Нитратные соли и фармацевтическая композиция на их основе - RU2238932C2
Код документа: RU2238932C2
Описание
Настоящее изобретение относится к композициям, которые используются в терапии и для предотвращения костных нарушений, таких как нарушения в костной ткани и суставах. В частности, оно относится к композициям, имеющим улучшенную терапевтическую активность и улучшенную желудочно-кишечную толерантность.
Из предшествующего уровня техники известно, что фармакологическое лечение костных нарушений подразумевает терапию, направленную на контроль патофизиологических процессов, которые связаны с костной тканью и суставами.
Среди соединений, известных из предшествующего уровня техники для фармакологического лечения нарушений скелетно-мышечной и суставной систем, могут быть упомянуты следующие (смотри “New Guide to Medicine & Drugs”, Brit. Med. Association, 1997, стр. 115; “Martindale, The Extra Pharmacopeia”, т. 31, 1996, стр. 11-13):
- дифосфонаты (алендронат, памидронат, ризедронат и т.д.);
- “оксикамы”, то есть пироксикам, теноксикам, аминопирин; томоксипрол, пеницилламин; метотрексат и т.д.
Они представляют собой лекарственные средства с ограниченной эффективностью и причиняют беспокойства, вызывая повреждения желудочно-кишечного тракта, в частности желудка. Дифосфонаты вызывают также повреждения в пищеводе.
Другими соединениями, используемыми в терапии костных нарушений, являются антилейкотриеновые лекарственные средства, например ибудиласт, пранлукаст и т.д.; аминосалицилаты.
Эффективность и желудочно-кишечная толерантность этих лекарственных средств не являются оптимальными.
Соединения, способные обойти недостатки, указанные для лекарственных средств, используемых при костных нарушениях, пока еще не были доступны из предшествующего уровня техники.
Ощущалась необходимость в доступных соединениях для использования их в лечении костных нарушений, обладающих улучшенными терапевтическими признаками и толерантностью и/или имеющих более высокую эффективность.
Заявитель неожиданно и с удивлением обнаружил соединения и композиции на их основе, обладающие улучшенными фармакотерапевтическими свойствам.
Объектом настоящего изобретения являются нитратные соли соединений, обладающих терапевтической активностью в отношении костных нарушений, или их фармацевтические композиции, причем упомянутые соединения отличаются тем, что они содержат, по крайней мере, одну реакционную группу, способную образовывать соли с азотной кислотой, и вышеуказанные соединения выбраны из следующих классов:
класс F1

соединение известно как алендронат динатрия;
класс F2A

известно как пироксикам;

известно как метотрексат;

известно как пеницилламин;

