Производные аминоуксусной кислоты в виде рацемата или d- и l-энантиомеров или их фармацевтически приемлемые соли, способ получения d- и l-энантиомеров производных аминоуксусной кислоты или их фармацевтически приемлемых солей и d- и l-энантиомеры аминоуксу - RU2111206C1
Код документа: RU2111206C1
Чертежи
Описание
Изобретение относится к новым производным аминоуксусной кислоты, которые можно применять для получения биологически активных пептидов, более конкретно к производным аминоуксусной кислоты общей
формулы
где
Z - остатки -O-, -S(O)n-, где n - означает число O - 2,
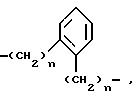
где
n имеет вышеуказанное значение,
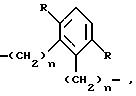
где
R и n имеют вышеуказанное значение,
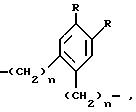
где
R и n имеют вышеуказанное значение,
-(CH2)n-C ≡ C-(CH2)n , ,
где
n имеет вышеуказанное значение,
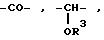
где
R3 имеет вышеуказанное значение, и
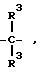
где
R3 имеет вышеуказанное значение,
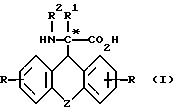
R - водород, метил, трифторметил, метокси, гидроксил, хлор, бром, фтор, йод, 2,4-дибром, 2,4-дихлор и 2,4-дифтор,
R1 - водород, алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, незамещенный или замещенный арил, арилалкил, незамещенный или замещенный гетероарил и флуоренилметил.
R2 - водород, бензилоксикарбонил, трет.-бутилоксикарбонил, флуоренилоксикарбонил, 1-адамантилоксикарбонил, 2-адамантилоксикарбонил и группа формулы
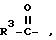
в виде рацемата или энантиомеров D и L и их фармацевтически приемлемых солей, при этом рацемическое соединение формулы (I), где Z означает -O-, R и R1, R2, - водород, исключено из притязаний.
Дополнительным объектом изобретения является способ
получения D- и L-энантиомеров производных аминоуксусной кислоты вышеприведенной общей формулы (I), где R2 означает водород, а R, R1 и Z имеют вышеуказанное значение, или их
фармацевтически приемлемой соли, который заключается в том, что
a) рацемическое соединение общей формулы
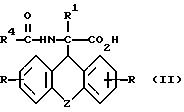
где
R, R1 и Z имеют вышеуказанное значение, а R4 означает низкий алкил, группу CX3, где X означает водород, галоид или арил,
подвергают взаимодействию с (-)цинхонидином в среде растворителя,
б) получаемое на стадии а) рацемическое соединение общей формулы
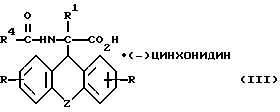
где
R, R1, R4 и Z имеют вышеуказанные значения,
разделяют фракционной кристаллизацией на D-энантиомер формулы
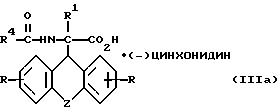
где
R, R1, R4 и Z имеют вышеуказанные значения, и L-энантиомер формулы
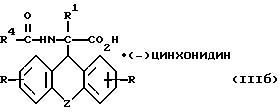
где
R, R1, R4 и Z имеют вышеуказанные значения,
в) получаемый на стадии (б) D- или L-энантиомер подвергают взаимодействию с кислотой в среде растворителя,
г) получаемый на стадии (в) D-энантиомер формулы (IIа) или L-энантиомер формулы (IIб)
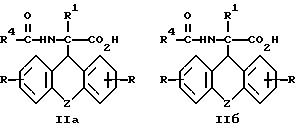
где
R, R1, R4 и Z имеют вышеуказанные значения,
подвергают взаимодействию с кислотой с последующим выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли.
Дальнейшим объектом изобретения являются D- и L-энантиомеры вышеприведенных формул (IIIа) и (IIIб), представляющие собой промежуточные продукты для синтеза производных аминоуксусной кислоты вышеприведенной общей формулы (I).
Под вышеприведенным термином "алкил" понимается разветвленный или неразветвленный углеводородный остаток с 1 - 12 атомами углерода. В качестве примеров можно назвать метил, этил н-пропил, изопропил, н-бутил, втор.-бутил, изобутил, трет. -бутил, н-пентил, н-гексил, н-гентил, н-октил, н-нонил, н-децил, ундецил, додецил.
Под термином "алкенил" понимается неразветвленный или разветвленный ненасыщенный углеводородный остаток с 2 - 12 атомами углерода. А качестве примеров можно назвать, например, этил, 2-пропенил, 1-бутенил, 2-бутенил, 1-пентенил, 2-пентенил, 3-метил-3-бутенил, 1-гексенил, 2-гексенил, 3-гексенил, 3-гептенил, 1-октенил, 1-ноненил, 1-деценил, 1-ундеценил, 1-додеценил.
Под термином "алкинил" понимается неразветвленный или разветвленный, имеющий тройную связь ненасыщенный углеводородный остаток с 2 - 12 атомами углерода. В качестве примеров можно назвать этинил, 2-пропинил, 1-бутинил, 2-бутинил, 3-бутинил, 1-пентинил, 3-пентинил, 1-гексинил, 2-гексинил, 3-гексинил, 3-гептинил, 1-октинил, 2-октинил, 1-нонинил, 2-нонинил, 3-нонинил, 4-нонинил, 1-децинил, 2-децинил, 2-ундецинил, 3-ундецинил, 3-додецинил.
Под термином "циклоалкил" понимается насыщенное углеводородное кольцо с 3 - 12 атомами углерода, такое как, например, циклопропил, циклобутил, циклопентил, циклогексил, адамантил.
Под термином "циклоалкилалкил" понимается насыщенное углеводородное кольцо, связанное с алкильной группой с 1 - 12 атомами углерода. При этом насыщенное углеводородное кольцо содержит 3 - 12 атомов углерода. В качестве примера можно назвать циклопропилметил, циклопентилметил, циклогексилметил, адамантилметил.
Под терминами "алкокси" и "тиоалкокси" понимается O-алкил или S-алкил, каждый с 1 - 12 атомами углерода.
Под термином "арил" понимается ароматический остаток, представляющий собой фенил, бензил, нафтил, бифенил, пиренил, антраценил, флуоренил и т.п. группы, незамещенные или замещенные 1 - 4 группами из числа алкила с 1 - 12 атомами углерода, алкоксила с 1 - 12 атомами углерода, тиоалкоксила с 1 - 12 атомами углерода, гидроксила, тиола, нитро, галоида, амино, группы
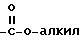
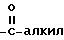
Под термином "арилалкил" понимается ароматический остаток, связанный с алкильной группой. При этом арил и алкил имеют вышеуказанное значение. В качестве примеров арилалкила можно назвать бензил, флуоренилметил.
Под термином "гетероарил" понимаются гетероароматический остаток, который представляет собой 2- или 3-тиенил, 2- или 3-фуранил, 2- или 3-пирролил, 2-, 4- или 5-имидазолил, 3-, 4- или 5-пиразолил, 2-, 4- или 5-тиазолил, 3-, 4- или 5-изотиазолил, 2-, 4- или 5-оксазолил, 3-, 4- или 5-изоксазолил, 3- или 5-(1,2,4-триазолил), 4- или 5-(1,2,3-триазолил), тетразолил, 2-, 3- или 4-пиридинил, 3-, 4- или 5-пиридазинил, 2-пиразинил, 2-, 4- или 5-пиримидинил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолинил, 2-, 3-, 4-, 5-, 6- или 7-индолил, 2-, 3-, 4-, 5-, 6- или 7-бензо[b] тиенил, 2-, 4-, 5-, 6- или 7-бензооксазолил, 2-, 4-, 5-, 6- или 7-бензимидазолил, 2-, 4-, 5-, 6- или 7-бензотиазолил, незамещенные или замещенные 1 или 2 остатками из группы, включающей алкил, арил, алкокси, тиоалкокси, каждый с вышеуказанным определением, гидроксил, тиол, нитро, галоид, формил, амино, группу
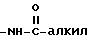
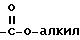
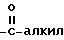
Под "галоидом" понимаются фтор, хлор, бром или йод.
В данном описании изобретения
используются следующие условные сокращения:
Asp - аспарагиновая кислота,
Bhg - 10,11-дигидро-5H-дибензо[a,d]-(циклогептен-5-ил)глицин или α -амино-10,11-дигидро-5H-дибензо[a,
d]циклогептен-5-уксусная кислота,
Ile - изолейцин,
Leu - лейцин,
Trp - триптофан,
Ac - ацетил,
Bzl - бензил,
Boc - трет-бутилоксикарбонил,
Eor - формил,
Z - бензилоксикарбонил.
Примечание: Если конфигурация аминокислоты отлична от L(S), то перед аминокислотой или условным сокращением указана соответствующая конфигурация, то есть D(R) или D(RS).
Энантиомеры D и L производных аминоуксусной кислоты вышеприведенной общей формулы (I) способны к образованию фармацевтически приемлемых кислотно-аддитивных солей и/или солей с основанием. Соединения вышеприведенных формул (IIа) и (IIб) могут образовать фармацевтически приемлемые соли с основанием. Все эти формы входят в объем данного изобретения.
Фармацевтически приемлемые кислотно-аддитивные соли энантиомеров D или L производных общей формулы (I) включают соли с нетоксичными органическими кислотами, такими как, например, хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, а также соли с нетоксичными органическими кислотами, такими как, например, алифатические моно- или дикарбоновые кислоты, замещенные фенилом алканкарбоновой кислоты, гидрокси-алканкарбоновые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты. Таким образом, солями являются сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрогенфосфат, дигидрогенфосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себецат, фумарат, малеат, соль миндальной кислоты, бензоат, хлорбензоат, метилбензоат, нитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат. Данным изобретением также охватываются соли с аминокислотами, такие как, например, аргинат, глюконат, галактуронат.
Кислотно-аддитивные соли основных соединений вышеприведенной общей формулы (I) получают путем контактирования свободного основания с достаточным количеством желаемой кислоты. Свободное основание можно снова получать путем контактирования соли с основанием и последующего выделения известными приемами. Основание несколько отличается от соответствующей соли по определенным физическим свойствам, таким как, например, растворимость в полярных растворителях. Но обе формы равноценно годятся для цели данного изобретения.
Фармацевтически приемлемые соли с основанием представляют собой соли с металлами или аминами, такими как, например, щелочные металлы или щелочноземельные металлы и органические амины. В качестве пригодных катионов металлов можно назвать натрий, калий, магний, кальций. В качестве подходящих аминов можно назвать N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метилглюкамин и прокаин.
Соли кислотных соединений вышеприведенной общей формулы (I) с основанием получаются путем контактирования свободной кислоты с достаточным количеством желаемого основания. Свободную кислоту можно снова получать путем контактирования соли с кислотой и последующего выделения свободной кислоты известными приемами. Свободная кислота несколько отличается от соответствующей соли по определенным физическим свойствам, таким как, например, растворимость в полярных растворителях. Но обе формы равноценно годятся для целей данного изобретения.
Некоторые соединения вышеприведенной общей формулы (I) могут иметься как в несольватированной форме, так и в сольватированной форме, включая гидратированные формы. В общем сольватированные формы включая гидратированные формы, эквивалентны несольватированным формам. Упомянутые формы также охватываются настоящим изобретением.
Как уже указывалось выше, энантиомеры формулы (I) пригодны в качестве гидрофобных производных естественных аминокислот для получения биологически активных пептидов, которые имеют повышенную активность и\или метаболическую стабильность.
Предпочтительными
являются соединения формулы (I), где
Z означает кислород, серу, группы
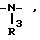
R - водород,
R1 - водород,
R2 - водород, бензилоксикарбонил, трет.-бутилоксикарбонил, флуоренилоксикарбонил, 1-адамантилоксикарбонил и 2-адамантилоксикарбонил.
