Способ торможения ретровирусных протеаз - RU2028155C1
Код документа: RU2028155C1
Чертежи



Описание
Изобретение относится к биохимии и касается способа торможения ретровирусных протеаз путем введения ингибитора протеаз.
Известно лишь небольшое количество ингибиторов ВИЧ-протеаза. Первым из них был открыт пепстатин А. IC50 которого равна примерно 0,5 ммоль. Позже было описано несколько других ингибиторов, обладающих от умеренной до высокой активностью.
Большие дозы пепстатина А оказывались в состоянии в процессе биосинтеза тормозить образование нуклеопротеина р24.
Был обнаружен новый класс соединений, тормозящих в ферментном тексте с высокой эффективностью ВИЧ-протеазу.
Согласно данному изобретению предлагается способ торможения ретровирусных протеаз путем введения ингибитора протеаз, отличающийся тем, что в
качестве ингибитора используют соединение общей формулы
R




-
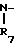
l = 0 или 1; m = 1;
А - радикал формулы IV;
A* - радикал формулы IV*
D - (Е)n - (F)o - (G)p- (IV)
D* - (E*)n* - (F*)o* - (G*)p*- (IV*) где D - радикал R1 или радикал формул V, VI или VII;
D* - радикал R*1 или радикал формулы V*, VI* или VIII*
R1-




R1-

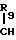


R1-O-


где n, n*, 0, 0*, P, P* - независимо друг от друга равны 0 или 1;
Е, Е*, F, F*, G и G* - независимо друг от друга означают аминокислоты из группы: Val, Lуs, Lуs (Z), Phe, Сhg, Ser, Asn, Gly, Ile, Тbg, Nva или Npg;
R1 и R*1 независимо друг от друга означают: водород, карбоксил, метилсульфонил, трет. бутилсульфонил, трет. -бутоксикарбонил, 2-гидроксиэтилсульфонил, 1,2,3-тригидроксипропил, 1,2,3-триацетоксипропил, бензилоксикарбонил, 4-метилфенилсульфонил, 4-хлорбензилтио, бензилсульфонил, 4-хлорбензилсульфонил, гексадецилсульфонил, 4-амино-1-пиперидинилсульфонил, N-трет. -бутоксикарбонил-4-амино-1-пиперидилсуль-фонил, 4-амино-1-пиперидил-карбонил, N-трет.бутоксикарбонил-4-амино-1-пиперидил-карбонил, 2-амино-3-фенил-пропил, N-трет.бутоксикарбонил-2-амино-3-фенилпропил, 2-амино-1-гидрокси-4-метилпентил, деоксифруктозил-1, маннофуранозил, 4-аминоциклогексилкарбонил, 2-пиридилацетил, 4-пиридилтиоацетил, 2-хинолилкарбонил, 1-нафтилацетил, 1-нафтилоксиацетил, 1-(4-пиридил)-этилсульфонил, 12-аминодеканоил, 4-(N-оксидопиридил), трет.-бутилтио, 4-z-н-трет.-бутоксикарбениламиноциклоге- ксилкарбонил, 4-пиридил, тетрадеканоил, фенил, амино- или трет.-бутоксикарбониламино-группа;
R2 и R*2 означают независимо друг от друга: водород, 2-(4-пиридил)-этил, изопропил, изобутил, н-пентил, бензил, 3, 4-метилендиоксибензил, 2,4-диметоксибензил, 4-трет.-бутилбензил, 2-фенилэтил или циклогексилметил;
R3 и R*3, R4 и R*4, R6 и R*6, R7, R10 и R*10 означают водород;
R5 и R*5 означают независимо друг от друга водород или гидроксил;
R8 и R*8 означают водород или вместе с R9 или R*9 и связанными с ними атомами образуют 1,2,3, 4-тетрагидрохинолиндиил-3,4;
R9 и R*9 независимо друг от друга означают водород, гидроксил, ацетокси, н-пропил, изопропил, изобутил, аминометил, 4-аминобутил, гидроксиметил, трет. -бутоксиметил, аминокарбонилметил, 2-бензилоксикарбонилэтил, трет. -бутоксикарбониламинометил, z-нафтилметил, трет.-бутилтиометил, 4-бензилоксикарбониламинобутил, N,No -ди-(бензилоксикарбонил)-гуанидинопропил, циклогексил, циклогексилметил, бензил, 2-фенилэтил, 4-гидроксибензил, 4-метоксибензил, 4-трет. -бутоксибензил, 1-нафтилметил, 2-тиенилметил, 1-имидазолилметил, 3-индолилметил, 4-пиридилметил, 4-(N-оксидопиридил)-метил, 2-метилтиоэтил, 2-метилсульфонилэтил, трет.-бутилсульфонилметил или 2-карбоксиэтил;
R11 и R*11 независимо друг от друга означают водород, гидроксил, ацетокси, причем в формуле I одна или несколько амидных групп (-CONH-) в основной цели могут быть заменены на группу -CH2NH- или -CН(OН)СH2-, или физиологически переносимые соли этих соединений.
Под солями соединений формулы I имеются в виду, в частности, фармацевтически приемлемые или нетоксичные соли. Такие соли могут быть, например, образованы соединениями формулы I, имеющими кислые группы, например карбокси-группы, и щелочными или щелочноземельными металлами, такими как, например Na, K, Mg и Са, а также физиологически приемлемыми органическими аминами, такими как, например, триэтиламин и трис-(2-окси-этил)-амин.
Соединения формулы I, имеющие основные группы, например амино- или гуанидино-группы, образуют соли с неорганическими кислотами, такими как, например, серная, соляная или фосфорная кислоты, или с органическими карбоновыми или сульфоновыми кислотами, такими как, например, уксусная, лимонная, бензойная, малеиновая, фумаровая, винная и n-толуолсульфоновая кислоты. Предпочтительными являются соединения формулы I, у которых остатки и символы с или без звездочек соответственно одинаковы. Предпочтительными далее являются соединения формулы I, симметричные относительно C2.
Соединения формулы I получают тем, что осуществляют сочетание фрагмента с концевыми карбоксильными группами или его реакционно-способного производного с соответствующим фрагментом со свободными аминными группами, отщепляют введение (при желании) временно для защиты других функциональных групп защитные группы и полученное соединение при желании переводят в его физиологически приемлемую соль.
Фрагменты соединения формулы I с
концевыми карбоксильными группами могут иметь, например, следующую формулу: D-OН (VIII) D-E-OН (IХ) D-F-OH (Х) D-G-OH (ХI) D-Е-F-OН (ХII)_ D-Е-G-OH (ХIII) D-F-G-OH (ХIV) D-Е-F-G-OН (ХIVа)
То
же самое относится и к аналогичным, помеченным звездочкой остаткам.
Фрагменты соединения формулы I с концевой амино-группой могут иметь, например, следующую формулу: Н-Z-Н (ХV) Н-G-Z-G*-Н (ХVI) Н-F-Z-F*-Н (ХVIa) Н-Е-Z-Е*-Н (ХVIв) H-F-G-Z-G*-F*-H (ХVII) H-E-G-Z-G*-E*-H (ХVIIa) Н-Е-F-Z-F* -Е*-Н (ХVIIв) H-E-F-G-Z-G*-F*-E*-Н (ХVIII) причем Z означает остаток формулы R




В случае несимметричных молекул целевого соединения можно использовать и другие, помимо формул ХV-ХVIII, фрагменты, концевая амино-группа которых может быть защищена.
Методы, которые могут быть использованы для получения амидной связи, широко описаны. Предпочтительно использовать следующие методы: метод активированного эфира с N-оксисукцинимидом, 1-оксибензотриазолом или 3-окси-4-оксо-3,4-дигидро-1,2,3-бензотриазином в качестве спиртового компонента, сочетание с карбодиимидом, например дициклогексилкарбодиимидом (DCC) или ангидридом н-пропанфосфоновой кислоты (РРA) и метод смешанных ангидридов с использованием пивалоилхлорида или этилового или изобутилового эфира хлормуравьиной кислоты, или реакцию сочетания с производными фосфония, например бензотриазол-1-ил-окси-трис- (диметиламинофосфонийгексафторфосфатом) (ВОP) или производными уровня, например, 2(1Н-бензотриазол-1-ил)-1,1,3,3- тетраметилуронийтетрафторбаратом (ТВТV).
Фрагменты формулы V или V* могут быть синтезированы обычными способами получения аминокислот;
формулы VI или VI* могут быть синтезированы, например, исходя из
соответствующих аминокислот, причем при этом сохраняется хиральный центр последних. Диазотирование при -20 ±50oС в среде разбавленных минеральных кислот приводит к образованию
α -бромкарбоновых кислот или через молочную кислоту к образованию α-трифторметансульфонилоксикарбоновых кислот, которые могут быть подвергнуты взаимодействию с содержащим остатки RI и RII, соответственно RI* и RII*, нуклеофилом, или получаются с использованием в качестве исходных соединений эфиров малоновой кислоты, алкилирование которых
приводит к образованию моно- или дизамещенных малоновых эфиров, которые после омыления путем декарбоксилирования переводятся в нужные производные;
формулы VII или VII*
синтезируются, исходя из соответствующих α -аминокислот, причем при этом сохраняется хиральный центр последних. Диазотирование при -20 ±50oC в среде разбавленных минеральных
кислот приводит к образованию молочных кислот, которые могут быть подвергнуты взаимодействию с электрофилом, содержащим заместитель RI или RI*.
Фрагменты формул IХ, Х, ХII и ХIII, ХIV и ХIVa cинтезируют известными методами, использующимися для получения аминокислот и пентидов.
Фрагменты формулы ХV получают, используя в качестве исходных соединений оптически активные α -аминокислоты или сахара или их производные. Так, например, для получения фрагментов с m = 1, l = 0, R5= R5* = OН и R6 = R6* = H аминокислоты известным образом переводят в N-защищенные альдегиды аминокислот, затем последние восстанавливают с помощью подходящих металлов, солей металлов или электрохимически в N-защищенные диаминодиолы. С этой целью N-защищенные альдегиды растворяют, например, в тетрагидрофуране, и при -30 ±60oC, предпочтительно при -10+30oC, путем добавления раствора йодида самария (II) в тетрагидрофуране переводят в N-защищенные диаминодиолы, при отщеплении защитных групп образуются соединения формулы (ХV). В результате получают смесь диастереомеров относительно центров с OН-группами, которую можно разделить на отдельные изомеры известными способами, например путем фракционной кристаллизации и/или с помощью хроматографии.
При синтезе с использованием в качестве исходных соединений сахаров или их производных хиральные центры исходного материала сохраняются или инвертируются. ОН-группы, которые необходимо сохранить, защищают соответствующим образом.
Остальные же активируют путем взаимодействия, например, с хлорангидридом сульфоновой кислоты, и могут быть заменены на нуклеофил. Целевые продукты образуются при этом в виде индивидуальных стереоизомеров.
Используя в качестве исходного соединения, например D-маннит, окси-группы пролиола в положении 3 и 4 защищают путем обработки смесью ацетона и серной кислоты и затем водного раствора уксусной кислоты, получая в результате ацетонит. В результате взаимодействия обеих концевых OH-групп со смесью хлорангидрида n-толуолсульфокислоты и пиридина и последующей обработки карбонатом калия в метаноле получают 1,2R-5R,6-диэпокси-3,4-0-изопропилиден-3R,4R-диол. Обработка этого диэпоксида купратами в среде, например, тетрагидрофурана приводит к раскрытию эпоксида и введению заместителей в положении 1 и 6. После активации OН-групп в положениях 2 и 5 путем взаимодействия с, например, хлорангидридом сульфоновой кислоты они обмениваются путем взаимодействия с азидом. Восстановление обеих азидных групп путем, например, каталитического гидрирования и отщепление ацетонидных защитных групп с помощью смеси HCl и метанола приводит к образованию производных остатка (ХV).
Фрагменты формулы ХV с m = 1, l = 1 и y-остатку формулы III получают путем взаимодействия N-защищенного альдегида аминокислоты (см. выше) в восстановительной среде (например NaBН3СN) c cоответствующим амином. Для этой цели альдегиды растворяют, например, в метаноле и подвергают взаимодействию, например, с ацетатом аммония, и, например, с цианоборгидридом натрия в качестве восстановителя. После отщепления защитных групп получают целевой фрагмент.
