Способ иодирования ароматических соединений - RU2506254C2
Код документа: RU2506254C2
Чертежи







Описание
Данное изобретение относится к способу получения поли-иодированных ароматических соединений. Точнее, оно относится к способу, включающему прямое иодирование 3,5-дизамещенных анилинов до соответствующих 3,5-дизамещенных-2,4,6-трииоданилинов, которые применимы как промежуточные продукты для синтеза контрастных сред для рентгеновских лучей и для приготовления самих контрастных сред.
Уровень техники
Иодированные контрастные средства являются хорошо известными соединениями, широко применяемыми в диагностических методиках с использованием фотоизображения в рентгеновских лучах. Подходящие примеры указанных соединений включают, например, диатризоат, иоталамат, иокситаламат, метризоат, иоксенол, иомепрол, иопамидол, иопентол, иопромид, иоверсол, иоксилан, йодиксанол, иосаркол, иогуламид, иоглюнид, иоглюамид, ацетризоат, йодамид, иоцетамид и метризамид, причем все имеют мономерную структуру, и иоксаглат, иотролан, иотасул, иодипамид, иокармат, иодоксамат, иотроксат, и тому подобное, которые вместо этого, являются димерами. Дополнительные примеры иодированных контрастных средств описаны, например, в WO 94/14478 (Бракко/Bracco).
В качестве общей особенности их химическая структура имеет трииодированное ароматическое ядро, которое обеспечивает повышенный контрастный эффект.
Упомянутые соединения могут быть приготовлены разными способами, которые обычно включают иодирование данных ароматических субстратов, например, подходящих 3,5-дизамещенных фенолов, которые претерпевают трииодирование в доступных 2, 4 и 6 положениях, таким образом, приводя к соответствующим 3,5-дизамещенным-2,4,6-трииодфенолам. Затем они, в свою очередь, могут быть дополнительно превращены и обработаны в рамках так называемой перегруппировки Смайлса до ожидаемых конечных соединений. Для общей ссылки по вышеприведенному синтетическому способу и перегруппировки Смайлса см., например, WO 88/09328, WO 97/05097 и WO 00/32561 (Бракко/Bracco).
В альтернативном случае, ароматическое иодирование может быть выполнено на подходящих анилинах, так чтобы получить соответствующие 2,4,6-трииоданилиновые производные, которые должны быть далее превращены и переработаны в конечное радиографическое средство, например, как раскрыто в US 5075502.
Стадия иодирования может быть выполнена с использованием различных методик.
В этом отношении в промышленных способах, используемых для получения вышеуказанных радиографических контрастных средств, иодирование ароматического кольца обычно выполняют с использованием растворов монохлорида иода (ICl) в концентрированной хлористоводородной кислоте (HCl) (44,5% I и 14% HCl) при высокой температуре (примерно 90°С) или, в альтернативном случае, при помощи аналогичных иодирующих средств, таких как, например, KICl2 или NaICl2 в водном растворе; см., для общей справки, WO 92/14695 (Guerbet), US 5013865 (Mallinckrodt), WO 96/37458 и WO 96/37459 (Fructamine).
Вышеуказанные методы имеют главные недостатки, обусловленные экстремально кислыми рабочими условиями, которые становятся более жесткими из-за HCl, получающейся в ходе реакции, и коррозионными свойствами и ограниченным периодом хранения иодирующих средств.
Кроме того, относящиеся к этому случаю проблемы в основном возникают из-за присутствия атомов хлора внутри самих иодирующих средств (получаемых при высокой температуре реакции, необходимой для полного иодирования анилиновых субстратов), что может приводить к образованию трудно удаляемых хлорированных побочных продуктов, которые могут таким образом влиять на выходы реакции и чистоту конечных соединений.
С другой стороны, и с иной точки зрения, существует все более и более ощущаемая потребность иметь промышленные процессы производства, которые могут объединить низкую стоимость производства, высокую эффективность производства и свести до минимума влияние на окружающую среду.
Таким образом, были предприняты попытки направить усилия на новые методы иодирования, основанные на применении иодирующих средств, альтернативных монохлориду иода или его производным.
Среди них имеются, например, способы электрохимического иодирования 3,5-дизамещенных анилинов или вышеназванных 3,5-дизамещенных фенолов, раскрытых в WO 96/37461 и WO 2009/103666, соответственно.
Кроме вышеприведенных подходов, также было испытано альтернативное иодирование ароматического ядра иодом, подходящим образом активированным окислителем. Например, иодирование данных фенольных производных, именуемых орто-гидрокси замещенными ароматическими карбонильными соединениями, в присутствии молекулярного иода, активированного сильным окислителем, включая иодноватую кислоту, было описано Patil et al. в Tetrahedron Letters 2005, 46, 7179-7181, и в ARKIVOC 2006, 104-108.
Данная статья, однако, умалчивает о возможности использования такого раскрытого синтетического подхода, а именно, совместного использования молекулярного иода и окислителя для иодирования или поли-иодирования анилина или анилиновых производных.
US 2007/0219396 раскрывает способ получения 2-амино-5-иодбензойной кислоты иодированием 2-аминобензойной кислоты, солюбилизированной в уксусной кислоте, иодом и в присутствии окислителя, особенно пероксида водорода.
Данная заявка, однако, не упоминает и не предлагает возможности использования раскрытой процедуры для получения полииодированных соединений и, в частности, трииодированных анилиновых производных, которые, несомненно, вряд ли были бы достигнуты при раскрытых условиях иодирования, как подтверждено сравнительным примером 1 в последующей экспериментальной части.
Применение иода и иодноватой кислоты для получения 3-амино-2,4,6-трииодбензойной и 3,5-диамино-2,4,6-трииодбензойной кислот также упомянуто в Chem. Ber., 1897, 30 (2), 1943-1948 и в Chem. Ber., 1896, 29 (3), 2833-2839, соответственно.
Данные ссылки являются совершенно недостаточными в отношении полного описания используемых условий иодирования, так что предотвращается возможность их точного воспроизводения.
В любом случае, раскрытые условия иодирования и количество иодирующего средства, в частности, иодноватой кислоты, оказывается совершенно недостаточным, чтобы осуществить трииодирование субстрата, по меньшей мере со значительным выходом и чистотой, как обсуждено более подробно в сравнительном примере 2 экспериментальной части ниже. Кроме того, в обеих процитированных статьях, полученный коричневый осадок должен быть промыт серной кислотой, растворен в разбавленном аммиаке и затем осажден серной кислотой, чтобы иметь продукт желаемой чистоты.
В этом отношении стоит отметить, что применение сильных окисляющих условий с анилином или даже галогенированными анилинами, как известно, ведет к образованию смесей окрашенных побочных продуктов, в основном азосоединений, полученных из-за реакций окислительной конденсации, затрагивающих ароматическую аминогруппу (см., например, Erich Baer and Anthony L. Tosoni, J. Am. Chem. Soc., 1956, 78 (12), 2857-2858), в то время, как вся вышеупомянутая статья не обращается к данной проблеме и даже не предполагает, как ее решить.
