8-замещенные-2-аминотетралиновые производные и способы из получения - RU2093507C1
Код документа: RU2093507C1
Чертежи


Описание
Изобретение относится к новым 8-карбониларилзамещенным 2-аминотетралинам, их энантиомерам и солям, способам их получения, лекарственным препаратам на их основе и использованию указанных соединений в терапии.
Цель настоящего изобретения создать соединения для терапевтического использования особенно соединений, проявляющих лечебное действие через центральную нервную систему. Другой целью является создание соединений с селективным действием на 5-гидрокситриптаминовые рецепторы млекопитающих, в том числе человека.
Терапевтически полезные производные тетралина, оказывающие действие на 5-гидрокситриптаминовые нейроны у млекопитающих, описаны в EP N 41488, 270947 и 272534.
Цель настоящего изобретения получить новые соединения, имеющие высокую степень сродства с 5-гидрокситриптаминовыми рецепторами центральной нервной системы с одновременным их действием в качестве агонистов, частичных агонистов или антагонистов к серотониновым рецепторам.
Таким образом, новые соединения формулы I предлагаемого изобретения, а также их энантиомеры и соли полезны для терапевтического лечения опосредованных 5-гидрокситриптамином состояний и расстройств, таких как депрессия, беспокойство, анорексия, старческое слабоумие, болезнь Альцгеймера, мигрень, нарушения терморегуляторных и половых функций. Дополнительные аспекты изобретения относятся к использованию указанных соединений, их энантиомеров и солей для снятия боли и модуляции сердечно-сосудистой системы.
Таким образом, настоящее изобретение предлагает соединения формулы:

в которой R означает водород или метил при условии, что C1- метильный заместитель находится в цис-конфигурации;
Z представляет водород или галоген;
Q представляет COR1 или фенил, фурил, тиенил, пиразолил, тиазолил, пиридил, хинолил, индоксазинил, индолил, бензофуранил, не обязательно замещенный галогеном, низшим алкилом, или низшим алкокси;
R1 представляет C1-6-алкил, C3-6-циклоалкил или фенил, не обязательно замещенный галогеном или низшим алкокси;
R2 представляет водород или C1-6-алкил;
R3 представляет группу C1-6-алкил или -(CH2)a-R4, в которой
a представляет 4;
R4 представляет -NR11R12;
R11 и R12 вместе с атомом азота образуют кольцо:

где n представляет 1 или 2,
и их энантиомеры и физиологически приемлемые соли.
C1-C6-алкил в формуле I означает неразветвленные или разветвленные алкильные группы с 1-6 атомами углерода, например метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил, трет-пентил, C3-C6-циклоалкил означает циклические алкильные группы с 3-6 атомами углерода, например циклопропил, циклобутил, циклопентил, метилциклопропил, этилциклопропил, метилциклобутил. В качестве предпочтительных алкильных групп используют C1-4-алкилы.
Низшая алкоксигруппа в формуле I представляет алкоксигруппу с 1-4 атомами углерода, например метокси, этокси, пропокси или бутокси, предпочтительно метокси и этокси.
Галоген в формуле I означает фтор, хлор, бром, йод, предпочтительно фтор, хлор и бром, особенно фтор.
Соединения предлагаемого изобретения содержат один или два асимметрических атома углерода. Когда R означает водород, заявленные соединения содержат асимметрический атом углерода у азота, то есть C2, и когда R означает метил, соединения содержат асимметрический атом углерода у азота и асимметрический атом углерода у метильной группы, то есть C1 и C2. Таким образом, предлагаемые соединения существуют в форме двух или четырех стереоизомеров, то есть энантиомеров и/или диастереоизомеров. В объем предлагаемого изобретения входят как чистые энантиомеры, так и рацемические смеси. Терапевтические свойства заявляемых соединений в большей или меньшей степени могут быть приписаны и рацемату и энантиомерам.
Было найдено, что C1-метилированные производные формулы I, в которых метильныЙ заместитель находится в цис-конфигурации относительно 2-амино заместителя с C2, являются сильными агонистами 5-гидрокситриптаминового рецептора. Предпочтительные соединения имеют 1S-, 2R-конфигурацию.
Для получения нетоксичных физиологически приемлемых аддитивных солей кислот предлагаемых соединений могут использоваться как органические, так и неорганические кислоты. В качестве примера можно привести серную, азотную, фосфорную, щавелевую, соляную, бромистоводородную, лимонную, уксусную, молочную, винную, памовую кислоты, этансульфокислоту, сульфаминовую, янтарную, циклогексилсульфаминовую, фумаровую, малеиновую и бензойную кислоты. Эти соли легко получают общеизвестными способами.
К предпочтительным соединениям относят такие, в которых Q представляет фенил, фторфенил, тиенил или фуранил или группы COR1, в которых R1 означает CH3, C2H5, C3H7, C4H9, C5H11, циклопропил, метилциклопропил, метилциклобутил, и R2 означает C1-6-алкил, а R означает водород.
Соединения предлагаемого изобретения можно получить одним из следующих способов, которые составляют еще один аспект изобретения.
а) Превращают соединение формулы II
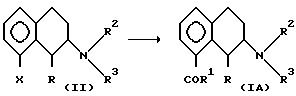
где X представляет уходящую группу, например трифторметансульфонатную (ТФ), фосфонатную, галогенидную, например Br или J, и R, R2 и R3 имеют вышеуказанные значения,
путем замещения группы X карбоксильной группой COR1 с получением соединения формулы IA.
Соединение II можно превратить в соединение IA с помощью следующего каталитического цикла. Металл (М) должен быть переходным металлом с нулевой валентностью М0, таким как Pd или Ni, который способен подвергаться окислительному присоединению к арил-X-связям, например арилгалогеновым связям. М0 может получаться in situ путем обработки М11 окисью углерода (CO). М1 должен быть таким металлом, как Sn, Mg, Zn, Zr, B, Al, Li который способен к трансметаллированию первоначально образованным карбонилированным σ-арил-металл-X-комплексом (например, s-арил-металгалогенидным комплексом).

Дополнительными реагентами являются окись углерода, амин, такой как триэтиламин, в инертном органическом растворителе, предпочтительно в полярном апротонном растворителе, таком как диметилформамид (ДМФ), диметилсульфоксид (ДМСО), ацетон, ацетонитрил и т.д. Реакцию обычно проводят при температуре в интервале от 40 до 120oC и давлении от 1 до 5 бар.
b) Соединение IA можно получить с помощью обратного процесса.
Реакцию осуществляют в виде каталитического цикла с использованием переходного металла нулевой валентности М0, например Pd или Ni, обладающего способностью окислительного присоединения к группе R1-X, где R1 имеет вышеуказанные в формуле IA значения, а X означает уходящую группу, такую как галогенид, осуществляют обработку окисью углерода с последующим добавлением соединения формулы III.
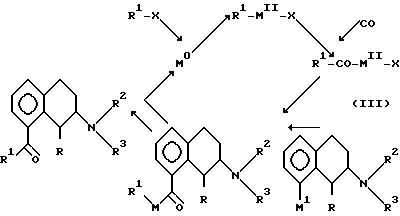
R1-CO-M11-X может также получаться непосредственно из R1-COCl. Условия реакции и реагенты аналогичны используемым в вышеуказанном способе "а".
с) превращают соединение формулы II

где X означает уходящую группу, такую как трифторметансульфонатная (ТФ), фосфатная, галогенидная, например Br, или J, а R, R2 и R3 имеют вышеуказанные значения, путем замещения группы Х 5- или 6-членным арилом, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и является или замещенным, или сконденсированным у двух соседних атомов углерода с арильным кольцом, как указано выше, с образованием соединения формулы IB.
Соединение формулы II может превращаться в соединение Ib с помощью реакции с переходным металлом с нулевой валентностью М0, таким как Pd или Ni, обладающим способностью к окислительному присоединению к арил-X-связи. Подходящий арил-заместитель может быть введен при использовании триалкил-арилстаннана.
Дополнительными реагентами являются амин, например триэтиламин, и соль лития, например хлористый литий. Реакцию предпочтительно проводят в полярном апротонном растворителе, например диметилформамиде, диоксане, ацетонитриле или диметилсульфоксиде, при температуре от 40 до 120oC.
d) превращают соединение формулы V
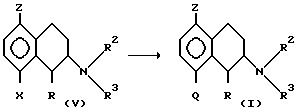
где X означает уходящую группу, такую как трифторметансульфонатную, Z означает галоген, а R, R2, R3 имеют вышеуказанные значения,
с помощью замещения группы X группой Q, которая означает группу COR1 или 5- или 6-членный арил, определенный выше.
е) превращают соединение
формулы IV (описанное в EP N 272534)

где R2 и R3 имеют вышеуказанные значения, путем замещения нитрила карбоксигруппой COR1 с образованием соединения формулы IA. Реакцию проводят с помощью обработки соответствующим металлоорганическим реагентом, предпочтительно литийорганическим соединением или реактивом Гриньяра в инертном органическим растворителе, предпочтительно в неполярном апротонном растворителе, таком как простые эфиры, например диэтиловый эфир, тетрагидрофуран, бензол, после чего осуществляют гидролиз промежуточного комплексного соединения с образованием целевого продукта.
Фармацевтические препараты
В соответствии с настоящим изобретением соединения формулы I вводятся, как правило, перорально, ректально или с помощью инъекций в форме фармацевтических препаратов, включающих активное вещество или в
виде свободного основания, или в виде фармацевтически приемлемой соли присоединения кислоты, например хлоргидрата, бромгидрата, лактата, ацетата, фосфата, сульфата, сульфамата, цитрата, тартрата,
оксалата и аналогичных, в фармацевтически приемлемой дозированной форме. Дозированная форма может быть представлена в виде жидкого, полужидкого или твердого препарата. Активное вещество обычно
составляет от 0,1 до 99 мас. препарата, более конкретно от 0,5 до 20 мас. для препаратов, предназначенных для инъекций, и от 0,2 до 50 мас. для препаратов, предназначенных для перорального приема.
Для приготовления фармацевтических препаратов, содержащих соединение формулы I в форме единичной дозы для перорального введения, выбранное соединение может смешиваться с твердым наполнителем, например лактозой, сахарозой, сорбитом, маннитом, крахмалами, например картофельным, кукурузным крахмалом или амилопектином, производными целлюлозы, связующим, например желатиной или поливинилпирролидоном, и смазывающим веществом, таким как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и т.д. и затем прессоваться в таблетки. Для получения таблеток с покрытием сердцевину, полученную вышеуказанным способом, можно покрывать концентрированным раствором сахара, который может включать, например, аравийскую смолу, желатин, диоксид титана и т.д. В альтернативном варианте таблетку можно покрывать любым известным специалисту полимером, растворенном в легколетучем органическом растворителе или смеси органических растворителей. Можно добавлять в указанные покрытия красящие вещества для упрощения различения таблеток, содержащих различные активные вещества или разные количества активных соединений.
Для получения капсул из мягкой желатины активное вещество можно смешивать, например, с растительным маслом или полиэтиленгликолем. Капсулы на основе твердой желатины могут содержать гранулы активного вещества с использованием вышеуказанных наполнителей для таблеток, например лактозы, сахарозы, сорбита, маннита, крахмалов (картофельного, кукурузного или амилопектина), производных целлюлозы или желатина. Твердые желатиновые капсулы можно также заполнять лекарственным препаратом в жидкой или полужидкой формах.
Единичные дозы для ректального применения могут быть в форме растворов или суспензий, либо их можно получить в виде суппозиториев, содержащих активное вещество в смеси с нейтральным жирным основанием, либо желатиновых капсул, содержащих активное вещество в смеси с растительным или парафиновым маслом.
Жидкие препараты для перорального использования могут быть в форме сиропов или суспензий, например растворов, содержащих примерно от 0,2 до 20 мас. активного вещества, описанного здесь, а остаток составляет сахар, и смесь этанола, воды, глицерина и пропиленгликоля. Такие жидкие препараты могут не обязательно включать красящие вещества, отдушки или вкусовые агенты, сахарин и карбоксиметилцеллюлозу в качестве загустителя или другие известные в данной области наполнители.
Растворы для парентерального применения в виде инъекций можно получить в форме водного раствора водорастворимой фармацевтически приемлемой соли активного вещества, предпочтительно в концентрации примерно от 0,5 до 10 мас. Указанные растворы могут также содержать стабилизаторы и/или буферные агенты. Для удобства указанные растворы могут производиться в ампулах с различной дозировкой для приема.
Подходящая суточная доза предлагаемых соединений при терапевтическом применении к человеку составляет примерно от 0,01 до 100 мг/кг массы тела при пероральном введении и от 0,001 до 100 мг/кг массы тела при парентеральном приеме.
Пример 1.
(+/-)-2-(Дипропиламино)-8-[(трифторметилсульфонил)окси]тетралин
Раствор ангидрида трифторметансульфокислоты (7,0 г, 24,8 ммоль) в дихлорметане (20 мл) добавляют к смеси карбоната калия (3,4 г,
24,8 ммоль) и 8-гидрокси-2- (дипропиламино)тетралина (3,06 г, 12,4 ммоль) в дихлорметане (300 мл), выдерживают смесь при -70oC. Охлажденную ванну удаляют, и перемешивание продолжают в
течение ночи. Смесь экстрагируют охлажденным льдом, насыщенным водным раствором карбоната калия. Органический слой сушат (карбонат калия), фильтруют и концентрируют. После очистки остатка на колонке
из глинозема при элюировании смесью простой эфир/петролейный эфир (1:8), получают 5,01 г масла, которое превращают в гидрохлорид. После перекристаллизации из смеси этанол/простой эфир получают 5,01 г
(97%) очищенного хлористоводородного 2-(дипропиламино)-8-[(трифторметилсульфонил)окси] тетралина. (+)-(R)-2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралин и
(-)-(S)-2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралин получают аналогичным способом из соответствующих энантиомеров 8-гидрокси-2-(дипропиламино)тетралина, которые можно получить с высоким
выходом и оптической чистотой.
Пример 2. Гидрохлорид (+/-)-8-ацетил-2-(дипропиламино)тетралина
Смесь, состоящую из 2-(дипропиламино)-8-[(трифторметилсульфонил)окси]тетралина
(455 мг, 1,2 ммоль), тетраметилолова (257 мг, 1,44 ммоль), хлорида лития (158 мг, 3,7 ммоль), дихлор-[1,1-бис-(дифенилфосфино)ферроцен]палладия (II) (PdCl2) (дффф); 61 мг, 0,07 ммоль),
молекулярных сит (4
Пример 3. Гидрохлорид (+/-)-метил-2-(дипропиламино)тетралин-8-карбоксилата
Смесь, состоящую из 2-(дипропиламино)-8-[(трифторметилсульфонил)окси] тетралина (3,5 г, 9,2
ммоль), триэтиламина (1,86 г, 18,4 ммоль), ацетата палладия (11) (62 мг, 0,28 ммоль), 1,11-бис(дифенилфосфино)ферроцена (306 мг, 0,55 ммоль), метанола (5,7 г, 184 ммоль) и
диметилсульфоксида (70 мл), перемешивают в течение ночи при положительном давлении окиси углерода. Реакционную смесь распределяют между насыщенным водным раствором хлористого натрия и эфиром.
Органический слой сушат (используя сульфат натрия) и концентрируют. Остаток очищают хроматографией на колонке глинозема с элюированием смесью простой эфир/петролейный эфир (1:16). Очищенные фракции
сливают и концентрируют. Оставшееся масло превращают в хлористоводородную соль. При перекристаллизации продукта из смеси диэтиловый эфир/хлороформ получают 2,08 г (92%) гидрохлорида метил
2-(дипропиламино)-тетралин-8-карбоксилата. Т. пл. 136-137oC.
Пример 4. (+/-)-8-Карбокси-2-(дипропиламино)тетралин
Раствор гидрохлорида
2-(дипропиламино)тетралин-8-карбоксилата (1,5 г, 4,6 ммоль), гидроокиси натрия (736 мг, 18,4 ммоль), метанола (25 мл) и воды (4 мл) перемешивают в течение ночи. Метанол выпаривают. Добавляют
концентрированную соляную кислоту до тех пор, пока pH не станет примерно 6. Затем раствор экстрагируют хлороформом. Органический слой сушат (сульфатом натрия) и концентрируют, получая 1,23 г (97%)
чистого 8-карбокси-2-(дипропиламино)тетралина в виде масла. Гидрохлорид плавят при температуре 245-247oC, его можно перекристаллизовывать из смеси метанол/диэтиловый эфир.
Пример 5. Гидрохлорид (+/-)-8-ацетил-2-(дипропиламино)тетралина
5%-ный раствор метиллития в простом эфире (0,6 мл, 0,96 ммоль) добавляют к резко охлажденной суспензии гидрохлорида
(+/-)-карбокси-2-(дипропиламино)тетралина (100 мг, 0,32 ммоль) в простом эфире. Смесь перемешивают при комнатной температуре в атмосфере азота в течение 3 дней. Осторожно добавляют воду, и смесь
экстрагируют простым эфиром. Органический слой сушат (карбонатом калия) и концентрируют. Остаток очищают хроматографией на колонке с глиноземом с элюированием смесью простой эфир/петролейный эфир (1:
4). Очищенные фракции сливают, концентрируют и превращают в гидрохлорид. Перекристаллизация из смеси ацетонитрил/простой эфир дает 55 мг (56%) чистого гидрохлорида
8-ацетил-2-(дипропиламино)тетралина.
Пример 6. (+)-Ацетил-2-(дипропиламино)тетралин
Смесь (+)-2-(пропиламино)-8-[(трифторметилсульфонил)окси]тетралина (300 мг, 0,79 ммоль),
тетраметилстаннана (167 мг, 0,95 ммоль), хлористого лития (104 мг, 2,5 ммоль), дихлор[1,11-бис(дифенилфосфино)ферроцен]палладия (II) (PdCl2) (дффф) (40 мг, 0,047 ммоль),
молекулярных сит (4