известно как трамадол;
класс F2B

известно как томоксипрол.
Предшественники для получения солей настоящего изобретения предпочтительно выбираются из следующих:
- класс F1: алендронат;
- класс F2A: пироксикам;
- класс F2B: томоксипрол.
В композициях согласно настоящему изобретению могут использоваться также изомеры соединений, принадлежащих к вышеупомянутым классам, если они доступны. Примерами изомеров являются цис-, транс-, оптические изомеры D и L или рацемические, энантиомеры. В основном одна изомерная форма имеет более высокую активность по сравнению с другой, например D форма по сравнению с L формой или наоборот.
Соли соединений, принадлежащих к вышеупомянутым классам, содержат, по крайней мере, один моль нитратных ионов/моль соединения. Предпочтительно соотношение между молями нитратных ионов и молями предшественника равно единице. Соли с более высоким молярным соотношением могут быть получены, когда в молекуле имеются другие основные аминогруппы, способные образовать соль.
Из солей настоящего изобретения могут быть получены соответствующие фармацевтические композиции в соответствии с хорошо известными методиками в данной области техники с использованием обычных наполнителей (смотри, например, книгу “Remington's Pharmaceutical Sciences, 15a Ed.”).
Дозы солей изобретения в их фармацевтических композициях являются такими же и в основном ниже, чем дозы их предшественников, относящихся к вышеупомянутым классам. Однако, так как они в целом более толерантны, их можно использовать в более высоких дозах, чем их предшественников, без возникновения при этом побочных действий, характерных для предшественников при высоких дозах.
Предшественников солей, относящихся к вышеупомянутым классам, получают согласно способам, описанным в следующих ссылках.
Класс F1:
алендронат динатрия, памидронат динатрия, ризедронат динатрия (смотри книгу "Index Merck 14а Ed.", приведенную здесь в качестве ссылки) ибандронат динатрия: ЕР 252502, соединение формулы (F1e): ЕР 325482, соединение формулы (F1f): ЕР 531253, соединение формулы (F1g): ЕР 592488, соединение формулы (F1h): ЕР 522576, соединение формулы (F1i): ЕР 546548, соединение формулы (F1l): ЕР 561296, соединение формулы (F1m): JP 93032684 (С.A. ref. Vol. 119 226194d), соединение формулы (F1n): JP 93222073 (С.А. ref. Vol. 120 134926m), соединение формулы (F1o): JP 92356496 (С.А. ref. Vol. 119 095828р), соединение формулы (F1p): WO 93/12122;
класс F2A:
пироксикам, метотрексат, пеницилламин, трамадол (смотри "Index Merck volume, 14a Ed.", приведенную здесь в качестве ссылки);
класс F2B:
томоксипрол (ЕР 12866); дроксикам: "Index Merck, 14a Ed."; целекоксиб (WO 94/2798), соединение формулы (F2BIV) (WO 95/15315); соединения формулы F2BV (7-нитроиндазол) и F2BVI (1,2-(трифторметилфенил)имидазол), коммерчески поставляются Lancaster Synthesis, Morecam-England.
Соли согласно настоящему изобретению получают согласно следующим способам.
Если соединение, которое используется для получения соли, доступно как свободное основание или как соответствующая соль, которые растворимы в органическом растворителе, который предпочтительно не содержит гидроксильных групп в молекуле, например в ацетонитриле, этилацетате, тетрагидрофуране и т.д., соль получают растворением этого вещества в растворителе при концентрации, предпочтительно равной или выше чем 10% (мас./об.), добавлением количества концентрированной азотной кислоты, соответствующего молям образующих соль аминогрупп, присутствующих в соединении. Азотную кислоту предпочтительно разводят в том же растворителе. Предпочтительно во время и после вышеупомянутого добавления смесь охлаждают до температур в диапазоне 20-0° С. Продукт в основном извлекают фильтрацией и промывают растворителем.
Когда, наоборот, вещество не очень растворимо или доступно как не очень растворимая соль в вышеупомянутых растворителях, могут использоваться соответствующие смеси гидроксилированных растворителей. Примерами таких растворителей являются метиловый спирт, этиловый спирт и вода. Осаждение может быть ускорено путем разбавления таким образом полученной смеси неполярным растворителем после добавления азотной кислоты.
