4-[2-(2-гидрокси-2-фенилэтиламино)этокси] фенилуксусная кислота или ее биопредшественник или фармацевтически приемлемая соль, способ ее получения, композиция на ее основе - RU2073666C1
Код документа: RU2073666C1
Чертежи


Описание
Предметом настоящего изобретения является 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусная кислота и ее биопредшественники. Предметом настоящего изобретения также являются способы и промежуточные соединения, предназначенные для их получения, применение этих соединений в лечебной практике и содержащие их фармацевтические составы. Соединения по настоящему изобретению представляют агонисты β3-адренорецептора и являются весьма полезными при лечении заболеваний, связанных с такими адренорецепторами. Введение соединений по настоящему изобретению теплокровным животным создает термогенный эффект, то есть в этом случае стимулируется термогенез, и введение этих соединений производится при лечении, например, ожирения и связанных с ним заболеваний, таких как юношеский диабет, возникающий вследствие ожирения. Кроме того, соединения по настоящему изобретению повышают толерантность к глюкозе у теплокровных животных и являются полезными для лечения заболеваний, при которых такое действие является благоприятным, например, они обладают гипогликемической активностью. Соединения по настоящему изобретению также можно использовать для лечения инсулиннезависимого сахарного диабета /NIDDM/ и заболеваний, при которых важное значение имеет резистентность к инсулину, таких как гипертензия, гиперлипемия и пониженный фибринолиз /синдром Ривена или синдром Х/.
Авторы настоящего изобретения провели глубокое исследование агонистов β3-адренорецепторов, в частности, их термогенного
действия. В выданном нам патенте США N 4772631 рассматриваются соединения формулы /A/:

в которой Ra представляет водород или фтор; Rb и Rc независимо друг от друга выбирают из водорода и С1-C3 алкил; а Z представляет гидроксиметил или группу формулы -CORd, в которой Rd является гидрокси, С1-C6 алкокси или амино.
В выданном нам патенте США N 4940168 рассматриваются
соединения формулы /B/:

в которой Ra представляет водород или фтор, а Re и Rf независимо друг от друга представляют водород или разнообразные группы, способствующие образованию вторичных и третичных амидов. Считается, что соединения формулы /A/, в которой Rd представляет алкокси или амино, и соединения формулы /B/ являются главным образом биопредшественниками, которые оказывают эффективное действие вместе с соответствующей оксиуксусной кислотой, то есть соединения формулы /A/, в которой R является гидрокси.
Авторы настоящего изобретения установили, что соединения формулы /A/, в которой Ra -Rc представляют водород, а Rd гидрокси, являются весьма интересными. Однако это соединение не демонстрировало идеальный профиль растворимости и поглощения. Поэтому авторы настоящего изобретения разработали биопредшественник этого соединения на основе вторичного амида. Такой вторичный амид вводили больным-добровольцам. К сожалению, при этом наблюдалось недостаточное действие на интенсивность обмена веществ в клинических условиях, а это фактически указывало на недостаточную эффективность свободной карбоновой кислоты и β3-адренорецептора.
Были произведены дальнейшие исследования, в результате которым нами было открыто соединение, которое обеспечивает значительное термогенное действие при дозах, вызывающих относительно небольшие побочные эффекты. Вполне понятно, что избирательность термогенного действия, например отсутствие побочных воздействий на сердце, является важным требованием, предъявляемым к терапевтическому средству, используемому для лечения, например, ожирения и связанных с ним заболеваний.
Соединение по настоящему изобретению представляет карбоновую кислоту и характеризуется значительным преимуществом по сравнению с вышеуказанным вторичным амидом и свободной карбоновой кислотой. В частности, было показано, что оно обладает высокой эффективностью в отношении β3-адренорецепторов, при этом было установлено, что соединение, ранее вводившееся больным, имело низкую эффективность. Кроме того, было продемонстрировано, что оно обладает удивительно хорошими характеристиками растворимости и поглощения.
В соответствии с настоящим изобретением предусматривается соединение формулы /I/

либо его биопредшественник, выбранный из амида или сложного эфира, или фармацевтически приемлемая соль.
Соединения формулы /I/ предпочтительно используются в форме карбоновой кислоты или ее фармацевтически приемлемой соли. 4-[2-2-Гидрокси-2-фенилэтиламино/этокси] фенилуксусная кислота и определенные биопредшественники являются амфотерными и могут использоваться в форме цвиттериона, в виде фармацевтически приемлемой соли присоединения кислоты или соли, образуемой основанием, что позволяет получить фармацевтически приемлемый катион. Отдельные примеры фармацевтически приемлемых солей присоединения кислоты включают, например, соли, образуемые неорганическими кислотами, такие как гидрогалогениды /в частности, гидрохлориды или гидробромиды/, сульфаты и фосфаты, и соли, образуемые органическими кислотами, такие как сукцинаты, цитраты, лактаты, тартраты, и соли, получаемые из кислотных водорастворимых полимеров. Определенные примеры солей, образуемых основаниями, которые позволяют получить фармацевтически приемлемый катион, включают, например, соли щелочных и щелочно-земельных металлов, такие как соли натрия, калия, кальция и магния, а также соли аммония и соли, образуемые приемлемыми органическими основаниями, такие как триэтаноламин.
Биопредшественниками являются такие фармацевтически приемлемые соединения, которые расщепляются в организме животного с образованием исходной кислоты. Такие соединения можно идентифицировать, например, путем перорального введения животному испытуемого соединения и последующего исследования жидкостей, содержащихся в организме этого животного.
Один класс биопредшественников составляют соответствующие сложно-эфирные биопредшественники карбоксильной группы 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты. Приемлемые сложные эфиры включают сложные С1-C6 алкиловые эфиры, например сложные метиловые и этиловые эфиры.
Другой класс биопредшественников образуют соответствующие сложно-эфирные биопредшественники гидроксильной группы /-CH/OH-/. Такие сложные эфиры включают в качестве ацильной группы ацетил, пропионил, пивалоил, С1-C4 алкоксикарбонил, например этоксикарбонид и фенилацетил.
Еще один класс биопредшественников образуют соответствующие амидные биопредшественники карбоксильной группы 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты. Приемлемые амиды включают, например, амиды формулы -CONR1R2, в которой R1 и R2 независимо друг от друга представляют водород, C1-C6 алкил, гидрокси-С2-C6 алкил /в котором гидроксильный заместитель не находится в положении альфа-углеродного атома/, С1-C4 алкокси-С1 -C6 алкил, фенилалкил, аллил, циклопропил или циклопентил, либо группу -NR1R2 представляет морфолино, пиперидино или пирролидино. Как правило, амиды, особенно те, у которых R1 и R2 независимо друг от друга представляют водород, прежде всего считаются промежуточными соединениями, предназначенными для получения соответствующей карбоновой кислоты или сложного эфира.
Предпочтительный класс биопредшественников составляют
соединения формулы (II)

