Производные {[2- (амино -3,4- диоксо -1- циклобутен -1-ил) амино] алкиловых}кислот - RU2039035C1
Код документа: RU2039035C1
Чертежи


Описание
Возбудительные аминокислоты такие, как глютаминовая кислота, как было установлено, являются важными нейропередатчиками (Джонсон, Р.Л. Кернер, Дж. Ф. J.Med.Chem. 1988, т. 31, стр. 2057), которые в избытке принимают участие в последовательности событий, приводящих к нейронным нарушениям после церебральной ишемии (Чой, Д.У. Trends Neurosci, 1988, т. II, стр. 465). Одним из важнейших подтипов рецептора возбудительной аминокислоты является НМДА-рецептор, который определяется выбранным агонистом, N-метил-D-аспартовой кислотой (NMDA). Блокирование действия эндогенного агониста выбранным антагонистом НМДА-рецептора, 4-(3-фосфонпропил)-2-пиперазинкарбоновой кислотой (СРР), как было установлено, предотвращает ишемические нарушения в головном мозге у песчанок (Боаст, К.А. и др. Brain Research 1988, т. 442, стр. 345). Кроме того НМДА-индуцированные конвульсии могут быть предотвращены при помощи СРР у мышей (Леман, Дж. и др. J. Pharmacol Exp. Ther. 1987, т. 240, стр. 737). Наконец, конкурирующие НМДА-антагонисты такие, как СРР, как было установлено предотвращают симптомы, напоминающие заболевание Паркинсона, индуцированные при помощи МРТР (1-метил-4-фенил-1,2,3,6-тетрагидропиридина) у крыс (Турский, Л. и др. Nature 1991, т. 349, стр. 414). По этим причинам НМДА-рецепторные антагонисты рассматриваются в качестве подходящих материалов для лечения эпилепсии, удара (Ингелсен, Б. Acta Neurol Scand 1986, т. 74, стр. 337) и нейтродегенеративных нарушений таких, как заболевание Элзхаймера (Марагос, У. Ф. и др. Trend Neurosci 1987, т. 10, стр. 65) и заболевание Паркинсона.
Химические материалы, известные в качестве конкурирующих антагонистов НМДА-рецепторов, содержат функциональности α-амино-карбоновой кислоты и алкилфосфиновой кислоты, которые разделены совокупностью блоков ("распорок"). "Непреукрашенным" примером является 2-амино-5-фосфонвалериановая кислота (АР5) [1] (Уоткинс, Дж.К. Эванс, Р.Х. Annu. Rev. Pharmacol. Toxicol 1981, т. 21, стр. 165), которая содержит насыщенную углеродную цепь. Более сложные примеры, которые содержат элементы, увеличивающие структурную жесткость и, следовательно, мощность воздействия, включают СРР (см. выше), цис-4-(фосфонметил)-2-пиперидинкарбоновую кислоту (СGS-19755) (Леман, Дж. и др. J. Pharmacol Exp. Ther. 1988, т. 246, стр. 65) [2] и (Е)-2-амино-4-метил-5-фосфон-3-пентеновую кислоту (СGP-37849) (Шмутц, М. и др. Abs. Soc. Neurosci. 1988, т. 14, стр. 864) [3] Хотя были предприняты усилия найти группы, которые являются биоизостерическими с группой алкилфосфоновой кислоты (Ченард, Б.Л. и др. J.Med.Chem. 1990, т. 33, стр. 1077), ни одного примера антагонистов НМДА-рецептора не появилось в литературе, который бы подтверждал биоизостерическое замещение функциональности α-аминокарбоновой кислоты.
В соответствии с изобретением
предлагается группа N-замещенных производных 3,4-диамино-3-циклобутен-1,2-диона, которые являются НМДА-антагонистами, которые "узнаются" соответствующими рецепторами и которые предотвращают
НМДА-индуцированную летальность ин виво. Эффективные при использовании в качестве противоконвульсивных агентов и нейрозащищающих агентов в случаях, сопровождающихся избыточным высвобождением
возбуждающих аминокислот, соединения, являющиеся предметом изобретения, представляются структурной формулой I

R2 является водородом, алкилом с 1-6 атомами углерода, алкенилом с 2-6 атомами углерода или фенилалкилом с 7-12 атомами углерода; или
R1 и R2, взятые вместе, образуют Z, которым являются -СН2СН2-, -СН2С(R6)(R7)CH2- или -CH2C(R8)(R9)-C(R10)(R11)CH2-, где R6, R8 и
R10являются независимо друг от друга водородом, алкилом с 1-6 атомами углерода, или гидроксилом, а R7, R9 и R11 являются независимо друг от друга водородом или алкилом, содержащим от 1 до 6 атомов углерода;
А является алкиленом с 1-6 атомами углерода или алкениленом с 2-6 атомами углерода;
Х является СО2R3 , в которой R3 является водородом или алкилом с 1-6 атомами углерода, Р(О)(OR4)OR5), в которой R4 и R5 являются независимо друг от друга водородом или алкилом с 1-6 атомами углерода, 3,5-диоксо-1,2,4-оксадиазолидин-2-илом или 5-тетразолилом; или их приемлемые с фармацевтической точки зрения соли.
Примерами алкила для R1-R5 и R6-R16 см. выше, являются линейные или разветвленные группы такие, как метил, этил, н-пропил, изопропил, бутил и гексил. Предпочтительные алкильные группы содержат от 1 до 4 углеродов. Примерами алкенила для R2 являются линейные или разветвленные группы такие, как винил, проп-1-енил, алкил, металлил, бут-1-енил, бут-2-енил и бут-3-енил.
Примерами фенилалкильных групп для R1 и R2 являются такие группы, в которых алкильная составляющая является линейной или разветвленной цепью такой, как бензил, фенэтил, 3-фенил-пропил, 4-фенил-бутил. В предпочтительном варианте алкильная составляющая такой группы содержит 1-4 атома углерода.
Предпочтительными значениями для R1 и R2 являются, независимо друг от друга, водород, метил, аллил, металлил и бензил, а когда R1 и R2, взяты вместе, как Z, они являются -СН2СН2-, -СH2C(R6)(R7)CH2- или -СН2С(R8)(R9)-C(R10)(R11)CH2-, где R6, R8 и R10 независимо являются водородом, алкилом с 1-6 атомами углерода или гидроксилом, а R7, R9 и R11 независимо друг от друга являются водородом или алкилом с 1 до 6 атомам и углерода. Примерами алкиленовых групп для А являются линейные или разветвленные группы, в предпочтительном варианте группы, содержащие от 1 до 4 атомов углерода, такие, как: -СН2-, -СН2СН2-, -СН(СН3)-СН2, -СН2СН(СН3)-, -(СН2)3- и -(СН2)4-. Примерами алкениловых групп для А являются цис и транс-группы, в предпочтительном варианте содержащие 2-4 атомов углерода такие, как -СН2-СН= СН-, -СН= С(СН3)-, -С(СН3)= СН, -СН=СН-СН2- и -СН2--СН=СН-СН2- и -СН2-СН= С(СН3)-. В предпочтительном варианте А является алкиленом с 1-4 атомами углерода или транс-2-бутиленом. Предпочтительными значениями для Х является карбоксил, фосфонил или 5-тетразолил.
Приемлемые с фармацевтической точки зрения соли соединений, являющихся предметом изобретения, включают соли щелочных металлов (натрий, калий или литий), щелочно-земельных металлов (кальций или магний) и соли аммония.
Кроме того, в соответствии с изобретением предлагается способ получения соединений формулы I.
Поэтому в соответствии с изобретением предлагается способ получения соединения формулы I, который включает:
а) взаимодействие соединения формулы II

HNR2-A-X, в которой R2, А и Х были определены выше, или его солью, или его защищенной формой, если необходимо удалить любую защищающую группу, чтобы получить соединение формулы I, где R2 был определен выше, если необходимо изолировать в форме приемлемой с фармацевтической точки зрения или
b) взаимодействие соединения формулы IV

Н2NR1, где R1 был определен выше, и удаление любой защищающей группы, чтобы получить соответствующее соединение формулы I или его соль; или
с) взаимодействие соединения формулы VI

H2N-Z-NH-A-X, где Х определен выше, или его солью, или его защищенной формой, Z и A уже были определены выше, если требуется удаление любой защищающей группы, чтобы получить соответствующее соединениe, имеющее формулу (I), в которой R1 и R2, взятые вместе, являются Z, А и Х, были определены выше; или
d) снятие защиты, селективное, если это необходимо, и циклизация соединения формулы VIII

е) взаимодействие соединения формулы IX
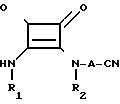
f) превращение соединения формулы (I), в которой R3, R4 и R5являются алкильными группами, чтобы получить соединения формулы (I), в которой R3, R4 и R5 являются водородом, или их соли; или
g) подкисление соли соединения формулы I, чтобы получить свободную кислоту или подщелачивание кислоты формулы I, чтобы получить ее соль; или
h) термолиз соединения формулы X
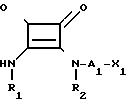
В некоторых из упомянутых реакций, когда желательно получить Х в форме карбокси или алкилфосфиновой кислоты, может оказаться необходимым или важным защитить такие группы во время реакции или использовать его производное С1-С6 алкилового сложного эфира, например, трет-бутилового сложного эфира, а затем их удалить. Примерами карбокси защищающих групп являются бензиловые сложные эфиры или сложные эфиры с 7 и более атомами углерода.
Аналогично, когда Х является 5-тетразолилом, эта группа может быть защищена при помощи стандартных приемов, например, используя тритиловую защищающую группу, которая может быть удалена подкислением или гидрогенизацией. Кроме того, когда R6, R8 и R10 являются окси, соответствующая защита во время реакции может быть осуществлена октильной, бензоильной, трет-бутильной, тритильной, а в предпочтительном варианте бензильной группой.
Что касается способа а), то вытеснение алкокси или аралкокси-заместителя из соединения формулы II может быть соответствующим образом осуществлено в отсутствие основания или с основанием таким, как гидрат окиси щелочного металла (например, NaOH) при нагревании или без него в присутствии инертного растворителя такого, как этанол. Когда Х является кислотной составляющей в соединении формулы III при щелочных реакционных условиях, продуктом будет соль, которая может быть выделена как таковая или превращена в свободную кислоту при помощи подкисления. Когда Х является функцией карбоновой кислоты, она может быть защищена, например, в форме сложного эфира или как соль с катионом.
Что касается способа б), то замещение OR12-группы может быть осуществлено с использованием аммиака или амина формулы Н2NR2 в присутствии инертного растворителя такого, как этанол с нагреванием или без него, группу Х защищают, если это необходимо.
Что касается способа с), то бициклические соединения формулы I могут быть получены при помощи осуществления двух реакций замещения, используя диамин формулы VII, в предпочтительном варианте при помощи нагревания инертного растворителя такого, как этанол. Если азот защищен, то реакцию можно осуществить в две стадии через промежуточное соединение формулы VIII с последующим снятием защиты и циклизацией, как это описано в способе d). Примерами амино-защищающих групп являются бензилоксикарбонил и его замещенные производные, удаляемые при помощи гидрогенизации.
Способ е) может быть осуществлен с использованием азида щелочного металла в инертном растворителе таком, как диметилформамид в присутствии хлорида аммония при нагревании, если оно необходимо. Если используют азид трибутилолова, то реакцию можно осуществить в инертном растворителе таком, как толуол, затем осуществляют подкисление.
Что касается процесса f), то когда любой из R3 или R4 и R5 является алкильной группой, то они могут быть удалены при помощи гидролиза и других известных приемов, чтобы получить соответствующие свободные кислоты или их соли.
Термолиз соединений формулы Х может быть осуществлен при помощи нагревания инертного растворителя с высокой температурой кипения такого, как толуол в присутствии бикарбоната натрия для того, чтобы получить соединения формулы I, в которой А является алкениленом. Аналогичным образом, этот способ может быть использован для того, чтобы получить исходные материалы, используемые в описанном способе, где это желательно, чтобы получить соединения, в которых А представляет алкенилен.
