Аминометилинданы - бензофураны и бензотиофены - RU2141474C1
Код документа: RU2141474C1
Чертежи


Описание
Настоящее изобретение относится к новому классу замещенных аминометилинданов, -2,3-дигидробензофуранам, -2,3-дигидробензотиофенам, -1,3-дигидробензофуранам и -1,3-дигидроизобензотиофенам, воздействующим на центральные рецепторы 5-HT1A. Поэтому эти аминометильные соединения полезны для лечения обычных психических и неврологических нарушений.
Предпосылки
изобретения
Предварительно в этой области были известны некоторые аминометилинданы и их производные.
Так, в Европейском патенте 0281261 раскрываются производные 1-аминометилиндана, 3-аминометилбензофурана и 3-аминометилбензотиофена с гидроксигруппой или с замещенной гидроксигруппой в положении 6 (индан) или в положении 5 (бензофуран, бензотиофен). Оказалось, что эти соединения обладают активностью в качестве центральных агонистов допамина, в особенности они оказывают влияние на пресинаптические рецепторы допамина.
Патент Великобритании N 2093837 касается класса производных 1-аминоалкил тетралина, имеющих один или более гидрокси- или алкоксизаместителей в положении 5, 6, и/или 7, и заявляется их адренергическое и допаминергическое влияние, что делает их полезными для лечения гипертензии. DeBernardis et al (J. Med. Chem., 1985, 28(10), 1398 - 1404) обсуждает такого рода влияние относительно гидрокси замещенных амино-метилтетралинов, -инданов и бензоциклобутенов.
В европейском патенте N 0402923 A2 раскрываются производные 2,5-диаминотетралина, которые, как заявляется, имеют агонистическую активность относительно допамина и разных соединений, взаимодействующих с различными функциональными рецепторами допамина, оказывая таким образом различные терапевтические эффекты, такие как воздействия на шизофрению, гипертензию и Паркинсонизм.
В патенте De N 3924365 A1 описывается класс производных 2-амино-7-карбамоилтетралина, которые обладают свойствами пресинаптического агониста допамина и, следовательно, имеют антигипертензивное действие, уменьшают частоту сердечных сокращений и воздействуют на центральную нервную систему.
В патенте США N 4500543 указано, что обычные 1-аминометилфталановые соединения демонстрируют адренергический эффект и, следовательно, антигипертезивные свойства и свойство уменьшать частоту сердечных сокращений. Этот патент охватывает соединения, имеющие заместителей в положении 5, 6 и/или 7.
Патент Франции N 2548146 относится к производным 3-аминометилтиофталида и 3-аминометилизобензофуранэтиона; заявлено, что эти соединения обладают аналгезирующим и/или антиконвульсивным действием.
Ни в одной из этих ссылок не обсуждается или не предполагается, чтобы любое из названных соединений обладало активностью относительно рецептора 5-HT1A.
Клинические испытания соединений, которые, как известно, обладают агонистической активностью относительно 5-HT1A, таких как 8-[4-[4-(2-пиримидил)бутил] -8-азаспиро[4,5]декан-7,9-дион, гепирон, 4,4-диметил-1-[4-[4-(2-пиримидил)-1-пиперазинил] бутил] -2,6-пиперидиндион и ипсапирон, 2-[4-[4-(2-пиримидил)-1-пиперазинил] бутил] -1,2-бензотиазол-3(2Н)-он-1,1-диоксид показали, что эти соединения полезны для лечения тревожных состояний, таких как общее тревожное состояние, паническое состояние и компульсивное навязчивое состояние (Glitz, D.A., Pohl, R., Drugs, 1991, 41, 11). Доклинические испытания показали также, что соединения, имеющие агонистическую активность относительно 5-HT1A, полезны при лечении названных выше нарушений, вызванных состоянием тревоги (Schipper, Human Psychopharm., 1991, 6, S53).
Существует также доказательство, и клиническое, и доклиническое, подтверждающее успешное влияние соединений, обладающих агонистической активностью относительно 5-HT1A, на лечение депрессий, а также и контролируемых нарушений и злоупотреблений алкоголем (van Herst, Psychopharm., 1992, 107, 474; Schipper et al. Human Psychopharm., 1991, 6, S53; Cervo et al, Eur. J. Pharm., 1988, 158, 53; Glitz, D.A., Pohl, R., Drugs, 1991, 41, 11).
Соединения с агонистической активностью относительно 5-HT1A ингибируют у мужских особей мышей агрессию, вызванную изоляцией, указывая, что эти соединения полезны для лечении агрессии (Sanchez et al., Psychopharmacology, 1992, в настоящее время находится в печати).
Более того, сообщалось, что соединения с агонистической активностью относительно 5-НТ1A демонстрируют на примере животных антипсихотическое воздействие (Wadenberg, Ahlenius, J. Neural Transm, 1991, 83, 43; Ahlenius Pharm. Tox. , 1989, 64, 3; Lowe et al., J. Med. Chem., 1991, 34, 1860; New et al., J. Med. Chem. , 1989, 32, 1147; Martin et al., J. Med. Chem., 1989, 32, 1052).
Недавние исследования также показали, что рецепторы 5-HT1A важны в серотонергической модуляции каталепсии, вызванной галоперидолом (Hicks, Life Science, 1990, 47, 1609). Предполагается, что агонисты 5-HT1A полезны для лечения побочных эффектов, вызванных общепринятыми антипсихотическими агентами, такими как, например, галоперидол.
Показано, что соединения, обладающие агонистической активностью относительно 5-HT1A, имеют нейропротекторные свойства (при моделировании локальной и церебральной ишемии на грызунах), и поэтому эти соединения могут быть полезны при лечении ишемических состояний (Prehn, Eur. J. Pharm., 1991, 203, 213). Как в моделях на животных, так и при клинических испытаниях было показано, что агонисты 5-HT1A оказывают антигипертензивный эффект посредством центрального механизма (Saxena, villalon. Trends Pharm, Sci., 1990, 11, 95; Gillis et al, J. Pharm. Exp. Ther., 1989, 248, 851). Таким образом, соединения, обладающие агонистической активностью относительно 5-HT1A, могут быть с успехом применены для лечения кардиоваскулярных нарушений.
Следовательно, соединения, обладающие агонистической активностью относительно 5-HT1A, будут полезны в терапии подобных состояний, и поэтому эти соединения крайне необходимы.
Резюме изобретения
В настоящее время найдено, что
некоторые новые аминометилинданы, -2,3-дигидробензофураны, -2,3-дигидробензотиофены, -1,3-дигидроизобензофураны и -1,3-дигидробензотиофены имеют агонистическое влияние на центральные рецепторы
5-HT1A. Следовательно, настоящее изобретение относится к новому классу соединений общей формулы I

где один из X и Y представляет собой CH2, а другой выбран из группы, содержащей CH2, О и S;
R1 - выбирают из группы, содержащей низший алкил, низший алкенил, низший алкинил, циклоалк(ен)ил, циклоалк(ен)ил-низший алк(ен/ин)ил, арил-низший алкил, ацил, низший алкил сульфонил, трифторметилсульфонил, арилсульфонил, R10ZCO, где Z представляет собой О или S, a R10 представляет собой алкил, алкенил, алкинил, циклоалк(ен)ил, циклоалк(ен)илалкил, или арил, или R11R12NCO, где R11 и R12 представляют собой независимо водород, алкил, алкенил, алкинил, циклоалк(ен)ил, циклоалк(ен)илалк(ен/ин)ил или арил;
R2 выбирают из группы, содержащей водород, низший алкил, низший алкенил, низший алкинил, циклоалк(ен)ил, циклоалк(ен)ил-низший алк(ен/ин)ил, арил-низший алкил;
R3 - R5 выбирают независимо из группы, содержащей водород, галоген, низший алкил, низший алкокси, ацил, низший алкилтио, гидрокси, низший алкилсульфонил, циано, трифторметил, циклоалкил, циклоалкилалкил или нитро;
R6 R7 каждый представляет собой водород или низший алкил или, связываясь друг с другом, они образуют 3 - 7-членный цикл;
R8 и R9 независимо представляют собой водород, алкил, алкенил, алкинил, циклоалк(ен)ил, цикло-алк(ен)ил-алк(ен/ин)ил, арилалкил или группу

где R13 представляет собой водород, низший алкил, низший алкенил, низший алкинил, циклоалк(ен)ил, циклоалк(ен)ил - низший алк(ен/ин)ил, арил - низший алкил или арил, W представляет собой О или S и r = 2 - 6;
R8 и R9 связаны, образуя при этом 3 - 7-членное кольцо, содержащее один атом азота;
любая из присутствующих алкил, циклоалкил или циклоалкилалкил групп, может быть по выбору замещена одной или двумя гидроксигруппами, которые вновь по выбору могут быть подвергнуты этерификации с алифатической или ароматической карбоновой кислотой, а любой из присутствующих арилзаместителей может быть по выбору замещен на галоген, низший алкил, низший алкокси, низший алкилтио, гидрокси, низший алкилсульфонил, циано, трифторметил, циклоалкил, циклоалкилалкил или нитро;
и фармацевтически приемлемую добавку из солей этих кислот.