в которой R3 представляет С1-C6 алкокси, например метокси.
Биопредшественники 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] -фенилуксусной кислоты сами по себе могут оказывать термогенное действие, и это является еще одним аспектом настоящего изобретения.
Особенно
предпочтительными соединениями по настоящему изобретению являются
/R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусная
кислота;
/R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетамид;
/R/-метил-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетат;
4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусная
кислота;
4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетамид; и
метил-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетат;
либо их фармацевтически приемлемые соли.
Весьма важно, если 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]-фенилуксусная кислота и ее биопредшественники содержат один или несколько асимметричных атомов углерода и могут существовать в виде оптически активных энантиомеров или оптически неактивных рацематов. В объем настоящего изобретения входят все энантиомеры, рацематы и/или /при наличии двух или большого числа асимметричных атомов углерода/ диастереоизомеры, которые при введении терапевтического количества создают термогенный эффект у теплокровных животных, при этом следует отметить, что в области химической технологии хорошо известны способы получения отдельных энантиомеров, например, путем повторного растворения рацемата или стереоспецифического синтеза, а также способы определения термогенных свойств, например, с помощью стандартных испытаний, которые описываются ниже. Предпочтение отдается соединениям по настоящему изобретению, имеющим абсолютную /R/ конфигурацию у группы -CH/OH- /в соответствии с правилами Кана Прелога Ингольда/.
C целью применения соединения по настоящему изобретению или его фармацевтически приемлемой соли для лечения теплокровных млекопитающих, включая человека, из него обычно изготавливают лекарственное средство в соответствии с общепринятой фармацевтической практикой, которое представляет фармацевтический состав.
Поэтому еще одним аспектом настоящего изобретения предусматривается фармацевтический состав, который включает 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусную кислоту, ее биопредшественник или фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Фармацевтические составы по настоящему изобретению можно вводить обычным путем, например перорально или парентерально. С этой целью при помощи известных методов из этих составов могут изготавливаться таблетки, капсулы, пилюли, порошки, водные или масляные растворы или суспензии, эмульсии и стерильные водные или масляные растворы или суспензии для инъекций.
Обычно предпочтение отдается составам, предназначенным для перорального введения.
Такие составы можно получить, используя стандартные наполнители и процедуры, хорошо известные в этой области. Стандартная лекарственная форма, например таблетки или капсулы, обычно содержит 0,1 500 мг активного ингредиента, более приемлемо 10 250 мг и предпочтительно 50 100 мг соединения по настоящему изобретению.
Эти составы также могут содержать другие активные ингредиенты, применяемые для лечения соответствующих заболеваний, например средства, понижающие аппетит, витамины, гипотензивные средства и полигликемические средства, такие как сульфонилмочевины, бигуаниды и тиазолидиндионы. Понятно, что такие составы охватывают приготовление лекарств совместного действия, одновременное и последовательное лечение двумя или большим числом активных ингредиентов.
В соответствии с одним аспектом настоящего изобретения из 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты или ее биопредшественника может быть изготовлено лекарственное средство пролонгированного действия, предназначенное для перорального введения, например матричные таблетки, включающие нерастворимый или разбухаемый полимерный наполнитель, либо драже с поверхностным покрытием.
При использовании с целью оказания термогенного действия на теплокровных животных, включая человека, 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусную кислоту, ее биопредшественник или фармацевтически приемлемую соль вводят в таком количестве, чтобы доза составляла 0,002 20 мг/кг, более приемлемо 0,02 10 мг/кг и предпочтительно 0,5 5 мг/кг при ежедневном введении в виде одной или нескольких разделенных доз от одного до трех раз в день. Однако специалистам в этой области понятно, что доза должна меняться в зависимости от сложности заболевания, возраста и пола больного, а также в соответствии с известными медицинскими принципами.
Кроме того, соединения по настоящему изобретению понижают уровни триглицерида и холестерина, а также повышают уровни альфа-липопротеинов высокой плотности /HDL/, и поэтому их можно использовать для лечения заболеваний, при которых такое понижение /и повышение/ указанных уровней считается благоприятным. Таким образом, эти соединения можно использовать для лечения гипертриглицеридемии, гиперхолестеринемии и заболеваний, связанных с низкими уровнями альфа-липопротеинов высокой плотности, помимо лечения атеросклероза, например коронарных, цереброваскулярных и периферических артерий, сердечно-сосудистых заболеваний и аналогичных болезней.
Еще одним аспектом настоящего изобретения предусматривается способ понижения уровней триглицерида и/или холестерина и/или увеличение уровней альфа-липопротеинов высокой плотности, который включает введение животному в случае необходимости терапевтически эффективного количества 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]-фенилуксусной кислоты, ее биопредшественника или фармацевтически приемлемой соли. Последующим аспектом настоящего изобретения предусматривается способ лечения атеросклероза, который включает введение животному в случае необходимости терапевтически эффективного количества 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты, ее биопредшественника или фармацевтически приемлемой соли. Эти составы изготавливают и вводят так же, как было детально описано выше для оказания термогенного действия. Они могут также содержать другие активные ингредиенты, применяемые для лечения атеросклероза и связанных с них заболеваний, например фибраты, такие как клофибрат, безафибрат и гемфиброзил; ингибиторы биосинтеза холестерина, такие как ингибиторы HMG-CoA редуктазы, например ловастатин, симвастатин и правастатин; ингибиторы поглощения холестерина, например бета-ситостерол, и ингибиторы /ацил СоА:холестерол-ацилтрансферазы/, например мелинамид; анионообменные смолы, например холестирамин, колестипол или диалкиламиноалкильное производное сшитого декстрана; никотиниловый спирт, никотиновую кислоту или ее соль; витамин Е и лекарственные средства, имитирующие гормоны щитовидной железы.
В соответствии с еще одним аспектом настоящего изобретения предлагаемые соединения стимулируют "нетипичные" β-адренорецепторы в желудочно-кишечном тракте и, таким образом, ингибируют двигательную функцию желудочно-кишечного тракта. Они могут быть полезны при лечении заболеваний, когда считается, что стимуляция "нетипичных" b-адренорецепторов в желудочно-кишечном тракте оказывает благоприятное воздействие, например, в случае заболеваний, требующих ингибирования двигательной функции желудочно-кишечного тракта. Например, их можно использовать для лечения воспалительных заболеваний кишечника /IBD/ /таких как болезнь Крона и неспецифический язвенный колит/, слизистый колит /IBS/, неспецифический понос и демпинг-синдром.
Таким образом, настоящим изобретением предусматривается способ стимуляции "нетипичных" b-адренорецепторов в желудочно-кишечном тракте, который включает введение животному в случае необходимости терапевтически эффективного количества соединения по настоящему изобретению.
Еще одним аспектом настоящего изобретения предусматриваются способы ингибирования двигательной функции желудочно-кишечного тракта, лечения воспалительных заболеваний кишечника, слизистого колита, неспецифического поноса и опорожнения желудка в случае демпинг-синдрома, который включает введение животному в случае необходимости терапевтически эффективного количества соединения по настоящему изобретению.
Еще одним аспектом настоящего изобретения предусматривается способ получения 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты, ее биопредшественника или
фармацевтически
приемлемой соли, который включает
a/ взаимодействие соединения формулы (III) или (IV) с соединением формулы (V)