Пример 7. Гидрохлорид (-)-8-ацетил-2-(дипропиламино)тетралина
Смесь
(-)-2-(дипропиламино)-8-[(трифторметилсульфонил)окси] тетралина (910 мг, 2,4 ммоль), тетраметилстаннана (514 мг, 2,88 ммоль), хлористого лития (315 г, 7,44 ммоль), дихлор-[1,11
-бис-(дифенилфосфино)ферроцен]палладия (II) (12 мг, 0,144 ммоль), молекулярных сит (4

Пример 8. Гидрохлорид
(+/-)-8-бензоил-2-(дипропиламино)тетралина
Смесь рацемата 2-(дипропиламино)-8-[(трифторметил)окси] тетралина (200 мг, 0,52 ммоль), фенилтриметилстаннана (154 мг, 0,64 ммоль), хлористого лития
(69 мг, 1,6 ммоль), дихлор[1,11-бис- (дифенилфосфино)ферроцен]палладия (II) (26 мг, 0,332 ммоль), 2,6-дитрет-бутил-4-метилфенола (в качестве катализатора) и молекулярных сит (4

Пример 9. (+/-)-8-(1-Оксопентил)-2-(дипропиламино)тетралин
Смесь гидрохлорида 2-(дипропиламино)-8-[(трифторметилсульфонил)окси]
тетралина (216 мг, 0,52 ммоль), тетрабутилстаннана (218 мг, 0,64 ммоль), триэтиламина (105 мг, 1,04 ммоль), хлористого лития (68 мг, 1,6 ммоль), дихлор-[1,11- бис-(дифенилфосфино)ферроцен]
палладия (26 мг, 0,03 ммоль), 2,6-дитрет-бутил-4-метилфенола (в каталитическом количестве) и молекулярных сит (4

Пример 10. Оксалат
(+/-)-8-фенил-2-(дипропиламино)тетралина
Смесь рацемического 2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралина (450 мг, 1,2 ммоль), триметилфенилстаннана (433 мг, 1,8 ммоль),
тетракис(трифенилфосфин)палладия (0) (69 мг, 0,06 ммоль), хлористого лития (153 мг, 3,6 ммоль) и 2,6-дитрет-бутил-4-метилфенола (катализатор) в 15 мл диоксана и 1,5 мл диметилформамида перемешивают
при 105oC в колбе с притертой пробкой в течение 3 дней. Полученную смесь фильтруют (целит), концентрируют и разделяют на слой насыщенного раствора бикарбоната калия и эфирный слой.
Органический слой сушат над карбонатом калия и концентрируют в вакууме. Остаток хроматографируют на колонке с глиноземом при элюировании вначале петролейным эфиром, а затем смесью простой
эфир/петролейный эфир (1:40), и после этого простым эфиром/петролейным эфиром (1: 20). Чистые фракции собирают и обрабатывают эфирной щавелевой кислотой, получая 232 мг (48%) оксалата
(+/-)-8-фенил-2-(дипропиламино)тетралина. Т. пл. 162-163oC. (+)-(R)-8-фенил(дипропиламино)тетралин и (-)-(S)-8-фенил-2-(дипропиламино)тетралин получают аналогично из (R)- и
(S)-2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралина, соответственно.
Пример 11. Оксалат (+/-)-8-(2-фуранил)-2-(дипропиламино)тетралина
Смесь рацемата
2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралина (100 мг, 0,26 ммоль), фуран-2-ил-триметилстаннана (75 мг, 0,32 ммоль), дихлор[1,1-бис-(дифенилфосфино)ферроцен] палладия (II) (12 мг, 0,014
ммоль), хлористого лития (69 мг, 1,6 ммоль), молекулярных сит (60 мг) и 2,6-дитрет-бутил-4-метилфенола (катализатор) в 3 мл диметилформамида перемешивают при температуре 90oC в герметичной
колбе в течение ночи. Смесь фильтруют (через целит) и распределяют между насыщенным раствором бикарбоната натрия и эфиром. Эфирный слой сушат (карбонатом калия), фильтруют и концентрируют в вакууме.
Остаток хроматографируют на колонке с глиноземом, используя смесь простой эфир/петролейный эфир в качестве элюента. Чистые фракции собирают, обрабатывают эфирной щавелевой кислотой, получая белый
порошок, после перекристаллизации которого из смеси MeOH/простой эфир получают 36 мг (36%) оксалата (+/-)-8-(фуран-2-ил)-2-(дипропиламино) тетралина. Т. пл. 113-114oC.
Пример 12. Оксалат (+/-)-8-(бензофуран-2-ил)-2-(дипропиламино)тетралина
Смесь рацемического 2-дипропиламино-8-(трифторметилсульфонилокси)тетралина (400 мг, 1,04 ммоль),
бензофуран-2-илтриметилстаннана (444 мг, 1,6 ммоль), тетракис(трифенилфосфин)палладия (0) (60 мг, 0,52 ммоль), хлористого лития (140 мг, 3,24 ммоль) и 266-дитрет-бутил-4-метилфенола (катализатор) в 12
мл 1,4-диоксана и 1,2 мл диметилформамида перемешивают при 105oC в герметичной колбе в течение 3 дней. Смесь фильтруют (через целит), концентрируют и распределяют между насыщенным раствором
бикарбоната калия и простым эфиром. Эфирный слой сушат (карбонатом калия), фильтруют и упаривают. Остаток хроматографируют на колонке с глиноземом при элюировании петролейным эфиром, затем смесью
простой эфир/петролейный эфир (1:40), далее смесью простой эфир/петролейный эфир (1: 20) и, наконец, простым эфиром. Очищенные фракции собирают, и после обработки эфирной щавелевой кислотой получают
220 мг (48%) оксалата (+/-)-8-(бензофуран-2-ил)-2-(дипропиламино)тетралина. Т. пл. - 168-170oC.
Пример 13. (1S,
2R)-1-Метил-2-(дипропиламино)-8- (трифторметилсульфонилокси)тетралин
Раствор гидрохлорида (1S,2R)-1-метил-8-метокси-2-(дипропиламино)тетралина (J. Med. Chem. 1987, 30, 2105-2109) в
свежеперегнанном 48%-ном растворе HBr перемешивают при 120oC в течение 3 часов. Реакционную смесь концентрируют и распределяют между охлажденным льдом насыщенным раствором бикарбоната
натрия и дихлорметаном. Органический слой сушат над сульфатом натрия, фильтруют и концентрируют. Остаток неочищенного (1S,2R)-8-гидрокси-1-метил-2-(дипропиламино)тетралина используют непосредственно
на следующей стадии. К смеси деметилированного исходного материала и карбоната калия (1,0 г, 6,6 ммоль) в 20 мл дихлорметана добавляют раствор ангидрида трифлововой (трифторметансульфоновой) кислоты
(1,3 г, 4,4 ммоль) в 10 мл дихлорметана при -78oC в течение 15 минут в атмосфере азота. Реакционную смесь выдерживают при комнатной температуре в течение ночи при перемешивании. Реакционную
смесь концентрируют и распределяют между насыщенным раствором карбоната калия и эфиром. Органический слой сушат карбонатом калия, фильтруют и концентрируют. Остаток хроматографируют на колонке с
глиноземом, используя в качестве элюента смесь простой эфир/петролейный эфир (1:8). Очищенные фракции собирают и концентрируют, получая 744 мг (86%) свободного основания трифлата.
Пример 14. (1S,2R)-8-Бензоил-1-метил(дипропиламино)тетралина гидрохлорид
Смесь, состоящую из (1S,2R)-1-метил-2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралина (100 мг, 0,25 ммоль)
(пример 13), фенилтриметилстаннана (80 мг, 0,33 ммоль), хлористого лития (33 мг, 0,77 ммоль), дихлор[1,1-бис-(дифенилфосфино)ферроцен]палладия (II) (13 мг, 0,015 ммоль), 2,
2-дитрет-бутил-4-метилфенола (катализатор) и молекулярных сит (4

Пример 15. Получение оксалатной соли 2-ди-п-пропиламино-8-пропионил-1,2,3,
4-тетрагидронафталина
2-Ди-п-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (8,5 г, 27,4 ммоль) растворялся в 80 мл тетрагидрофурана и охлаждался до -78oC, после чего добавлялось 25,
7 мл н-бутиллития (1,6 М в гексане). Смесь перемешивалась при -78oC в течение одного часа, после чего добавлялось 2,4 мл (32,9 ммоль) пропиональдегида. Смесь нагревалась до комнатной
температуры, затем выливалась в воду и экстрагировалась метиленхлоридом. Экстракт сушился над сульфатом натрия и упаривался, давая 9,1 г желтого масла.
Масло помещалось на силикагельную колонку и элюировалось смесью 3%-ного метанола в метиленхлориде, содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 6,5 (82,0%) 2-ди-н-пропиламино-8-(1'-гидроксипропил)-1,2,3,4-тетрагидронафталина в виде светлого масла.
Вышеупомянутый продукт растворялся в 250 мл метиленхлорида, и добавлялось 17 г (78,7 ммоль) пиридинийхлорформата (PCC) наряду с 30 г молекулярных сит 4

Смесь перемешивалась в течение 3 часов при комнатной температуры, после чего добавлялось 250 мл простого эфира и целита. Смесь выливалась на короткую силикагельную колонку и элюировалась простым эфиром. Для растворения коричневой суспензии добавлялся метанол, и после добавления простого эфира к реакционной смеси осаждался осадок. Данное вещество добавлялось на колонку и элюировалось 10% -ным метанолом в метиленхлориде. Элюент концентрировался, давая коричневое масло, которое затем очищалось хроматографией на колонке с использованием смеси гексан/простой эфир (2:1), а затем чистого эфира в качестве растворителя. Фракции, содержащие продукт, объединялись и концентрировались, давая 4,7 г продукта. Образовывалась оксалатная соль 2,5 г данного вещества и перекристаллизовывалась три раза из смеси этанол/простой эфир, давая продукт в виде белого твердого вещества (1,5 г). Т. пл. - 114,5-115oC.
Элементный анализ:
вычислено: C 66,82, H 8,29, N 3,71;
найдено: C 67,07, H 8,20, N 4,
00.
Пример 16. Получение гидробромидной соли 2-ди-н-пропиламино-8-бутаноил- 1,2,3,4-тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (5,0 г, 16,1
ммоль) растворялся в 50 мл тетрагидрофурана, и смесь охлаждалась до -78oC, после чего добавлялся 21 мл н-бутиллития (0,92 М в гексане). Смесь перемешивалась в течение 30 минут и добавлялась
в 1,85 мл (21,0 ммоль) бутиральдегида. Смесь оставлялась нагреваться до комнатной температуры и перемешивалась в течение ночи, после чего она выливалась в воду и экстрагировалась метиленхлоридом.
Экстракт сушился над сульфатом натрия и упаривался, давая 6,4 г остатка. Остаток помещался на силикагельную колонку и элюировался смесью 2%-ного метанола в метиленхлориде, содержащей следы
гидрооксидаммония. Соответствующие фракции объединялись, давая 4,8 г 2-ди-н-пропиламино-8-(1'-гидроксибутил)-1,2,3,4-тетрагидронафталин в виде густого масла.
Масло (4,0 г, 13,2 ммоль) растворялось в 200 мл метиленхлорида и добавлялись 4