Наиболее предпочтительными являются соединения формулы (I), где Z означает кислород, серу или группы
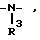
В частности, предпочитаются следующие
соединения:
D- α -амино-9H-ксантен-9-уксусная
кислота,
L- α -амино-9H-ксантен-9-уксусная кислота,
D- α -амино-9H-тиоксантен-9-уксусная кислота,
L- α -амино-9H-тиоксантен-9-уксусная кислота,
DL- α -амино-9H-тиоксантен-9-уксусная кислота,
D- α -амино-10,11-дигидро-5H-дибензо[a, d]-циклогептен-5-уксусная
кислота,
L- α -амино-10,11-дигидро-5H-дибензо[a,
d]-циклогептен-5-уксусная кислота,
D, L- α -амино-10,11-дигидро-5H-дибензо[a,d]-циклогептен-5-уксусная кислота,
D- α -амино-5H-дибензо[a,d]-циклогептен-5-уксусная
кислота,
L- α -амино-5H-дибензо[a,d]-циклогептен-5-уксусная кислота и
D,L- α -амино-5H-дибензо[a,
d]-циклогептен-5-уксусная кислота.
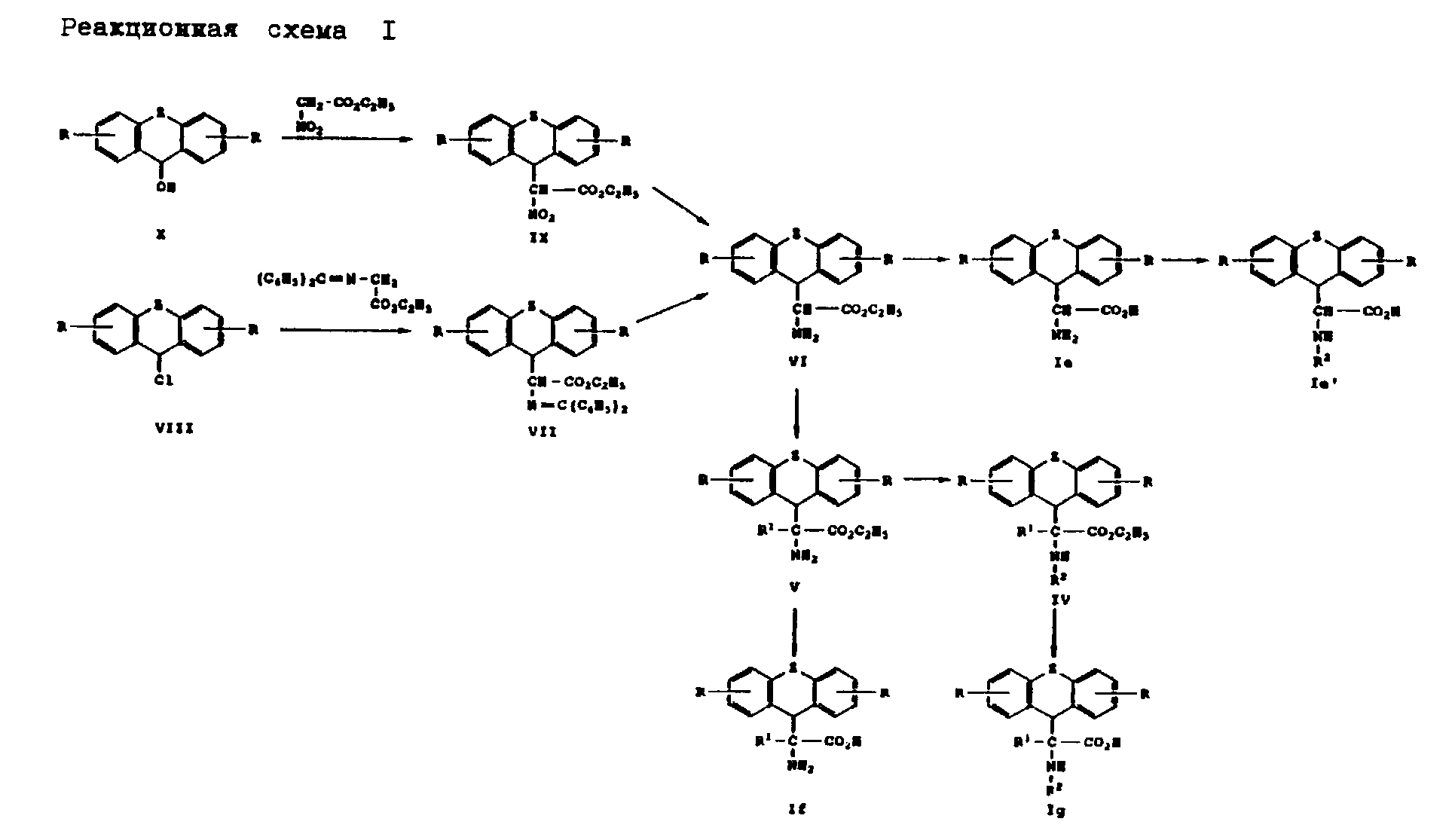
Получение производных аминоуксусной кислоты вышеприведенной общей формулы (I) представлено на реакционной схеме I, приведенной в конце описания.
Согласно реакционной схеме I спирт формулы (X), где R и Z имеют вышеуказанные значения, и сложный этиловый эфир нитроуксусной кислоты нагревают до температуры примерно 100-110o C в соответствии с методом, описанным В.И. Рябой и О. Ф. Гинзбург, Журнал органической химии 1, стр. 2069-2071, 1965. Получаемое при этом соединение формулы (IX), где Z и R имеют вышеуказанное значение, подвергают восстановлению водородом в присутствии катализатора, такого как, например, палладий на угле, в среде растворителя, такого как, например, этанол, и в присутствии кислоты, такой как, например, хлористоводородная кислота. При этом получают соединение формулы (VI). В соединении формулы (IX), где Z означает ненасыщенный остаток, нитрогруппу избирательно восстанавливают гидридом металла, таким как, например, боран натрия, в среде растворителя, такого как, например, метанол, в присутствии хлористого кобальта или т.п. вещества. При этом получают соединение формулы (VI), где Z означает ненасыщенный остаток, а R имеет вышеуказанное значение.
Соединение формулы (VI), где Z и R имеют вышеуказанные значения, можно также получать путем взаимодействия соединения формулы (VIII), где Z и R имеют вышеуказанные значения, с бензофенонимином сложного этилового эфира глицина. Получаемое при этом соединение формулы (VII), где Z и R имеют вышеуказанные значения, превращают до соединения формулы (VI) в соответствии с методом, описанным М. И. О'Донелл и Т.М. Экрих, Tetrahedron Letters, стр. 4625-4628, 1978; М. И. О'Донелл и Р.Л. Польт, Journal of Organic Chemistry 47, стр. 2663 - 2666 (1982), и М.И. О'Донелл и др. Journal of the American Chemical Society III, стр. 2353 - 2355 (1989).
Соединение формулы (V), где Z, R и R1 имеют вышеуказанные значения, получают путем алкилирования соединения формулы (VI) известными приемами. Соединение формулы (IV), где Z, R, R1 и R2 имеют вышеуказанные значения, получают путем введения в соединение формулы (V) защитных групп для азота известными приемами. Соединения формул (Ie), (If) и (Ig), где Z, R, R1 и R2 имеют вышеуказанные значения, получают путем гидролиза соответствующего соединения формулы (VI), (V) и (IV). Соединение формулы (Ie'), где Z, R и R2 имеют вышеуказанное значение, получают в соответствии с методом, применяемым для получения соединения формулы (IV) из соединения формулы (V).
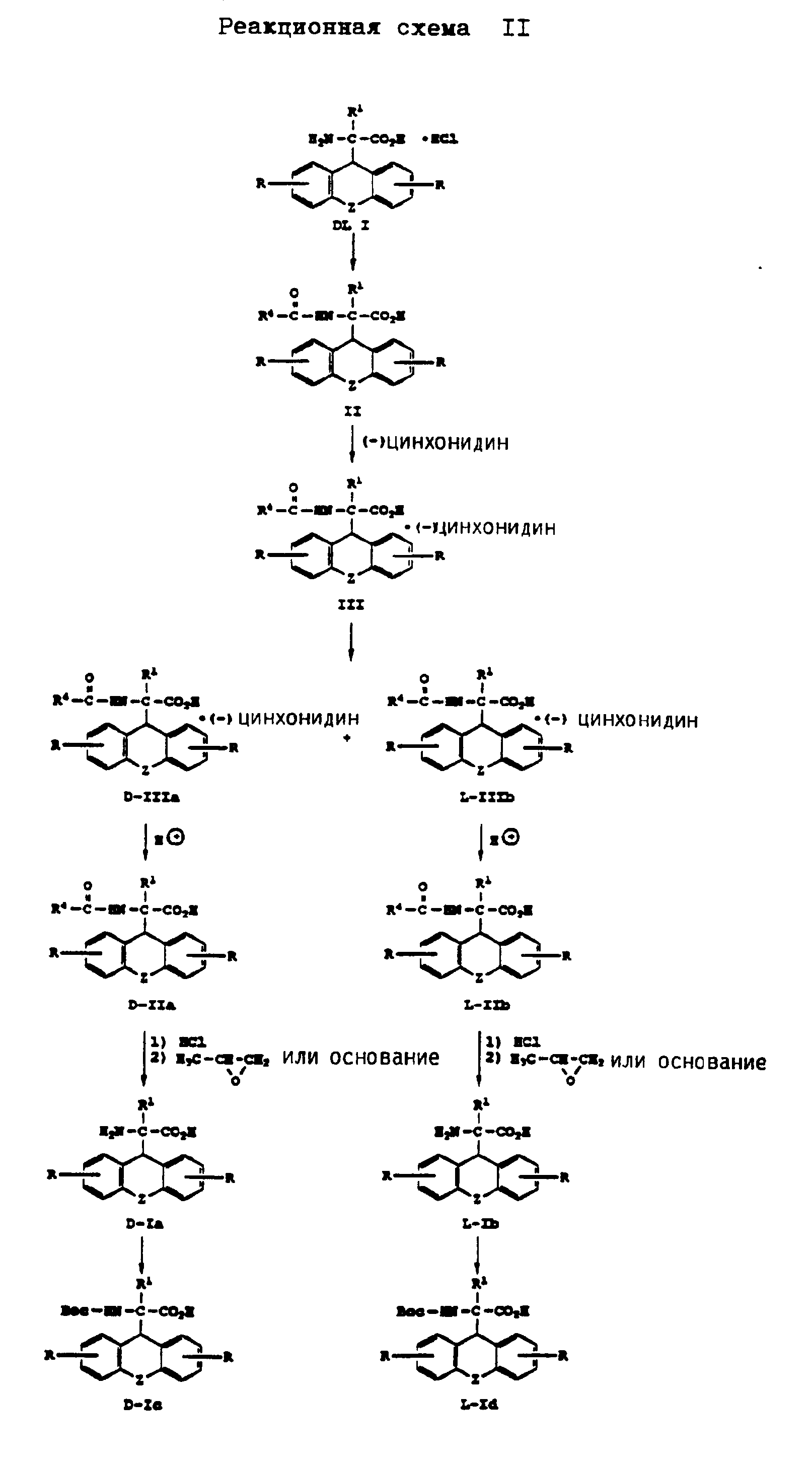
Получение энантиомеров D и L производных аминоуксусной кислоты формулы (I) представлено на реакционной схеме II, приведенной в конце описания.
Согласно реакционной схеме II соединение формулы DL I в виде гидрохлорида, которое представляет собой рацемическую смесь изомеров, подвергают ацетилированию соединением формулы
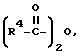
Кислотно-аддитивную соль соединения формулы D-Ia или формулы L-Id подвергают обработке основанием, таким как, например, гидроокись аммония, с получением соединения формулы D-Ia или формулы L-Ib в качестве свободной аминокислоты. Кислотно-аддитивную соль соединения формулы D-Ia или формулы L-Ib можно также получать путем обработки пропиленоксидом в соответствии с методом, описанным У. Шоллкопф и др., Synthesis, стр. 966 - 969, 1981. При этом также получают соединения формулы D-Ia или формулы L-Ib в качестве свободной аминокислоты.