Когда исходный продукт образует соль с соляной кислотой, можно получить соль с азотной кислотой, непосредственно добавляя нитрат серебра к раствору соединения. После отфильтровывания хлорида серебра раствор концентрируют и охлаждают для выделения нитратной соли.
Когда исходным продуктом является соль, высвобождать соответствующее основание можно также обработкой насыщенным раствором карбоната или бикарбоната натрия или калия, или разбавленным раствором гидроксида натрия или калия. Основание затем экстрагируют подходящим органическим растворителем (например, галогенированными растворителями, сложными эфирами, простыми эфирами) и обезвоживают. Органический растворитель выпаривают и затем следуют вышеупомянутым способам получения, растворяя основание в ацетонитриле или в других упомянутых растворителях.
Следующие примеры приведены только с целью проиллюстрировать данное изобретение, и не ограничивают его.
ПРИМЕР 1
Получение нитратной соли пироксикама
Раствор пироксикама (3 г, 9,05 ммолей) в ацетонитриле (30 мл) и тетрагидрофуране (50 мл) обрабатывают при 4° С 65% азотной кислотой (0,63 мл), растворенной в ацетонитриле (5 мл). Смесь перемешивают и оставляют стоять. Через 30 минут при 4° С ее фильтруют и собирают осадок, который промывают этиловым эфиром и высушивают под вакуумом.
Получают белое твердое вещество (3,23 г), имеющее температуру плавления 120-123° С.
Элементный анализ
Рассчитано, %: C 45,68; Н 3,58; N 14,12; S 8,13.
Обнаружено, %: С 45, 76; Н 3,54; N 14,11; S 8,16.
ПРИМЕР 2
Получение нитратной соли алендроната
Раствор алендроната (0,92 г, 3,7 ммолей) в 50% азотной кислоте (2 мл) капают при температуре 4° С в этиловый эфир (30 мл). Смесь перемешивают и оставляют стоять. Через 40 минут при 4° С отфильтровывают осадок, промывают этиловым эфиром и высушивают под вакуумом. Получают белое аморфное твердое вещество.
Элементный анализ
Рассчитано, %: С 15,39; Н 4,52; N 9,01.
Обнаружено, %: С 15,41; Н 4,50; N 8,99.
ПРИМЕР 3
Получение нитратной соли пеницилламина
(L)-Пеницилламин (5 г, 33,5 ммоля) в метаноле (50 мл) обрабатывают при +4° С 65% азотной кислотой (2,5 мл), растворенной в ацетонитриле (7 мл). Смесь перемешивают и оставляют стоять. Через 15 минут при 4° С добавляют этиловый эфир, отфильтровывают осадок и высушивают под вакуумом. Получают аморфное твердое вещество (3,2 г).
Элементный анализ
Рассчитано, %: С 28,30; Н 5, 65; N 13,19; S 15,11.
Обнаружено, %: С 28,29; Н 5, 66; N 13,22; S 15,08.
ПРИМЕР 4
Получение нитратной соли метотрексата Раствор метотрексата (5 г, 11,00 ммоля) в метаноле (60 мл) обрабатывают при 4° С при перемешивании 65% азотной кислотой (0, 82 мл), растворенной в ацетонитриле (5 мл).
Через 30 минут при 4° С обрабатывают этиловым эфиром. Отфильтровывают осадок и промывают этиловым эфиром. Высушивают под вакуумом. Получают аморфное твердое вещество (2,2 г).
Элементный анализ
Рассчитано, %: С 46,42; Н 4,44; N 24,35.
Обнаружено, %: С 46,44; Н 4,40; N 24,39.
ПРИМЕР 5
Получение нитратной соли томоксипрола
Раствор томоксипрола (2 г, 6,32 ммоля) в ацетонитриле (50 мл) и метаноле (18 мл) обрабатывают при 4° С 65% азотной кислотой (0,432 мл), растворенной в ацетонитриле (10 мл). Смесь перемешивают и оставляют стоять при 4° С. Через 30 минут раствор обрабатывают этиловым эфиром.
Сформировавшийся осадок отфильтровывают, промывают этиловым эфиром и высушивают под вакуумом. Получают белое твердое вещество с температурой плавления 197-199° С.
Элементный анализ
Рассчитано, %: С 65,81; Н 5,52; N 11,01.
Обнаружено, %: С 65,78; Н 5,59; N 11,02.
ПРИМЕР 6
Получение (4-нитрокси)бутилового эфира 5-амино-2-гидроксибензойной кислоты (5-ASA-NO2)
ПРИМЕР 6а
Получение 5-трет-бутилоксикарбониламино-2-гидроксибензойной кислоты