Масло помещалось на силикагельную колонку и элюировалась смесью 3%-ного метанола и метиленхлорида, содержащей следы гидроокиси аммония.
Соответствующие фракции объединялись, давая масло, которое растворялось в простом эфире, вызывая образование коричневого осадка. Осадок удалялся фильтрованием, фильтрат упаривался, давая 3,0 г светло-коричневого масла в виде свободного основания целевого соединения.
Один грамм масла превращался в гидробромидную соль и перекристаллизовывался из смеси метанола и этилацетата, давая 0,9 г целевого соединения в виде рыжевато-коричневых кристаллов. Т. пл. 122-123o C. После второй перекристаллизации получали 750 мг. Т. пл. 125-126,5oC.
Элементный анализ:
вычислено C 62,82, H 8,43, N 3,66;
найдено: C 63,09, H 8,22, N 3,
66.
Пример 17. Получение гидробромидной соли 2-ди-п-пропиламино-8(α-метилпропионил)-1,2,3,4-тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (1,
0 г, 3,2 ммоль) растворялся в 10 мл тетрагидрофурана и охлаждался до -78oC, после чего добавлялось 3,5 мл (1,0 М в гексане) н-бутиллития. Спустя 30 минут к полученной смеси добавлялось 0,41
мл (3,5 ммоль) метилизобутирата, смесь перемешивалась в течение 30 минут при -10oC и затем выливалась в 10%-ный водный раствор соляной кислоты, промывалась простым эфиром, и значение pH
доводилось до 10. Смесь затем экстрагировалась метиленхлоридом, и экстракт сушился над сульфатом натрия и упаривался, давая 0,72 г остатка.
Остаток помещался на силикагельную колонку и элюировался последовательно смесью гексана и простого эфира (4:1), содержащей следы гидроокиси аммония, и затем смесью гексана и простого эфира (3:1), содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 190 мг свободного основания целевого соединения.
Соединение превращалось в его гидробромидную соль и перекристаллизовывалось из этилацетата, давая 80 мг целевого соединения в виде рыжевато-коричневых кристаллов. Т. пл. -175-176,5oC.
Элементный анализ:
вычислено C 62,82, H 8,43, N 3,66;
найдено:
C 62,54, H 8,53, N 3,44.
Пример 18. Получение гидробромидной соли 2-ди-н-пропиламино-8-(b-метилбутирил)-1,2,3,4-тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1,2,3,
4-тетрагидронафталин (1,0 г 3,2 ммоль) растворялся в 10 мл тетрагидрофурана и охлаждался до -78oC, после чего добавлялось 3,5 н-бутиллития (1,0 М в гексане). Спустя 20 минут добавлялось 0,
53 мл (3,5 ммоль) этилизовалерата, и смесь нагревалась до -10oC и оставлялась стоять в течение 30 минут. Смесь затем выливалась в разбавленную кислоту, промывалась простым эфиром, и
показатель pH доводился до 10. Смесь экстрагировалась метиленхлоридом, и экстракт сушился над сульфатом натрия и упаривался, давая 0,83 г остатка.
Остаток помещался на колонку из силикагеля и элюировался последовательно смесью гексана и простого эфира (4:1), содержащей следы гидроокиси аммония, и затем смесью гексана и простого эфира (3:1), содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 50 мг свободного основания целевого соединения.
Свободное основание превращалось в гидробромидную соль, которая перекристаллизовывалась из смеси этилацетата и гексана, давая 30 мг целевого соединения в виде рыжевато-коричневого порошка. Т. пл. 131-132oC.
Элементный анализ:
вычислено C 63,63, H 8,64, N
3,53;
найдено: C 63,35, H 8,42, N 3,83.
Пример 19. Получение гидробромидной соли 2-ди-н-пропиламино-8- диметилпропионил-1,2,3,4-тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (1,0 г 3,2 ммоль) растворялся в 20 мл тетрагидрофурана и охлаждался до -78oC, после чего добавлялось 4,7 мл н-бутиллития (0,82 М в
гексане). Смесь перемешивалась в течение 30 минут при -78oC, после чего добавлялось 0,56 мл (4,2 ммоль) метилтриметилацетата. Смесь оставлялась нагреваться до комнатной температуры и затем
выливалась в воду и экстрагировалась метиленхлоридом. Экстракт сушился над сульфатом натрия и упаривался, давая 1,6 г остатка.
Остаток помещался на силикагельную колонку и элюировался смесью гексана и простого эфира (3:1), содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 140 мг свободного основания целевого соединения.
Свободное основание превращалось в гидробромидную соль и перекристаллизовывалось из смеси метанол/этилацетат, давая 80 мг целевого соединения. Т. пл. 157-158oC.
Элементный анализ:
вычислено C 63,63, H 8,65, N 3,53;
найдено C 63,39, H 8,46, N 3,43.
Пример 20. Получение оксалатной соли 2-ди-н-пропиламино-8- циклогексанкарбонил-1,2,3,
4-тетрагидронафталина
Способ A: 2-ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (1,0 г, 3,2 ммоль) растворялся в 10 мл тетрагидрофурана и охлаждался до -78oC, после чего
добавлялось 2,8 мл н-бутиллития (1,27 М в гексане). Смесь перемешивалась при -78oC в течение 45 минут, после чего добавлялось 0,59 мл (3,5 ммоль) этилциклогексанкарбоксилата. Смесь
нагревалась до комнатной температуры и затем выливалась в 10% -ный раствор соляной кислоты, промывалась простым эфиром, величина pH доводилась до 10 с помощью гидроокиси аммония, и смесь
экстрагировалась метиленхлоридом. Экстракт сушился над сульфатом натрия и упаривался, давая 0,8 г остатка.
Остаток помещался на силикагельную колонку и элюировался смесью гексана и простого эфира (3:1), содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 0,36 г целевого соединения.
Способ B: К раствору 8-бром-2-ди-н-пропиламино-1,2,3, 4-тетрагидронафталина (1,0 г, 3,2 ммоль) в тетрагидрофуране (10 мл) добавлялся бутиллитий (1,2 М в гексане 3,0 мл, 3,5 ммоль) при температуре -78oC, и смесь перемешивалась в течение 45 минут. Добавлялся циклогексанкарбоксальдегид (0,47 мл, 3,9 ммоль). Реакционная смесь перемешивалась при -78oC в течение 5 минут, нагревалась до комнатной температуры, выливалась в разбавленный раствор соляной кислоты и промывалась простым эфиром. Водный слой подщелачивался NH4OH и экстрагировался метиленхлоридом. Экстракт сушился (сернокислый натрий) и концентрировался, давая 1,1 г неочищенного сырого продукта. Сырой продукт растворялся в метиленхлориде (50 мл), и добавлялись молекулярные сита и пиридинийхлорформат (1,4 г, 6,4 ммоль). Реакционная смесь перемешивалась при комнатной температуре в течение двух часов. Добавлялся метанол (50 мл), и реакционная смесь концентрировалась, давая шлам. Шлам растворялся в метиленхлориде (50 мл), добавлялось достаточное количество простого эфира, давая мутный раствор. Это вещество добавлялось на колонку из силикагеля и элюировалось простым эфиром.
Силикагельная колонка элюировалась 10%-ным метанолом в метиленхлориде, и элюент концентрировался, давая маслянистый остаток. Данное вещество растиралось с метанолом и фильтровалось через целит. Фильтрат объединялся с эфирным раствором, полученным выше, и концентрировался. Вещество растворялось в метиленхлориде. Добавлялся простой эфир до тех пор, пока раствор не становился мутным, и затем раствор фильтровался через флорисил. Фильтрат концентрировался, давая 560 мл масла, которое очищалось мгновенной хроматографией на силикагеле с использованием смеси гексан/простой эфир (3:1), содержащей следы NH4OH, в качестве растворителя. Соответствующие фракции объединялись и концентрировались, давая 350 мг желаемого соединения. Образовывалась оксалатная соль и кристаллизовывалась из смеси этилацетат/гексан, давая 370 мг белого твердого вещества. Т. пл. - 98,5-100oC.
Элементный анализ:
вычислено C 69,58, H 8,64, N 3,35;
найдено C 69,28, H 8,
86, N 3,00.
Пример 21. Получение 2-ди-н-пропиламино-8-(п-хлорбензоил)-1,2,3,4- тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (1,0 г, 3,2 ммоль)
растворялся в 10 мл ТГФ и охлаждался до -78oC, после чего добавлялось 3,5 мл н-бутиллития (1,0 М в гексане). Смесь перемешивалась в течение одного часа при -78oC, после чего
добавлялось 680 мг (1,5 эквивалента) 4-хлорбензальдегида в ТГФ. Смесь перемешивалась в течение 15 минут при -78oC, а затем оставлялась подогреваться до комнатной температуры. Смесь
выливалась в 10%-ный водный раствор соляной кислоты, промывалась эфиром, величина pH доводилась до 10 с помощью гидроокиси аммония, и раствор экстрагировался метиленхлоридом. Экстракт сушился над
сульфатом натрия и выпаривался, давая 1,5 г остатка.
Остаток помещался на силикагельную колонку и элюировался смесью гексана и этилацетата(1: 1), содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 1,3 г по существу чистого 2-ди-п-пропиламино-8-(альфа-метил-4'-хлорбензил)-1,2,3,4-тетрагидронафталина.
Вышеуказанный продукт (3,2 ммоль) растворялся в 50 мл метиленхлорида, и добавлялось 30 г 4
MС (FD) m/e 369.
Пример 22. Получение п-толуолсульфонокислотной соли 2-ди-н-пропиламино-8-(о-фторбензоил)-1,2,3,4-тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (1,0 г, 3,22 ммоль) растворялся в ТГФ (25 мл) и охлаждался до -78oC и добавлялось 2,5 мл н-бутиллития (1,27 М в гексане). Спустя один
час добавлялся о-фторбензоилхлорид (0,38 г, 3,22 ммоль). Смесь перемешивалась в течение 10 минут при -78oC, после чего реакция гасилась добавлением воды при -78oC. Реакционная
смесь выливалась в разбавленный раствор HCl и экстрагировалась метиленхлоридом. Водный слой подщелачивался гидроокисью натрия и экстрагировался метиленхлоридом. Основной экстракт сушился (сульфатом
натрия) и концентрировался, давая 200 мг остатка, который по данным ЯМР не содержал продукта. Экстракт из кислотного материала сушился (сульфатом натрия) и концентрировался, давая 2,0 г остатка.
Очистка данного материала с помощью флэш-силикагельной хроматографии с использованием смеси эфир/гексан (1:1), содержащей следы гидроокиси аммония в качестве растворителя, давала свободное основание
целевого соединения (340 мг). Соль 130 мг данного вещества с п-толуолсульфокислотой приготавливалась и кристаллизовалась из смеси этилацетат/эфир, давая 118 мг целевого соединения. Т. пл. 107-109oC.
Элементный анализ:
вычислено C 68,55, H 6,90, N 2,66;
найдено: C 68,41, H 7,02, N 2,65.
Пример 23. Получение
2-ди-циклопропилметиламино-8-ацетил-1,2,3,4-тетрагидронафталина
а. 2-Ди-циклопропилметиламино-8-бром-1,2,3,4-тетрагидронафталин
Раствор 8-бром-2-тетралона (9,0 г, 40 ммоль),
дициклопропилметиламина (10 г, 80 ммоль) и птолуолсульфоновой кислоты (0,1 экв. 4 ммоль, 760 мг) в толуоле (200 мл) нагревался до температуры дефлегмации в течение 5 часов, при этом вода собиралась в
ловушку Дина-Старка. Реакционный раствор затем концентрировался в вакууме, и получающийся в результате остаток брался в тетрагидрофуран (200 мл), и добавлялся цианоборгидрид натрия NaCNBH3
(50 ммоль, 3 г). Добавлялся газообразный HCl до тех пор, пока реакционный раствор не насыщался. Раствор затем перемешивался при комнатной температуре на протяжении ночи, выливался в ледяную воду и
подщелачивался раствором гидроокиси натрия. Получающийся раствор экстрагировался метиленхлоридом, экстракт сушился (сульфатом натрия) и затем концентрировался в вакууме, давая коричневое масло. Данное
масло растворялось в тетрагидрофуране (200 мл), и добавлялся триэтиламин (25 мл). Получающийся раствор нагревался до дефлегмации и фильтровался через слой основной окиси аммония. Окись алюминия
прополаскивалась метиленхлоридом. Объединенные фильтраты концентрировались, давая 11,8 г неочищенного соединения в виде масла. Очистка данного неочищенного вещества с помощью флэш-хроматографии
(эфир/гексан (2:1), гидроокись аммония) давала 8,6 г целевого продукта в виде желтого масла.
1ЯМР (CDCl3): δ 7,4 (д, 1 H), 7,03 (д, 1 H), 6,98 (т, 1 H), 3, 42-3,29 (м, 1 H), 3,16-3,00 (м, 1 H), 3,00-2,78 (м, 2 H), 2,78-2,44 (м, 5 H), 2,15-1,97 (м, 1 H), 1,67-1,45 (м, 1 H), 10,4-0,87 (м, 2 H), 0,64-0,48 (м, 4 H), 0,28-0,10 (м, 4 H).
MC (FD) m /e 333 (100, M+), 355 (97, M+).
Элементный анализ для C18H24 Br:
вычислено C 64,67, H 7,24, N 4,19;
найдено C 64,47,
H 7,32, N 4,28.
b. 2-Дициклопропилметиламино-8-(1-гидроксиэтил)-1,2,3,4- тетрагидронафталин
Раствор н-бутиллития в гексане (1,37 М, 5,65 мл, 7,7 ммоль) добавлялся к раствору
2,0 г (6,45 ммоль) соединения, полученного на стадии "a", в тетрагидрофуране при -78oC. Получающийся раствор затем перемешивался при -78oC в течение 20 минут, и добавлялся
ацетальдегид (12 ммоль, 530 мг). После перемешивания при -78oC в течение 10 минут реакционный раствор подогревался до комнатной температуры, выливался в воду, а затем экстрагировался
метиленхлоридом. Экстракт сушился (сульфатом натрия) и концентрировался в вакууме, давая 2,3 г оранжевого масла. Очистка данного масла с помощью флэш-хроматографии (эфир/гексан (2:1), гидроокись
аммония) давала 1,53 г целевого продукта.
1H ЯМР (CDCl3): d 7,40 (т, 1 H), 7,20 (т, 1 H), 7,04 (д, 1 H), 5,24-5,10 (м, 1 H), 3,55-3,30 (м, 1 H), 3,20-2,40 (м, 7 H), 2,18-2,03 (м, 1 H), 1,80-1,40 (м, 5 H), 1,30 (шир.с, 1 H), 1,18-0,80 (м, 2 H), 0,65-0,48 (м, 4 H), 0,27-0,10 (м, 4 H).
с. 2-Дициклопропилметиламино-8-ацетил-1,2,3,
4-тетрагидронафталин
Реактив Джонса (1,9 мл) добавлялся к раствору 1,5 г (4,36 ммоль) соединения, полученного на стадии "b", в смеси ацетона (50 мл) и 2 М серной кислоты (16 мл) со скоростью,
медленной настолько, чтобы поддерживалась температура реакционного раствора ниже 30oC. Получающийся раствор перемешивался при комнатной температуре в течение 20 минут, а затем медленно
гасился добавлением изопропанола (0,5 мл). Образовавшийся осадок отфильтровывался, и фильтрат добавлялся к воде. Водный раствор экстрагировался смешанным раствором изопропанол/хлороформ (1: 3),
подщелачивался раствором гидроокиси натрия, а затем экстрагировался метиленхлоридом. Объединенные органические слои фильтровались через целит, промывались водой, сушились (сульфатом натрия), а затем
концентрировались, давая 1,5 г серого масла. Данное масло очищалось с помощью флэш-хроматографии (5%-ный метанол в метиленхлориде,гидроокись аммония), давая 1,2 г желтого масла. Дополнительная очистка
с помощью флэш-хроматографии (эфир/гексан (4:1), гидроокись аммония) давала 1,0 г целевого продукта.
1H ЯМР (CDCl3): d 7,48 (д, 1 H), 7,24-7,16 (м, 2 H), 3,35-3,18 (м, 1 H), 3,16-3,03 (м, 1 H), 3,03-2,84 (м, 3 H), 2,64-2,46 (м, 3 H), 2,57 (с, 3 H), 2,16-1,95 (м, 1 H), 1,75-1,55 (м, 2 H), 1,02-0,88 (м, 2 H), 0,57-0,47 (м, 4 H), 0,22-0,08 (м, 4 H).
Пример 24. Получение хлоргидрата диметил-8-(о-метоксибензоил)-1,2,3,4-тетрагидронафталина
a. 2-Диметил-8-(-гидрокси-2-метоксибензил)-1,2,3,4- тетрагидронафталин
Раствор н-бутиллития
в гексане (1,42 М, 14,54 мл, 20,65 ммоль) добавлялся к раствору 3,5 г (13,77 ммоль) 2-диметил-8-бром-1,2,3,4-тетрагидронафталина в тетрагидрофуране (300 мл) при -78oC. Получающийся раствор
затем перемешивался при -78oC в течение 10 минут, и добавлялся о-анисальдегид (27,54 ммоль, 3,75 г). После перемешивания при -78oC в течение 60 минут реакционный раствор
подогревался до комнатной температуры, выливался в воду, а затем экстрагировался смесью хлороформ/изопропанол (3:1). Экстракт сушился (сульфатом натрия) и концентрировался в вакууме, давая 7,83 г
желтовато-белого твердого вещества. Очистка данного твердого вещества с помощью флэш хроматографии (10% метанол в метиленхлориде (гидроокись аммония)) давала 2,54 г целевого продукта в виде белого
твердого вещества.
b. 2-Диметил-8-(о-метоксибензоил)-1,2,3,4-тетрагидронафталихлоргидрат
Реактив Джонса (3,0 мл) добавлялся к раствору 2,54 г соединения полученного выше в
смеси ацетона (100 мл) и 2 М серной кислоты (24,48 мл) со скоростью, достаточно медленной для поддержания температуры реакционного раствора ниже 30oC. Получающийся раствор перемешивался при
комнатной температуре до тех пор, пока тонкослойная хроматография не указывала на то, что реакция завершилась, и затем гасился добавлением изопропанола (2 мл). Получающийся раствор выливался в воду, а
затем подщелачивался раствором гидроокиси натрия. Основной раствор экстрагировался смесью изопропанол/хлороформ (1:3) в виде раствора, сушился над сульфатом натрия, а затем концентрировался в вакууме,
давая 2,599 г целевого неочищенного соединения. Данный неочищенный продукт очищался с помощью флэш-хроматографии (5%-ный метанол в метиленхлориде, гидроокись натрия), давая 1,781 г целевого масла.
Данное масло растворялось в метаноле, а затем при перемешивании добавлялось 5,8 мл 1 н. раствора соляной кислоты. Получающийся раствор затем концентрировался в вакууме, давая белую пену. Данная пена
повторно растворялась в гексане. После того, как пена полностью растворялась, добавлялся этилацетат медленно, вызывая осаждение (после обработки ультразвуком) белого осадка. Данный осадок выделялся
фильтрованием, давая 1,46 г целевого соединения Т. пл. 170-172oC.
Анализ, вычисленный для C20H23NO2•HCl:
вычислено: C 69,45,
H 6,99, N 4,05;
найдено: C 69,20, H 7,08, N 4,07.
Пример 25. 2-Ди-н-пропиламино-8-(1-метилпиразол-3-ил)-1,2,3,4- тетрагидронафталин, малеатная соль и
2-ди-н-пропиламино-8-(1-метилпиразол-5-ил)-1,2,3,4- тетрагидронафталин, бромгидратная соль
Раствор -н-бутиллития (1,6 М в гексане 15,1 мл, 24,2 ммоль) добавлялся к раствору
