Способ получения динитродиазаалканов и промежуточных продуктов - RU2274637C2
Код документа: RU2274637C2
Описание
Уже в течение нескольких лет известны порохи для боевых зарядов, которые в качестве энергетического пластификатора, называемого также нитроглицерином, содержат динитродиазаалканы, а именно, в частности, 2,4-динитро-2,4-диазапентан, один или в смеси с другими соответствующими алканами (патенты США 4476322 и 4457791).
В природе динитродиазаалканов заложено то, что полученные с их участием порохи для боевых зарядов обладают почти независимым от температуры свойством сгорать. Это очень важное свойство. Оно означает, что развившееся в системе при сгорании боевого заряда давление газа только в незначительной или очень незначительной степени зависит от температуры окружающей среды. Порохи для боевых зарядов с независимым от температуры свойством сгорать способствуют использованию максимального потенциала мощности системы в соответственно широкой области температур.
До настоящего времени широкому применению динитродиазаалканов для получения порохов для боевых зарядов с соответственно сбалансированной температурной характеристикой препятствовало то, что их трудно получать, и соответственно этому они дороги.
По одному известному способу получения (патент США 4476322, там же и другие ссылки) 2,4-динитро-2,4-диазапентан получают из диметил- или диэтилмочевины. Мочевину нитруют азотной кислотой и продукт нитрования гидролизуют в метил- или этилнитрамин. Полученные нитрамины с помощью параформальдегида и серной кислоты конденсируют в 2, 4-динитро-2,4-диазапентан. Аналогичным способом можно также получать 2,4-динитро-2,4-диазагексан и 3,5-динитро-3,5-диазагептан, а также смеси трех упомянутых алканов (Tartakofsky и другие, Russian Chemical Bulletin, 1993, 42, 1916 и далее). Способ получения из мочевины дает только сравнительно небольшой общий выход, а применяемая диэтилмочевина очень дорогая. Кроме того, нитрованное соединение мочевины представляет собой крайне нестабильный, чувствительный к температуре и кислоте, взрывоопасный промежуточный продукт.
Кроме того, известно получение вышеупомянутую смесь из трех динитродиазаалканов путем взаимодействия метил- или этиламина со сложным эфиром хлор-муравьиной кислоты с использованием раствора едкого натра с образованием промежуточного продукта (см. М. Curry J.P. Mason, J. Am. Chem. Soc. 1951, 73, стр.5043 и след.), который затем нитруют азотной кислотой (см. Leon Goodman, J. Am. Chem. Soc, 1953, 75, стр.3019 и след.). Продукт нитрования действием аммиака и этанола при кипении превращают в метил- или этилнитрамин, который затем конденсируют в динитродиазаалканы, как в вышеуказанном способе. Стадия получения нитрамина является очень дорогостоящей и длительной, так что в промышленном масштабе ее нельзя реализовать.
Задачей изобретения является создание не связанного с особыми затратами и не имеющего риска для безопасности осуществляемого в промышленном масштабе способа получения динитродиазаалканов.
Поставленная задача решается описанным в формуле изобретения способом.
В способе согласно изобретению исходят из сложного диалкилового эфира дикарбоновой кислоты, предпочтительно из сложного диэтилового эфира щавелевой кислоты. Его подвергают взаимодействию с первичным алифатическим амином, предпочтительно этиламином, с образованием соответствующего диалкиламида. При этом реакция происходит в водной среде. Температура реакции лежит между 0 и 80°С. Диалкиламиды выпадают в виде осадка и их можно отфильтровывать. Следующая реакционная схема воспроизводит первую стадию способа согласно изобретению:
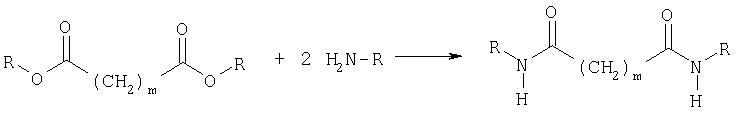
R=CnH2n+1c n=1, 2, ..., 10
m=0, 1, ..., 10
При этом данная схема показывает вариант, когда в случае первичного алифатического амина образуется диалкилдиамид дикарбоновой кислоты. Однако вместо диамидов с алифатическими остатками могут также образовываться диамиды дикарбоновых кислот с циклическими или ароматическими остатками, что регулируют путем выбора соответствующего циклического или ароматического амина.
