей 0,005 М буфер NaH2PO4/H2O, pH которого доводили до 3 добавлением H3PO4. Данные спектров1H ЯМР приведены в миллионных долях сдвига в слабое поле относительно тетраметилсилана; “с” означает синглет, “д” означает дублет, “т” означает триплет, “кв” означает квартет, “м” означает мультиплет, “дд” означает дублет дублетов, “дт” означает дублет триплетов, “ушир.с” означает уширенный синглет, и “ушир.м” означает уширенный мультиплет.
Справочный пример 1
Получение 5-хлор-2-[(этоксикарбонил)амино]-3-метилбензойной кислоты (соединение формулы 4)
В 2-л реактор, снабженный верхней мешалкой и термопарой, загружали 150 г (0,672 моль) 2-[(этоксикарбонил)амино]-3-метилбензойной кислоты (прибл. 98% чистоты) и уксусную кислоту (500 г). Полученную суспензию нагревали до 35-40°C, с получением раствора, который охлаждали до 30°C и затем добавляли хлористоводородную кислоту (37%, 150 г, 1,5 моль, 2,2 экв). Поддерживая температуру смеси на уровне 30°C, добавляли водный раствор пероксида водорода (30%, 96 г, 0,85 моль, 1,25 экв) в течение примерно 1 ч. Затем смесь нагревали до 35°C и выдерживали при этой температуре в течение примерно 1 ч. В течение примерно 30 минут добавляли приблизительно 600 мл воды, поддерживая температуру на уровне 30-35°C. Смесь охлаждали до 10°C, отделяли продукт фильтрованием и влажный осадок на фильтре промывали водой (3×100 мл); при третьей промывке испытывали воду на отрицательную реакцию с помощью индикаторной бумаги KI-крахмал. Влажный осадок сушили до постоянной массы в вакуумной печи при 50°C. Выход неочищенного вещества составлял примерно 150 г (примерно 84%, исходя из измеренной чистоты 2-[(этоксикарбонил)амино]-3-метилбензойной кислоты 98% и измеренной чистоты продукта 95%). Часть неочищенного продукта подвергали первичной перекристаллизации из толуола и затем повторно перекристаллизовывали из водного метанола, получая образец для анализа с температурой плавления 124-126°C.
1H ЯМР (ДМСО-d6) δ 1,19 (т, 3Н), 2,22 (с, 3Н), 4,05 (кв, 2Н), 7,54 (м, 2Н), 8,9 (ушир.с, 1Н), 13,1 (ушир.с, 1Н).
Пример 1
Получение 6-хлор-8-метил-2H-3,1-бензоксазин-2,4(1H)-диона (соединение формулы 2)
В 1-л трехгорлую колбу, снабженную капельной воронкой, термометром, обратным холодильником, устройством для барботирования азота и поглотителем щелочи, загружали 5-хлор-2-[(этоксикарбонил)амино]-3-метилбензойную кислоту (т.е. продукт справочного примера 1) (74,0 г, 0,288 моль) и толуол (300 мл). Смесь нагревали до 60-65°C и при этой температуре в течение примерно 60 минут добавляли трибромид фосфора (39 г, 0,144 моль). Смесь нагревали при 65°C в течение примерно 30 минут, в результате чего согласно данным ВЭЖХ-анализа в реакционной смеси оставалось не более 0,2% промежуточно образовавшегося 6-хлор-2-этокси-8-метил-4H-3,1-бензоксазин-4-она. В смесь барботировали азот для удаления бромистого водорода и этилбромида и затем охлаждали до 20°C. Продукт отделяли фильтрованием и осадок на фильтре последовательно промывали толуолом (30 мл) и водой (2×100 мл), и затем сушили при отсасывании. Сушка выделенного твердого вещества в вакуумной печи до постоянной массы приводила к получению указанного в заголовке соединения (59 г, чистота примерно 97% по данным анализа ВЭЖХ). Часть высушенного продукта перекристаллизовывали путем растворения в N,N-диметилформамиде (4 объема) при 60°C и охлаждения до 20°C, с получением образца с чистотой 99%, плавящегося при температуре >250°C.