Для получения фрагментов формулы ХV с m = 0, l = 0, R5 = OH, R6 = Н соответствующие нитро-производные депротонируют путем взаимодействия с основаниями, например тетраметилгуанидином, и затем проводят реакцию присоединения к N-защищенному альдегиду аминокислоты (см. выше). После восстановления нитро-группы с помощью, например, никеля Ренея и отщепления защитных групп получают соединения формулы ХV в виде диастереомеров, которые разделяют вышеописанным образом.
Фрагменты формулы ХVI, ХVIa, ХVIв, ХVII, ХVIIа, ХVIIв и ХVIII cинтезируют общеизвестными методами получения аминокислот и пептидов.
Пептидные аналоги этого типа могут быть получены известными способами.
Необходимые для получения соединений формулы I предварительные и заключительные операции, такие как введение и отщепление защитных групп, являются известными. Соли соединений формулы I, имеющих солеобразующие группы, получают известным образом, например путем взаимодействия соединения формулы I с основной группой со стехиометрическим количеством подходящей кислоты или путем взаимодействия соединения формулы I c кислой группой со стехиометрическим количеством подходящего основания.
Смесь стереоизомеров, в частности смеси диастереомеров, которые могут образовываться при синтезе соединений формулы I, могут быть разделены известными способами, путем фракционной кристаллизации или с помощью хроматографии.
Соединения формулы I в соответствии с данным изобретением оказывают тормозящее действие на ферменты, в частности они тормозят действие ретровирусных аспартилпротеаз, например ВИЧ-протеаз. Эту их ингибирующую активность, проявляющуюся в пределах концентраций от милли- до наномолярных, можно определить следующим образом.
Принцип испытаний.
В качестве субстрата ВИЧ-протеазы ранее использовался, в частности гептапептид Н-ser-phe-asn-phe-pro-gln-Ile-OH. ВИЧ-протеаза расщепляет указанный субстрат между вторым phe и pro.
Совершенно неожиданно было обнаружено, что замена в этой последовательности пролина на 5-оксапролин дает субстрат, который может расщепляться ВИЧ-протеазой значительно быстрее, что дает возможность проводить анализ быстрее и с меньшим расходом фермента.
Пропись для проведения испытаний ингибиторов ВИЧ-протеаз
а) Приготовление
раствора субстрата
2 мг H-ser-phe-Asn-phe-opr-gln-Ile-OН (Н-opr-OН=5-оксапропил) растворяют в 1 мл буфера MGТЕ15 (возможно применение при этом ультразвука) и фильтруют полученный раствор
через стерильный фильтр (0,45 мкм).
б) Приготовление раствора ингибитора.
Берут навеску ингибитора в количестве, в 2,5 раза превышающем нужную молярность в 1 мл раствора, и растворяют ее в ДМСО (10% от конечного объема). Разбавляют полученный раствор буфером MGТЕ15 до конечного объема и фильтруют его через стерильный фильтр (0,45 мкм).
в)
Приготовление раствора протеазы
5 мкл раствора ВИЧ-протеазы разбавляют до нужной концентрации буфером MGТЕ25.
г) Проведение испытаний.
Отбирают пипеткой до 10 мкл раствора субстрата в пробирки (16х100) с завинчивающейся пробкой. Для слепого опыта в пробирку отбирают пипеткой 10 мкл буфера MGТЕ15, содержащего 10% ДМСO. В остальные пробирки добавляют до 10 мкл раствора ингибитора. Пробирки выдерживают в течение 5-10 мин при 37oC, после чего к каждой пробирке добавляют по 5 мкл раствора протеазы. После протекания реакции в течение 2 ч при 37oC из каждой пробы отбирают пипеткой по 10 или 20 мкл (в зависимости от чувствительности прибора для высокопроизводительной жидкостной хроматографии), заливают их в микропузырьки и разбавляют 120 мкл используемой для высокопроизводительной жидкостной хроматографии подвижной фазы.
д) Условия проведения высокопроизводительной жидкостной хроматографии Подвижная фаза: 80% 0,1 М фосфорной кислоты, рН 2,5 20% (мас.%) ацетонитрила Колонка: Merck

Используемый растворитель
1) буфер MGТЕ15:
20 мМ морфолиноэтансульфокислоты (MES)
15% (W/V)
глицерина
0,1 об.% тритона х100
5 мМ ЭДТА
0,5 М NaСl
1 мМ фенилметилсульфонилфторида (PMSF)
2) буфер MGТЕ25
Состав такой же, как и буфера MGТЕ15,
с той разницей, что он содержит 25% (W/V) глицерина и дополнительно 1 мМ дитиотрейта (DТТ).
В колбу Эрленмейера загружают MES, ЭДТА, NaCl, ДТТ и PMSF, растворяют их в небольшом количестве воды и устанавливают рН раствора равным 6. В мерную колбу вносят соответствующую навеску глицерина и отмеривают пипеткой

3) Подвижная фаза для высокопроизводительной жидкостной хроматографии.
Готовят из ортофосфорной кислоты (FLUKA, ч.д.а.) 0,1 М раствор. С помощью триэтиламина (FLUKA, ч.д.а) рН этого раствора устанавливают равным 3,5. Раствор взвешивают и добавляют к нему соответствующее количество ацетонитрила (под тягой). Смесь хорошенько перемешивают и продувают в течение примерно 5 мин гелием для удаления газов.
ж) Оценка
При выбранных условиях гептапептиды отделяются от образующегося в результате ферментативного расщепления тетрапептида с концевым N-атомом. Процентное содержание пиков тетрапептида по отношению к сумме
тетрапептида и гептапептида характеризует скорость расщепления.
В табл.1 приведены значения IC50, представляющие собой те концентрации ингибитора, при которых скорость расщепления снижается в два раза.
Целевой пептид получали ступенчато по Fmoc-методу, с использованием этерифицированной Fmoc-Ile-OH n-бензилоксикарбонзилово-спиртовой смолы фирмы Novabiochem (загрузка около 0,5 ммоль/г cмолы). Загрузка смолы составляла 1 г. Синтез проводили по программе синтеза, модифицированной для Fmos-метода.
Использовали следующие производные аминокислот: OH, Fmoc-opr-OH, Fmoc-phe-oobt, Fmoc-asn-OH и Fmoc-ser/трет-бут/-oobt. Для синтеза Fmoc-opr-OH синтезировали H-opr-OtBu по методу Vasella и др. (J.С.S. Сhem. Сomm. 1981, с. 97-98) и подвергали его затем взаимодействию с Fmoc-osu в смеси диоксана и воды при соотношении 1:1 в присутствии NaHCO3. При последующем расщеплении третбутилового эфира трифторуксусной кислотой образуется Fmoc-opr-OН.
В патроны реактора загружены по 1 ммоль производного аминокислоты со свободной карбоксильной группой и 0,95 ммоль НOObt. Предварительная активация этих аминокислот осуществлялась непосредственно в патронах путем растворения в 4 мл ДМФ и добавлением 2 мл 0,55 М раствора диизопропилкарбодиимида в диметил. НOObt - эфиры других аминокислот растворяли в 6 мл NMP и затем так же, как и в случае предварительно активированных in situ аминокислот, осуществляли сочетание с предварительно деблокированной 20%-ным раствором пиперидина в ДМФ смолой. После окончания синтеза проводили отщепление пептида (при одновременном удалении защитных групп боковых цепей) путем обработки трифторуксусной кислотой с использованием тиоанизола и этандитиола для связывания катионов. Остаток после отгонки трифторуксусной кислоты многократно настаивали с уксусным эфиром и центрифугировали.
Остаток после центрифугирования подвергали хроматографическому разделению на декстрановом геле с использованием в качестве подвижной фазы 10% -ной уксусной кислоты. Содержащую чистый пептид фракцию упаривали и высушивали с помощью сублимационной сушки.
Масс-спектр (FAB): 854 (М+Н+)
Аминокислотный анализ: asp: 0,98, сеr: 0,80, glu: 1,00, Ile: 1,05, phe: 2,10, NH3: 1,76.
Изобретение относится к применению соединений формулы I в качестве лекарственных средств и фармацевтических препаратов, содержащих эти соединения. Предпочтительным является их применение для лечения приматов, в частности человека.
Фармацевтические препараты содержат эффективное количество активного вещества формулы I в комбинации с неорганическим или органическим фармацевтическим приемлемым носителем. Предлагаемые препараты можно вводить в нос, внутривенно, подкожно или перорально. Дозировка активного вещества зависит от вида теплокровного, его веса, возраста и способа введения. Фармацевтические препараты в соответствии с изобретением получают известными способами: путем растворения, смешения, гранулирования, таблетирования.
Для получения оральных форм активные соединения смешивают с используемыми обычно для этих целей вспомогательными добавками, такими как носители, стабилизаторы или инертные разбавители, и с помощью обычных методов формируют из смесей нужные формы, например таблетки, драже, капсулы, водные, спиртовые или масляные суспензии или водные, спиртовые или масляные растворы. В качестве инертных носителей можно использовать, например, гуммиарабик, оксид магния, карбонат магния, фосфат калия, молочный сахар, глюкозу, стеарилфумарат магния или крахмалы, в частности кукурузный крахмал. При этом композиции можно получать как в виде сухого, так и влажного гранулята. В качестве масляных носителей или растворителей можно использовать, например, растительные или животные масла, такие как подсолнечное масло и рыбий жир.
Для подкожного или внутривенного введения активные соединения или их физиологически приемлемые соли, при желании в комбинации с обычно использующимися для таких целей материалами, такими как агенты растворения, эмульгаторы или другие вспомогательные добавки, готовят в виде растворов, суспензий или эмульсий. В качестве растворителей можно использовать воду, физиологические растворы поваренной соли или спирты, например, этанол, пропандиол или глицерин, а также растворы сахаров, таких как глюкоза или маннит, или смеси различных из перечисленных растворителей.
Возможно также использование ретардных композиций для инъекций. В качестве лекарственных препаратов можно использовать, например, кристаллические суспензии, микрокапсулы, палочки или имплантаты, причем последние могут быть выполнены из ткущихся полимеров, в частности биоразлагающихся полимеров, например на базе сополимеров полимолочной и полигликолевой кислот, или из человеческого альбумина.
Сокращения, использующиеся для обозначения аминокислот, соответствуют принятому в химии пептидов трехбуквенному коду (описанному, например, в Еur. J. Biochem. 138. 1984, 9-37). Если это не оговорено, упоминаемые аминокислоты имеют α -конфигурацию.
Другие сокращения:
FAB - бомбардировка быстрыми атомами
М
- молекулярный пик
БОК - трет.-бутоксикарбонил
П р и м е р 1. N,N'-бис-(третбутоксикарбонил-L-фенилаланил-L- валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
100 мг N,N'-бис-(L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорида, 111мг N-третбутоксикарбонил-L-фенилаланина, 0,57 мл N-этилморфолина и 60 мг оксибензотриазола растворяли в 1,5 мл диметилформамида. После добавления к раствору 85 мг ЕDAC при 0oC перемешивание продолжали в течение еще часа при 0oC и затем в течение ночи при комнатной температуре. После этого растворитель отгоняли в вакууме, остаток растворяли в ЕЕ и проводили экстракцию насыщенным раствором KHCO3, 10%-ным раствором KHSO4 и водой. Органическую фазу высушивали безводным NaSO4 и упаривали. Остаток перекристаллизовывали из смеси этанола и воды. Выход составлял 92 мг.
MS (FAB): 993 (М+H)+
, 975, 893, 793
ЯМР (270 мГц, ДМСО
П р и м е р 2. N,N'-бис-(L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диолдигидрохлорид
220 мг N, N'-бис-(третбутоксикарбонил-L-валил)-2S, 5S-диамино-1,6-дифенил-3,4-0-изопропилиденгексан-3R, 4R-диола растворяли в 10 мл примерно 3 н. раствора НСl в смеси диоксана и
метанола при соотношении 1:1 и перемешивали раствор в течение часа при комнатной температуре. Летучие компоненты раствора отгоняли в вакууме и высушивали остаток в глубоком вакууме. Полученный
материал использовали без дополнительной очистки на последующей стадии. Выход 184 мг.