Напротив, необходимость сбора промежуточных продуктов процесса и конечных соединений с высокой степенью чистоты имеет весьма высокую важность, чтобы оптимизировать в значительной степени стадии очистки, требуемые для конечного агента, который должен соответствовать жесткому профилю чистоты и пределам, установленным Фармакопеей, в частности, для продуктов, предназначенных для введения.
Например, аналитические спецификации, установленные Фармакопеей ЕР для 5-амино-2,4,6-трииодизофталевой кислоты, представляют собой:
Потери при сушке: ≤3,5%
Степень чистоты: 98,0-102%
Золы: ≤1,0%
Общие родственные соединения: ≤1% (предполагается как сумма всех известных и неизвестных примесей, в основном представленных частично иодированными соединениями и хлорированными соединениями), из которых сумма хлорированных примесей должна составлять ≤0,35%.
Авторы в настоящей работе установили, что трииодирование подходящих 3,5-дизамещенных анилинов может быть выполнено преимущественно с высокими выходами и чистотой при использовании системы для иодирования, содержащей молекулярный иод и окислитель, преодолевающие вышеупомянутые основные недостатки.
Предмет изобретения
Данное изобретение относится к способу трииодирования 3,5-дизамещенных анилинов иодом, активированным подходящим образом, а также к способу получения контрастных средств для рентгеновских лучей, включающему вышеупомянутую стадию иодирования.
Точнее, первый предмет данного изобретения представлен способом получения 5-амино-2,4,6-трииодизофталевой кислоты формулы (II)

и этот способ включает иодирование 5-аминоизофталевой кислоты формулы (I) или ее соли

молекулярным иодом в присутствии подходящего окислителя.
Способ изобретения является особенно полезным, так как он предоставляет возможность для полного трииодирования 5-аминоизофталевой кислоты формулы (I) или соответствующей ее соли, и приводит к соответствующей 5-амино-2,4,6-трииодизофталевой кислоте формулы (II) с высокими выходами и чистотой.
Является замечательным и отличным от предшествующей информации по способности к окислению анилинов, что на вышеупомянутый способ не влияет присутствие побочных продуктов, получающихся вследствие частичного иодирования ароматического кольца или окислительной конденсации, происходящей на аминогруппе.
Преимущественно, поэтому, способ изобретения не требует какой-либо стадии очистки полученного трииодированного соединения, которое выделено из неочищенного раствора фильтрованием и, удовлетворяя аналитическим спецификациям для промышленно полученного промежуточного продукта, может, следовательно, быть использовано как таковое на следующей стадии реакции с иодированным средством, представляющим интерес.
Кроме того, за счет потребления эффективным образом всего добавленного молекулярного иода и образования воды как единственного побочного продукта реакции, согласно деталям ниже, необходимость последующих стадий для выделения и повторного использования не прореагировавшего иода и для обработки промышленных циркулирующих потоков, может быть сведена к минимуму в очень значительной степени.
Как сообщено ранее, в способе настоящего изобретения реакция иодирования, приводящая к образованию 5-амино-2,4,6-трииодизофталевой кислоты формулы (II), происходит с молекулярным иодом (I2) в присутствии подходящего окислителя по хорошо известному механизму электрофильного замещения.
Для данного случая эффективное иодирующее средство может быть представлено катионами иода (I+), по меньшей мере часть которых сначала генерируется молекулярным иодом (I2), тогда как не прореагировавшие противоионы иодида (I-), полученные таким образом, легко окисляются окислителем обратно до молекулярного иода, или даже до катионов иода с более высокой степенью окисления, что делает их все еще пригодными для иодирования ароматического кольца.
Исходя из вышеизложенного, если иначе не указано, подходящие окислители для применения по способу изобретения, представляют собой окислители, обычно используемые в промышленном масштабе, и которые способны к окислению иодидных ионов до более высокого состояния окисления, активного для иодирования, как детализировано в последующих параграфах.
Подходящие примеры окислителей, таким образом, включают, например, азотную кислоту, серную кислоту, иодноватую кислоту, триоксид серы, пероксид водорода, озон, и тому подобное. Вообще говоря, выбор окислителя будет зависеть от нескольких факторов, среди которых есть, например, условия процесса, позволяющие им должным образом проявить их окислительную функцию в течение хода реакции, так чтобы привести к образованию требуемого соединения, а также их доступность.
Как таковой, и согласно первому варианту осуществления способа изобретения, окислитель предпочтительно выбран между пероксидом водорода и иодноватой кислотой, причем последняя является даже более предпочтительной.
Когда молекулярный иод используется в присутствии иодноватой кислоты (HIO3), фактически, не прореагировавшие ионы иодида, образованные в реакции иодирования, превращаются обратно в молекулярный иод по так называемой реакции Душмана/Dushman, согласно следующей схеме 1 реакции:
IO3-+5I-+6H+→3I2+3H2O
Замечательно, что данная реакция кроме того приводит к легкому восстановлению иодатных ионов (IO3-) в молекулярный иод, еще доступный для иодирования ароматического кольца (см., например, Furuichi, R. and Liebhafsky, H.A. Radioactive iodine exchange and the Dushman reaction. Bull. Chem. Soc. Japan 1973, 46, 2008-2010 и Bull. Chem. Soc. Japan 1975, 48, 745-750).
В результате этого достигается полное трииодирование 5-аминоизофталевого субстрата, с получением преимущественно требуемого соединения формулы (II) с высокими выходами и чистотой, потреблением стехиометрического количества иодирующих средств, которое рассчитано в виде суммы добавленных как I2, так и HIO3, согласно следующей общей схеме реакции 2.

Иначе говоря, объединенное использование иода и иодноватой кислоты, согласно предпочтительному варианту осуществления изобретения, предоставляет возможность для полного иодирования ароматического субстрата формулы (I) с устранением, с одной стороны, необходимости в каком-либо избытке иодирующего средства, особенно молекулярного иода, и с другой, образования побочных продуктов, особенно не реакционно-способных полииодидных ионов, например, ионов I3, получающихся главным образом от комбинации I2 c ионами иодида.
В этом отношении квалифицированному специалисту ясно, что эквивалентное отношение между субстратом 5-аминоизофталевой кислоты и рассмотренным иодирующим средством, которое упомянуто в виде суммы как I2, так и HIO3, должно быть равно по меньшей мере 1:3 согласно предшествующей общей схеме 2.
При допустимости данной точки зрения, в способе настоящего изобретения трииодирование 5-аминоизофталевого субстрата иодом и иодноватой кислотой будет выполнено с использованием по меньшей мере одного моля молекулярного иода для каждого моля 5-аминоизофталевого субстрата формулы (I). Предпочтительно, молярное соотношение между иодом и 5-аминоизофталевым субстратом (I) [I2/(I)] будет изменяться от 1 до 1,5, предпочтительнее от 1 до 1,3; еще предпочтительнее, трииодирование 5-аминоизофталевого субстрата иодом и иодноватой кислотой будет выполнено с использованием только 1,2 моля иода на моль субстрата (I).
С другой стороны, из-за стехиометрии вовлеченной реакции, молярное отношение между иодом и иодноватой кислотой будет равно по меньшей мере 1:0,5, тогда как молярное отношение между 5-аминоизофталевым субстратом (I) и иодноватой кислотой будет равно по меньшей мере 1:0,6.
