Арил-конденсированные 2,4-диазепиновые соединения, промежуточные продукты, антиаритмический агент и композиция - RU2114833C1
Код документа: RU2114833C1
Чертежи

















Описание
Изобретение относится к новым 4,5-дигидро-1H-2,4-ариловым диазепинам и бензодиазоцинам соответствующих диаминов и аминоамидов, к способам их получения и способам и композициям для лечения аритмии у млекопитающих с применением упомянутых 4,5-дигидро-1H-2,4-ариовых диазепинов и бензодиазоцинов.
В патенте США 3693093 раскрывается один 3, 4-бензодиазепин двойного замещения: 3,4-диметил-4,5-дигидро-1H-2,4-бензодиазепин гидрохлорид.
Раскрываются также 4,5-дигидро-1H-2,4-бензодиазепины, монозамещенные в третьей позиции бензилом, диметил-аминоэтилом, амино, 1-пиперидинилметилом, и фенилом. Утверждается, что соединения могут использоваться в качестве кардиоваскулярных агентов, например, в лечении или контроле различных форм повышенного кровяного давления или заболеваний, связанных с застойными явлениями. В патенте не раскрываются антиаритмические свойства, и единственный пример бензодиазепина двойного замещения не был выявлен как активный в качестве антиаритмического агента, когда он проходил опробование по протоколу для оценки соединений настоящего изобретения.
В заявке Японии N 59//013766 (CA 101:23612м) раскрывается серия 1,2,4-трехзамещенных-тетрагидробензодиазепинов с общей
структурой
в которой
R1 - низший алкил или фенэтил (произвольно замещенный низшим алкокси). О соединениях говорится, что они обезболивающие.
Elslager и др. [J. Het. Chem. 5, 609-613 (1968) описывает синтез ряда тетрагидротиазоло-[3,2-в] [2,4]
бензодиазапинов. Авторы заявляют, что "ни одно из соединений не обладает сколько-нибудь существенной биологической активностью". Так промежуточные соединения в синтезе они раскрывают
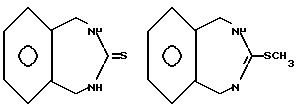
1,2,4,5-метрагидро-3H-2,4-бензодиазепин-3-тион и 2,5-дигидро-3-(метилтио)-1H-2,4-бензодиазепин гидройодид
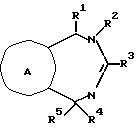
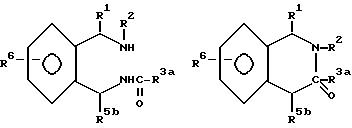
Настоящее соединение относится к соединениям с формулой XXXVI
в которой
A - кольцо, выбранное из группы, состоящей из фенила, тиенила, фуранила, нафтила, пиридинила, циклогексила и фенила с одним или двумя заместителями, выбранным из группы, состоящей из амино, низшего алкила, низкого алкокси, галогена, нитро и низшего алкилсульфонамида;
R1 - водород, низший алкил, бензил, нафтил, тиенил, пиридинил, фенил или фенил с одним или двумя заместителями, выбранными из группы, состоящей из низшего алкила и низкого алкокси;
R2 - водород; низший алкил; бензил; фенил; фенил, замещенный галогеном, низшим алкилом или низшим алкокси; или R2-CH2CH2R7, где R7 - низший алкокси; бензил; ди-низший алкил/амино, пирролидино; пиперидино; морфолино; фенил; или фенил, замещенный амино, нитро или низшим-алкильным сульфонамидо;
R3 - Jp - (CH2)m - Xn - R8
в котором
Y- -NH-, -O-, -S-,
или

где
p - нуль или единица;
m - целое число от 0 до 7;

n - 0 и 1; и
R8 - водород; низший алкил; фенил; фуранил; тиенил; приридинил; фенил с одним или двумя заместителями, выбранными из группы, состоящей из галогена, низшего алкила, нитро, гидрокси, низшего алкокси, низшего алкиламино, низшего алкилсульфонамида, динизшего-алкиламиносульфонила и амино; или с 5-членным гетероциклом, содержащим один или несколько атомов кислорода; или если n - 0 и m не 0, R8 - дополнительно галоген; бензил/низший алкил/-амино; ди-(низший алкил)амино; или 5- или 6-членный гетероцикл, содержащий 1 или 2 атома азота, причем упомянутый гетероцикл незамещен или замещен одной низшей (алкильной)группой; или X и R8 взятые вместе - циклогексилидин;
R4 - водород, низший алкил, аллил, низший (алкокси-низший алкил, ацетил, низший) алкокси-карбонил, низший-алкил-карбокси-алкил; или α -гидрокси-низший-алкил; и
R5 - водород, низший алкил, нафтил, тиенил, пиридинил, бензил, фенил, или фенил с одним или двумя заместителями, выбранными из группы, состоящей из низшего алкила, низшего алкокси, галогена, гидроксида, амино, ди-(низшего-алкил)амино, низшего алкилсульфонамида и низшего ациламина;
с оговоркой, что общее число атомов углерода в R1 плюс R2 плюс R4 плюс R5 должно быть 5 или больше.
Соединения с формулой XXXVI полезны в качестве антиаритмических агентов.
Низший алкил, используемый здесь представляет собой линейные, разветвленные или циклические насыщенные углеродные цепи из 8 или нескольких атомов углерода: низший алкокси, используемый здесь, представляет линейные или разветвленные алкокси заместители, содержащие 8 или несколько атомов углерода; галоген - бром, хлор или фтор.
В последующем тексте заместители R определены как они представлены в начале и сохраняют это определение далее.
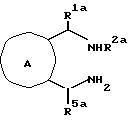
Изобретение далее относится к соединениям с формулой III