В целом было обнаружено, что соединения настоящего изобретения могут ингибировать связь тритированного 8-гидрокси-2-дипропиламинотетралина (8-OH-DPAT) и рецепторов 5-HT1A in vitro. Более того, было доказано, что настоящие соединения демонстрируют активность относительно 5-HT1A in vivo и что при использовании животных в качестве моделей они обнаруживали предсказанную антипсихотонические и анксиолитические свойства соответственно. Следовательно, соединения настоящего изобретения считаются полезными в качестве лекарственных средств для лечения психозов, тревожных состояний, таких как общее тревожное состояние, паническое состояние, злоупотребление алкоголем, компульсивное навязчивое состояние, депрессия, контролируемые нарушения, агрессия, ишемические болезни и побочные эффекты, вызванные общепринятыми антипсихотическими средствами, или кардиоваскулярные нарушения.
Следовательно, другим аспектом изобретения является обеспечение фармацевтической композицией, содержащей по меньшей мере одно новое соединение согласно настоящему изобретению, как это было определено выше, или фармацевтически приемлемую добавку солей этих кислот в терапевтически эффективных количествах и в комбинации с одним или более фармацевтически приемлемыми носителями или разбавителями.
Дальнейшим аспектом настоящего изобретения является обеспечение использования соединения согласно настоящему изобретению или фармацевтически приемлемой соли этого соединения для получения фармацевтического препарата для лечения психоза, тревожных состояний, депрессий, контролируемых нарушений, злоупотребления алкоголем, агрессии, ишемических заболеваний, побочных эффектов, вызванных общепринятыми антипсихотическими средствами или кардиоваскулярных нарушений.
Детальное описание изобретения
Некоторые
соединения общей формулы I могут существовать в виде своих оптических изомеров, и такие оптические изомеры также включаются в настоящее изобретение. Употребляемый здесь термин "алкил" относится к
прямой или разветвленной C1 - C20-группе, аналогично термин "алкенил" и "алкинил" означает прямую или разветвленную цепь углеводородов C2 - C20, имеющую
одну или более двойных или тройных связей соответственно. Термин "циклоалкил" обозначает карбоциклическое кольцо, включающее 3 - 8 атомов углерода или бициклическое или трициклическое карбоциклическое
кольцо, такое как например адамантил. Термины "низший алкил", "низший алкокси", "низший алкилтио" и т. д. относятся к таким разветвленным или неразветвленным группам, которые включают 1 - 6 атомов
углерода. Примерами таких групп являются метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил, 2-метил-1-пропил, метокси, этокси, 1-пропокси, метилтио, этилтио, 1-пропилтио, 2-пропилтио,
метилсульфонил, этилсульфонил и им подобные. Аналогично термины "низший алкенил" и "низший алкинил" относится к таким группам, которые включают 2 - 6 атомов углерода и одну или более двойных или
тройных связей.
Термин "алк(ен/ин)ил" означает что эта группа может представлять собой алкил, алкенил- или алкинилгруппу. Термин "арил" относится к моно- или бициклической или гетероциклической ароматической группе, такой как фенил, индолил, тиенил, фуранил, тиазолил, бензофуранил, бензотиенил, бензотиазолил и бензизоксазолил.
"Галоген" означает фторо-, хлоро-, бромо или иодо.
Термин "ацил", используемый здесь, относится к формил, алкилкарбонил, алкенилкарбонил, алкинилкарбонил, арилкарбонил, арилалк(ен/ин)илкарбонил, циклоалкилкарбонил или циклоалкалк(ен/ин)илкарбонильной группе.
В формуле I X является предпочтительно CH2, О или S, а Y представляет собой CH2.
R1 является предпочтительно арил-низший алкил группой или ацил группой, низшей алкилсульфонил группой или группой R11R12N-CO-, где R11 представляет собой водород или низший алкил, a R12 представляет собой водород, алкил, арил или циклоалкил. Наиболее предпочтительно R1 представляет собой бензил или замещенный бензил, формил, алкилкарбонил, в особенности ацетил, арилкарбонил, в особенности бензоил или замещенный бензоил или группу R11R12N-CO-, где R11 представляет собой водород или низший алкил, a R12 представляет собой водород, низший алкил, фенил, замещенный фенил или C5-6 циклоалкил.
R2 предпочтительно является галогеном или низшим алкилом, предпочтительно, чтобы каждый R3, R4, R5 представляли собой водород или галоген и чтобы и R6 и R7 оба представляли собой водород.
R8 предпочтительно является водородом или низшим алкилом, a R9 предпочтительно является низшим алкилом, арил - низшим алкилом, циклоалкил - низшим алкилом или группой имеющей формулу Iа, где W представляет собой О, a R13 представляет собой водород, низший алкил, циклоалкил или арил, или же R8 и R9 связаны между собой, образуя C3 - C7-членное кольцо, содержащее один атом азота. Наиболее предпочтительно, чтобы R9 представлял собой фенил - низший алкил, замещенный фенил - низший алкил, индолил - низший алкил, циклогексил - низший алкил или группу, имеющую формулу Iа, где W представляет собой О, а R13 представляет собой водород или низший алкил, циклоалкил, фенил или замещенную фенильную группу, или R8 и R9 связаны, образуя пирролидиновое или пиперидиновое кольцо.
Соли кислот по настоящему изобретению представляют собой фармацевтически приемлемые соли соединений формулы I и нетоксичных кислот. Примерами таких органических солей являются соли малеиновой, фумаровой, бензойной, аскорбиновой, янтарной, щавелевой, бис-метиленсалициловой, метансульфоновой, этандисульфоновой, уксусной, пропионовой, винной, салициловой, лимонной, глюконовой, молочной, миндальной, коричной, цитраконовой, аспартиновой, стеариновой, пальмитиновой, итаконовой, гликоновой, п-аминобензойной, глютаминовой, бензолсульфоновой и теофиллин уксусной кислот, а также 8-галотеофиллинов, например, 8-бромотеофиллин. Примерами таких неорганических солей служат соли соляной, бромистоводородной, серной, сульфамидной, фосфорной и азотной кислот.
Фармацевтические соединения или соединения, полученные согласно настоящему изобретению, можно использовать любым подходящим способом, например, орально в виде таблеток, капсул, порошков, сиропов и т.д. или парентерально в виде растворов для инъекции. Для получения таких соединений могут быть применены известные в этой области методы и любые приемлемые фармацевтические носители, разбавители, или иные добавки, обычно используемые в этой области.
Обычно соединения настоящего изобретения вводятся в единичной дозировочной форме, содержание указанных соединений составляет от ≈ 0.01 до 50 мг.