в которой -COR4 представляет карбокси или его биопредшественник, а L является замещаемой группой; или
b/ гидролиз соединения формулы (VI)
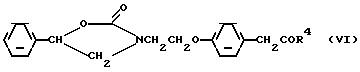
с/ гидролиз соединения формулы (VII) с целью получения 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты
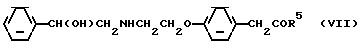
в которой R5 представляет гидролизуемую группу;
d/ взаимодействие соединения формулы (VIII) c соединением формулы (IX)


в которой -COR4 имеет указанные выше значения, а L' представляет замещенную группу;
е/ удаление защитной группы из соединения формулы (X):

в которой R6 представляет защищенное производное группы -COR4;
f/ превращение 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты в биопредшественник или наоборот, либо превращение биопредшественника 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты в другой биопредшественник 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты;
g/ восстановление соединения формулы (XI)

в которой -COR4 имеет указанные выше значения;
h/ восстановление соединения формулы (XII)

в которой -COR4 имеет указанные выше значения;
i/ восстановление соединения формулы (XIII)

в которой COR4 имеет указанные выше значения;
и в которой любая функциональная группа может быть необязательно защищена, после чего при необходимости производят:
/I/ удаление любых защитных групп;
/II/ образование фармацевтически приемлемой соли.
Защитные группы обычно выбирают из любых групп, описанных в литературе или известных квалифицированному химику, которые подходят для защиты указанной группы и могут быть введены обычными способами.
Защитные группы могут удаляться любым способом, описанным в литературе или известным квалифицированному химику, который подходит для удаления рассматриваемой защитной группы, причем такие методы выбирают с учетом того, чтобы удаление защитной группы вызывало минимальное нарушение других групп в молекуле.
В качестве примера защитной группы можно привести гидрогенолизируемую группу, находящуюся в положении у атома азота группы -CH/OH/CH2NCH2CH2O-. Приемлемой защитной группой является бензил или замещенная бензильная группа. Такую защитную группу можно удалить обычным образом, используя методы каталитической гидрогенизации, например, в присутствии катализаторов на основе паллидированного угля в атмосфере водорода. Необходимую обработку производят при нормальных и повышенных температурах и давлениях в таком растворителе, как С2-C6 алканол, например этанол или пропан-2-ол. Соединения, соответствующие формуле (1), защищенные гидрогенолизируемой группой, находящейся в положении у атома азота, можно получить в соответствии с методами, аналогичными описанным выше для получения соединения формулы (1).
Взаимодействие между соединением формулы (III) или (IV) и соединением формулы (V) может осуществляться в приемлемом растворителе, например в таком спирте, как этанол или пропан-2-ол, при температуре в интервале от 10 до 110oC, а в лучшем случае при температуре кипения реакционной смеси или близкой к ней. В соединении формулы (IV) L может представлять, например, галоген, такой как хлор или бром, либо арилсульфонилоксигруппу, такую как толуолсульфонилокси, или алкансульфонилоксигруппу, такую как метансульфонилокси.
Соединения формулы (V) получают любым способом, известным специалисту в этой области. Например, эти соединения можно получить путем взаимодействия соединения (XIV) с соединением формулы (XV)
NH2CH2CH2OH, (XIV)

Например, такое взаимодействие можно осуществлять с помощью реакции Мицунобу с диэтилазодикарбоксилатом и трифенилфосфином. Во время этой реакции желательно обеспечить защиту функциональной аминогруппы /и функциональной карбоксильной группы, если она присутствует/ и последующее снятие защиты обычным образом. Примеры приемлемой защитной группы для функциональной аминогруппы включают фталоильную и трет.-бутоксикарбонильную группы. Соединения формулы (XV) можно получить в соответствии со способами, известными в этой области.
Соединение формулы (VI) можно подвергать гидролизу с образованием 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты или ее биопредшественника при создании условий, известных в области получения блокаторов бета-адренорецепторов, например, путем щелочного гидролиза в приемлемом растворителе.
Соединения формулы (VI) можно получить в результате взаимодействия соединения формулы (XV) c соединением формулы (XVI)

в которой R7 представляет группу -CH2CH2OH. Эта реакция может осуществляться любым известным способом, например, при помощи метода, в соответствии с которым производится взаимодействие соединений формул (XIV) и (XV). В качестве альтернативы соединения формулы (VI) можно получить путем взаимодействия соединения формулы (XVI), в которой R7 представляет водород, с соединением формулы (IX), как это описывалось выше. В качестве другой альтернативы соединения формулы (VI) можно получить в результате взаимодействия соединения формулы (III) c соединением формулы (XVII)