где
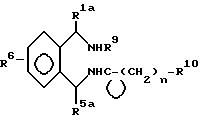
R1a - водород, низший алкил или фенил;
R5a - водород, фенил, фенил с одним или двумя заместителями, выбранными из группы, состоящей из галогена, низшего алкила и низшего-алкокси, нафтила; тиенил, пиридинил, бензил;
R9 - водород, низший алкил, бензил, фенэтил или ди-(низший алкил)амино низший алкил;
R10 - водород, низший алкил, фенил, фенил, замещенный галогеном, низший -алкил, низший алкилсульфонамидо или низший-алкокси; фенокси; фенокси-замещенный галогеном, низшим-алкилом или низшим-алкокси; бензил; или R10 - 5 или 6-членный гетероцикл, содержащий 1 и 2 атома азота, как вариант, замещенный низший алкильной группой; и n - 0 и 1.
Все соединения формулы III полезны как промежуточные соединения в синтезе соединений с формулой I (см. ниже) и как антиаритмические агенты.
Далее изобретение относится к
соединениям с
формулой XXXVII
где
R2a - низший-алкил, бензил, фенил, фенил, замещенный галогеном, низшим алкилом или низшим алкокси, или
R2a- -CH2CH2R7, где R7 - низший-алкокси, фенил, бензил, ди-(низший-алкил)амино, пирролидино, пиперидино или морфолино, и по меньшей мере один из R1a и R5a - фенил, замещенный фенил, бензил, нафтил, тиенил или пиридинил.
Соединения с формулой XXXVII полезны в качестве промежуточных соединений в синтезе соединений с формулой XXXVI, а так же как антиаритмические агенты.
Изобретение также
относится к способам получения
бензодиазепинов c формулой V

где
R3a -(Ya)p-(CH2)-(xa)-R8, в которой Ya- -O-, -S- или
и
xa, в -SO2-, -O- или -CH = CH-
при конденсации диаминов с формулой VId

с аминоэфирами с формулой R3aC(OR12)NH,
где R12 - метил или этил, с ортоэфирами с формулой R3aC(OR12)3 или эфирами с формулой R3aC(OR12)3 или эфирами с формулой
Изобретение также относится к способу получения соединения с формулой V

которое включает реакцию соединения с формулой XXVII или XXVIII с триалкилалюминием.
Соединение также относится к способу получения соединения с формулой 1a

где
R4b - низший-алкил, аликл, низший/алкоксинизший-алкил, ацетил, низший-алкокскарбонил низший алкилкарбоксиалкил или -гидроксинизшийакил; и
R5c - фенил, фенил с одним или двумя заместителями, выбранными из группы, состоящей из низшего (алкила, низшего-алкокси и галогена; нафтил, тиенил, или пиридил, который включает реакцию соединения с формулой Va с сильным основанием и затем с подходящим электрофилом.
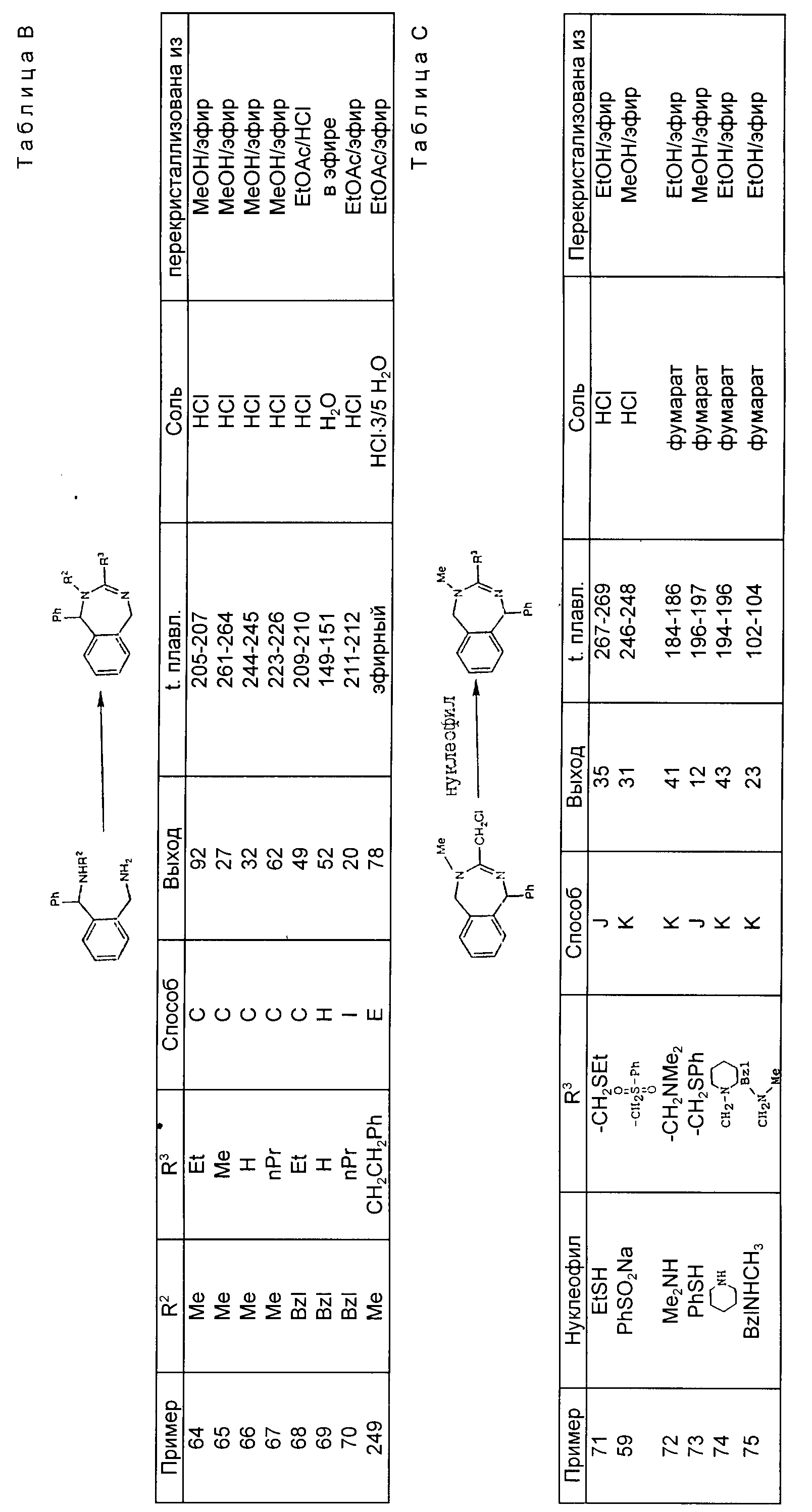
Изобретение также
относится к способу получения соединения с формулой VII