К суспензии 5-амино-2-гидроксибензойной кислоты (15 г, 98 ммолей) в диоксане (105 мл) и воде (150 мл) добавляют триэтиламин (24,6 мл, 176 ммолей). К полученному раствору добавляют ди-трет-бутилдикарбонат (25,65 г, 118 ммолей). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 4 дней. В конечном счете, раствор концентрируют под вакуумом до объема примерно 150 мл, охлаждают льдом и подкисляют 5% соляной кислотой, экстрагируя затем этилацетатом. Отбирают органическую фазу и промывают водой. Органическую фазу обезвоживают сульфатом натрия. Упаривая растворитель под вакуумом, получают продукт в виде аморфного твердого вещества (20,8 г).
ПРИМЕР 6b
Получение 4-бромбутилового эфира 5-трет-бутилоксикарбониламино-2-гидроксибензойной кислоты
К раствору 5-трет-бутилоксикарбониламино-2-гидроксибензойной кислоты (20 г, 85,7 ммолей) в тетрагидрофуране (200 мл) добавляют трифенилфосфин (44,9 г, 171 ммоля), а затем тетрабромид углерода (56,7 г, 171 ммоля). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 24 часов и затем выпаривают под вакуумом. Полученный остаток очищают колоночной хроматографией на силикагале (элюент: н-гексан/этилацетат, 8/2, об./об.), проверяя содержание собранных фракций тонкослойной хроматографией. Продукт получают, собирая головные фракции после элюирования из колонки реактива СВr4, использованного в синтезе. Фракции собирают и высушивают. Получают белое твердое вещество (21,16 мг), имеющее температуру плавления 108-111° С.
ПРИМЕР 6с
Получение 4-нитроксибутилового эфира 5-трет-бутилоксикарбониламино-2-гидроксибензойной кислоты
Раствор 4-бромбутилового эфира 5-амино-2-гидроксибензойной кислоты (21, 16 г, 57,6 ммолей) в ацетонитриле (150 мл) нагревают при 80° С в темноте в течение 5 часов с нитратом серебра. После охлаждения до комнатной температуры отфильтровывают твердое вещество и отбирают растворитель, который затем выпаривают досуха, получая остаток, который очищают колоночной хроматографией на силикагеле, используя в качестве элюента н-гексан/этилацетат (7/3 об./об.), проверяя содержание собранных фракций тонкослойной хроматографией. Головные фракции, содержащие соединение, упаривают досуха, получая 12,6 г белого твердого вещества, имеющего температуру плавления 107-109° С.
ПРИМЕР 7 (сравнительный)
Получение гидрохлоридной соли 4-нитроксибутилового эфира 5-аминосалициловой кислоты (5-ASA-NO2-HCl).
Раствор 4-нитроксибутилового эфира 5-трет-бутилоксикарбониламино-2-гидроксибензойной кислоты (10 г, 28,6 ммоля) растворяют в этилацетате (8 мл) и охлаждают до 0° С. 3М этилацетат/НСl (30 мл), полученный при барботировании газообразным НС1 этилацетата, добавляют при перемешивании до тех пор, пока не будет получена требуемая молярная концентрация НСl. Смесь доводят до комнатной температуры и оставляют перемешиваться в течение 2 часов.
Формируется твердое вещество, которое отфильтровывают, промывают этиловым эфиром и высушивают под вакуумом. Полученный продукт (7,1 г) является белым твердым веществом, имеющим температуру плавления 136-140° С.
Элементный анализ
Рассчитано, %: С 43,09; Н 4,89; N 9,13; Cl 11,56.
Обнаружено, %: С 43,05; Н 4,88; N 9, 10; Cl 11,54.
ПРИМЕР 8
Получение нитратной соли 4-нитроксибутилового эфира 5-амино-2-гидроксибензойной кислоты (5-АSА-NO2 ·НNО3 )
К раствору гидрохлоридной соли 4-нитроксибутилового эфира 5-аминосалициловой кислоты (2 г, 7 ммолей) в смеси ацетонитрила (50 мл) и тетрагидрофурана (15 мл) добавляют нитрат серебра (1,19 г, 7 ммолей). Через 10 минут отфильтровывают сформировавшуюся соль (AgCl). Раствор оставляют стоять на 30 минут, затем отфильтровывают осадок, промывают этиловым эфиром и высушивают под вакуумом. Получают соль (1,27 г) в виде беловатого твердого вещества, имеющего температуру плавления 123-128° С.
Элементный анализ
Рассчитано, %: С 39,66; Н 4, 50; N 12,61.
Обнаружено, %: С 39,70; Н 4,53; N 12,67.
ФАРМАКОЛОГИЧЕСКИЕ ТЕСТЫ
ПРИМЕР 9
Острая токсичность
Группе из 10 крыс весом 20 г вводили однократную дозу, равную 100 мг/кг, нитратной соли пироксикама (пример 1) перорально через канюлю в карбоксиметилцеллюлозной водной суспензии (2% мас./об.).
За животными наблюдали в течение 14 дней. Ни у одного из животных в группе не было обнаружено наличия признаков токсичности.
ПРИМЕР 10
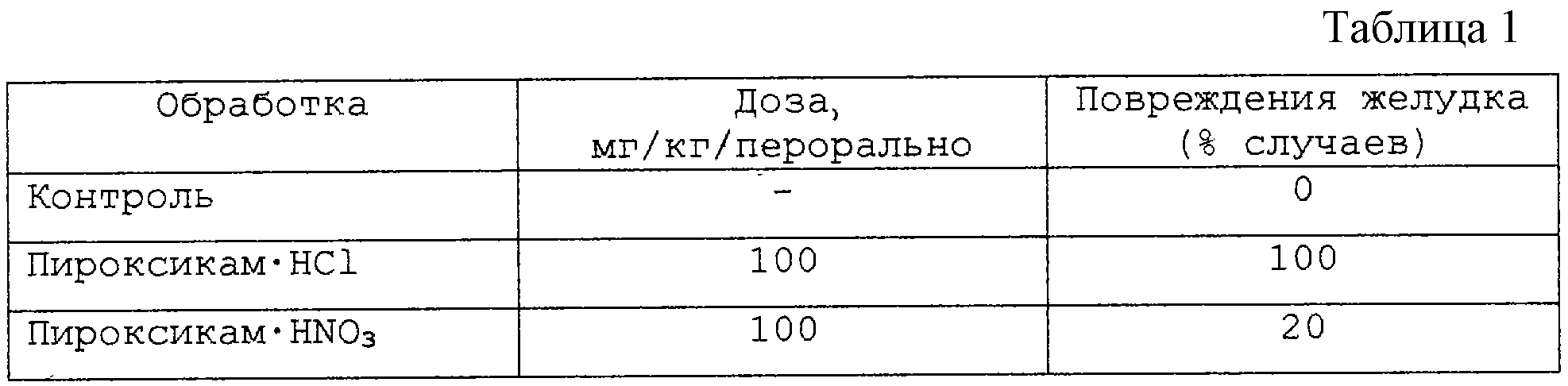