В соответствии с этим, в особенно предпочтительном варианте осуществления изобретения трииодирование 5-аминоизофталевого субстрата иодом и иодноватой кислотой будет выполняться с использованием молярного отношения 5-аминоизофталевый субстрат (I):иод:иодноватая кислота, равного 1:1,2:0,6.
Однако, небольшой избыток свыше минимального стехиометрического количества иодноватой кислоты над молекулярным иодом может быть, необязательно, использован с одинаково хорошими результатами, как показано в экспериментальной части.
В соответствии с этим, в одном другом варианте осуществления изобретения, будет использовано молярное отношение иода к иодноватой кислоте, изменяющееся от 1:0,5 до примерно 1:1 и, предпочтительнее, от 1:0,5 до примерно 1:0,8.
В этом отношении, в конечную реакционную среду может быть добавлено, например, минимальное количество бисульфита натрия, чтобы разложить любые необязательные остаточные иодирующие вещества. В данном случае, оптимальное количество может, например, быть определено потенциометрически как минимальное количество бисульфита, приводящее к окислительно-восстановительному потенциалу конечной смеси предпочтительно ниже, чем 250 мВ.
Реакция иодирования изобретения, включающего применение иодирующей системы I2/HIO3, которая представлена выше, предпочтительно проводится в присутствии полярного растворителя, предпочтительно протонного растворителя, и в кислых условиях.
Не ограничивающие примеры подходящих растворителей могут, таким образом, включать, например, воду или водные растворители, включающие водные физиологические растворы, низшие спирты С1-С4, например, метанол или этанол и их водно-спиртовые смеси, диоксан, гликоли, такие как, например, диэтиленгликоль, триэтиленгликоль, и полиэтиленгликоли, подобные PEG 600, PEG 1000 или PEG 2000 или их смеси, и их водные смеси.
Предпочтительные растворители представляют собой воду или водные растворы, метанол, этанол и диоксан, а также их смеси с водой или водным раствором.
В особенно предпочтительном варианте осуществления изобретения способ иодирования выполнен в воде или водных растворителях, которые в значительной степени вносят вклад в снижение цены и во влияние на окружающую среду данного способа.
В еще одном наиболее предпочтительном варианте осуществления изобретения способ иодирования проводят с неочищенным водным раствором, полученным от промышленного процесса для получения исходного 5-аминоизофталевого субстрата, например, выполненного как описано в WO 96/37459, необязательно разбавленного водой и подходящим образом подкисленного.
Специфические кислотные условия достигаются в присутствии подходящей кислоты включающей, например, фосфорную, метансульфоновую или серную кислоту, например, 96% H2SO4. Предпочтительно, подходящие кислотные условия получают при использовании 96% H2SO4, например, в количестве, изменяющемся в интервале от примерно 0,5 до 2 моль и, предпочтительно, от 0,7 до 1,5 моль H2SO4 на моль субстратного соединения (I).
Для данного случая и согласно предпочтительному варианту осуществления, реакция иодирования выполняется при рН (реакционной смеси) меньше чем 3,5, предпочтительно заключенного в интервале от 1 до 3,0, еще предпочтительнее, от 1,5 до 2,5, предпочтительно достигаемого при использовании концентрированной H2SO4.
В этом отношении стоит отметить, что в случае подкисления до данного упомянутого последним интервала значений серной кислотой, рН реакции преимущественно поддерживается сам по себе на уровне от 1,5 до 2,5 на протяжении времени реакции, тогда как для сохранения рН около 3 является необходимым добавление основания, например, разбавленного NaOH.
Интересно, что вопреки тому факту, что для вышеприведенных условий рН известно, что они эффективно дезактивируют любое электрофильное замещение на анилиновых субстратах, данные условия, очевидно неблагоприятные, позволяют получить 5-амино-2,4,6-трииодизофталевую кислоту с очень высокими выходами, в основном не загрязненную побочными продуктами частичного иодирования или окрашенными примесями.
Вместо этого, при более высоком рН, например, больше чем 4, может быть получен требуемый иодированный продукт, но с меньшими выходами и чистотой, так что требуется дополнительная очистка, чтобы достигнуть аналитических спецификаций, установленных для промышленно производимого промежуточного продукта.
При обработке в таких кислых условиях, ароматический субстрат, претерпевающий трииодирование, представлен 5-аминоизофталевой кислотой формулы (I), либо используемой в качестве исходного продукта способа либо, в альтернативном случае, образованной in situ (на месте) из соответствующей соли.
Эта последняя, если иначе не указано в данном описании, предпочтительно выбрана из солей 5-аминоизофталевой кислоты с щелочными или щелочноземельными металлами такими как, например, соли натрия, лития, калия, кальция или магния.
Особенно предпочтительна из них натриевая соль 5-аминоизофталевой кислоты, которая может быть использована как таковая, т.е. как чистое соединение или, в альтернативном случае, как содержащаяся в неочищенном растворе, непосредственно полученном на предыдущей стадии способа для приготовления трииодированных контрастных средств, например, иопамидола.
Интересно, что согласно вышеприведенным условиям обработки, т.е. в присутствии кислой водной окружающей среды, 5-амино-2,4,6-трииодизофталевая кислота неожиданно получается с высокими выходами и чистотой, вопреки практической нерастворимости исходного ароматического субстрата, предназначенного для иодирования.
Когда 5-аминоизофталевая кислота используется в качестве исходного продукта, фактически, надлежащее количество субстратного соединения сначала суспендируется и таким образом сохраняется в реакционной среде, прежде чем происходит реакция иодирования. В альтернативном случае, когда используется водный раствор соответствующей соли, например, исходя из промышленного водного раствора соответствующей натриевой соли, кислая окружающая среда является такой, чтобы стимулировать осаждение нерастворимой кислоты формулы (I), которая поддерживается в суспензии с помощью обычных средств, например, при магнитном или механическом перемешивании.
То же самое относится к иоду, который загружают в виде твердого вещества в суспензию изофталевого субстрата, подкисленного надлежащим образом, как указано.
Для данного случая надлежащее количество иодноватой кислоты может быть, затем добавлено к полученной суспензии сразу или, в альтернативном случае, постепенно, либо непрерывно на протяжении всего периода, либо частями согласно обычным способам, тем самым вызывается постепенная частичная солюбилизация 5-аминоизофталевого субстрата, который таким образом постепенно превращается в требуемый трииодированный продукт.
Точнее, и согласно последующей экспериментальной части, иодноватая кислота может быть добавлена быстро, например, за несколько минут или даже сразу в реакционную суспензию, подкисленную более умеренно, например, до рН≥2,5, т.е. около 3. Вместо этого, при обработке в более сильнокислых условиях, т.е. при рН примерно 2 или даже ниже, предпочитают медленное добавление иодноватой кислоты, которое может быть осуществлено в продолжение периода времени, например, за период вплоть до 6 часов, и предпочтительно, за период от 2 до 6 часов.
В этом отношении, c успехом может быть использован окислитель в интервале концентраций, например, от 8 до 50% (мас./мас.).
Реакция иодирования проводится при температуре, изменяющейся в интервале от 50°С до 85°С.