На второй стадии способа согласно изобретению полученные диалкиламиды нитруют с помощью обычного нитрующего средства в соответствующие диалкилдинитроамиды. Следующая реакционная схема это показывает.

а)HNO3/H2 SO4
б) HNO3 /ангидрид уксусной кислоты
в) N2O5
R=CnH2n+1c n=1, 2, ..., 10
m=0, 1, ..., 10
Если на второй стадии исходят из диамидов с циклическими или ароматическими остатками, то получают динитроамидные соединения с соответствующими остатками. Нитрование производится с помощью обычных нитрующих средств, предпочтительно с помощью смеси азотной и серной кислот, азотной кислоты, уксусного ангидрида или пентаоксида азота, с растворителем или без него. Во время добавления нитрующего средства температура должна находиться в области от -20 до +20°C. В случае жидких динитросоединений образуются две фазы, твердые динитросоединения можно отфильтровывать.
На следующей, третьей стадии, диалкилдинитроамиды подвергают взаимодействию с метил- и/или этиламином в воде. При этом в качестве промежуточного продукта образуются диметил- и/или диэтилдиамиды, которые после нитрования можно снова использовать для получения метил- и этилнитрамина, как и после подкисления алкилнитраминов, алкильный остаток которых соответствует остатку динитроамида. Следующая реакционная схема изображает третью стадию:
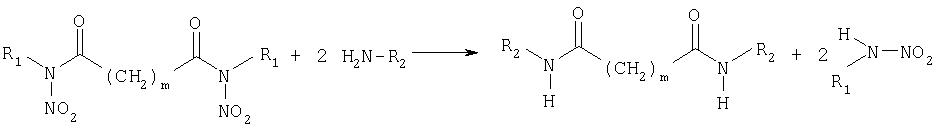
R1=CnH2n+1c n=1, 2, ..., 10
m=0, 1, ..., 10
R2=метил-, этил-
Выделение алкилнитраминов происходит предпочтительно экстракцией с помощью органического растворителя, такого, как, например, простой диэтиловый эфир, дихлорметан, простой метил-трет, бутиловый эфир, этилацетат или толуол, причем предпочтительным является простой эфир.
На четвертой стадии выделенные алкилнитрамины конденсируют в динитродиазаалканы само по себе известным способом. Пригодные для этого методы описаны в патенте США 4476322 и у Tartakofsky и др., как указано выше. Предпочтительный способ конденсации описан в пункте 10, причем применяют 50-98%-ную серную кислоту. Применяемый при этом растворитель может быть таким же, какой уже использовали на третьей стадии, и без удаления в конце тот же самый растворитель используют далее. Следующая схема воспроизводит четвертую стадию:
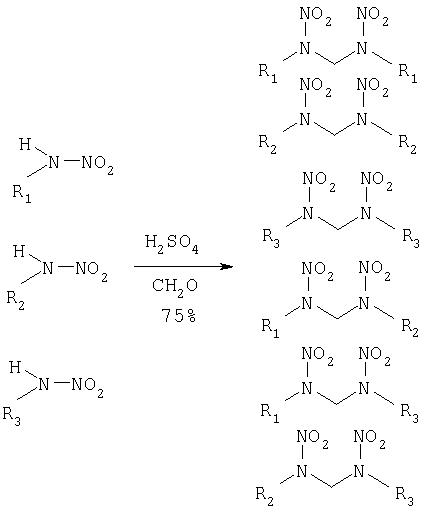
R1, R2, R3=CnH2n+1 с n=1, 2, ..., 10
Изобретение, в частности, направлено на получение уже рассмотренной в начале смеси их трех соединений: 2,4-динитро-2,4-диазапентана, 2,4-динитро-2,4-диазагексана и 3,5-динитро-3, 5-диазагептана, названной здесь ДНДА 57, потому что эта смесь представляется особенно пригодной для получения порохов для боевых зарядов со сбалансированными температурными свойствами.
Желаемый состав смеси можно регулировать при конденсации путем относительных количеств различных применяемых нитраминов.