Общая дневная доза обычно составляет от ≈ 0.05 до 500
мг, а наиболее предпочтительно от ≈ 0.1 до 20 мг активного соединения по данному изобретению. Кроме того, изобретение касается метода получения новых аминометильных производных инданов, 2,
3-дигидробензофуранов, 2,3-дигидробензотиофенов, 1,3-дигидроизобензофуранов и 1,3-дигидроизобензотиофенов формулы I, включающего:
а) ацилирование аминопроизводного, имеющего формулу II

где R2 - R9, X и Y такие, как это было определено выше, ацилирующий агент типа галогенида карбоновой кислоты R1'CO-hal, причем R1'CO представляет собой ацил группу, заключающую в себе определения R1, a hal представляет собой галоген, ан гидрид карбоновой кислоты или смешанный ангидрит кислоты R1' CO-OCOR, причем R представляет собой алкил, арил или алкокси, изоцианат, изотиоцианат, или аналогичное активированное ацилированное производное, хорошо известное в этой области;
b) алкилирование аминопроизводного формулы II для получения соединения формулы I, в котором R1 представляет собой низший алкил, низший алкенил, низший алкинил, циклоалк(ен)ил-низший алк(ен/ин)ил или арил-низший алкил, в качестве алкилирующих агентов используют алкилгалогениды типа R1''-hal, мезилат R1''OSO2CH3, тозилат R1''OSO2C6H4-CH3 или аналогичный алкилирующий реагент с подходящими отщепляющими группами, причем R1'' представляет собой низший алкил, низший алкенил, низший алкинил, циклоалк(ен)ил, циклоалк(ен)ил - низший алк(ен/ин)ил или арил-низший алкил;
c) восстановление двойной связи в соединении формулы III

где R1-R9, X и Y такие, как это было определено выше, по крайней мере один из X и Y представляет собой CH2, а одна или две пунктирные линии обозначают двойную связь; или
d) алкилирование аминопроизводного формулы IV

где R1-R8, X и Y таковы, как это было определено выше, с помощью алкилирующего агента, такого как алкилгалогенид R9-hal, мезилат R9OSO2CH3, тозилат R9OSO2C6H4 или аналогичные алкилирующие реагенты с подходящей отщепляющей группой, где R9 такое, как это было определено выше; или
е) восстановление амидного производного, имеющего формулу V:

где R2-R9, X и Y такие, как это было определено выше, а группа R1''' CH2 содержит группу R1 с целью получения соединения формулы I, где R1 представляет собой ранее определенную группу, имеющую, однако, в положении I относительно аминогруппы метильную группу; или же
f) введение заместителей R3, R4 или R5 восстановлением соединения формулы VI

где по меньшей мере один из R3' - R5' представляет собой водород, а остальные таковы, как это было определено выше для R3, R4 или R5, a R6 -R9, X и Y такие, как это было определено выше, с использованием реагентов типа галогена или галогенирующего агента, сульфонирующего агента, нитрующего агента, или агента, генерирующего ионы карбония (RCO+, R+), где R представляет собой алкил, алкенил, алкинил, арил, циклоалкил или циклалк(ен/ин)ил.
Алкилирование в разделе а) обычно проводят при низких температурах (например ниже комнатной температуры) в инертном растворителе типа ацетона, дихлорметана, тетрагидрофурана или диметоксиэтана при использовании в реакции хлорангидридов карбоновых кислот, изоцианатов или изотиоцианатов. Формилированные амины получают из соответствующих аминов при взаимодействии в муравьиной кислоте с эфирами муравьиной кислоты или при взаимодействии со смешанным ангидридом муравьиной кислоты, полученным in situ. Обычно температуры протекания реакции находятся между 0oC и температурой кипения соединений, являющихся предшественниками формила.
Алкилирование согласно разделам b) и d) обычно проводят при нагревании в колбе с обратным холодильником в подходящем растворителе, таком как ацетон, метилизобутил кетон, тетрагидрофуран, диоксан, этанол или 2-пропанол в присутствии основания, такого как триэтиленамин или карбонат калия.
Восстановления двойных связей согласно разделу с) обычно проводят при каталитическом гидрировании при низком давлении (менее 3 атм) в аппарате Парра.
Восстановления амидов согласно разделу е) проводят обычно с использованием LiFlH4, AlH3 или диборана в инертном растворителе, таком как диэтиловый эфир, тетрагидрофуран или диоксан при комнатной или при незначительно повышенной температуре. Галогенирование согласно разделу f) обычно проводят при использовании хлора, брома или N-хлорсукцинамида, N-бромсукцинамида или другого молекулярного предшественника галогена в присутствии катализатора, такого как ионы Fe или минеральная кислота.
Далее настоящее изобретение проиллюстрировано некоторыми примерами, которые, однако, не могут рассматриваться в качестве лимитирующих.
Пример 1.
1-(N,N-дипропиламинометил)-6-формиламиноиндан, оксалат 1a (метод а).
Исходное соединение, 1-инданкарбоновая кислота, было получено согласно процессу, изложенному V. Asham и W.H.Linnell, J.Chem. Soc., 1954, 4691 - 4693. Смесь инданкарбоновой кислоты (32.2 г), тионилхлорида (50 мл) и двух капель диметилформамида в дихлорметане (100 мл) нагревали в течение 3 часов в колбе с обратным холодильником. Летучее соединение выпарилось, а оставшийся сырой продукт использовали без дальнейшей очистки. К раствору N, N-дипропиламина (50 мл) в дихлорметане, который держали при 0 - 5oC, по каплям добавляли раствор сырого ангидрида карбоновой кислоты в дихлорметане (200 мл). Температуру постепенно повышали до комнатной, а реакционную смесь продолжали перемешивать в течение всей ночи. Растворители упаривали в вакууме, а оставшееся вязкое масло очищали фильтрованием через силикагель (элюируя диэтиловым эфиром и дихлорметаном 1:1). Выход N,N-дипропил-1-инданкарбоксамида 32 г. К хорошо перемешанному раствору инданкарбоксамида (20 г) в концентрированной H2SO4 (120 мл), которую держали при -10oC, по каплям добавили охлажденную смесь 100% HNO3 (6 г) и концентрированной H2SO4 (40 мл) при температуре от -15 до -10oC. Допускался подъем температуры до 5oC (не выше для предотвращения образования динитросоединения) и смесь затем выливали на измельченный лед (2 кг). Органическое соединение экстрагировали диэтиловым эфиром (дважды по 200 мл), смешанную органическую фазу промывали разбавленным раствором Na2CO3 (3 х 50 мл). Органическую фазу высушивали (безводный MgSO4) и обрабатывали активированным углем. После выпаривания диэтилового эфира в вакууме был получен в виде масла N,N-дипропил-6-нитро-1-инданкарбоксамид (15 г). Все это масло было растворено в 90% этаноле (250 мл). Раствор нагревали в колбе с обратным холодильником и в течение 1 часа при энергичном перемешивании порциями к нему добавляли порошок железа (10 х 2 г) и 6 М водную соляную кислоту (10 х 0.2 мл). Смесь нагревали еще в течение 1 часа в колбе с обратным холодильником. Неорганическое соединение отфильтровывали, пока смесь еще была горячей. Раствор обрабатывали активированным углем, а растворители затем выпаривали в вакууме. Оставшееся твердое соединение перекристаллизовывали из смеси диэтилового и изопропилового эфира 1:1, получив при этом 9.5 г 6-амино-N, N-дипропил-1-инданкарбоксамида с температурой плавления 100oC. К суспензии LiAlH4 (4 г) в сухом тетрагидрофуране по каплям добавили весь раствор 6-аминоинданкарбоксамида. Смесь нагревали в колбе с обратным холодильником в течение 2 часов, а затем охлаждали льдом. Смесь вода/тетрагидрофуран была добавлена осторожно, чтобы не разрушить избыток LiAlH4. Неорганические соли отфильтровывали, а фильтрат тщательно промывали дихлорметаном. Растворители выпаривали в вакууме. Оставшееся масло растворили в толуоле, а избыток воды удалили выпариванием толуола, при этом в виде масла был получен 6-амино-1-(N,N-дипропиламинометил)индан. Выход 7.0 г. К муравьиной кислоте (98%, 18 мл) добавили 3.1 мл ангидрида уксусной кислоты при температуре чуть выше 0oC. Весь 6-амино-1-(N,N-дипропиламинометил)индан растворили в дихлорметане (15 мл) и добавляли по каплям при 0 - 5oC. После перемешивания в течение 1 часа при 5oC добавили этилацетат (100 мл) и водный аммиак (100 мл). Органическую фазу отделили и обработали так же, как остающееся и указанное в заголовке соединение 1а в виде масла. Оксалатную соль кристаллизовали из ацетона. Выход 7.4 г. Температура плавления 150 - 152oC.
Аналогичным путем были получены следующие N,N-замещенные-6-формило-1-амино-метилинданы:
1-(N,
N-диметиламинометил)-6-формиламиноиндан, оксалат 1b. Температура плавления 163 - 164oC.