в которой

R4C - низший-алкил, аллил или низший-алкоксинизший-алкил.
R14
- водород или метил,
который включает реакцию соединения с формулой VIIa

с сильным основанием с последующей реакцией с электрофилом, выбранным из группы, состоящей из R8COOR12, R8CHNR12, R8CHO, R8SSR8 и R8(Xb)n(CH2)m-1Z, где Z группа, подлежащая нуклеофильному замещению.
Изобретение также относится к способу получения соединения
с формулой VIa

который включает реакцию соединения с формулой VIII или IX

с избытком диборана.
Изобретение также относится к способу получения соединения с формулой
VIb

который включает реакцию соединения с формулой X или XI

или
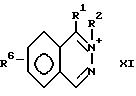
с избытком диборана.
Изобретение также относится к способу
получения соединения с формулой IVa

который включает восстановление соединения с формулой VIIIa

последовательно с гидридом алюминия, водородом в присутствии катализатора благородного металла и водородом в присутствии никелевого катализатора.
Изобретение также относится к способу получения
соединения с формулой IIa

который включает реакцию диамина с формулой VIc

с W-галоортоэфиром с формулой ZCH2(CH2)qCH2C(OR12 )3 или ZCH2(CH2)qCH2 C(OR12)NH
Другие аспекты изобретения включают способы использования диазепинов изобретения для лечения сердечной аритмии и композиции, содержащие эти соединения. Бензолметанамины и S-аминоамиды изобретения полезны и как промежуточные соединения и сами по себе в способах и композициях для лечения аритмии.
Общий синтез соединений изобретения с общей формулой XXXXVI показан в схеме A (см. в конце текста). Синтез более подробно показан в схеме B (см. в конце текста), где A - фенил или замещенный фенил.
Замещенная γ-оксо-кислота с формулой XIII реагирует с замещенным гидразином для образования фталазинона (VIII). В этом случае, где нужно, чтобы R1 был не водородом, фталазинон реагирует с небольшим избытком подходящего алкила или ариллитиевого соединения в инертном растворителе, предпочтительно THF(ТГФ) при -78o - 0oC, предпочтительно около -65oC, и полученный продукт присоединения восстанавливается как описано ниже без изоляции. В том случае, если R1 - водород, фталазинон (VIII) восстанавливается непосредственно до диамина (VI) с 3.5 - 9.0 эквивалентами диборана в инертном растворителе, предпочтительно THF* - ТГФ при 20 - 100o, а лучше 67oC. Можно добавить каталитическое количество борогидрата натрия и немного диглима.
Диамин (VI) можно конденсировать одним из трех способов для получения бензодиазепина (J, R4=H)
(I) свободное основание диамина в уксусной кислоте обрабатывается 5-7 эквивалентами
соответствующего ортоэфира R3C(OR12)3 при 0-50oC, предпочтительно
25oC, или двукислотная соль диамина, предпочтительно дигидрохлоридная соль в
инертном растворителе обрабатывается 5-7 эквивалентами соответствующего ортоэфира плюс 1-2 эквивалента слабого
основания, ацетата натрия или калия; (2) двукислотная соль диамина (VI), предпочтительно
дигидрохлоридная соль, в инертном растворе, предпочтительно метаноле, обрабатывается двумя-тремя эквивалентами
соответствующего гидрохлорида иминоэфира и двум эквивалентами слабого основания,
предпочтительно ацетата натрия при 0 - 60oC, предпочтительно 25oC, или свободное основание
диамина (VI) в инертном растворителе, предпочтительно метаноле, обрабатывается 2-3
эквивалентами соответствующего гидрохлорида аминоэфира и 2-3 эквивалентами слабой кислоты, предпочтительно уксусной
кислоты при 0-60oC, предпочтительно 25oC, или (3) диамин или
двукислотная соль диамина, предпочтительно дигидрохлоридная соль в инертном растворителе, предпочтительно толуоле,
обрабатывается несколько большим количеством, чем 2 эквивалента триметилалюминия при
-30o - +110oC с последующей обработкой 1-1,5 эквивалентами низше-олкильного эфира
соответствующей кислоты (R3COOR12)
В том случае, если желательно,
чтобы R4 был не водородом, диазепин может реагировать с сильным основанием, таким как
бутиллитий и полученный анион реагирует с соответствующим электрофилом.
Реакции, описанные для соединений с формулой XXVI, в которых A - фенильное кольцо, также применимы к соединениям, в которых A - не фенил.
Соединения с формулой V, подрядбензодиазепинов с формулой I можно получить закрытием кольца аминоамидов. Моносоль аминоамида XXVII или XXVIII (см. в конце текста), предпочтительно моногидрохлорид в инертном растворителе, предпочтительно толуоле, обрабатывается небольшим избытком, предпочтительно около 1.1 эквивалента, триметилалюминия при 0-150o, лучше 110oC для получения бензодиазепина с формулой V (см. в конце текста).
Аминоамиды с формулой XXVII можно получать как описано в примерах 177-184
неполной циклизацией или, как описано в общем способе V гидролитическим расщеплением бензодиазепинов.
Аминоамиды XXVII или XXVIII или их смеси можно также получить процедурами хорошо известными в
области технически для конденсирования кислот с формулой R3aCOOH с аминами с формулой VI