Масс-спектр (FAB): 499 (М+Н)+, 481, 463.
П р и м е р 2.1. N,N'-бис-(трет.-бутоксикарбонил-L-валил)-2S, 5S-диамино-1,6-дифенил-3,4-0-изопропилиденгексан-3R, 4R-диол.
136 мг 2S, 5S-диамино-1,6-дифенил-3,4-0-изопропилиденгексан-3R, 4R-диола, 0,54 мг NEM и 260 мг N-третбутоксикарбонил-L-валина растворяли в 2 мл сухого этилацетата. При -10oC к приготовленному раствору добавляли 0,97 мл 50%-ного раствора ангидрида н-пропил фосфоновой кислоты и этилацетата. Смесь перемешивали в течение часа при 0oC и затем в течение ночи при комнатной температуре. После этого раствор разбавляли этилацетатом и последовательно экстрагировали насыщенным раствором NaHCO3 10%-ным раствором KHSO4 и водой. Органическую фазу высушивали над безводным MgSO4, упаривали в остаток, подвергали очистке с помощью хроматографии на силикагеле (подвижная фаза: смесь дихлорметана и метанола при соотношении 97:3). Выход целевого соединения 230 мг.
Масс-спектр (FAB): 739 (М+Н)+, 681, 639, 569, 539.
П р и м е р 2.2. 2S, 5S-диамино-1,6-дифенил-3,4-0-изопропилиденгексан-3R, 4R-диол.
2,3 г 2S, 5S-диазидо-1,6-дифенил-1,6-0-изопропилиденгексан-3R, 4R-диола растворяли в 50 мл метанола и приготовленный раствор подвергали гидрированию в течение 2 ч при нормальном давлении в присутствии примерно 0,2 г палладия на угле (10%). Катализатор затем отфильтровывали и после упаривания раствора остаток подвергали хроматографическому разделению на силикагеле (подвижная фаза: смесь дихлорметана и метанола при соотношении 99:1). Выход 1,33 г.
Масс-спектр (FAB): 341 (М+Н)+
ЯМР (270 мГц, ДМСО
П р и м е р 2.3. 2S, 5S-диазидо-1,6-дифенил-3,4-0- изопропилиденгексан-3R, 4R-диол
8,5 г 2R, 5R-ди-(4-нитрофенилсульфонилокси)-1,
6- дифенил-3,4-0-изопропилиденгексан-3S, 4S-диола растворяли в 300 мл ДМФ и приготовленный раствор нагревали в течение 4 ч при 50oC примерно 9,2 г NaN3 и 6,3 г 18-крон-6. Большую
часть растворителя отгоняли затем в вакууме, остаток растворяли в эфире и проводили экстракцию водным раствором NaHCO3. После промывки водой органическую фазу высушивали и упаривали.
Остаток подвергали хроматографическому разделению на силикагеле (подвижная фаза: cмесь толуола и н-гектана в соотношении 2:5-2:3). В результате получали 2,37 г целевого соединения.
ЯМР
(270 мГц, ДМСО
П р и м е р 2.4. 2R, 5R-ди-(4-нитрофенилсульфонилокси) -1,6-дифенил-3,4-0-изопропилиденгексан-3S, 4S-диол.
5,6 г 2R, 5R-диокси-1,6-дифенил-3,4-0-изопропилиденгексан-3R, 4R-диола, 7,9 г 4-диметиламинопиридина растворяли в 300 мл хлороформа. К приготовленному раствору добавляли при комнатной температуре 14,5 г п-нитробензолсульфонилхлорида и перемешивали смесь в течение 3 ч при 50oС. После этого ее разбавляли метиленхлоридом и экстрагировали последовательно растворами бикарбоната, KHSO4 и NaCl. После высушивания органической фазы ее упаривали. Выход: 11,8 г.
Масс-спектр (FAB): 713 (М+Н)+, 697, 510.
ЯМР (270 мГц, ДМСО < D6 >): 1,42 (с, 6Н); 2,87 (двойной д, 15 Гц, 9 Гц, 2Н); 3,11 (двойной д, 15 Гц, 3 Гц, 2Н); 4,41(c,2H); 5,07 (дм, 9 Гц, 2Н); 6,95-7,11 (м, 10Н); 7,73 (д, 9 Гц, 4Н); 8,18 (д, 9 Гц, 4Н).
П р и м е р 2.5. 2R, 5R-диокси-1,6-дифенил-3, 4-0- изопропилиден-3R, 4R-диол 1,12 г 1,2R-5R,6-диэпокси-3,4-0-изопропилиден-3R-4R-диола добавляли в атмосфере аргона при -78oC к раствору 36 ммоль (С6Н5)2 СuLi в 60 мл сухого эфира. После этого удаляли охлаждающую баню и при перемешивании нагревали смесь до комнатной температуры. Затем ее смешивали с 250 мл этилацетата и трижды экстрагировали смесью 25%-ного аммиака и хлорида аммония. Фазу этилацетата промывали раствором NaCl, высушивали и упаривали. Остаток подвергали очистке с помощью хроматографии на силикагеле (подвижная фаза: смесь дихлорметана и этилацетата при соотношении (97:3)-(90:10). В результате получали 1,86 г целевого соединения.
Масс-спектр (FAB): 343 (М+Н)+, 327, 285, 267.
ЯМР (270 мГц,
ДМСО
П р и м е р ы 3-5. 3) N,N'-бис-(трет.-бутоксикарбонил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
4) N,N'-бис-(третбутоксикарбонил)-2S, 5S-диамино-1, 6-дифенилгексан-3S, 4S-диол.
5) N,N'-бис-(трет.-бутоксикарбонил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4S-диол.
17 г третбутоксикарбонил-L-фенилаланина растворяли в 500 мл сухого тетрагидрофурана и охлаждали раствор до 0oС в атмосфере аргона. Затем к нему добавляли в течение примерно 20 мин 1 л 0,1 М. раствора SmI2 в тетрагидрофуране и перемешивали смесь в течение 30 мин при комнатной температуре, подкисляли 0,1 н. водным раствором НСl до рН 1-2, разбавляли ЕЕ, отделяли органическую фазу и проводили из нее экстракцию 0,1 н. НСl, дважды раствором Na2S2O3 и дважды водой. После высушивания над MgSO4 ее упаривали и подвергали хроматографии на силикагеле (подвижная фаза: смесь этилацетата и петролейного эфира при соотношении 1:2).
Фракцию, содержащую 3R, 4R-изомер, перекристаллизовывали из смеси этанола и воды.
Из фракции, содержавшей 3S, 4S- и 3R, 4S-изомеры, 3S, 4S-изомер можно было получать путем кристаллизации из смеси дихлорметана, изопропилового эфира и гептана. Для получения 3R, 4S-изомера меточный раствор подвергали хроматографическому разделению на силикагеле R Р18 (подвижная фаза; смесь ацетонитрила и воды при соотношении 4:6).
Выход: 3R, 4R-изомер 1,61 г; 3S, 4S-изомер 1,00 г; 3R, 4S-изомер 0,71 г.
Rf: силикагель, смесь ЕЕ и гексана при соотношении 1:2; 3R, 4R-изомер 0,18; 3S, 4S-изомер 0,41; 3R, 4S-изомер 0,39.
Масс-спектр (FAB): 501 (М+Н)+, 401, 345, 327, 301 - 3R, 4R-изомер; 501 (М+Н)+, 401, 345, 327, 301 - 3S, 4S-изомер; 501 (М+Н)+, 401, 345, 327 - 3R, 4S-изомер.
1Н-ЯMP (270 мГц,
ДМСO
Идентификацию 3R, 4S-изомера осуществляли по двойному набору сигналов, 3R, 4R- и 3S, 4S-изомеры различали путем сравнения с синтетическими образцами, полученными из D-маннита (см. пример 3.1). Определение констант сочетания после отщепления третбутоксикарбонильных групп и перевода изомеров с помощью фосгена в двойную 2-оксазолидиноновую систему давало согласующие результаты.
П р и м е р 3.1. Определение стереоконфигурации изомеров в соответствии с примерами 3-5.
N, N'
-бис-(трет.-бутоксикарбонил)-2S, 5S-диамино-1,6-дифенил-гексан-3R, 4R-диол
140 мг 2S,5S-диамино-1,6-дифенил-3,4-0- изопропилиденгексан-3R,4R-диола растворяли в смеси 5 мл 1 н. НСl в метаноле
и 5 мл 5 н. НСl в диоксане и перемешивали раствор в течение 4 ч при комнатной температуре. Летучие компоненты отгоняли в вакууме. Остаток высушивали в глубоком вакууме и полученный 2S, 5S-диамино-1,
6-дифенилгексан-3R, 4R-диолдигидрохлорид (масс-спектр (FAB): 301 (М+Н)+ cвободного основания) без дополнительной очистки использовали на последующей стадии.
45 мг 2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диолдигидрохлорида растворяли в 5 мл сухого дихлорметана и перемешивали в течение 3 ч при комнатной температуре с 40 мкл триэтиламина и 75 мг дитретбутилового эфира пироугольной кислоты. Смесь затем разбавляли дихлорметаном и подвергали ее экстракции растворами KHSO4, NаHCO3 и NaCI. После высушивания над безводным Na2SO4 органическую фазу упаривали и остаток очищали с помощью хроматографии на силикагеле, используя в качестве подвижной фазы смесь ацетонитрила и DCM в соотношении 1:8. Выход 23 мг.
Масс-спектр (FAB): 501 (М+H)+, 401, 345, 327, 301.
Полученное соединение было идентичным наиболее полярному из изомеров в соответствии с примерами 3-5.
П р и
м е р 6. N,N'-бис-(третбутоксикарбонил-L-фенилаланил-L- валил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол
38 мг N,N'-бис-(третбутоксикарбонил-L-валил)-2S, 5S-диамино-1,
6-дифенилгексан-3S, 4S-диола обрабатывали в течение 30 мин 5 н. HCl в диоксане. Летучие компоненты удаляли в вакууме и остаток высушивали. Полученный таким образом N, N'-бис-(L-валил)-2S,
5S-диамино-1,6-дифенилгексан-3S, 4S-диол-дигидрохлорид вместе с 40 мг третбутоксикарбонилфенилаланина, 22 мг гидроксибензотриазола и 51 мг тетрафторбората 2-(1Н-бензотриазолил-1)-1,1,3,
3- тетраметилуронил (ТБТУ) растворяли в 1 мл сухого диметилформамида. К раствору добавляли 60 мкл этилдиизопропиламина и перемешивали смесь в течение 15 мин при комнатной температуре. Диметилформамид
отгоняли, остаток растворяли в этилацетате и экстрагировали последовательно растворами KHSO4, NaHCO3 и водой. После высушивания над MgSO4 органическую фазу
концентрировали, причем при этом происходило выпадение кристаллического осадка, который отфильтровывали, промывали эфиром и получали в результате 30 мг целевого соединения.
Масс-спектр (FAB): 1015 (М+Na)+, 993 (М+H)+, 893, 793.
ЯМР (270 мГц, ДМСО < D6 >): 0,79 (м, 12Н); 1,28 (с, 18Н); 1,85 (м, 2Н); 2,68-2,82 (м, 4Н); 2, 85-3,03 (м, 4Н); 3,37 (м, 2Н); 4,00-4,13 (м, 4Н); 4,21 (м, 2Н); 4,66 (д, 7 Гц, 2Н); 7,03 (д, 7Гц, 2Н); 7,05-7,34 (м, 2OН); 7,62 (д, 7Гц, 2Н); 7,68 (д, 8Гц, 2Н).
П р и м е р 7. N,N'-бис-(третбутоксикарбонил-L- фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4S-диол.
Целевое соединение синтезировали по способу, аналогичному описанному в примере 6, используя в качестве исходного соединения N,N'-бис-(требутоксикарбонил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4S-диол.
Масс-спектр (FAB), 1015 (М+Na)+, 993 (М+H)+, 893, 793.