N- α -трет. -бутилоксикарбонильные производные соединения формулы D-Ia или L-Ib, имеющие формулу D-Ic или L-Id, получают обработкой соединения формулы D-Ia или L-Ib в соответствии с методом, описанным в патенте США N 4766109. Но эти производные можно также получать другими известными методами. Другие N-защищенные производные соединения формулы (I), где R2 имеет вышеуказанное значение, за исключением водорода, можно получать путем обработки соединения формулы D-Ia или L-Ib известными приемами.
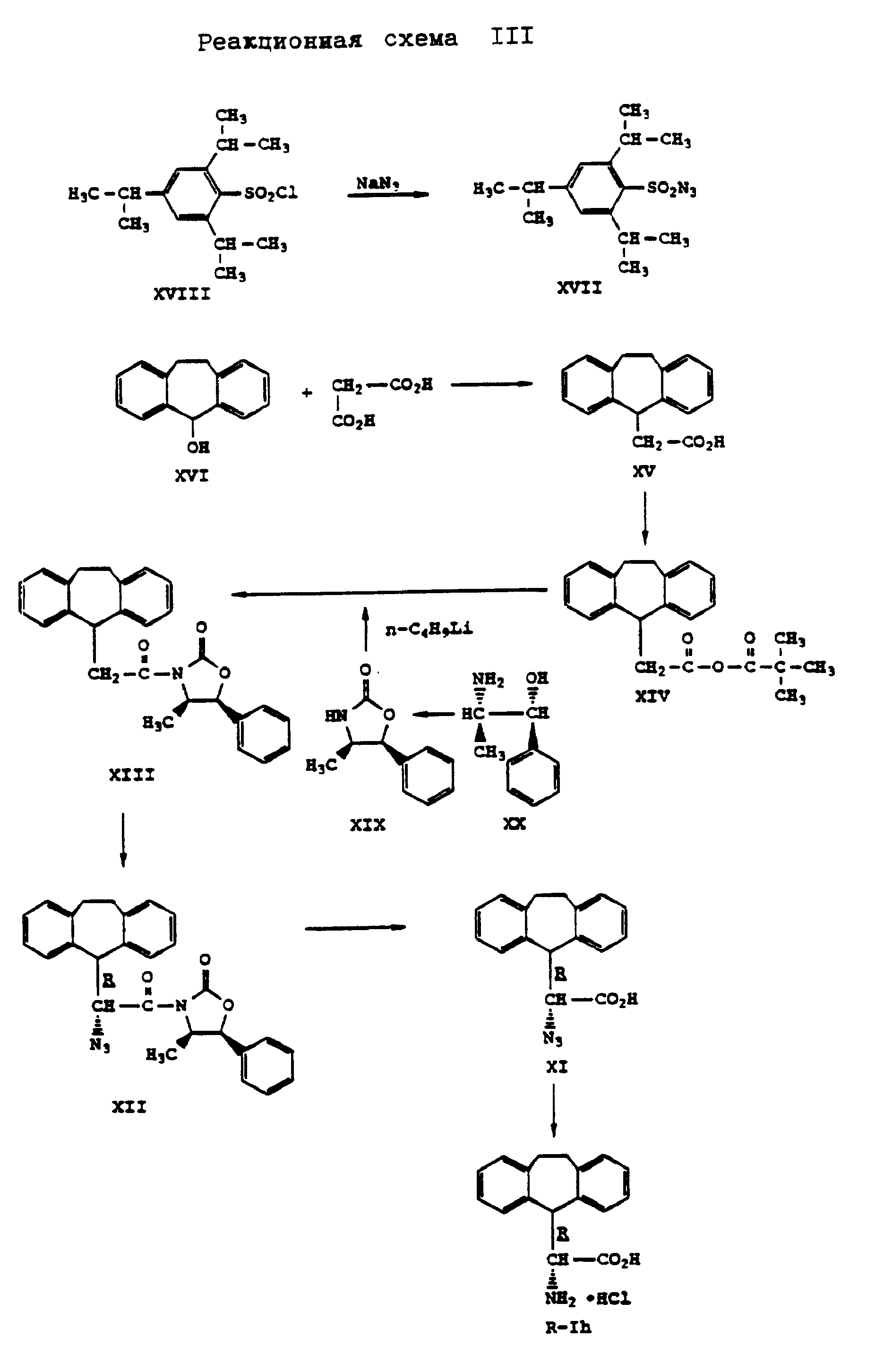
Конфигурацию соединения формулы (Ia) или (Ib) определяют путем хирального синтеза D- и L-10,11-дигидро-5H-дибензо[a, d]-(циклогептен-5-ил)глицина в соответствии с методом, описанным Д.А. Эвенс и др. J. Am. Soc. 112, стр. 411 - 4030, 1990 и Д.А. Эвенс и др. J. Am. Chem. Soc. 11, стр. 1063-1072, 1989, что представлено на реакционных схемах III и IV, приведенных в конце описания, где энантиомеры обозначены буквой R (вместо D) и S (вместо L).
Согласно реакционной схеме III дибензосуберол формулы (X) подвергают взаимодействию с малоновой кислотой при этой температуре примерно 160oC. Раствор получаемый при этом 10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты формулы (XV) в таком растворителе, как, например, этиленгликольдиметиловый эфир, подвергают взаимодействию с пивалоилхлоридом в присутствии основания, такого как, например, N,N-диизопропилэтиламин, с получением смешанного ангидрида формулы (XIV).
Азид натрия добавляют к раствору 2,4,6-триизопропилбензолсульфонилхлорида формулы (XVIII) в таком растворителе, как, например, этанол, с получением азида формулы (XVII). (1S,2R)-норэфедрин формулы (XX) подвергают взаимодействию с диэтилкарбонатом в присутствии основания, такого как, например, карбонат калия, с получением (4R, 5S)-4-метил-5-фенил-2-оксазолидинона формулы (XIX). Раствор смешанного ангидрида формулы (XIV) в таком растворителе, как, например, этиленгликольдиметиловый эфир, добавляют к раствору литийсодержащего оксазолидинона в таком растворителе, как, например, тетрагидрофуран (литийсодержащий оксазолидинон получают путем обработки соединения формулы (XIX) н-бутиллитием), с получением ацилоксазолидинона формулы (XIII). В результате депротонирования соединения формулы (XIII) обработкой бис(триметилсиллил)-амидом калия в среде растворителя, такого как, например, тетрагидрофуран, и последующего последовательного добавления раствора азида формулы (XVII) в таком растворителе, как например, тетрагидрофуран, и кислоты, такой как, например, уксусная кислота, получают азидо-оксазолидинон формулы (XII). В результате гидролиза азидо-оксазолидинона формулы (XII) гидроксидом лития в среде перекиси водорода получают азидокислоту формулы (XI). В результате обработки азидокислоты формулы (XI) водородом в присутствии катализатора, такого как, например, палладий на угле, в среде тетрагидрофурана и 1 н. хлористоводородной кислоты получают гидрохлорид (R)- α -амино-10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты формулы (R-Ih).
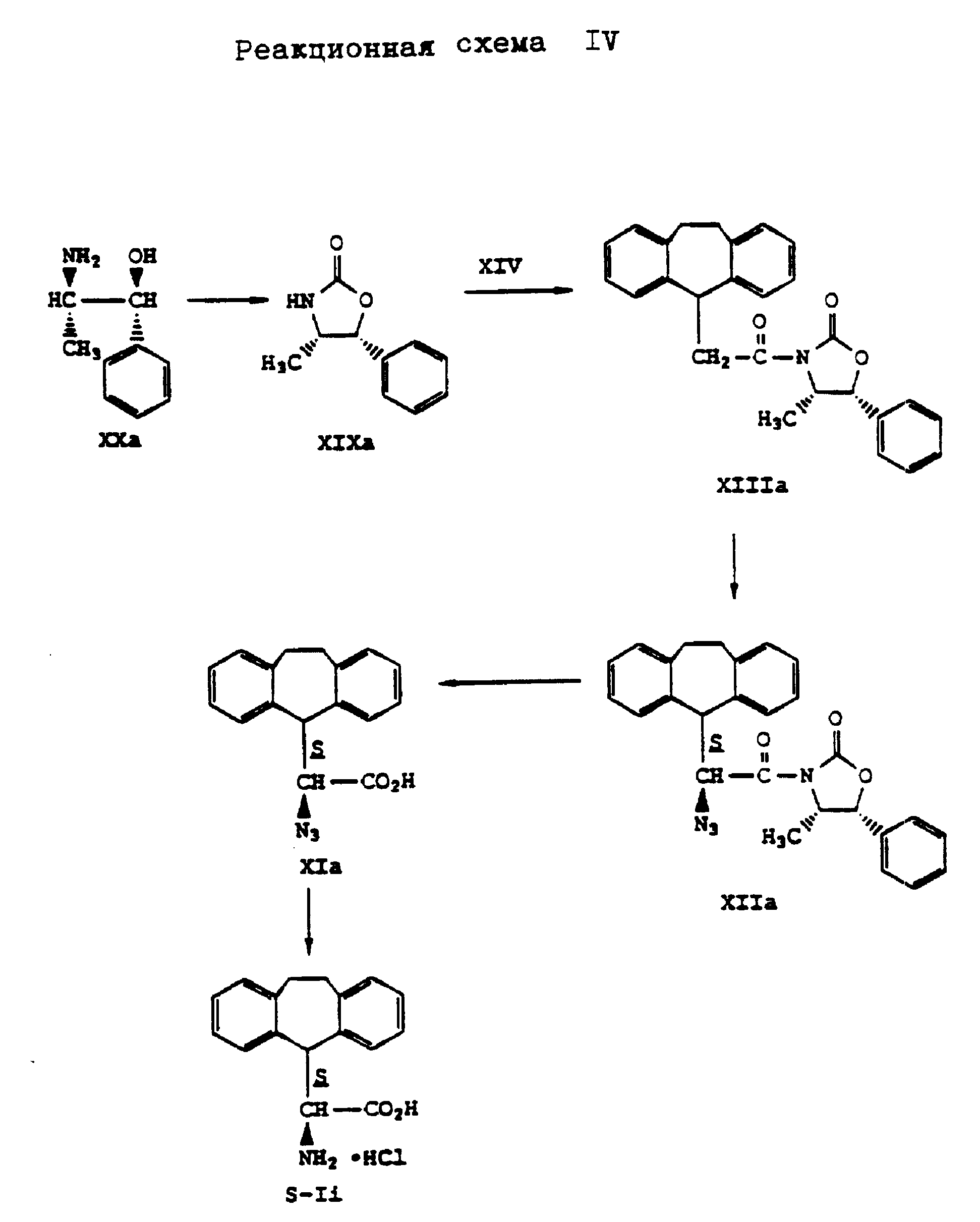
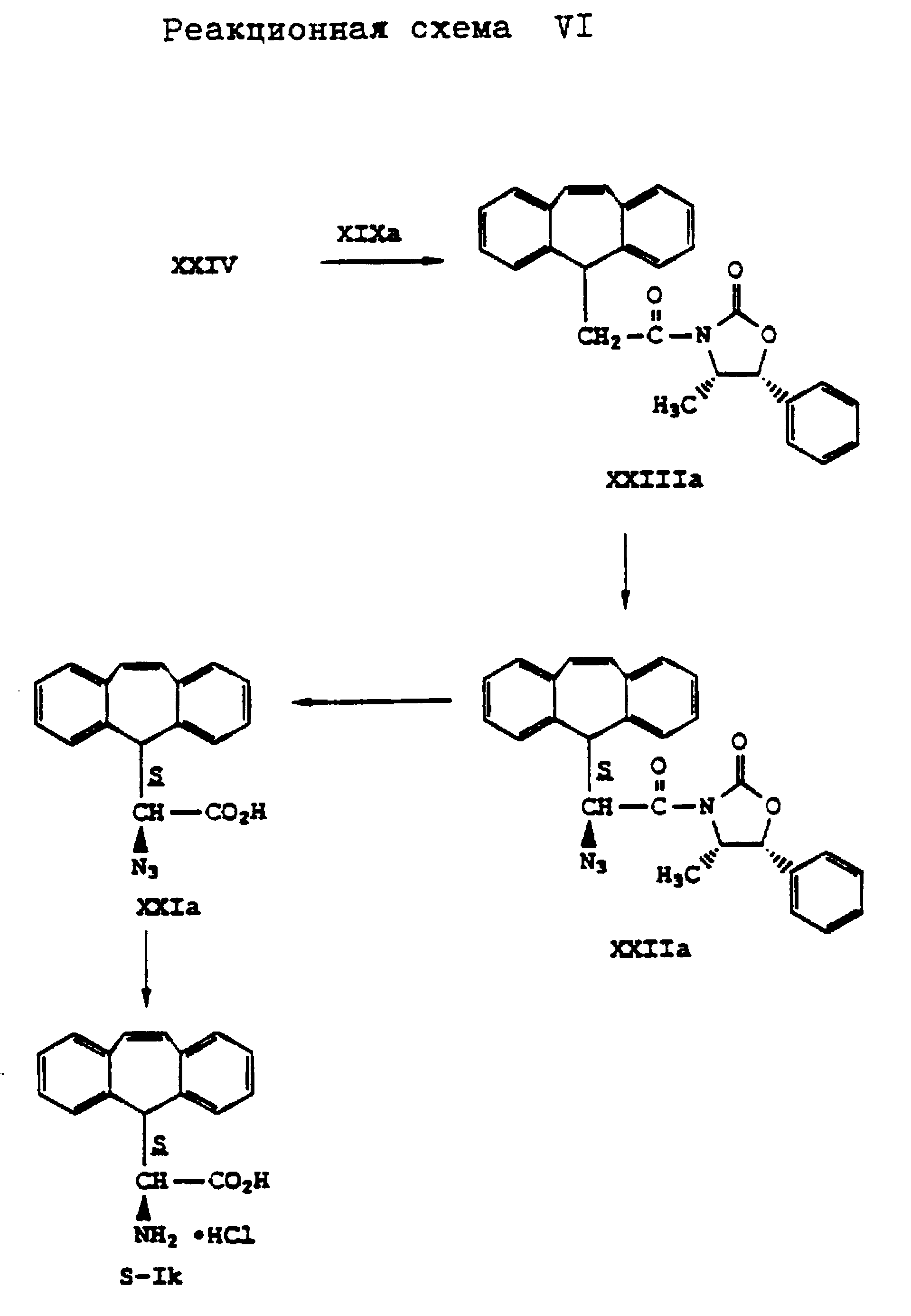
Аналогично гидрохлориду (S)- α -амино-10,11-дигидро-5H-дибензо[a,d]-циклогептен-5-уксусной кислоты получают также (S)- α -амино-10,11-дигидро-5H-дибензо[a, d] -циклогептен-5-уксусную кислоту в виде гидрохлорида формулы (S-Ii). При этом вместо (4R,5S)-4-метил-5-фенил-2-оксазолидинона формулы (XIX) применяют (4S,5R)-5-метил-5-фенил-2-оксазолидинон формулы (XIXa), что поясняется реакционной схемой IV, приведенной в конце описания.