в которой COR4 имеет указанные выше значения, а R8O- представляет отщепляемую группу, например R8O- является С1 -C4 алкокси.
Соединение формулы (XVI), в которой R7 представляет -CH2CH2OH, можно получить, например, в результате взаимодействия соединения формулы (III) c N-алкоксикарбонильным производным соединения формулы (XIV), в которой гидроксильная группа может быть необязательно защищена простым тетрагидропираниловым эфиром тетра-бутоксикарбониламиноэтанола. Соединения формулы (XVI), в которой R7 представляет водород, получают обычным способом. Соединения формул (IX) и (XVII) можно получить путем алкилирования соединений формулы (XV) обычным способом.
Взаимодействие между соединениями формул (VIII) и (IX) обычно осуществляют в условиях, аналогичных тем, которые создают при взаимодействии соединения формулы (IV) c соединением формулы (V). L' может иметь такие же значения, которые были приведены выше для L.
В соединениях формулы (VII) примеры гидролизуемых групп R5 включают C1-C6 алкокси и группы -NR1R2, так что -COR5 представляет сложный С1-C6 алкиловый эфир или функциональную амидную группу. Такие группы можно подвергать гидролизу /кислотному, щелочному, ферментативному/ c образованием группы -CO2H в обычных условиях. Превращения, в соответствии с которыми R5 становится гидролизуемой частью в живом организме, также включают примеры взаимопревращений биопредшественника 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты в 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусную кислоту. Приемлемые кислотные условия обеспечиваются, например, сильной минеральной кислотой, такой как хлористоводородная, серная или фосфорная кислота, обычно при температуре в интервале от 20 до 110oC и в полярном растворителе, таком как вода, С1 -C4 алканол /например, метанол или этанол/ или уксусная кислота. В таких случаях обычным способом можно выделить соответствующую соль 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты, образуемую минеральной кислотой. Альтернативно можно применять основные условия, создаваемые с помощью гидроксида лития, натрия или калия, обычно в приемлемом растворителе или разбавителе, таком как водный С1-C4 алканол, при температуре в интервале от 10 до 110oC; или галогенидом щелочного металла, например хлоридом лития в полярном растворителе, таком как диметилсульфоксид. В качестве других альтернатив в том случае, если -COR5 представляет трет-бутоксикарбонил, можно производить разложение, например, путем термолиза при температуре в интервале от 100 до 220oC без добавления или в присутствии приемлемого разбавителя, такого как простой дифениловый эфир.
Соединения формулы (VII) можно получить в соответствии с методами, которые были описаны выше для получения 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты или ее биопредшественника, при обеспечении необязательной защиты функциональной аминогруппы, например, с помощью бензильной группы.
4-[2-/2-Гидрокси-2-фенилэтиламино/этокси]фенилуксусную кислоту и ее амидные биопредшественники можно превратить в ее сложно-эфирные предшественники. Необходимую обработку производят в процессе нагрева с обратным холодильником в соответствующем алканоле при создании кислотных условий, например, посредством добавления концентрированной серной кислоты в качестве катализатора.
Восстановление соединений (XI), (XII) и (XIII) можно производить с помощью обычных химических или каталитических методов, таких как химическое восстановление с использованием борогидрида натрия или каталитическая гидрогенизация в присутствии таких катализаторов, как палладированный уголь или платина.
Восстановление борогидридом натрия обычно осуществляется в спирте, таком как метанол, причем эта реакция выполняется при температуре 0 20oC.
Каталитическое восстановление обычно производится в обычном растворителе для гидрогенизации, таком как спирт, например в этаноле. Гидрогенизацию, как правило, выполняют в атмосфере газообразного водорода при давлении от 1 до 10 атмосфер и при комнатной или повышенной температуре.
Соединения формулы (XI) можно получить при взаимодействии соединения формулы (V) c соединением формулы
(XVIII)

в которой L" представляет замещаемую группу.
Взаимодействие между соединением формулы (XVIII) и соединением формулы (V) можно производить в приемлемом растворителе, таком как спирт или простой эфир, например метанол или простой диэтиловый эфир, при температуре в интервале от -10 до 110oC, а в лучшем случае при комнатной температуре. В соединениях формулы (XVIII) может представлять, например, галоген, такой как хлор или бром.
Полученные соединения формулы (XI) можно превратить на месте в 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусную кислоту или в ее биопредшественники.
Соединения формулы (XVIII) можно получить в соответствии со способами, известными в этой области.
Соединения формулы (XII) можно получить в результате взаимодействия соединения (XIX) c соединением (V)

Взаимодействие между соединением (XIX) и соединением формулы (V) можно производить в приемлемом растворителе, таком как спирт, например этанол, при температуре 0 80oC, а лучше всего при комнатной температуре. Полученные соединения формулы (XII) можно на месте превратить в 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусную кислоту или в ее биопредшественники.
Соединения формулы (XIII) можно получить в результате взаимодействия соединений формулы
(XX) с соединением формулы (VIII)

в которой COR4 имеет указанные выше значения.
Взаимодействие между соединением формулы (XX) и соединением (VIII) можно производить в приемлемом растворителе, таком как спирт, например этанол, при температуре в интервале 0 80oC, а в лучшем случае при комнатной температуре. Полученные соединения формулы (XIII) можно превратить на месте в 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусную кислоту или в ее биопредшественники.
Соединения формулы (XX) можно превратить путем
гидролиза в соединение формулы (XXI)

в которой COR4 имеет указанные выше значения, а R9 и R10 независимо друг от друга представляют водород или С1-C4 алкил. Гидролиз производится в кислотных условиях, создаваемых, например, сильной минеральной кислотой, такой как хлористоводородная или серная кислота, обычно при температуре в интервале 20 110oC в приемлемом растворителе, таком как тетрагидрофуран, дихлорметан или простой диэтиловый эфир.
Соединения формулы (XXI) можно получить в соответствии со стандартными
методами, известными в этой области. Например, в результате взаимодействия соединения
формулы (XXII) c соединением формулы (XV) в присутствии слабого основания