В соответствии с изобретением
предлагаются также промежуточные соединения для получения соединений формулы (I). К этим промежуточным соединениям относятся соединения, имеющие формулы IV, VIII, которые приведены и определены выше,
и, кроме того, промежуточные соединения для соединений формулы IV, которые являются промежуточными соединениями, имеющими формулу XI
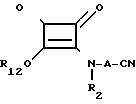
Эти промежуточные соединения могут быть представлены одной общей формулой XII

где Х2 представляет СN, СООR14, где R14 является водородом, алкилом с 1-6 атомами углерода или карбоксильной защищающей группой, -РО(OR15)(OR16), в которой R15 и R16 являются водородом или алкилом с 1-6 атомами углерода, 3,5-диоксо-1,2,4-оксадиазолидинилом или, возможно, защищенным 5-тетразолилом, R12 и А уже были определены выше, а R13представляет водород, алкил с 1-6 атомами углерода, алкенил с 2-6 атомами углерода, фенилалкил с 7-12 атомами углерода или -Z-NHP, где Р является амино-защищающей группой; Z определен выше.
Кроме того, в соответствии с изобретением предлагается способ получения промежуточных соединений
формулы VI, которая определена выше, с соединением формулы XIII
HR13N-A-X2 где А, R13 и Х2 определены выше, чтобы получить соответствующее
соединение формулы XII, или
N-алкилирование соединения формулы

Исходные материалы, используемые в любом из упомянутых способов для получения финальных продуктов и промежуточных соединений, являются соединениями или они могут быть получены при помощи приемов, аналогичных для известных соединений.
Соединения
изобретения получают при помощи известных приемов, включающих (I) замещение алкокси-заместителя 3-амино-4-алкокси-3-циклобутен-1,2-диона аминокислотой (Н2NAX) в присутствии эквивалентной
массы гидрата окиси натрия, чтобы получить производные, в которых R2 является водородом;
(2) взаимодействие 3,4-диалкокси-3-циклобутен-1,2-диона с амином (Н2NR2) с последующим N-алкилированием с необходимым трет-бутилгалокарбоксилатом (галоАСО2-трет-бутилом) в диметилформамиде, используя гидрид натрия в качестве основания с последующим
замещением оставшегося алкокси-заместителя при помощи необходимого амина (Н2NR1) и снятием защиты трет-бутилового сложного эфира кислотой такой, как муравьиная кислота и (3)
аналогичным образом, 3,4-диалкокси-3-циклобутен-1,2-дион может взаимодействовать с соответствующей аминокислотой (H2N-A-PO3Et2), чтобы заместить одну алкокси группу, а
это промежуточное соединение может быть N-алкилировано в диметилформамиде с соответствующим алкилгалоидом (R2-гало) в присутствии гидрида натрия и/или другой алкокси-заместитель может быть
замещен соответствующим амином (Н2NR1). Полученный в результате сложный эфир фосфоната превращают в свободную кислоту при помощи бромтриметилсилана. Используя диамин такой, как
Н2N-(СН2)n-NHCH2CH2PO3Et2 (n= 2-4), с 3,4-диэтокси-3-циклобутен-1,2-дионом получают сложные эфиры
диоксодиазабициклоэтилалкилфосфоновой кислоты такие, которые проиллюстрированы в примере 7, см. ниже. Сложный эфир фосфоната превращают в свободную кислоту при помощи известного приема. Исходные
диаминовые реагенты получают в результате взаимодействия N-защищенных алкилендиаминов с диэтиловым сложным эфиром (2-оксоэтил/алкилфосфоновой кислоты в присутствии цианоборогидрида натрия в метаноле
при рН ≈ 6,5 с последующим удалением N-защищающей группы такой, как бензилоксикарбонил при помощи гидрогенизации.
Кроме того, (4) исходные моно-защищенные диаминфосфонатные
реагенты получают при помощи взаимодействия N-защищенных алкилендиаминов с Br-A--PO3Et2 в присутствии карбоната натрия в этаноле. Монозащищенный диаминфосфонат такой, как
(защищающая группа (-NH-(CH2)n-NH-A-PO3Et2(n=2-4) взаимодействует с 3,4-диалкокси-3-циклобутен-1,2-дионом, чтобы вытеснить одну алкоксигруппу. Удаление
бензилоксикарбонил N-защищающей группы при помощи гидрогенизации или трет-бутилоксикарбонил N-защищающей группы при помощи обработки муравьиной кислотой приводит к циклизации. Полученные в результате
сложные эфиры диоксодиазабициклоалкилалкилфосфоновой кислоты подвергают гидролизу в свободную кислоту при помощи бромтриметилсилана;
(5) моно N-бензилоксикарбонил диамин может быть
алкилирован при помощи Br-A-CO2-трет-Bu в присутствии диизопропилэтиламина в диметилформамиде. Промежуточное соединение, СВZ-NH-(СН2)n-NH-A-CO2-трет-Bu,
взаимодействует с 3,4-диалкокси-3-циклобутен-1,2-дионом, чтобы вытеснить алкокси группу. Удаление N-защищающей группы при помощи гидрогенизации промежуточного соединения приводит к циклизации.
Трет-бутиловую группу расщепляют при помощи муравьиной кислоты, чтобы получить свободную кислоту; и (6) моно N-трет-бутилоксикарбонилдиамин может быть алкилирован с Вr-А-СN при помощи известного
приема. Промежуточное соединение ВОС-NH-(СН2)n-NH-A-CN взаимодействует с 3,4-диалкокси-3-циклобутен-1,2-дионом, чтобы вытеснить одну алкоксигруппу. Удаление N-защищающей группы
при помощи обработки муравьиной кислотой приводит к циклизации. Нитрил превращают в тетразол при помощи нагревания с азидом натрия и хлоридом аммония в диметилформамиде. Если алкиленовый мостик
диамина содержит гидроксильную группу, ее можно защитить на эту реакцию.
Во всех упомянутых реакциях реагенты являются либо известными, либо доступными на рынке, либо их можно получить легко при впомощи приемов, известных каждому специалисту в области медицинской химии.
Соединения изобретения могут содержать один или несколько асимметричных атомов углерода (например, когда любой из R6-R10 является алкилом или любой из R6, R8 и R10 является гидроксилом), поэтому предлагаемые соединения могут существовать в различных стереоизомерических формах. Все стереоизомерические формы включены в область притязании изобретения. Такие соединения могут быть, например, рацематами оптически активных форм. Оптически активные формы могут быть получены в результате разделения рацематов или при помощи асимметрического синтеза.
П р и м е р 1. N-(2-амино-3,4-диоксо-1-циклобутен-1-ил)бета-аланин.
Раствор 3-амино-4-этокси-3-циклобутен-1,2-диона (2,0 г, 14 ммолей) в этаноле (100 мл) обрабатывали β-аланином (1,26 г, 14 ммолей), растворенным в 1 н. растворе гидрата окиси натрия
(14 мл, 14 ммолей). Спустя 5,5 ч при комнатной температуре образовавшийся желтый твердый материал фильтровали, промывали этанолом и концентрировали под высоким вакуумом, чтобы получить чистое
соединение из заголовка примера в виде соли натрия, полугидрата (2,6 г, 86% от теоретического, температура точки плавления 280-282оС);
ИК (КВr, см-1): 1810; МС (-FAB)
205 (М-Н, 13), 183 (М-Na, 44), 175 (17), 148 (100);1Н-ЯМР (D2O, 400 МГц): δ 3,59 (широкий синглет, 2Н), 2,31 (триплет, J=6 Гц, 2Н);
13С-ЯМР (D2О, 400 МГц): долей на миллион 182,01, 181,61, 179,30, 168,94, 168,54, 41,18, 37,97.
Вычислено, С 39,08; Н 3,75; N 13,02.
С7Н7NaN2
O4 1/2 H2O
Найдено, C 38,78; Н 3,48; N 12,86.
а) При помощи такой же процедуры, конденсируя стехиометрические количества 3-амино-4-этокси-3-циклобутен-1, 2-диона с этилгликоколатом, получают этиловый сложный эфир N-(2-амино-3,4-диоксо-1-циклобутенил)гликокола, температура точки плавления 231-233оС.
Вычислено, С 48,49; Н 5,09; N 14,14.
С8Н10N2O4
Найдено, С 48,40; Н 4,90; N 14,02.
b) Замена гликоколом β -аланина в процедуре из примера 1 дает N-(2-амино-3,4-диоксо-1-циклобутенил)гликокол в виде соли натрия, 4/3 Н2О, температура точки плавления 210-215оС (разлож.).
Вычислено, С 33,34; Н 3,58; N 12,96.
С6Н5NaN2O4 4/3 Н2О
Найдено, С 33,36; Н 3,26; N 13,12.
с) Аналогичным образом, замена 4-аминобутановой кислотой β -аланина в процедуре из примера 1 дает 4-[(2-амино-3,4-диоксо-1-циклобутенил/амино]бутановую кислоту в виде соли натрия, частичного гидрата, температура точки плавления 240-243оС (разлож.).
Вычислено, С 41,67; Н 4,44; N 12,15.
С8Н9NaN2O4 0,58 Н2О
Найдено, С 41,27; Н 4,04; N 12,03.
П р и м е р 2. 2-[2-[(2-Амино-3,4-диоксо-1-циклобутен-1-ил)амино]этил] 1,2,4-оксади- азолидин-3,5-дион.
Раствор
3-амино-4-этокси-3-циклобутен-1,2-диона (0,56 г, 4,0 ммоля) в этаноле (20 мл) добавляли в бромгидрат 2-(2-аминоэтил)-1,2,4-оксадиазолидин-3,5-диона
(0,90 г, 4,0 ммолей) в этаноле (100 мл).
Реакционную смесь обрабатывали 1 н. раствором гидрата окиси натрия (8 мл, 8 ммолей) и перемешивали в течение 24 ч при комнатной температуре. Полученный осадок фильтровали, растворяли в воде и
пропускали через ионообменную колонну (АГ 50W-Х2, 100-200 меш (0,149-0,074 мм), Н+-форма), элюируя водой. Элюент сушили замораживанием, в результате чего получали соединение из заголовка
примера в виде твердого вещества кремового цвета, частичного гидрата (0,45 г, 45% температура точки плавления 225оС (разлож.),
ИК (КВr, см-1) 3300, 3140, 1820, 1740,
1720, 1640; МС (+FAB) 241 (МН+);1Н ЯМР (ДМСО, 400 МГц): δ 12,4 (широкий синглет, NH), 7,5 (широкий синглет, 3NH), 4,0-3,5 (мультиплет, 4Н);13С-ЯМР (ДМСО, 400
МГц): долей на миллион 183,72, 183,63, 170,06, 168,96, 158,17, 152,72, 50,41, 41,68.
Вычислено, C 38,70; Н 3,61; N 22,56.
С8Н8N4О5 ˙0,45 Н2О
Найдено, 39,10; Н 3,24; N 22,19.
а) Те же реакционные условия, что описаны в примерах 1 и 2, примененные к β аланину и 4-этокси-3-метиламино-3-циклобутен-1,2-диону, давали N-[2-(метиламино)-3,4-диоксо-1-циклобутенил] бета-аланин в виде соли натрия, 1/4 Н2О, температура точки плавления 310оС (разлож.).
Вычислено, С 42,77; Н 4,26; N 12,47.
С8Н9NaN2O4 ˙1/4H2О
Найдено, С 42,77; Н 3,88; N 12,
53.
b) Аналогично, 3-бензиламино-4-этокси-3-циклобутен-1,2-дион, когда он взаимодействовал с β -аланином, дает 3[[3,4-диоксо- 2[(фенилметил)амино]-1-циклобутенил] амино] пропановую кислоту в виде натриевой соли, 1/2 Н2О, температура точки плавления 298-302оС (разлож.).
Вычислено, С 55,08; Н 4,62; N 9,18.
С14Н13NaN2O4 ˙1/2 Н2О
Найдено, C 54,74; Н 4,53; N 9,06.
П р и м е р 3. N-(2-Амино-3, 4-диоксо-1-циклобутен-1-ил)-N-(2-пропенил)-гликокол.
Раствор 3,4-диэтокси-3-циклобутен-1,2-диона (5,2 г, 31 ммолей) в этаноле (80 мл) обрабатывали при комнатной температуре аллиламином
(2,3 мл, 31 ммоль), который растворяли в этаноле (40 мл), в течение 2 ч. Реакционную смесь концентрировали под вакуумом, чтобы получить сырой 1-(2-пропениламино)-2-этокси-3,4-диоксо-1-цик- лобутен в
виде светло-желтого твердого вещества (5,6 г). Сырое промежуточное соединение растворяли в безводном диметилформамиде (50 мл) и по каплям добавляли в суспензию 60% гидрида натрия (1,5 г, 37 ммолей) в
безводном диметилформамиде (50 мл) в атмосфере азота. Анион быстро охлаждали трет-бутил бромацетатом (6,0 мл, 37 ммолей) и реакционную смесь перемешивали 1,5 ч, сливали в воду (500 мл), экстрагировали
этилацетатом (2х200 мл) и сушили (сульфат магния), чтобы получить 1,1-диметилэтиловый сложный эфир N-(2-этокси-3,4-диоксо-1-циклобутен-1-ил)-N-(2-пропенил)- гликокола. Очистку осуществляли при помощи
оперативной хроматографии (диаметр 10 см, элюирование 20% этилацетатом в петролейном эфире), после чего получали желтое масло (4,56 г, 50%);
1Н-ЯМР (CDCl3, 300 МГц):
δ 5,88-5,72 (мультплет, 1Н), 5,35-5,22 (мультплет, 2Н), 4,80-4,68 (мультплет, 2Н), 4,35, 4,08 (дублет, J=7 Гц, 2Н), 4,28, 3,95 (синглет, 2Н), 1,48 (синглет, 9Н), 1,45 (триплет, J=7 Гц, 3Н).