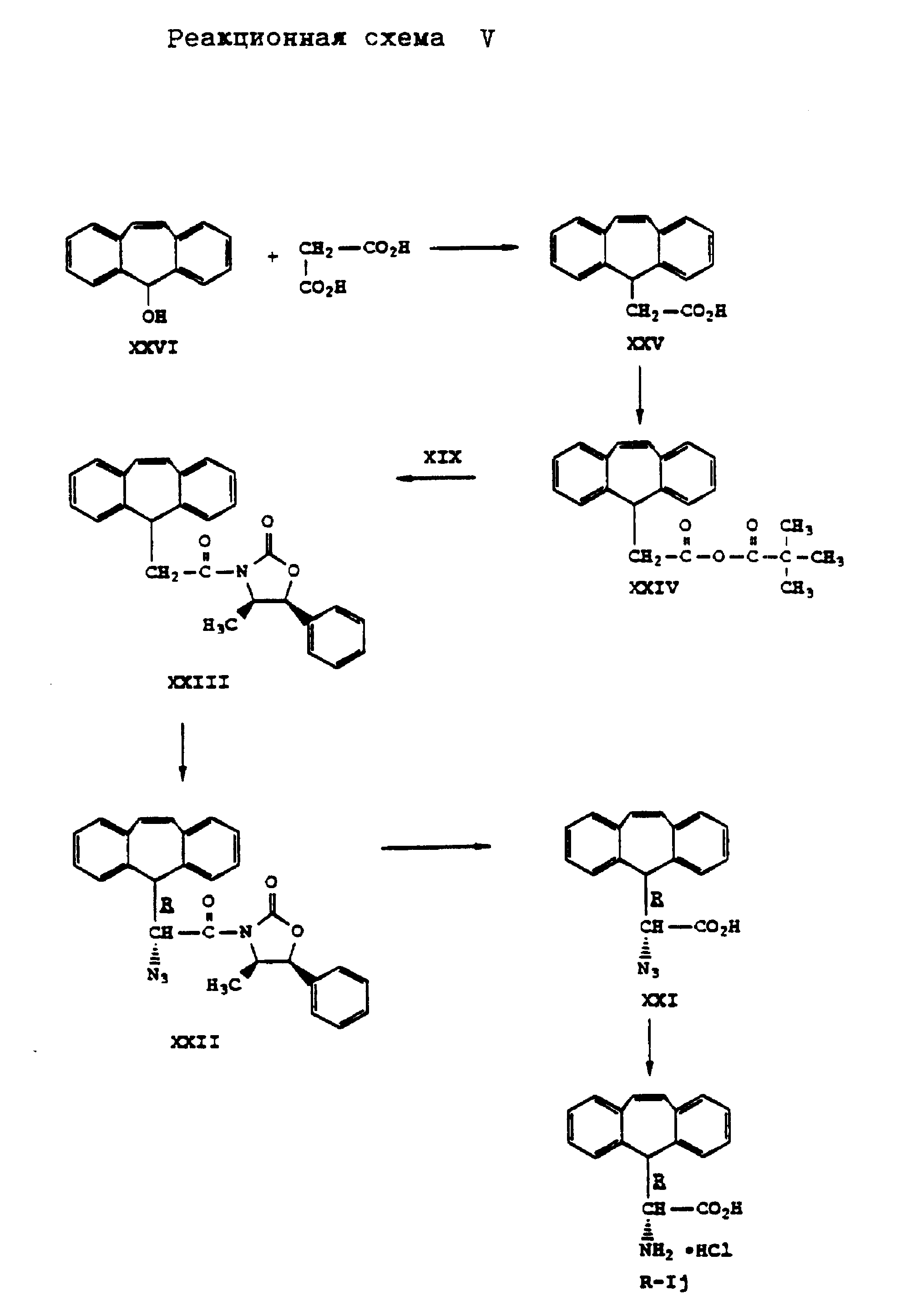
Аналогично способу по реакционной схеме III осуществляют синтез D- α -амино-5H-дибензо[a, d] -циклогептен-5-уксусной кислоты формулы (R-Ij), представленный на реакционной схеме V, приведенной в конце описания. Восстановление азида кислоты формулы (XXI) лучше осуществлять химическим путем, например, обработкой двуххлористым оловом в среде растворителя, такого как, например, метанол. Таким путем можно предотвратить восстановление двойной связи, которая может иметь место в условиях каталитического гидрирования.
Аналогично способу по реакционной схеме V получают L- α -амино-5H-дибензо[a, d] -циклогептен-5-уксусную кислоту формулы (S-Ik), что представлено на реакционной схеме VI, приведенной в конце описания, где вместо (4R,5S)-4-метил-5-фенил-2-оксазолидинона формулы (XIX) применяют (4S, 5R)-4-метил-5-фенил-2-оксазолидинон формулы (XIXa).
Нижепредставленные примеры поясняют настоящее изобретение и применение α -амино-10,11-дигидро-5H-дибензо[a, d] циклогептен-5-уксусной кислоты для получения ацетил-D-Bhg-Leu-Asp-Ile-Ile-Trp, которые представляют собой антагонист эндотелина, который можно применять для лечения гипертензии, инфаркта миокарда, метаболических, эндокринологических и неврологических расстройств, застойной сердечной недостаточности, эндотоксинового бактериально-токсического шока, субарахноидальной почечной недостаточности, аритмии, астмы, преэклампсии и диабета.
Пример 1. α -амино-9H-ксантен-9-уксусная кислота.
Стадия А. Получение сложного этилового эфира α -нитро-9H-ксантен-9-уксусной кислоты.
9,9 г (50 ммоль) 9-гидроксиксантена и 6,1 мл (55 ммоль) сложного этилового эфира нитроуксусной кислоты перемешивают и нагревают при температуре 100oC (на масляной бане) в течение 90 мин. После охлаждения воскообразную смесь суспендируют в 40 мл абсолютного этанола, добавляют 40 мл 1,25 М этанольного раствора гидроокиси калия. Получаемый раствор перемешивают в течение 5 мин при комнатной температуре, после чего этанол отгоняют, оставшееся твердое вещество суспендируют в воде и экстрагируют диэтиловым эфиром. Водную фазу подкисляют до pH 2-3 добавлением 85%-ной фосфорной кислоты, получаемую белую суспензию охлаждают в течение ночи, получаемый остаток собирают путем фильтрации и сушат в вакуумной печи при температуре 50oC. Получают 9,18 г целевого продукта в качестве белого твердого вещества с т. пл. 89-91oC.
Стадия Б. Получение гидрохлорида сложного этилового эфира α -амино-9Н-ксантен-9-уксусной кислоты.
7,5 (24 ммоль) сложного этилового эфира α -нитро-9Н-ксантен-9-уксусной кислоты растворяют в 150 мл этанола и 2 мл концентрированной хлористоводородной кислоты и подвергают гидрированию на взятом в количестве 1,0 г 20%-ном палладии на угле в течение 53 ч при давлении 3, 5858 кг/см2. После фильтрации растворитель отгоняют и остаток промывают дихлорметаном. Получают 4,83 г целевого соединения. Кроме того, в результате охлаждения промытого дихлорметаном продукта получают еще 0,61 г целевого соединения.
Стадия В. Получение гидрохлорида α -амино-9Н-ксантен-9-уксусной кислоты.
К 5,33 г (17 ммоль) гидрохлорида сложного этилового эфира α -амино-9Н-ксантен-9-уксусной кислоты добавляют 45 мл абсолютного этанола и 27 мл 1,25 М этанольного раствора гидроокиси калия (2 эквивалента). За ходом реакции следят при помощи тонкослойной хроматографии на силикагеле с применением в качестве элюента смеси гексана и 2-пропанола в соотношении 3:7, содержащей 1% уксусной кислоты. По истечении 13 ч и после добавления дополнительных двух эквивалентов раствора гидроокиси калия тремя порциями исходное соединение в основном прореагировалось. Этанол отгоняют, остаток смешивают с 100 мл воды и экстрагируют диэтиловым эфиром. Водную фазу подкисляют до pH 2 добавлением 0,5 н. хлористоводородной кислоты (примерно 100 мл), после чего охлаждают в течение ночи, осадок собирают путем фильтрации, последовательно промывают холодной водой и диэтиловым эфиром и сушат при температуре 50oC и давлении 10 мм рт. ст. При этом получают 3,18 г целевого соединения с точкой плавления 264-269oC (разл.).
Стадия Г. Получение α - амино-9Н-ксантен-9-уксусной кислоты.
Гидрохлорид α -амино-9Н-ксантен-9-уксусной кислоты растворяют в воде, pH получаемого раствора доводят до 7 путем добавления разбавленного раствора гидроокиси аммония, получаемый осадок собирают путем фильтрации, промывают водой, кипятят в присутствии воды короткое время, охлаждают до комнатной температуры и собирают. Получают вышеуказанный целевой продукт.
Пример 2. α -амино-9Н-тиоксантен-9-уксусная кислота.
Стадия А. Получение 9-гидрокситиоксантена.