8-бром-2-ди-н-пропиламино-1, 2,3,4-тетрагидронафталина (5,0 г, 16,1 ммоль) в ТГФ (50 мл) при -48oC, и реакционная смесь перешивалась при -78oC в течение одного часа. Через
реакционную смесь барботировалась газообразная двуокись углерода при -78oC до тех пор, пока не исчезал темно-фиолетовый цвет, который образуется. Добавлялся метиллитий (1,4 М в эфире, 23
мл). Реакционная смесь перемешивалась при -78oC в течение 30 минут и подогревалась до комнатной температуры. Реакционная смесь перемешивалась в течение дополнительных десяти минут при
комнатной температуре, за это время розовая окраска терялась. Добавлялось дополнительно 10 мл раствора метиллития, и реакционная смесь снова становилась розовой. Спустя 15 минут розовый цвет терялся,
и добавлялось дополнительно 10 мл раствора метиллития. Реакционная смесь выливалась на лед, подкислялась соляной кислотой и экстрагировалась эфиром. Водный слой подщелачивался и экстрагировался
метиленхлоридом. Основные экстракты сушились (сульфатом натрия) и концентрировались, давая 3,8 г неочищенного продукта. Очистка с помощью силикагельной флэш-хроматографии с использованием смеси
гексан/эфир (2:1), содержащей следы гидроокиси аммония, давала 2-ди-н-пропиламино-8-ацетил-1,2,3,4-тетрагидронафталин в виде желтого масла (2,7 г, 61%).
2-Ди-н-пропиламино-8-ацетил-1,2, 3,4-тетрагидронафталин (3,0 г, 11,0 ммоль) растворялся в 125 мл толуола, после чего добавлялось 4,6 мл (27,5 ммоль) трис-(диметиламино)метана. Смесь нагревалась до 80oC на протяжении ночи, после чего она выпаривалась, и остаток растворялся в 100 мл метанола. Добавлялся метилгидразин (2,9 мл, 54,9 ммоль). Смесь нагревалась с обратным холодильником в течение шести часов, а затем перемешивалась при комнатной температуре на протяжении ночи. Смесь затем выливалась в воду, и водная смесь экстрагировалась метиленхлоридом. Метиленхлоридный экстракт сушился над сульфатом натрия и выпаривался, давая 3,7 г остатка, который содержал оба целевых соединения.
Остаток помещался на силикагельную колонку и элюировался смесью 2%-ного метанола в метиленхлориде, содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 2,1 г основного изомера 2-дин-н-пропиламино-8-(1-метилпиразол-3-ил)-1,2,3,4-тетрагидронафталин (Rf 0,31 в 2%-ном метаноле в метиленхлориде, содержащем следы гидроокиси аммония). Данное вещество превращалось в малеатную соль, и соль перекристаллизовывалась из смеси этанола и эфира, давая 2,3 г белых кристаллов. Т. пл. 139,5-140,5oC. MC(FD) 311 (100).
Анализ:
вычислено: C 67,42, H 7,78, N 9,83;
найдено: C 67,62, H 7,81, N 9,80.
Соответствующие фракции объединялись, давая 165 мг второстепенного изомера.
2-Дин-н-пропиламино-8-(1-метилпираэол-5-ил)-1,2,3,4- тетрагидронафталин (Rf 0,27 в 2%-ном метаноле в метиленхлориде, содержащем следы гидроокиси аммония). Образовывалась бромгидратная соль данного вещества и перекристаллизовывалась из смеси метанола и этилацетата, давая 30 мг твердого вещества. Т. пл. 203-204oC. MC(FD) 311 (100).
Анализ:
вычислено: C 50,76, H 6,60, N 8,88;
найдено: C 50,09, H 6,61, N 8,65.
Пример 26. Получение
2-ди-н-пропиламино-8-(5-метоксипиразол-3-ил)-1,2,3,4-тетрагидронафталина
Раствор 20 ммоль диазометана получался путем добавления 4,29 (29 ммоль) 1-метил-3-нитро-1-нитрозогуанидина к 25%-ному
раствору KOH (10 мл) и Et2O на ледяной бане. Эфирная фаза декантировалась в раствор 25 мл метанола, содержащего 400 мг (1,28 ммоль) 2-ди-н-пропиламино-8-(5-гидрокипиразол-3-ил)-1,2,3,
4-тгидронафталина. Целевое соединение изолировалось путем концентрации реакционной смеси и очистки 500 мг коричневого масла (сырого) мгновенной хроматографией на колонке, элюирование смесью
метиленхлорида в метаноле (9:1).
Пример 27. Получение хлоргидратной соли 2-ди-п-пропиламино-8-(тиазол-2-ил)-1,2,3,4-тетрагидронафталина
Тиазол (0,46, 6,5 ммоль) растворялся в
10 мл тетрагидрофурана, и смесь охлаждалась до -78oC, после чего добавлялся н-бутиллитий (6,5 ммоль, 1,0 М в гексане). Смесь нагревалась до -20oC и затем охлаждалась снова до
-78oC. Полученная в результате смесь медленно добавлялась к дефлегмирующей смеси 1,0 г (3,2 ммоль) 2-ди-п-пропиламино-8-бром-1,2,3,4-тетрагидронафталина и 370 мг (0,3 ммоль) Pd (Ph3P)4 в 50 мл толуола. К смеси добавлялись дополнительные 10 мл тетрагидрофурана для достижения полного перехода веществ в реакционную смесь. Если реакция и была заметна, то очень
незначительно. Чувствовалось, что анион тиазола или вообще не образовывался, или разлагался. Еще два эквивалента тиазола обрабатывались N-бутиллитием в 20 мл ТГФ при -78oC в течение 30
минут. Получающаяся смесь светло-желтая суспензия добавлялась к дефлегмирующей реакционной смеси, и смесь нагревалась с обратным холодильником в течение 45 минут и перемешивалась на протяжении ночи
при комнатной температуре. Реакционная смесь, теперь черная, выливалась в 10%-ную водную соляную кислоту и промывалась простым эфиром. Водный слой отделялся, подщелачивался гидроокисью аммония и
экстрагировался метиленхлоридом. Экстракт сушился над сульфатом натрия и выпаривался, давая 1,6 г темно-коричневого масла.
Масло помещалось на силикагельную колонку и элюировалось градиентной смесью 3:1 до 1:1 гексана и эфира, содержащей следы гидроокиси аммония. Фракции 12-16 объединялись, давая 120 мг коричневого масла.
Масло превращалось в бромгидратную соль, и соль перекристаллизовывалась из смеси метанола и этилацетата, давая 45 мг целевого соединения в виде рыжевато-коричневых кристаллов.
Анализ:
вычислено: C 47,91, H 5,93, N
5,88;
найдено: C 47,62, H 6,10, N 6,15.
Пример 28. Получение 2-ди-н-пропиламино-8-(тиазол-4-ил)-1,2,3,4- тетрагидронафталин, п-толуолсульфатной соли
К 5 мл 85%-ной
фосфорной кислоты добавлялось 0,87 г (2,6 ммоль) 2-ди-п-пропиламино-8-(2-аминотиазол-4-ил)-1,2,3,4-тетрагидронафталина (очень незначительно растворенного). Растворитель затем менялся добавлением 20 мл
35% -ной серной кислоты. Смесь охлаждалась до 0oC, и с помощью шприца ниже поверхности реакционной смеси добавлялся концентрированный водный раствор 460 мг (6,6 ммоль) нитрита натрия. Смесь
медленно добавлялась к 20 мл 50%-ной гипофосфорной кислоты при 0oC, и реакционная смесь затем подогревалась до комнатной температуры в течение одного часа. Смесь выливалась на лед, и
величина pH доводилась до 11 с помощью гидроокиси аммония. Смесь экстрагировалась метиленхлоридом, и экстракт сушился над сульфатом натрия и выпаривался, давая 0,7 г остатка.
Остаток помещался на силикагельную колонку и элюировался смесью гексана и эфира (2:1), содержащей следы гидроокиси аммония. Соответствующие фракции объединялись, давая 350 мг масла. Данное вещество превращалось в п-толуолсульфонатную соль и перекристаллизовывалось из смеси этилацетата и гексана, давая 220 мг рыжевато-коричневого твердого вещества. Т. пл. - 143-144oC. (FD) 315 (100).
Анализ (1/3 H2O):
вычислено: C 63,38, H 7,09, N 5,69;
найдено: C 63,29, H 7,01, N 5,67.
Пример 29. Получение
2-ди-н-пропиламино-8-(хинол-2-ил)-1,2,3,4- тетрагидронафталина
2-Ди-н-пропиламино-8-бром-1, 2, 3, 4-тетрагидронафталин (1 г, 3,22 ммоль) добавлялся к дефлегмирующей смеси магниевых стружек
(120 мг, 4,9 ммоль) в 50 мл ТГФ. Образование реактива Гриньяра инициировалось добавлением 0,2 мл 1,2-дибромэтана (0,002 ммоль). Спустя 1,5 часа добавлялось 220 мг (0,33 ммоль) Ni(PPh3)2Cl2 в 10 мл ТГФ с последующим добавлением 800 мг (4,89 ммоль) 2-хлорхинолина. Раствор продолжал нагреваться с обратным холодильником в течение 15 минут, после чего он охлаждался,
выливался в воду и экстрагировался три раза метиленхлоридом. Экстракт сушился сульфатом натрия и концентрировался, давая 2,1 г черного масла. Неочищенное вещество очищалось с использованием двух
колонок для флэш-хроматографии. Первая элюировалась смесью гексан/простой эфир (1:1), а вторая смесью метилен хлорид/метанол (20:1). В качестве продукта собиралось 130 мг целевого соединения. После
кристаллизации в этаноле и воде выделялось 66 мг твердого вещества. Т. пл. 82oC.
Анализ (1/3 H2O):
вычислено: C 82,37, H 8,48, N 7,68;
найдено:
C 82,33, H 8,44, N 7,73.
Пример 30. Получение 2-ди-н-пропиламино-8-(хинол-3-ил)-1,2,3,4- тетрагидронафталина, бромгидратной соли
К дефлегмирующей смеси 520 мг (21 ммоль)
магния в 50 мл ТГФ добавлялось 5 г (16,13 ммоль) 2-ди-н-пропиламино-8- бром-1,2,3,4-тетрагидронафталина. Реактив Гриньяра образовался спустя 2 часа после нагревания с обратным холодильником, и затем с
помощью шприца добавлялось 1,05 г (1,6 ммоль) Ni(PPh3)2Cl2 в 25 мл ТГФ. Спустя 5 минут добавлялся 3-бромхинодин (3 мл, 21 ммоль) в 25 мл ТГФ. Реакционная смесь
продолжала нагреваться с обратным холодильником в течение одного часа.
После охлаждения раствор выливался в водный раствор бикарбоната натрия и экстрагировался три раза метиленхлоридом. После сушки сульфатом натрия и концентрирования получалось 7,2 г черного масла. Данное вещество очищалось с помощью флэш-хроматографии на колонке с элюированием смесью эфир/гексан (1: 1).
Часть вещества (500 мг) использовалась для получения HBr соли. После кристаллизации из метанола и эфира выделялось 423 мг. Т. пл. 200oC.
Анализ для 2
HBr соли:
вычислено: C 57,71, H 6,23, N 5,38;
найдено: C 57,75, H 6,27, N 5,34.
Пример 31. Получение 2-ди-н-пропиламино-8-(пирил-3-ил)-1,2,3,4- тетрагидронафталина,
оксалатной соли
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (5 г, 16,12 ммоль) в 50 мл ТГФ медленно добавлялся к дефлегмирующей смеси 500 мг (20 ммоль) магния в 50 мл ТГФ.
Инициирование образования реактива Гриньяра приводилось добавлением капли 1,2-дибромэтана. Нагревание с обратным холодильником продолжалось в течение 2 часов перед добавлением 1,05 г (1,6 ммоль)
Ni(PPh3)2Cl2 в 25 мл ТГФ и 3 мл (31,7 ммоль) 3-бромпиридина в 25 мл ТГФ. После нагревания с обратным холодильником в течение дополнительных 15 минут смесь охлаждалась,
выливалась в бикарбонат натрия и экстрагировалась метиленхлоридом. Экстракты сушились сульфатом магния и концентрировались, давая 10,2 г черного масла.
Неочищенный продукт очищался с помощью флэш-хроматографии на колонке с использованием смеси эфир/гексан (1:1) в качестве растворителя, давая 1,05 г свободного основания целевого соединения.
Образовывалась соль щавелевой кислоты и кристаллизовалась из смеси ацетон/эфир, давая 173 мг продукта. Т. пл. 135oC.
Анализ:
вычислено: C 69,32, H 7,59, N 7,03;
найдено: C 68,
94, H 7,62, N 6,90.
Пример 32. Получение 2-ди-н-пропиламино-8-(пирил-2-ил)-1,2,3,4- тетрагидронафталина, оксалатной соли
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин
(5 г, 16,72 ммоль) в 50 мл ТГФ медленно добавлялся к дефлегмирующей смеси 500 мг (20 ммоль) магниевых стружек в 50 мл, ТГФ. Инициирование образования реагента Гриньяра производилось добавлением капли
1,2-диброметана. Нагревание с обратным холодильником продолжалось в течение 2 часов перед добавлением 1,05 г (1,6 ммоль) Ni(PPh3)2Cl2 ТГФ и 3 мл (31,7 ммоль)
2-бромпиридина в 25 мл ТГФ. Реакционная смесь нагревалась с обратным холодильником в течение дополнительных 15 минут, перед тем как она выливалась при охлаждении в воду, и экстрагировалась
метиленхлоридом. Экстракты сушились сульфатом натрия и концентрировались, давая 10,5 г черного масла.
Очистка проводилась с помощью флэш-хроматографии на колонке с элюированием смесью гексан/эфир (1: 1), давая 1,5 г свободного основания целевого соединения. Образовывалась соль щавелевой кислоты и кристаллизовались из смеси ацетон/эфир, давая 215 мг коричневого порошка. Т. пл. 135oC.
Анализ:
вычислено C 69,32, H 7,59, N 7,03;
найдено: C 69,12, H 7,64, N 6,88.
Пример 33. Получение
2-ди-н-пропиламино-8-(пирид-4-ил)-1,2,3,4- тетрагидронафталина, оксалатной соли
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (5 г, 16,12 ммоль) в 50 мл ТГФ медленно добавлялся к
дефлегмирующей смеси 500 мг (20 ммоль) магниевых стружек в 50 мл ТГФ. Инициирование образования реагента Гриньяра вызывалось каплей 1,2-дибромэтана. Нагревание с обратным холодильником продолжалось в
течение 2 часов перед добавлением 105 г (1,6 ммоль) Ni(PPh3)2Cl2 в 25 мл ТГФ и 3 мл (31,2 ммоль) 4-хлорпиридина в 25 мл ТГФ. Реакционная смесь нагревалась с обратным
холодильником в течение дополнительных 1,5 часов перед охлаждением, выливанием в воду и экстрагированием метиленхлоридом. Экстракты сушились сульфатом магния и концентрировались, давая 9,7 г черного
масла.
Очистка проводилась с помощью флэш-хроматографии на колонке при элюировании смесью гексан/эфир (1:1). Свободное основание целевого продукта (850 г) выделялось. Получалась соль щавелевой кислоты и кристаллизовалась в смеси метанол/эфир. Т. пл. 191oC.
Анализ:
вычислено: C 69,32, H 7,59, N 7,03;
найдено: C 69,47, H 7,55, N 6,99.
Пример 34. Получение 2-ди-н-пропиламино-8-(индоксазин-3-ил)-1,2,3,4- тетрагидронафталина, хлоргидратной соли
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (1,0 г, 3,22
ммоль), растворенный в ТГФ (25 мл), охлаждался до -78oC, и добавлялось 2,5 мл н-бутиллития (1,27 М в гексане). Спустя один час добавлялся о-фторбензоилхлорид (0,38 мл 3,22 ммоль). Смесь
перемешивалась в течение 10 минут при -78oC, после чего реакционная смесь гасилась добавлением воды при -78oC. Реакционная смесь подщелачивалась гидроокисью натрия и
экстрагировалась три раза метиленхлоридом. Основной экстракт сушился (сульфатом натрия) и концентрировался, давая 2,0 г неочищенного остатка. Очистка данного вещества с помощью флэш-хроматографии на
силикагеле с использованием смеси эфира и гексана (1: 1), содержащей следы гидроокиси аммония, в качестве растворителя давала 2-ди-н-пропиламино-8-(2-фторбензоил)-1,2,3,4-тетрагидронафталин (340
мл).
К 47,5 мг ацетон-оксима (0,65 ммоль) в 25 мл ТГФ добавлялось 73 мг (0,66 ммоль) трет-бутилата калия. Смесь перемешивалась при комнатной температуре в течение одного часа, после чего с помощью шприца добавлялось 210 мг 2-ди-н-пропиламино-8-(2'-фторбензоил)-1,2,3,4-тетрагидронафталина в ТГФ. Получающаяся смесь нагревалась с обратным холодильником в течение 3 часов, после чего она охлаждалась и выливалась в водный раствор хлористого аммония. Смесь затем экстрагировалась эфиром, и экстракт сушился и концентрировался, давая 343 мг 2-ди-н-пропиламино-8-[2-[(изопропилиденамино)окси]бензол]- 1,2,3,4-тетрагидронафталина.
Предыдущее соединение перемешивалось при нагревании с обратным холодильником в течение 2,5 часов в смеси 10 мл 5%-ной соляной кислоты и 10 мл этанола, после чего она перемешивалась при комнатной температуре на протяжении ночи. Смесь затем выливалась в воду, подщелачивалась бикарбонатом натрия и экстрагировалась метиленхлоридом. Экстракт сушился над сульфатом магния и концентрировался, давая 219 мг нечистого продукта.
Остаток очищался с помощью флэш-хроматографии на колонке с использованием смеси метиленхлорида и метанола (10:1) в качестве элюента, давая 121 мг свободного основания целевого соединения.
Свободное основание превращалось в хлоргидратную соль, которая перекристаллизовывалась из смеси этилацетата и эфира, давая 65 мг твердого вещества. Т. пл. 186oC.
Пример 35. Получение
2-ди-н-пропиламино-8-(пиразол-8-ил)-1,2,3,4- тетрагидронафталина, оксалатной соли
2-Ди-н-пропиламино-8-(1-оксо-3-диметиламино)-пропенил)-1,2,3,4-тетрагидронафталин, полученный, как описано в
примере 8, (0,75 г, 2,29 ммоль) растворялся в 10 мл метанола. К смеси добавлялось 0,16 мл гидразина, и смесь перемешивалась при комнатной температуре в атмосфере азота в течение 18 часов, после чего
летучие вещества удалялись в вакууме, давая темно-оранжевый остаток. Остаток растворялся в эфире и помещался на флэш-колонку из двуокиси кремния. Колонка элюировалась эфиром, содержащим следы
гидроокиси аммония.
Фракции 9-13 собирались, объединялись и концентрировались в вакууме, давая 0,39 г бесцветного вязкого масла. Масло превращалось в оксалатную соль, которая кристаллизовалась из смеси этанола и эфира, давая 0,26 г целевого соединения в виде бесцветных кристаллов. Т. пл. -143-150oC.
Анализ:
вычислено: C 65,10, H 7,54, N
10,84;
найдено: C 65,33, H 7,44, N 10,83.
Пример 36. Получение 8-(индол-3-ил)-2-дипропиламино-тетралиноксалата
A. 1-Фенилсульфонилиндол
Гидроокись натрия
(50 г, 1,25 ммоль), кислый сульфат тетрабутиламмония (3,5 г, 10,3 ммоль) и индола (46,75 г, 0,4 ммоль) растворялись в 500 мл охлажденного (0oC) метиленхлорида. Затем добавлялся медленно
раствор фенилсульфонилхлорида (63,8 мл, 0,5 ммоль), растворенного в 300 мл метиленхлорида. Получающийся раствор оставлялся подогреваться до комнатной температуры, а затем перемешивался в течение 4
часов. Спустя 4 часа осадки, которые образовались во время реакции, удалялись с помощью фильтрования, и получающийся фильтрат концентрировался в вакууме, давая оранжевое масло. Данное масло
растворялось в горячем метаноле, и получающийся раствор оставлялся охлаждаться до комнатной температуры на протяжении ночи. На следующее утро оранжевые твердые вещества, которые выпали в осадок из