Желудочная токсичность нитратной соли пироксикама (пример 1) в сравнении с пироксикамом
Трем группам по 10 крыс в каждой, у которых желудок был пустым в течение 24 часов, перорально вводили:
- 5 мл/кг 2% карбоксиметилцеллюлозной водной суспензии,
- количество нитрата пироксикама, соответствующее 100 мг/кг пироксикама, в 5 мл/кг 2% карбоксиметилцеллюлозной водной суспензии,
- количество гидрохлорида пироксикама, соответствующее 100 мг/кг пироксикама, в 5 мл/кг 2% карбоксиметилцеллюлозной водной суспензии.
Через шесть часов животных умерщвляли и оценивали степень желудочных повреждений. Результаты представлены в таблице 1, они показывают, что крысы, обработанные нитратом пироксикама, показывают улучшенную желудочную толерантность по сравнению с гидрохлоридом пироксикама.
ПРИМЕР 11
Миелопероксидазная активность 4-нитроксибутилового эфира 5-аминосалициловой кислоты по сравнению с соответствующими гидрохлоридом и нитратом, исследованная на модели острого колита.
Были сформированы 4 группы по 6 животных в каждой. Эти группы обрабатывали соответственно носителем (1% водный раствор карбоксиметилцеллюлозы), 100 мг/кг 5-ASA, количеством 5-ASA-NO2·HCl, соответствующим 100 мг/кг 5-ASA, количеством 5-ASA-NO2·НNО3, соответствующим 100 мг/кг 5-ASA.
Животных обрабатывали ректальным путем вышеупомянутыми соединениями в нулевое время. Через час животных обрабатывали 0,5 мл раствора 60 мг/мл 2,4,6-тринитробензолсульфоновой кислоты в 50% этиловом спирте. Через два часа животных снова обрабатывали ректальным путем теми же соединениями и с 12-часовыми интервалами до суммарного количества 6 введений.
Измеряли тканевые уровни фермента миелопероксидазы (МРО), который является маркером воспалительного процесса в различных тканях, в том числе в костно-суставной ткани (С. Rathakrishnan и соавт., "Release of oxygen radicals by articular chondrocytes: A study of luminol-dependent chemiluminescence and hydrogen peroxide secretion", J. Bone Miner. Res. 7/10, 1139-1148, 1992).
Миелопероксидазную активность измеряли, используя модифицированную версию экспериментальной модели, описанной Bradley и соавт., J. Invest. Dermatol., 78, 206-209, 1982. Из каждого животного отбирали образцы кишечной ткани, которые затем суспендировали в 0,1% бромиде гексадецилтриметиламмония (50 мг/мл) при рН 6 и гомогенизировали в течение 15 секунд (Polytron® РТ-7 генератор). Образцы замораживали и размораживали три раза перед центрифугированием (9000 g) в течение 2 минут в центрифуге Eppendorf® Benchtop. Миелопероксидазную активность измеряли, добавляя 7 мкл супернатанта к 200 мкл реагента о-дианизидина (Sigma) и измеряя изменение оптической плотности при 450 нм за время 2 минуты в Microtitre Multiscan®. Реагент содержал 0,0005% пероксида водорода в качестве субстрата для фермента миелоперксидазы. Одна единица миелопероксидазной активности определялась как единица, способная превращать микромоль пероксида водорода в воду за одну минуту при 22° С. Результаты представлены в таблице 2 и выражены в числе единиц миелопероксидазной активности/мг ткани (влажной).
Из таблицы 2 видно, что в группе, обработанной S-ASA-NO2·HNO3, миелопероксидазная активность ниже, чем у других групп.
ПРИМЕР 12
Предотвращение повреждения желудка, вызванного аспирином, введением алендроната по сравнению с нитратной солью алендроната.
Три группы по 5 крыс в каждой обрабатывали перорально, как показано ниже.
I группу обрабатывали алендронатом (Ален.) дозой 80 мг/кг,
II группу - за 1 час до дозы 80 мг/кг алендроната вводили перорально аспирин (Асп.) дозой 125 мг/кг,
III группу - за 1 час до нитрата алендроната (Ален· НNО3) в количестве, соответствующем 80 мг/кг дозы алендроната, вводили перорально аспирин дозой 125 мг/кг.
Оценку желудочных повреждений проводили, умерщвляя животных через три часа после обработки алендронатом или нитратом алендроната.
Результаты представлены в таблице 3, они показывают, что введение нитрата алендроната уменьшает уровень желудочных повреждений в 4-5 раз по сравнению с этим значением после введения алендроната.