10 г (47 ммоль) тиоксантен-9-она растворяют в 150 мл метанола, получаемый раствор охлаждают на ледяной бане и порциями добавляют 5 г (132 ммоль) борана натрия. Ледяную баню удаляют, реакционную смесь перемешивают при комнатной температуре в течение 15 мин до завершения реакции. Метанол удаляют в вакууме и остаток промывают диэтиловым эфиром. Получают 9,58 г целевого соединения, которое без дальнейшей очистки используют на следующей стадии.
Стадия Б. Получение сложного этилового эфира α -нитро-9-тиоксантен-9-уксусной кислоты.
9,58 г (45 ммоль) сырого 9-гидрокситиоксантена смешивают с 5,5 мл (49 ммоль) сложного этилового эфира нитроуксусной кислоты и смесь нагревают при температуре 100oC в течение 1 ч. Согласно данным жидкостной хроматографии на силикагеле с применением в качестве элюента смеси гексана и 2-пропанола в соотношении 3:1 реакция завершена через 45 мин. Реакционную смесь охлаждают, после чего обрабатывают 100 мл этанола и 39 мл 1,25 М этанольного раствора гидроокиси калия. Этанол отгоняют и остаток смешивают с водой и экстрагируют диэтиловым эфиром. Водный слой подкисляют до pH 2-3 добавлением 85%-ной фосфорной кислотой и на ночь оставляют в холодильнике. Кристаллическое твердое вещество собирают путем фильтрации и сушат при температуре 50oC и давлении 10 мм рт. ст. Последние операции повторяют еще два раза. Получают всего 8,4 г целевого соединения с точкой плавления 111-115oC.
Стадия В. Получение гидрохлорида сложного этилового эфира α -амино-9Н-тиоксантен-9-уксусной кислоты.
4,4 г (13,4 ммоль) сложного этилового эфира α -нитро-9Н-тиоксантен-9-уксусной кислоты подвергают гидрированию в среде 200 мл этанола и в присутствии 13,5 мл концентрированной хлористоводородной кислоты на взятом в количестве 1 г 20%-ном палладии на угле в течение 28 ч при давлении 3,5858 кг/см2. После фильтрации растворитель удаляют, остаток промывают дихлорметаном и сушат при температуре 45oC и давлении 2 мм рт.ст. При этом получают 3,66 г целевого соединения с точкой плавления 238 - 240oC. В результате охлаждения маточного раствора получают еще 0,41 г целевого соединения с точкой плавления 233 - 236oC.
Стадия Г. Получают гидрохлорид α -амино-9H-тиоксантен-9- уксусной кислоты.
3,5 г (10 ммоль) гидрохлорида сложного этилового эфира α -амино-9H-тиоксантен-9-уксусной кислоты растворяют в 30 мл абсолютного этанола, после чего добавляют 33 мл 1,25 М этанольного раствора гидроокиси калия (4 эквивалента). Реакционную смесь перемешивают при комнатной температуре в течение 22 ч. Затем согласно данным жидкостной хроматографии на силикагеле с применением в качестве элюента смеси гексана и 2-пропанола в соотношении 3 : 1 реакция завершена. Этанол отгоняют в вакууме, остаток подают в 65 мл воды, экстрагируют диэтиловым эфиром, водный раствор подкисляют до pH 7 добавлением 1 н. хлористоводородной кислоты, получаемый осадок собирают путем фильтрации, промывают водой и сушат над пятиокисью фосфора при температуре 45oC и добавлении 2 мм рт. ст. При этом получают 1,65 г целевого соединения с точкой плавления 244 - 246oC. Маточный раствор сгущают и в результате охлаждения до температуры 3oC получают еще 0,57 г целевого соединения в качестве свободного основания, которое смешивают с 50 мл воды и подкисляют до pH 1 путем добавления 1 н. хлористоводородной кислоты. Получаемую смесь сгущают досуха в вакууме и остаток сушат при температуре 50oC и давлении 2 мм рт. ст. Получают целевое соединение с точкой плавления 269 - 273oC (разл.).
Стадия Д. Получение α -амино-9H-тиоксантен-9-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид α -амино-9H-тиоксантен-8-уксусной кислоты. При этом получают вышеуказанный целевой продукт.
Пример 3. α -амино-10,11-дигидро-5H-дибензо[a, d]циклогептен- 5-уксусная кислота.
Способ А.
Стадия А. Получение сложного этилового эфира α -[(дифенилметилилен)-амино]-10,11-дигидро-5H-дибензо[a, d]циклогептен- 5-уксусной кислоты.
12 г (45 ммоль) сложного этилового эфира N-(дифенилметилен)глицина и 12,32 г (54 ммоль) 5-хлордибензосуберана растворяют в 250 мл дихлорметана, после чего добавляют 17,6 г (55 ммоль) бромида тетрабутиламмония и 47 мл 50%-го раствора гидроокиси натрия и получаемую смесь механически перемешивают при комнатной температуре в течение 4 ч. Разбавляют 75 мл дихлорметана и водой, получаемые слои отделяют, органический слой промывают водой, сушат над сульфатом магния, фильтруют и перегоняют в вакууме с получением коричнево-оранжевого масла, которое подают в 250 мл диэтилового эфира и 100 мл воды. Эфирный раствор дополнительно промывают водой, сушат над сульфатом магния и перегоняют с получением примерно 21 г прозрачного красно-оранжевого клейкого масла, которое без дальнейшей очистки используют на следующей стадии.
Стадия Б. Получение гидрохлорида α -амино-10,11-дигидро- 5H-дибензо[a,d] циклогептен-5-уксусной кислоты.
Сырой сложный этиловый эфир α -[(дифенилметилен)амино]- 10,11-дигидро-5H-дибензо[a, d] циклогептен-5-уксусной кислоты смешивают с 250 мл 6 н. раствора хлористоводородной кислоты и нагревают с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры твердое вещество собирают путем фильтрации, последовательно промывают водой и диэтиловым эфиром, сушат сначала на воздухе и затем при температуре 50oC и давлении 2 мм рт. ст. При этом получают 6,43 г целевого соединения с точкой плавления >> 280oC.
Стадия В. Получение α -амино-10, 11-дигидро-5H- дибензо[a,d]циклогептен-5-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид 2-амино-10,11-дигидро-5H-дибензо[a, d] циклогептен-5-ил- уксусной кислоты в виде гидрохлорида.
Способ Б.
Стадия А. Получение гидрохлорида сложного этилового эфира α -амино-10,11-дигидро-5H-дибензо[a, d]циклогептен-5-уксусной кислоты.
13 г (40 ммоль) сложного этилового эфира α -нитро-5H- дибензо[a, d]циклогептен-5-уксусной кислоты (см. стадию А примера 4) растворяют в 150 мл этанола, 40 мл воды и 3,5 мл концентрированной хлористоводородной кислоты, после чего гидрируют на взятом в количестве 2 г 20%-ном палладии на угле в течение 30 ч при давлении 3,6561 кг/см2 . После фильтрации растворитель отгоняют и получаемое белое твердое вещество промывают диэтиловым эфиром и сушат при температуре 50oC и давлении 2 мм рт. ст. Получаемый при этом сырой продукт без дальнейшей очистки используют на следующей стадии.
Стадия Б. Получение гидрохлорида α -амино-10,11-дигидро- 5H-дибензо[a,d] циклогептен-5-уксусной кислоты.
13 г (39 ммоль) гидрохлорида сложного этилового эфира α -амино- 10,11-дигидро-5H-дибензо[a, d] циклогептен-5-уксусной кислоты перемешивают в среде 400 мл 1 М этанольного раствора гидроокиси калия при комнатной температуре в течение ночи, после чего этанол отгоняют, а остаток подают в воду, экстрагируют диэтиловым эфиром и подкисляют до pH 1 добавлением 1 н. хлористоводородной кислоты. Получаемый осадок собирают путем фильтрации, промывают диэтиловым эфиром и сушат при температуре 5oC и давлении 2 мм рт. ст. Получают 8,81 г целевого соединения с точкой плавления > 285oC.
Стадия В. Получение α -амино-10,11-дигидро-5H- дибензо[a, d]циклогептен-5-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид 2-амино-10,11-дигидро-5H-дибензо[a, d] циклогептен-5- ил-уксусной кислоты.
Пример 4. α -амино-5H-дибензо[a,d]циклогептен-5-уксусная кислота.
Стадия А. Получение сложного этилового эфира α -нитро- 5H-дибензо-[a,d] циклогептен-5-уксусной кислоты.
42 г (0,2 моль) дибензосуберенола смешивают с 25 мл (0,23 моль) сложного этилового эфира нитроуксусной кислоты и получаемую смесь нагревают при температуре 120oC (на масляной бане) до получения расплава, который поддерживают еще при температуре 110oC в течение 150 мин. Масляную баню удаляют, охлажденную реакционную смесь растворяют в 50 мл метанола и подают в 350 мл кипящего диэтилового эфира. Получаемый темный раствор обрабатывают активным углем, фильтруют и охлаждают в течение ночи. Получаемый осадок собирают путем фильтрации, последовательно промывают гексаном и диэтиловым эфиром, сушат в вакууме при комнатной температуре. Получают 26,7 г целевого соединения с точкой плавления 101 - 103oC.
Стадия Б. Получение гидрохлорида сложного этилового эфира α -амино-5H-дибензо[a, d]циклогептен-5-уксусной кислоты.
1 г (3,4 ммоль) сложного этилового эфира α -нитро- 5H-дибензо[a,d]циклогептен-5-уксусной кислоты и 1,6 г (6,8 ммоль) гексагидрата хлористого кобальта (II) растворяют в 20 мл абсолютного этанола, после чего порциями добавляют в течение 20 мин 1,28 г (34 ммоль) борана натрия. Добавляют еще 15 мл этанола для обеспечения растворения всего борана натрия, после чего перемешивание продолжают в течение 1 ч, добавляют 30 мл 3 н. хлористоводородной кислоты и раствор перемешивают до того, пока он не становится пурпурным. Нерастворимое вещество в реакционной смеси фильтруют, этанол отгоняют в ротационном испарителе, водный раствор фильтруют, получаемое при этом твердое вещество голубого-белого цвета интенсивно промывают 6 н. хлористоводородной кислотой и затем еще раз промывают диэтиловым эфиром. Получаемое при этом белое твердое вещество сушат при температуре 50oC и давлении 2 мм рт. ст. Получают 0,65 г целевого соединения с точкой плавления 253 - 256oC (разл.).
Стадия В. Получение гидрохлорида α -амино-5H- дибензо[a,d]циклогептен-5-уксусной кислоты.
0,21 г (0,6 ммоль) гидрохлорида сложного этилового эфира α -амино-5H-дибензо[a, d] циклогептен-5-уксусной кислоты растворяют в 10 мл абсолютного этанола и раствор перемешивают в среде 3 мл 1,25 M, этанольного раствора гидроокиси калия до завершения реакции, определяемого путем тонкослойной жидкостной хроматографии на силикагеле с применением в качестве элюента смеси гексана и 2-пропанола в соотношении 3 : 1 (через 2 ч). Растворитель отгоняют, остаток подают в воду, экстрагируют дихлорметаном, водный слой подкисляют до pH 1 добавлением 6 н. хлористоводородной кислоты и сгущают досуха. Получаемый остаток обрабатывают 5 мл кипящей воды, охлаждают до комнатной температуры и затем на ночь подают в холодильник. Получаемое твердое вещество собирают и промывают диэтиловым эфиром. При этом получают 0,05 г целевого соединения с точкой плавления 225oC (разл.).
Стадия Г. Получение α -амино-5H-дибензо[a,d]циклогептен-5- уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид α -амино-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
Пример 5. D- α -амино-10, 11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусная кислота.
Стадия А. Получение α -ацетиламино-10,11-дигидро-5H- дибензо[a,d]циклогептен-5-уксусной кислоты.