Например, в одном варианте, температура реакции в ходе способа может поддерживаться постоянной до величины, заключенной от примерно 60°С до 85°С и, предпочтительно, от примерно 65°С до 80°С, посредством обработки согласно обычным методам.
В альтернативном случае, все реагенты могут быть внесены при комнатной температуре, таким образом образуется смесь, которую затем нагревают до температуры, находящейся в интервале от 65°С до 80°С, или, в свою очередь, иодирующие средства (I2 и HIO3) могут быть добавлены в суспензию, нагретую до примерно 45°С и затем следует поднимать и поддерживать температуру реакции от 65°С и 80°С, согласно следующей экспериментальной части.
Время реакции может меняться согласно выбранным условиям обработки и обычно может находиться в интервале от примерно 2 до примерно 10 часов, предпочтительнее от 5 до 8 часов.
Обычно обработкой при ранее данных температурах, в рамках способа можно достигать точки кипения растворителей, особенно когда используются более низкокипящие растворители, подобные метанолу. Кроме того, также может происходить частичная сублимация иода, даже если сублимированное количество остается незначительным, когда температуру реакции сохраняют внутри первого интервала величин.
Тем не менее, может, использоваться, например, стандартное охлаждающее или конденсирующее оборудование, чтобы конденсировать как растворитель, так и сублимированный иод, который затем возвращают обратно в реакционную смесь согласно общепринятым методам, например, добавлением небольших количеств свежего растворителя.
В этом отношении стоит отметить, что хотя использование, рекомендуемое цитируемой публикацией (US 2007/0219396), уксусной кислоты в качестве растворителя реакции, решает проблему солюбилизации иода, это, наоборот, не вносит вклада в повышение солюбилизации 5-аминоизофталевой кислоты, которая остается нерастворимой, даже нагретая до 80°С. Кроме того, что неблагоприятно, это не создает возможности для простого выделения продукта иодирования, а именно, 5-амино-2,4,6-трииодизофталевой кислоты, которая не осаждается количественно из уксусной кислоты, не охлажденной даже до комнатной температуры, если ее надлежащим образом не разбавить водой.
К тому же, растворимость HIO3 в уксусной кислоте является очень низкой. Поэтому, когда окислитель добавлен в уксусную реакционную среду не разбавленную надлежащим образом водой, согласно условиям, рекомендуемым цитируемой публикацией, это приводит к образованию негомогенной фазы, которая значительно снижает его эффективность в активации иода, как подтверждено данным сравнительным примером 1 последующей экспериментальной части.
Вышеописанные недостатки могут быть разрешены при обработке в условиях иодирования согласно статьям Chem. Ber., которые вместо этого рекомендуют применение кислой водной среды, разбавленной так чтобы достигать, с одной стороны, требуемой солюбилизации исходного субстрата, но, с другой стороны, вносить в наибольшей степени возможный вклад в то, чтобы препятствовать осаждению трииодированного соединения по изобретению, а именно, 5-амино-2,4,6-трииодизофталевой кислоты, которая не осаждается из неочищенного раствора, даже охлажденного до комнатной температуры, как подтверждено представленным сравнительным примером 2 последующей экспериментальной части.
Кроме того, цитируемая статья Chem. Ber., 1987, 30(2), 1943-1948 рекомендует применение иодирующего раствора, приготовленного частично солюбилизацией твердого I2 в водном КОН (или NaOH) c последующим добавлением твердой HIO3 и последующим разбавлением водой.
Для данного случая кроме того, процитированная статья не указывает ни на объем используемого водного раствора КОН, ни на его концентрацию; стоит отметить, что предлагаемое количество HIO3, используемой, чтобы приготовить упомянутую иодирующую смесь, является недостаточным, чтобы обратно превратить все иодидные ионы, образованные в реакции иодирования. Данная недостаточность означает, с одной стороны, необходимость использования избытка иода выше минимального стехиометрического количества, требуемого, в противоположность этому, в способе иодирования данного изобретения. С другой стороны, это дополнительно приводит к нежелательной аккумуляции иодидных ионов в реакционной среде, что вероятно может влиять на чистоту продукта иодирования и его соответствие аналитическим спецификациям для промышленно полученного промежуточного продукта.
Квалифицированному специалисту ясно, что альтернативные иодирующие системы из числа опубликованных ранее и включающие молекулярный иод в присутствии окислителя иного, чем иодноватая кислота например, пероксид водорода, и условия их обработки, также должны рассматриваться, как включенные в объем изобретения.
Из всего вышеизложенного квалифицированному практику должно быть ясно, что способ данного изобретения по существу включает: получение суспензии 5-аминоизофталевой кислоты в водном растворителе, подкисленном надлежащим образом, а именно имеющим рН ниже, чем 3,5, и добавление твердого I2 и HIO3 к упомянутой суспензии.
Более детально и согласно практическому предпочтительному варианту осуществления изобретения, надлежащее количество 5-аминоизофталевого субстрата суспендируют или солюбилизируют, в зависимости от обстоятельств, в водном растворителе, обычно в воде. Полученные раствор/суспензию сначала разбавляют до концентрации субстрата, изменяющейся в интервале от 8% до 3% (мас./мас.) и, предпочтительно, от 5% до 3%, и затем подкисляют до рН ниже, чем 3,5, предпочтительно около 2, подходящим количеством кислоты, например, 96% H2SO4.
Предпочтительно, в качестве исходного продукта используют сырой раствор, непосредственно полученный из промышленного процесса и включающий 5-аминоизофталевый субстрат в виде натриевой (главным образом, мононатриевая, хотя не исключена и динатриевая) соли в концентрации, обычно составляющей около 7-8%. Данный сырой раствор затем разбавляют, обычно водой, до представленного выше интервала концентраций и затем подкисляют до вышеупомянутых значений, например, посредством 96% H2SO4. Твердый I2 затем добавляют в полученную суспензию 5-аминоизофталевой кислоты, которую поддерживают при перемешивании и нагревают при значениях температуры, указанных ранее.
Надлежащее количество водного раствора HIO3 медленно добавляют в суспензию, что вызывает постепенное превращение 5-аминоизофталевого субстрата в требуемый триоидированный продукт.
Посредством обработки путем добавления HIO3 вплоть до завершения, образовавшаяся 5-амино-2,4,6-трииодизофталевая кислота осаждается из реакционной смеси в виде твердого вещества. В этот момент подкисление неочищенной реакционной смеси, например, с помощью 96% H2SO4 при рН около 1, и охлаждение смеси до комнатной температуры, способствует почти полному осаждению трииодированного соединения. Кроме того, добавление минимального количества бисульфита натрия (к конечной неочищенной смеси) позволяет гарантированно разложить любое необязательное остаточное иодирующее средство и получить еще более чистый твердый продукт, который фильтруют и сушат. Отфильтрованное соединение является чистым и легко может быть использовано на следующих стадиях для приготовления требуемого контрастного средства без необходимости какой-либо дополнительной очистки.
С другой стороны, только что полученная 5-амино-2,4,6-трииодизофталевая кислота формулы (II) затем может быть легко превращена в требуемое контрастное средство для рентгеновских лучей обработкой согласно известным методам.
В этом отношении, способ, являющийся предметом данного изобретения, имеет общую применимость и дает очень выгодный путь для получения иодированных контрастных средств, исходя из промежуточной 5-амино-2,4,6-триоидизофталевой кислоты.