метанольного раствора, удалялись фильтрованием. Данные твердые вещества снова растворялись в горячем метаноле. Метанольный раствор снова оставлялся охлаждаться на протяжении ночи, в течение которой в
осадок выпадали белые твердые вещества. Эти вещества выделялись фильтрованием, давая 50 г целевого продукта в виде белых кристаллов.
B. 1-Фенилсульфонил-З-броминдол
Часть
соединения, полученного выше (14,77 г, 57,1 ммоль), растворялась в 150 мл метиленхлорида. Затем добавлялся к индольному раствору на протяжении 15 минут бром (8,2 г, 51,38 ммоль), растворенный в 50 мл
метиленхлорида. Получающийся раствор перемешивался в течение 4 часов, после обработки получалось 17,3 г белого порошка. Данный белый порошок анализировался как целевое соединение. Т. пл. 120oC.
C. 8-(1-Фенилсульфонилиндол-3-ил)-2-дипропиламинотетралин
Реакция Гриньяра начиналась путем добавления раствора 8-бром-2-дипропиламинотетралина (5,0 г, 16,13 ммоль)
в 50 мл тетрагидрофурана к раствору дефлегмирующего тетрагидрофурана (50 мл), содержащего магний. После 2 часов перемешивания получающегося раствора при нагревании с обратным холодильником к
дефлегмирующему раствору добавлялся раствор NiCl2(PPh3)2 тетрагидрофурана с последующим добавлением раствора 10,8 г (32,26 ммоль) соединения, полученного на стадии B,
в 15 мл тетрагидрофурана. Получающийся раствор затем перемешивался при нагревании с обратным холодильником в течение дополнительного часа и охлаждался до комнатной температуры. После охлаждения
реакционная смесь гасилась выливанием реакционного раствора в воду, и получающийся водный раствор экстрагировался метиленхлоридом. Органический экстракт промывался насыщенным солевым раствором,
сушился над сульфатом натрия, а затем концентрировался досуха в вакууме, давая 13 г твердого материала, который по данным анализа представлял неочищенное целевое соединение. Данный неочищенный
материал очищался с помощью хроматографии с использованием 3%-ного метанола в метиленхлориде в качестве элюента, давая 1,35 г целевого соединения в виде коричневого масла.
D.
8-(Индол-3-ил)-2-дипропиламинотетралиноксалат
Масло, полученное на стадии C, растворялось в 100 мл метанола и 40 мл 1 н. раствора гидроокиси натрия. Получающийся раствор затем нагревался до
дефлегмации и перемешивался в условиях дефлегмации в течение 6 часов. Спустя 6 часов реакционный раствор охлаждался до комнатной температуры и выливался в воду. Получающийся в результате водный
раствор экстрагировался метиленхлоридом. Органический экстракт сушился над сульфатом натрия, а затем концентрировался досуха в вакууме, давая 1,2 г желтого масла, которое анализировалось как
неочищенное целевое соединение. Данное неочищенное вещество очищалось с помощью флэш-хроматографии с использованием смеси гексан/диэтиловый эфир (1: 1) в качестве элюента, давая желтое масло, которое
по данным анализа представляло чистую форму свободного основания целевого продукта.
Оксалатная соль свободного основания целевого соединения получалась путем растворения масла, полученного выше, в щавелевой кислоте. Твердые вещества, которые выпадали в осадок, отделялись фильтрованием, затем очищались с помощью перекристаллизации из метанол/диэтилофоэфирного раствора, давая 174 мг целевого соединения. Т. пл. 144oC.
Анализ для C24H30N2•C2H2O4:
вычислено: С 71,54,
H 7,39, N 6,42;
найдено: С 71,79, H 7,43, N 6,736.
Пример 37. Получение дихлоргидрата 8-(индол-2-ил)-2-дипропиламинотетралина
A.
8-(1-Фенилсульфонилиндол-2-ил)-2-дипропиламинотетралин
Раствор 1-фенилсульфонил-2-броминдола (1,7 г, 6,45 ммоль), полученного по способу, в основном аналогичному описанному в примере 36, A и
B, в 30 мл тетрагидрофурана охлаждался до -78oC. После охлаждения добавлялось 5,7 мг 1,13 М раствора н-бутиллития (6,45 ммоль). Получающийся раствор затем подогревался до -20oC
после перемешивания при -78oC в течение 15 минут. После перемешивания реакционного раствора при -20oC в течение 30 минут добавлялся холодный раствор индола к дефлегмирующей смеси
8-бром-2- дипропиламинотетралина (2,0 г, 6,45 ммоль) и 7,44 мг (0,645 ммоль) Pd(PPh3)4 в 40 мл толуола. Получающаяся смесь перемешивалась при нагревании с обратным холодильником
в течение одного часа, а затем оставлялась охлаждаться до комнатной температуры. Перемешивание реакционной смеси продолжалось при комнатной температуре на протяжении ночи. На следующее утро
реакционная смесь охлаждалась выливанием реакционного раствора в воду. Получающийся водный раствор экстрагировался метиленхлоридом. Органический экстракт промывался насыщенным солевым раствором,
сушился над сульфатом натрия, а затем концентрировался в вакууме, давая б мг коричневого масла, которое по данным анализа представляло сырое целевое соединение. Данное масло очищалось с помощью
флэш-хроматографии на колонке с использованием 3%-ного метанола в метиленхлориде в качестве элюента, давая 230 мг вещества, которое по данным анализа представляло, по существу, чистое целевое
соединение.
B. 8-(Индол-2-ил)-2-дипропиламинотетралин-дихлоргидрат Соединение, полученное на стадии A, растворялось в растворе 20 мл 10%-ной гидроокиси калия в метаноле. Получающийся в результате раствор нагревался до температуры дефлегмации, а затем перемешивался при этой температуре на протяжении ночи. На следующее утро реакционная смесь гасилась выливанием реакционного раствора в воду. Получающийся водный раствор экстрагировался три раза метиленхлоридом, и органические экстракты, полученные таким образом, объединялись и сушились над сульфатом натрия, а затем концентрировались в вакууме, давая неочищенное целевое соединение. Данное неочищенное вещество очищалось с использованием флэш-хроматографии на колонке (3%-ного метанол и метиленхлорида в качестве элюента), давая 126 мг очищенного целевого соединения (форма свободного основания) в виде желтого масла.
Дихлоргидратная соль свободного основания целевого соединения получалась с помощью растворения масла, полученного выше, в соляной кислоте. Твердые вещества, которые выпадали в осадок, отделялись фильтрованием, а затем очищались с помощью перекристаллизации из смешанного этилацетат/гексанового раствора с последующей перекристаллизацией из этилацетат/гексанового раствора, содержащего небольшое количество метанола, давая 23 мг целевого соединения. Т. пл. более 160oC.
Анализ для C24H30N2•HCl:
вычислено: C 75,27, H 8,16, N 7,31;
найдено: C 74,98, H 8,10, N 7,29.
Пример 38. Хлоргидрат (-)-5-фтор-8-(трифторметансульфонил)оксид-2-(дипропиламино)тетралина
К раствору (-)-6-F-8-OH-2-(дипропиламино)тетралина (500 мг, 1,88 ммоль) и 2,4,6-триметилпиридина
(912 мг, 7,5 ммоль) в дихлорметане (10 мл) добавлялся ангидрид трифторметансульфокислоты (849 мг, 3 ммоль) в дихлорметане (3 мл) при -78oC, и смесь перемешивалась при данной температуре в
течение одного часа. Охлаждающая баня удалялась затем, и добавлялся диэтиловый эфир. Эфирный раствор промывался насыщенным раствором карбоната калия, а затем водой, сушился (карбонатом калия) и
выпаривался. Хроматография остатка на окиси алюминия с использованием E/PE (1:20) в качестве элюента давала целевое соединение. Очищенное основание превращалось в хлоргидрат (580 мг, 71% выход). Т.
пл. -145-146oC.
Пример 39. Хлоргидрат (-)-5-фтор-8-(2-фурил)-2-(дипропиламино)тетралина
Способ 1
К смеси хлоргидрата
(-)-5-фтор-8-(трифторметансульфонио)оксид-2-(дипропиламино)тетралина (400 мг, 1 ммоль), (Ph3P)4Pd (23,1 мг, 0,02 ммоль), хлористого лития (127 мг, 3 ммоль) в ДМФ (10 мл)
добавлялся трибутил (2-фурил) станнан (428,5 мг, 1,2 ммоль) в ДМФ (6 мл). Колба заполнялась азотом и герметизировалась. Колба погружалась в масляную баню при 120oC. Реакция завершалась,
когда смесь становилась черной, приблизительно через 5 минут. Катализатор отфильтровывался через целит, растворитель выпаривался, и сырой остаток растворялся в эфире, эфирный раствор промывался
насыщенным раствором карбоната калия, сушился (карбонатом калия) и выпаривался. Хроматография остатка на окиси алюминия с использованием E/PE (1:20) в качестве элюента давала целевое соединение в виде
чистого основания, которое превращалось в хлоргидрат и перекристаллизовывалось из смеси MeOH/эфир (265 мг, 75% выход). Т. пл. 159-160oC. (MeOH/эфир).
Способ 2
Смесь (-)-5-фтор-8-(трифторметансульфонио)оксида, -2-(диипропиламино)тетралина, хлоргидрата (100 мг, 0,25 ммоль) (Ph3P)4Pd (7,28 мг, 6,3•10-3 ммоль), хлористого
лития (21,36 мг, 0,504 ммоль), 2 М раствора карбоната натрия (0,38 мл) в ДМЕ (3 мл) и EtOHabc (0,75 мл) и 2-фуранбороновой кислоты (36,2 мг, 0,38 ммоль) нагревалась с обратным холодильником
в течение 3 часов. Растворитель выпаривался, и остаток растворялся в эфире, раствор промывался насыщенным раствором карбоната калия и солевым раствором, сушился (карбонатом калия) и выпаривался.
Хроматография на окиси алюминия с использованием E/PE (1:30) в качестве элюента позволила получить целевое соединение в виде чистого основания, которое превращается в хлоргидрат (57 мг, 65% выход). Т.
пл. -160-161oC (MeOH/эфир).
Пример 40. Хлоргидрат (-)-5-фтор-8-(2-ацетил)-2-(дипропиламино)тетралина
Раствор триэтиламина (20,37 мг, 2,013 ммоль), Pd(OAc)2 (5,65 мг, 0,025 ммоль) и oppp (дифенилфосфинпропан) (11,42 мг, 0,027 ммоль) в ДМФ (1,3 мл) добавлялся к (-) 1 C 20 (200 мг, 0,503 ммоль). К получающемуся раствору добавлялся бутилвиниловый
эфир (504 мг, 5,03 ммоль). Реакционная колба заполнялась азотом, герметизировалась и погружалась в масляную баню, предварительно нагретую до 90oC. После нахождения в течение одного часа при
данной температуре реакция завершалась (отсутствие исходного вещества по данным ТСХ). Затем к реакционной смеси добавлялась 5%-ная HCl (2 мл), и смесь оставлялась стоять при комнатной температуре в
течение 15 минут, затем она подщелачивалась бикарбонатом натрия, водный раствор экстрагировался эфиром, органические вещества промывались водой до нейтральной величины pH, сушились (карбонатом калия)
и выпаривались. Неочищенный остаток очищался с помощью флэш-хроматографии на окиси алюминия с использованием E/PE (1:8) в качестве элюента. Целевое соединение в виде полученного чистого основания,
превращалось в хлоргидрат и перекристаллизовывалось из смеси этилацетат/эфир (140 мг, 85% выход). Т. пл. 106-107oC (AcOEt/эфир).
Пример 41. Хлоргидрат
(-)-5-[фтор-8-](2-тиенил)-2-(дипропиламино)тетралина
Трифенилфосфинпалладий (13,78 мг, 0,0119 ммоль) и хлорид лития (50,55 мг, 1,19 ммоль) в ДМФ (1,5 мл) добавлялись к хлоргидрату
(-)-5-фтор-8-(трифторметансульфонио)оксид, 2-(дипропиламино)тетралина (158 мг, 0,397 ммоль), к данной смеси добавлялся трибутил-(2-тиенил)станнан (222,5 мг, 0,596 ммоль) в ДМФ (1,5 мл). Реакционная
колба помещалась на баню, предварительно нагретую до 120oC в атмосфере азота. Через 10 минут смесь становилась черной, и по данным ТСХ исходного вещества не оставалось. Реакционная смесь
разбавлялась эфиром и промывалась насыщенным раствором карбоната калия и солевым раствором, сушилась (карбонатом калия) и выпаривалась. Мгновенная (флэш) хроматография с использованием окиси алюминия
и смеси эфир/петролейный эфир (1:20) в качестве элюента давала целевое соединение в виде чистого основания, которое превращалось в хлоргидрат и перекристаллизовывалось из смеси MeOH/эфир (106 мг, 73%
выход). Т. пл. -157-159oC.
Пример 42. Хлоргидрат (+/-)-8-ацетил-2-N-циклопропиламинотетралина
(+/-)-2-N-циклопропиламино-8-[(трифторметилсульфонил)оксид]тетралин
(0,4 г, 1,3 ммоль), хлорид лития (0,2 г, 4,7 ммоль) и ДМФ (6 мл) смешивались в трехгорлой круглодонной колбе. Колба вакуумировалась с последующим вводом окиси углерода (процедура повторялась 5 раз, CO
газ в бюретке с водой). Добавлялся тетраметилстаннан (0,3 мл, 2,2 ммоль), дихлор [1,1-бис- (дифенилфосфино)ферроден] палладий (II) (PdCl2) dppf (40 мг, каталитическое количество) и 2,
6-ди-третбутил-4-метилфенол (каталитическое количество), и реакционная смесь затем перемешивалась в атмосфере CO при 100oC в течение 3 часов перед разбавлением диэтиловым эфиром и
фильтровалась через целит. Фильтрат промывался бикарбонатом натрия и сушился (сульфатом магния). Растворитель удалялся в вакууме, давая неочищенный масляный остаток, который очищался с помощью
мгновенной хроматографии /двуокись кремния, этилацетат (0,5% аммиака)/. Чистые фракции сливались и концентрировалось. Получающееся масло превращалось в хлоргидрат, который затем перекристаллизовывался
из смеси этанол/диэтиловый эфир, давая 60 мг (20%) чистого хлоргидрата (1/-)-8-ацетил-2-N-циклопропиламинотетралина. Т. пл. -196-198oC.
Пример 43. Хлоргидрат
(+/-)-2-(N-изопропил-N-н-пропиламино-8-трет- бутилкарбонилтетралина
(+/-)-2-(N-изопропил-N-н-пропиламино)-8-(2,2'-диметил-1- пропанол)тетралин (0,6 г, 1,9 ммоль) растворялся в метиленхлориде
(сушился молекулярными ситами, 3
Пример 44. Хлоргидрат (+/-)-8-ацетил-2-(N-циклопропил-N-н-пропиламино)тетралина
(+/-)-2-(N-циклопропил-N-н-пропил)-8-[(трифторметилсульфонил)оксид] тетралин (1,5-3,8 ммоль), хлорид лития (0,7 г, 1,5 ммоль), ДМФ (15 мл, содержащих 1% воды), дихлор-[1,
1'-бис-(дифенилфосфино)ферроцен]палладий (II) [PdCl2(dppf)] (0,2 г каталитическое количество), 2,6-дитрет-бутил-4-метилфенол (каталитическое количество) и тетраметилстаннан (0,8 г, 4,6
ммоль) перемешивались в атмосфере окиси углерода в течение 7 часов при 110oC. Растворитель удалялся в вакууме, и остаток растворялся в смеси диэтиловый эфир/аммиак. Слои разделялись, и
продукт (эфирная фаза) затем подвергался кислотно-основной обработке и сушился (сернокислым магнием). Растворитель удалялся в вакууме, давая сырой маслянистый остаток, который очищался мгновенной
хроматографией (SiO2, CH2Cl2/EtOAc, 4:1). Чистые фракции объединялись и концентрировались. Полученное в результате масло превращалось в хлоргидрат, который затем
перекристаллизовывался из смеси EtOAc/диэтиловый эфир, давая 140 мг 12%-ного хлоргидрата (+/-)-8-ацетил-2-(N-циклопропил-N-н-пропиламино)тетралина. Т. пл. - 126-128oC.
Пример 45. Хлоргидрат (+/-)-8-ацетил-2-(N-изопропил-N-н-пропиламино)тетралина
(+/-)-2-(N-изопропил-N-н-пропиламино)-8-[(трифторметилсульфонил)оксид] тетралин (0,85 г, 2,2 ммоль),
тетраметилстаннан (0,48 г, 2,7 ммоль), хлорид лития (0,38 г, 9,0 ммоль), дихлор-[1,1'-(бисфенилфосфино)ферроцен]палладий (II) [PdCl2(dppf)] (120 мг, каталитическое количество), 2,
6-дитрет-бутил-4-метилфенол (каталитическое количество) в ДМФ (35 мл, содержащих 1% воды) перемешивались в атмосфере окиси углерода (2 бар) в течение 6 часов при 110oC. Растворитель
удалялся в вакууме, и остаток растворялся в диэтиловом эфире, промывался аммиаком и сушился (сернокислым магнием). Растворитель удалялся в вакууме, давая маслянистый остаток, который очищался
мгновенной хроматографией (SiO2, EtOAc). Чистые фракции объединялись и концентрировались. Хлоргидрат осаждался из диэтилового эфира (0oC) и перекристаллизовывался из смеси
EtAOc/диэтиловый эфир, давая 130 мг (19%) хлоргидрата (+/-)-8-ацетил-2-(N-изопропил-N-н-пропиламино)тетралина. Т. пл. 154-156oC.
Пример 46. Хлоргидрат
(+/-)-8-циклопентилкарбонил-2-(N-изопропил-N-н-пропиламино)тетралина
(+/-)-8-(1-Циклопентилметанол)-2-(N-изопропил-N-н-пропиламино)тетралин (0,44 г, 1,3 ммоль) растворялся в метиленхлориде
(высушенном молекулярными ситами, 3
Пример 47
Хлоргидрат (+/-)-8-ацетил-2-N-изопропиламинотетралина
(+/-)-2-N-изопропиламино-8-[(трифторметансульфонил)оксид] тетралин (1,0 г, 3,0
ммоль), тетраметилстаннан (0,6 г, 3,6 ммоль), хлорид лития (0,5 г, 11,8 ммоль), дихлор-[1,1'-бис-(дифенилфосфино)ферроцен] палладий (II) [PdCl2(dppf)] (150 мг), 2,
6-дитрет-бутил-4-метилфенол (каталитическое количество) в ДМФ (30 мл, содержащих 1% воды) перемешивались в атмосфере окиси углерода (2 бар) при 110oC в течение 5 часов. Растворитель удаляли
в вакууме, и остаток растворялся в этилацетате, промывался аммиаком и сушился (сернокислым магнием). Растворитель удалялся в вакууме, давая маслянистый остаток, который очищался мгновенной
хроматографией [SiO2, EtOAc/EtOH (10:1) (+0,5% NH3)] Чистые фракции объединялись и концентрировались. Полученное в результате масло превращалось в хлоргидрат (осажденный из
диэтилового эфира), давая 100 мг хлоргидрата (+/-)-8-ацетил-2-N-изопропиламинотетралина. Т. пл. 241-243oC.
Пример 48. Хлоргидрат (S)-8-(2-фурил)-2-[N[4-(8-азаспиро[4,5]
декан-7,9-дион-8-ил)бутил]пропиламина]тетралина
а) Хлоргидрат (S)-8-метокси-2-[N[4-(8-азаспиро[4,5] декан-7,9-дион-8-ил)бутил]пропиламино]тетралина
Раствор
(S)-8-метокси-2-(пропиламино)тетралина (300 мг, 1,37 ммоль), 8-(4-бромбутил)-8-азаспиро[4,5] декан-7,9-диона (455 мг, 1,5 ммоль) и карбоната калия (563 мг, 4,11 ммоль) в 20 мл ацетонитрила
перемешивался при 85oC в атмосфере азота в течение 6 дней. Реакционная смесь разбавлялась простым эфиром и фильтровалась для удаления твердого карбоната калия. Фильтрат концентрировался, и
остаток хроматографировался на колонке из окиси алюминия, элюировался смесью простой эфир/легкий петролейный эфир (2:1). Чистые фракции объединялись, концентрировались и обрабатывались эфирной соляной