6-формиламино-1-(1-пиперидинометил)индан, фумарат 1с. Температура плавления 188 - 190oC.
6-формиламино-1-(1-пирролидинометил)индан, оксалат 1d. Температура плавления 176 - 179oC.
Пример 2.
6-ацетиламино-1-(N, N-дипропиламинометил)индан, гидрохлорид 2а (метод а).
К раствору 6-амино-1-(N,N-дипропиламинометил)индана (9г), полученному, как в примере 1, в дихлорметане (50 мл) добавили триэтиламин (4,2 г). Раствор ацетилхлорида (3,3 г) в дихлорметане (10 мл) добавляли по каплям при 0 - 10oC. Потом смесь перемешивали при комнатной температуре в течение 1/2 часа, а затем вылили в разбавленный водный NH4OH (100 мл). Органическую фазу отделили и получили при этом 11.0 г сырого продукта 2а, указанного в заглавии, в виде масла. Гидрохлоридную соль кристаллизовали из ацетона. Выход 974 г, температура плавления 212 - 216oC.
Аналогичным способом были получены следующие N,N-бизамещенные 6-ациламино-1-аминометил инданы:
6-ацетиламино-1-(N,N-диметиламинометил)индан, гидрохлорид 2b. Температура плавления 236 - 238oC;
1-(N, N-дипропиламинометил)-6-(4-фторбензоиламино) индан гидрохлорид 2с.
Температура плавления 217 - 220oC;
6-ацетиламино-7-хлор-1-1 (N,N-дипропиламинометил)индан, оксалат 2f. Температура плавления 77-79oC (содержит 20% 6-амино-7-хлор-1-1-(N,
N-дипропиламинометил)индан, диоксалат);
1-(N,N-дипропиламинометил)-6-(метилсульфониламино)индан, оксалат 2g. Температура плавления более 300oC;
7-хлор-1-(N,
N-дипропиламинометил)-6- (метилсульфониламино)индан, оксалат 2h. Температура плавления 80 - 84oC;
5-ацетиламино-3-(1-пиперидинометил)-2,3-дигидробензотиофен 2i. Температура
плавления 126 - 128oC.
Пример 3.
1-(N,N-дипропиламинометил)-6-уреидо-индан 3а (метод а).
К раствору 6-амино-1-(N,N-дипропиламинометил)индана (5г), полученного, как в примере 1, в метаноле (10 мл), добавили уксусной кислоты (7.3 г). Раствор KOCN (3.3 г) в воде (5 мл) добавляли по каплям при 0 - 10oC. Затем смесь перемешивали при комнатной температуре в течение ночи. Добавили воду (200 мл) и этилацетат (50 мл), органическую фазу отделили и обработали. Фумаратовую соль соединения 3а, указанного в заглавии, кристаллизовали из этанола. Свободное основание в виде кристаллического продукта выделили из фумарата. Выход 1.1 г. Температура плавления 101oC.
Пример 4
1-(N,
N-дипропиламинометил)-6-(3-фенил-1-уреидо)индан 4а (метод а).
К раствору 6-амино-1-(N, N-дипропиламинометил)индана (4.9 г), полученного, как в примере 1, в дихлорметане (100 мл), добавили фенилизоцианат (3 г). Смесь нагревали в колбе с обратным холодильником в течение 1.5 часа. В процессе реакции соединение 4а, указанное в заглавии, кристаллизовалось. Смесь охладили с помощью льда, а осажденный продукт отфильтровали. Выход 4.0 г. Температура плавления 198 - 201oC.
Аналогичным способом были получены следующие N,N-бизамещенные
6-уреидо-1-аминометилинданы:
1-(N, N-дипропиламинометил)-6-(3-метил-1-уреидо)индан, гидрохлорид 4b. Температура плавления 180 - 182oC;
6-(3,3-диметил-1-уреидо)-1-(N,
N-дипропиламинометил)индан, гидрохлорид 4с;
1-(N, N-дипропиламинометил)-6-(3-нонил-1-уреидо)индан, гидрохлорид 4d. Температура плавления 148 - 150oC;
6-(3-циклопентил-1-уреидо)-1-(N, N-дипропиламинометил)индан 4е. Температура плавления 136 - 138oC.
Пример 5.
3-(N,N-дипропиламинометил)-5-формиламино-2, 3-дигидробензотиофен, оксалат 5а (метод а).
Исходное вещество, 5-амино-2,3-дигидро-N,N-дипропил-3- бензотиофенкарбокамид, было получено согласно методу, изложенному в Европейском патенте N 88-301073 CA110(9): 75302у (1988), J. Amer. Chem. Soc., 1948, 70, 1955 и J. Chem. Soc. (с), 1967, 1899. К раствору карбоксамида (10 г) в тетрагидрофуране (200 мл) добавили таблетки LiAlH4 (3 х 1 г) и смесь осторожно нагревали в колбе с обратным холодильником в течение 2 часов. Избыток LiAlH4 осторожно разрушили, добавив 10% раствор воды в тетрагидрофуране (25 мл) при 20oC. Неорганические соли отфильтровали, а растворители выпаривали в вакууме. Оставшееся масло (7.0 г) использовали без дальнейшей очистки. К полученному таким образом сырому 5-амино-3-(N, -N-дипропиламинометил) 2,3-дигидробензотиофену в толуоле (50 мл) добавили 98% муравьиную кислоту (20 мл). Толуол/муравьиную кислоту постепенно разгоняли, пока не была получена температура 130 - 140oC. Затем смесь вылили в разбавленный водный NH4OH (250 мл) и добавили этилацетат (100 мл). Органическую фазу отделили и обрабатывали. Указанное в заглавии соединение 5а очищали колоночной хроматографией на силикагеле (элюируя 3% триэтиламином в смеси дихлорметана и этилацетата 1:1). Кристаллическую соль оксалата получали из 15% раствора этанола в ацетоне. Выход 2.4 г. Температура плавления 135 - 136oC.
Аналогичным путем были получены следующие N,N,-бизамещенные 3-аминометил-2,3-дигидробензотиофены:
3-[N-(2-фенилэтил)-N-пропиламинометил] -5- формиламинометил-2,3-дигидробензотиофен 5b, выделенный в виде масла;
6-ацетиламино-1-(1-пиперидинометил)индан, оксалат 5с, температура плавления 158
- 160oC.
Пример 6.
1-(N, N-дипропиламинометил)-6-(4-фторобензиламино)индан, оксалат 6а (метод е).
К раствору 6-амино-N, N-дипропил-1-инданкарбоксамида (10 г), полученному, как в примере 1, и триэтиленамина (4.2 г) в дихлорметане (75 мл), который хранили при -5oC, по каплям добавляли раствор 4-фторбензоилхлорида (6 г) в дихлорметане (30 мл). Температуру слабо повышали до комнатной. Добавили воду (200 мл), органическую фазу обработали, получив 15 г сырого производного 4-фторбензоиламино в виде масла. Небольшой образец очистили и кристаллизовали. Температура плавления 135oC. К суспензии LiAlH4 (1 г) в сухом тетрагидрофуране добавили очищенное 4-фторбензоиламино производное (3 г) и смесь осторожно нагревали в колбе с обратным холодильником в течение 1 часа. После охлаждения избыток LiAlH4 разрушили, осторожно добавив 10% раствор воды в тетрагидрофуране (15 мл) при 20oC. Неорганические соли отфильтровали, фильтрат тщательно промыли дихлорметаном (2 х 50 мл), растворители выпарили, получив сырой продукт 6а, указанный в заглавии, в виде масла. Оксалатную соль (1.5 моль щавелевой кислоты/моль соединения, указанного в заглавии) кристаллизовали из ацетона. Выход 1.1 г. Температура плавления 135 - 140oC.
Пример 7.
5-ацетиламино-3-(1-пиперидинометил)-2,3-дигидробензофуран, оксалат 7а (метод а).