В случае соединений с формулой I, где p (в R3)-1 и Yb- -NH-, -S- или -O-, соединения можно получить другим путем из диамина VI, показанным на схеме C ( см. в конце текста).
Диамин (VI) реагирует с карбонилдиимидазолом (КДИ) в инертном растворителе, предпочтительно хлороформе, при температуре окружающей среды для получения бензодиазепин-3-она, который обрабатывается с большим избытком, предпочтительно около 13 эквивалентов фосфорного оксихлорида и предпочтительно около 0,25 эквиваленте фосфорного пентоксида для получения 3-хлорбензодиазепина (XIV) (см. в конце текста). Затем 3-хлорбензодиазепин реагирует, обычно без изоляции, с подходящими нуклеофилом, R2Xn(CH2)m(Yb)H для получения бензодиазепинов со структурой Yb.
Альтернативно диамин VI реагирует с предпочтительно около 1 эквивалента дисульфида углерода в инертном растворителе, предпочтительно 2-пропанола, при 0-100o, предпочтительно при 20-35oC, и полученная карбамодитионовая кислота обрабатывается каталитическим количеством кислоты, предпочтительно соляной кислоты, в инертном растворителе, предпочтительно этаноле, при 0-100oC, предпочтительно при 78oC, для получения тетрагидроазепин-3-тиона. Тион окисляется чуть больше, чем тремя эквивалентами 30% пероксида водорода согласно способу Maryanaff и др. J.Org. Chem. 51, 1882 (1986)) для получения сульфокислоты XXIX (см. в конце текста). Сульфонат может быть затем замещен подходящим нуклеофилом, как и раньше, в инертном растворителе при 0 - 100oC. Пример 153 иллюстрирует возможный, с низким выходом, преобразованием тиона и соединениями с формулой Vb.
В случае, когда R2, R4 и R5 не являются водородом, подвид соединений по формуле VII в которой R3 присоединен бензоазепиновому кольцу через углерод (т.е. R3 p=0 и R3 - это -CH2) (CH2 )m-1XnR8 или p=1 и
может быть получен, как вариант

обработкой соединения с формулой VIIa, в которой R14 - водород или метил в инертном растворителе, предпочтительно THF, при -78 - +25oC с небольшим избытком, предпочтительно от 10 до 25% сильного основания, предпочтительно H-бутиллития с последующей обработкой небольшим избытком, предпочтительно 10-50%, подходящего электрофила. Электрофил этот может быть формы R8(Xa)n-(CH2)m-1-Z, где Z - группа, которая легко замещается анионом, таким как галоген, сульфид, сульфонат, эстер и т.д., или в случае, когда m-1=0, это может быть альдегид, кетон или амин, так что добавление аниона к электрофилу, сопровождаемое охлаждением протоновым источником, приводит к общему добавлению элементов VIIa к электрофилу.
В случаях, когда R3 присоединен с бензодиазепину через метиленовую гетероатомную цепь (XV), то есть R3 - формы -CH2(Xc)nR8, где Xc -S- -O- или -SO2- т.е. в формуле XXXVI X - это -S-, -O- или -SO2-, p = 0, m = 1 и n = 1, когда R8 низший алкил, фенил или замещенный фенил; n = 0, когда R8 - амино или N - присоединенный гетероцикл), соединения могут удобно синтезироваться из соответствующих хлорметиловых видов (XVI).