Этаноловый раствор аммиака (25 мл) добавляли в колбу, содержащую 1,1-диметилэтиловый сложный эфир N-(2-этокси-3,4-диоксо-1-циклобутен-1-ил)-N-(2-пропенил)- гликокола (2,5 г, 8,5 ммолей), при комнатной температуре. Спустя 5 ч реакционную смесь концентрировали и подвергали очистке при помощи оперативной хроматографии (5 см диаметром, элюирование 5% метанолом в дихлорметане), чтобы получить 1,1-диметил-этиловый сложный эфир N-(2-амино-3,4-диоксо-1-циклобутен-1-ил)-N-(2-пропенил)гликокола в виде белого твердого вещества (1,6 г, 71% температура точки плавления 175-176о С).
ИК (КВr, см-1): 3300, 3140, 1810, 1740, 1670, 1650; МС (Е1): 266 (М+, 34), 210 (24), 165 (100), 109 (54), 95 (89), 68 (68);1Н-ЯМР (ДМСО, 400 МГц): δ 6,70 (широкий синглет, NH2), 5,84-5,77 (мультиплет, 1Н), 5,26 (дублет, J=17 Гц, 1Н), 5,19 (дублет, J=10 Гц, 1Н), 4,3-4,0 (широкий мультиплет, 4Н), 1,39 (синглет, 9Н).
Снятие защиты с 1,1-диметил этилового сложного эфира N-(2-амино-3,4-диоксо-1-циклобутен-1-ил)-N-(2-пропенил)гликола (1,6 г, 6,0 ммолей) осуществляли при помощи перемешивания в муравьиной кислоте (20 мл) в течение 24 ч. Реакционную смесь концентрировали, подвергали азеотропии дихлорметаном и сопровождающейся трудностями рекристаллизации (при этом несколько раз удаляли масло), чтобы получить соединение из заголовка примера, 1/4 гидрат, в виде не совсем белого твердого вещества (0,80 г, 62% температура точки плавления 172-175оС).
ИК (КВr, см-1 ): 3330, 3180, 1810, 1720, 1640; МС (Е1): 210 (М+, 75), 165 (34), 109 (41), 95 (100), 68 (63).
1Н-ЯМР (ДМСО, 400 МГц): δ 12,94 (широкий синглет, ОН), 7,70 (синглет, NH2), 5,86-5,77 (мультиплет, 1Н), 5,26 (дублет, J=17 Гц, 1Н), 5,19 (дублет, J=10 Гц, 1Н), 4,3-4,0 (широкий мультиплет, 4Н).
Вычислено, С 50,35; Н 4,93; N 13,05.
С9Н10N2O4 ˙1/4 Н2О
Найдено, С 50,13; Н 4,82; N 12,86.
а) Следуя процедуре из примера 3, за тем исключением, что 2-метилаллиламин использовали в качестве исходного реагента, получали N-(2-амино-3,4-диоксо-1-циклобутенил)-N-(2-метил-2-пропенил)гликокол, температура точки плавления 184-186оС.
Вычислено, С 53,14; Н 5,44; N 12,39.
C10Н12N2О4 ˙0,1 Н2О
Найдено, C 53,09; Н 5,38; N 12,
16.
b) Аналогичным образом, используя бензиламин в качестве исходного реагента, по процедуре из примера 3 получали N-(2-амино-3,4-диоксо-1-циклобутенил)-N-(фе- нилметил)гликокол, температура точки плавления 177-179оС.
Вычислено, С 60,00; Н 4,65; N 10,76.
С13Н12N2O4
Найдено, С 59,74;
Н 4,60; N 10,61.
П р и м е р 4. [2-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино]этил] фосфиновая кислота.
В раствор 3,4-диэтокси-3-циклобутен-1,2-диона (4,00 г, 23,5 ммолей) в этаноле (100 мл) добавляли диэтиловый сложный эфир 2-аминоэтилалкилфосфиновой кислоты (5,43 г, 30,0 ммолей) в этаноле (100 мл) в течение 1 ч. После выдерживания в течение ночи реакционную смесь предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (5,5 см диаметр, градиентное элюирование при помощи 2,5-10% изопропанола в дихлорметане), чтобы получить диэтиловый сложный эфир [2-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)амино] этил] фосфиновой кислоты в виде масла, которое отверждается после выдерживания (3,98 г, 55% температуре точки плавления 66-68оС).
ИК (КВr, см-1): 3400, 3180, 1800, 1700, 1600; МС (+FAB): 306 (МН+, 100), 278 (14), 137 (14), 109 (35).
1 Н-ЯМР (СDСl3, 400 МГц): δ 6,58, 6,46 (широкий синглет, NH), 4,75 (широкий мультиплет, 2Н), 4,21-4,07 (мультиплет, 4Н), 4,00, 3,75 (широкий мультиплет, 2Н), 2,08 (дублет триплетов, J=17,5 и 6,5 Гц, 2Н), 1,46 (широкий мультиплет, 3Н), 1,35 (триплет, J=7 Гц, 6Н).
Раствор диэтилового сложного эфира [2-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)амино] этил] фосфиновой кислоты (1,69 г, 5,5 ммолей) в 100% этаноле (100 мл) помещали в колбу, снабженную воронкой и входным отверстием для азота. Насыщенный этано- ловый раствор аммиака (190 мл) помещали в капельную воронку и добавляли в течение 1 ч. Реакционную смесь перемешивали при комнатной температуре всего в течение 24 ч, а затем концентрировали под вакуумом. Полученное в результате твердое вещество подвергали рекристаллизации из метанола в этилацетате, чтобы получить диэтиловый сложный эфир [2-[(2-амино-3,4-диоксо-1-циклобутен-1-ил(амино] этил] фосфи- новой кислоты в виде желтого твердого вещества (27г, 82% температура точки плавления 150-152оС (разлож.)).
ИК (КВr, см-1): 1805, 1650; МС (+FAB): 277 (MH+, 100), 182 (20), 109 (15).
1Н-ЯМР (ДМСО, 400 МГц): δ 7,5 (широкий синглет, 3 NH), 4,1-3,9 (мультиплет, 4Н), 3,7-3,6 (мультиплет, 2Н), 2,11 (дублет триплетов, J=17,5 и 7,5 Гц, 2Н), 1,22 (триплет, 6Н).
Вычислено, С 42,92; Н 6,27; N 10,01.
C10H17N2O5P ˙1/5 H2O
Найдено, С 42,90; Н 6,05; N 10,00.
Cуспензию диэтилового сложного эфира [2-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино] этил] фосфиновой кислоты, одна пятая гидрат (0,90 г, 3,2 ммолей) в сухом 1,2-дихлорэтане (47 мл) помещали в колбу, которая снабжена дефлегматором и из которой предварительно откачивали воздух и заполняли азотом. В колбу через шприц добавляли бромтриметилсилан (2,6 мл, 19,8 ммолей) и реакционную смесь дефлегмировали 10 мин. Затем смесь концентрировали под вакуумом, чтобы получить твердое вещество цвета ржавчины, которое растворяли в деионизированной воде (80 мл). Воду промывали диэтиловым простым эфиром (2х100 мл) и концентрировали под вакуумом. Полученное в результате твердое вещество цвета ржавчины подвергали рекристаллизации из метанола и воды в этилацетате, чтобы получить соединение из заголовка примера в виде темно-желтого твердого вещества (0,360 г, 50% температура точки плавления 230-239оС (разлож.)).
ИК (КВr, см-1): 1790.
1Н-ЯМР (ДМСО, 400 МГц): δ 7,5 (широкий синглет, 3 NH), 3,67 (широкий синглет, 2Н), 1,85 (дублет триплетов, J=7,5 Гц, 2Н).
Вычислено, С 32,21; Н 4,24; N 12,52.
С6Н9N2O5P ˙1/5 H2O
Найдено, С 32,20; Н 4,00; N 12,46.
а) Следуя процедуре из примера 4 за тем исключением, что диэтиловый сложный эфир аминометилфосфиновой кислоты использовали в качестве реагента, получали [[(2-амино-3,4-диоксо-1-циклобутенил)ами- но]метил]фосфиновую кислоту в форме три четверти гидрата, температура точки плавления 220-250оС (разлож.).
Вычислено, С 27,35; Н 3,90; N 12,76.
С5Н7N2O5Р
˙3/4 Н2О
Найдено, С 27,72; Н 3,39; N 12,39.
b) Снова используя диэтиловый сложный эфир 3-аминопропилалкилфосфиновой кислоты в качестве реагента в процедуре из примера 4, получали [3-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино]пропил]фосфи- новую кислоту, температура точки плавления 220-230оС (разлож.).
Вычислено, С 36,91; Н 4, 74; N 11,96.
С7Н11N2O5P
Найдено, С 35,94; Н 4,57; N 11,76.
с) С диэтиловым сложным эфиром 4-аминобутилалкилфосфиновой кислоты в качестве реагента в процедуре из примера 4 получали [4-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино] бутилфосфиновую кислоту, 0,25 гидрат в качестве продукта, температура точки плавления 220-242оС (разлож.).
Вычислено, С 38,03; Н 5,38; N 11,09.
С8Н13N2O5P ˙1/4
Н2О
Найдено, С 38, 09; Н 5,01; N 11,09.
П р и м е р 5. [(Е)-4-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино]-2-бутенил] фосфиновая кислота.
Раствор
диэтилового сложного эфира [(Е)-4-(N-фталимидо)-2-бутен-1-ил] алкилфосфиновой кислоты (8,58 г, 25,4 ммолей) в оболочке азота получали по способу Коннела и др. J.Org.Chem. т. 54, стр. 3359 (1989) в
этаноле (75 мл), затем обрабатывали 85% гидрата гидразина (5 мл) и доводили до дефлегмации на 15 мин. Фактически твердую реакционную смесь концентрировали и разделяли между 2,5 н. раствором гидрата
окиси натрия (250 мл) и дихлорметаном (150 мл) при перемешивании в течение 30 мин. После разделения водный слой снова экстрагировали дихлорметаном (2х150 мл) и соединенные органические слои сушили
(сульфат натрия) и концентрировали, чтобы получить диэтиловый сложный эфир [(Е)-4-амино-2-бутен-1-ил]фосфиновой кислоты (5,32 г, 25 ммолей); этот материал растворяли в этаноле (100 мл) и добавляли в
течение 1,5 ч в этаноловый раствор (100 мл) 3,4-диэтокси-3-циклобутен-1,2-диона (4,32 г, 25,4 ммолей). После выдерживания в течение ночи реакционную смесь концентрировали и подвергали очистке при
помощи оперативной хроматографии (7,5 см диаметр, элюирование 3%-ным этанолом в дихлорметане), чтобы получить диэтиловый сложный эфир [(Е)-4-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)амино]-2-бутенил]
фосфиновой кислоты в виде светло-желтого масла (7,15 г, 85%);
1Н-ЯМР (СDCl3, 200 МГц): δ 6,5 (широкий синглет, NH), 5,75-5,66 (мультиплет, 2Н), 4,77 (квартет, J=7
Гц, 2Н), 4,23 (широкий синглет, 2Н), 4,2-4,0 (мультиплет, 4Н), 2,61 (дублет дублетов, J=22 и 7 Гц, 2Н), 1,46 (триплет, J=7 Гц, 3Н), 1,33 (триплет, J=7 Гц, 6Н).
Этаноловый раствор аммиака (235 мл) соединяли с диэтиловым сложным эфиром [(Е)-4-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)амино] -2-бутенил] фосфиновой кислоты (7,15 г, 21,6 ммолей) и раствор перемешивали 3 дня, а затем выпаривали. В результате рекристаллизации из метанола в этилацетате удаляли желтые примеси, но оперативная хроматография (7,5 см диаметр, элюирование 5% метанолом в дихлорметане) требовалась для того, чтобы исключить более полярные примеси. После рекристаллизации этого материала с метанолом в этилацетате (финальный объем 200 мл) получали диэтиловый сложный эфир [(Е)-4-[2-амино-3, 4диоксо-1-циклобутен-1-ил)амино]-2-бутенил]алкилфосфиновой кислоты в виде хлопьеобразного твердого вещества (4,43 г, 68% температура точки плавления 145-147оС).