3-бензофуранкарбоновая кислота: К раствору бензофурана (75 г) в хлороформе (600 мл) по каплям добавляли раствор брома (41 мл) в хлороформе (150 мл) при -10oC. Температура слабо повышалась, пока не достигла комнатной, затем хлороформ выпаривали в вакууме, получив при этом сырой 2,3-дибромо-2,3-дигидробензофуран в виде кристаллического продукта, который использовали без дальнейшей очистки. Выход 171 г. К дибромпроизводному (147 г) в этаноле (600 мл) добавили раствор КОН (59 г) в этаноле (200 мл). Смесь нагревали в колбе с обратным холодильником в течение 2.5 часов. После охлаждения до комнатной температуры смесь вылили в воду и экстрагировали этилацетатом (2 х 300 мл). Смешанные органические фазы обрабатывали, а 3-бромбензофуран затем очищали, промывая через силикагель, при этом в качестве элюента использовали н-гептан. В результате получили 51 г полукристаллического продукта. Раствор всего полученного таким образом 3-бромбензофурана и CuCN (33 г) в N-метил-2-пирролидоне (350 мл) нагревали при 190oC под N2. Через часовые интервалы добавляли CuCN (3 х 4.5 г). Смесь вылили в раствор FeCl3 х 6Н2О в воде (610 мл) и концентрированной соляной кислоты (156 мл) пока он был еще горячим. Оставшуюся смесь перемешивали в течение 1 часа при 60oC, а затем вылили в лед/вода (5 л). В результате экстрагирования диэтиловым эфиром (3 х 300 мл) и обработки смешанных органических фаз получили сырой кристаллический 3-цианобензофуран, плавящийся при 76 - 82oC. Выход 34 г. Полученный 3-цианобензофуран растворили в 660 мл смеси уксусная кислота - концентрированная серная кислота - вода 1:1:1 и нагревали в колбе с обратным холодильником в течение 3 часов. После охлаждения добавили воду и провели окончательную экстракцию 3-бензофуранкарбоновой кислоты диэтиловым эфиром (3 х 200 мл) и ее обработку. Выход 37 г. Температура плавления 152 - 156oC.
3-(1-пиперидилкарбонил)бензофуран:
Смесь 3-бензофуранкарбоновой кислоты (15 г), N,
N-диметилформамида (2 мл) и тионилхлорида (25 мл) в дихлорметане (200 мл) нагревали в течение 5 часов в колбе с обратным холодильником. Летучие соединения удаляли выпариванием в вакууме, а избыток
тионилхлорида удаляли проводя дважды выпаривание с толуолом. Затем полученный таким образом хлорангидрид 3-бензофуранкарбоновой кислоты растворили в дихлорметане (100 мл) и добавляли по каплям к
раствору пиперидина (21.4 г) в метиленхлориде (100 мл) при 0 - 5oC. Затем в течение часа смесь перемешивали при комнатной температуре. После промывания водой и соляным раствором
органическую фазу обрабатывали так, как это было описано выше. Сырое пиперидиновое производное затем очищали колоночной хроматографией на силикагеле (элюируя смесью этилацетат - гептан 3:1), при этом
было получено 6.7 г чистого соединения, указанного в заглавии, в виде масла.
3-(1-пиперидилкарбонил)-2,3-дигидробензофуран:
К раствору 3-(1-пиперидилкарбонил)бензофурана (6.7
г) в метаноле (150 мл), сохраняемому при 35 - 50oC, маленькими порциями в течение 5 часов добавляли Mg (всего 3 г). После перемешивания в течение последующего часа при 50oC смесь
вылили в водный раствор NH4Cl. Водный раствор экстрагировали дихлорметаном (2 х 200 мл). Смешанные органические фазы обработали, получив при этом 6.7 г 2,3-дигидробензофуранового
производного в виде масла.
5-нитро-3-(1-пиперидилкарбонил)-2,3-дигидробензофуран:
Все полученное ранее 2,3-дигидробензофурановое производное растворили в трифторуксусной
кислоте (35 мл) и охладили до 10oC. Раствор, который стал черным, немедленно вылили на лед и экстрагировали этилацетатом (2 х 50 мл). Смешанные органические фазы тщательно промывали сначала
водным раствором Na2CO3 (2 х 25 мл), а в заключение - соляным раствором. После обработки органической фазы было получено 6.4 г сырого 5-нитропроизводного в виде масла. Дальнейшая
очистка колоночной хроматографией на силикагеле (элюируя этилацетатом - гептаном 3:1) дала 3.2 г чистого 5-нитро-3-(1-пиперидилкарбонил)- 2,3-дигидробензофурана, который кристаллизовался при
отстаивании. Температура плавления 108 - 114oC.
5-амино-3-(1-пиперидилкарбонил)-2,3-дигидробензофуран:
К раствору всего 5-нитробензофурана в 90% этаноле (50 мл),
сохраняемому при нагревании в колбе с обратным холодильником, небольшими порциями в течение 10 мин добавляли порошок Fe (всего 2.5 г) и концентрированную HCl (всего 0.1 мл). Смесь нагревали в колбе с
обратным холодильником еще в течение 1 часа. Неорганические осадки отфильтровали, а смесь вылили в соляной раствор и этилацетат (250 мл). После обработки органической фазы был получен 1 г
кристаллического производного 5-аминобензофурана.
5-ацетиламино-3-(1-пиперидинометил)-2,3-дигидробензофуран, оксалат 7а:
Производное 5-аминобензофурана (1 г), растворенное в
сухом тетрагидрофуране, добавляли по каплям к раствору LiAlH4 (0.5 г) в сухом тетрагидрофуране (30 мл). Смесь нагревали в течение 2 часов в колбе с обратным холодильником. После охлаждения
на водяной бане избыток LiAlH4 гидролизовали добавлением водного раствора NaOH (0.5 мл 15%). Неорганические соли отфильтровали. Тетрагидрофуран выпаривали в вакууме, а оставшееся вязкое
масло растворили в дихлорметане (100 мл). После высушивания (безводный MgSO4) дихлорметан выпарили, при этом получилось 0.7 г сырого 5-амино-3-(1-пиперидинометил)-2,3-дигидробензофурана,
который использовали без дальнейшей очистки. 5-амино-группу ацетилировали согласно методу в примере 2. Соединение, указанное в заглавии, кристаллизовали из ацетона в виде оксалатной соли. Выход 0.15
г. Температура плавления 144 - 150oC.
Пример 8.
Разделение соединений
(-)-6-ацетиламино-1-(N,N-дипропиламинометил)индан, гидрохлорид 8а.
К раствору 6-ацетамино-1-(N,N-дипропиламинометил)индана (36.5 г), полученного так же, как в примере 2, в ацетоне добавили (S)-(+)-бинафтил-2,2'-диилгидрофосфат ((+)-BNP) (21.2 г) при нагревании в колбе с обратным холодильником. Смесь охладили и на ночь оставили в холодильнике. Осадившийся кристаллический продукт отфильтровали, а раствор использовали для получения другого энантиомера 8b. Перекристаллизацией из смеси 2:3 метанола и этанола получили 30 г, (+)-BNP соли. Температура плавления 257 - 260oC, [α]D = +293.1o. Было выделено свободное основание соединения, указанного в заглавии 8а ([α]D = -84.9o), и затем выкристаллизовано в виде гидрохлоридной соли из ацетона. Температура плавления 236 - 238oC, [α]D = -51.3o.
(+)-6-ацетиламино-1-(N,N-дипропиламинометил)индан, гидрохлорид 8b.
Ацетоновый раствор, полученный выше, вылили в воду (300 мл) и подщелочили, добавив водной разбавленной NaOH. Экстрагировали этилацетатом (2 х 150 мл) и затем кристаллизовали (-)-BNH соль из смеси ацетон/метанол 1:1. Выход 24.0 г, температура плавления 257 - 260oC, [α]D = +54.3o.
Соединение 2b разделяли аналогичным способом, используя O,O-дитолуилвинную кислоту в качестве
расщепляющего агента:
(-)-6-aцетилaминo-1-(N, N-диметилaминoметил)индaн, выделенный в виде масла, 8с;
(+)-6-ацетиламино-1-(N, N-диметиламинометил)индан, выделенный в виде масла,
8d.
Соединение 2i было разделено аналогичным путем, используя O,O-дитолуилвинную кислоту в качестве расщепляющего агента:
(-)-6-ацетиламино-1-(1-пиперидинометил)индан;
(+)-O, O-ди-(4-толуил)-D-тартрат 8е. Температура плавления 144 - 147oC, [α]D = +59.2o (свободное основание выделяли, как описано выше, [α]D =
-46.8o).