3-хлорметил-2, 4-бензодиазепин (XVI) в инертном растворителе, предпочтительно хлороформе, если гетероатомом является азот, и метаноле или ацетонитриле, если гетероатомом является сера, обрабатывается 1-3 эквивалентными соединениями вида R8(Xc)nH при 0 - 100oC, предпочтительно при 25 - 56oC. Хлорметиловый бензодиазепин XVI может синтезироваться непосредственно из диамина VI конденсацией с этил-2-хлорэтанимидатом или в случае, когда R2, R4 и R5 не является водородом, хлорметил бензодиазепин (XVI) может синтезироваться из соответствующего 3-метил-бензодиазеприна образованием аниона, как описано в предыдущем параграфе и охлаждаться 1,1 эквивалентами гексахлорэтана.
Соединения формулы III можно синтезировать из соответствующих соединений с формулой I гидролизом в присутствии 1-5 эквивалентов водного основания, предпочтительно гидроокиси калия в совмещающемся растворителе, предпочтительно метаноле, при 0-70oC, лучше 25-30oC.
Соединения с формулой VI можно получить восстановлением и расщеплением фталазинонов и фталазинов, как показано на схеме F (см. в конце текста).
Фталазинон с формулой VIII вступает в реакцию с 3,5 - 9,0 эквивалентами диборана в инертном растворителе, предпочтительно THF, при 20-100oC, лучше 67oC, чтобы получить диамины с формулой VId, в которой R' - водород. Можно добавить каталитическое количество борогидрата натрия в диглим. Если нужно, чтобы был не водородом, фталазины с формулами XXII, XXIII и XXV можно восстановить аналогичным образом. Фталазины XXV и XXIII можно получить из соответствующих фталазинонов реакцией с подходящим алкиллитием или ариллитием в инертном растворителе, предпочтительно THF при -73 - 0oC, предпочтительно около -65oC. Полученный фталазин может существовать как гидроксифталазин XXII или при удалении элементов воды образовать виды фталазина XXIII. Фталазины с формулой XXV могут быть синтезированы конденсацией соответствующего гидразина γ - галокетоном, предпочтительно γ - бромокетоном.
В том случае, если в формуле VIII R2 - водород, простое восстановление дибораном, описанное выше, проходит настолько медленно, что представляет собой небольшую практическую пользу, нежели трехступенчатое восстановление, показанное на схеме G, приведенной в конце текста.
Фталазинон VIIa обрабатывается двумя эквивалентами восстанавливающего агента гидрида алюминия, предпочтительно литий алюминий гидрида в инертном растворе, предпочтительно THF, при 20-120oC, желательно 65oC. Полученный дигидро-фталазин реагирует с водородом в инертном растворе, предпочтительно низшем алканоле, лучше этаноле в присутствии палладиевого катализатора при 20-60oC, предпочтительно 40-50oC при давлении в 3 атм. Полученный тетрагидрофталазин (XXVI) реагирует с водородом в инертном растворе, предпочтительно метаноле, в присутствии никелевого катализатора Ранея при 20-80oC, предпочтительно около 65oC и при давлении в 3 атм.
Многие из соединений изобретения асимметричны в C-1 бензодиазепина или бензодиазоцина. В некоторых случаях дает преимущество использования одного или другого энантиомера для лечения аритмии. Одинарные энантиомеры могут синтезироваться из хиральных исходных материалов или рацематы могут быть разделены способами, известными в области техники, такими как хроматографии на хиральной среде или перекристаллизация диастереометрических солей.
Соединения изобретения полезны как в виде свободного основания, так и в виде солей кислотного добавления, и оба вида выключены в объем изобретения. Соли кислотного дополнения в некоторых случаях более удобны для использования, и в практике использование этой формы в конечном итоге ведет к использованию формы основания. Кислоты, которые можно использовать для получения солей кислотного добавления, включают такие кислоты, которые при комбинации со свободным основанием дают фармакологически приемлемые соли, то есть соли, анионы которых безвредны для организма животного в медицинских дозах солей, с тем, чтобы полезные свойства, присущие свободному