ЯМР (270 мГц, ДМСО < D6 >): 0,68-0,85 (м, 12Н); 1,28 (с, 9Н); 1,30 (с, 9Н); 1,75-2,03 (м, 2Н); примерно 2,5-3,30 (м, 8Н); примерно 3,3-3,51 (м, 2Н); 4,05-4,30 (м, 5Н); 4,43 (м, 1Н); 4,74 (д, 4 Гц, 1Н); 5,32 (д, 7 Гц, 1Н); 6,93-7,35 (м, 22Н); 7,61 (д, 8 Гц, 1Н); 7,67 (д, 7 Гц, 1Н); 7,85 (д, 8 Гц, 1Н); 7,92 (д, 7 Гц, 1Н).
П р и м е р 8. N,NI-бис-(требутоксикарбонил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол.
164 мг N, N'-бис-(требутоксикарбонил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диола обрабатывали в течение 1,5 ч при комнатной температуре 10 мл 5 н. НСl в диоксане. Летучие компоненты затем удаляли в вакууме и остаток высушивали. Полученный таким образом 2S, 5S-диамино-1,6-дифенил-гексан-3S, 4S-диол дигидрохлорид растворяли вместе с 178 мл третбутоксикарбонил-L-валина и 0,56 мл NEM в 15 мл сухого диметилформамида. К полученному раствору добавляли при -5oC 0,53 мл 50%-ного раствора ангидрида н-пропил-фосфоновой кислоты (АПФ) в этилацетате, перемешивали смесь в течение часа при 0oC и затем в течение ночи при комнатной температуре, растворитель отгоняли, в ротационном испарителе, остаток растворяли в этилацетате и подвергали экстракции водой, растворами NaHCO3 и KHSO4 и снова водой. После высушивания над безводным Na2SO4 органическую фазу упаривали в вакууме. При обработке остатка диэтиловым эфиром выпадал кристаллический осадок продукта, который перекристаллизовывали из смеси этанола и воды. Выход 59 мг.
Масс-спектр (FAB): 699 (М+H)+, 599, 499.
П р и м е р 9. N,N' -бис-(третбутоксикарбонил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R,4S-диол.
Синтез осуществляли таким же образом, как и в примере 8, используя в качестве исходного соединения N, N'-бис-(третбутоксикарбонил)-2S, 5S-диамино-1,6-дифенилгексан-3R,4S-диол.
Масс-спектр (FAB): 699 (М+H)+, 599, 499.
П р и м е р 10. N,N'-бис-(L-лизил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол-тетрагидрохлорид.
Синтез осуществляли таким же образом, как и в примере 2, из соединения в соответствии с примером 11.
Масс-спектр (FAB, LiI), 761 (М+Li)+, 755 (М+Н)+, 737.
П р и м е р 11. N,N'-бис-(Nα < третбутоксикарбонил>-L-лизил-L-валил) 2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол-дигидрохлорид.
36 мг N,N'-бис-(
Масс-спектр (FAB, LiI): 961 (М+Li)+
ЯМР (270 мГц, ДМСО < D6 >): 0,75 (д, 5 Гц, 6Н); 0,78 (д, 5 Гц, 6Н);
примерно 1,13-1,60 (м, примерно 12Н); 1,38 (с, 18Н); 1,88 (м, 2Н); примерно 2,50-2,68 (м, 2Н); 2,72-2,94 (м, 6Н); 3,72 (м, 2Н); 4,22 (м, 2Н); 4,37 (м, 2Н); 4,41-4,55 (м, 4Н);4,72 (м, 2Н); 6,76 (м, 2Н);
7,05-7,23 (м, 16Н); 7,66 (д, 8 Гц, 2Н); 8,15 (д, 9 Гц, 2Н).
П р и м е р 12. N,N'-бис-(Nα <третбутоксикарбонил-L-фенилаланил>-L-лизил)-2S, 5S-диамино-1, 6-дифенилгексан-3R, 4R-диол-дигидрохлорид.
Синтез осуществляли таким же образом, как и в примере 11.
Масс-спектр (FAB): 1051 (М+H)+, 951.
П р и м е р 13. N,N'-бис-<[2S-(1,1-диметилэтилсульфонилметил) -3-(1-нафтил)-пропионил]-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
57 мг N,N'-бис-(L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорида, 95 мг [2S-(1,1-диметилэтилсульфонилметил)-3-(1-нафтил)]-пропионовой кислоты, 41 мг гидроксибензотриазола, и 96 мг ТБТУ растворяли в 1 мл сухого диметилформамида. При комнатной температуре к раствору добавляли 0,11 мл N-этилдиизопропиламина и смесь перемешивали в течение часа. Растворитель затем отгоняли в ротационном испарителе, остаток растворяли в 30 мг этилацетата и подвергали экстракции растворами бисульфата, бикарбоната и водой. После высушивания над Na2SO4 и концентрирования полученный продукт подвергали очистке с помощью хроматографии на силикагеле (подвижная фаза: смесь дихлорметана и метанола при соотношении 97:3). В результате получали 31 мг целевого соединения.
Масс-спектр (FAB), 1153 (М+ Na)+, 1131 (М+H)+, 716.
ЯМР (270 мГц, DMCO < D6 >: 0,69 (д, 7 Гц, 6Н); 0,76 (д, 7 Гц, 6Н); 1,10 (с, 18Н); 1,86 (м, 2Н); 2,63-2,87 (м, 6Н); 3,08 (м, 2Н); примерно 3,25-3,44 (м, примерно 2Н); 3,52-3,63 (м, 2Н); 4,08 (м, 2Н); 7,32 (д, 8Гц, 2Н); 7,38-7,48 (м, 4Н); 7,47-7,62 (м, 4Н); 7,81 (м, 2Н); 7, 92 (м, 2Н); 8,12-8,25 (м, 4Н).
П р и м е р 14. N,N'-бис-(L-серил-L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R,4R-диолдигидрохлорид.
Синтез осуществляли таким же образом, как это описано в примере 16.
Масс-спектр (FAB, LiI): 973 (М+Li)+, 967 (M+H)+.
П р и м е р 15. N,N'
-бис[третбутоксикарбонил-L- (0-третбутилсерил)-L-фенилаланил-L-валил]-2S,5S-диамино-1,6-дифенил-гексан-3 R,4R
52 мг N,N'-бис-(L-фенилаланил-L-валил)-2S, 5S-диамино-1,
6-дифенилгексан-3R, 4R-диол дигидрохлорида, 18 мг гидроксибензотриазола, 15,3 мкл N-этилморфолина и 35 мл О-трет.-бутил-N-третбутоксикарбонил-L-серина растворяли в 1 мл сухого ДМФ и смешивали
полученный раствор при 0oC с 25,3 мг гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (ХДКИ). Смесь перемешивали в течение часа при 0oC и затем в течение ночи при
комнатной температуре. Растворитель затем отгоняли в ротационном испарителе, остаток растворяли в этилацетате и подвергали экстракции растворами бисульфата, бикарбоната и водой. Органическую фазу
высушивали над безводным сульфатом натрия и концентрировали. Остаток подвергали очистке с помощью хроматографии на силикагеле. В результате получали 28 мг целевого соединения.
Масс-спектр (FAB): 1301 (М+ Na)+, 1279 (М+H)+, 1261, 1179, 1079.
ЯМР (270 мГц, DMCO < D6): 0,78 (д, 7 Гц, 6Н); 0,81 (д, 7 Гц, 6Н); 1,06 (с, 18Н); 1,38 (с, 18Н); 1,82 (м, 2Н); 2,61-2,98 (м, 8Н); примерно 3,15-3,45 (м, примерно 6Н); 3,92 (м, 2Н); 4,11 (двойной д, 8 Гц, 6 Гц, 2Н); 4,47 (м, 2Н); 4,63 (м, 4Н); 6,58 (д, 8 Гц, 2Н); 7,04-7,25 (м, 2ОН); 7,46 (д, 9 Гц, 2Н); 7,77 (д, 8 Гц, 2Н); 7,83 (d, 8 Гц, 2Н).
П р и м е р 16. N,N-бис-(L-фенилаланил-L-авлил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорид.
100 мг N,N'-бис-(третбутоксикарбонил-L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол (пример 1) обрабатывали в течение 30 мин при комнатной температуре смесью 2 мл 5 н НСl диоксана и 1 мл НСl в метаноле. Летучие компоненты удаляли в вакууме, остаток промывали эфиром и высушивали в глубоком вакууме. Выход 59 мг.
Масс-спектр (FAB): 793 (М+H)+, 775.
П р и м е р 17. N,N'-бис-(L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диолдигидрохлорид.
Синтез осуществляли с помощью способа, аналогичного описанному в примере 16.
Масс-спектр (FAB): 793 (M+H)+, 775.
П р и м е р 18. N,N'-бис-(L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4S-диол дигидрохлорид.
Синтез осуществляли с помощью способа, аналогичного описанному в примере 16.
Масс-спектр (FAB): 793 (М+H)+, 775.
П р и м е р 19. N,N'-бис-(L-серия-L-фенилаланил- L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол-дигидрохлорид.
Синтез осуществляли по способу, аналогичному описанному в примере 14.
Масс-спектр (FAВ): 967 (М+H)+.
П р и м е р 20. N,N'-бис-(L-серил-L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4S-диол дигидрохлорид.
Синтез осуществляли по способу, аналогичному описанному в примере 14.
Масс-спектр (FAB): 967 (М+H)
П
р и м е р 21. Бис-[N-(L-фенилаланил-L-валил)-2S- амино-3-фенилпропил] -аминтригидрохлорид.
Синтез осуществляли по способу, аналогичному описанному в примере 16, из соединения в соответствии с примером 22.
Масс-спектр (FAB): 776 (M+H)+.
П р и м е р 22. Бис-[(N-третбутоксикарбонил-L-фенилаланил -L-валил)-2S-амино-3-фенилпропил]-амин.
Синтез осуществляли по способу, аналогичному описанному в примере 6, из соединения в соответствии с примером 23.
Масс-спектр (FAB, LiI): 982 (M+Li)+, 976 (M+H)+.
ЯМР (270 мГц, DMCO < D6 >): 0,81 (м, 12Н); 1,29 (с, 18Н); 1,89) (м, 2Н); примерно 2,45-2,98 (м, примерно 12Н); 3,97 (м, 2Н); 4,05-4,25 (м, 4Н); 7,03 (д, 9 Гц, 2Н); 7,10-7,31 (м, 20Н); 7,65 (д, 8 Гц, 2Н); 7,84 (д, 8 Гц, 2Н).
П р м е р 23. Бис-[N-(L-валил)-2S-амино-3-фенилпропил]-амин тригидрохлорид.
Синтез осуществляли способом, аналогичным описанному в примере 16, из соединения в соответствии с примером 24.
Масс-спектр (FAB): 482 (М+H)+.
П р и м е р 24. Бис-[N-(трибутоксикарбонил-L-валил)-2S-амино-3- фенил-пропил]-амин
Синтез осуществляли с помощью способа,
аналогичного описанному в примере 16 из примера 25.
Масс-спектр (FAB): 682 (M+H)+.
ЯМР (270 МГц, DMCO < D6 >): 0,73 (д, 6 Гц, 6Н); 0,77 (д, 6 Гц, 6Н); 1,38 (с, 18Н); 1,65 (м, 1Н); 1,82 (м, 2Н); 2,42 - примерно 2,53 (м, примерно 4Н); 2,64 (двойной д, 14 Гц, 8 Гц, 2Н); 2,84 (двойной д, 14 Гц, 6 Гц, 2Н); 3,68 (м, 2Н); 3,93 (м, 2Н); 6,50 (д, 9 Гц, 2Н); 7,12-7,28 (м, 10Н); 7,62 (д, 8 Гц, 2Н).
П р и м е р 25. Бис-(N-третбутоксикарбонил-2S-амино-3- фенилпропил)-аминогидрохлорид.
9,6 третбутоксикарбонил-L-фенилаланинала, 30,5 г NH4OAc и 1,7 г NaBH3CN растворяли в 300 мл метанола и перемешивали раствор в течение 6 ч при комнатной температуре. С помощью концентрированной НСl раствор подкисляли до рН < 2. При этом продукт выпадал в осадок, который настаивали с диэтиловым эфиром и водой и высушивали в глубоком вакууме. В результате получали 3,1 г целевого соединения.