В этой связи особенно предпочтительным является дальнейшее развитие изобретения согласно пункту 12. При этом метилнитрамин и этилнитрамин получают совместно в одном и том же процессе и к тому же их соотношение можно устанавливать с самого начала соответственно желаемому составу ДНДА 57, так что оба нитрамина можно сразу, без дополнительной предварительной обработки конденсировать на четвертой стадии в ДНДА 57.
Способ согласно изобретению по сравнению с изложенными в начале способами синтеза имеет различные преимущества: расходы на исходные материалы незначительны и исходные материалы имеются в распоряжении в большом количестве. Выходы относительно высокие. Получают выделяемые промежуточные продукты. Способ сравнительно просто выполним в промышленном масштабе. И, наконец, его можно осуществлять без ущерба для окружающей среды, потому что большая часть реакций происходит в водной среде и все отходы хорошо биологически расщепляются.
Кроме того, объектом изобретения в качестве новых веществ являются диалкилдинитроамиды высшей дикарбоновой кислоты в соответствии с пунктом 14, кроме того, динитроамиды дикарбоновой кислоты, у которой алкильный остаток согласно пункту 14 замещен на циклический или ароматический остаток. Такие вещества получают в качестве промежуточного продукта при осуществлении способа согласно изобретению в конце второй стадии, т.е. путем нитрования, причем остаток предопределяют путем соответствующего выбора применяемого на первой стадии амина. Предпочтительное применение этих новых веществ, особенно диалкилдинитроамидов согласно пункту 14, заключается в их использовании в качестве промежуточного продукта для получения алкилнитраминов или динитродиазаалканов, например, с помощью стадии 3 или стадии 3 и 4 способа согласно изобретению. Диметилдинитроамид щавелевой кислоты уже известен из Chemical Abstract, Zitat 46: 904 G, однако без указания на вышеупомянутую возможность применения.
В дальнейшем изобретение более подробно поясняется с помощью примеров для синтеза метил- и этилнитрамина и для получения образующейся из них энергетической смеси пластификаторов ДНДА 57.
1) Синтез N,N'-диметилдиамида щавелевой кислоты из диэтилового эфира щавелевой кислоты и метиламина
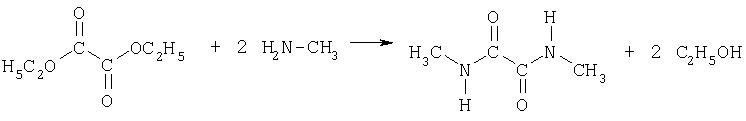
К 292 г (2.0 моль, d=1.08, 270 мл) диэтилового эфира щавелевой кислоты прибавляют по каплям при перемешивании 389 г (5.0 моль, d=0.90, 432 мл) 40%-ного раствора метиламина. При этом температура не должна превышать 80° С. Через час после окончания реакции бесцветное твердое вещество отфильтровывают, промывают небольшим количеством воды и сушат. Выход: 125 г (1.1 моль, 54%).
2) Синтез N, N'-диэтилдиамида щавелевой кислоты из диэтилового эфира щавелевой кислоты и этиламина
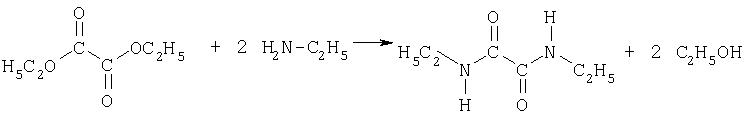
К 292 г (2.0 моль, d=1.08) диэтилового эфира щавелевой кислоты прибавляют по каплям при перемешивании 387 г (5.0 моль, d=0.81) 70%-ного раствора этиламина. После окончания прибавления перемешивают еще один час при комнатной температуре. Бесцветное твердое вещество отфильтровывают, промывают небольшим количеством воды и сушат. Выход: 154 г (1.1 моль, 53%).