1H ЯМР (ДМСО-d6) δ 2,33 (с, 3Н), 7,67 (дд, 1Н, J=2,5 и 0,6 Гц), 7,72 (д, 1Н, J=2,4 Гц), 11,2 (ушир.с, 1Н).
Пример 2
Получение 2-амино-5-хлор-N,3-диметилбензамида (соединения формулы 1)
В 300-мл колбу, снабженную термометром и устройством для барботирования азота, загружали этилацетат (100 мл) и 12,6 г (0,21 моль) уксусной кислоты. Под поверхность жидкой смеси, которую охлаждали, поддерживая температуру ниже 35°C, вводили безводный метиламин (6,3 г, 0,20 моль). Затем порциями добавляли 6-хлор-8-метил-2H-3,1-бензоксазин-2,4-(1H)-дион (21 г, 0,10 моль) (т.е. продукт примера 1), поддерживая температуру реакционной смеси 35-40°C. После завершения добавления 6-хлор-8-метил-2H-3,1-бензоксазин-2,4-(1H)-диона температуру поддерживали при 40-45°C и следили за ходом реакции с помощью ВЭЖХ-анализа. Примерно через 20 минут, когда оставалось не более 0,5% 6-хлор-8-метил-2H-3,1-бензоксазин-2,4-(1H)-диона, добавляли воду (50 мл). Присоединяли насадку для отгонки растворителя, создавали умеренный вакуум и отгоняли этилацетат при внутренней температуре примерно 46-60°C и давлении примерно 30-50 кПа. Для восполнения объема, удаленного отгонкой этилацетата, добавляли воду до исходного объема жидкости в реакционном сосуде. Когда начинали отгоняться значительные объемы воды, водную суспензию охлаждали до 10°C. Твердое вещество отделяли фильтрованием и сушили при 60°C и 13,3 кПа, с получением указанного в заголовке соединения в виде белого кристаллического твердого вещества (19 г, выход приблизительно 95%, чистота >98%, определена по площади пиков на хроматограмме ВЭЖХ).
Пример 3
Вторая методика получения 2-амино-5-хлор-N,3-диметилбензамида
В 250-мл колбу, снабженную термометром и устройством для барботирования азота, загружали 6-хлор-8-метил-2H-3,1-бензоксазин-2,4-(1H)-дион (9,0 г, 43 ммоль) (т.е. продукт примера 1), этилацетат (50 мл) и уксусную кислоту 3,8 г (63 ммоль). Смесь нагревали до 50°C и под поверхность смеси вводили безводный метиламин (1,6 г, 50 ммоль), поддерживая температуру при 48-52°C. Смесь выдерживали в течение 1 часа при 50°C, добавляли воду (65 мл) и удаляли этилацетат отгонкой. Когда температура в колбе достигла 83°C, в раствор вводили затравку для кристаллизации и полученную суспензию охлаждали в течение 3 часов до 10°C. Твердое вещество отделяли фильтрованием и сушили, с получением указанного в заголовке соединения в виде белого твердого кристаллического вещества (7,94 г, чистота >98,5 мас.% по данным ВЭЖХ, выход 93%), температура плавления 143-145°C.
1H ЯМР (ДМСО-d6) δ 2,08 (с, 3Н), 2,72 (д, 3Н, J=4,5 Гц), 6,36 (с, 2Н), 6,36 (с, 2Н), 7,13 (д, 1Н, J=2,1 Гц), 7,40 (д, 1Н, J=2,1 Гц), 8,33 (кв, 1Н, J=4,5 Гц).