Масс-спектр (FAB): 484 (М+H)+, 428, 372.
ЯМР (270 мГц, DMCO < D6): 1,33 (с, 18Н); 2,55-2,90 (м, 8Н); 3,82 (м, 2Н); 6,75 (м, 2Н); 7,12-7,325 (м, 20Н).
П р и м е р 26. N,N'-бис-(5S-амино-4S-окси-7-метилоктаноил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол дигидрохлорид.
Синтез осуществляли по способу, аналогичному описанному в примере 16.
Масс-спектр (FAB): 643 (M+H)+, 625.
ЯМР (270 мГц, DMCO < D6 > ): 0,92 (М, 12Н); 1,43 (м, 4Н); 1,60 (м, 4Н); 1,74 (м, 2Н); 2,15 (м, 2Н); 2,26 (м, 2Н); 2,72 (двойной д, 14 Гц, 11 Гц, 2Н); 2,93 (м, 2Н ); 3,12 (дм, 2Н); 3,44 (м, 4Н); 4,03 (м, 2Н); примеpно 4,85 (м, примерно 4Н); 7,13-7,38 (м, 20Н); 7,82 (м, 6Н); 8,13 (д, 9 Гц, 2Н).
П р и м е р 26.1. N,N' -бис-(N-третбутоксикарбонил-5S- амино-7-метил-4S)-трет-бутилдиметилсилил(окси-октаноил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол.
88,5 мг N,N' -бис-третбутилоксикарбонил-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диола обрабатывали в течение 30 мин при комнатной температуре 2 мл 5 н НСl в диоксане. Летучие компоненты отгоняли в вакууме, а остаток высушивали в глубоком вакууме. Полученный 2S, 5S-диамино-1,6-дифенилгексан-3S, 5S-диол дигидрохлорид, растворяли вместе с 211 мг N-третбутилоксикарбонил-5S-амино-7-метил-4S-(третбутилдиметилсилил) -оксиоктановой кислоты, синтезированной из (5S)-5-<(1S)-1-(N-БОК-амино)-3-метилбутил> дигидрофуран-2-(3Н)-она, 72 мг гидроксибензотриазола (ГБТ) и 28,5 микротров N-этилморфолина (N-ЭМ) в 5 мл сухого диметилформамида. При 0oC к полученному раствору добавляли 101 мг ХДКИ, раствор перемешивали при этой температуре в течение часа и затем в течение ночи при комнатной температуре. Растворитель затем отгоняли в ротационном испарителе, остаток растворяли в этилацетате и подвергали экстракции растворами KHSO4, NaHCO3 и NaCl. После высушивания органическую фазу концентрировали и остаток подвергали очистке с помощью хроматографии на силикагеле (подвижная фаза: смесь дихлорметана и ацетонитрила при соотношении 5:1).
Выход 129 мг.
Масс-спектр (FAB): 1093 (M+H)+, 1071 (M+H)+, 971,871.
ЯМР (270 мГц, DMCO < D6 >): 0,02 (с, 6Н); 0,08 (с, 6Н); 0,77-0,93 (м, 3OH); примерно 1,1-1,4 (м, примерно 6Н); 1,45-1,63 (м, 4Н); 1,91 (м, 2Н); 2,02-2,16 (м, 2Н); 2,67 (двойной д, 11 Гц, 14 Гц, 2Н); 3,36 (м, 2Н); 3,42-3,56 (м, 4Н); 3,95 (м, 2Н); 4,81 (д, 6 Гц, 2Н); 6,44 (д, 8 Гц, 2Н); 7,08-7,30 (м, 10Н); 7,79 (д, 9 Гц, 1Н).
П р и м е р 27. N,N' -бис-(третбутоксикарбонил-L-фенилаланил-L- валил)-3S, 6S-диамино-1,8-ди-(4-пиридил)-октан-4R, 5R-диол.
Синтез осуществляли по способу, аналогичному описанному в примере 6, используя в качестве исходного соединения 3S, 6S-диамино-1,8-ди-(4-пиридил)-октан-4R, 5R-диол тетрагидрохлорид.
ЯМР (270 мГц, DMCO < D6 >): 0,85 (д, 6 Гц, 12Н); 1,20 (с, 18 н); 1,66 (с, 2Н); 1,78 (м, 2Н); 2,00 (м, 2Н); примерно 2,48 (м, 4Н); 2,74 (м, 2Н); 2,98 (м, 2Н); примерно 3,31 (м, 2Н); 4,08 (м, 2Н); 4,19 (м, 2Н); 4,30 (м, 2Н); 4,68 (м, 2Н); 7,01 (д, 8 Гц, 2Н); 7, 10-7,30 (м, 14Н); 7,62 (д, 8 Гц, 2Н); 7,74 (д, 8 Гц, 2Н); 8,43 (д, 4,8 Гц, 4Н).
Масс-спектр (FAB): 1023 (M+H)+ ; 923, 823.
П р и м е р 27.1. 3S, 6S-диамино-1,8-ди-(4-пиридил) -октан-4R,5R-диол тетрагидрохлорид.
Синтез осуществляли аналогичным описанному в примере 2, 2.2, 2.3 и 2.4 способом, используя в качестве исходных соединений 1,2R-5R,6-диэпокси-3,4-0-изопропилиден-3R,4R-диол и 4-пиколиллитий.
ЯМР (270 мГц, DMCO < D6 >): 1,87-2,20 (м, 4Н); 3,10 (м, 4Н); 3,29 (м, 2Н); 3,34 (д, 6 Гц, 2Н); примерно 3,3-4,5 (широкая, примерно 4Н); 8,07 (д, 7 Гц, 4Н); 8,18 (м, 6Н); 8,88 (д, 7 Гц, 4Н).
Масс-спектр (FAB): 331 (M+H)+.
П р и м е р 28. N,N'-бис-(2S-<2S-амино-3-фенил-пропил> -амино-3-метил-бутаноил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол тетрагидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAB): 765 (M+H)+.
П р и м е р 29. N,N'-бис-(2S-<2S-третбутоксикарбониламино
-3-фенилпропил>-амино-3-метилбутаноил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол
Во взятом в количестве 50 мг N,N'-бис-(третбутоксикарбонил) -2S,5S-диамино-1,
6-дифенилгексан-3S, 5S-диоло отщепляли аналогичным описанному в примере 8 способом защитные группы. Образующийся в результате 2S, 5S-диамино-1,6-дифенилгексан-3S, 5S-диол дигидрохлорид вместе с 70 мг
2S-(2S-третбутоксикарбониламино-3-фенилпропил)-амино-3-метилбутановой кислоты, синтезированной путем восстановительного сочетания третбутоксикарбонил-L-фенилаланинала и гидрохлорида L-валинметилового
эфира с NaBH3CN и последующего обычного расщепления метилового эфира, 41 мг гидроксибензотриазола и 12,6 мкг N-ЭМ растворяли в 5 мл сухого диметилформамида. При 0oС к полученному
раствору добавляли 57 мг ХДКИ и перемешивали смесь в течение часа при 0oC и затем в течение ночи при комнатной температуре. После этого диметилформамид отгоняли в вакууме, остаток
растворяли в дихлорметане и промывали растворами KHSO4, NaHCO3 и NaCl. После высушивания органической фазы и концентрирования остаток растирали с диэтиловым эфиром.
Выход 33 мг.
Масс-спектр (FAB): 965 (M+H)+, 865, 765.
ЯМР (270 мГц, DMCO < D6 >): 0,74 (д, 7 Гц, 6Н); 0,78 (д, 6 Гц, 6Н); 1,33 (с, 18Н); 1,63 (м, 2Н); 1,94-2,16 (м, 4Н); примерно 2,5 (м, примерно 4Н); 2,64 (м, 2Н); 2,81 (двойной д, 14 Гц; 5 Гц, 2Н). 3,13 (дв.м, 14 Гц, 2Н); 3,42 (м, 2Н); 3,56 (м, 2Н); 4,10 (м, 2Н); 4, 90 (м, 2Н); 6,58 (д, 9 Гц, 2Н); 7,05-7,30 (м, 2OН); 7,85 (д, 8 Гц, 2Н).
П р и м е р 30. N,N'-бис-(L-фенилаланил-L-валил)-2R, 5R-диамино-1,6-дифенилгексан-3R, 4R-диол-дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAB): 793 (M+H)I
П р и м е р 31 N,N'-бис-(L-фенилаланил-L-валил)-2R,5R- диамино-1,6-дифенилгексан-3S, 4S-диод дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAВ): 793 (M+H)+
П р и м е р 32. N,N'-бис-(L-фенилаланил-L-валил)-2R, 5R-диамино-1,6-дифенилгексан-3R, 4S-диол дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAB): 793 (M+H)+.
П р и м е р 33. N,N' -бис-(третбутоксикарбонил-L- фенилаланил-L-валил)-2R, 5R-диамино-1,6-дифенилгексан-3R, 4R-диол.
Масс-спектр (FAB): 993 (M+H)+, 893, 793.
ЯМР (270 мГц, DMCO < D6 >): 0,48 (д, 7 Гц, 6Н); 0,54 (д, 6 Гц, 6Н), 1,25 (с, 18 Н); 1,70 (м, 2Н); 2,60 (т, 13 Гц, 2 Гц, 2Н); 2,74 (двойной д, 14 Гц, 11 Гц, 2Н); 2,96 (двойной д, 13 Гц, 4 Гц, 2Н); 3,13 (двойной м, 14 Гц, 2Н); 3,39 (м, 2Н); 4,02-4,25 (м, 6Н); 4,88 (д, 4 Гц, 2Н); 7,02 (д, 9 Гц, 2Н); 7,07-7,33 (м, 2OH); 7,60 (д, 9 Гц, 2Н); 8,24 (д, 9 Гц, 2Н).
П р и м е р 34. N, N'-бис-(третбутоксикарбонил-L-фенилаланил -L-валил)-2R,5R-диамино-2,6-дифенилгексан-3S, 4S-диол.
Синтез осуществляли аналогичным описанному в примере 6 способом.
Масс-спектр (FAB): 993 (M+Н)+, 893, 793.
П р и м е р 35. N,N'-бис-(третбутоксикарбонил-L- фенилаланил-L-валил)-2R, 5R-диамино-1,6-дифенилгексан-3R, 4S-диол.
Синтез осуществляли аналогичным описанному в примере 6 способом.
Масс-спектр (FAB): 993 (M+H)+, 893, 793.
П р и м е р ы 36-38.
36) N,N'-бис-(третбутоксикарбонил)-2R,5R-диамино -1,6-дифенилгексан-3R, 4R-диол.
37) N,N'-бис-(третбутоксикарбонил)-2R,5R-диамино -1,
6-дифенилгексан-3S, 4S-диол
38) N,N'-бис-(третбутоксикарбонил)-2R,5R-диамино -1,6-дифенилгексан-3R, 4S-диол.
Синтез осуществляли аналогичным описанному в примерах 3-5 cпособом, используя в качестве исходного соединения третбутоксикарбонил-D-фенилаланинал. Масс-спектры и характеристики ЯМР соответствуют аналогичным характеристикам их энантиомеров в соответствии с примерами 3-5.
П р и м е р 39. N,N'-бис-[L-(1-нафтил)аланил-L-валил]-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAB. LiI): 899 (M+Li)+, 893 (M+H)+, 875.
П р и м е р 40. N,N' -бис-[третбутоксикарбонил-L- (1-нафтил)аланил-L-валил]-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
Синтез осуществляли аналогичным описанному в примере 6 способом.
Масс-спектр (FAB): 1093 (M+H)+, 993.
ЯМР (270 мГц, DMCO < D6 >): 0,76 (м, 12Н); 1,23 (с, 18Н); 1,89 (м, 2Н); 2,60-2,87 (м, 4Н); 3,12 (двойной д; 14 Гц, 10 Гц, 2Н); примерно 3,33 (м, 2Н); 3,52 (двойной м, 4 Гц, 2Н); 4,16-4,35 (м, 4Н); 4,44 (м, 2Н); 4,70 (с, 2Н); 7,00-7,27 (м, 12Н); 7,37-7,44 (м, 4Н); 7,46-7,68 (м, 8Н); 7,79 (м, 2Н); 7,92 (д, 8 Гц, 2Н); 8,13 (д, 8 Гц, 2Н).