Приемлемые условия взаимодействия включают нагрев в соответствующем растворителе, таком как дихлорметан, в присутствии слабого основания, например карбоната натрия. В соединении формулы (XXII) L''' может быть галогеном, таким как бром.
Соединения формулы (VI), (VII), (X), (XI), (XII) и (XIII) обладают признаками новизны и образуют еще один аспект настоящего изобретения.
Сложно-эфирные биопредшественники гидроксильной группы можно получить обычным способом, например, в результате взаимодействия гидроксильной группы с активированным производным кислоты при создании условий, известных в области синтеза блокаторов бета-адренорецепторов.
Фармацевтически приемлемые соли можно получить в результате взаимодействия 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты или ее биопредшественника с соответствующей кислотой или основанием, производимого обычным образом. Альтернативно, при необходимости получения галоидоводородной соли ее можно получить путем гидрогенизации свободного основания вместе со стехиометрическим количеством соответствующего бензилгалогенида.
Это изобретение иллюстрируется следующими биологическими испытаниями, данными и примерами.
Термогенное действие.
Термогенное действие соединений формулы (I) и их биопредшественников
можно продемонстрировать с помощью одного или нескольких следующих стандартных испытаний:
/a/ Крыс в течение 5
дней адаптировали к холоду при температуре 4oC c целью увеличения их
способности к термогенезу. Затем их переводили в теплые условия содержания при температуре 25oC, в которых
они находились в течение 2 дней. На следующий день крысам вводили испытуемое
соединение подкожно или перорально. Через час животных умерщвляли и удаляли межлопаточное скопление бурой жировой ткани
/BAT/. Митохондрию бурой жировой ткани получали путем дифференциального
центрифугирования, после чего определяли связывание ГДФ /гуанозин-5'-дифосфат/ /Холловей и др. International Journal of Obesity
1984, 8, 295/ в качестве меры термогенной активации. В каждом испытании
участвовала контрольная группа, которой вводили только носитель, представляющий раствор или суспензию, и положительная
контрольная группа, которой вводили изопреналин /в виде сульфата/ в дозе 1
мг/кг-1. Испытуемое соединения вводили в соответствии с определенным режимом в дозе 0,1; 0,3; 1,0; 3,0 и 10
мг/кг-1, а результаты выражали в виде воздействия на связывание ДГФ,
характерного для положительной контрольной группы. Исходя из этих результатов, по методу выравнивания кривой высчитывали
дозу /50%-ная эффективная доза /ED50//, необходимую для достижения
50%-ного эффекта изопреналина. При выполнении этого испытания соединения считались активными, если они вызывали
значительное увеличение связывания ГДФ по сравнению с контрольными группами.
/b/ Крыс адаптировали к термонейтральным условиям содержания /29oC/ в течение 2 недель с целью снижения их способности к термогенезу без возникновения озноба, который определяется бурой жировой тканью. На протяжении последних 5 дней животных приучали пользоваться аппаратом для неинвазивного измерения частоты сердечных сокращений с помощью прикрепляемых к лапам электродов, соединенных c интегратором электрокардиографа, позволяющего получать непрерывное считывание частоты сердечных сокращений. Испытуемое соединение вводили подкожно или перорально в виде ED50, установленной в испытании /a/, после чего в течение 15 30 минут определяли частоту сердечных сокращений. Эту процедуру повторяли в последующих испытаниях при увеличении ED50, определенной в испытании /a/, до тех пор, пока частота сердечных сокращений /HP/ не достигала или не превышала 500 сокращений в минуту, либо до тех пор, пока вводимая доза не достигала 100-кратного размера ED50, определенной в испытании /a/. Определяли дозу, необходимую для достижения частоты сердечных сокращений, равной 500 сокращениям в минуту /доза D500 /.
Отношение D500 к ED50, полученной в испытании /a/, определяется как индекс избирательности /SI/ и представляемому избирательности соединения в отношении бурой жировой ткани по сравнению с сердечно-сосудистой системой. Считается, что соединения обладают значительной избирательностью, если у них индекс избирательности больше 1 /S1 > 1/. Неизбирательные соединения имеют индекс избирательности < 1 /например, изопреналин 0,06/.
/c/ Крыс содержали при температуре 23oC в течение по крайней мере двух дней, затем на протяжении ночи их лишали пищи. На следующий день определяли основной обмен животных, используя для этого аппарат для измерения поглощения кислорода замкнутого типа, описанный Арунделем и др. 1984, J. Appl. Physiol. Respirat Environ. Exercise Physiol. 1984, 57/5/ 1591 1593. Затем крысам вводили /перорально/ испытуемое соединение в дозе 1 мг/кг-1 в виде раствора или суспензии в полисорбате 80 /0,5 мл/100 г/ при 0,025% отношении веса к объему. После этого определяли интенсивность обмена веществ, выполняя эту процедуру в течение по крайней мере одного часа после введения дозы. При выполнении этого испытания соединения считались активными, если они вызывали значительное увеличение интенсивности обмена веществ по сравнению с контрольными животными /t-критерий Стьюдента: p < 0,05/, которым вводили только носитель, представляющий раствор или суспензию.
В рассмотренных выше испытаниях
соединения формулы (I) обычно оказывали действие
следующего порядка без вызывания явной токсичности:
испытание /a/: подкожное или пероральное введение ED50 для связывания ГДФ в
митохондрии бурой жировой ткани при дозе 0,01 10
мг/кг-1;
испытание /b/: демонстрирует индекс избирательности > 50; и
испытание /c/: демонстрирует поглощение 2 9 мл
О2 мин-1/кг0,75
/-1 при дозе 1 мг/кг-1, вводимой перорально.
В целях иллюстрации следует отметить, что соединение, описанное в
прилагаемом примере 1, оказывает следующее
действие в вышеописанных испытаниях:
/a/ пероральное введение ED50, равной 0,55 мг/кг-1;
/b/ индекс избирательности
> 50 /пероральное введение/;
/c/ 6,53 мл О2 мин-1/кг0,75/-1 при пероральном введении 1 мг/кг-1.
Испытание на толерантность к глюкозе при пероральном введении.
Самцов крыс /125 150 г/ лишали пищи в течение 24 часов. После голодания анестезировали группу из шести крыс и брали у них пробу крови из сердца. Затем крысам из других групп вводили соединение по примеру 1 /5,0 мг/кг перорально/, растворенное в водном растворе 0,025% полисорбата. Контрольным крысам вводили только раствор полисорбата. Объем раствора составлял 0,5 мл/100 г веса тела. Через 60 минут после введения указанного средства анестезировали шесть контрольных и шесть получавших соединение крыс и брали у них пробы крови из сердца. Остальным крысам перорально вводили глюкозу /1 г/кг/ в виде 20% раствора. D-глюкозы /0,5 мл/100 г/. Затем анестезировали по шесть крыс из контрольной и получавшей соединение групп и брали у них пробы крови через 20, 60 и 120 минут после введения глюкозы. Содержание глюкозы и инсулина в плазме определяли стандартными методами (табл. 1).