Фармакологические тесты с нитратными солями томоксипрола и трамадола
Пример 13
Ингибиторная активность нитратной соли томоксипрола на повреждении бычьей эндотелиальной клетки, вызванном куменгидропероксидом.
В этом тесте потенциальный эффект томоксипрола оценивался на нарушение костной ткани путем определения его активности для эндотелиальной защиты при оксидативном стрессе. Этот эксперимент относится к хорошо известным внутренним зависимостям между оксидативным стрессом, эндотелиальной дисфункцией и нарушением костной системы, как описано в Free Radical. Biol. Med., 1997, 22, 269-85; Ann. Biomed. Eng., 1999 июль-август, 27 (4), 508-16; Re. Rhum. Engl. Ed., Mar 1999, 66(3), 158-166; Exp. Naphrol., ноябрь-декабрь 1996, 4(6), 314-21, и N.Y.Acad.Sci., 26, 673, 120-5.
В следующей экспериментальной модели изучался ингибиторный эффект нитрата томоксипрола VS гидрохлорида томоксипрола на повреждении бычьей эндотелиальной клетки, вызванном куменгидропероксидом. Адаптированная процедура была описана Fiorucci и др. Aliment. Pharmacol. Ther., 13, 421-35, 1999. Куменгидропероксид (270 μ M в этаноле) выращивался с бычьими эндотелиальными клетками, как описано Korubane и др., Jpn.J. Pharmacol., 68, 263-9, 1995, и фрагменты DNA были взяты как значение повреждения клетки. Полученные результаты выражались в % фрагмента DNA с контрольным сравнением и представлены в таблице 4. Ясно, что нитрат томоксипрола имеет большую цитозащитную активность, чем соответствующий гидрохлорид.