П р и м е р 41. N,N'-бис-[(2-(2-оксиэтилсульфонилметил) -3-фенилпропионил)-L-валил]-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол.
Синтез осуществляли аналогичным описанному в примере 13 способом.
Масс-спектр (FAB): 1007 (M+H)+.
П р и м е р 42. N,N-бис-[L-фенилаланил-L-валил]-2S, 5S-диамино-1,6-дициклогексилгексан-3R, 4R-диол-дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAB): 805 (M+Н)+, 787.
П р и м е р 43. N,N'-бис-[L-фенилаланил-L-валил]-2S, 5S-диамино-1,6-дициклогексилгексан-3S, 4S-диол-дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16 способом.
Масс-спектр (FAB): 805 (M+H)+, 787.
П р и м е р 44. N,N' -бис-(третбутоксикарбонил-L-фенилаланил-L-2S, 5S-диамино-1,6-дициклогексилгексан-3R, 4R-диол.
Синтез осуществляли аналогичным описанному в примере 6 способом.
Масс-спектр (FAB): 1005 (M+H)+, 987, 905, 805.
П р и м е р 45. N,N'-бис-(третбутоксикарбонил-L- фенилаланил-L-валил)-2S, 5S-диамино-1,6-дициклогексилгексан-3S, 4S-диол.
Синтез осуществляли аналогичным описанному в примере 6 способом.
Масс-спектр (FAB): 1005 (M+H)+ ; 987; 905; 805.
ЯМР (270 мГц,
DMCO
П р и м е р 46. N,N'-бис-(третбутоксикарбонил)-2S, 5S-диамино-1,6-дициклогексилгексан-3S,4S-диол.
200 мг N, N'-бис-(третбутоксикарбонил)-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диола растворяли в 25 мл ледяной уксусной кислоты и осуществляли гидрирование в течение 18 ч при 60oC и 120 бар в присутствии 100 мг диоксида платины в качестве катализатора. Катализатор затем отфильтровывали, растворитель отгоняли в вакууме и остаток перекристаллизовывали из смеси этанола и воды.
Выход 150 мг.
Масс-спектр (FAB): 535 (M+Na)+ , 513 (M+H)+, 413.
ЯMR (270 МГц, DMCO
П р и м е р 47. N,N'-бис-(третбутоксикарбонил)-2S, 5S-диамино-1,6-дициклогексилгексан-3R, 4R-диол.
Синтез осуществляли аналогичным описанному в примере 46 способом.
Масс-спектр (FAB): 513 (M+H)+, 413.
ЯМР (270 мГц, DMCO
П р и м е р 48. N,N' -бис-(третбутоксикарбонил)-2S, 5S-диамино-1,6-дициклогексилгексан-3R, 4S-диол.
Синтез осуществляли аналогичным описанному в примере 46 способом.
Масс-спектр (FAB): 513 (M+H)+, 413.
П р и м е р 49. N,N'-бис-(4Z-аминоциклогексанкарбонил) -L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенил-3S, 5S-диол дигидрохлорид.
Синтез осуществляли аналогичным описанному в примере 16, соответственно 6, способом.
Масс-спектр (FAB): 1043 (M+H)+, 1025.
П р и м е р 50. N,
N'-бис-(4Z-N-третбутоксикарбониламино)- циклогексанкарбонил-L-фенилаланил-L-валил)-2S, 5S-диамино-1,6-дифенил-3S, 5S-диол дигидрохлорид
Синтез осуществляли аналогичным описанному в
примере 6 способом.
Масс-спектр (FAB): 1243 (М+H)+, 1143, 1043.
П р и м е р 51. N,N'-бис-(2S-(1,1-диметилэтилсульфонилметил
-3-(1-нафтил)-пропионил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол
Синтез осуществляли аналогичным описанному в примере 13 способом.
Масс-спектр (FAB): 1131 (M+H)+, 716.
ЯМР (270 мГц, DMCO
П р и м е р 52 N,N'-бис-<(2S-(1,1-диметилэтилсульфонилметил)-3- фенил-пропионил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
Синтез осуществляли аналогичным описанному в примере 13 способом.
Масс-спектр (FAB): 1053 (М+Na)+, 1031 (M+H)+.
ЯМР (270 мГц, DMCO
П р и м е р 53. N,N' -бис-<(3-(1,1-диметилэтилсульфонил)- пропионил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
Синтез осуществляли аналогичным описанному в примере 13 способом.
Масс-спектр (FAB): 873 (M+Na)+ , 851 (M+H)+:
ЯМР (270 мГц, DMCO
П р и м е р 54. N,N'-бис-<(2R-(1,1-диметилэтилсульфонилметил) -3-(2-тионил)-пропионил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
Синтез осуществляли аналогичным описанному в примере 13 способом.
Масс-спектр (FAB), 1065 (M+Na)+, 1049 (M+Li)+.
ЯМР (270 мГц, DMCO
П р и м е р 55. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 56 способом. Масс-спектр (FAB): 725 (M+H)+. П р и м е р 56. N,N'-бис-<(третбутоксикарбонил-L- фенилаланил-L-валил)-4S, 7S-диамино-2,9-диметилдекан-5,6-диол дигидрохлорид. Синтез осуществляли
аналогичным описанному в примере 6, соответственно в примерах 3-5, способу. Масс-спектр (FAB): 925 (M+H)+, 826, 725. ЯМР (270 мГц, DMCO П р и м е р 57. N,N'-бис-<(2S-(1,
1-диметилэтилсульфонилметил) -3-фенил-пропионил)-L-валил>-4S, 7S-диамино-2,9-диметилдекан-3,4-диол. Синтез осуществляли аналогичным описанному в примере 13, соответственно в
примерах 3-5, cпособом. Масс-спектр (FAB): 985 (М+Na)+ , 963 (M+H)+. ЯМР (270 мГц, DMCO П р и м е р 58. N,N'-бис-<
(2-пиридил)-ацетил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. 74 мг 2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорида и 68 мг гидрохлорида
2-пиридилуксусной кислоты растворяли в 2 мл диметилформамида и затем смешивали приготовленный раствор с 53 мг гидроксибензотриазола, 125 мг ТБТУ и 0,221 мл диизопропилэтиламина. Смесь перемешивали в
течение 2 ч при комнатной температуре и затем перерабатывали обычным образом. После хроматографии на силикагеле (подвижная фаза: смесь DCM и MeOH при соотношении (95:5)-(90:10) получали 68 мг целевого
соединения. Масс-спектр (FAB): 759 (M+Na)+ , 737 (M+H). ЯМР (270 МГц, DMCO П р и м е р 59. N,N'-бис-<4-пиридилтио-ацетил-L-валил)-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. 74
мг 2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорида и 66 мг 4-пиридилмеркаптоуксусной кислоты растворяли в 2 мл диметилформамида и смешивали приготовленный раствор с 53 мг ОН-бензотриазола,
125 ТБТУ 0,177 мл диизопропиламина. Смесь перемешивали в течение 2 ч при комнатной температуре, отгоняли растворитель в вакууме и смешивали остаток в течение 30 мин с этилацетатом и раствором
NaHCO3. Нерастворившуюся часть отфильтровывали и промывали этилацетатом и водой. Сырой продукт растворяли в теплом диметилформамиде, раствор фильтровали и смешивали с этилацетатом. Осадок
отсасывали и высушивали. Выход 76 мг. Масс-спектр (FAB): 801 (M+H)+. ЯМР (270 мГц, DMCO П р и м е р 60. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 793 (M+H)+
. П р и м е р 61. N,N'-бис- Синтез осуществляли
аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 793 (M+H)+. П р и м е р 62. N,N'-бис-<третбутоксикарбонил-L-фенилаланил
-D-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 993 (M+H)+, 893, 793. ЯМР (270 мГц, DMCO П р и м е р 63. N,
N'-бис-<третбутоксикарбонил-D-фенилаланил -L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 1015 (M+Na)+, 993 (M+H)+, 893, 793. ЯМР (270 мГц, DMCO П р и м е р 64. N,N'-бис-< L-фенилаланилглицин>-2S, 5S-диамино-1,6-дифенилгексан-2S, 4S-диол дигидрохлорид. Синтез осуществляли аналогичным
описанному в примере 16 способом. Масс-спектр (FAB): 709 (M+H)+. П р и м е р 65. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 821 (M+H)+. П р и м е р 66. N,N'-бис- Синтез осуществляли аналогичным описанному
в примере 20 способом. Масс-спектр (FAB): 641 (M+H)+. П р и м е р 67. N,N'-бис-<третбутоксикарбонил-L- фенилаланилглицин>2S,
5S-диамино-1,6-фенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 931 (M+Na)+ ; 909 (M+Н)+; 809, 709. ЯМР (270 мГц, DMCO П р и м е р 68. N,N'-бис-<
третбутоксикарбонил-L-фенилаланил -L-изолейцил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом.
Масс-спектр (FAB): 1021 (M+ H)+, 921, 821. ЯМР (270 мГц, DMCO П р и м е р 69. N,
N'-бис-<трeтбутоксикарбонил-L- лейцилглицил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 863 (M+Na)+, 841 (M+H)+, 741, 641. ЯМР (270 мГц, DMCO П р и м е р 70. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 769 (M+H)+.
П р и м е р 71. N,N'-бис-<5S-амино-4S-окси-7-метил-2R- пропилоктаноил-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол дигидрохлорид. 56 мг дигидрохлорида 2S,
5S-диамино-1,6-дифенилгексан-3S, 4S-диаола и 134 мг N-третбутоксикарбонил-5S-амино-метил-2R- пропил-4S-(третбутилдиметилсилилокси)-октановой кислоты растворяли в 3 мл диметилформамида и приготовленный
раствор смешивали с 43 мг НОbt, 101 мг ТБТУ и 155 мг диизопропилэтиламина. Смесь перемешивали в течение 4 ч при комнатной температуре, удаляли растворитель в вакууме и распределяли остаток между
дихлорметаном и водой. Органическую фазу подвергали экстракции растворами KHSO4, NaHCO3 и водой. После высушивания над безводным сульфатом натрия органическую фазу
концентрировали и остаток подвергали хроматографии на силикагеле (подвижная фаза: смесь циклогексана и этилацетата при соотношении 3: 1). В результате получали 157 мг N,N'-бис-<
N-третбутоксикарбонил-5S-амино-7-метил-2R- пропил-4S-(третбутилдиметилсилилокси)-октаноил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол дигидрохлорида. Обработкой его НСl в диоксане таким же
образом, как это описано в примере 16, получали целевое соединение. N-третбутоксикарбонил-5S-амино-7-метил-2R-пропил-4S- (требутилдиметилсилилокси)-октановую кислоту, второй компонент
реакции сочетания, получали аналогичным описанному в примере 27 способом. Для этой цели исходное соединение (5S)-5-<(1S)-1-(N-Boc-амино) -3-метилбутил>
дигидрофуран-2(3Н)-oн дополнительно алкилировали аллилбромидом и затем гидрировали (таким же образом, как при получении соединения 11, описанного Fray и др.). Масс-спектр (FAB): 727
М+H)+. ЯМР (270 мГц, DMCO П р и м е р 72. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 873 (M+H)+
. П р и м е р 73. N,N'-бис-<третбутоксикарбонил-L-фенилаланил -L-циклогексилглицил>2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. Синтез
осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 1073 (M+H)+ , 973, 873. ЯМР (270 мГц, DMCO П р и м е р 74. N,N'-бис- Синтез осуществляли
аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 761 (M+H)+. П р и м е р 75. N,N'-бис-<третбутоксикарбонил-L-метионил
-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): (M+H)+,
861, 761. ЯМР (270 мГц, DMCO П р и м е р 76. N,N'-бис-<(0-метилтирозил)-L-валил>2S, 5S-диамино-1,
6-дифенилгексан-3S, 4S-диол дигидрохлорид. Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 853 (M+H)+.