5 г (16,5 ммоль) гидрохлорида α -амино-10,11-дигидро- 5H-дибензо[a,d] циклогептен-5-уксусной кислоты растворяют в 70 мл 1 н. гидроокиси натрия и 50 мл воды, охлаждают на ледяной бане и обрабатывают 2,3 мл (25 ммоль) уксусного ангидрида. При этом pH среды поддерживают при 10 - 10,5. Реакционную смесь перемешивают в течение дополнительных 20 мин, pH смеси доводят до 3 - 4 путем добавления концентрированной хлористоводородной кислоты, получаемый осадок собирают, последовательно промывают водой и диэтиловым эфиром и сушат в вакуумной печи при температуре 5oC и давлении 10 мм рт. ст. Получаемое твердое вещество перекристаллизовывают из смеси этилацетата и гексана с получением 2,41 г целевого соединения с точкой плавления 220 - 223oC.
Стадия Б. Получение цинхонидиниевой соли α -ацетиламино-10, 11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
2,25 г (7,3 ммоль) α -ацетиламино-10,11-дигидро-5H-дибензо[a, d]-циклогептен-5- уксусной кислоты растворяют в 10 мл метанола и получаемый раствор смешивают с раствором 2,36 г (8 ммоль) (-)-цинхонидина в 15 мл кипящего метанола. Реакционной смеси дают охлаждаться до комнатной температуры, после чего подают в холодильник (+2oC). Получают 0,77 г энантиомера с [α]D= -38° и 0,27 г энантиомера с [α]D= -41°, которые объединяют и применяют для получения D-энантиомера. Растворитель удаляют из маточного раствора и получаемый при этом остаток перекристаллизовывают из метанола. Получают 2,4 г цинхонидиниевой соли с [α]D= -74,5° применяют для получения L-энантиомера.
Стадия В. Получение D- α -ацетиламино-10, 11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
0,32 г (0,5 ммоль) цинхонидиниевой соли D- α -ацетиламино-10,11- дигидро-5H-дибензо [a,d]-циклогептен-5-уксусной кислоты смешивают с 6 мл сложного этилового эфира уксусной кислоты и 2 мл 1н., хлористоводородной кислоты и получаемую реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Органический слой отделяют, последовательно промывают водой и солевым раствором и сушат над сульфатом магния. После фильтрации растворитель отгоняют. Получают при этом 0,12 г целевого соединения, которое без дальнейшей очистки используют на следующей стадии.
Стадия Г. Получение гидрохлорида D- α -амино-10,11-дигидро-5H-дибензо[a, d]циклогептен-5-уксусной кислоты.
Смесь 0,13 г D- α -ацетиламино-10, 11-дигидро-5H-дибензо[a,d]циклогептен-5- уксусной кислоты и 10 мл 6 н. хлористоводородной кислоты нагревают с обратным холодильником в течение 6 ч, после чего охлаждают, получаемый осадок собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакуумной печи при температуре 5oC и давлении 10 мм рт. ст. При этом получают 0,09 г целевого соединения с точкой плавления ≥ 280oC. [α]D= -47°± 1° (c = 1% в метаноле).
Стадия Д. Получение D- α -амино-10,11-дигидро-5H-дибензо[a, d]циклогептен-5- уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид D- α -амино-10, 11-дигидро-5H-дибензо[a,d]-циклогептен-5- уксусной кислоты.
Пример 6. L- α -амино-10,11-дигидро-5H-дибензо[a,d]- циклогептен-5-уксусная кислота.
Стадия А. Получение L- α -ацетиламино-10, 11-дигидро-5H- дибензо[a,d]циклогептен-5-уксусной кислоты.
2 г (3,3 ммоль) цинхонидиниевой соли L- α -ацетиламин-10, 11- дигидро-5H-дибензо-[a, d] циклогептен-5-уксусной кислоты по примеру 5 смешивают с 45 мл этилацетата и 16 мл 1 н. хлористоводородной кислоты и получаемую смесь перемешивают при комнатной температуре в течение 1 ч. В результате переработки описанным в примере 5 образом получают 0,27 твердого вещества, которое собирается путем фильтрации. В результате перегонки маточного раствора получают еще 0,77 г целевого соединения, которое без дальнейшей очистки применяют для получения L-энантиомера.
Стадия Б. Получение гидрохлорида L- α -амино-10, 11- дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
0,75 г L- α -ацетиламино-10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты смешивают с 30 мл 6 н. хлористоводородной кислоты и получаемую смесь нагревают с обратным холодильником в течение 6 ч. После охлаждения реакционную смесь на ночь хранят в холодильнике. Получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат при температуре 5o C и давлении 10 мм рт. ст. При этом получают 0,57 г целевого соединения с [α]D= +36° (c = 1% в метаноле).
Стадия В. Получение L- α -амино-10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид L- α -амино-10, 11-дигидро-5H-дибензо[a, d]циклогептен-5-уксусной кислоты.
Пример 7. (R)- α -амино-10,11-дигидро-5H-дибензо[a, d]циклогептен-5-уксусная кислота.
Стадия А. Получение 10,11-дигидро-5H-дибензо-[a,d]циклогептен- 5-уксусной кислоты.
В снабженную перегонным аппаратом круглодонную колбу подают 25 г (0,119 моль) дибензосуберола и 61,9 г(0,595 моль) малоновой кислоты. Реакционную смесь нагревают на масляной бане с температурой 160oC. Твердое вещество расплавляется. Выделяются газы и воду и уксусную кислоту отгоняют через конденсатор. По истечении 30 мин реакционную смесь охлаждают до комнатной температуры и растворяют в этилацетате. Органический раствор промывают солевым раствором и сушат над сульфатом магния. В результате вакуумной отгонки растворителя получают беловатое твердое вещество, которое перекристаллизовывают из смеси гексана и этилацетата в соотношении 1: 1. При этом получают 28,8 г целевого соединения в качестве белых кристаллов с точкой плавления 164 - 165oC.
Стадия Б. Получение (4R-цис)-3-[(10, 11-дигидро-5H-дибензо[a,d]циклогептен-5-ил) ацетил]4-метил-5-фенил-2-оксазолидинона.
Раствор 12 г (47,56 ммоль) 10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты в 60 мл безводного этиленгликольдиметилового эфира охлаждают в атмосфере азота до температуры -20oC, после чего последовательно добавляют 9,9 мл (57,07 ммоль) свеже-дистиллированного диизопропилэтиламина и 6,4 мл (52,52 ммоль) пивалоилхлорида. Получаемую мутную суспензию перемешивают при температуре -20oC в течение 30 мин, получаемое белое твердое вещество фильтруют, промывают 10 мл безводного этиленгликольдиметилового эфира и фильтрат охлаждают до температуры -68oC в атмосфере азота. В отдельной колбе 9,3 г (52,32 ммоль) (4R)-метил-(5S)-фенил-2-оксазолидинона растворяют в 100 мл свеже-дистиллированного тетрагидрофурана и получаемый раствор охлаждают до температуры -78oC в атмосфере аргона. В качестве индикатора добавляют несколько кристаллов трифенилметана. К раствору добавляют 34,3 мл (54,93 ммоль) 1,6 М раствора н.бутиллития в гексане. Получаемый красный раствор перемешивают при температуре -78oC в течение 5 мин, после чего при помощи канюли добавляют полученный выше раствор смешанного ангидрида. Получаемый при этом светло-желтый раствор перемешивают при температуре -78oC в течение 15 мин, после чего нагревают до комнатной температуры в течение 2 ч, реакцию прекращают путем добавления водного хлористого аммония, тетрагидрофуран удаляют в вакууме и остаток три раза экстрагируют дихлорметаном, взятым в количестве по 200 мл. Органические экстракты объединяют, последовательно промывают водным раствором бикарбоната натрия и солевым раствором, сушат над сульфатом магния и сгущают в вакууме. Получаемое желтое твердое вещество перекристаллизовывают из смеси гексана и этилацетата в соотношении 4 : 1. Получают 16,2 г целевого соединения в качестве белого твердого вещества с точкой плавления 133 - 134oC. [α]D= +14,0° (c = 1% в хлороформе).
Стадия В. Получение [4R-[3(R*), 4α, 5α]]-3-[азидо-(10,11-дигидро-5H-дибензо[a, d]-циклогептен-5-ил)ацетил]-4- метил-5-фенил-2-оксазолидинона.
К раствору 15,6 г (37,9 ммоль) (4R-цис)-3-[(10, 11-дигидро- 5H-дибензо)[a, d]циклогептен-5-ил)ацетил]-4-метил-5-фенил-2- оксазолидинона в 505 мл свеже-дистиллированного тетрагидрофурана в атмосфере аргона прикапывают 83,4 мл (41,7 ммоль) 0,5 М толуольного раствора бис(триметилсилил)амида калия при температуре -78oC. Получаемый желтый раствор перемешивают при температуре -78oC в течение 1 ч, после чего добавляют раствор 15,3 г (49,3 ммоль) 2,4,6- триизопропилбензолсульфонилазида по нижепредставленному примеру 16 в 10 мл сухого тетрагидрофурана, продолжают перемешивать при температуре -78oC в течение 10 мин, быстро добавляют 9,1 мл (159,2 ммоль) уксусной кислоты, реакционную смесь нагревают на паровой бане до температуры 30oC и затем перемешивают при комнатной температуре в течение 2 ч. Реакционная смесь становится мутной. Тетрагидрофураны удаляют в вакууме, получаемую беловатую пасту растворяют в 600 мл дихлорметана, последовательно промывают полунасыщенным солевым раствором и полунасыщенным раствором бикарбоната натрия, сушат над сульфатом магния, растворитель упаривают в вакууме и получаемое при этом беловатое твердое вещество перекристаллизовывают из смеси гексана и этилацетата в соотношении 1,5 : 1. Получают 14,3 г целевого соединения в качестве белых кристаллов с точкой плавления 171oC. [α]D= -178,5° (c = 1% в хлороформе).
Стадия Г. Получение (R)- α -азидо-10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
В атмосфере азота к раствору 13,7 г (30,27 ммоль) [4R-[3(R*),4α, 5α-]] -3-[азидо(10,11-дигидро-5H-дибензо [a, d]циклогептен-5-ил)ацетил]-4-метил-5-фенил-2-оксазолидинона в 300 мл смеси тетрагидрофурана и воды в соотношении 4: 1 прикапывают при температуре 0oC раствор 2,5 г (60,54 ммоль) гидроокиси лития в 17,2 мл (151,35 ммоль) 30%-ной водной перекиси водорода. Получаемую мутную смесь перемешивают при температуре 0oC в течение 1 ч., после чего добавляют 50 мл водного сульфита натрия, основное количество тетрагидрофурана упаривают в вакууме, водный раствор охлаждают на ледяной бане и подкисляют до pH 1 добавлением 6 н. хлористоводородной кислоты. Получаемое более твердое вещество экстрагируют пять раз дихлорметаном, сушат над сульфатом магния, растворитель упаривают в вакууме с получением желтого масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси гексана, этилацетата и уксусной кислоты в соотношении 100:50:1. Получают 1,3 г целевого соединения в качестве белого твердого вещества с точкой плавления 101 - 102oC. [α]D= -38,7° (c = 1% в метаноле). Применяемый в качестве хирального вспомогательного вещества (4R)-метил-(5S)-фенил-2-оксазолидинон рекуперируют в качестве белого твердого вещества в количестве 5,2 г, точка плавления 120 - 122oC.
Стадия Д. Получение гидрохлорида (R)- α -амино-10,11-дигидро-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
К раствору 3,5 г (1,9 ммоль) (R)- α -азидо-10, 11-дигидро-5H-дибензо[a, d] циклогептен-5-уксусной кислоты в 75 мл тетрагидрофурана, 10 мл воды и 1 мл концентрированной хлористоводородной кислоты добавляют 0,5 г 20%-ного палладия на угле. Реакционную смесь встряхивают при температуре 25oC и давлении водорода, равном 3,6561 кг/см2, в течение 6 ч, после чего твердое вещество фильтруют, фильтраты сгущают в вакууме и получаемое светло-блестящее зеленое твердое вещество перекристаллизовывают из 3 н. хлористоводородной кислоты, содержащей активный уголь. Получают 2,8 кг целевого соединения в качестве белого твердого вещества с точкой плавления 313 - 314oC (разл.). [α]D= -47,6° (c = 1% в метаноле).