Приведенные результаты представляют среднее значение ± стандартная ошибка наблюдений для шести крыс в каждой группе. Для проверки значимости разности между контрольной и получавшей испытуемое соединение группами использовали t-критерий Стьюдента. Соединения по примеру 1 обладают ярко выраженной гипогликемической активностью.
Влияние на уровни глюкозы в крови у инсулинрезистентных парных мышей.
Мышей С57BL/KSY /парных/ делили на две группы и обеспечивали им свободный доступ к контрольному рациону или рациону, содержащему соединение по примеру 1 с концентрацией 50 мг/кг рациона. В эксперименте также участвовала группа контрольных /+/+/ мышей. Через 16 дней скармливания мышам указанного рациона у них брали пробы крови с целью определения уровней глюкозы в крови (табл. 2).
Полученные результаты представляют среднее значение ± стандартная ошибка наблюдений в группах, состоящих из 15 мышей. Для определения значимости разности между контрольной /+/+/ и получавшей испытуемое соединение /парные мыши/ группами использовали t-критерий Стьюдента. Соединение по примеру 1 нормализует уровни глюкозы в крови у животных с подобной моделью резистентности к инсулину.
Испытание на липолиз адипоцитов у крыс.
У самцов крыс иссекали жировую ткань эпидидимиса и путем разложения коллагеназой получали адипоциты. Клетки отделяли с помощью флотации и четырежды промывали бикарбонатным буфером Кребса Рингера /KRB/, а в конце промывали буфером KRB, содержащим 2% бычьего сывороточного альбумина /KRB/BSA/. Аликвоты клеточной суспензии инкубировали в присутствии различных концентраций испытуемого соединения в общем объеме, равном 1 мл KRB/2% BSA, содержащем 0,1 мг/мл аскорбата, в атмосфере, состоящей из 95% О2 и 5% CO2. Инкубацию также производили в присутствии концентрации изопреналина /3 x 10-6 ммоля/, которая, как известно, оказывает максимальное влияние на липолиз. Контрольную инкубацию производили в KRB/2% BSA, содержащем аскорбат. Инкубации заканчивали через 90 минут, помещая пробирки на лед, и удаляли аликвоты внутренней жидкости для анализа свободных жирных кислот, содержание которых измеряли с помощью набора для выполнения анализов WAKO NEFA-C /Альфа лабораториз/. Липилитическую активность соединений оценивали путем определения степени увеличения концентрации свободных жирных кислот, вызываемого указанными соединениями по сравнению с контрольными. Максимальное воздействие /эффективность/ соединений определяли и выражали в виде процентного значения от максимального воздействия изопреналина (табл. 3).
Эффективность определяется как максимальное воздействие испытуемого соединения на липолиз, выраженное в виде процентного значения от максимального воздействия изопреналина.
Сравнительное испытание на активность в отношении ГДФ.
В другом сравнительном испытании активность соединения по примеру 1 в представленном выше испытания а/ сравнивали с эталонным соединением (табл. 4).
Настоящее изобретение далее иллюстрируется следующими примерами, в которых, если нет
специального
указания:
a/ хроматография выполнялась на кизельгеле /артикул 9385, 230 400 меш/, полученном из фирмы "Е. Мерк", Дармштадт, Федеративная Республика Германии/.
b/ выпаривания производили при пониженном давлении с использованием роторного испарителя.
с/ температуры плавления не корректировались.
Пример 1.
/R/-4-[2-/2-Гидрокси-2-фенилэтиламино/этокси]фенилуксусная кислота.
/R/-4-[2-/2-Гидрокси-2-фенилэтиламино/этокси] фенилацетамидгидрохлорид /3,5 г/ нагревали в паровой бане в 2 н. растворе HCl в течение двух часов /100 мл/. Реакционную смесь фильтровали, охлаждали и путем фильтрования собирали твердое вещество. Полученное твердое вещество кристаллизовали из воды с образованием /R/-4-[2/2-Гидрокси-2-фенилэтиламино/этокси]фенилуксусной кислоты в виде хлористоводородной соли /2,5 г/, температура плавления 207 209oC.
Микроанализ.
Обнаружено: C 61,5; H 6,5; N 3,8; Cl 10,1%
Необходимо для C18H22ClNO4: C 61,5; H 6,3; N 4,0; Cl 10,1%
[α]= -27,3° /c 0,99 в метаноле/.
Хлористоводородную соль продукта /1 г/ растворяли в дистиллированной воде /50 мл/ при комнатной температуре, после чего показатель рН осторожно доводили до рН 6,7 посредством добавления 2 н. раствора NaOH. Выделенное твердое вещество представляло /R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусную кислоту /0,8 г/, температура плавления 209 211oC.
Микроанализ.
Обнаружено: C 68,4; H 6,8; N 4,
5%
Необходимо для C18H21NO4: C 68,6; H 6,7; N 4,5%
[α]= -30,1° /c 1,0 в уксусной кислоте/.
В качестве альтернативы этот продукт можно получить с помощью кислотного гидролиза /R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилацетамида, как это описывалось выше, с перекристаллизацией из воды.
Пример 2.
/R/-4-[2-/2-Гидрокси-2-фенилэтиламино/этокси]фенилацетамид
/R/-4-[2-/N-Бензил-N-/2-гидрокси-2-фенилэтил/амино/этокси] фенилацетам- ид /12,9 г/ растворяли в этаноле /150 мл/ и ледяной
уксусной
кислоте /50 мл/. Полученный раствор гидрогенизировали в присутствии палладированного угля /1,0 г/ с 10%-ным соотношением массы при давлении 20 бар и температуре 60oC в течение 24
часов. Эту
смесь охлаждали, фильтровали и фильтрат выпаривали при пониженном давлении. 1,2 г полученного таким образом остаточного масла /9,8 г/ растворяли в этилацетате и обрабатывали раствором
простого эфира,
насыщенным хлороводородом. Осажденное твердое вещество кристаллизовали из метанола с образованием /R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетамидгидрохлорида /0,8 г/,
температура плавления
255 257oC.
Микроанализ.
Обнаружено: C 61,3; H 6,3; N 8,0; Cl 10,2%
Необходимо для C18H23ClN2
O3: C 61,6;
H 6,6; N 8,0; Cl 10,1%
[α]= -19,5° /c 1,
0 в DMCO/.
Эту реакцию также можно выполнять в водном растворе изопропанола, содержащем ледяную уксусную кислоту /1 эквивалент/ при нормальных температуре и давлении.
Исходный материал был получен следующим образом.
Cмесь 4-[2-/бензиламино/этокси] фенилацетамида /OLS 2135678/ /14,0 г/, /R/-стиролоксида /5,92 г/ и пропан-2-ола /200 мл/ нагревали с обратным холодильником в течение 72 часов. Полученную смесь охлаждали, а растворитель выпаривали при пониженном давлении. Остаток очищали посредством испарительной хроматографии на "сухих" колонках. Элюирование 10% метанолом в дихлорметане позволило получить /R/-4-[2-/N-бензил-N-/2-гидрокси-2-фенилэтиламино/этокси] фенилацетамид в виде масла /12,9 г/. Эту реакцию также можно выполнять в трет-амиловом спирте при нагревании с обратным холодильником.