Следовательно, дополнительный предмет данного изобретения представляет собой способ получения соединений формулы (III) ниже

в которой:
R и R', одинаковые или отличные друг от друга, означают группу, выбираемую из карбокси (-СООН), карбоксисложноэфирной (-COOR1) и карбоксамидо (-CONH2, -CONHR1 или -CONR2R3), где R1, R2 и R3, одинаковые или отличные друг от друга, представляют собой прямую или разветвленную С1-С4алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами, и
R4 и R5, одинаковые или отличные друг от друга, представляют собой водород или прямую или разветвленную С1-С6алкильную группу, необязательно замещенную одной или несколькими гидроксильными или С1-С6алкоксигруппами,
причем упомянутый способ включает получение промежуточного соединения формулы (II) по способу настоящего изобретения.
Предпочтительнее, упомянутый способ включает:
а) получение 5-амино-2,4,6-триоидизофталевой кислоты формулы (II)
иодированием 5-аминоизофталевой кислоты формулы (I) или ее соли
молекулярным иодом в присутствии подходящего окислителя;
b) превращение соединения формулы (II) в соответствующий дихлорангидрид кислоты, и
с) применение дихлорангидрида в качестве промежуточного соединения для получения требуемых соединений формулы (III).
Согласно упомянутому способу, стадия иодирования а) проводится, как широко обсуждалось в предыдущих разделах, в то время, как все последующие стадии, вся совокупность экспериментальных условий обработки и их дополнительные варианты должны быть выполнены согласно общим методам, опубликованным в данной области и включающим, в основном, превращение 5-амино-2,4,6-трииодизофталевой кислоты (II) в соответствующий дихлорангидрид кислоты известными методами, например, в присутствии тионилхлорида; его последующую конденсацию с хлорангидридом 2-[(ацетилокси)]пропионовой кислоты, чтобы образовать соответствующее 5-карбоксамидопроизводное, наконец, конденсацию данного последнего соединения с серинолом и последующую обработку, включающую любое возможное расщепление защитных групп, чтобы получить ожидаемое конечное соединение.
Предпочтительно, данный способ может быть применен для приготовления соединения формулы (III), в которой как R так и R' представляют собой группу -CONH-CH(CH2OH)2, R4 представляет собой водород и R5 представляет собой метильную группу, общеизвестную как иопамидол, или согласно равно предпочтительному варианту осуществления, для приготовления соединения формулы (III), в которой как R так и R' представляют собой группу -CONH-СН2-СН(ОН)CH2OH, R4 представляет собой метил, и R5 представляет собой водород, общеизвестный как иомепрол.
В соответствии с этим, дополнительный предмет данного изобретения относится к способу получения иопамидола или иомепрола, который отличается тем, что в нем исходным является соединение формулы (II), полученное по способу настоящего изобретения.
В упомянутом способе, указанное получение исходного соединения формулы (II) выполняется, как широко описано выше, в то время как последующие стадии, вся совокупность экспериментальных условий обработки и их дополнительные варианты выполнены согласно общим методам и условиям обработки, например, раскрытым в WO 96/037459, WO 96/037460, US 5362905, WO 97/047590 и WO 98/24757, EP 0026281.
Дополнительные подробности, относящиеся к способу изобретения, даны в последующей экспериментальной части с единственной целью лучше проиллюстрировать данное изобретение, без наложения какого-либо ограничения на него.
Краткое описание графиков
Фиг.1: Анализ с помощью ВЭЖХ иодированного продукта примера 3.
Фиг.2: Анализ с помощью ВЭЖХ иодированного продукта примера 4.
Фиг.3: Анализ с помощью ВЭЖХ неочищенного раствора сравнительного примера 1, спустя 3 часа при 22°С.
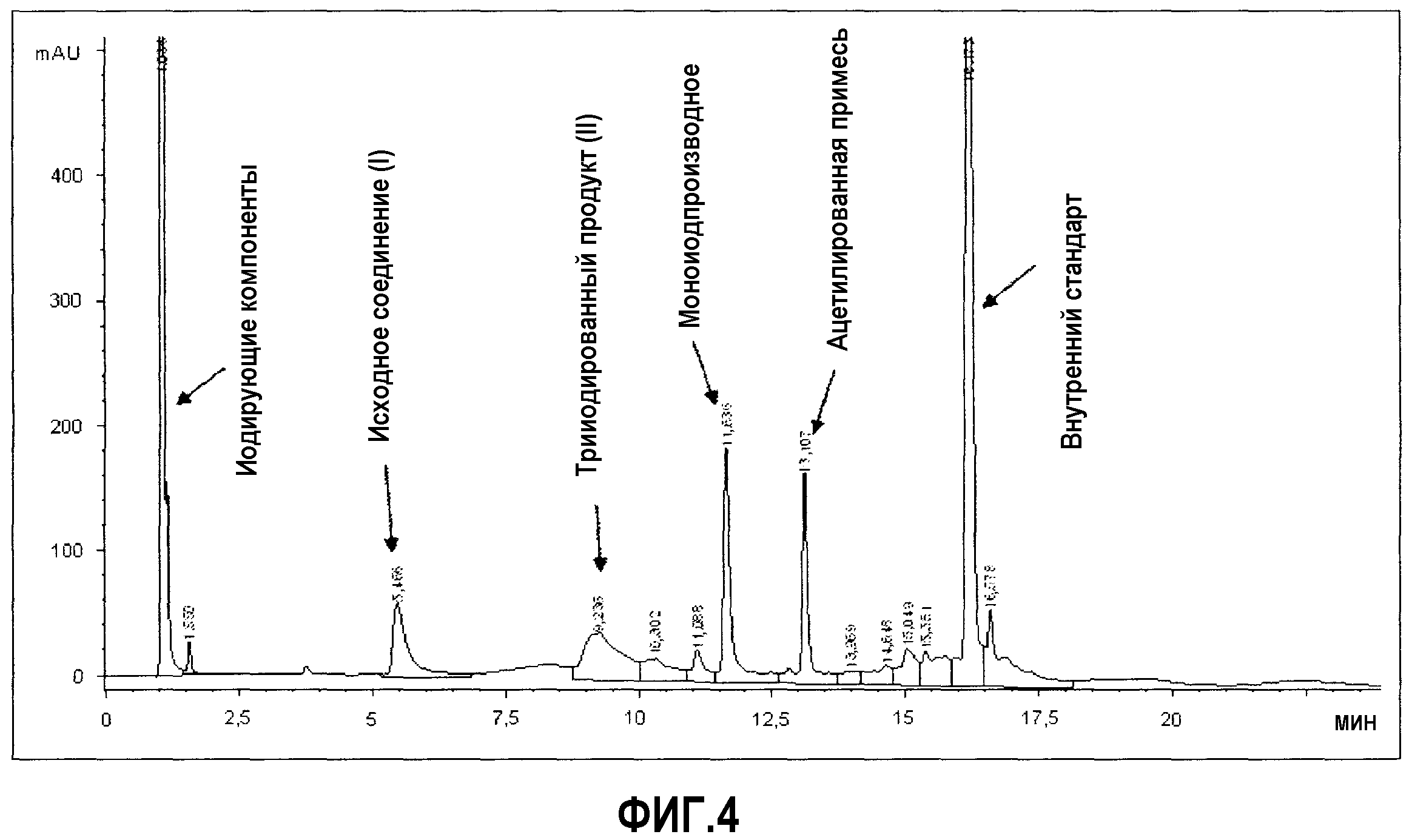
Фиг.4: Анализ с помощью ВЭЖХ неочищенного раствора сравнительного примера 1, спустя 3 часа при 22°С и дополнительных 6 часов при 60°С.