3) Синтез бис-[метилнитроамида] щавелевой кислоты
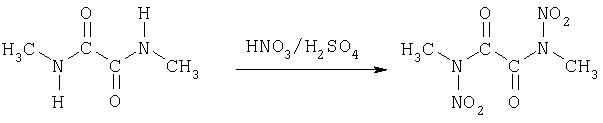
Растворяют 10.0 г (0.09 моль) бис-[метиламида] щавелевой кислоты в 25 мл 96%-ной азотной кислоты и, не допуская сильного нагревания (25-45°С), смешивают с 50 мл серной кислоты. Образующуюся густую массу выливают на лед, фильтруют, промывают водой до нейтральной реакции и сушат. Выход: 14.8 г (0.07 моль, 79%), т.пл. 124°С из этанола.
4) Синтез бис-[этилнитроамида] щавелевой кислоты
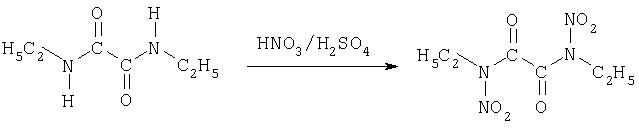
Растворяют 26.0 г (0.18 моль) бис-[этиламида] щавелевой кислоты в 50 мл 96%-ной азотной кислоты и, не допуская сильного нагревания (25-45°С), смешивают с 100 мл серной кислоты. Образуются две фазы. Органическую фазу отделяют, промывают водой и насыщенным раствором карбоната натрия. Выход: 30,6 г (0.13 моль, 89%).
5) Синтез метилнитрамина
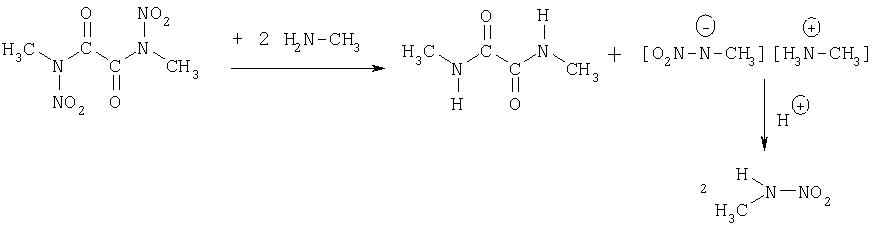
10.0 г (0.05 моль) Бис-[метилнитроамида] щавелевой кислоты смешивают маленькими порциями с 17.5 мл (0.23 моля) 40%-ного раствора метиламина. Смесь разогревается и постепенно изменяется. Приблизительно через час отфильтровывают от вновь образовавшегося бис-[метиламида] щавелевой кислоты и промывают небольшим количеством воды. Водную фазу подкисляют серной кислотой, при этом образуется метилнитрамин и метиламинсульфат. Экстрагируют 3 раза по 50 мл диэтилового эфира. После сушки над сульфатом магния эфир удаляют. Выход: 6.2 г (0.08 моль, 82%).
6) Синтез этилнитрамина
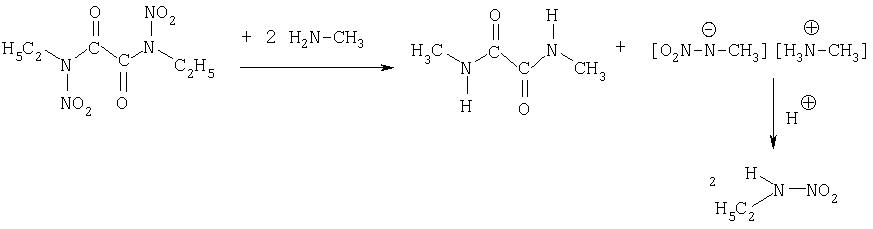
11.7 г (0.05 моль) Бис-[этилнитроамида]щавелевой кислоты смешивают маленькими порциями с 17.5 мл (0.23 моль) 40%-ного раствора метиламина. Смесь разогревается и постепенно изменяется. Приблизительно через час отфильтровывают от образующегося бис-[метиламида] щавелевой кислоты и промывают небольшим количеством воды. Водную фазу подкисляют серной кислотой, при этом образуется этилнитрамин и метиламинсульфат. Экстрагируют 3 раза по 50 мл диэтилового эфира. После сушки над сульфатом магния эфир удаляют. Выход: 8.9 г (0.10 моль, 99%).