Пример 3.
/R/-Метил-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетат.
/R/-4-[2-/2-Гидрокси-2-фенилэтиламино/этокси] фенилацетамидгидрохлорид /0,45 г/ нагревали с обратным холодильником в метаноле /20 мл/, содержащем концентрированную серную кислоту /0,5 мл/, в течение 18 часов. Эту смесь охлаждали, а растворитель выпаривали при пониженном давлении. Остаток растворяли в дихлорметане /30 мл/ и последовательно промывали водой /20 мл/, 5% раствором NaHCO3 /50 мл/ и водой /20 мл/, сушили над MgSO4, после чего удаляли растворитель при пониженном давлении. Остаток растворяли в метилацетате /20 мл/ и обрабатывали раствором простого эфира, насыщенным хлороводородом. Осажденное твердое вещество кристаллизовали из смеси метанола и метилацетата с образованием /R/-метил-4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилацетатгидрохл- орида /0, 25 г/, температура плавления 181 183oC.
Микроанализ.
Обнаружено: C 62,6; H 6,6; N 3,8; Cl 9,9%
Необходимо для C19H24
ClNO4: C 62,4; H 6,6; N 3,8; Cl 9,7%
[α]= -25,3° /c
0,99 в
метаноле/.
Пример 4.
4-[2-/2-Гидрокси-2-фенилэтиламино/этокси]фенилуксусная кислота.
4-[2-/2-Гидрокси-2-фенилэтиламино/этокси] фенилацетамидгидрохлорид /2,8 г/ нагревали в паровой бане в 4 н. растворе HCl /60 мл/ в течение 4 часов. Реакционную смесь фильтровали, охлаждали, при этом твердое вещество, собранное фильтрованием, представляло 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусную кислоту в виде хлористоводородной соли /1,4 г/, температура плавления 183 184oC /размягчается при температуре около 178oC/.
Микроанализ.
Обнаружено: C 61,4; H 6,3; N 4,1; Cl 10,3% необходимо для C18H22ClNO4: C 61,5; H 6,3; N 4,0; Cl 10,
1%
Пример 5.
4-2-/2-Гидрокси-2-фенилэтиламино/этокси фенилацетамид.
Заранее полученный раствор 4-[2-/N-бензил-N-/2-гидрокси-2-фенилэтил/амино/этокси] фенилацетамида в пропан-2-оле и ледяной уксусной кислоте /см. ниже/ гидрогенизировали в присутствии палладированного угля /1,0 г/ с 10% -ным соотношением массы при давлении 20 бар и температуре 60oC в течение 12 часов. Эту смесь охлаждали, фильтровали и выпаривали фильтрат при пониженном давлении. Полученное таким образом остаточное масло растворяли в этилацетате и обрабатывали раствором простого эфира, насыщенным хлороводородом. Осажденное твердое вещество кристаллизовали из метанола с образованием 4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилацетамидгидрохлорида /6,8 г/, температура плавления 250 251oC /размягчается при температуре около 248oC/.
Микроанализ:
Обнаружено: C 61,2; H 6,5; N 8,3; Cl 10,3%
Необходимо
для C18H23ClN2O3: C 61,6; H 6,6; N 8,0; Cl 10,1%
Исходный материал был получен следующим образом:
Смесь
4-/2-N-бензиламиноэтокси/фенилацетамид /OLS2135678/ /5,68 г/, стиролоксида /2,4 г/ и пропан-2-ола /100 мл/ нагревали с обратным холодильником в течение 72 часов. Полученный таким образом
4-[2-/N-бензил-N-/2-гидрокси-2-фенилэтиламино/этокси] фенилацетамид охлаждали и разбавляли пропан-2-олом /30 мл/ и ледяной уксусной кислотой /20 мл/.
Альтернативный метод получения
указанного в заголовке соединения представляет
2-Гидрокси-2-фенилэтиламино /1,91 г/, 4-/2-бромэтокси/фенилацетамид /3,59 г/ и триэтиламин /1,41 г/ в этаноле /450 мл/ нагревали с обратным
холодильником в течение 24 часов, после чего раствор охлаждали и фильтровали с целью удаления небольшого количества нерастворимого материала /0,2 г/. Растворитель удаляли из фильтрата при пониженном
давлении, остаточное твердое вещество растирали в порошок с небольшим количеством воды, отделяли фильтрованием и сушили. Твердое вещество /3,1 г/ растворяли в метаноле /100 мл/ и обрабатывали
раствором простого эфира, насыщенным хлороводородом. Осажденное твердое вещество кристаллизовали из метанола с образованием 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетамидгидрохлорида,
температура плавления соединения и температура смеси 250 251oC.
Другой альтернативный метод получения указанного в заголовке соединения представляет следующее.
Фенацилбромид /0,49 г/, 4-/2-аминоэтокси/фенилацетамид /0,49 г/, карбонат калия /0,35 г/ и метанол /20 мл/ нагревали с обратным холодильником в течение 2 часов, охлаждали при комнатной температуре и перемешивали при одновременном добавлении борогидрида натрия /1,0 г/, который вводили небольшими порциями в течение 1 часа. Перемешивание продолжали в течение 18 часов, после чего растворитель выпаривали при пониженном давлении. Остаток разделяли между дихлорметаном /20 мл/ и водой /10 мл/. Водный слой отделяли и экстрагировали из дихлорметана /20 мл/. Соединенные экстракты дихлорметана промывали водой /20 мл/, сушили над MgSO4 и выпаривали. Остаточную смолу растворяли в этилацетате и обрабатывали раствором простого эфира, насыщенным хлороводородом. Осажденное твердое вещество кристаллизовали из смеси метанола и этилацетата с образованием 4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетамидгидрохлорида, температура плавления соединения и температура плавления смеси 248 250oC.
Еще один альтернативный метод получения указанного в заголовке соединения представляет следующее.
Фенилглиоксальгидрат /0,57 г/ и 4-/2-аминоэтокси/фенилацетамид /0,49 г/ в метаноле /15 мл/ нагревали в паровой бане с образованием светлого раствора. Этот раствор затем охлаждали в ледяной бане и при перемешивании в течение 1 часа небольшими порциями добавляли борогидрид натрия /1 г/. После добавления 100 мг борогидрида натрия начинало отделяться белое твердое вещество, которое вновь растворяли путем добавления дополнительного количества метанола /35 мл/. Полученную смесь перемешивали при комнатной температуре в течение 18 часов, после чего растворитель выпаривали при пониженном давлении. Остаточное твердое вещество обрабатывали дихлорметаном /20 мл/ и водой /20 мл/, а твердое вещество, которое не растворилось, отделяли фильтрованием. Это твердое вещество растворяли в метаноле /20 мл/ и обрабатывали раствором простого эфира, насыщенным хлороводородом. Основное количество растворителя выпаривали, а затем добавляли этилацетат /20 мл/ с целью осаждения указанного в заголовке соединения в виде хлористоводородной соли, температура плавления соединения и температура плавления смеси 250 251oC.
Пример 6.
Метил-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]фенилацетат.