основанию, не сводились на нет побочными действиями, приписываемыми анионам. В осуществлении настоящего изобретения удобно получить соли гидрохлорида, фумарата, толуолсульфоната, кислого фильфата, метаносульфоната или малеат. Однако другими подходящими фармацевтически приемлемыми солями в объеме изобретения являются соли, полученные из других минеральных солей и органических кислот. Соли кислотного основания основных соединений готовятся либо растворением свободного основания в водном спиртовом растворе, содержащем соответствующую кислоту и изоляцией соли испарением раствора, либо реакцией свободного основания и кислоты в органическом растворителе, когда соль отделяется непосредственно, осаждается со вторым органическим растворителем или может быть получена концентрацией в растворе. Хотя предпочитаются фармакологически приемлемые соли основных соединений, все соли кислотного дополнения входят в объем настоящего изобретения. Все соли кислотного дополнения полезны как источники формы свободного основания, даже если определенная соль как таковая нужна только в качестве промежуточного продукта, как например, когда соль образуется только для целей очистки или идентификации, или когда она используется в качестве промежуточного продукта в получении фармакологически приемлемой соли ионообменными процессами.
Структуры соединений изобретения определялись видом синтеза, элементарным анализом, ИК, ЯМР и масспектроскопией. Ход реакций, идентичность и гомогеничность продуктов определялись тонкослойной хроматографией (ТСХ) и жидкостной хроматографией с высоким давлением (ЖХВД). Исходные материалы - имеющиеся в продаже или готовились способами хорошо известными специалистам.
В следующих процедурах точки плавления даются в градусах C и не скорректированы.
В последующих примерах Me - метил, Et - этил, Ph - фенил, BZl - бензил, iPr - изопропил, tBu-t- - бутил, OAl ацетил, THF - тетрагидрофуран, hex - гексан, IPA - изопропиламин, DMF - диметилформамид, TMS - триметилсилил.
Далее изобретение будет описано со ссылкой на следующие примеры (см. табл. A-M в конце текста).
Соответствующий диамин и 5-7 эквивалентов соответствующего триэтилортоэфира перемешивались при комнатной температуре и одновременно одной порцией вводились 0,4-0,5 мл уксусной кислоты на Мм диамина. Смесь перемешивалась при нагревании с обратным холодильником в течение 2-72 ч. Реакция разбавлялась этилацетатом, промывалась 2N гидроокисью натрия и экстрагировались тремя порциями 2N HCl. Экстракты HCl комбинировались, промывались дважды эфиром, делались основными избытком 35% гидроокиси натрия и экстрагировались в три порции эфиром. Эфирные экстракты соединялись, высушивались на сульфате магния и растворитель удаляли in vacuo. Свободное основание или соль рекристаллизуются, как показано в табл. A (см в конце текста).
Общий способ B
Диамин добавлялся к двум
эквивалентам ацетата калия или каталитическому количеству ацетата калия
в 0,8-1,2 мл ацетокусусной кислоты на Мм диамина. Смесь перемешивалась при комнатной температуре и добавлялись от 2 до 5
эквивалентов соответствующего триэтилортоэфира. Реакционная смесь
перемешивалась при комнатной температуре 18-72 ч и отгонялась in vacuo. Продукт дорабатывался как описано в общем способе A.
Общий способ C
Дигидрохлорид диамина растворяли в
1-3 мл уксусной кислоты на мМ диамина и добавлялись около 2,0-2,5 эквивалентов ацетата натрия. Смесь перемешивалась в течение 10
мин при комнатной температуре и затем добавлялись 3-5 эквивалентов
соответствующего триэтилортоэфира. Смесь перемешивалась при комнатной температуре 2-48 ч и отгонялась in vacuo. Далее она
дорабатывалась как описано в общем способе A.
Общий способ D.
Дигидрохлорид диамина и 2-3 эквивалента соответствующего гидрохлорида метоксиимина растворялись в 2-6 мл метанола на мМ диамина. Смесь перемешивалась и добавлялись 2 эквивалента ацетата натрия. После 2-18 ч растворитель удалялся и продукт дорабатывался как описано в общем способе E.