ИК (КBr, см-1): 3280, 3100, 1800, 1640; МС (+FAB): 303 (МН+, 100), 135 (62).
1Н-ЯМР (ДМСО, 400 МГц): δ 7,5 (широкий синглет, 3 NH), 5,74-5,68 (мультиплет, 1Н), 5,57-5,48 (мультиплет, 1Н), 4,09 (широкий синглет, 2Н), 4,02-3,91 (мультиплет, 4Н), 2,64 (дублет дублетов, J=21,5 и 7 Гц, 2Н), 1,19 (триплет, J=7 Гц, 6Н).
Вычислено, С 47,68; Н 6, 34; N 9,27.
С12Н19N2O5P
Найдено, C 47,46; Н 5,95; N 9,21.
Раствор диэтилового сложного эфира [(E)-4-[(2-амино-3, 4-диоксо-1-циклобутен-1- ил)амино] -2-бутенил] фосфиновой кислоты (1,0 г, 3,3 ммоля) и бромтриметилсилана (4,6 мл, 35 ммолей) в безводном 1,2-дихлорэтане (30 мл) в атмосфере азота подвергали дефлегмации 20 мин, затем охлаждали и выпаривали. Остаток растворяли в воде (150 мл) и промывали диэтиловым простым эфиром (2х75 мл). Полученный в результате материал после концентрации водного слоя подвергали рекристаллизации из метанола в этилацетат (финальный объем 100 мл), чтобы получить [(E)-4-[(2-амино-3,4-диоксо-1-циклобутен-1-ил)амино]-2-бутенил] фосфи- новую кислоту, одна третья гидрат в виде желтого твердого вещества (0,59 г, 71% температура точки плавления 220-230оС (разлож.)).
ИК (КВr, см-1): 3280, 3100, 1800, 1640; МС (-FAB): 245 (M-Н),
1Н-ЯМР (ДМСО, 400 МГц): δ 7,47 (широкий синглет, 3NH), 5,63-5,58 (мультиплет, 2Н), 4,10 (широкий синглет, 2Н), 2,38 (дублет дублетов, J=21 и 6 Гц, 2Н).
Вычислено, С 38,11; Н 4,66; N 11,11.
С8Н11N2O5P ˙1/3 Н2О
Найдено, С 38,10; Н 4,46; N 11,00.
П р и м е р 6. [2-[(2-Амино-3,4-диоксо-1-циклобутен-1-ил)метиламино] этил]фосфи-новая кислота.
Холодную (0оС) суспензию 60% гидрида натрия (500 мг, 12,5 ммолей) в безводном диметилформамиде (15 мл) в азоте обрабатывали раствором диэтилового сложного эфира [2-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)амино]этил] алкилфосфиновой кислоты (3,23 г, 10,6 ммолей), полученным, как в примере 4, в диметилформамиде (20 мл) в течение 30 мин. Вводили иодметан (0,78 мл, 12,5 ммолей) и ледяную ванну снимали на 30 мин, а затем снова привлекали для введения 1 н. раствора хлористоводородной кислоты (20 мл). Реакционную смесь сливали в воду (200 мл), экстрагировали дихлорметаном (2х200 мл), сушили (сульфат магния) и концентрировали под высоким вакуумом (1 мм) при 40оС. Сырой материал подвергали очистке с использованием оперативной хроматографии (7,5 см диаметр, элюирование 2,5% метанолом в дихлорметане), чтобы получить диэтиловый сложный эфир [2-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)метиламино]этил]алкилфосфиновой кислоты в виде масла (3,00 г, 89%).
ИК (чистый, см-1) 1805, 1715, 1620; МС (+FAB): 320 (MH+, 100), 109 (20).
1Н-ЯМР (СDСl3, 400 МГц): δ 4,78-4,74 (мультиплет, 2Н), 4,16-4,09 (мультиплет, 2Н), 4,16-4,09 (мультиплет, 4Н), 3,94, 3,68 (мультиплет, 2Н), 3,35, 3,19 (синглет, 3Н), 2,15-2,09 (мультиплет, 2Н), 1,48-1,44 (мультиплет, 3Н), 1,34 (триплет, J=7 Гц, 6Н).
Этаноловый раствор (40 мл) диэтилового сложного эфира [2-[(2-этокси-3,4-диоксо-1-циклобутен-1-ил)метиламино] этил] фосфи- новой кислоты (3,00 г, 9,40 ммолей) соединяли с этаноловым раствором аммиака (70 мл) и выдерживали 18 ч. После концентрирования под вакуумом твердое вещество подвергали рекристаллизации дважды из метанола в этилацетате (финальный объем 50 мл), чтобы получить диэтиловый сложный эфир [2-[(2-амино-3, 4-диоксо-1-циклобутен-1-ил)метиламино] этил] алкилфосфино- вой кислоты в виде твердого вещества (2,10 г, 77% температура точки плавления 130-132оС).
ИК (КBr, см-1 ): 3320, 3160, 1800, 1670, 1650, 1640; МС (+FAB): 291 (МН+, 100), 196 (22), 109 (20).
1Н-ЯМР (ДМСО, 400 МГц): δ 7,61 (широкий синглет, NH2), 4, 02-3,94 (мультиплет, 4Н), 3,74 (широкий синглет, 2Н), 3,13 (широкий синглет, 3Н), 2,13 (дублет триплетов, J=18 и 7,5 Гц, 2Н), 1,22 (триплет, J=7 Гц, 6Н).
Вычислено, С 45,52; Н 6,60; N 9,65.
С11Н19N2О5Р
Найдено, С 45,41; Н 6,55; N 9,65.
Суспензию диэтилового сложного эфира [2-[(2-амино-3, 4-диоксо-1-циклобутен-1-ил)метиламино]этил]фосфиновой кислоты (660 мг, 2,3 ммолей) в безводном 1,2-дихлорэтане (20 мл) в атмосфере азота обрабатывали бромтриметилсиланом (2,0 мл, 15 ммолей), нагревали до дефлегмации на 10 мин. Желтый раствор концентрировали и полученное в результате твердое вещество растворяли в воде (75 мл), промывали диэтиловым простым эфиром (2х50 мл) и выпаривали. Твердое вещество растворяли в кипящем метаноле, фильтровали и концентрировали при добавлении этилацетата до финального объема 75 мл, чтобы получить соединение из заголовка примера в виде желтого твердого вещества (310 мг, 58% температура точки плавления 230-260оС (разлож.)).
ИК (КВr, см-1): 3340, 1800; МС (-FAВ): 233 (М-Н, 32), 148 (100);1Н-ЯМР (ДМСО, 400 МГц): δ 7,62 (широкий синглет, NH2), 3,68 (широкий синглет, 2Н), 3,16 (широкий синглет, 3Н), 1,90 (дублет триплетов, J=18 и 7,5 Гц, 2Н).
Вычислено, С 35,91; Н 4, 74; N 11,96.
С7Н11N2О5Р
Найдено, С 35,52; Н 4,79; N 11,83.
а) Следуя процедуре в примере 6 за тем исключением, что диэтиловый сложный эфир аминометилалкилфосфиновой кислоты использовали в качестве исходного реагента, получали [[(2-амино-3,4-диоксо-1-циклобутен-1-ил)метиламино] метил]фосфи- новую кислоту, температура точки плавления 245-250оС (разлож.).
Вычислено, C 32,74; Н 4,12; N 12,73.
С6Н9N2O5P
Найдено, С 32,62; Н 4,15; N 12,87.
П р и м е р 7. [2-(7,8-диоксо-2,5-диазабицикло[4,2,0]окт-1(6)-ен-2-ил)этил]фосфоновая кислота.
Раствор фенилметилового сложного
эфира (2-аминоэтил)карбаминовой кислоты (3,06 г, 16 ммолей), диэтилового сложного эфира (2-оксоэтил)алкилфосфиновой кислоты (2,88 г, 16 ммолей) и цианоборогидрида натрия (1,00 г, 16 ммолей) в сухом
метаноле (90 мл) получали в атмосфере азота. Метаноловый раствор хлористого водорода добавляли до тех пор, пока раствор не станет слабо кислым (рН 6,5). Спустя 3 ч вводили дополнительное количество
цианоборогидрида натрия (0,25 г, 4,0 ммолей) и реакцию оставляли на ночь. После подкисления до рН 1,5 при помощи концентрированной хлористоводородной кислоты метанол удаляли под вакуумом, а остаток
разбавляли водой (25 мл). После промывки диэтиловым простым эфиром (3х25 мл) водный слой подщелачивали до рН 10 при помощи 1 н.раствора гидрата окиси натрия, насыщали твердым хлоридом натрия, а затем
экстрагировали хлороформом (3х50 мл). Высушенный (сульфат натрия) органический слой предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (3 см диаметр,
градиентное элюирование 5-10% метанолом в дихлорметане), чтобы получить фенилметиловый сложный эфир [2-[[2-диэтоксифосфинил)этил]амино]этил] карбаминовой кислоты в виде бледно-желтого масла (2,90 г,
51%),
ИК (чистый, см-1): 3300, 1715; МС (+FAB): 359 (MH+, 100), 91 (70).
1Н-ЯМР (СDСl3, 400 МГц): δ 7,36 (мультиплет, 5Н), 5, 46 (широкий синглет, NH), 5,10 (синглет, 2Н), 4,16-4,02 (мультиплет, 4Н), 3,29 (квартет, J=5,5 Гц, 2Н), 2,89 (дублет триплетов, J=17 и 7 Гц, 2Н) триплет, J=5,5 Гц, 2Н), 1,95 (дублет триплетов, J=18 и 7 Гц, 2Н), 1,82 (широкий синглет, NH), 1,31 (триплет, J=7 Гц, 6Н).
Вычислено, С 51,55; Н 7,73; N 7,51.
С16Н27N2O5P ˙
4/5 Н2О
Найдено, С 51,69; Н 7,83; N 7,53.
В колбу, содержащую 10% палладий на углероде (3,79 г) в атмосфере азота, добавляли фенилметиловый сложный эфир [2-[[2-(диэтоксифосфинил)этил]амино] этил]карбаминовой кислоты (3,79 г, 10 ммолей) в этаноле (50 мл), затем добавляли 1,4-циклогексадиен (10,4 мл, 110 ммолей). После перемешивания суспензии в течениe ночи, смесь фильтровали через Целит

1Н-ЯМР (CDCl3, 400 МГц): δ 4,18-4,04 (мультиплет, 4Н), 2,91 (дублет триплетов, J=15 и 7 Гц, 2Н), 2,80 (триплет, J=5,5 Гц, 2Н), 2,68 (триплет, J=5,5 Гц, 2Н), 1,98 (дублет триплетов, J=18 и 7 Гц, 2Н), 1,68 (широкий синглет, 3 NH), 1,33 (триплет, J=7 Гц, 6Н).
Растворы (10 мл каждого) 3,4-диэтокси-3-циклобутен-1,2-диона (1,27 мл, 8,6 ммолей) и диэтилового сложного эфира [2-[(2-аминоэтил/амино]этил]алкилфосфи- новой кислоты (1,92 г, 8,6 ммолей) в этаноле отдельно впрыскивали через шприц в дефлегмирующий этанол (22 мл) в течение 3 ч. После дефлегмации в течение ночи красно-коричневый раствор предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (7 см диаметр, градиентное элюирование 2,5-10% метанолом в этилацетате) и подвергали рекристаллизации (метанол в этилацетате), чтобы получить диэтиловый сложный эфир [2-(7,8-диоксо-2,5-диазабицикло[4,2,0]окт-1(6)-ен-2-ил/этил]фосфиновой кислоты в виде бежевого твердого вещества (0,78 г, 30% температура точки плавления 115-116оС).
ИК (КВr, см-1): 3170, 1780, 1660; МС (+FAB): 303 (MH+, 100), 109 (38).
1Н-ЯМР (СDCl3, 400 МГц): δ 7,64 (широкий синглет, NH), 4,16-4,06 (мультиплет, 4Н), 3,80-3,73 (мультиплет, 2Н), 3,63-3,60 (мультиплет, 2Н), 3,48 (триплет, J=5 Гц, 2Н), 2,31 (дублет триплетов, J=18 и 7,5 Гц, 2Н), 1,33 (триплет, J=7 Гц, 6Н).