4-[2-/2-Гидрокси-2-фенилэтиламино/этокси] фенилацетамидгидрохлорид /2,5 г/ нагревали с обратным холодильником в метаноле /50 мл/, содержащем концентрированную серную кислоту /1,5 мл/, в течение 24 часов. Полученную смесь охлаждали и выпаривали растворитель при пониженном давлении. Остаток разделяли между дихлорметаном /150 мл/ и 5% раствором NaHCO3 /150 мл/. Органический слой отделяли и последовательно промывали 5% раствором NaHCO3 /20 мл/ и водой /20 мл/, сушили над MgSO4, а затем удаляли растворитель при пониженном давлении. Остаток растворяли в метилацетате /40 мл/ и обрабатывали раствором простого эфира, насыщенным хлороводородом. Осажденное твердое вещество кристаллизовали из смеси метанола и метилацетата с образованием метил-4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилацетатгидрохлорида /1,8 г/, температура плавления 169 171oC.
Микроанализ.
Обнаружено: C 62,5; H 6,6; N 3,8; Cl 9,7%
Необходимо для C19H24ClNO4: C 62,4; H 6,6; N 3,8; Cl 9,7%
Пример 7.
/R/-4-[2-/2-Ацетокси-2-фенилэтиламино/этокси]фенилуксусная кислота.
Раствор N-трет-бутоксикарбонил-/R/-4-[2-/2-ацетокси-2-фенилэтиламино/этокси] ф- енилуксусной кислоты /500 мг/ в смеси дихлорметана /5 мл/ и трифторуксусной кислоты /5 мл/ оставляли для выстаивания в течение 90 минут при температуре 20oC. Растворитель удаляли при пониженном давлении, а остаток растворяли в этаноле /5 мл/. Раствор охлаждали до -20oC и добавляли раствор хлорводорода в эфире, что позволило получить белые кристаллы указанного в заголовке соединения в виде хлористоводородной соли /300 мг/, температура плавления 158 160oC; [α]= -35, 4° /c 1,0 в метаноле/.
Микроанализ.
Обнаружено: C 60,2; H 6,5; N 3,4; H2O 0,9%
Необходимо для C20H24
ClNO5•H2O: C 60,3; H 6,2; N 3,5; H2O 1,1%
Исходный материал был получен следующим образом:
a/ Ди-трет-бутилкарбонат /1,25 г/ в трет-бутаноле
(15 мл) добавляли к перемешиваемому раствору хлористоводородной соли /R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси] фенилуксусной кислоты /2,0 г/ в 1 н. водном растворе гидроксида натрия /30 мл/.
Реакционную смесь перемешивали в течение 90 минут при температуре 20oC. Растворитель удаляли при пониженном давлении. К остатку добавляли воду /20 мл/ и раствор подкисляли 2 н. водным
раствором лимонной кислоты. Образовавшийся продукт экстрагировали введением в 5% раствор метанола в дихлорметане /5 x 20 мл/. Экстракты сушили, а растворитель удаляли при пониженном давлении с
образованием N-трет-бутоксикарбонил-/R/-4-[2-/2-гидрокси-2-фенилэтиламино/этокси]ф- енилуксусной кислоты в виде стекла /1,3 г/; [α]= +6,2° /c 1,0 в метаноле/.
b/ Раствор продукта, полученного на стадии а/, описанной выше, в пиридине /2,5 мл/ и уксусном ангидриде /2,5 мл/ оставляли для выстаивания в течение 16 часов при температуре 20oC. Смесь растворителей удаляли при пониженном давлении. Остаток растворяли в дихлорметане /20 мл/. Раствор промывали водой /3 x 10 мл/, сушили и удаляли растворитель при пониженном давлении с образованием вязкого масла /1,3 г/. Раствор масла /1,3 г/ в смеси диоксана /10 мл/ и воды /6 мл/ перемешивали при нагревании с обратным холодильником в течение 2 часов. Растворитель удаляли при пониженном давлении, а остаток очищали с помощью хроматографии при использовании в качестве элюента 10% раствора метанола в дихлорметане. Соответствующие фракции собирали с образованием N-трет-бутоксикарбонил-/R/-4-[2-/2-ацетокси-2-фенилэтиламино/этокси]ф- енилуксусной кислоты в виде вязкого масла /500 мг/; [α]= -16,8° /c 2,0 в дихлорметане/.
Пример 8.
Как указывалось выше, приемлемые фармацевтические составы, включающие соединения формулы (I), можно получить в соответствии со стандартными методами изготовления лекарственных средств.
Типичный состав в виде таблеток, предназначенный для перорального введения теплокровным животным, включает в качестве активного ингредиента соединение формулы (I) или его фармацевтически приемлемую соль, рассмотренные выше /например, в соответствии с одним из предшествующих примеров/ и может быть получен путем водной грануляции или прямого прессования с измельченной лактозой, содержащей стандартный дезинтегратор и/или смазывающее вещество. При необходимости получения таблеток с небольшим содержанием активного ингредиента /например, 0,5 10 мг/ можно использовать метод прямого прессования, при осуществлении которого активный ингредиент смешивают с лактозой в соотношении 1: 10 массовых частей и/или с микрокристаллической целлюлозой, содержащей 0,5 мас. смазывающего вещества /такого как стеарат магния/ и 5 мас. дезинтегратора /такого как сшитая натрий-карбоксиметилцеллюлоза или гликолят натриевого крахмала/. Типичные таблетки, полученные путем водной грануляции, содержат активный ингредиент /50 100 мг/, лактозу /230 мг/, маисовый крахмал /80 мг/, желатин /2,2 мг/, стеарат магния /4 мг/ и кроскармеллозный натрий /7 мг/.
Реферат
Использование: в качестве биологически активных соединений для стимуляции термогенеза и/или повышения толерантности к глюкозе. Сущность изобретения: продукт - 4-[2-(2-гидрокси-2-фенилэтиламино)этокси] фенилуксусная кислота, 4-[C6H5CH(OH)CH2NHCH2 CH2O] C6H4CH2COOH или ее биопредшественник, выбранный из амида или сложного эфира, или фармацевтически приемлемая соль и фармацевтическая композиция. Реагент 1: стиролоксид. Реагент 2: 4-(H2NCH2CH2O) C6H4CH2COR4, COR4 - амидная группа. Условия реакции: при нагревании с последующим при необходимости гидролизом продукта для получения кислоты и превращением последней при необходимости в сложный эфир и выделением продукта при необходимости в виде фармацевтически приемлемой соли. 5 с. и 7 з. п. ф-лы, 4 табл.
Формула

или ее биопредшественник, выбранный из амида или сложного эфира, или фармацевтически приемлемая соль.

в которой R4 (C1-C6)-алкокси, или его фармацевтически приемлемую соль.

или ее биопредшественника, выбранного из группы: амид или сложный эфир, или фармацевтически приемлемой соли, отличающийся тем, что осуществляют взаимодействие соединения формулы

с соединением формулы

где COR4 амидная группа,
с последующим, при необходимости, гидролизом продукта для получения кислоты и превращением последней, при необходимости, в сложный эфир и выделением продукта, при необходимости, в виде фармацевтически приемлемой соли.

где COR5 амидная группа.
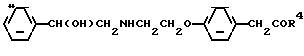
отличающийся тем, что осуществляют взаимодействие соединения формулы

с соединением формулы

где COR4 амидная группа;
L' замещаемая группа.

12. Соединение по п.1, или его биопредшественник, или фармацевтически приемлемая соль для стимуляции термогена и/или повышения толерантности к глюкозе.
Комментарии