Пример 4
Третья методика получения 2-амино-5-хлор-N,3-диметилбензамида
В 250-мл колбу, снабженную термометром и устройством для барботирования азота, загружали 6-хлор-8-метил-2H-3,1-бензоксазин-2,4-(1H)-дион (21,0 г, 0,099 моль) (т.е. продукт примера 1), этилацетат (100 мл) и уксусную кислоту 12,6 г (0,21 моль). Смесь перемешивали при 22°C и под поверхность смеси порциями в течение 45 минут вводили безводный метиламин (4,3 г, 0,14 моль), поддерживая температуру при 22-41°C. Смесь выдерживали в течение 2 часов при 40°C, добавляли воду (150 мл) и удаляли этилацетат отгонкой. Когда температура в колбе достигла 83°C, в раствор вводили затравку для кристаллизации и суспензию продукта охлаждали в течение примерно 2 ч до 10°C. Твердое вещество отделяли фильтрованием и сушили, с получением указанного в заголовке соединения в виде белого твердого кристаллического вещества (18,38 г, чистота >97,4 мас.% по данным ВЭЖХ, выход 94%), температура плавления 143-145°C.
Пример 5
Получение 2-амино-5-хлор-N,3-диметилбензамида с использованием водного раствора метиламина
Модифицировали методику примера 3, используя вместо безводного метиламина водный раствор метиламина (40%, 10,75 г, 0,138 моль). После сбора твердого вещества и сушки получали 18,57 г неочищенного продукта, ВЭЖХ-исследование которого показало лишь 92,4 мас.% содержание указанного в заголовке соединения, что соответствует выходу 87,3%. По данным ВЭЖХ неочищенный продукт содержал также примерно 3,4 мас.% 6-хлор-3,8-диметил-2,4(1H,3H)-хиназолиндиона - побочного продукта, полученного в результате циклизации 5-хлор-3-метил-2-[[(метиламино)карбонил]амино]бензойной кислоты, а также примерно 1,7% продукта гидролиза 2-амино-5-хлор-3-метилбензойной кислоты. Данный пример демонстрирует, что вода оказывает неблагоприятное воздействие на выход и чистоту продукта.
В таблице 1 показаны конкретные химические превращения, приводящие к получению соединений формулы 1 согласно способу настоящего изобретения. При проведении указанных реакций карбоновая кислота чаще всего представляет собой уксусную кислоту. В таблице 1 и последующих таблицах t означает третичный, s означает вторичный, n означает нормальный, i означает изо, c означает цикло, Me означает метил, Et означает этил, Pr означает пропил и Bu означает бутил. Последовательности групп сокращаются аналогичным образом; например, «c-PrCH2» означает циклопропилметил.

В таблице 2 показаны конкретные химические превращения, которые ведут к получению соединений формулы 2, согласно способу настоящего изобретения.
Соединения формулы 4, перечисленные в таблице 3, могут быть получены способами и методиками, описанными в настоящем описании в сочетании со способами, известными в данной области. В частности, данные соединения являются полезными промежуточными соединениями, которые могут быть получены способом, приведенным на схеме 3, и они являются исходными веществами для получения соединений формулы 2 по способу схемы 2.

Соединения формулы 1, полученные способом по схеме 1 настоящего описания, полезны в качестве промежуточного соединения для получения соединений формулы 5.

где
X представляет собой Cl или Br;
Z представляет собой CR7 или N;
R1 представляет собой H, С1-С4 алкил, циклопропил, циклопропилметил или метилциклопропил;
R2 представляет собой CH3 или Cl;
R4 представляет собой Cl, Br, CF3, OCF2H или OCH2CF3;
R5 представляет собой F, Cl или Br;
R6 представляет собой H, F или Cl; и
R7 представляет собой H, F, Cl или Br.
Соединения формулы 5 полезны в качестве инсектицидов, что описано, например, в патентных публикациях PCT WO 2003/015518 и WO 2006/055922. Возможен ряд способов получения соединения формулы 5, исходя из соединения формулы 1. В одном из способов, показанном на схеме 4, соединение формулы 5 получают путем взаимодействия соединения формулы 1, карбоновой кислоты формулы 6 и сульфонилхлорида, следуя общей методике, изложенной в патентной публикации PCT WO 2006/062978, которая во всей полноте включена в настоящее описание посредством ссылки.