Общий способ E.
Дигидрохлорид диамина, 1,3-3,0 эквивалента соответствующего триметил- или триэтилортоэфира и 1,0-1. 3 эквивалента ацетата натрия комбинировались в 3-6 мл изопропилового ацетата на мМ диамина. Смесь подвергалась нагреванию с обратным холодильником 3-18 ч. Затем она охлаждалась, промывалась двумя порциями 2N гидроокиси натрия и высушивалась на сульфате натрия. Растворитель удалялся in vacuo и из остатка очищалась либо соль, либо свободное основание как показано в табл. A (см. в конце текста).
Общий способ Z.
К гидрохлориду диамина, суспендированному в 3 мл толуола на мМ диамина добавлялось по каплям при 0o в азоте 2.1 эквивалента 2M триметилалюминия в толуоле. Реакционная смесь выстаивалась при комнатной температуре и перемешивалась 2 ч. Затем добавлялись 1,25-1,50 эквивалентов соответствующего метилового или этилового эфира. Далее все нагревалось с обратным холодильником 2 ч, охлаждалось и еще охлаждалось последовательным добавлением льда, метанола, дихлорметана и 2N NaOH. Соли алюминия отфильтровывались, слои разделялись, промывались еще дихлорметаном, высушивались на сульфате натрия, отгонялись и кристаллизовывались как показано в табл. A. Иногда перед кристаллизацией была необходима хроматография на кремниевом геле с MeOt, возможно с содержанием до 2% изопропиламина.
Общий способ G
Свободное основание диамина, 3 эквивалента
уксусной кислоты и 3 эквивалента при эфире или соответствующего триэтилортоэфира или гидрохлорида соответствующего
этоксиимина и 2-4 мл метанола на мМ диамина перемешивались при комнатной температуре
18 ч. Растворитель удалялся in vacuo. Продукт обрабатывался как описано в общем способе A.
Общий
способ H
Гидрохлорид диамина, 2.2 эквивалента ацетата натрия и 1,5
эквивалента триэтилортоэфира соединялись в 1,2 мл изопропилового ацетата на мМ диамина и нагревались с обратным
холодильником 4 дн. Растворитель удалялся in vacuo, остаток забирался дихлорметаном и
дихлорметан промывался 2 раза 2N гидроокисью натрия и высушивался на сульфате магния. Растворитель удалялся in
vacuo и продукт хроматографировался на силикагеле с разбавлением 49:49:2 этилацетатом
(дихлорметаном) диэтиламином. Гидрохлорид очищенного свободного основания готовился растворением свободного
основания в этилацетате и добавлением HCL в эфир.
Общий способ I
Дигидрохлорид диамина, 2,1 эквивалента ацетата натрия, 3 эквивалента триметилортоэфира и 5 эквивалентов
уксусной кислоты перемешивались вместе 7 дн при комнатной температуре. Доработка была та же,
что описана для общего способа H.
Общий способ J
3-хлорметилбензодиазепин
растворялся в 3 мл ацетонитрила) на мМ бензодиазепина и добавлялся к 1,0 - 1,7 эквивалентам триола
плюс 2,3 эквивалентам измельченного карбоната калия в 3 мл ацетонитрила на мМ бензодиазепина. Раствор
перемешивался при комнатной температуре 18 ч и фильтровался. Ацетонитрил удалялся in vacuo,
продукт дорабатывался как описано в общем способе A.
Общий способ K
3-хлорметилбензодиазепин и 3 эквивалента соответствующего амина или сульфинат натрия комбинировались в 1-5
мл растворителя на мМ бензодиазепина и нагревались с обратным холодильником 3-5 ч. Реакции с
аминами проводились в хлороформе; реакции с сульфинатом - в метаноле. Продукт дорабатывался как описано в
общем способе A.
Общий способ L
Бензодиазепин в 3-7 мл THF на мМ
бензодиазепина перемешивался при -78oC - -42oC, в течение чего в азоте добавилось 1,1
эквивалент N-бутиллития. Раствор перемешивался в течение 1 ч при -78oC,
добавлялось 1,1-1,3 эквивалента соответствующего электрофила и реакция выстаивалась при комнатной температуре. Затем
содержимое выливалось в 1N HCl, промывалось водой, делилось основным и
экстрагировалось в эфир. Соединенные эфирные слои высушивались в карбонате калия, фильтровались и растворитель удалялся in
vacuo. Свободное основание рекристаллизовалось или получалась соль как
показано в таблице.
Пример 265.
1-/4-хлорфенил/-3-[2-/4-хлорфенил/этил] -4,5-дигидро-1,
4-диметил-1H-2,4-бензодиазепин