Фиг.5: Анализ с помощью ВЭЖХ твердого вещества а1.
Фиг.6: Анализ с помощью ВЭЖХ твердого вещества а2.
Фиг.7: Анализ с помощью ВЭЖХ маточных растворов а2.
Фиг.8: Анализ с помощью ВЭЖХ реакционной смеси b1.
Экспериментальная часть
Характеристика полученного соединения
Чистота полученной 5-амино-2,4,6-трииодфталевой кислоты была определена с помощью ВЭЖХ сравнением со стандартом (чистое соединение) или использованием бензойной кислоты в качестве внутреннего стандарта.
Общая процедура
Хроматографический метод ВЭЖХ

Пример 1
В 250 мл трехгорлую круглодонную колбу, снабженную термометром, конденсатором и магнитной мешалкой, помещали раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 3,86% (мас./мас.) кислоты (129,42 г раствора; 27,6 ммоль) и подкисляли до рН примерно 1 с помощью 96% H2SO4 (2 мл; 35,3 ммоль). Затем добавляли твердый I2 (8,42 г; 33,2 ммоль), смесь нагревали при 72°С с помощью масляной бани, и 18,65% (мас./об.) раствор HIO3 в Н2О (20 мл; 21,2 ммоль) добавляли к нагреваемой смеси в течение 5,2 ч через впрыскивающий насос.Спустя дополнительный 1 ч при 72°С (общее время реакции 6,2 ч) реакционную смесь охлаждали при комнатной температуре и фильтровали; твердое вещество промывали с помощью Н2О и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (12,74 г; 22,8 ммоль) в виде бледно-розового твердого вещества. Выход 82,6%. Продукт, проанализированый с помощью ВЭЖХ, при сравнении со стандартом, отвечал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Пример 2
В 250 мл трехгорлую круглодонную колбу, снабженную термометром, конденсатором и магнитной мешалкой, добавляли раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 3,86% (мас./мас.) кислоты (129,42 г раствора; 27,6 ммоль) и подкисляли до рН примерно 1 с помощью 96% H2SO4 (2 мл; 35,3 ммоль); затем добавляли твердый I2 (5,26 г; 21,5 ммоль) и смесь нагревали при 85°С с помощью масляной бани. 3,08% (мас./об.) раствор H2O2 в Н2О (25 мл; 22,6 ммоль) медленно добавляли в течение 8,5 ч через впрыскивающий насос; в конце добавляли дополнительный твердый I2 (5,26 г; 21,5 ммоль). Соответственно спустя 0,5 ч, 2,5 ч и 6 ч при 85°С три порции 7% (мас./об.) раствора H2O2 в Н2О (3×10 мл; в целом 61,7 ммоль) медленно добавляли в течение 1,7 ч для каждой порции через впрыскивающий насос.Реакционную смесь выдерживали при 85°С в течение дополнительного 1 ч, затем охлаждали при комнатной температуре и фильтровали; твердое вещество промывали с помощью Н2О и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (12,41 г; 22,2 ммоль) в виде бледно-коричневого твердого вещества. Выход 80,4%. Продукт анализировали с помощью ВЭЖХ сравнением со стандартом, и он соответствовал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Пример 3
В 3 л реактор с рубашкой, снабженный термометром, конденсатором и механической мешалкой, помещали раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 6,7% (мас./мас.) кислоты (1194 г раствора; 0,442 моль), разбавляли с помощью Н2О (636 мл) и подкисляли (до рН 2,8) с помощью 50% H2SO4 (73,63 г; 0,375 моль). Затем смесь нагревали до 45-50°С и добавляли твердый I2 (134,5 г; 0,530 моль). 50% (мас./мас.) раствор HIO3 в H2O (93,22 г; 0,265 моль) добавляли за 15 мин, полученную смесь нагревали до 75°С и выдерживали при данной температуре в течение 4 часов, в течение которых рН смеси поддерживался сам по себе в интервале от 2,5 до 2,2. Дополнительную 50% H2SO4 (430 г; 2,190 моль) затем добавляли к неочищенной суспензии за 1,5 ч (до рН<1) и полученную суспензию охлаждали при комнатной температуре 2 ч. При перемешивании добавляли 18% (мас./мас.) раствор бисульфита натрия (13,48 г; 0,023 моль). Затем твердое вещество фильтровали, промывали с помощью Н2О (200 мл) и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (228,9 г; 0,409 моль) в виде бледно-розового твердого вещества. Выход 92,6%. Продукт анализировали с помощью ВЭЖХ сравнением со стандартом, и он соответствовал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Пример 4
В 1,5 л реактор с рубашкой, снабженный термометром, конденсатором и механической мешалкой, помещали раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 6,7% (мас./мас.) кислоты (597 г раствора; 0,221 моль), разбавляли с помощью Н2О (318 мл) и подкисляли с помощью 50% H2SO4 (30,32 г; 0,155 моль). Затем смесь нагревали до 45-50°С и добавляли I2 (67,26 г; 0,265 моль). 50% (мас./мас.) раствор HIO3 в H2O (46,60 г; 0,132 моль) добавляли за 15 мин (рН полученной смеси: примерно 3) и смесь нагревали до 75°С в течение 4 ч, (в течение которых рН смеси падает до примерно 2). Затем добавляли 50% H2SO4 (222 г; 1,13 моль) (до рН<1) за 2 ч и суспензию охлаждали до 25°С в течение 2 ч. Добавляли 18% (мас./мас.) раствор бисульфита натрия (5,91 г; 0,010 моль), смесь выдерживали при перемешивании, затем твердое вещество фильтровали, промывали с помощью Н2О (150 мл) и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (109,8 г; 0,196 моль) в виде беловатого твердого вещества. Выход 88,9%. Продукт анализировали с помощью ВЭЖХ сравнением со стандартом, и он соответствовал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Пример 5
В 1 л реактор с рубашкой, снабженный термометром, конденсатором и механической мешалкой, помещали при комнатной температуре раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 6,7% (мас./мас.) кислоты (373 г раствора; 0,138 моль), Н2О (200 мл), 50% (мас./мас.) раствор HIO3 в H2O (29,12 г; 0,083 моль), 50% H2SO4 (15,71 г; 0,080 моль) и I2 (42,03 г; 0,166 моль). Смесь нагревали до 60°С за 30 мин, подкисляли с помощью 50% H2SO4 (7,64 г; 0,039 моль), и затем нагревали до 75°С в течение 3 ч (рН 1,9). Полученную суспензию затем дополнительно подкисляли (до рН<1) с помощью 50% H2SO4 (120 г; 0,612 моль), медленно добавляли в течение 2 ч и охлаждали до 25°С в течение 2 ч. Затем к смеси добавляли 18% (мас./мас.) раствор бисульфита натрия, при перемешивании, вплоть до окислительно-восстановительного потенциала <250 мВ. Затем твердое вещество фильтровали, промывали с помощью Н2О (100 мл) и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (64,61 г; 0,116 моль) в виде беловатого твердого вещества. Выход 83,8%. Продукт анализировали с помощью ВЭЖХ сравнением со стандартом, и он соответствовал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Пример 6