Схема 4
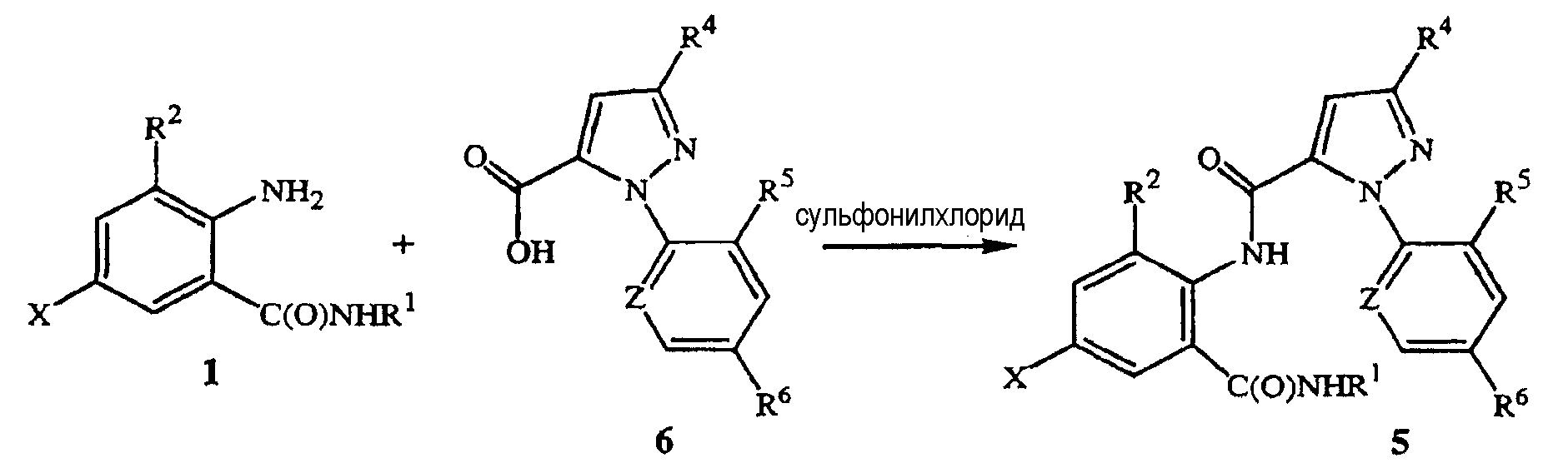
Как указано в WO 2006/062978, в данном способе возможны различные условия проведения реакции. Как правило, сульфонилхлорид добавляют к смеси соединений формул 1 и 6 в присутствии растворителя и основания. Сульфонилхлориды по существу соответствуют формуле RS(O)2Cl, где R является углеродсодержащим радикалом. Как правило, в данном способе R представляет собой C1-C4 алкил, C1-C2 галогеналкил или фенил, необязательно замещенный 1-3 заместителями, выбранными из группы, состоящей из галогена, C1-C3 алкила и нитро. Коммерчески доступные сульфонилхлориды включают метансульфонилхлорид (R представляет собой CH3), пропансульфонилхлорид (R представляет собой (CH2)2CH3), бензилсульфонилхлорид (R представляет собой фенил) и п-толуолсульфонилхорид (R представляет собой 4-метилфенил). Особенно подходящим является метансульфонилхлорид по причине более низкой стоимости, легкости добавления и/или меньших отходов. Для завершения превращения со стехиометрической точки зрения необходим по меньше мере один мольный эквивалент сульфонилхлорида на моль соединения формулы 6. Как правило, мольное соотношение сульфонилхлорида и соединения формулы 6 не превышает примерно 2,5, преимущественно не превышает примерно 1,4.