7) Превращение смеси из метил- и этилнитрамина в ДНДА 57
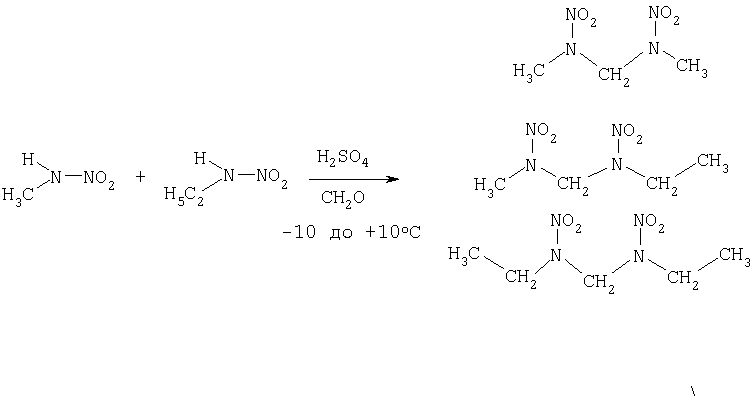
Для синтеза ДНДА 57 помещают 2.3 г параформальдегида в 40 мл 75%-ной серной кислоты и охлаждают до 0°С. Смесь 7.2 г (95 ммоль) метилнитрамина и 4.5 г (50 ммоль) этилнитрамина прибавляют по каплям с такой скоростью, чтобы температура реакционного раствора не поднималась выше 5°С. Через час после окончания реакции выливают в ледяную воду и водную фазу экстрагируют в совокупности около 50 мл дихлорметана. Объединенные органические фазы промывают насыщенным раствором карбоната натрия и сушат над сульфатом магния. После удаления растворителя получают ДНДА 57 с выходом 10.3 г (83%). При этом соотношение трех компонентов представляет собой следующее:
Общая схема синтеза ДНДА
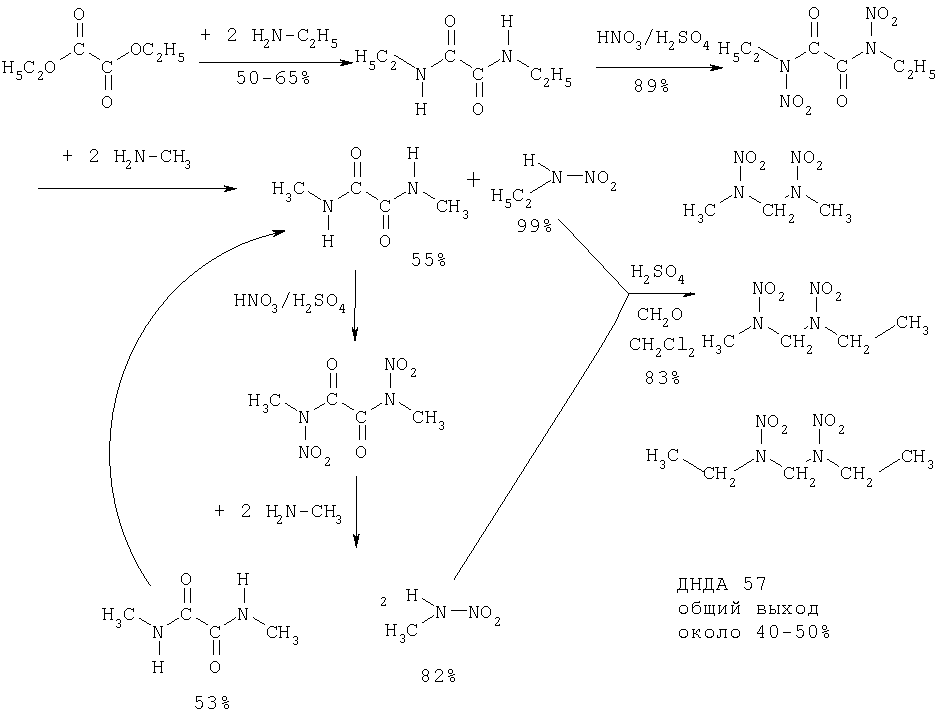
Вышеуказанная общая схема синтеза ДНДА показывает, как отдельные описанные ранее стадии объединяются в единый способ получения ДНДА 57. Сначала исходя из сложного диэтилового эфира щавелевой кислоты и этиламина синтезируют диэтилдиамид щавелевой кислоты и из него нитрованием с помощью азотной и серной кислот получают диэтилдинитроамид щавелевой кислоты. Последний подвергают взаимодействию с метиламином с образованием диметилдиамида щавелевой кислоты в качестве побочного продукта и этилнитрамина. Побочный продукт нитруют, как раньше, в результате чего получают диметилдинитроамид щавелевой кислоты. Реакцией этого побочного продукта с метиламином снова получают - в качестве побочного продукта - диметилдиамид щавелевой кислоты, который снова подвергают нитрованию, а также метилнитрамин. Оба полученные нитрамины совместно превращают в целевую смесь ДНДА 57 трех динитродиазаалканов конденсацией с помощью серной кислоты и параформальдегида. В общей схеме приведены выходы, достигнутые на отдельных стадиях. Общий выход составляет около 40-50%.