Вычислено, С 47,68; Н 6,34; N 9,27.
С12Н19N2O5P
Найдено,С 47,39; Н 6,32; N 9,22
Раствор диэтилового сложного эфира [2-(7,8-диоксо-2,5-диазабицикло[4.2.0] окт-1(6)-ен-2-ил)этил] фосфиновoй кислоты (0,78 г, 2,6
ммолей) и бромтриметилсилана (2,1 мл, 16 ммолей) в сухом 1,2-дихлорэтане (30 мл) подвергали дефлегмации в азоте в течение 20 мин. Охлажденную реакционную смесь концентрировали под вакуумом и остаток
растворяли в воде (100 мл) и промывали диэтиловым простым эфиром (3х50 мл). После концентрирования водного слоя остаток подвергали рекристаллизации из воды (25 мл) и метанола (300 мл), чтобы получить
твердые бежевые примеси, которые удаляли фильтрацией. Фильтрат концентрировали и подвергали рекристаллизации из воды в изопропанол, чтобы получить соединение из заголовка примера в виде желтого
твердого вещества (0,37 г, 58% температура точки плавления 220-270оС (разлож.));
ИК (КВr, см-1): 1800; МС (-FAВ): 245 (М-Н);1Н-ЯМР (ДМСО) капля DCl, 400
МГц): δ 3,58-3,51 (мультиплет, 2Н), 3,40-3,33 (мультиплет, 4Н), 2,07-1,98 (мультиплет, 2Н).
Вычислено, С 39,04; Н 4,50; N 11,38.
С8Н11
N2O5P
Найдено, С 38,60; Н 4,30; N 11,11.
П р и м е р 8. [2-(8,9-диоксо-2,6-диазабицикло[5.2.0]нон-1(7)-ен-2-ил)этил]фосфиновая кислота.
Раствор фенилметилового сложного эфира (3-аминопропил)карбаминовой кислоты (6,11 г, 29 ммолей), диэтилового сложного эфира (2-оксоэтил)алкилфосфиновой кислоты (5,24 г, 29 ммолей) и цианоборогидрида
натрия (2,73 г, 43 ммолей) в сухом метаноле (100 мл) получали в атмосфере азота. Метаноловый раствор хлористого водорода добавляли до тех пор, пока раствор не оставался слабо кислым (рН 6,5). После
того, как в течение нескольких часов реакцию поддерживали кислой (рН 1,5) при помощи концентрированной хлористоводородной кислоты, метанол удаляли под вакуумом и остаток разбавляли водой (25 мл).
После промывки диэтиловым простым эфиром (3х30 мл) водный слой подщелачивали до рН 10 при помощи 10 н.раствора гидрата окиси натрия, насыщали твердым хлоридом натрия, а затем экстрагировали
хлороформом (3х50 мл). Высушенный (сульфат натрия) органический слой предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (7 см диаметр, градиентное
элюирование 5-20% метанолом в дихлорметане), чтобы получить фенилметиловый сложный эфир [3-[[2-(диэтоксифосфинил)этил]амино] про-пил] карбаминовой кислоты в виде восковидного твердого вещества (3,86 г,
36%),
ИК (чистый, см-1): 3300, 1720, 1250, 1030; МС (+FAB): 373 (МН+, 100), 91(90).
1Н-ЯМР (СDCl3, 400 МГц): δ 7,36-7,29 (мультиплет, 5Н), 5,58 (широкий синглет, NH), 5,09 (синглет, 2Н), 4,15-4,04 (мультиплет, 4Н), 3,29 (квартет, J=6 Гц, 2Н), 2,91 (дублет триплетов, J=16 и 7 Гц, 2Н), 2,71 триплет, J=6 Гц, 2Н), 1,98 (дублет триплетов, J=18 и 7 Гц, 2Н), 1,70 (пентет, J=6 Гц, 2Н), 1,31 (триплет, J=7 Гц, 6Н).
Раствор фенилметилового сложного эфира [3-[[2-диэтоксифосфинил/этил]амино]пропил]карбаминовой кислоты (3,17 г, 8,5 ммолей) в абсолютном этаноле (40 мл) добавляли в течение 45 минут в 3,4-диэтокси-3-циклобутен-1,2-дион (2,3 мл, 16 ммолей), растворенный в этаноле (55 мл). После выдерживания в течение ночи реакционную смесь предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (7 см диаметр, градиентное элюирование 2,5-10% метанолом в дихлорметане), чтобы получить фенилметиловый сложный эфир [3-[[2-(ди-этоксифосфинил)этил](2-этокси-3,4-диоксоцик- лобутен-1-ил)амино]пропил]карбаминовой кислоты в виде вязкого масла (3,75 г, 89% ).
1Н-ЯМР (СDCl3, 400 МГц): δ 7,35 (мультиплет, 5Н), 5,45 (широкий мультиплет, NH), 5,09 (синглет, 2Н), 4,80-4,71 (мультиплет, 2Н), 4,16-4,09 (мультиплет, 4Н), 3,90-3,48 (мультиплет, 4Н), 3,23-3,20 (мультиплет, 2Н), 2,16-2,04 (мультиплет, 2Н), 1,85-1,79 (мультиплет, 2Н), 1,47, 1,41 (триплет, J=7 Гц, 3Н), 1,34 (триплет, J=7 Гц, 6Н).
В колбу, содержащую 10% палладий на углероде (2,11 г), в атмосфере азота добавляли фенилметиловый сложный эфир [3-[[2-(диэтоксифосфинил)этил](2-этокси-3, 4-ди- оксо-1-циклобутен-1-ил) амино]пропил]карбаминовой кислоты (2,11 г, 4,2 ммолей) в абсолютном этаноле (180 мл), затем 1,4-циклогексадиен (4,3 мл, 45 ммолей). После перемешивания в течение 5 ч реакционную смесь фильтровали через Целит

ИК (KВr, см-1): 1810.
1Н-ЯМР (СDСl3, 400 МГц): δ 7,50 (широкий синглет, NH), 4,18-4,10 (мультиплет, 4Н), 4,09-4,00 (мультиплет, 2Н), 3,51-3,46 (мультиплет, 4Н), 2,19 (дублет триплетов, J=18 и 7,5 Гц, 2Н), 2,10-2,04 (мультиплет, 2Н), 1,34 (триплет, J=7 Гц, 6Н).
Вычислено, С 47,99; Н 6,82; N 8,61.
С13Н21N2O5P ˙1/2 H2O
Найдено, С 48,23; Н 6,57; N 8,52.
Раствор диэтилового сложного эфира [2-(8,9-диоксо-2, 6-диазабицикло[5,2,0] нон-1(7)-ен-2-ил)этил] фосфиновой кислоты (1,41 г, 4,5 ммолей) и бромтриметилсилина (4,2 мл, 32 ммолей) в сухом 1,2-дихлорэтане (50 мл) подвергали дефлегмации 20 мин в атмосфере азота. Охлажденную реакционную смесь концентрировали под вакуумом, а остаток растворяли в воде (100 мл) и промывали диэтиловым простым эфиром (3х75 мл). Воду удаляли под вакуумом, а полученное в результате твердое вещество подвергали рекристаллизации из воды и изопропанола, чтобы получить [2-(8,9-диоксо-2,6-диазабицикло[5,2,0]-нон-1/7-ен-2-ил/ /этил] фосфиновую кислоту, одна пятая гидрат в виде желтого твердого вещества (0,91 г, 78% температура точки плавления 260-278оС, (разлож.).
ИК (КВr, см-1): 1800.
1Н-ЯМР (ДМСО и 1 капля DCl, 400 МГц): 3,85-3,79 (мультиплет, 2Н), 3,35-3,32 (мультиплет, 2Н), 3,25-3,23 (мультиплет, 2H), 1,97-1,87 (мультиплет, 4Н).
Вычислено, С 40,98; Н 5,12; N 10, 68.
С9Н13N2О5Р ˙1/5 Н2О
Найдено, С 40,98; Н 4,98; N 10,38.
П р и м е р 9. [2-(4-окси-8,9-диоксо-2, 6-диазабицикло[5,2,0]нон-1(7)-ен-2-ил)этил]фос- финовая кислота.
Раствор 1,3-диамино-2-оксипропана (27,0 г, 0,300 молей) в сухом ацетонитриле (270 мл) поддерживали при окружающей температуре в водяной ванне и обрабатывали ди-трет-бутил дикарбонатом (23,0 мл, 0,100 молей) в ацетонитриле (90 мл) в течение 2 ч при энергичном механическом перемешивании, а затем выдерживали в течение ночи. Реакционную смесь концентрировали под вакуумом и остаток растворяли в соляном растворе (250 мл), в который добавляли бромкрезольную зелень. Смесь обрабатывали 1 н. раствором хлористоводородной кислоты до тех пор, пока он не превратится в желтый, промывали дихлорметаном (3х250 мл), а затем превращали в щелочной (рН 12) добавлением 2,5 н.раствора гидрата окиси натрия. Продукт экстрагировали в хлороформе (15х250 мл), сушили сульфатом натрия и концентрировали, чтобы получить 1,1-диметилэтиловый сложный эфир (3-амино-2-оксипропил)карбаминовой кислоты в виде белого твердого вещества (7,76 г, 41% температура точки плавления 77-79оС).
ИК (КВr, см-1): 3360, 1680, МС (+FAВ): 191 (МН+, 42), 135 (100), 58 (78).
1Н-ЯМР (СDCl3, 400 МГц): δ 5,04 (широкий синглет, NH), 3,65-3,60 (мультиплет, 1Н), 3,34-3,22 (мультиплет, 1Н), 3,12-3,05 (мультиплет, 1Н), 2,83 (дублет дублетов, J=13 и 4 Гц, 1Н), 2,63 (дублет дублетов, J=13 и 7,5 Гц, 1Н), 1,44 (синглет, 9Н).
Раствор 1,1-диметилэтилового сложного эфира (3-амино-2-оксипропил)карбаминовой кислоты (7,73 г, 41 ммоль), карбоната натрия (6,52 г, 62 ммоля) и диэтил 2-бромэтилфосфоната (11,8 мл, 65 ммолей) в абсолютном этаноле (150 мл) получали в атмосфере азота. Смесь подвергали дефлегмации в течение ночи, а затем концентрировали под вакуумом. Остаток растворяли в хлороформе (150 мл), а затем предварительно адсорбировали на силикагеле и подвер- гали очистке при помощи оперативной хроматографии (7 см диаметром, градиентное элюирование 2,5-20% метанолом в дихлорметане, чтобы получить 1,1-диметилэтиловый сложный эфир [3-[[2-(диэтоксифосфинил) этил]амино]-2-оксипропил]карбаминовой кис- лоты в виде бесцветного масла (11,16 г, 77%).
ИК (чистый, см-1): 3320, 1710, 1250, 1170, 1040; МС (+FAB): 355 (MH+, 68), 255 (90), 58 (100).
1Н-ЯМР (СDСl3, 400 МГц): δ 5,23 (широкий синглет, NH), 4,16-4,05 (мультиплет, 4Н), 3,77 (мультиплет, 1Н), 3,32-2,90 (мультиплет, 4Н), 2,73 (дублет дублетов, J=12 им 3,5 Гц, 1Н), 2,58 (дублет дублетов, J=12 и 8,5 Гц, 1Н), 2,00 (дублет триплетов, J= 18 и 7 Гц, 2Н), 1,43 (синглет, 9Н), 1,32 (триплет, J=7 Гц, 6Н).
В раствор 3,4-диэтокси-3-циклобутен-1,2-диона (5,0 мл, 34 ммоля) в абсолютном этаноле (180 мл), полученный в атмосфере азота, добавляли раствор 1,1-диметилэтилового сложного эфира [3-[[2-(диэтоксифосфинил)этил]амино]-2-оксипропил] карбаминовой кислоты (12,11 г, 34 ммоля) в этаноле (60 мл) в течение 1 ч. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали под вакуумом. Остаток растворяли в хлороформе (150 мл), предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (7 см диаметр, градиентное элюирование 2,5-15% метанолом в дихлорметане), чтобы получить 1, 1-диметилэтиловый сложный эфир [3-[[2-(диэтоксифосфинил)этил](2-этокси-3,4-ди-оксо-1-циклобутен-1-ил)амино] -2-оксипропил]карбаминовой кислоты в виде прозрачного вязкого масла (11,4 г, 70%).
ИК (чистый, см-1): 3350, 1800, 1700.