Соединение формулы 5 образуется в том случае, когда исходные соединения формул 1 и 6 и сульфонилхлорид подвергают взаимодействию в объединенной жидкой фазе, в которой каждый из компонентов растворим по меньше мере частично. В частности, поскольку исходные соединения формул 1 и 6, как правило, являются твердыми веществами при обычной комнатной температуре, данный способ наиболее удовлетворительно реализуется при использовании растворителя, в котором исходные вещества растворимы в значительной степени. Таким образом, описываемый способ чаще всего реализуют в жидкой фазе, включающей растворитель. В некоторых случаях карбоновая кислота формулы 6 может обладать лишь незначительной растворимостью, но ее соль, образованная добавлением основания, может обладать более высокой растворимостью в растворителе. Подходящие для данного способа растворители включают нитрилы, такие как ацетонитрил и пропионитрил; сложные эфиры, такие как метилацетат, этилацетат и бутилацетат; кетоны, такие как ацетон, метилэтилкетон (MEK) и метилбутилкетон; галогеналканы, такие как дихлорметан и трихлорметан; простые эфиры, такие как этиловый эфир, бутил-трет-метиловый эфир, тетрагидрофуран (ТГФ) и п-диоксан; ароматические углеводороды, такие как бензол, толуол, хлорбензол и дихлорбензол; третичные амины, такие как триалкиламины, диалкиламины и необязательно замещенные пиридины; а также смеси перечисленных соединений. Особенно подходящие растворители включают ацетонитрил, пропионитрил, этилацетат, ацетон, MEK, дихлорметан, бутил-трет-метиловый эфир, ТГФ, п-диоксан, толуол и хлорбензол. Особенно предпочтительным растворителем является ацетонитрил, т.к. он часто обеспечивает наиболее высокий выход и/или чистоту продукта.
Поскольку в реакции по описываемому способу в качестве побочного продукта образуется хлористый водород, способ лучше всего реализовывать в присутствии по меньше мере одного добавленного основания, поскольку хлористый водород иначе будет связываться с основными центрами соединений формул 1, 5 и 6. Основание может также содействовать необходимому для осуществления способа взаимодействию карбоновой кислоты с сульфонилхлоридом и антраниламидом. Взаимодействие добавленного основания с карбоновой кислотой формулы 6 приводит к образованию соли, которая может иметь большую растворимость в реакционной среде, чем сама карбоновая кислота. Хотя основание можно добавлять одновременно, попеременно или даже после добавления сульфонилхлорида, как правило, основание добавляют до сульфонилхлорида. Некоторые из растворителей, например третичные амины, также могут действовать в качестве оснований, и если их используют в качестве растворителей, они будут присутствовать в большом стехиометрическом избытке, в качестве основания. Если основание не используют в качестве растворителя, обычное мольное соотношение введенных в реакцию основания и сульфонилхлорида составляет, обычно, примерно от 2,0 до 2,2 и предпочтительно примерно от 2,1 до 2,2. Предпочтительными основаниями являются третичные амины, включающие замещенные пиридины. Более предпочтительно основания включают 2-пиколин, 3-пиколин, 2,6-лутидин и пиридин. Особенно предпочтительным в качестве основания является 3-пиколин, причем его соли с карбоновыми кислотами формулы 6 часто обладают высокой растворимостью в растворителях, таких как ацетонитрил.
Продукт N-фенилпиразол-1-карбоксамидных соединений формулы 5 может быть выделен из реакционной смеси методами, известными специалистам в данной области, включая кристаллизацию, фильтрование и экстракцию. В WO 2006/062978 раскрыты конкретные примеры, относящиеся к способу по схеме 4.
Соединения пиразолкарбоновой кислоты формулы 6 могут быть получены с применением способов синтеза гетероциклических соединений, известных в литературе, включая патентные публикации PCT WO 1998/57397, WO 2003/015519, WO 2006/055922 и WO 2006/062978, а также источники, входящие в следующий перечень: Rodd's Chemistry of Chemistry of Carbon Compounds, Vol.IVa-IVl, S. Coffey editor, Elsevier Scientific Publishing, New York, 1973; Comprehensive Heterocyclic Chemistry, vol.1-7, A.R. Katritzky and C.W. Rees editors, Pergamon Press, New York, 1984; Comprehensive Heterocyclic Chemistry, vol.1-9, A.R. Katritzky and C.W. Rees, and E.F. Scriven editors, Pergamon Press, New York, 1996; и серии, The Chemistry of Heterocyclic Compounds, E.C. Taylor, editor, Wiley, New York.