(+)-6- ацетиламино-1-(1-пиперидинометил)индан;
(-)-O, O-ди-(4-толуил)-L-тартрат 8f. Температура плавления 141 - 146oC, [α]D =
-56.3o (свободное основание выделяли, как описано выше, [α]D = +50.7o).
Пример 9.
(-)- и (+) 6-амино-1-(N, N-дипропиламинометил)индан 9а и 9b.
Соединения 8а и 8b гидролизовали до соответствующих стереоизомеров 6-аминоинданов, соответственно:
Раствор (-) -6-ацетамидо-1-(N,
N-дипропиламинометил)индана (8,4 г) в 10% концентрированной соляной кислоте в метаноле (100 мл) нагревали в колбе с обратным холодильником в течение 8 часов. Растворители упаривали, а оставшееся масло
добавили в разбавленной водной NH4OH (pH больше 9) и экстрагировали этилацетатом (2 х 100 мл). Органическую фазу обрабатывали, получая (-)-соединение 9а в виде масла. Выход 6.5 г, [α
]D = -81.3o.
Аналогичным путем из 8b был получен:
(+)-6-амино-1-(N,N-дипропиламинометил)индан 9b; [α]D = +81.4o.
Аналогичным путем соединения 8с, 8d, 9e и 8f гидролизовали до:
(-)-6-амино-1-(1-пиперидинометил)индана 9d, выделенного в виде масла;
(-)-6-амино-1-(N,
N-диметиламинометил)индана 9с, выделенного в виде масла;
(-)-6-амино-1-(1-пиперидинометил)индана 9d, выделенного в виде масла;
(+)-6-амино-1-(1-пиперидинометил)индана 9f, выделенного
в виде масла.
Пример 10.
Следующие разделенные производные были получены из соединений 9а - 9f с использованием обычного процесса ацетилирования, описанного выше:
(-)-1-(N, N-дипропиламинометил)-6-формиламиноиндан, оксалат 10а. Температура плавления 149 - 151oC, [α]D = -44o;
(+)-1-(N,
N-дипропиламинометил)-6-формиламиноиндан, оксалат 10b. Температура плавления 152 - 154oC, [α]D = +43.9o;
(-)-6-(3,3-диметил-1-уреидо)-1-(N,
N-дипропиламинометил)индан, гидрохлорид 10с. Температура плавления 180 - 182oC, [α]D = -49.2o;
(+)-6-(3,3-диметил-1-уpеидо)-1-(N,
N-дипpoпилминoметил)индан, гидрохлорид 10d. Температура плавления 180 -182oC, [α]D= +48.8o;
(-)-6-[3(4-фторфенил)-1-уреидо] -1-(N,
N-дипропиламинометил)индан, 10е. Температура плавления 200oC, [α]D = -68.5o;
(+)-6-[3(4-фторфенил)-1-уреидо] -1-(N, N- дипропилминометил)индан, 10f.
Температура плавления 200oC, [α]D = +69.8o;
(-)-1-(N,N-диметиламинометил)-6-формиламиноиндан, оксалат 10g. Температура плавления 167 - 175oC,
[α]D = -57.4o;
(+)-1-(N,N-диметилaминoметил)-6-фopмилaминoиндaн, оксалат 10h. Температура плавления 161 - 171oC, [α]D = +59.5o;
(-)-6-формиламино-1-(1-пиперидинометил)индан, оксалат 10i. Температура плавления 153 - 160oC, [α]D = -47.3o;
(+)-6-формиламино-1-(1-пиперидинометил)индан, оксалат 10j. Температура плавления 161 - 164oC, [α]D= +47.4o.
Пример 11.
(i) 6-ацетиламино-5-хлор-1-(N, N-дипропиламинометил)индан оксалат 11a (метод f).
К раствору 6-aцетилaминo-1-(N,N-дипpoпилaминoметил)индана 2а (3,0 г) в уксусной кислоте (15 мл) добавили
одной порцией SO2Cl2 (1.5 г). Температура поднялась до 65oC. После перемешивания в течение 2 часов при комнатной температуре
смесь вылили в разбавленный
водный NH4OH и экстрагировали диэтиловым эфиром (2 х 50 мл). Смешанные органические фазы обработали, а соединение 11а, указанное в заглавии, очищали колоночной хроматографией на силикагеле
(элюируя этилацетатом/гептаном/триэтиламином 40:60:3). Соль щавелевой кислоты кристаллизовали из ацетона. Выход 1.3 г. Температура плавления 172 - 174oC.
Энантиомеры
соединения 11а получали из ацетильных производных 8а и 8b соответственно при хлорировании, как это было описано выше:
(-)6-ацетиламино-5-хлор-1-(N, N-дипропиламинометил)индан оксалат 11b.
Температура плавления 188 - 194oC, [α]D = -35.4o;
(+)6-ацетиламино-5-хлор-1-(N, N-дипропиламинометил)индан оксалат 11c. Температура плавления 190
- 195oC, [α]D = +39.7o.
(ii) 6-ацетиламино-5-бром-1-(N,N-дипропиламинометил)индан оксалат 11d.
К раствору 6-ацетиламино-1-(N, N-дипропиламиноэтил)индана 2а (5.0 г) в уксусной кислоте (50 мл) по каплям добавляли раствор Br2 (1.2 мл) в уксусной кислоте при 50-55oC. Смесь перемешивали в течение ночи при комнатной температуре. Затем ее вылили в воду/этилацетат, органическую фазу отделили и промыли Na2SO3 (1% водный раствор). После обработки органической фазы, в оставшемся масле содержалось довольно значительная часть непрореагировавшего исходного вещества, а соединение, указанное в заглавии, выделяли из смеси колоночной хроматографией на силикагеле (элюируя этилацетатом - гептаном - триэтиламином 40:60:3). Соль щавелевой кислоты соединения, указанного в заглавии 11d, кристаллизовали из ацетона. Выход 2.0 г. Температура плавления 156 - 159oC.
Пример 12.
6-ацетиламино-1-(N-метиламинометил)индан, фумарат 12а (метод g).
К раствору 6-ацетиламино-1-(N, N-диметиламинометил)индана 2b (54 г) в диоксане (1 л) по каплям при 50oC добавляли раствор 2,2,2,-трихлорэтил хлорформиата (48.7 г) в диоксане (200 мл). Затем смесь выдерживали в течение 0.5 часа при 50 - 60oC. Растворитель выпарили, а карбамат очищали фильтрованием через силикагель (элюируя этилацетатом). Выход сырого карбамата в виде масла составил 89.7 г. К раствору полученного таким образом карбамата (45 г) в 90% водной уксусной кислоте (575 мл) небольшими порциями добавляли цинк (84 г) при 30 - 40oC. После перемешивания в течение 3 часов избыток цинка и соли цинка отфильтровали. Добавили воду (2 л) и, экстрагируя диэтиловым эфиром (2 х 200 мл), удалили нейтральные продукты. Охлажденную льдом водную фазу подщелочили (pH более 10), добавив осторожно NaOH. В результате экстракции дихлорметаном (4 х 150 мл) и последующей обработки было получено 14 г соединения 12а, указанного в заглавии, в виде масла. Фумаратовую соль кристаллизовали из этанола. Температура плавления 176 - 178oC.
Пример 13
6-ацетиламино-1-[N(-4-циклогексилбутан-1-ил)-N-метиламинометил] индан, сесквиоксалат 13а (метод d).
Смесь 6-ацетиламино-1-(N-метиламинометил)индана (1.5 г), 4-циклогексилбутан-1-ол мезилата (2.5 г), карбоната калия (1.4 г) и кристаллы иодида калия в MIBK (80 мл) нагревали в колбе с обратным холодильником в течение 5 часов. После охлаждения неорганические соли отфильтровали, а растворители выпарили в вакууме. Оставшееся масло обрабатывали с помощью колоночной хроматографии на SiO2 (элюируя этилацетатом - гептаном - триэтиламином 80: 20:4). Свободное основание соединения, указанного в заглавии, выделили в виде масла. Сесквиоксалат 13а кристаллизовали из ацетона. Выход 1.6 г. Температура плавления 145 - 155oC.