кислотой, давая 440 мг (67%-ный выход) целевого соединения (48, a).
Хлоргидрат (R)-8-метокси-2-[N-[4(8-азаспиро[4,5] декан-7,9-дион-8-ил)бутил] пропиламино] тетралина получался в соответствии со способом, описанным в примере 48, a.
b) Хлоргидрат (S)-8-гидрокси-2-[N[4(8-азаспиро[4,5] декан-7,9-дион-8-ил)бутил]пропиламино]тетралина
К раствору трибромида
бора (2,445 г, 9,76 ммоль) в 15 мл хлороформа добавлялся раствор соединения примера 48 a (538 мг, 1,22 ммоль) в 10 мл хлороформа при комнатной температуре в атмосфере азота в течение 10 минут. Реакция
завершалась после перемешивания при комнатной температуре в течение 1,5 часа и прерывалась добавлением насыщенного водного раствора бикарбоната натрия. Водный раствор экстрагировался 3 порциями по 30
мл хлороформа, и органический слой собирался, сушился (сульфат натрия), фильтровался и концентрировался при пониженном давлении. Маслянистый остаток хроматографировался на силикагеле, элюировался
смесью хлороформ/метанол (30:1). Чистые фракции объединялись и обрабатывались эфирной соляной кислотой, давая 550 мг (97%-ный выход) целевого соединения 48, b.
Хлоргидрат (R)-8-гидрокси-2-[N[4(8-азаспиро[4,5] декан-7,9-дион-8-ил)бутил]пропиламино]тетралина получался в соответствии с методом, описанным в примере 48, b.
c) Хлоргидрат
(S)-8-(трифторметансульфонил)окси-2-[N[4-(8-азаспиро[4,5] декан-7,9-дион-8-ил)бутил]пропиламино]тетралина
Раствор трифторметансульфонового ангидрида (410 мг, 1,46 ммоль) в 20 мл дихлорметана
добавлялся по каплям к смеси соединения 48, b (413 мг, 0,97 ммоль), коллидина (176 мг, 1,46 ммоль) и карбоната калия (199 мг, 1,46 ммоль) в 10 мл дихлорметана при -78oC в атмосфере азота в
течение 10 минут. Реакция завершалась после перемешивания в течение 2 часов и прерывалась путем добавления насыщенного водного раствора карбоната калия. Реакционная смесь распределялась между CH2Cl2 и раствором K2CO3. Органический слой сушился (K2CO3), фильтровался и концентрировался. Полученный в результате остаток
хроматографировался на колонке из окиси алюминия, элюировался смесью простой эфир/легкий петролейный эфир (1:1) и затем простым эфиром. Чистые фракции объединялись и превращалась в соответствующий
хлоргидрат обработкой эфирной HCl, давая 540 мг (97%-ный выход) целевого соединения 48, c.
Хлоргидрат (S)-8-(трифторметансульфонил)окси-2-1[N[4(8- азаспиро[4, 5] декан-7, 9-дион-8-ил)бутил] пропиламино]тетралина получался в соответствии со способом 48, c.
d) Хлоргидрат (S)-8-(2-фурил)-2-[N[4(8-азаспиро[4,5]декан-7,
9-дион-8-ил)бутил]пропиламино]тетралина
В круглодонную колбу, заполненную смесью соединения 48, c (100 мл, 0,18 ммоль), водного хлорида лития (61 мг, 1,44 ммоль), трифенилфосфина (19 мг, 0,
072 ммоль) и хлорида бис-(трифенилфосфино)палладия в диметилформамиде, добавлялись (2-фурил)трибутилстаннан (129 мг, 0,36 ммоль) и кристалл радикального ингибитора (2,6-дитрет-бутил-4-метилфенола).
Колба заполнялась током азота, герметизировалась и нагревалась при 120oC в течение 20 часов. ДМФ удалялся в вакууме, и полученный в результате остаток распределялся между простым эфиром и
водой. Эфирный слой сушился (K2CO3), фильтровался и концентрировался. Остаток хроматографировался на колонке из окиси алюминия с использованием смеси простой эфир/легкий
петролейный эфир (2:1) в качестве элюента. Чистые фракции объединялись и обрабатывались эфирной соляной кислотой, давая 70 мг (76%-ный выход) целевого соединения 48, d. [α] - 36,8oC
Хлоргидрат (R)-8-(2-фурил)-2-[N[4 (8-азаспиро[4,5]
декан-7,
9-дион-8-ил)бутил]пропиламино]тетралина получали в соответствии со способом 48, d.
Пример 49. Хлоргидрат цис-1S,2R-метил-8-ацетил-2-[N[4(8-азаспиро[4,5]декан-7,
9-дион-8-ил)бутил]пропиламино]тетралина
a) Хлоргидрат цис-1S,2R-1-метил-8-метокси-2-[N[4(8-азаспиро[4,5]декан-7,9-дион-8-ил)бутил]пропиламино]тетралина
Раствор цис-1S,
2R-1-метил-8-метокси-2-(пропиламино)тетралина (300 мг, 1,28 ммоль), 8-(4-бромбутил)-8-азаспиро[4,5] декан-7,9-диона (427 мг, 1,42 ммоль) и карбоната калия (263 мг, 1,92 ммоль) в 10 мл ацетонитрила
нагревался с обратным холодильником при 85oC в атмосфере азота в течение 5 дней. Реакционная смесь разбавлялась простым эфиром, фильтровалась и концентрировалась. Остаток
хроматографировался на силикагеле, элюировался смесью хлороформ/метанол (15:1). Чистые фракции объединялись и обрабатывались эфирной соляной кислотой, давая 591 мг (9%-ный выход) целевого соединения
49, a.
b) Хлоргидрат цис-1S, 2R-1-метил-8-гидрокси-2-[N[4(8-азаспиро[4,5] декан-7,9-дион-8-ил)бутил]пропиламино]тетралина
К раствору трехбромистого бора (1,63 г, 6,52 ммоль)
в 15 мл хлороформа добавлялся раствор соединения 49, a (370 мг, 0,81 ммоль) в 10 мл CHCl3. Реакционная смесь перемешивалась при комнатной температуре в атмосфере азота в течение 10 часов.
Реакция завершалась и прерывалась добавлением 10%-ного водного раствора NaHCO3 Органический слой после распределения между дихлорметаном и водным раствором сушился (над сульфатом натрия),
фильтровался и концентрировался. Остаток хроматографировался на силикагеле, элюировался смесью хлороформ/метанол (15:1). Чистые фракции объединялись и обрабатывались эфирной соляной кислотой, давая
370 мг (95%-ный выход) целевого соединения 49, b.
c) Хлоргидрат цис-1S, 2R-1-метил-8-[трифторметансульфонил(окси)] -2-[N[4-(8-азаспиро[4,5]декан-7,
9-дион-8-ил)бутил]пропиламино]тетралина
Раствор трифлового ангидрида (178 мг, 0,63 ммоль) в 5 мл дихлорметана добавлялся к смеси продукта примера 49, b (185 мг, 0,42 ммоль), коллидина (76 мг,
0,63 ммоль) и карбоната калия (86 мг, 0,63 ммоль) в 10 мл CH2Cl2. Реакция завершалась после перемешивания при -78oC в течение 2 часов и прерывалась добавлением
насыщенного водного раствора K2CO3. Экстрагирование тремя порциями по 30 мл дихлорметана давала органический слой, который сушился (над карбонатом калия), фильтровался и
концентрировался. Остаток хроматографировался на окиси алюминия, элюировался смесью простой эфир/легкая нефть (1:1), затем простым эфиром. Чистые фракции объединялись и обрабатывались эфирной соляной
кислотой давая 217 мг (84%-ный выход) целевого соединения 49, c.
d) Хлоргидрат цис-1S,2R-1-метил-8-ацетил-2-[N[4(8-азаспиро[4,5]декан-7,9-дион-8-ил)бутил]пропиламино]тетралина
Реакционная смесь 49 c (120 мг, 0,21 ммоль), трибутилэтоксивинилолова (83 мг, 0,23 ммоль), бис-(трифенилфосфино)палладийхлорида (PdCl2(Ph3P)2) (18 мг, 0,025 ммоль),
трифенилфосфина (22 мг, 0,084 ммоль), хлорида лития (71 мг, 1,88 ммоль) и кристалла радикального ингибитора (2,6-3-трет-бутил-4-метилфенола) перемешивалась при 120oC в течение 18 часов в
герметизированной колбе, заполненной заранее азотом. Смесь концентрировалась в вакууме для удаления ДМФ и перемешивалась вместе с 10%-ным водным раствором соляной кислоты при комнатной температуре в
течение 15 минут. Раствор подщелачивался 1 М NaOH и экстрагировался тремя порциями простого эфира по 30 мл каждая. Эфирная фаза сушилась (K2CO3_ фильтровалась и
концентрировалась. Остаток хроматографировался на колонке из окиси алюминия с использованием смеси простой эфир/легкая нефть (2:1) в качестве элюента. Чистые фракции объединялись и обрабатывались
эфирной соляной кислотой, давая 80 мг (76%-ный выход) целевого соединения 49, d. [α] +2,
2oC.
Пример 50. N-циклопропил-N-пропил-8-фенил-2-аминотетралин
N-циклопропил-N-пропил-8-трифторметансульфонилокси-2 -аминотетралин (2,50 г, 6,35 ммоль),
фенилбороновая кислота (0,90 г, 7,30 ммоль), хлористый литий (0,54 г, 12,7 ммоль), Na2CO3 (2 М 9 мл), этанол (29 мл) и толуол (63 мл) смешивались в 250 мл трехгорлой круглодонной
колбе. Колба затем откачивалась с последующим вводом азота (дважды), перед тем как добавлялся Pd(PPh3)4 (240 мг, 0,2 ммоль). Реакционная смесь нагревалась при 95oC в
течение 12 часов. Растворитель затем упаривался до объема 70 мл. Остаток разбавлялся этилацетатом и промывался дважды 1 М NaOH, обрабатывался солевым раствором и сушился (сульфатом магния).
Растворитель удалялся в вакууме, давая маслянистый желто-коричневый остаток, который очищался мгновенной хроматографией (SiO2, CH2Cl2 этилацетат 100:2 -->
10:1).
N-циклопропил-N-пропил-8-фенил-2-аминотетралин выделялся с выходом 72% (1,4 г) в виде бесцветного масла. Масло растворялось в эфире. Раствор охлаждался на ледяной бане, и по каплям добавлялась соляная кислота (2 М в простом эфире). Фильтрование и перекристаллизация из смеси этилацетат/простой эфир давали бесцветные кристаллы целевого соединения. Т. пл. 153-156o C.
Пример 51. Оксалат (+/-)-2-(дипропиламино)-8-(4'-фторфенил)тетралина
Смесь 2-(дипропиламино)-8-((трифторметансульфонил)окси)тетралина (200 мг, 0,51 ммоль),
тетракис(трифенилфосфин)палладия (0)(30 мг, 0,026 ммоль), 4-фторфенилтрибутилстаннана (232 мг, 0,6 ммоль, см. пример 53), хлорида лития (67 мг, 1,6 ммоль) в диметилформамиде (3 мл) и 1,4-диоксана (3
мл) нагревалась при температуре дефлегмации в течение ночи. Фильтрование и концентрирование давали маслянистый остаток, который хроматографировался на колонке из окиси алюминия, элюировался смесью
простой эфир/легкая нефть (1:20). Чистые фракции объединялись и обрабатывались эфирной щавелевой кислотой, давая 145 мг (68%-ный выход) целевого соединения. Т. пл. -197-199oC.
Оба энантиомера оксалата (±)-2- (дипропиламино)-8-(4'- фторфенил)тетралина синтезировались тем же путем, но с использованием разделенных исходных материалов:
оксалат
(+)-2-(дипропиламино)-8-(4'-фторфенил)тетралина. Выход: 59% (α) 136,8oC.
оксалат (-)-2-(дипропиламино)-8-(4'-фторфенил) тетралина. Выход: 65% (a) -27,2o C.
Пример 52. Хлоргидрат (±)-2-(дипропиламино)-8-(4'-метоксифенил)тетралина
Смесь 2-(дипропиламино)-8-(трифторметилсульфонилокси)тетралина (1 г, 2,5 ммоль),
три-н-бутил-(4-метоксифенил)станнана (1,2 г, 3,02 ммоль, приготовленного, как описано в работе: Wardell I.I. Ahmed S.I. //Organomet. Chem. 1971, 78, 395), тетракис(трифенилфосфин)палладия (0), (87 мг,
0,075 ммоль), хлорида лития (330 мг, 7,75 ммоль) в 1,4-диоксане (15 мл) и диметилформамиде (2 мл) перемешивалась при 120oC в герметизированной колбе в течение 20 часов. Катализатор
отфильтровался (целит), и фильтрат концентрировался. Полученный в результате остаток очищался хроматографией на колонке из окиси алюминия с использованием смеси простой эфир/легкая нефть в качестве
элюента (1: 20). Чистые фракции объединялись и концентрировались. Эфирный раствор продукта обрабатывался эфирной HCl и оставлялся стоять при комнатной температуре в течение ночи, давая 793 мг (85%-ный
выход) целевого соединения в виде белого твердого вещества.
Пример 53. Три-н-бутил-(4-фторфенил)станнан
Раствор н-фторбромбензола (5 г, 28,6 ммоль) в эфире (15 мл) добавлялся
к вновь измельченному магнию при перемешивании в атмосфере азота. Реакционная смесь нагревалась до температуры дефлегмации до тех пор, пока не растворится магний. К реактиву Гриньяра добавлялся
раствор трибутилоловохлорида (7,8 г, 2,4 ммоль) в простом эфире (10 мл), и реакция прерывалась после трехдневного нагревания с обратным холодильником. Реакционная смесь разбавлялась водным насыщенным
раствором хлористого аммония и распределялась между простым эфиром и водой. Органический слой сушился (сульфат магния), фильтровался и концентрировался. Остаток перегонялся в вакууме, давая 7,1 г
(68%) три-н-бутил-(4-фторфенил)станнана в виде жидкости (117-134oC) 1 мм Hg.
Проверка фармакологического действия
Лечение депрессии у человека
Имеется
информация о том, что у больных депрессией передача нервных сигналов по центральной нервной системе (ЦНС) может прерываться. По-видимому, эти нарушения связаны с нейромедиаторами норадреналином (NA) и
5-гидрокситриптамином (5-HT). Лекарственные препараты, наиболее часто применяемые при лечении депрессии, оказывают действие, как полагают, за счет улучшения передачи нервных импульсов любым или обоими
указанными физиологическими агонистами. Имеющиеся результаты свидетельствуют о том, что при усилении 5-гидрокситриптаминовой нейросекреции, по существу, устраняются депрессия и психоз, в то время как
при усилении норадреналиновой нейросекреции скорее происходит ослабление симптомов, появляющихся у больных с депрессивными состояниями. В последние годы было предпринято множество попыток по созданию
новых лекарственных препаратов с высокой избирательностью для улучшения 5-HT-проведения нервных сигналов по ЦНС.
Механизм действия лекарственных препаратов, используемых в настоящее время при лечении депрессии, не связан с прямым их действием, то есть они оказывают действие при блокировании повышенного содержания нейромедиаторов (NA и/или 5-HT), высвобождаемых из нервных окончаний в ЦНС, повышая тем самым концентрацию указанных медиаторов в синаптической щели, а в результате восстанавливая соответствующую передачу нервных импульсов.
Фундаментально другой способ регуляции проведения нервных импульсов в 5-HT-нейронах ЦНС состоит в использовании непосредственных 5-HT-рецепторных агонистов/антагонистов. Для минимизации побочных эффектов в таком случае предпочтительно избирательное действие для такого типа рецепторов.
Неожиданно было установлено, что группа соединений формулы I обладает избирательным, прямым стимулирующим действием на ЦНС или блокирует подгруппу 5-HT-рецепторов. Установлено также, что некоторые из указанных соединений имеют прекрасную биологическую доступность при пероральном применении. Для оценки средства указанной подгруппы 5-HT- рецепторов проведено исследование in vitro влияния на различные рецепторы мозга крысы при использовании анализа связывания с рецепторами (К нМ).
in
vitro тест. Анализ связывания рецептора
Тест на связывание 5-HT1A. Кору головного мозга вместе с гиппокампом от каждой крысы отделяют и гомогенизируют в 15 мл охлажденного льдом 50
мМ трис-HCl буфера, 4,0 мМ CaCl2 и 5,7 мМ аскорбиновой кислоты, pH 7,5, на гомогенизаторе Ultra Turrax (Janke and Kunkel, Staufen, ФРГ) в течение 10 секунд. После центрифугирования в
течение 12,5 минут при скорости 17000 об/мин (39800 • об. в центрифуге Бекмана с ротором A-17) Bechman, Paolo Alto, CA, USA, гранулы повторно суспендируют в том же самом буфере и гомогенизируют,
и центрифугируют еще раз. К каждой грануле добавляют 5 мл охлажденного льдом 0,32 М раствора сахарозы и гомогенизируют в течение 5 секунд. Указанные образцы выдерживают в замороженном состоянии при
температуре -79oC. При использовании их разбавляют буферным раствором в расчете 8 мг ткани/мл и гомогенизируют в течение 10 секунд.
Тканевые гомогенаты инкубируют в течение 10 минут при температуре 37oC, затем вводят 10 мкМ паргилина с последующей повторной инкубацией в течение 10 минут.
Проводимый далее анализ на связывание рецептора описан в: Peroutka J. //Neurochem, 1986, 47, 529-540. Инкубационная смесь (2 мл), содержит3H-8-OH-PAT (от 0,25 до 8 нМ), 5 мг/мл тканевого гомогената в 50 мМ трис-HCl буфере, содержащем 4,0 мМ CaCl2 и 5,7 мМ аскорбиновой кислоты, pH 7,5. Проанализировано 6 разных концентраций3H-8-OH-PAT. Тест на связывание начинают с введения тканевого гомогената с последующим инкубированием в течение 10 минут при температуре 37oC. Инкубационные смеси фильтруют через стеклянные фильтры типа Whatman IB при использовании клеточного харвестера Брандела (Gaithersburg, MD, USA). Фильтры промывают дважды 5 мл охлажденного льдом 50 мМ трис-HCl буферного раствора, pH 7,5, и подсчитывают клетки при использовании 5 мл готового раствора HP (Beckman) в сцинтилляционном счетчике модели Beckman LS 3801. Неспецифическое связывание определяют при добавлении 10 мкМ 5-HT к реакционной смеси. Результаты теста на связывание обработаны анализом на ЭВМ с использованием метода нелинейных меньших квадратов (Munson and Rodbard Anal. Biochem, 1980, 107, 220-239).
Результаты оценки различных соединений настоящего изобретения помещены в таблицу. В этой таблице указана структура соединения с учетом формулы, помещенной в заголовке таблицы. В колонке указаны соли исследованных соединений, и в последней колонке дана концентрация соединения в наномолях, необходимая для 50%-ного подавления связывания3H-8-OH-DPAT в виде показателя 1C50.
Реферат
Предложены соединение формулы I, где Z означает водород или галоген, Q означает COR1 или 5- или 6-членный арил, который может содержать 1 или 2 гетероатома из N, O или S, возможно замещенный или конденсированный, где R означает водород или C1-метил (цис-конфигурация), R1 означает C1-6-алкил или ароматическое кольцо, которое может содержать гетероатомы O или S, и может быть замещено или сконденсировано с возможно замещенным бензольным кольцом, R2 означает атом водорода или C1-6-алкил, а R3 может иметь различные значения, указанные в пункте 1 формулы, и его энантиомеры или соли, способы получения указанных соединений, лекарственные препараты на их основе, а также способ лечения заболеваний центральной нервной системы при использовании указанных соединений. 3 с. и 12 з.п.ф-лы, 1 табл.
Формула