Способ, показанный на схеме 4, является иллюстрацией только одного из многих способов преобразования аминного соединения формулы 1 в соответствующий карбоксамид формулы 5. В данной области известно множество общих способов получения карбоксамидов из карбоновых кислот и аминов. Для ознакомления с общим обзором см. M.North, Contemporary Org.Synth. 1995, 2, 269-287. Конкретные способы включают взаимодействие соединения формулы 1 с соединением формулы 6 в присутствии дегидратирующих сшивающих агентов, таких как 1,3-дициклогексилкарбодиимид, 1,1'-карбонилимидазол, хлорангидрид бис(2-оксо-3-оксазолидинил)фосфиновой кислоты или гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония, или их сшитых с полимерами аналогов, например сшитого с полимером дициклогексилкарбодиимида, как правило, в инертном растворителе, таком как дихлорметан или N,N-диметилформамид, что по существу раскрыто в патентной публикации PCT WO 2003/15518. Кроме того, в приведенной ссылке раскрыты альтернативные способы получения ацилхлоридного производного соединения формулы 6, например, путем взаимодействия тионилхлорида или оксалилхлорида в присутствии каталитического количества N,N-диметилформамида, и затем взаимодействие полученного ацилхлорида с соединением формулы 1 в присутствии поглотителя кислоты, такого как аминное основание (например, триэтиламин, N,N-диизопропилэтиламин, пиридин и их аналоги на полимерных подложках) или гидроксид или карбонат (например, NaOH, KOH, Na2CO3, K2CO3), обычно в инертном растворителе, таком как тетрагидрофуран, 1,4-диоксан, этиловый эфир или дихлорметан. Продукт соединений формулы 5 может быть выделен из реакционной смеси методами, известными специалисту в данной области, включая кристаллизацию, фильтрование и экстракцию.
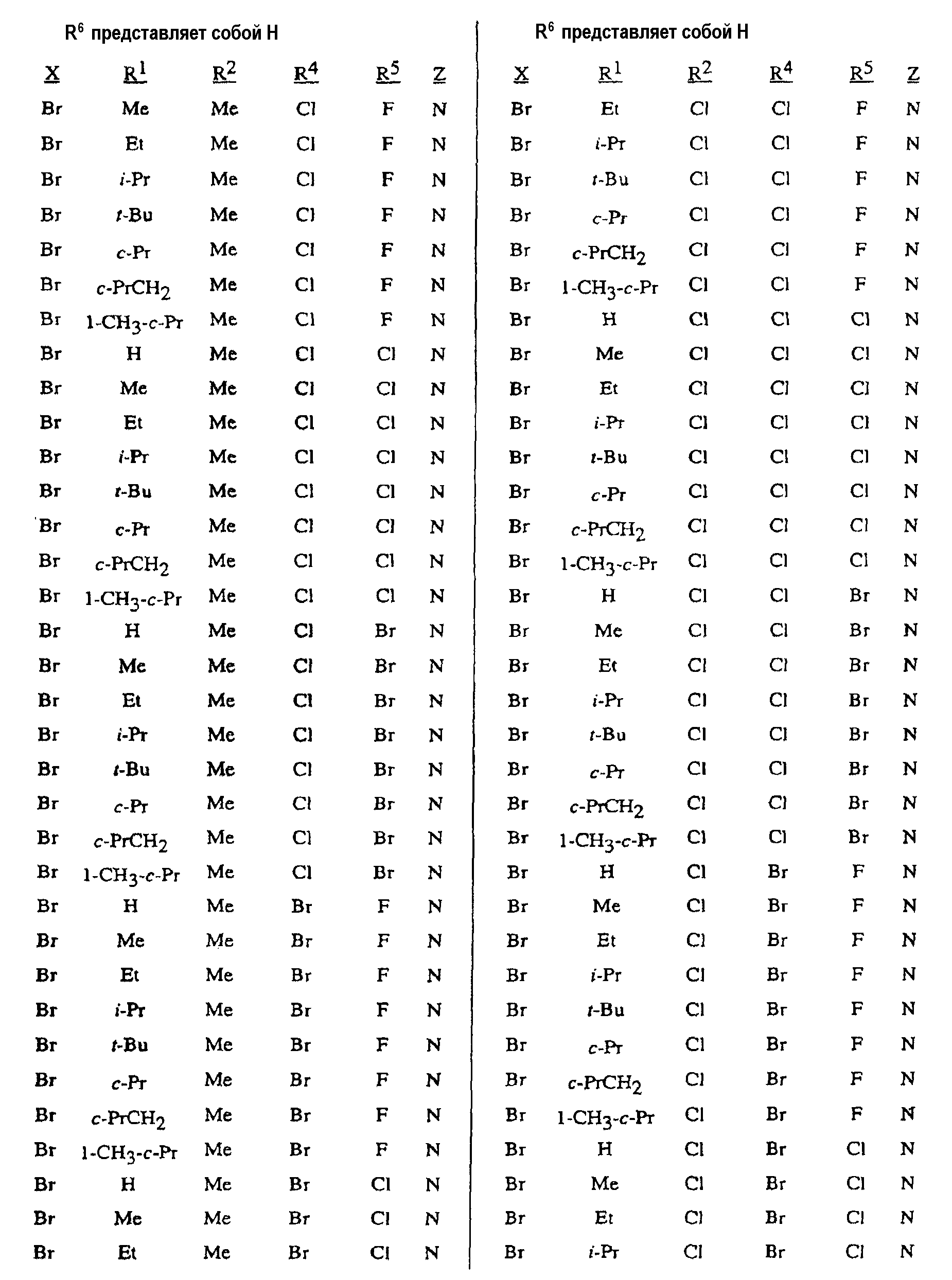
Таблица 4 иллюстрирует конкретные химические преобразования, позволяющие получить соединения формулы 5 из соединений формул 2 и 3, согласно способу настоящего изобретения. Преобразование соединения формулы 1 в соединение формулы 5 может быть выполнено, например, способом, приведенным на схеме 4, с использованием сульфонилхлорида, например метансульфонилхлорида в присутствии растворителя, например, ацетонитрила и основания, например, 3-пиколина.