Стадия Е. Получение (R)- α -амино-10, 11-дигидро-5H-дибензо [a, d]циклогептен-5-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид (R)- α -амино-10,11-дигидро-5H-дибензо[a, d]циклогептен- 5-уксусной кислоты. Свободная аминокислота имеет т.пл. 187 - 190oC.
Пример 8. N-трет. -бутилоксикарбонил-D- α -амино-10,11-дигидро- 5H-дибензо[a, d]циклогептен-5-уксусная кислота.
К прозрачному бесцветному раствору 0,5 г (1,65 ммоль) гидрохлорида (R)- α -амино-10,11-дигидро-5H-дибензо[a, d] циклогептен- 5-уксусной кислоты по примеру 7 в 15 мл метанола последовательно добавляют диизопропилэтиламин и ди-трет. -бутилдикарбонат. Получаемый прозрачный раствор перемешивают при комнатной температуре в течение 18 ч, после чего сгущают в вакууме и получаемое бесцветное масло подвергают флеш-хроматографии на силикагеле с применением в качестве элюента смеси гексана, этилацетата и уксусной кислоты в соотношении 100:50:1. Получают 0,58 г целевого соединения в качестве белого твердого вещества с точкой плавления 179 - 180oC (разл.). [α]D= +27,2° (c = 1,0% в метаноле).
Аналогично примеру 7 получают соединение примера 9.
Пример 9. (S)- α -амино-10,11-дигидро-5H-дибензо[a,d]-циклогептен-5-уксусная кислота.
Стадия А. Получение 10,11-дигидро-5H-дибензо-[a,d]циклогептен-5-уксусной кислоты с точкой плавления 167 - 168oC.
Стадия Б. Получение (4S-цис)-3-[(10, 11-дигидро-5Н-дибензо[a,d] циклогептен-5-ил)ацетил] -4-метил-5-фенил-2-оксазолидинона с точкой плавления 130 - 131oC. [α]D= -14,0° (c = 1% в хлороформе).
Стадия В. Получение [4S-[3(S*),4 α , 5 α ]-3-[азидо- (10,11-дигидро-5Н-дибензо[a, d]циклогептен-5-ил)ацетил]-4-метил-5-фенил- 2-оксазолидинона с точкой плавления 169 - 171oC. [α]D= +179,4° (c = 1% в хлороформе).
Стадия Г. Получение (S)- α -азидо-10,11-дигидро-5Н-дибензо [a, d]циклогептен-5-уксусной кислоты с точкой плавления 99 - 101oC. [α]D= +37,8° (c = 1% в метаноле).
Стадия Д. Получение (S)-α -амино-10,11-дигидро-5Н-дибензо[a,d]циклогептен- 5-уксусной кислоты в виде гидрохлорида с точкой плавления 316 - 318oC (разл.). [α]D= +45,6° (c = 1% в метаноле).
Стадия Е. Получение (S)- α -амино-10,11-дигидро-5Н-дибензо [a,d]циклогептен-5-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид (S)- α -амино-10,11-дигидро-5Н-дибензо[a,d]циклогептен- 5-уксусной кислоты.
Аналогично примеру 8 получают соединение примера 10.
Пример 10. N-трет.-бутилоксикарбонил-L- α -амино-10,11- дигидро-5Н-дибензо[a, d] циклогептен-5-уксусная кислота с точкой плавления 178 - 179oC. [α]D= -31,4° (c = 1% в метаноле).
Пример 11. N-трет.-бутилоксикарбонил-DL- α -амино 10,11-дигидро-5Н-дибензо[a,d]циклогептен-5-уксусная кислота.
1,70 г (5, 43 ммоль) гидрохлорида DL - Вgh суспендируют в 150 мл смеси п-диоксана и воды в соотношении 2 : 1 при комнатной температуре, после чего к перемешиваемому раствору добавляют 1,40 г (6,42 ммоль) ди-трет.-бутилдикарбоната. pH получаемого раствора доводят до > 9 добавлением 1 н. гидроокиси натрия, после чего pH среды поддерживают между 9 и 10 добавлением аликвотных количеств 1 н. гидроокиси натрия до постоянства величины pH. Раствор сгущают под пониженным давлением примерно до 75 мл, на него подают 50 мл этилацетата и подкисляют до величины pH примерно 2,5 добавлением 10%-ной хлористоводородной кислоты. Органический слой отделяют, последовательно промывают два раза 10%-ной водной хлористоводородной кислотой, взятой в количестве по 50 мл, два раза солевым раствором, взятым в количестве по 50 мл, и три раза водой, взятой в количестве по 50 мл, сушат над сульфатом магния, фильтруют и сгущают под пониженным давлением. Получаемое масло перекристаллизовывают из смеси этилацетата и гептана, взятой в количестве 1,82 г. Получаемое белое твердое вещество идентифицируют методами протонного ЯМР, масс-спектрометрии с применением бомбардировки быстрыми атомами (M+1=368), а также элементным анализом.
Пример 12. (R)- α -амино-5Н-дибензо[a,d]циклогептен-5-уксусная кислота.
Стадия А. Получение 5Н-дибензо[a,d]циклогептен-5-уксусной кислоты.
В снабженную перегонным аппаратом круглодонную колбу подают 10 г (48,01 ммоль) дибензосуберенола и 25 г (2,08 ммоль) малоновой кислоты, после чего смесь нагревают на масляной бане с температурой 160oC. Твердое вещество расплавляется, выделяется газ, воду и уксусную кислоту отгоняют через конденсатор. Через 90 мин смесь охлаждают до комнатной температуры и растворяют в этилацетате. Получаемый органический раствор промывают солевым раствором, сушат над сульфатом магния, растворитель удаляют в вакууме и получаемое при этом беловатое твердое вещество перекристаллизовывают из смеси гексана и этилацетата в соотношении 1:1. Получают 10,2 г целевого соединения в качестве белых кристаллов с точкой плавления 167 - 168oC.
Стадия Б. Получение (4R-цис)-3-[(5H-дибензо[a, d] -циклогептен-5-ил)ацетил]-4-метил-5-фенил- 2-оксазолидинона.
Раствор 6 г (23,97 ммоль) полученной на стадии A кислоты в 30 мл безводного этиленгликольдиметилового эфира охлаждают до температуры - 20oC, после чего в атмосфере азота последовательно добавляют 5 мл (28,77 ммоль) свеже-дистиллированного диизопропилэтиламина и 3,2 мл (26,37 ммоль) пивалоилхлорида (который медленно добавляют). Получаемую мутную суспензию перемешивают при температуре -20oC в течение 30 мин, белое твердое вещество фильтруют, промывают 5 мл безводного этиленгликольдиметилового эфира и фильтрат смешанного ангидрида охлаждают до температуры -78oC в атмосфере азота. В отдельной колбе 4,7 г (26, 37 ммоль) (4R)-метил-(5S)-фенил-2-оксазолидинона растворяют в 53 мл свеже-дистиллированного тетрагидрофурана и раствор охлаждают до температуры -78oC в атмосфере аргона, после чего в качестве индикатора добавляют несколько кристаллов трифенилметана. К получаемому раствору добавляют 17,3 мл (27,81 ммоль) 1,6 М раствора н.-бутиллития в гексане, получаемый красный раствор перемешивают при температуре -78oC в течение 5 мин и при помощи канюли добавляют полученный выше раствор смешанного ангидрида. Получаемый светло-желтый раствор перемешивают при температуре -78oC в течение 15 мин и затем нагревают до комнатной температуры в течение 2 ч. Реакцию прекращают путем добавления водного хлористого аммония, тетрагидрофуран удаляют в вакууме, остаток три раза экстрагируют дихлорметаном, взятым в количестве по 100 мл, органические экстракты объединяют, последовательно промывают водным раствором бикарбоната натрия и солевым раствором, сушат над сульфатом магния и сгущают в вакууме. Получаемое желтое твердое вещество перекристаллизовывают из смеси гексана и этилацетата в соотношении 2 : 1. Получают 6,3 г целевого соединения в качестве белого твердого вещества с точкой плавления 172-173oC. [α]D= +10,1° (c=1% в хлороформе).
Стадия В. Получение [4R-[3(R*),4a, 5S]]-3-[азидо-(5H-дибензо[a,d]циклогептен-5- ил)ацетил]-4-метил-5-фенил-2-оксазолидинона.
К раствору 5 г (12,21 ммоль) полученного на стадии Б ацилоксазолидинона в 162 мл свеже-дистиллированного тетрагидрофурана прикапывают в атмосфере аргона при температуре -78oC 26,9 мл (13,43 ммоль) 0,5 М раствора бис(триметилсилил)амида калия, получаемый желтый раствор перемешивают при температуре 78oC в течение 30 мин, после чего добавляют раствор 4,9 г (15,87 ммоль) полученного в нижепредставленном примере 16 2,4,6-триизопропилбензолсульфонилазида в 10 мл сухого тетрагидрофурана. Через 2 мин при температуре 78oC быстро добавляют 2,5 мл (43,96 ммоль) уксусной кислоты, получаемую смесь нагревают на водяной бане до температуры 30o C и затем перемешивают при комнатной температуре в течение 2 ч. Из получаемой мутной смеси тетрагидрофуран удаляют в вакууме, получаемую при этом беловатую пасту растворяют в 400 мл дихлорметана, последовательно промывают полунасыщенным солевым раствором и полуненасыщенным раствором бикарбоната натрия, сушат над сульфатом магния и растворитель упаривают в вакууме. Получаемое при этом беловатое твердое вещество перекристаллизовывают из смеси гексана и этилацетата в соотношении 1,5:1. Получают 4,7 г целевого соединения в качестве белых кристаллов с точкой плавления 175o C. [α ]D= -119,4° (c=1% в хлороформе).
Стадия Г. Получение (R)- α -азидо-5H-дибензо[a,d]-циклогептен-5-уксусной кислоты.
В атмосфере азота к раствору 0,87 г (1,93 ммоль) полученного на стадии В азида в 20 мл смеси тетрагидрофурана и воды в соотношении 4 :1 при температуре 0oC прикапывают раствор 0,16 г (3,86 ммоль) гидрата гидроокиси лития в 1,10 мл (9,65 ммоль) 30%-ной водной перекиси водорода. Получаемую мутную реакционную смесь перемешивают при температуре 0oC в течение 1 ч, после чего добавляют 10 мл водного сульфида натрия, основное количество тетрагидрофурана упаривают в вакууме, получаемый водный раствор охлаждают на ледяной бане и подкисляют до pH 1 добавлением 6 н. хлористоводородной кислоты. Получаемое белое твердое вещество экстрагируют пять раз дихлорметаном, сушат над сульфатом магния, растворитель упаривают в вакууме и получаемое желтое масло очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси гексана, этилацетата и уксусной кислоты в соотношении 100 : 50 : 1. Получают 0,5 г целевого соединения в качестве белого твердого вещества с точкой плавления 119-120oC. [α]D= -40,6° (c=1% в метаноле). Используемое хиральное вспомогательное вещество рекуперируют в качестве белого твердого вещества с точкой плавления 120-122oC в количестве 0,31 г.
Стадия Д. Получение гидрохлорида (R)- α -амино-5H-дибензо[a,d]-циклогептен-5-уксусной кислоты.
К суспензии 0,41 г (1,80 ммоль) хлорида олова (II) в 5 мл метанола добавляют 0,35 г (1,20 ммоль) полученной на стадии Г азидо-кислоты. Реакция протекает экзотермически. Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, после чего подкисляют до pH 1 добавлением 6 н. хлористоводородной кислоты, растворитель удаляют в вакууме и получаемую при этом суспензию очищают на йоните марки ДОВЕКС 50 • 8 - 100. Получают 0,25 г беловатого твердого вещества, представляющего собой свободную аминокислоту. 0,15 г этой кислоты перекристаллизовывают из 3 н. хлористоводородной кислоты с получением 0,21 г целевого гидрохлорида в качестве беловатого твердого вещества с точкой плавления 229-232oC (разл.). [α]D= +24,7° (c=1% в метаноле).