1Н-ЯМР (СDCl3, 400 МГц): δ 5,37, 5,19 (широкий синглет, NH), 4,82-4,72 (мультиплет, 3Н), 4,16-3, 11 (мультиплет, 11Н), 2,24-2,14 (мультиплет, 2Н), 1,47-1,43 (мультиплет 12Н), 1,32 (триплет, J=7 Гц, 6Н).
Раствор 1,1-диметилэтилового сложного эфира [3-[[2-диэтоксифосфинил)этил] (2-этокси-3,4-диоксо-1-циклобутен-1-ил)ами-но] -2-оксипропил]карбаминовой кислоты (11,34 г, 24 ммоля) в 96% муравьиной кислоте (100 мл) получали в атмосфере азота и перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем концентрировали под вакуумом, чтобы получить липкое желтое масло, которое растворяли в абсолютном этаноле (120 мл) и по каплям добавляли в течение 1, 5 ч в раствор диизопропилэтиламина (16,7 мл, 96 ммолей) в абсолютном этаноле (360 мл). После дефлегмации в течение ночи реакционную смесь концентрировали под вакуумом, а остаток растворяли в хлороформе (150 мл), предварительно адсорбировали на силикагеле, и подвергали очистке при помощи оперативной хроматографии (9 см диаметр, градиентное элюирование 5-20% метанолом в дихлорметане), чтобы получить диэтиловый сложный эфир [2-(4-окси-8,9-диоксо-2,6-диазабицикло[5,2,0]нон-1(7)-ен-2-ил/этил]фосфиново й кислоты в виде белого твердого вещества (5,94 г, 74% температура точки плавления 169-171оС).
ИК (КВr, см-1): 3330, 3200, 1800, 1250, 1030; МС (+FAB): 333 (MH+, 100), 185 (50), 179 (78).
1Н-ЯМР (СDCl3, 400 МГц): δ 7,18 (широкий синглет, NH), 4,52-4,44 (мультиплет, 1Н), 4,20 (мультиплет, 1Н), 4,14-4,04 (мультиплет, 4Н), 3,79-3,70 (мультиплет, 2Н), 3,57-3,54 (мультиплет, 2Н), 3,36 (дублет, J=14 Гц, 1Н), 2,33-2,13 (мультиплет, 2Н), 1,32 (дублет триплетов, J=13 и 7 Гц, 6Н).
Вычислено, С 46,89; Н 6,37; N 8,43.
С13Н21N2O6P
Найдено, С 47,07; Н 6,11; N 8,37.
В раствор диэтилового сложного эфира [2-(4-окси-8,9-диоксо-2,6-диазабицикло[5,2,0] нон-1(7)-ен-2-ил)этил]фосфиновой кислоты (2,5 г, 7,5 ммолей) в сухом 1,2-дихлорметане (100 мл) в атмосфере азота добавляли бромтриметилсилан (9,2 мл, 60 ммолей). Реакционную смесь подвергали дефлегмации 20 мин, а затем концентрировали под вакуумом, чтобы получить желто-оранжевую пену, которую растворяли в воде (100 мл). Воду промывали простым эфиром (3х100 мл), а затем концентрировали под вакуумом, чтобы получить твердое вещество, которое подвергали рекристаллизации из воды в изопропанол, чтобы получить [2-(4-окси-8,9-диоксо-2,6-диазабицикло[5,2,0] -нон-1(7)-ен- -2-ил)этил]фосфиновую кислоту, одна шестая гидрат в виде бледно-желтого твердого вещества (1,79 г, 86% температура точки плавления 268-270оС, разлож.).
ИК (КBr, см-1): 3480, 3220, 1820; МС (-FAB): 275 (M-Н, 22), 148 (100).
1Н-ЯМР (ДМСО, 1 капля DCl, 400 МГц): δ 3,90 (мультиплет, 1Н), 3,85-3,74 (мультиплет, 2Н), 3,46 (дублет, J=13 Гц, 1Н), 3,34 (дублет дублетов, J=13 и 7 Гц, 1Н), 3,28 (мультиплет, 2Н), 1,98-1,89 (мультиплет, 2Н).
Вычислено, C 38,72; Н 4,81; N 10,03.
С9Н13N2O6P
˙1/6 H2O
Найдено, С 38,46; Н 4,59; N 10,03.
а) Следуя процедуре из примера 9 за тем исключением, что 2,2-диметил-1,3-пропандиамин использовали в качестве реагента, получали [2-(4,4-диметил-8,9-диоксо-2,6-диазабицикло[5,2,0] нон-1(7)-ен-2-ил)этил] фосфиновую кислоту, моногидрат, температура точки плавления 265-282оС (разлож.).
Вычислено, С 43,14; Н 6,25; N 9,15.
С11Н17N2O5Р ˙Н2О
Найдено, С 42,98; Н 6,04; N 9,02.
b) Следуя процедуре из примера 9 за тем исключением, что 1,3-пропандиамин и диэтил 3-бромпропилфосфонат являются реагентами, получали [3-(8,9-диоксо-2,6-диазабицикло[5,2,0] нон-1-(7)-ен-2-ил)пропил] фосфиновую кислоту, температура точки плавления 244-248оС.
Вычислено, С 43,93; Н 5,42; N 10,18.
С10Н15N2O5P
Найдено, С 43,93; Н 5,42; N 10,18.
с) Следуя процедуре из примера 9 за тем исключением, что 1,3-пропандиамин и диэтил (Е)-(3-хлор-1-пропенил)фосфонат являются реагентами, получали [(Е)-3-(8,9-диоксо-2,6-диазабицикло[5,2,0]нон-1(7)-ен-2-ил)-1-пропенил]фосфи новукислоту, одна шестая гидрат, температура точки плавления 255-275оС (разлож.).
Вычислено, С 43,64; Н 4,88; N 10,18.
С10Н13N2O5P ˙1/6 H2O.
Найдено, C 43,69; Н 4,68; N 10,02.
П р и м е р 10. 8,9-Диоксо-2,6-диазабицикло[5,2,0]нон-1(7)-ен-2-уксусная кислота.
Раствор фенилметилового сложного эфира (3-аминопропил)карбаминовой кислоты (7,08 г, 34 ммолей) и N,N-диизопропилэтиламина (4,5 мл, 26 ммолей) в безводном диметилформамиде (100 мл) в атмосфере азота охлаждали до 10оС, и обрабатывали трет-бутил бромацетатом (2,80 мл, 17 ммолей) в течение 5 мин. Спустя 1 ч ванну снимали и реакционную смесь перемешивали в течение ночи, сливали в воду (500 мл) и превращали в щелочной (рН 12) 2,5 н. раствором гидрата окиси натрия. Водный слой экстрагировали дихлорметаном (2х250 мл), который сушили над сульфатом натрия и концентрировали под вакуумом при нагревании, чтобы удалить деметилформамид. Остаток подвергали очистке при помощи оперативной хроматографии (9 см диаметр, градиентное элюирование 2,5-3,5% метанолом в дихлорметане), чтобы получить фенилметиловый сложный эфир [3-[[(1,1-диметилэтилоксикарбонил)метил] амино] про- пил]карбаминовой кислоты в виде бледно-желтого масла (4,50 г, 82%).
1Н-ЯМР (СDCl3, 200 МГц): δ 7,35 (мультиплет, 5Н), 5,43 (широкий синглет, NH), 5,09 (синглет, 2Н), 3,35-3,24 (мультиплет: 4Н), 2,68 (триплет, J=6,5 Гц, 2Н), 1,68 (пентет, J=6,5 Гц, 2Н), 1,46 (синглет, 9Н).
Этаноловый раствор (60 мл) фенилметилового сложного эфира [3-[[(1,1-диметилэтилоксикарбонил/метил] амино] пропил]карба- миновой кислоты (4,49 г, 13,9 ммолей) в атмосфере азота добавляли в 3,4-диэтокси-3-циклобутен-1,2-дион (2,0 мл, 13 ммолей) в том же растворителе (60 мл) в течение 1 ч. Спустя еще 3,5 ч, реакционную смесь предварительно адсорбировали на силикагеле и подвергали очистке при помощи оперативной хроматографии (7,5 см диаметр, градиентное элюирование 30-60% этилацетатом в петролейном эфире), чтобы получить фенилметиловый сложный эфир [3-[[(1,1-диметилэтилоксикарбонил)метил]-(2-этокси-2, 4-диоксо-1-циклобутен- 1-ил)амино]пропил]карбаминовой кислоты в виде вязкого бесцветного масла (5,26 г, 91%); МС (+Сl): 447 (МН+, 100), 391 (38).
1Н-ЯМР (СDСl3, 400 МГц): δ 7,35 (мультиплет, 5Н), 5,55, 4,97 (широкий мультиплет, NH), 5,10 (синглет, 2Н), 4,75, 4,73 (квартет, J=7 Гц, 2Н), 4,29, 3,99 (синглет, 2Н), 3,74, 3,49 (триплет, J=6,5 Гц, 2Н), 3,29-3,22 (мультиплет, 2Н), 1,84-1,75 (мультиплет, 2Н), 1,48, 1,47 (синглет, 9Н), 1,45-1,40 (мультиплет, 3Н).
В охлажденную колбу (20оС), содержащую 10% палладий на углероде (5,25 г) в атмосфере азота, добавляли фенилметиловый сложный эфир [3-[[(1,1-диметилэтилоксикарбонил)метил]-(2-этокси-3,4-диоксо-1-циклобутен- 1-ил)амино]пропил] карбаминовой кислоты (5,25 г, 11,8 ммолей) в этаноле (500 мл), затем 1,4-циклогексадиен (11 мл, 0,12 моля) в течение 5 мин. Спустя 5 ч суспензию фильтровали через Целлит

ИК (КВr, см-1): 3200, 1800, 1720; МС (Е1): 266 (М+, 42), 210 (33), 166 (37), 165 (100), 154 (58), 138 (37), 70 (58).
1Н-ЯМР (ДМСО, 400 МГц): δ 8,62 (широкий синглет, NH), 4,38 (синглет, 2Н), 3,36-3,27 (мультиплет, 4Н), 1,91 (мультиплет, 2Н), 1,40 (синглет, 9Н).
Вычислено, C 58,63; Н 6,81; N 10,52.
С13Н18N2O4
Найдено, С 58,57; Н 6,78; N 10,40.
Этаноловый (86 мл) раствор 1,1-диметилэтилового сложного эфира (8,9-диоксо-2,6-диазабицикло[5,2,0] нон-1(7)-ен-2-ил[уксус- ной кислоты) (2,29 г, 8,60 ммолей) обрабатывали при комнатной температуре 2, 5 н.раствором гидрата окиси натрия (3,5 мл, 8,7 ммолей) и перемешивали в течение ночи. Суспензию фильтровали, промывали этил ацетатом, чтобы получить твердое вещество, которое подвергали рекристаллизации из метанола (воды) изопропанола (финальный объем 50 мл), чтобы получить гидрат соли натрия (8,9-диоксо-2,6-диазабицикло[5,2,0]нон-1(7)-ен-2-ил)уксусной кислоты (1,43 г, 66% температура точки плавления 280-300оС, разлож.); МС (-FAB): 231 (М-Н, 37), 209 (М-Na, 100).
1Н-ЯМР (ДМСО, 1 капля DCl, 400 МГц): 4,40 (синглет, 2Н), 3,36-3,25 (мультиплет, 4Н), 1,89 (мультиплет, 2Н).
Вычислено, С 43,21; Н 4,43; N 11,20.
С9Н9N2NaO4 ˙H2O
Найдено, С 43,29; Н 4,38; N 11,32.
а) Следуя процедуре из примера 10 за тем исключением, что этил 3-бромпропионат использовали в качестве реагента, получали полуторный гидрат натриевой соли 3-(8,9-диоксо-2,6-диазабицикло[5,2,0]нон-1(7)-ен-2-ил]пропановой кислоты, температура точки плавления 310оС (разлож.).
Вычислено, С 44,04; Н 4,99; N 10, 27.
С10Н11N2NaO4 ˙1,5 Н2О
Найдено, С 44,28; Н 5,15; N 10,24.
П р и м е р 11.
2-[(1Н-тетразол-5-ил)метил]-2,6-диазабицикло[5,2,0] нон-1(7)-ен-8,9- дион
Раствор 1,1-диметилэтилового сложного эфира (3-аминопропил)карбаминовой кислоты (6,00 г, 34 ммолей), полученный с
использованием процедуры Саари и др. J.Med.Chem. т. 33, стр. 97 (1990), в абсолютном этаноле (90 мл), обрабатывали карбонатом натрия (3,96 г, 37 ммолей), затем раствором бромацетонитрила (2,6 мл, 37
ммолей) в этаноле (30 мл) в течение 45 мин. После перемешивания при комнатной температуре в течение ночи содержимое колбы фильтровали, предварительно адсорбировали на силикагеле и подвергали очистке
при помощи оперативной хроматографии (7 см диаметр, элюирование 2,5% метанолом в дихлорметане), чтобы получить 1,1-диметилэтиловый сложный эфир [3-(цианометиламино)пропил] карбаминовой кислоты в виде
желтого масла (3,99 г, 55%).