Вариантом к представленным выше синтезам 3)-6) является одновременный синтез метил- и этилнитрамина из обоих диалкилдиамидов щавелевой кислоты
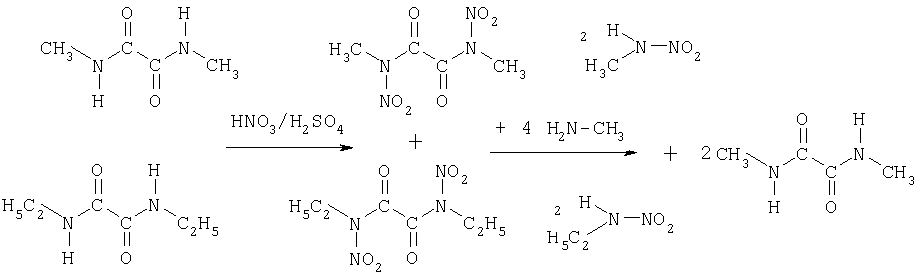
Растворяют 15.2 г (0.13 моль) бис-[метиламида]щавелевой кислоты и 6.5 г (0.04 моль) бис-[этиламида] щавелевой кислоты в 50 мл 9б%-ной азотной кислоты и, не допуская сильного разогревания (25-45°С), смешивают с 100 мл серной кислоты. Образующуюся густую массу выливают на лед, фильтруют и промывают водой до нейтральной реакции. Образовавшиеся диамиды щавелевой кислоты смешивают маленькими порциями, в общей сложности, приблизительно с 60 мл (0.79 моль) 40%-ного раствора метиламина. Смесь разогревается и постепенно изменяется. Приблизительно через час отфильтровывают от вновь образующегося бис-[метиламида]щавелевой кислоты и промывают небольшим количеством воды. Водную фазу подкисляют серной кислотой, при этом образуются метилнитрамин, этилнитрамин и метиламинсульфат. Экстрагируют 3 раза по 50 мл диэтилового эфира. После сушки над сульфатом магния эфир удаляют. Получают оба нитрамина в желаемом соотношении с выходом 61%, и их можно сразу использовать на следующих стадиях синтеза без дальнейшей обработки.
Реферат
Изобретение относится к способу получения динитродиазаалканов, которые могут быть использованы для получения порохов для боевых зарядов. Описывается способ получения динитродиазаалканов из алкиламинов и сложных эфиров, включающий комбинацию следующих приемов: 1. сложный диэфир дикарбоновой кислоты подвергают взаимодействию с алкиламином в водной среде с образованием соответствующего диалкилдиамида дикарбоновой кислоты, 2. полученный диалкилдиамид нитруют с помощью обычного нитрующего средства в соответствующий диалкилдинитроамид дикарбоновой кислоты, 3. полученный диалкилдинитроамид превращают в соответствующий алкилнитрамин тем, что диалкилдинитроамид смешивают в водной среде с метил- и/или этиламином, образующийся при этом диметил- и/или диэтилдиамид дикарбоновой кислоты отделяют, остающийся продукт подкисляют и экстрагируют алкилнитрамин, 4. выделенный алкилнитрамин конденсируют в динитродиазаалканы само по себе известным способом. Технический результат - создание промышленного способа получения динитродиазаалканов, который осуществляется легко, без особых затрат и большого риска. 2 н. и 11 з.п. ф-лы.
Комментарии