Аналогичным способом были получены следующие N,N-бизамещенные производные:
6-ацетиламино-1-[N(-метил-N-4-фенилбутан-1-ил)-аминометил]индан, сесквиоксалат 13b. Температура плавления 121
- 123oC;
6-ацетиламино-1-[N-4-(индол-3-ил)бутан-1-ил)-N-метиламинометил] индан, оксалат 13с. Температура плавления 111 - 113oC;
6-ацетиламино-1-[N-2-(2-имидозолинон-1-ил)-этил] -N- метиламинометил] индан, сесквиоксалат 13d. Температура плавления 154 - 158oC:
6-ацетиламино-1-[N-2-[3-(4-фторфенил)-2-имидозолинон-1-ил] - этил]-N-метиламинометил]индан, сесквиоксалат 13е. Температура плавления 109 - 114oC;
6-ацетиламино-1-[-N-[4-(3-циклогексил-2-имидозолинон-1-ил] -1-бутил]-N-метиламинометил]индан, сесквиоксалат 13f. Температура плавления 98 - 101oC;
6-ацетиламино-1-[N-метил-N-[3-(3-фенил-2-имидозолинон-1-ил] - 1-пропил] -аминометил]индан, оксалат 13g. Температура плавления 191 - 194oC;
6-ацетиламино-1-[N-[4-[3(4-фторфенил)-2-имидозолинон-1-ил] -1- бутил]-N-метиламинометил]индан, оксалат 13h. Температура плавления 102 - 106oC;
6-ацетиламино-1-[N-[3-(3-циклогексил-2-имидозолинон-1-ил] -1-пропил]-N-метиламинометил] индан, сесквиоксалат, полугидрат 13i. Температура плавления 108 - 115oC;
6-ацетиламино-1-[N-[6-(3-циклогексил-2-имидозолинон-1-ил] -1-гексил]-N-метиламинометил] индан, сесквиоксалат, полугидрат 13j. Температура плавления 87 - 93oC;
6-ацетиламино-1-[N-[4-(2-имидозолинон-1-ил]-1-бутил]-N- метиламинометил] индан, полуфумарат 13k. Аморфный замерзающий сухой порошок;
6-ацетиламино-1-[N-[2-(3-(2-пропил)-2-имидозолинон-1-ил]-1-пропил]-N-метиламинометил]индан, сесквиоксалат 13l. Температура плавления 151 - 154oC.
Пример 14.
6-(N-ацетил-N-этиламино)-1-(N, N-дипропиламинометил)индан, оксалат 14а (метод а).
Раствор свободного основания соединения 2а (5.0 г) в сухом тетрагидрофуране по каплям добавляли к суспензии 1 г LiAlH4 в 50 мл сухого тетрагидрофурана при 20 - 25oC. Смесь нагревали в колбе с обратным холодильником в течение 2 часов, избыток LiAlH4 разрушали осторожно добавив 2 мл разбавленного водного раствора NaOH. Неорганические соли отфильтровали, а сырой 6-этиламино-1-(N,N-дипропиламинометил)-индан выделили как вязкое масло при упаривании растворителей. Выход 3.0 г. Все полученное таким образом производное этиламиноиндана растворили в 60 мл дихлорметана и добавили 2.3 мл триэтиламина. Смесь охладили до 0oC и затем добавили по каплям при 5 - 10oC раствор 1 мл ацетилхлорида в 10 мл дихлорметана. Смесь еще в течение 1 часа перемешивали при комнатной температуре. Добавили воду (100 мл), и органическую фазу отделили и обработали. Оставшееся масло растворили в ацетоне и добавили щавелевую кислоту. Кристаллизованную оксалатную соль соединения, указанного в заглавии 14 а, отфильтровали и высушили в вакууме. Выход 2.5 г. Температура плавления 129 - 130oC.
Следующее соединение было получено аналогичным способом, используя соединение 1а в качестве исходного:
6-(N-ацетил-N-метиламино)-1-(N, N-дипропиламинометил)индан, оксалат 14b. Температура плавления 126 - 129oC.
Фармакология
Соединения формулы I испытывали согласно
надежным и общепринятым фармакологическим методам для определения их сродства к рецептору 5-HT1A и для определения In vivo агонистического воздействия этих соединений относительно
указанного рецептора. Эти испытания были следующими.
Ослабление связи3H-8-OH-DPAT с рецепторами 5-HT1A серотонина в мозгу крыс in vitro
С помощью этого
метода определяли in vitro ослабление лекарственными средствами связи3H-8-OH-DPAT (1 nM) с рецепторами 5-HT1A в мембранах из мозжечка крыс. Следовательно, этот тест является
определением сродства к 5-HT1A рецептору.
Процедура
Мужские особи крыс умерщвлялись, а их мозг иссекали и взвешивали. Ткань мозжечка гомогенизировали в 10 мл
имеющего температуру льда 50 nM трис-буфера (pH 8.0. 25oC), содержащем 120 nM NaCl, 4 nM CaCl2 и 4 nM MgCl2. Гомогенат центрифугировали при 20000g и 4oC в
течение 10 мин. Таблетку гомогенизировали в 10 мл буфера и инкубировали в течение 10 мин при 37oC. Гомогенат центрифугировали, как это описано выше, а таблетку гомогенизировали в 100
объемах буфера, имеющего температуру льда и содержащего 10 μм паргилина.
Трубочки для инкубирования сохраняли на льду в 100 μм лекарственного раствора в воде (или просто в воде для общей связи) и 1000 μм суспензии ткани (конечное содержание ткани соответствует 10 mg исходной ткани). Эксперимент инициировался добавлением 100 μм3H-8-OH-DPAT (конечная концентрация 1 nM) и помещением трубочек в водяную баню с температурой 37oC. После инкубирования в течение 15 мин образцы фильтровали под вакуумом (0.50 мбар) через 25 мм фильтр из ватмана. Трубочки промыли 5 мл 0.9% NaCl, имеющего температуру льда, а затем поместили на фильтры. Затем фильтры промывали 0.9% NaCl (2 х 5 мл). Фильтры поместили в счетную склянку и добавили 4 мл подходящей сцинтиляционной жидкости (например Pi-cofluor ТМ 15). После встряхивания в течение 1 часа и выдержки в течение 2 часов в темном месте с помощью жидкостного сцинтиляционного счетчика определяли радиоактивную концентрацию. Специфическое связывание получали при уменьшении неспецифической связи в присутствии 10 μм 5-HT1A.
Для определения ослабления связывания были использованы пять концентраций лекарственных средств в течение 3 декад.
Измеренные значения концентраций и концентрации лекарственного средства были отложены на полулогарифмической бумаге, и затем построили кривую, которая больше всего соответствовала S-форме. Величина IC50 определяется как концентрация, при которой связывание составляет 50% от суммарного связывания в контрольных образцах за вычетом неспецифического связывания в присутствии 10 μм 5-НТ.3H-8-OH-DPAT были получены от Amersham international plc., England. Специфическая активность составила примерно 200 Ci/mmol.
8-ОН DPAT Стимулятор агонизма у крыс
Эту модель испытаний использовали для определения агонистического влияния
испытываемых соединений на 5-HT1A рецепторы in vivo. Соответствующий метод описан в работах Tricklrbank, M.D., et al, Eur. J. Pharmacol., 1987, 133, 47 - 56; Arnt, J. Pharmacology &
Toxicology, 1989, 64, 165.
Процедура
Мужские особи крыс обучались распознавать 8-ОН DPAT (0.4 мг/кг, например 15-минутная предварительная обработка) и физиологический раствор
в камерах, оборудованных двумя рукоятками для ответной реакции. Между этими рукоятками располагалась емкость, в которой в качестве вознаграждения помещали воду (0.1 мл). Крыс не допускали к воде в
течение по меньшей мере 24 часов, они работали в фиксированном режиме.
Введение 8-ОН DPAT осуществлялось только предназначенной для этого ("лекарственной") рукояткой, в то время как действие на противоположную рукоятку оставалась без последствий. Введение физиологического раствора управлялось рукояткой, противоположной "лекарственной" рукоятке. Испытания с лекарством и физиологическим раствором чередовались по дням. Уровень точности распознавания выражается в процентах ответной реакции на лекарственное средство и вычисляется как число правильных откликов, умноженное на 100 и деленное на сумму правильных и неправильных откликов вплоть до первого вознаграждения. Время до первого вознаграждения также фиксируют как меру времени реакции. При достижении стабильной точности (средняя правильная ответная реакция 90%, индивидуальная ответная реакция составляет по меньшей мере 75% от ее величины) испытания проводят между днями обучения. Контрольный опыт оканчивают, когда сделано в общей сложности 32 отклика на каждый рукоятку или по истечение 20 мин. Никакого вознаграждения теперь крысам не дают и они имеют свободный доступ к воде в течение 20 - 30 мин после испытания. Воздействие выражают в процентах лекарственной реакции. В результаты анализа включены только результаты, полученные от крыс, давших по меньшей мере 10 "ответов" на каждую рукоятку. Более того, включены только те серии испытаний, в которых по меньшей мере половина крыс дала "ответ".