ИК (чистый, см-1): 3330, 2240, 1700; МС (+Сl): 214 (МН+, 20), 187 (76), 158 (100), 131 (44).
1Н-ЯМР (СDCl3, 400 МГц): δ 4,80 (широкий синглет, NH), 3,61 (синглет, 2Н), 3,22 (квартет, J= 6,5 Гц, 2Н), 2,79 (триплет, J=6,5 Гц, 2Н), 1,69 (пентет, J=6,5 Гц, 2Н), 1,45 (синглет, 9Н).
Раствор 3,4-диэтокси-3-циклобутен-1,2-диона (2,6 мл, 18 ммолей) в абсолютном этаноле (90 мл) обрабатывали 1,1-диметилэтиловым сложным эфиром [3-(цианометиламино)пропил] карбаминовой кислоты (3,90 г, 18 ммолей) в этаноле (30 мл) в течение 90 мин. Спустя 40 ч реакционную смесь выпаривали, растворяли в дихлорметане, предварительно адсорбировали на силикагель и подвергали очистке при помощи оперативной хроматографии (7 см диаметр, градиентное элюирование 0-5% метанолом в дихлорметане), чтобы получить 1,1-диметилэтиловый сложный эфир [3-[(цианометил)-(2-этокси-3, 4-диоксо-1-циклобутен-1-ил)амино]пропил]карбами новокислоты в виде желтого масла (5,48 г, 90%).
ИК (чистый, см-1): 3360, 1800; МС (+FAB): 282 (76), 238 (98), 158 (100),
1Н-ЯМР (СDСl3, 400 МГц): δ 4,81 (квартет, J=7 Гц, 2Н), 4,75 (широкий синглет, NH), 4,45, 3,66 (синглет, 2Н), 3,84, 3,60 (широкий мультиплет, 2Н), 3,26-3,15
(мультиплет, 2Н), 1,91 (пентет, J=6,5 Гц, 2Н), 1,50 (триплет, J=7 Гц, 3Н), 1,45 (синглет, 9Н).
Раствор 1,1-диметилэтилового сложного эфира [3-[(цианометил)-(2-этокси-3, 4-диоксо-1-циклобутен-1-ил)амино] пропил] карбаминовой кислоты (5,48 г, 16 ммолей) в 96% муравьиной кислоте (40 мл) получали в атмосфере азота. Спустя 24 ч растворитель удаляли, а остаток растворяли в изопропаноле и концентрировали несколько раз. Добавляли этанол (40 мл) и суспензию перемешивали в течение ночи, затем фильтровали, чтобы получить (8,9-диоксо-2,6-диазабицикло[5,2, 0]-нон-1(7)-ен-2-ил)ацетонитрил, одна восьмая гидрат в виде белого твердого вещества (1,50 г, 48% температура точки плавления 213-218оС).
ИК (КВr, см-1): 3220, 1810; МС (DEI): 191 (М+, 57), 135 (41), 70 (41), 43 (70), 41 (100).
1Н-ЯМР (ДМСО, 400 МГц): δ 8,83 (широкий синглет, NH), 4,84 (синглет, 2Н), 3,40-3,30 (мультиплет, 4Н), 1,95 (мультиплет, 2Н).
Вычислено, 55,93; Н 4,69; N 21,74.
С9Н9N3O2 ˙1/8 H2O
Найдено, С 55,99; Н 4,76; N 21,90.
В раствор (8,9-диоксо-2,6-диазабицикло[5,2,0]нон-1(7)-ен-2-ил)ацетонитрила, одна восьмая гидрат (1,54 г, 8,0 ммолей) в сухом диметилформамиде (35 мл) в атмосфере азота добавляли азид натрия (0,79 г, 12 ммолей) и хлорид аммония (0,65 г, 12 ммолей). Реакционную смесь медленно нагревали до 125оС, поддерживали при этой температуре 2 ч, охлаждали до комнатной температуры, а затем фильтровали. Концентрированный фильтрат растворяли в воде и выпаривали. Остаток подвергали рекристаллизации из метанола, чтобы получить 2-[(1Н-тетразол-5-ил)метил]-2,6-диазабицикло [5,2,0]нон-1(7)-ен-8,9-дион, одна третья гидрат в виде не совсем белых кристаллов (1,18 г, 61% температура точки плавления 264-267оС).
ИК (КВr, см-1): 3200, 1810;1Н-ЯМР (ДМСО, 400 МГц): δ 8,70 (синглет, NH), 5,24 (синглет, 2Н), 3,34-3,28 (мультиплет, 4Н), 1,92 (мультиплет, 2Н).
Вычислено, С 45,00; Н 4,48; N 34,98.
С9Н10N6O2 ˙1/3 H2O
Найдено, С 45,28; Н 4,21; N 34,76.
а) Следуя процедуре из примера 11 за тем исключением, что 3-бромпропионитрил использовали в качестве реагента, получали 2-[2-(1Н-тетразол-5-ил)этил] -2,6-диазабици- кло[5,2,0] нон-1(7)-ен-8,9-дион, температура точки плавления 255-262оС (разложение).
Вычислено, С 48,38; Н 4,87; N 33,85.
С10Н12N6О2
Найдено, С 48,10; Н 4,80; N 34,19.
П р и м е р 12. [2-(9,10-Диоксо-2,7-диазабицикло[6,2,0]дец-1(8)-ен-2-ил)этил]фосфи-новая кислота.
Раствор 1,4-диаминобутана (30 мл, 0,30 моля) в сухом тетрагидрофуране (90 мл) поддерживали при 0оС и обрабатывали ди-трет-бутил дикарбонатом (23 мл, 0,10 моля) в тетрагидрофуране (90 мл) в течение 1,5 ч при энергичном механическосм перемешивании, а затем выдерживали в течение ночи. Реакционную смесь концентрировали под вакуумом и остаток растворяли в воде (100 мл), в которую добавляли бромкрезольную зелень. Эту смесь обрабатывали 1 н.раствором хлористоводородной кислоты до тех пор, пока она не станет желтой, промывали дихлорметаном (2х250 мл), а затем превращали в щелочную (рН 12) при помощи добавления 2,5 н.раствора гидрата окиси натрия. Продукт экстрагировали в хлороформе (6х250 мл), сушили сульфатом натрия и концентрировали, чтобы получить 1,1-диметилэтиловый сложный эфир (4-аминобутил)карбаминовой кислоты в виде желтого масла (13,4 г, 71%).
ИК (чистый, см-1): 3350; 1700; МС (Е1): 188 (М+, 4), 132 (56), 115 (33), 70 (100).
1Н-ЯМР (СDСl3, 400 МГц): δ 4,76 (широкий синглет, NH), 3,11 (квартет, J= 6 Гц, 2Н), 2,70 (триплет, J=6 Гц, 2Н), 1,58-1,45 (мультиплет, 4Н), 1,43 (синглет, 9Н).
Раствор 1, 1-диметилэтилового сложного эфира (4-аминобутил)карбаминовой кислоты (13,3 г, 71 ммолей), карбоната натрия (11,25 г, 106 ммолей) и диэтил 2-бром-этилфосфоната (20 мл, 104 ммолей) в абсолютном этаноле (150 мл) получали в атмосфере азота. Эту смесь подвергали дефлегмации в течение ночи, а затем концентрировали под вакуумом. Остаток растворяли в хлороформе (150 мл), а затем предварительно адсорбировали на силикагель и подвергали очистке при помощи оперативной хроматографии (7 см диаметр, градиентное элюирование 5-30% метанолом в дихлорметане), чтобы получить 1,1-диметилэтиловый сложный эфир [4-[[2-(диэтоксифосфинил)этил] амино]бутил]карбаминовой киcлоты в виде бесцветного масла (13,7 г, 55%); МС (+FAB): 353 (МН+, 100), 150 (68), 56 (83).
1Н-ЯМР (СDCl3, 400 МГц): δ 5,01 (широкий синглет, NH), 4,17-4,03 (мультиплет, 4Н), 3,15-3,08 (мультиплет, 2Н), 2,92 (дублет триплетов, J=15 и 7,5 Гц, 2Н), 2,84 (широкий синглет, NH), 2,66 (широкий триплет, J=7 Гц, 2Н), 2,02 (дублет триплетов, J=18 и 7,5 Гц, 2Н), 1,56-1,52 (мультиплет, 4Н), 1,43 (синглет, 9Н), 1,33 (триплет, J=7 Гц, 6Н).
В раствор 3,4-диэтокси-3-циклобутен-1, 2-диона (5,8 мл, 39 ммолей) в абсолютном этаноле (200 мл), полученный в атмосфере азота, добавляли раствор 1,1-диметилэтилового сложного эфира [4-[[2-(диэтоксифосфинил)этил]амино] бутил] карбаминовой кислоты (13,7 г, 39 ммолей) в этаноле (75 мл) в течение 1,75 ч. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали под вакуумом. Остаток растворяли в хлороформе (150 мл), предварительно адсорбировали на силикагеле и подвергали очистке на оперативной хроматографии (9 см диаметр, элюирование 2,5% этанолом в дихлорметане), чтобы получить 1, 1-диметилэтиловый сложный эфир [4-[[2-(диэтоксифосфинил)этил] (2-этокси-3,4-диоксо-1-циклобутен-1-ил) амино]бутил] карбаминовой кислоты в виде желтого вязкого масла (16,8 г, 90).
Раствор 1,1-диметилэтилового сложного эфира [4-[[2-(диэтоксифосфинил) этил] (2-этокси-3,4-диоксо-1-циклобутен-1-ил)амино] бутил]карбаминовой кислоты (16,8 г, 35 ммолей) в 96% муравьиной кислоте (100 мл) получали в атмосфере азота и перемешивали при комнатной температуре в течение ночи. Реакционную смесь затем концентрировали под вакуумом, чтобы получить липкое желтое масло, которое растворяли в абсолютном этаноле (125 мл) и по каплям добавляли в течение 1,5 ч в раствор диизопропилэтиламина (17 мл, 98 ммолей) в абсолютном этаноле (375 мл). После дефлегмации в течение ночи реакционную смесь концентрировали под вакуумом, а остаток растворяли в хлороформе (150 мл), предварительно адсорбировали на силикагеле, и подвергали очистке на оперативной хроматографической колонне (9 см диаметр, градиентное элюирование 5-10% метанолом в дихлорметане), чтобы получить диэтиловый сложный эфир [2-(9,10-диоксо-2,7-диазабицикло[6,2,0] дец-1(8)-ен-2-ил)этил] фосфино- вой кислоты в виде бледно-желтого твердого вещества (9,65 г, 83% температура точки плавления 103-105оС).
ИК(КВr, см-1): 3440, 3230, 1795; МС (+FAB): 331 (МН+, 100), 109 (31).
1Н-ЯМР (СDСl3, 400 МГц): δ 7,14 широкий синглет, NH), 4,16-4,05 (мультиплет, 6Н), 3,39-3,36 (мультиплет, 4Н), 2,17 (дублет триплетов, J=19 и 7,5 Гц, 2Н), 1,74-1,68 (мультиплет, 4Н), 1,33 (триплет, J=7 Гц, 6Н).
Вычислено, С 50,91; Н 7,02; N 8,48.
С24Н23N2O5P
Найдено, С 50,76; Н 6,82; N 8,37.
В раствор диэтилового сложного эфира [2-(9,10-диоксо-2,7-диазабицикло[6,2,0]дец- -1(8)-ен-2-ил)этил]алкилфосфиновой кислоты (7,60 г, 23 ммолей) в сухом 1,2-дихлорэтане (300 мл) в атмосфере азота добавляли бромтриметилсилан (25 г, 160 ммолей). Реакционную смесь подвергали дефлегмации 25 мин, а затем концентрировали под вакуумом, чтобы получить темно-рыжее масло, которое растворяли в воде (250 мл). Воду промывали простым эфиром (3х250 мл), а затем концентрировали под вакуумом, чтобы получить желто-оранжевое твердое вещество, которое подвергали рекристаллизации из воды (100 мл), чтобы получить [2-(9,10-диоксо-2,7-диазабицикло[6,2,0]дец-1(8) -ен-2-ил)этил]фосфиновую кислоту, гидрат в форме рыжевато-коричневого твердого вещества (3, 80 г, 60% температура точки плавления 266-271оС, разлож.).