П р и м е р 77. N,N'-бис-<третбутоксикарбонил- (0-метилтирозил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогично
описанному в примере 16 способом. Масс-спектр (FAB): 1053 (M+H)+ , 953, 853. ЯMP (270 мГц, DMCO П р и м е р 78. N,N'-бис- Синтез осуществляли аналогичным
описанному в примере 16 способом. Масс-спектр (FAB): 825 (M+H)+. П р и м е р 79. N,N'-бис-<(N-третбутоксикарбонил-0-третбутил
-L-тирозил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 1137
(M+H)+; 1037; 937. ЯМР (270 мГц, DMCO П р м е р 80. N,
N'-бис- Синтез
осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB/LiI): 1229 (M+H)+. П р и м е р 81. N,N'-бис- Синтез осуществляли аналогичным описанному в
примере 6 способом. Масс-спектр (FAB): 1319 (M+H)+; 1219, 1185. ЯМР (270 мГц, DMCO П р и м е р
82. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16
способом. Масс-спектр (FAВ/LiI): 763 (M+Li)+, 757 (M+H)+. ЯМР (270 мГц, DMCO П р и м е р 83. N,N'-бис-<третбутоксикарбонил-L- глютамил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол.
Синтез осуществляли, используя в качестве исходного материала соединение в соответствии с примером 84, путем каталитического гидрирования на палладиевом катализаторе на угле в смеси ледяной уксусной
кислоты и воды при соотношении 9:1. Масс-спектр (FAB): 979 (M+Na)+ , 958 (M+H)+. ЯМР (270 мГц, DMCO П р и м е р 84. N,N'-бис-<(N-третбутоксикарбонил-0-бензил -L-глютамил)-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диод. Синтез
осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 1159 (M+Na)+, 1137 (M+H)+, 1037. ЯМР (270 мГц, DMCO П р и м е р 85. N,N'-бис-<глицил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R,
4R-диол дигидрохлорид. Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 635 (M+Na)+ , 613 (M+H)+. П р и м е р 86. N,N'-бис-<третбутоксикарбонилглицин-L- валил>2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. Синтез осуществляли аналогичным
описанному в примере 6 способом. Масс-спектр (FAB): 835 (M+Na)+ ; 813 (M+Н)+. ЯМР (270 мГц, DMCO П р и м е р 87. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 747 (M+Na)+ , 725 (M+H)+. П
р и м е р 88. N,N'-бис-<третбутоксикарбонил-L-лейцил-L- валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол. Синтез осуществляли аналогичным описанному в
примере 6 способом. Масс-спектр (FAB): 947 (M+Na)+ ; 925 (M+Н)+; 825, 725. ЯМР (270 мГц, DMCO П р и м е р 89. N,N'-бис- Синтез осуществляли аналогичным описанному в примере 16 способом. Масс-спектр (FAB): 847 (M+Nа)+, 825(M+H)+ Синтез
осуществляли аналогичным описанному в примере 6 способом. Масс-спектр (FAB): 1074 (M+Na)+. ЯМР (270 мГц, DМСO П р и м е р 91. N,N'-бис-<[2S-(1,1-диметилэтилсульфонилметил) -3-фенилпропионил] -L-третбутилглицил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. Синтез осуществляли аналогичным описанному в примере 13 способом. Масс-спектр (FAB): 1081 (M+Na)+, 1059 (M+H)+. ЯМР (270 мГц, DMCO
П р и м е р 92. N,N'-бис-<[2S-(1,
1-диметилэтилсульфонилметил) -3-фенилпропионил] -L-неопентилглицил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 5R-диол. Синтез осуществляли аналогичным описанному в примере 13
способом. Масс-спектр (FAB): 1109 (M+Na)+, 1087 (M+Н)+. ЯМР (270 мГц, СDCl3), 0,86 (с, 18Н); 1,08 (двойной, д, 8 Гц, 14 Гц, 2Н); 1,35
(с, 18Н); 1,58 (двойной д, 14 Гц, 2Н); 2,75-3,45 (м, примерно 8Н); 3,80 (м, 2Н); 4,12 (м, 2Н); 5,80 (д, 8 Гц, 2Н); 6,27 (д, 8 Гц, 2Н); 7,10-7,36 (м, примерно 10Н). П р и м е р 93. N,
N'-бис-<(2-S-окси-3-фенилпропионил) -L-валил>-2S, 5S-диамино-1,6-дифенилгексан-2S, 4S-диол. К 0,065 ммоль N,N'-бис-<-L-валил>-2S,
3S-диамино-1,6-дифенилгексан-3S, 4S-диол гидрохлорида и 33 мг S-фенилмолочной кислоты в 4 мл диметилформамида добавляли 27 мг OН-bt, 64 мг ТБТУ и затем медленно 0,088 мл диизопропилэтилена. Через 15
мин удаляли в вакууме при комнатной температуре диметилформамид, растворяли остаток в этилацетате и осуществляли экстракцию растворами KHSO4 и NaHCO3 и водой. Органическую фазу
высушивали MgSO4 и концентрировали, остаток растирали с эфиром и отсасывали. Выход 43 мг. Масс-cпектр (FAB): 795 (M+H)+. ЯМР (270 мГц, DMCO
П р и м е р 94. N,N'-бис-<(2S-окси-4-фенилбутирил) -L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S,
4S-диол. Синтез осуществляли аналогичным описанному в примере 93 способом. Масс-спектр (FAB): 345 (M+Na)+ , 323 (M+H)+. ЯМР
(270 МГц, DMCO П р и м е р 95. N,N'-бис-<
[2-(1-имидазолилметил)-3- фенилпропионил]-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3S, 4S-диол (исходя из "диастереомера 1"). 35,8 мг 2S, 5S-диамино-1,6-дифенилгексан-3S,
4S-диол дигидрохлорида и 90 мг 2-(1-имидазолилметил)-3-фенилпропионил-L- валина ("диастереомер 1") растворяли в 2 мл диметилформамида и смешивали приготовленный раствор при комнатной температуре с 32
мг OH-bt, 77 мг ТБТУ и затем с 0,163 мл диизопропилэтиламина. Смесь перемешивали в течение 3 ч, растворитель удаляли в вакууме и остаток распределяли между этилацетатом и раствором NaHCO3.
Органическую фазу промывали полуконцентрированным раствором NaCl, высушивали и концентрировали. Остаток растирали с диэтиловым эфиром, отсасывали и подвергали хроматографии на силикагеле (подвидная
фаза: смесь ЕЕ и MeOH при соотношении 85:15). В результате получали 57 мг целевого соединения. Масс-спектр (FAB): 923 (M+H)+.
2(1-имидазолилметил)-3-фенилпропи-онил-L-валин получали следующим образом: 1,53 г эфира бензилакриловой кислоты и 550 мг имидазола растворяли в 30 мл ЕtOH и смешивали полученный раствор в атмосфере
аргона при комнатной температуре с 40 мг NaH. Через 7 дней реакционный раствор выливали в 50 мл раствора KH2PO4 и трижды проводили экстракцию 50 мл метилтретбутилового эфира. Из
органической фазы дважды проводили экстракцию NaHSO4, водную фазу подщелачивали K2СO3 и снова дважды подвергали экстракции 50 мл метилтретбутилового эфира. В
результате после концентрирования получали 390 мг этилового эфира 2-бензол-3-(1-имидазолил)пропановой кислоты, который омыляли NaOH и затем подвергали сочетанию с валинметиловым эфиром через АПФ.
Диастереомеры разделяли с помощью смеси этилацетата и MeOH при соотношении 10: 1. 0,34 = диастереомер 1 0,18 = диастереомер 2 П р и м е р 96. N,N'-бис-<[2-(1-имидазолилметил)-3- фенилпропионил]-L-валил->2S, 5S-диамино-1,
6-дифенилгексан-3S, 4S-диол (исходя из "диастереомера 2"). Получение см. пример 95. Mасс-спектр (FAB): 923 (M+H)+. П р и м е р 97. N,N'
-бис-<3-(4-амино-1-пиперидилсульфонил) -2-бензилпропионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. Синтез осуществляли аналогичным описанному в примере 16
способом. Macc-спектр (FAB): 1115 (M+H)+. П р и м е р 98. N,N'-бис-<2-бензил-3(4-третбутоксикарбониламино
-1-пиперидилсульфонил)-пропионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. 57 мг N,N'-бис-
Масс-спектр (FAB): 1337 (M+Na)+, 1315 (M+H)+, 1237, 1215, 1137, 1115. 2-бензил-3(4-третбутоксикарбонила- мино-1-пиперидилсульфонил)- пропионовую кислоту
синтезировали аналогичным описанному в примере 13 способом. На промежуточной стадии путем взаимодействия эфира бензилакриловой кислоты тиоуксусной кислоты получали бензиловый эфир
3-ацетил-тио-2-бензилпропионовой кислоты. Последующее окисление его хлором приводило к образованию бензилового эфира 2-бензил-3-хлорсульфонилпропионовой кислоты, который путем сочетания с
4-третбутоксикарбониламинопиперидином и последующего гидрирования переводили в вышеуказанный компонент для реакции сочетания. П р и м е р 99. N,N'-бис-<
3-(4-амино-1-пиперидилкарбонил) -2R-бензилпропионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорид. Синтез осуществляли аналогичным описанному в примере 16
способом. Масс-спектр (FAB): 1043 (M+H)+. П р и м е р 100. N,N'-бис-<
2R-бензил-3-(4- третбутоксикарбониламино-1-пиперидилкарбонил)-пропионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорид. 57 мг N,N'-бис-<
L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорида и 129 мг 2R-бензил-3-(4-трет.-бутоксикарбонила- мино-1-пиперидилкарбонил)- пропионовой кислоты, синтезированной путем
сочетания 4-третбутоксикарбониламинопиперидина и бензилового эфира 2-R-бензил-3-карбоксипропионовой кислоты, растворяли в 1 мл диметилформамида и смешивали, полученный раствор с 41 мг OН-bt, 96 мг
ТБТУ и затем медленно добавляемого 0,135 мл диэтилизопропиламина. Через 20 мин растворитель удаляли в вакууме, остаток растворяли в ЕЕ и проводили экстракцию растворами KHSO4 и NaHCO3 и водой. Органическую фазу высушивали над MgSO4 и концентрировали. Остаток растворяли в небольшом количестве DCM, высаживали продукт диэтиловым эфиром и отфильтровывали. Выход 64
мг. Mасс-спектр (FAB): 1265 (M+Na)+,1243 (M+H)+. П р и м е р 101. N,N'-бис-<(2R-бензил-3-карбоксил)- пропионил-L-валил>
-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. Синтез осуществляли исходя из соединения в соответствии с примером 102, путем обработки его трифторуксусной кислотой.
Масс-спектр (FAB): 901 (M+Na)+ , 879 (M+H)+. П р и м е р 102. N,N'-бис-<(2R-бензил-3- третбутоксикарбонил)-пропионил-L-валил>-2S,
5S-диамино-1,6-дифенилгексан-3R, 4R-диол. 45 мг N,N'-бис- Масс-cпектр (FAB): 1013 (M+Na)+,
991 (M+H)+. ЯМР (270 мГц, DMCO П р и м е р 103. N,
N'
-бис-<(3-амино-2-бензил)-пропионил -L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорид (исходя из "диастереомера 1") Mасс-спектр (FAB): 843 (M+Na)+ , 821 (M+Н)+. П р и м е
р 104. N,N'-бис-<(3-амино-2-бензил)-пропионил -L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорид (исходя из "стереомера 2"). Синтез
осуществляли аналогичным описанному в примере 16 способом из соединения в соответствии с примером 106. Mасс-спектр (FAB): 843 (M+Na)+ , 821 (M+H)+. П р и м е р 105. N,N'-бис-<(2-бензил-3- третбутоксикарбониламино)-пропионил-L-валил>-2S, 5S-диамино-1,6- дифенилгексан-3R, 4R-диол (исходя из "диастереомера 1"). 37 мг 2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол дигидрохлорида сочетали с 98 мг N,N'-бис-<(2-бензил-3-трет- бутоксикарбониламино)-пропионил-L-валина по ТБТУ-методу.