Пример 14
Антисептическая активность нитрата трамадола VS гидрохлорида трамадола.
Остеопорозис у пациентов может вызывать очень часто боли, и лечение такой болезни вызывает необходимость проведения терапии и интервенций не только для того, чтобы предотвратить дальнейшее разрушение костей и уменьшить риск переломов, но также и облегчить боли. Антисептическая активность соединений после их внутривенного введения определялась в соответствии с методом, описанным M.Bianchi и др., Brain Res., 797, 163-6, 1998.
Были использованы крысы Sprague Dawley (мужские особи) весом 200-250 г. Животные помещались в прозрачную пластиковую камеру и акклиматизировались в течение 5 минут перед тестом. Свет галогенной колбы 8В-50Вт направлялся на подошвенную поверхность задних лап крыс через донышко пластикового ящика. Луч имел диаметр примерно 12 мм. Измерялось время отдергивания левой задней лапы. Опыты проводились только на крысах, у которых основное скрытое состояние остается стабильным (±2,0 S) для таких последовательных измерений; значение, полученное при последнем измерении, рассматривалось как основное скрытое состояние.
Нитрат и гидрохлорид трамадола вводились внутривенно в 0,9% физиологическом растворе дозой 16 мг/кг. Антисептическая активность определялась через 30 и 120 минут после введения соединений. Результаты приведены в таблице 5 и выражаются как разница времени скрытого состояния до и после обработки животных в указанное время (30 или 120 минут после введения соединений).

Реферат
Изобретение относится к новым нитратным солям соединений формул (I)-(VI), которые могут быть использованы в медицине для лечения костных нарушений, таких как нарушения в костной ткани и суставах. Указанные соединения имеют повышенную терапевтическую активность и улучшенную желудочно-кишечную толерантность по сравнению с основаниями формул (I)-(VI). 3 н. и 2 з.п. ф-лы, 5 табл.
Нитратные соли соединений, выбранных из следующих классов:
класс F1

алендронат динатрия;
класс F2A

пироксикам;

метотрексат;
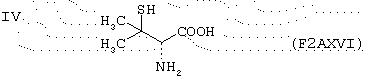
пеницилламин;

трамадол;
класс F2B

томоксипрол.
Формула





Комментарии