ИК (КВr, см-1): 3350, 3260, 1800; МС (-FAB): 273 (М-Н).
1Н-ЯМР (ДМСО, 400 МГц): δ 8,32 (широкий синглет, NH), 3,94-3,87 (мультиплет, 2Н), 3,31-3,28 (мультиплет, 2Н), 3,22-3,18 (мультиплет, 2Н), 1,96-1,87 (мультиплет, 2Н), 1,61-1,49 (мультиплет, 4Н).
Вычислено, С 41,10;Н 5,86; N 9,59.
С10Н15N2О5Р ˙H2O
Найдено, С 41,22; Н 5,76; N 9,65.
При
помощи процедуры, аналогичной процедурам из примеров 7 и 8, получали следующие соединения:
Пример Соединение 13 [(E)-3-(7,8-диоксо-2,5-
диазабицикло[4,2,0]окт-1(6)-
ен-2-ил)-1-пропенил] фосфи-
новая кислота 14 [3-(7,8-диоксо-2,5-
диазабицикло[4.2.0]окт-
1(6)-ен-2-ил)пропил]фос-
финовая кислота
Устанавливали, что
соединения, являющиеся предметом настоящего изобретения, являются конкурирующими НМДА-антагонистами; это делали при помощи определения их способности ингибировать связывание
3-(2-карбоксипиперазинил-4-ил)пропил-1-алкилфосфиновой кислоты (СРР), известного конкурирующего НМДА-антагониста, с НМДА-рецепторами в гомогенате клеток головного мозга крысы. При помощи процедуры,
аналогичной процедуре из работы Мерфи и др. I. Pharmacol. Exp. Therap. т. 240 (3), стр. 778 (198), гомогенаты клеток головного мозга, из которых предварительно удаляли эндогенный глютамат,
инкубировали с (3Н) СРР (8 нМ), соединением, подлежащим испытанию, а затем буфером, причем с каждым 15 мин при 23оС, чтобы получить финальный объем в 2 мл. Количество радио-лиганда,
демонстируемое испытываемым соединением, выражали в процентах от радио-лиганда, показываемого НМДА. Ингибиторную концентрацию испытываемого соединения, которая будет вытеснять 50% меченного СРР,
определяли из оценок типа доза-ответ.
Свойства этих соединений, кроме того, устанавливали непосредственно при помощи подтверждения свойств НМДА-антагониста представителей соединений на самцах мышей Суисс-альбино (штамм СД-1, Чарлз Ривер) весом 18-22 г через 18 ч после лишения корма, которых помещали в камеры для наблюдения на 30 мин. Мышей предварительно обрабатывали представителями испытываемых соединений, затем, через тридцать минут, НМДА (195 мг/кг, внутрибрюшинным способом, ЕД90-доза для общего миоклонуса). Затем за мышами наблюдали 30 мин, фиксируя скрытое начало общего миоклонуса (неконтролируемое царапание задними лапами конечностей и/или внезапное подергивание мышц туловища с потерей рефлекса подъема на задние лапы) и гибель. Из последнего определяли ЕД50 для выживания. В этой стандартной экспериментальной процедуре испытаний испытывали конкретные соединения и их активность, при помощи которой определяли активность для всех соединений, приведенных в таблице.
Когда смещение СРР-связывания выражается в виде процентов, М-концентрация задается в виде концентрации в круглых скобках. Аналогично, когда ингибирование указано в процентах летальности, доза содержится в круглых скобках.
Таким образом, соединения, являющиеся предметом изобретения, подтверждают способность противостоять индуцированной НМДА летальности ин виво у мышей. Они конкурируют с 3-(2-карбоксипиперазинил-4-ил)-пропил-1-фосфиновой кислотой (СРР), известным конкурирующим НМДА-антагонистом, за использование сайта связывания. Соединения изобретения, таким образом, являются конкурирующими НМДА-антагонистами.
Соединения по изобретению являются активными против индуцированной НМДА летательности и являются эффективными при лечении расстройств ЦНС (центральной нервной системы) таких как судороги, разрушение клеток головного мозга и подобные нейродегенеративные расстройства, включая старческое слабоумие, заболевание Алзхаймера, хорею Хантингдона, удар, гипогликемию, церебральный паралич, церебральную ишемию, эпилепсию и оливопонто мозжечковую атрофию.
Следовательно, наряду с новыми соединениями, (см, выше), предлагается способ предотвращения нейродегениративных расстройств, индуцированных перевозбуждением НМДА-рецепторов в головном мозге и спинном мозге возбуждающими аминокислотами, который содержит применение к млекопитающему, страдающему таким дегеративным заболеванием, противоконвульсивного, нейрозащищающего количества НМДА-антагониста, являющегося предметом настоящего изобретения.
Как таковые, соединения по изобретению могут быть применены, чистыми или с фармацевтическим носителем, к пациенту в случае необходимости. Фармацевтический носитель может быть твердым или жидким.
Поэтому в соответствии с изобретением предлагается также фармацевтическая композиция, содержащая соединение формулы 1, которая была приведена и определена выше, или его приемлемую с фармацевтической точки зрения соль, и приемлемый с фармацевтической точки зрения носитель.
Твердый носитель может включать один или несколько материалов, которые могут также действовать, как ароматизирующие агенты, смазочные агенты, агенты, увеличивающие растворимость, суспендирующие агенты, наполнители, агенты, увеличивающие скольжение, вспомогательные агенты для прессовки, связывающие агенты или агенты, разрыхляющие таблетки; это может быть также образующий капсулу материал. В порошках носителем может быть тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным ингредиентом. В таблетках, активный ингредиент смешивают с носителем, обладающим необходимыми свойствами прессовки, в соответствующих пропорциях, а затем смеси придают искомые форму и размер. Порошки и таблетки в предпочтительном варианте содержат до 99% активного ингредиента. Соответствующие твердые носители, например, включают фосфат кальция, стеарат магния, тальк, сахар, лактозу, декстрин, крахмал, желатин, целлюлозу, метил целлюлозу, натрий карбоксиметилцеллюлозу, поливинилпирролидин, воски с низкой температурой плавления и ионообменные смолы.
Жидкие носители используют при получении растворов, суспензии, эмульсии, сиропов, эликсиров и композиции, находящихся под давлением. Активный ингредиент может быть растворен или суспендирован в приемлемых с фармацевтической точки зрения жидких носителях таких, как вода, органический растворитель, смесь того и другого, или приемлемых с фармацевтической точки зрения маслах или жирах. Жидкий носитель может содержать другие, приемлемые с фармацевтической точки зрения, добавки такие, как агенты, увеличивающие растворимость, эмульгаторы, буферы, консервирующие агенты, вкусовые, ароматизирующие агенты, суспендирующие агенты, загущающие агенты, окрашивающие агенты, регуляторы вязкости, стабилизаторы или осмо-регуляторы. Соответствующие примеры жидких носителей для стоматического и парентерального применения включают воду (частично содержащую добавки, упомянутые выше, например, производные целлюлозы, в предпочтительном варианте раствор натрий карбоксиметил целлюлозы), спирты (включая одноатомные спирты и многоатомные спирты, например, гликоли) и их производные, и масла (например, фракционированное кокосовое масло и арахисовое масло). Для парентерального применения носителем могут быть также маслянистые сложные эфиры такие, как этил олеат и изопропил миристат. Стерильные жидкие носители используют в композициях стерильных жидких форм для парентерального применения. Жидкий носитель для композиции, содержащихся под давлением, может быть галогенизированным углеводородом или другим, приемлемым с фармацевтической точки зрения, распыляющим материалом.
Жидкие фармацевтические композиции, которые являются стерильными растворами или суспензиями, могут быть использованы, например, для внутримышечных, внутрибрюшинных или подкожных инъекций. Стерильные растворы могут быть также применены внутривенно. Эти соединения могут быть также применены стоматическим способом в виде композиции либо в форме жидкости, либо твердого вещества.
В предпочтительном варианте, фармацевтическая композиция находится в форме единичной дозы, например, таблеток или капсул. В такой форме композицию подразделяют на единичную дозу, содержащую соответствующие количества активного ингредиента; формы единичных доз могут быть упакованными композициями, например, упакованными порошками, пузырьками, ампулами, предварительно заполненными шприцами или упаковками, содержащими жидкость. Формой единичной дозы может быть, например, сама капсула или таблетка, или это может быть соответствующее число любых таких композиций в упакованной форме.
Чтобы определить эффективное количество соединения, которое необходимо применить для облегчения при дегенеративной дисфункции ЦНС, врачу необходимо только оценить воздействия данных НМДА-антагонистов у пациента при помощи постепенного увеличения стоматической дозы от примерно 1 мг/кг до примерно 20 мг/кг до тех пор, пока не будет достигнут целевой уровень симптоматического воздействия. Режим последующих доз может быть затем модифицирован, чтобы добиться целевогоо результата, с областью изменения от примерно 1 до 100 мг/день. Аналогичные приемы можно использовать при определении области эффективных доз при внутривенном или внутримышечном применении. Если соединения используют профилактически с целью прекращения снижения функции узнавания, например, при заболевании Алзхаймера, можно использовать более предметный подход, например, при помощи связывания дозы препарата с улучшенной реакцией памяти или аналогичными целевыми реакциями, которые могут быть связаны со смягчением перевозбуждения под воздействием возбуждающих аминокислот.
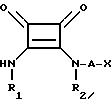
Реферат
Использование: в медицине, в частности в качестве противоконвульсивных и нейрозащищающих веществ в случаях, сопровождающихся избыточным высвобождением возбуждающих аминокислот. Сущность изобретения: продукт ф-лы I, где R1 водород, C1-C6 алкил или фенилалкил с C7-C12 R2 - водород C1-C6 алкил, C2 -C6 алкенил или фенилалкил с C7-C12 или R1 и R2 вместе группа: CH2-CH2-(CH2)-CR6R7-CH2 или -(CH2)-CR8R9-CR10R11-CH2- в которых R6 R8 R10 независимы друг от друга водород, C1-C6 алкил или гидроксил, R7, R9, R11 независимы друг от друга водород или C1-C6 алкил; А- C1-C6 -алкилен или C2-C6 -алкенилен; Х группа -C(O)OR3 при R3 водород, C1-C6 -алкил или P(O)(OR4)(OR5) при R4, R5 независимы друг от друга водород, C1-C6 -алкил, 3,5-диоксо-1,2,4-оксадиазолидин-2-ил или 5-тетразолил, или их фармацевтически приемлемые соли. Реагент 1: соединения ф-лы II, где R1 см. выше, R12 -алкокси, аралкоксизамещенная группа, содержащая C1-C5 или C7-C12 Реагент 2: соединение ф-лы: HNR2 -A-X где R2 А и Х см. выше. Условия реакции: в среде растворителя при комнатной температуре или при нагревании. Структура соединений ф-лы I и II:

Формула

где R1 водород, С1 С6-алкил, или фенил-С1 - С7-алкил;
R2 водород, С1 С6-алкил, С2 - С6 -алкенил, или фенил-С7 С12-алкил,
или R1 и R2 вместе представляют -CH2CH2 - CH2C(R6)(R7) CH2- или -CH2C(R8)(R9) - C(R10)(R11)CH2-, где R6, R8 и R10 независимо друг от друга водород, С1 С6-алкил или гидроксил, R7, R9 и R11 независимо друг от друга - водород или С1 С6-алкил;
A С1 С6-алкилен или С2 С6-алкенилен;
X группа CO2R3, где R3 водород или С1 - С6-алкил; P(O)(OR4)(OR5), где R4 и R5 независимо друг от друга водород, С1 С6-алкил, 3,5-диоксо-1,2, 4-оксадиазолидин-2-ил или 5-тетразолил,
или их фармацевтически приемлемые соли.
22.01.91 при R1 водород, С1 С6-алкил, или фенил-С7 С12-алкил; R2 водород, С1 - С6-алкил, С2 С6-алкенил, или фенил-С7 - С12-алкил, или R1 и R2 вместе представляют -CH2CH2CH2C(R6)(R7) CH2- или -CH2 C(R8)(R9) - C(R10)(R11)CH2-, где R6 R11 водород, A и X имеют указанные значения;
17.12.91 при R1 и R2 вместе представляют -CH2CH2CH2C(R6)(R7) CH2- или -CH2C(R8)(R9) C(R10)(R11) - CH2-, где R6, R8 и R10 С1 - С6-алкил или гидроксил; R7, R9 и R11 С1 С6-алкил, A и X имеют указанные значения.
Комментарии