Реферат
В изобретении раскрыт способ получения соединения формулы 1 путем взаимодействия соединения формулы 2, где R1 представляет собой Н, C1-C4 алкил, циклопропил, циклопропилметил или метилциклопропил, R2 представляет собой CH3 или Cl; и X представляет собой Cl или Br, включающий взаимодействие соединения формулы 2 с R1-NH2 в присутствии карбоновой кислоты в, по существу, безводной реакционной среде, включающей подходящий органический растворитель, а также способ получения соединения формулы 2, где R2 представляет собой СН3 или Cl; и X представляет собой Cl или Br; включающий взаимодействие соединения формулы 4, где R3 представляет собой C1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом, с трибромидом фосфора. Также заявляется соединение 4, где R2 представляет собой CH3 или Cl; R3 представляет собой C1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом; и X представляет собой Cl или Br; при условии когда R2 и X каждый представляет собой Cl, тогда R3 отличен от CH3. 3 н. и 9 з.п. ф-лы, 6 пр., 4 сх., 4 табл.
Формула

где R1 представляет собой Н, С1-С4 алкил, циклопропил, циклопропилметил или метилциклопропил;
R2 представляет собой CH3 или Cl; и X представляет собой Cl или Br;
включающий взаимодействие соединения формулы 2

с соединением формулы 3

в присутствии карбоновой кислоты в, по существу, безводной реакционной среде, включающей подходящий органический растворитель.

где R3 представляет собой C1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом;
с трибромидом фосфора.
где R2 представляет собой CH3 или Cl; и X представляет собой Cl или Br;
включающий
взаимодействие соединения формулы 4

где R3 представляет собой C1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом;
с трибромидом фосфора.

где R2 представляет собой CH3 или Cl;
R3 представляет собой C1-С6 алкил или С3-С6 алкенил, каждый из которых необязательно замещен не более чем 3 атомами галогена и не более чем 1 фенилом; и
X представляет собой Cl или Br;
при условии, когда R2 и X, каждый представляет собой Cl, тогда R3отличен от CH3.