В 1 л реактор с рубашкой, снабженный термометром, конденсатором и механической мешалкой, помещали раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 7,2% (мас./мас.) кислоты (277,7 г раствора; 0,110 моль), разбавляли с помощью Н2О (220 мл) и подкисляли с помощью 96% H2SO4 (8,8 мл; 0,159 моль). Затем добавляли этанол (73 мл) и I2 (33,6 г; 0,132 моль). Смесь нагревали до 80-82°С и 32,6% (мас./мас.) раствор HIO3 в H2O (35,62 г; 0,066 моль) добавляли по каплям в течение 3 ч (рН смеси: 1,8). Смесь выдерживали при выше представленной температуре в течение дополнительных 4 ч, затем подкисляли до рН<1 с помощью 50% H2SO4 (44 мл; 0,314 моль) и охлаждали до 25°С в течение 2 ч. Добавляли при перемешивании бисульфит натрия (0,820 г; 4,31 ммоль), затем твердое вещество фильтровали, промывали с помощью Н2О (100 мл) и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (51,36 г; 0,092 моль) в виде бледно-розового твердого вещества. Выход 83%. Продукт анализировали с помощью ВЭЖХ сравнением со стандартом, и он соответствовал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Пример 7
В 1 л реактор с рубашкой, снабженный термометром, конденсатором и механической мешалкой, помещали раствор натриевой соли 5-аминоизофталевой кислоты (I) в Н2О, соответствующий 6,7% (мас./мас.) кислоты (313,1 г раствора; 0,138 моль), разбавляли с помощью Н2О (200 мл) и подкисляли с помощью 50% H2SO4 (41,15 г; 0,210 моль). Затем добавляли твердый I2 (42,03 г; 0,166 моль) при комнатной температуре и полученную смесь затем нагревали при 75°С.0,66 М раствор HIO3 в H2O (140,0 г; 0,0833 моль) добавляли по каплям за 1 ч и полученную смесь поддерживали при перемешивании при 75°С в течение дополнительных 4 часов. В течение всего нагревания, добавления HIO3 и последующего времени окончания (4 часа) рН реакционной смеси поддерживали при 3,0 добавлением 2 М NaOH. Суспензию окончательно подкисляли до рН=1 с помощью 50% H2SO4 (143 г; 0,729 моль), медленно добавляли в течение 1,5 ч, охлаждали до 25°С в течение 2 часов. 18% (мас./мас.) раствор бисульфита натрия добавляли вплоть до окислительно-восстановительного потенциала суспензии <250 мВ. Затем твердое вещество фильтровали, промывали с помощью Н2О (100 мл) и сушили с получением 5-амино-2,4,6-трииодизофталевой кислоты (II) (66,0 г; 0,118 моль). Выход 85%. Продукт анализировали с помощью ВЭЖХ сравнением со стандартом, и он соответствовал аналитическим спецификациям для промышленно изготовленной 5-амино-2,4,6-трииодизофталевой кислоты.
Сравнительный пример 1
Данный тест проводили, чтобы оценить использование условий иодирования, раскрытых в US 2007/0219396, подходящим образом адаптированных по количеству средства для иодирования, чтобы обеспечить трииодированное производное.
В 25 мл трехгорлую круглодонную колбу, снабженную термометром, конденсатором и магнитной мешалкой, добавляли твердую 5-аминоизофталевую кислоту (I) (1 г; 5,5 ммоль), твердый I2 (1,61 г; 6,34 ммоль) и уксусную кислоту (15 мл) и перемешивали при 22°С. Затем добавляли 70% (мас./мас.) раствор HIO3 в H2O (0,96 г; 3,8 ммоль) в течение 0,5 ч.
В этом отношении стоит отметить, что из-за очень слабой растворимости иодноватой кислоты в уксусной кислоте добавление окислителя в концентрации, рекомендуемой процитированной статьей, а именно, 70% (мас./мас.), ведет к негомогенной смеси.
Полученную смесь поддерживали при данной температуре в течение 3 часов и затем анализировали с помощью ВЭЖХ. Полученная хроматограмма (фиг.3) показывает полное отсутствие любой детектируемой конверсии в иодированное соединение.
Для чисто экспериментальных целей, не предлагаемых процитированной заявкой, реакционную смесь затем нагревали при 60°С в течение дополнительных 6 часов (общее время реакции 9 ч). Затем полученную темную смесь охлаждали при комнатной температуре, без обеспечения любой кристаллизации или осаждения требуемой 5-амино-2,4,6-трииодизофталевой кислоты (II).
Затем смесь анализировали с помощью ВЭЖХ и результаты, показанные на фиг.4, указывают на присутствие очень небольшого количества трииодированного производного, и, наоборот, значительного количества примеси, идентифицированной как N-ацетил-5-аминоизофталевая кислота формулы

Сравнительный пример 2
Данный сравнительный пример выполняли, чтобы тестировать условия иодирования, раскрытые упомянутыми статьями Chem. Ber., особенно, статьей Chem. Ber., 1897 30(2), 1943-1948, которая дает несколько больше экспериментальных подробностей, допускающих попытку для их воспроизведения.
В соответствии с этим, авторы сначала тестировали условия иодирования, рекомендуемые процитированной статьей на том же самом субстрате, а именно, на 3-аминобензойной кислоте, и использованием раскрытого количества иодирующих средств, т.е. стехиометрического количества, требуемого для гипотетического полного дииодирования субстратного соединения.
В этом отношении, однако, стоит отметить, что молярное отношение I2:HIO3, используемое и рекомендуемое процитированной статьей, а именно 2.8, является нецелесообразным для полного переноса добавленного иода (рассматриваемого как сумма из I2 и HIO3) в ароматический субстрат.Фактически, и как упомянуто ранее, чтобы иметь полный перенос, молярное отношение между иодом и иодноватой кислотой должно составлять 2 (теоретическое отношение) или меньше.
Только чтобы иметь лучшее представление по используемым условиям иодирования, рН реакционной смеси был проверен в различные периоды реакции.
а. Иодирование 3-аминобензойной кислоты
Иодирующий раствор готовили растворением I2 (4 г; 15,74 ммоль) в 20% водного КОН (9,5 мл) с получением суспензии белого твердого вещества в бледно-желтом растворе, который превращался в прозрачный раствор при разбавлении Н2О (30 мл); затем добавляли раствор HIO3 (1 г; 5,69 ммоль) в Н2О (10 мл) и конечный темный раствор разбавляли до 250 мл Н2О.