где R водород или метил при условии, что C1-метильный заместитель находится в цис конфигурации;
Z водород или галоген;
Q COR' или фенил, фурил, тиенил, пиразолил, тиазолил, пиридил, хинолил, индоксазинил, индолил, бензофуранил, необязательно замещенный галогеном или низшим алкокси;
R1 C1 C6-алкил или фенил, необязательно замещенный галогеном или низшим алкокси, или C3-C6-циклоалкил;
R2 водород или C1 C6-алкил;
R3 группа C1 C6-алкил или -(CH2)a-R4, где a 4, R4 - -NR11R12, R11 и R12 вместе с атомом азота образуют кольцо

n 1 или 2,
или их энантиомеры и физиологически приемлемые соли.
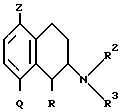
в которой R водород или метил при условии, что C1-метильный заместитель находится в цис конфигурации;
Z водород или галоген;
Q COR1;
R1 C1 C6-алкил или фенил, необязательно замещенный галогеном или низшим алкокси, или C3 C6-циклоалкил;
R2 водород или C1 C6-алкил;
R3 группа C1 C6-алкил или -(CH2)a-R4, где a 4, R4 - -NR11R12, R11 и R12 вместе с атомом азота образуют кольцо;

n 1 или 2,
или их энантиомеров и физиологически приемлемых солей, отличающийся тем, что соединение формулы II