После обычной обработки и хроматографии (подвижная фаза: смесь DCM и метанола при соотношении (98:2)-(95:5) получали 28 мг целевого соединения. Масс-cпектр (FAB): 1043 (M+Na)+, 1021, (M+H)+, 921, 821. Компонент N,N'-бис-<(2-бензил-3-третбутоксикарбониламино) -пропионил-L-валин получали следующим образом: 2,3 г натрия
растворяли в 170 мл EtOH и смешивали образующийся раствор с 32 мг этилового эфира циануксусной кислоты. При перемешивании добавляли к смеси по каплям 11,5 мл бензилхлорида. Раствор оставляли стоять на
ночь при комнатной температуре, после чего отфильтровывали осадок NaCl и отгоняли растворитель. Остаток растворяли в этилацетате и подвергали его подвергали его экстракции водой. Органическую фазу
концентрировали и остаток перегоняли в вакууме (0,5 мм рт. ст., 120-125oC). Выход 8,1 г. Полученный этиловый эфир бензилциануксусной кислоты растворяли в 200 мл EtOH и
гидрировали в присутствии никеля Ренея. После отсасывания катализатора и концентрирования получали 8,2 г маслянистой жидкости и после хроматографии на силикагеле (этилацетат после смеси этилацетата и
MeOH при соотношении 5: 1) 5,5 г этилового эфира 3-амино-2-бензилпропионовой кислоты. Полученное соединение подвергали взаимодействию с Вос2O с образованием в результате этилового эфира
2-бензил-3-(третбутоксикарбониламино)- пропионовой кислоты, который омыляли и подвергали сочетанию с H-Val-OMе по АПФ-методу. Полученные диастереомеры разделяли с помощью хроматографии (подвижная
фаза: смесь толуола и диизопропилового эфира при соотношении 1: 1). Rf = 0,140 = диастереомер 1 П р и м е р 106. N,N'
-бис-<(2R-бензил-3- третбутоксикарбониламино)-пропионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол (исходя из диастереомера 2). Синтез осуществляли
аналогичным описанному в примере 105 способом. Масс-спектр (FAB): 1043 (M+Na)+, 1021 (M+H)+, 921, 821. П р и м е р 107. N,N'
-бис-<0-(D-маннофуранозил)-2S-окси-3- фенилпропионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. 20 мг соединения в соответствии с примером 108 перемешивали в
течение 30 мин при комнатной температуре с метанольным раствором соляной кислоты. Летучие компоненты затем отгоняли в вакууме, остаток настаивали с диэтиловым эфиром, отсасывали и высушивали. Выход 13
мг. ЯМР (270 МГц, DMCO П р и м е р 107а. N,N'-бис-<-0-(2,3-5,6-диизопропилиден-D- маннофуранозил-2S-окси-3-фенилпропионил-L-валил>-2S,
5S-диамино-1,6-дифенилгексан-3R, 4R-диол. 57 мг N,N'-бис- Масс-спектр
(FAB): 1279 (M+H)+, 1261, 1221. ЯМР (270 мГц, DMCO 405 мг (0-)-2,3-5,6-диизопропилиден-D-маннофуранозил трихлорацетимидата и 194 мг этилового эфира фенилмасляной кислоты
растворяли в 15 мл абсолютного СH2Сl2. Раствор охлаждали до 0oC и добавляли к нему 100 мкл 1М раствора BF3 эфирата в СH2Сl2. Раствор перемешивали
в течение часа при 0oC, выливали его в 100 мл раствора NaHCO3 и экстрагировали CH2Сl2. Органическую фазу высушивали Na2SO4 и
концентрировали. После хроматографии на силикагеле "подвижная фаза" (смесь метилтретбутилового эфира и гептана при соотношении 1:1) получали 195 мг целевого соединения. П р и м е р
108. N,N'-бис-(L-фенилаланил-L-валил)-3S, 6S-диамино-1,8-ди-(4-пиридил)-октан-4R, 5R-диол тетрагидрохлорид. Синтез осуществляли аналогичным описанному в примере 16 способом
из соединения в соответствии с примером 27. Mасс-спектр (FAB): 823 (M+H)+. П р и м е р 109. N,N'-бис- 69 мг N,N'-бис- Выход 71 мг. Масс-спектр (FAB): 1138
(M+Na)+, 1117 (M+H)+. П р и м е р 110. N,N'-бис- Синтез: соединение в соответствии с примером 111 обрабатывали насыщенным раствором аммиака в метаноле. Масс-спектр (FAB): 1171 (M+Na)+. П р и м
е р 111. N,N'-бис-<2,3,4,5,6-пента-0-ацетил-D-ацетил-D- глюконил-L-фенилаланил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол. Cинтез осуществляли
путем сочетания 2,3,4,5,6-пента-0-ацетил-D- глюконовой кислоты с N,N'-бис- Mасс-спектр (FAB): 1569 (M+H)+ . П р и м е р 112. N,N'-бис-<третбутоксикарбонил-L-фенилаланил-L- валил>-1,4-диаминобутан-2R, 3R-диол. Cинтез осуществляли аналогичным описанному в примере 6 способом, исходя из 1,4-диаминобутан-2R,3R-диол дигидрохлорида. Масс-спектр (FAB): 835 (M+Na)+ , 813
(M+H)+, 713, 613. ЯМР (270 мГц, DMCO П р и м е р 113. 1,4-диаминобутан-2R,3R-диол дигидрохлорид.
Синтез осуществляли аналогичным описанному в примерах 2, 2.2 и 2.3 способом, исходя из (+)-1,4-ди-0-тозил-2,3-0-изопропилиден-D-треитола. Масс-спектр (DCl): 121 (M+H)+,
104. ЯМР (60 мГц, DMCO П р и м е р 114. N,
N'-бис-(третбутоксикарбонил)-2S, 4-диамино-1,5-дифенилпентан-3-ол. 2,3 г Третбутоксикарбонил-L-фенилаланала растворяли в 10 мл этанола. После добавления при 0oC
0,05 мг тетраметилгуанидина и раствора 2,42 г 2-нитро-1-фенилэтана в 2 мл этанола смесь нагревали до комнатной температуры и перемешивали в течение ночи. Раствор затем упаривали, а остаток в виде
светлой маслянистой жидкости (4,8 г) использовали непосредственно на последующей стадии. 4,7 г полученной маслянистой жидкости растворяли в 70 мл этанола и после добавления 0,1 мл
ледяной уксусной кислоты и 1 г никеля Ренея раствор встряхивали в течение 16 ч при 50oC и 25 ат в стеклянной емкости в автоклаве. Катализатор затем отфильтровывали и элюат упаривали до
получения маслянистой жидкости. Остаток растворяли в смеси воды и 1 н. НСl и четырежды экстрагировали уксусным эфиром. Эфирный экстракт упаривали (2,6 г) и непосредственно использовали на последующей
стадии. 2,57 г полученного амино-производного растворяли в 25 мл диоксана при комнатной температуре. После добавления 0,86 мл триэтиламина и 1,7 г дитретбутилдикарбоната раствор
перемешивали в течение еще 30 мин и затем концентрировали, а остаток смешивали в ледяной водой, уксусным эфиром и раствором KHSO4 до рН 2. Уксусно-эфирную фазу промывали водным раствором
NaCl, высушивали над безводным Nа2DSO4 и упаривали. В результате получали 3,3 г маслянистой жидкости, которую подвергали очистке с помощью хроматографии на силикагеле (подвижная
фаза: смесь CH2Cl2, метанола и ледяной уксусной кислоты при соотношении 100:3:0,3). В результате получали 2,2 г целевого продукта в виде смеси диастереомеров.
Масс-спектр (FAB): 471 (M+H)+, 371, 315.
П р и м е р 90. N,N'-бис-<третбутоксикарбонил-L-(S-диоксо) метионил-L-валил>-2S, 5S-диамино-1,6-дифенилгексан-3R, 4R-диол.
При омылении NaOH в смеси диоксана и воды получали компоненты для
реакции сочетания, использующиеся в примерах 95 и 96.
Cинтез осуществляли
аналогичным описанному в примере 16 способом из соединения в соответствии с примером 105.
Rf = 0,097 = диастереомер 2
Омыление NaOH в
смеси диоксана и воды приводит к получению компонентов для реакции сочетания, использовавшихся в качестве исходных соединений в примерах 105 и 106.
Реферат
Изобретение относится к биохимии. Сущность изобретения: для торможения ретровирусных протеаз вводят соединение общей формулы


Формула

где Y - остаток общей формулы

l = 0 или 1;
m = 1;
A - радикал формулы
D - (E)n - (F)o - (G)p;
и A* - радикал формулы
D*-(E*)n *-(F*)o* - (G*)p* ,
где D - радикал R1 или радикал указанных формул;
D* - радикал R или радикал общих формул


n, n*, o, o*, p, p* независимо друг от друга равны 0 или 1;
E, E*, F, F*, G, G* независимо друг от друга - аминокислоты из группы Val, Lys, Lys (Z), Phe, Chg, Ser, Asn, Gly, Jle, Tbg, Nva;
или R1 и R независимо друг от друга - водород, карбоксил, метилсульфонил, трет-бутилсульфонил, трет-бутоксикарбонил, 2-гидроксиэтилсульфонил, 1,2,3-тригидроксипропил, 1,2,3-триацетоксипропил, бензилоксикарбонил, 4-метилфенилсульфонил, 4-хлорбензилтио, бензилсульфонил, 4-хлорбензилсульфонил, гексадецилсульфонил, 4-амино-1-пиперидинил-сульфонил, N-трет-бутоксикарбонил-4-амино-1-пиперидилсульфонил, 4-амино-1-пиперидилкарбонил, N-трет-бутоксикарбонил-4-амино-1-пиперидилкарбонил, 2-амино-3-фенил-пропил, N-трет-бутоксикарбонил-2-амино-3-фенилпропил, 2-амино-1-гидрокси-4-метилпентил, деоксифруктозил-1, маннофуранозил, 4-аминоциклогексилкарбонил, 2-пиридилацетил, 4-пиридилтиоацетил, 2-хинолилкарбонил, 1-нафтилацетил, 1-нафтилоксиацетил, 1-(4-пиридил)-этилсульфонил, 12-аминододеканоил, 4-(N-оксидопиридил), трет-бутилтио, 4-Z-н. трет-бутоксикарбонил, аминоциклогексилкарбонил, 4-пиридил, тетрадеканоил, фенил, амино- или трет-бутоксикарбониламиногруппа;
R2 и R независимо друг от друга - водород, 2-(4-пиридил)-этил, изопропил, изобутил, н.пентил, бензил, 3,4-метилендиоксибензил, 2,4-диметоксибензил, 4-трет-бутилбензил, 2-фенилэтил или циклогексилметил;
R3 и R R4 и R R6 и R R7, R10, R0 - водород;
R5 и R независимо друг от друга - водород или гидроксил;
R8 и R - водород или вместе с R9 или R/ и связанными с ними атомами образуют 1,2,3,4-тетрагидрохинолиндиил-3,4;
R9 и R независимо друг от друга - водород, гидроксил, ацетокси, н.пропил, изопропил, изобутил, аминометил, 4-аминобутил, гидроксиметил, трет-бутоксиметил, аминокарбонилметил, 2-бензилоксикарбонилэтил, трет-бутоксикарбониламинометил, Z-нафтилметил, трет-бутилтиометил, 4-бензилоксикарбониламинобутил, N,N0-ди(бензилоксикарбонил)-гуанидинопропил, циклогексил, циклогексилметил, бензил, 2-фенилэтил, 4-гидроксибензил, 4-метоксибензил, 4-трет-бутоксибензил, 1-нафтилметил, 2-тиенилметил, 1-имидазолилметил, 3-индолилметил, 4-пиридилметил, 4-(N-оксидопиридил)-метил, 2-метилтиоэтил, 2-метилсульфонилэтил, трет-бутилсульфонилметил или 2-карбоксиэтил;
R11 и R1 независимо друг от друга - водород, гидроксил, ацетокси,
причем в общей формуле I одна или несколько амидных групп (-CONH-) в основной цепи могут быть заменены на группу -CH2NH- или -CH(OH)CH2-,
или физиологически переносимые соли этих соединений.
Комментарии