Раствор, полученный таким образом, добавляли по каплям в течение 3 ч к кислому раствору 3-аминобензойной кислоты (2,5 г; 18,23 ммоль) в смеси Н2О (500 мл) и 36-38% водного HCl (50 мл) (рН раствора: около 0), нагретому при 30°С.После того, как добавление завершали (рН 0,25), начинало кристаллизоваться твердое вещество. Реакционную смесь затем перемешивали при комнатной температуре в течение 12 ч, затем твердое вещество фильтровали, промывали с помощью Н2О (15 мл) и сушили с получением коричневатого твердого вещества а1 (2,3 г). В соответствии с описанием, дополнительный иодирующий раствор, приготовленный, как описано выше, (125 мл; 7,87 ммоль I2; 2,85 ммоль HIO3), добавляли по каплям в течение 2 ч к маточному раствору, поддерживаемому при примерно 30°С, благоприятствуя тем самым осаждению другого твердого вещества. Через 12 ч при комнатной температуре данное твердое вещество фильтровали, промывали с помощью Н2О и сушили с получением коричневатого твердого вещества а2 (2,2 г). Анализ с помощью ВЭЖХ двух полученных твердых веществ, (фиг.5 и 6, соответственно), показывает, что оба осадка соответствуют смеси из двух содержащихся компонентов, в двух случаях, с разным % отношением площади ВЭЖХ, как представлено в таблице 1 ниже.
Сравнением интегралов спектров1Н-ЯМР двух твердых веществ с относительным ВЭЖХ-содержанием двух компонентов, авторы могли идентифицировать компонент с временем удерживания 25,1 мин, как один из трех возможных дииодпроизводных, и компонент с временем удерживания 25,4 мин, как трииодпроизводное.
С другой стороны, анализ конечного маточного раствора с помощью ВЭЖХ показывает, что не прореагировавший 3-аминобензойный субстрат еще присутствует в растворе, наряду с компонентом с временем удерживания 25,1 мин и тремя неизвестными компонентами, (фиг.7), что таким образом подтверждает, что конверсия иодирования была не полной (выход полученного трииодпроизводного мог быть примерно оценен как приблизительно 30% от теоретического), и полученный продукт не был чистым.
b. Иодирование 5-аминоизофталевой кислоты
Те же самые условия иодирования, надлежащим образом адаптированные для того, чтобы получить требуемое трииодированное соединение, затем оценивали на субстратном соединении настоящего изобретения.
В соответствии с этим, иодирующий раствор готовили, как описано выше (114 мл, 7,18 ммоль I2; 2,55 ммоль HIO3) и добавляли по каплям к кислому раствору 5-аминоизофталевой кислоты (I)(1 г; 5,52 ммоль) в смеси Н2О (150 мл) и 36-38% водного HCl (15 мл) (рН раствора: около 0), нагретому при 30°С. В соответствии с рекомендацией предыдущей статьи, смесь затем поддерживали при перемешивании при 30°С в течение 19 ч без наблюдения любой кристаллизации или осаждения. Смесь (рН 0,33) таким образом, охлаждали до комнатной температуры и анализировали с помощью ВЭЖХ. Наблюдаемые результаты, представленные на фиг.8, подтверждают, что конверсия исходного продукта не была полной и значительное количество исходного субстрата еще присутствует в неочищенном растворе.
Количественное определение полученной 5-амино-2,4,6-трииодизофталевой кислоты, сделанное по сравнению с внутренним стандартом, показывает выход 28,2%.
Реферат
Изобретение относится к способу получения иодированных анилинов, в частности оно относится к способу, включающему прямое иодирование 3,5-дизамещенных анилинов до соответствующих 3,5-дизамещенных-2,4,6-трииоданилинов, которые применимы как промежуточные продукты для синтеза контрастных сред для рентгеновских лучей и для приготовления самих контрастных сред. Способ получения 5-амино-2,4,6-трииодизофталевой кислоты формулы (II) включает иодирование 5-аминоизофталевой кислоты формулы (I), проводимое в полярном растворителе и в кислых условиях, или ее соли молекулярным иодом и в присутствии йодноватой кислоты, где молярное соотношение между молекулярным иодом и 5-аминоизофталевым субстратом (I) составляет от 1 до 1,5, и молярное отношение иода к йодноватой кислоте составляет от 1:0,5 до 1:1, и эквивалентное соотношение между 5-аминоизофталевым субстратом (I) и иодирующим компонентом, рассматриваемым в виде суммы Iи HIO, составляет по меньшей мере 1:3. Изобретение также относится к способам получения соединений формулы (III), в которой R и R', одинаковые или отличные друг от друга, означают группу, выбираемую из карбокси (-СООН), карбоксисложноэфирной (-COOR) и карбоксамидо (-CONH, -CONHRили -CONRR), где R, Rи R, одинаковые или отличные друг от друга, представляют собой прямую или разветвленную С-Салкильную группу, необязательно замещенную одной или несколькими гидроксильными группами, и Rи R, одинаковые или отличные друг от друга, представляют собой водород или прямую или разветвленную С-Салкильную группу, необязательно замещенную одной или несколькими гидроксильными или C-Салкоксигруппами. 3 н. и 12 з.п. ф-лы, 8 ил., 9 пр., 1 табл.
Формула
включающий иодирование 5-аминоизофталевой кислоты формулы (I), проводимое в полярном растворителе и в кислых условиях
или ее соли молекулярным иодом и в присутствии иодноватой кислоты, где молярное соотношение между молекулярным иодом и 5-аминоизофталевым субстратом (I) составляет от 1 до 1,5, и молярное отношение иода к иодноватой кислоте составляет от 1:0,5 до 1:1, и эквивалентное соотношение между 5-аминоизофталевым субстратом (I) и иодирующим компонентом, рассматриваемым в виде суммы I2 и HIO3, составляет по меньшей мере 1:3.
в которой:
R и R', одинаковые или отличные друг от друга, означают группу, выбираемую из карбокси (-СООН), карбоксисложноэфирной (-COOR1) и карбоксамидо (-CONH2, -CONHR1 или -CONR2R3), где R1, R2 и R3, одинаковые или отличные друг от друга, представляют собой прямую или разветвленную С1-С4алкильную группу, необязательно замещенную одной или несколькими гидроксильными группами, и
R4 и R5, одинаковые или отличные друг от друга, представляют собой водород или прямую или разветвленную C1-С6алкильную группу, необязательно замещенную одной или несколькими гидроксильными или С1-С6алкоксигруппами,
причем упомянутый способ включает:
a) получение промежуточного соединения формулы (II) согласно способу по любому из пп.1-11; и дополнительно,
b) взаимодействие соединения формулы (II) с тионилхлоридом с образованием соответствующего дихлорангидрида кислоты,
c) получение требуемого соединения формулы (III) из образованного дихлорангидрида.
а) получение 5-амино-2,4,6-трииодизофталевой кислоты формулы (II)
иодированием 5-аминоизофталевой кислоты формулы (I),
или ее соли молекулярным иодом и в присутствии иодноватой кислоты согласно способу по п.1,
b) взаимодействие соединения формулы (II) с тионилхлоридом с образованием соответствующего дихлорангидрида кислоты,
c) конденсацию дихлорангидрида с хлорангидридом 2-[(ацетилокси)]пропионовой кислоты с образованием соответствующего 5-карбоксамидопроизводного,
d) конденсацию полученного производного с серинолом и удаление необязательных защитных групп с получением иопамидола.
Комментарии