Ослабление в процентах лекарственного "ответа", полученного от каждой дозы испытуемого соединения, используют для вычисления величины ЭД50.
Полученные результаты приведены ниже в таблице. Известные лиганды рецептора 5-HT1A 8-OH-DPAT и бушпирон также включены в испытания для сравнения.
Из приведенной ниже таблицы очевидно, что соединения настоящего изобретения являются лигандами рецептора 5-HT1A, ингибирующими связывание тритированного 8-гидрокси-2-дипропиламинотетралина (8-OH-DPAT) с рецепторами 5-HT1A in vitro, многие из которых имеют потенциал более 50 nM, и даже в диапазоне от 0.5 до 10 nM. Видно также, что обычно эти соединения и in vivo имеют свойства агонистов 5-HT1A.
Кроме того, изучалось сродство соединений настоящего изобретения к рецептору допамина D2 определением его способности ослаблять связывание3H-спироперидола с рецепторами D2 методом Hyttel et al, J. Neurochem., 1985, 44, 1615.
Соединения были также подвергнуты тесту Methylphenidate согласно работе Pedersen, Christensen, Acta Pharmacol. et Toxicol., 31, 488 - 496 (1972).
Следующее испытание, оценка каталепсии, было проведено согласно методу Sanchez, C. et al.; Drug Dev.Res. 1991, 22, 239 - 250.
Испытанные соединения настоящего изобретения, как было показано, не обладают значительным сродством к рецепторам допамина D2 и не имеют никаких каталептогенных воздействий в самой высокой дозе, в то время как многие из них оказываются эффективными в Methylphenidate тесте при значениях ЭД50 в диапазоне μмол/кг. Результаты этих испытаний показывают, что соединения настоящего изобретения обладают антипсихотическими свойствами и не демонстрируют экстрапирамидальных побочных эффектов.
В заключение соединения настоящего изобретения были испытаны на наличие анксиолитических свойств при измерении их способности ослабить ультразвуковые сигналы, вызванные звуком шагов. Взрослые крысы испускают ультразвуковые сигналы бедствия как "ответ" на вызывающий отвращение раздражитель, которого нельзя избежать, такой как шок от шагов. Это предлагается в качестве испытательной модели тревожного состояния (Tonoue Т. et al. , Psychoneuroendocrinology. 1986. 11, N2, 177 - 184).
Испытанные соединения продемонстрировали потенциальное
анксиолитическое воздействие, причем величина ЭД50 обычно находилась в пределах от 0.03 до 1.0 μмол/кг.
Примеры прописей
Фармацевтические прописи настоящего
изобретения могут быть получены общепринятыми способами, известными в этой области.
Например, таблетки можно получить, смешивая активный ингредиент с обычными адъювантами и/или разбавителями и затем прессуя смесь в обычной таблетировочной машине. Примеры адъювантов или растворителей включают: картофельный крахмал, кукурузный крахмал, тальк, стеарат магния, желатин, лактозу, смолу и им подобные. Любые другие адъюванты или добавки, используемые обычно для этих целей, такие как красители, корригенты, консерванты и т.п., можно использовать при условии, что они совместимы с активными ингредиентами.
Растворы для инъекций могут быть получены растворением активного ингредиента и возможных добавок в части растворителя, используемого для инъекций, предпочтительно в стерильной воде, доведением раствора до требуемого объема, его стерилизацией и заполнением подходящих ампул или склянок. Может быть добавлена любая подходящая, используемая обычно в этой области добавка, такая как тонизирующие агенты, консерванты, антиоксиданты и т.п.
Типичными примерами фармацевтических прописей согласно настоящему изобретению являются
следующие:
1) Таблетки, содержащие 5.0 мг соединения 1а в расчете на свободное основание:
Соединение 1а - 5.0 мг
Лактоза - 60 мг
Кукурузный крахмал - 30 мг
Гидроксипропилцеллюлоза - 2.4 мг
Микрокристаллическая целлюлоза 19.2 мг
Кроскармеллоз натрия тип A - 2.4 мг
Стеарат магния - 0.84 мг
2) Таблетки, содержащие 0.5 мг
соединения 2b в расчете на свободное основание:
Соединение 2b - 0,5 мг
Лактоза - 46.9 мг
Кукурузный крахмал - 23,5 мг
Повидон - 1,8 мг
Микрокристаллическая
целлюлоза - 14,4 мг
Кроскармеллоз натрия тип A - 1,8 мг
Стеарат магния - 0,63 мг
3) Сироп, содержащий в 1 мл:
Соединение 1с - 2,5 мг
Сорбитол - 500 мг
Гидроксипропилцеллюлоза - 15 мг
Глицерол - 50 мг
Метил-парабен - 1 мг
Пропил-парабен - 0,1 мг
Этанол - 0,005 мл
Ароматизатор - 0,05 мг
Сахарин - 0,5 мг
Вода - До 1 мл
4) Раствор для инъекций, содержащий в 1 мл:
Соединение 10j - 0,5 мг
Сорбитол - 5,1 мг
Уксусная кислота - 0,08 мг
Вода для инъекций - До 1 мл
Проведенные испытания заявленного вещества в терапевтически приемлемых количествах не выявили вредного влияния вещества на пациента.
Реферат
Аминометилинданы - бензофураны и
бензотиофены общей формулы I

где один из радикалов Х или Y представляет СН2, а другой выбран из СН2, О или S; R1 - низший алкил, фенилнизший алкил, ацил, алкилсульфонил или R11R12NC(O)-, R11, R12 - Н, низший алкил, С3-8-циклоалкил, фенил, возможно замещенный галогеном; R2 - Н, низший алкил; R3 - R5 - Н или галоген; R6, R7 - Н; R8, R9 - Н, низший алкил, С3-8-циклоалкил - (низший) алкил, фенил - низший алкил, индолил - низший алкил или группа формулы A
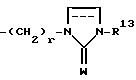
R13 - Н, низший алкил, С3-8-циклоалкил, фенил, фторфенил; r = 2-6; W = O; или R8, R9 связаны вместе с образованием 3-7-членного кольца, содержащего один атом азота и их фармацевтически приемлемые соли, воздействуют на аминометильные рецепторы 5-НТ1A, поэтому эти соединения полезны для лечения обычных психических и неврологических нарушений. 2 с. и 5 з.п.ф-лы, 1 табл.
Формула

где один из радикалов X или Y представляет собой CH2, а другой выбран из группы, содержащей CH2, O, S;
R1 представляет собой низший алкил, фенил - низший алкил, ацил, низший алкилсульфонил или R11R12NC(O)-, где R11 и R12 независимо представляют собой водород, низший алкил, C3-C8-циклоалкил, фенил, возможно, замещенный галогеном;
R2 представляет собой водород или низший алкил;
R3 - R5 независимо выбирают из группы, содержащей водород или галоген;
R6, R7 - водород;
R8, R9 независимо представляет собой водород, низший алкил, C3-C8-циклоалкил-низший алкил, фенил - низший алкил, индолил - низший алкили или группу;
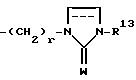
где R13 представляет собой водород, низший алкил, C3-C8-циклоалкил, фенил, фторфенил;
r = 2-6;
w = 0,
или R8, R9 связаны вместе с образованием 3-7-членного кольца, содержащего один атом азота и их фармацевтически приемлемые соли.
R11 представляет собой водород, низший алкил, а R12 представляет собой водород, низший алкил, фенил, возможно замещенный галогеном или C3-C8-циклоалкил.

где w представляет собой О;
R13 представляет собой водород, низший алкил, C3-C8-циклоалкил, фенил или фторфенил.
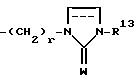
где w означает О;
R13 означает водород, низший алкил, C3-C8-циклоалкил, фенил или фторфенил.
Комментарии