в которой X представляет уходящую группу и R, R2 и R3 имеют значения, определенные выше для соединения формулы I, подвергают превращению с помощью каталитического цикла с использованием переходного металла с нулевой валентностью М0, который подвергается окислительному присоединению к арил-X-связи, обработке окисью углерода с последующим трансметаллированием между R1 M1, где M1 металл и R1 имеет значения, определенные для соединения формулы I, и первоначально образовавшимся карбонилированным о-арил-металл-X комплексом с образованием соединения формулы I, после чего в случае необходимости соединение, полученное в виде основания, превращают в физиологически приемлемую кислотно-аддитивную соль или соединение, полученное в виде соли, превращают в основание или в иную физиологически приемлемую кислотно-аддитивную соль, полученную изомерную смесь в случае необходимости разделяют на чистые изомеры.

где R водород или метил при условии, что C1-метильный заместитель находится в цис конфигурации;
Z водород или галоген;
Q фенил, фурил, тиенил, пиразолил, тиазолил, пиридил, хинолил, индоксазинил, индолил, бензофуранил, необязательно замещенный галогеном или низшим алкокси;
R2 водород или C1 C6-алкил;
R3 группа C1 C6-алкил или -(CH2)a-R4, где a 4, R4 -NR11R12, R11 и R12 вместе с атомом азота образуют кольцо
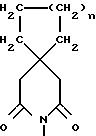
n 1 или 2,
или их энантиомеров и физиологически приемлемых солей, отличающийся тем, что соединение формулы II

где X представляет уходящую группу, и R, R2 и R3 имеют значения, определенные выше для соединения формулы I, подвергают превращению с помощью реакции с использованием переходного металла с нулевой валентностью M0 и подходящего арил-заместителя, такого как триалкил-арилстаннан с образованием соединения формулы I, после чего в случае необходимости соединение, полученное в виде основания, превращают в физиологически приемлемую кислотно-аддитивную соль или соединение, полученное в виде соли, превращают в основание или в иную физиологически приемлемую кислотно-аддитивную соль, полученную изомерную смесь в случае необходимости разделяют на чистые изомеры.
Комментарии