Стадия Е. Получение (R)- α -амино-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
Повторяют стадию Г примера 1 с той лишь разницей, что применяют гидрохлорид (R)- α -амино-5H-дибензо[a,d]циклогептен-5-уксусной кислоты.
Свободная аминокислота имеет т.пл. 194-196oC.
Пример 13. N-трет.-бутилоксикарбонил-(R)- α -амино-5H-дибензо[a,d]циклогептен-5-уксусная кислота.
К суспензии 0,10 г (0,37 ммоль) полученного на стадии D примера 12 гидрохлорида в 5 мл метанола добавляют 0,07 мл (0,37 ммоль) N,N'-диизопропилэтиламина и 0,16 г (0,74 ммоль) ди-трет.-бутилдикарбоната, после чего реакционную смесь перемешивают при комнатной температуре в течение 18 ч, сгущают в вакууме и получаемое желтое твердое вещество флеш-хроматографии на силикагеле с применением в качестве элюента смеси гексана, этилацетата и уксусной кислоты в соотношении 100 : 50 : 1. При этом получают 0,12 г целевого соединения в качестве белого твердого вещества с точкой плавления 150 - 151oC (разл.). [α]D= +27,3° (c=1% в метаноле).
Пример 14. Ac-D-Bhg-Leu-Asp-Ile-Ile-Trp.
Линейный гексапептид получают путем стандартного твердофазного синтеза пептидов с применением трет.
-бутилоксикарбонильных и
бензильных защитных групп (см. И.М. Стьюарт и И.Д. Яанг. Solid Phase Peptide Synthesis. Пирс Кемикель Ко. Рокфорд, штат Илинойс, США, 1984). Все защищенные аминокислоты и
реагенты представляют собой
торговые продукты за исключением пептида N- α Boс-DL-Bhg (см. пример 11), которые далее не очищаются. Защищенную пептидную смолу получают в реакторе типа "Applied
Biosystems 430 А Peptide
Synthesizer" с соблюдением режима, предусмотренного для осуществления реакции сочетания в присутствии дицилкогексилкарбодиимида (стандартный метод 1.0, версия 1.40). Исходя
из 0,710 г (0,70 мэкв/г)
N- α Boc-Trp на 4-(оксиметил)-фенилацетамидометильной смоле (0,497 мэкв/г Boc-Trp(For)) защищенный пептид получают путем последовательного сочетания со следующими
аминокислотами (а порядке
добавления): N- α -Boc-Ile•0,5H2O, N- α -Boc-Ile•0,5H2O, N- α -Boc-Asp(Bzl), N- α -Boc-Leu•H2O и N- α
Boc-DL-Bhg. Типичный режим сочетания с отдельным аминокислотным остатком заключается в следующем (согласно справочнику ABI):
1) 33%-ная трифторуксусная кислота в
дихлорметане в течение 80 с,
2) 50%-ная трифторуксусная кислота в дихлорметане в течение 18,5 мин,
3) трехкратная промывка дихлорметаном,
4) 10%-ный N,
N'-диизопропилэтиламин в диметилформамиде в
течение 1 мин,
5) 10%-ный N,N'-диизопропилэтиламин в диметилформамиде в течение 1 мин,
6) пятикратная промывка диметилформамидом.
7) осуществление сочетания,
8) пятикратная промывка дихлорметаном.
После завершения сочетания N- α -Boc-DL-Bhg защитную трет.-бутилоксикарбонильную группу снимают вместе с концевой аминогруппой. Получаемый пептид отделяют от твердого носителя и защитную группу карбоксилата аспарагиновой кислоты снимают путем обработки 9 мл безводного фтористого водорода, 0,5 мл анизола и 0,5 диметилсульфида при температуре 0oC в течение 60 мин. После удаления фтористого водорода в потоке азота получаемую смолу три раза промывают диэтиловым эфиром, взятым в количестве по 30 мл, и последовательно экстрагируют три раза 20%-ной уксусной кислотой в воде, взятой в количестве 30 мл, и два раза ледяной уксусной кислотой, взятой в количестве по 30 мл. Водные экстракты объединяют, сгущают под пониженным давлением и лиофилизуют. Получают 360 мг сырого пептида, который растворяют в 4 мл смеси 50%-ной трифторуксусной кислоты и воды, фильтруют и подвергают хроматографии на имеющей размеры 2,2 • 25,0 см колонке марки Выдак 218ТП 1022 в следующих условиях: скорость подачи 15 мл/мин, элюент А: 0,1%-ная трифторуксусная кислота в воде, элюент Б: 0,1%-ная трифторуксусная кислота в ацетонитриле, градиент: 0% Б в течение 10 мин, 10 - 40% Б в течение 120 мин. Две отдельные фракции собирают и на основании анализа высокопроизводительной жидкостной хроматографии объединяют. Объединенные фракции отдельно сгущают под пониженным давлением, разбавляют 50 мл воды и лиофилизуют. Получают 40 мг рацемата, который разделяют на два диастереомера (изомеры А и Б) в указанных условиях (время удерживания: изомер А 15,63 мин, изомер Б 16,79 мин). Позднее стекающие пиковые фракции (изомер Б) повторно очищают в тех же условиях с соблюдением градиента 30 - 50% элюента Б в течение 120 мин при скорости подачи 15 мл/мин. 20 мг изомера Б подвергают ацетилированию в 90%-ной уксусной кислоте с последующим добавлением 5 мл уксусного ангидрида и перемешиванием в течение ночи. В результате упаривания и сушки получают Ac-D-Bhg-Leu-Asp-Ile-Ile-Trp со степенью чистоты 99% (согласно данным высокопроизводительной жидкостной хроматографии на вышеупомянутой колонке при соблюдении следующих условий: скорость подачи 15 мл/мин, элюент А: 0,1%-ная трифторуксусная кислота в ацетонитриле, градиент: 20 - 86% элюента Б в течение 22 мин). При этом время удерживания составляет 18,66 мин. Гомогенность и структура получаемого пептида подтверждены аналитической высокопроизводительной жидкостной хроматографией, а также анализом H1-ЯМР и масс-спектроскопией (бомбардировка быстрыми атомами): М + 1 972,0; М Na+ 995,9.
Получение исходных соединений.
Пример 15. (4R, 5S)-4-метил-5-фенил-2-оксазолидинон.
В круглодонную колбу, снабженную перегонным аппаратом, подают 18,16 г (99,1 ммоль) (1S,2R)-норэфедрина, 27,6 мл (228 ммоль) диэтилкарбоната и 28, 9 г (209 ммоль) карбоната калия, после чего реакционную смесь нагревают при температуре 160oC на масляной бане. Температуру верха перегонного аппарата держат примерно при 80oC, когда этанол собирают в приемной колбе, которую охлаждают на ледяной бане. Когда температура верха снижается до 60oC (примерно через 5 ч), масляную баню удаляют и реакционную смесь охлаждают до комнатной температуры, разбавляют дихлорметаном, два раза промывают водой и сушат над сульфатом магния. В результате сгущения в вакууме получают 17,88 г беловатого твердого вещества, которое перекристаллизовывают из смеси гексана и этилацетата в соотношении 1 : 1,5. При этом получают 15,2 г вышеуказанного целевого соединения в качестве белых кристаллов с точки плавления 120 - 121oC. [α]D= 171,4° (c = 2,042% в хлороформе).
Пример 16. 2,4,6-триизопропилбензолсульфонилазид.
К перемешиваемому раствору 18,2 г (60 ммоль) 2,4,6-триизопропилбензолсульфонилхлорида в 70 мл ацетона добавляют раствор 4,2 г (60 мкмоль) азида натрия в 40 мл смеси этанола и воды в соотношении 1 : 1. При этом температура реакционной смеси повышается от 21 до 29oC. После перемешивания при комнатной температуре в течение 2 ч реакционную смесь распределяют между дихлорметаном и полунасыщенным солевым раствором. Водный раствор три раза экстрагируют дихлорметаном, объединенный органический раствор промывают полунасыщенным солевым раствором и сушат над сульфатом магния. В результате удаления растворителя в вакууме получают бесцветное масло, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси гексана и этилацетата в соотношении 4 : 1. При этом получают 18,5 г вышеуказанного целевого соединения в качестве белого твердого вещества с точкой плавления 44 - 44,5oC (согласно данным Д. Эвенс и др. Journal of the American Chemical Society, 112 : 4011, 1990, точка плавления составляет 43 - 44o C).
Пример 17. Полученное по примеру 6 на стадии Б твердое вещество растворяют в 3 мл метанола с температурой 60oC, после чего добавляют 1 мл 6 н. соляной кислоты. Получаемому раствору дают охлаждаться до комнатной температуры и подают в холодильник, имеющий температуру 2oC. Получаемый осадок, представляющий собой рацемат, фильтруют. Из фильтрата отгоняют растворитель и получаемый остаток сушат в вакууме 10 мм рт.ст. при температуре 50oC. В результате повторного осуществления описанной перекристаллизации получают 0,15 г целевого продукта с показателем вращения [α]D= +45° (c = 1% в метаноле).
Реферат
D- и L-энантиомеры производных α -амино-10, 11-дигидро-5H-дибензо [a, d] циклогептен-5-уксусной кислоты получают взаимодействием рацемата с (-)цинхонидином в среде растворителя, полученный продукт разделяют фракционной кристаллизацией на D- и L-энантиомеры. Полученные новые соединения можно применять для получения биологически активных пептидов, которые представляют собой антагонист эндотелина, применяемый для лечения гипертензии, инфаркта миокарда, метаболических, эндокринологических и неврологических расстройств. 3 с. и 8 з.п. ф-лы.
Формула
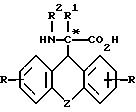
где Z - остатки -O-, -S(O)n-, где n = 0 - 2, -(CH2)m-, где m = 1 - 4, -(CH2)n - CH = CH(CH2)n-, где n имеет указанное значение;
R - водород, метил, трифторметил, метокси, гидроксил, хлор, бром, фтор, йод, 2,4-дибром, 2,4-дихлор или 2,4-дифтор;
R1 - водород, алкил; R2 - водород, бензилоксикарбонил, трет.-бутилоксикарбонил, флуоренилоксикарбонил, 1-адамантилоксикарбонил, 2-адамантилоксикарбонил,
в виде рацемата или энантиомеров D и L или их фармацевтически приемлемые соли, за исключением рацемического соединения формулы I, где Z означает -0-, R, R1 и R2 - водород.
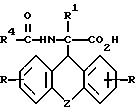
где R, R1 и Z имеют значения, указанные в п.1, а R4 - низший алкил, группа СХ3, где Х - водород, галоид или арил,
подвергают взаимодействию с (-) цинхонидином в среде растворителя, полученное соединение общей формулы III
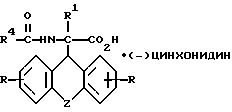
где R, R1, R4 и Z имеют указанные значения,
разделяют фракционной кристаллизацией на D-энантиомер формулы IIIa
где R, R1, R4 и Z имеют указанные значения,
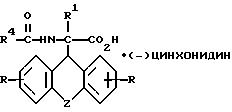
и L-энантиомер формулы IIIб
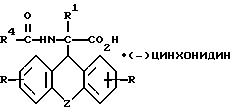
где R, R1, R4 и Z имеют указанные значения,
затем D- или L-энантиомер подвергают взаимодействию с кислотой в среде растворителя с получением D-энантиомера формулы IIa или L-энантиомера формулы IIб
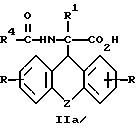
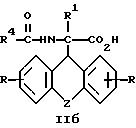
где R, R1, R4 и Z имеют указанные значения, которые подвергают взаимодействию с кислотой с последующим выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли.
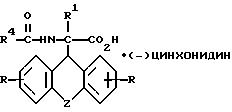
где R, R1 и Z имеют значения, указанные в п.1;
R4 - низший алкил, группа СХ3, где Х - водород, галоид или арил,
в виде цинхонидиновых солей в качестве промежуточных соединений для синтеза производных аминоуксусной кислоты формулы I по п.1.
Комментарии