(формула Ia: R1, R6
=H, R2a, R4b=Me
В соответствии с общим способом L, 14,5 г 1-(4-хлорфенил)-3[2-/4- хлорфенил/этил]-4,5-дигидро-1,4-диметил-1H-2,4-бензодиазепина в виде его соли фумарата было получено из 17,3 г соединения примера 228 и 7,6 г метил йодида. Соль была перекристаллизована на EtOH/эфира, т.пл. 173-175oC.
Общий способ M
Была повторена
процедура по общему способу L, за исключением того, что использовались 2
эквивалента N-бутиллития и 2 эквивалента альдегида.
Общий способ N
Бензодиазепин в 3-7 мл THF на мМ
бензодиазепине перемешивался при -78oC, и в это время в азоте
добавлялась 1,1 эквивалента бутиллития. Раствор перемешивался в течение 1 ч при -78oC, добавлялось 1,1-1,3
эквивалента гексахлорэтана и содержимое перемешивалось в течение получаса при
-78oC. Реакционная смесь вылилась в 1N HCl промывалось три раза эфиром, делалась основной 35% гидроокиси
натрия, фильтровалась и десорбировалась. Полученное масло коричневого цвета
фильтровалось через кремнезем этил ацетатом, десорбировалось и использовалось непосредственно для следующей стадии,
3-хлорметил бензодиазепин либо растворялся в хлороформе и обрабатывался 3-5
эквивалентами соответствующего амина, либо растворялся непосредственно в большом избытке амина. Раствор нагревался с
обратным холодильником 1-20 ч. Растворитель уделялся и продукт кристаллизовался как
показано в табл. E (см. в конце текста).
Общий способ O
Процедура аналогична процедуре
общего способа L, за исключением того, что здесь наоборот к 1,5 эквивалентам хлорэфира
добавлялась литиевая соль бензодиазепина.
Общий способ P
Процедура по сути аналогична
процедуре общего способа, за исключением того, что литий диизопропиламид, полученный из
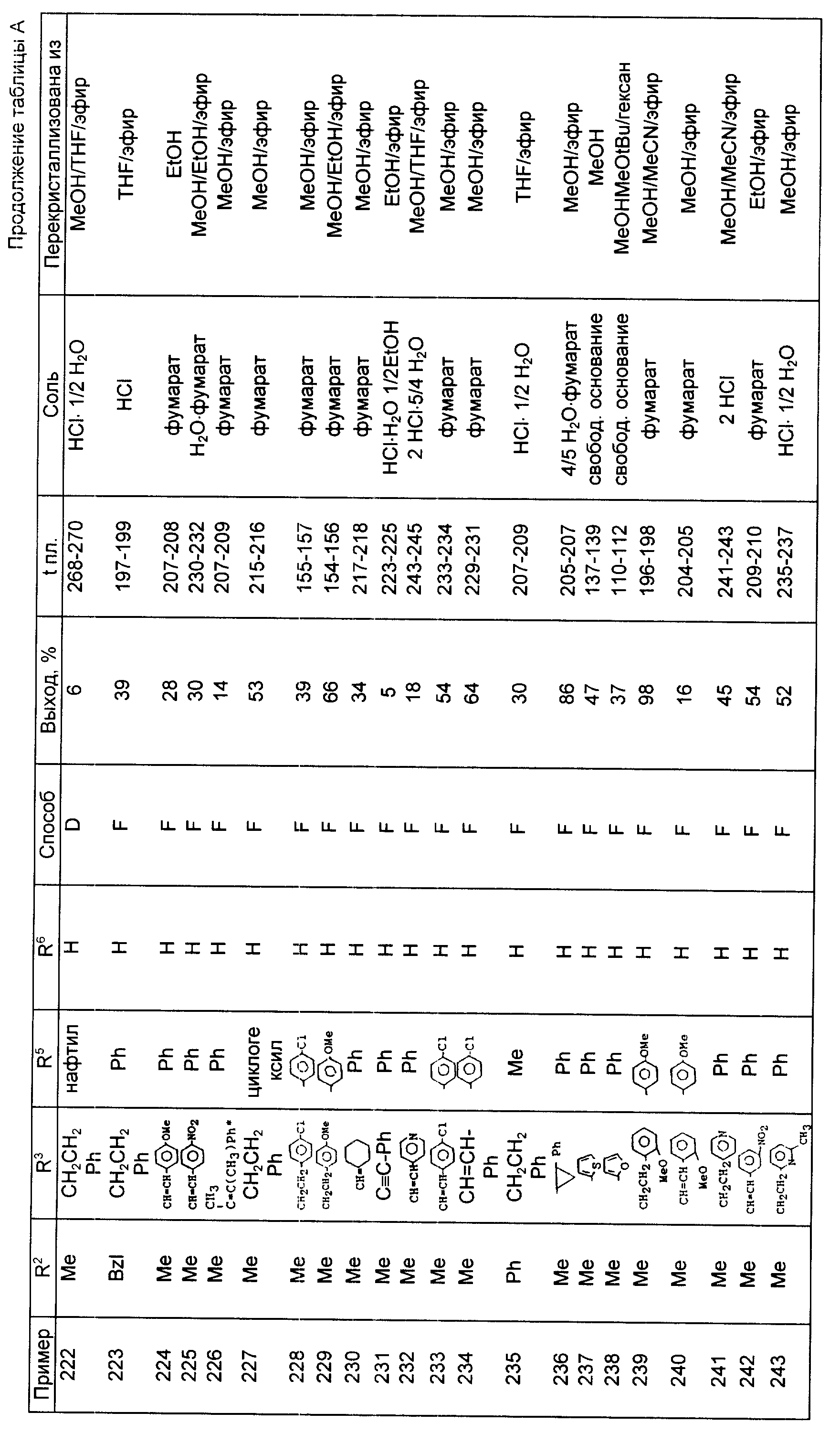
бутиллития и диизопропиламина, использовался в качестве основания и реакция проводилась при 0oC.
Общий способ Q
Процедура аналогична процедуре общего способа L, за
исключением того, что реакционная смесь охлаждалась льдом после часового перемешивания при -55oC с
добавлением небольшого избытка уксусной кислоты в THF.
Общий способ R
Бензодиазепин-3-он растворялся в 13-14 эквивалентах фосфорного оксихлорида и добавлялось 1/4 эквивалента
фосфорного пентоксида. Смесь перемешивалась при комнатной температуре в азоте, затем
нагревалась 18 ч при 90oC. Раствор десорбировался in vacuo и остаток обрабатывался 4-9 эквивалентами
соответствующего амина и перемешивался 2 ч при комнатной температуре. Избыточный амин
десорбировался in vacuo и остаток кристаллизовался как показано на табл. F (см. в конце текста).
Общий способ S
Соответствующий фталазин или фталазинон обрабатывались 4-8
эквивалентами диборана в ТГФ и смесь нагревалась с обратным холодильником 2-5 дн в азоте. В ходе реакции 4-8
эквивалентов диборана обычно добавлялись двумя или тремя порциями. Реакционная смесь
охлаждалась до комнатной температуры и в азоте аккуратно добавлялась избыточная водная или спиртовая соляная
кислота. Далее проводилось нагревание с обратным холодильником, ТН F (ТН F - ТГФ) удалялся
in vacuo и остаток делался основным 35% водной гидроокисью натрия. Продукт экстрагировался в этилацетате,
высушивался на сульфате натрия, концентрировался и либо очищался как соль гидрохлорида, как
показано в табл. G (см. в конце текста), либо, более привычно использовался без дальнейшей очистки в
качестве свободного основания. В табл. G римская цифра IX указывает, что исходным материалом был
соответствующий фталазин: римская цифра VIII указывает, что исходным материалом был соответствующий
фталазинон.
Общий способ T
Процедура практически аналогична процедуре
способа S за исключением того, что добавлялись 0,1-0,5 эквивалента борогидрида натрия и 0,7-1,5 из
диглима на мМ фталазинона.
Пример 149
4,
5-дигидро-3-этил-4-метил-1-фенилметил-1H-2,4-бензодиазепин
(формула I: R1, R4, R6=H; R2=Me; R3=Et; R5=BZl)
Раствор
из 12,5 г (50 мМ) 2-[(1-амино-2-фенил)-этил]-N-метилбензолметанамина в 150 ил изопропилацетата обрабатывался 4,1 г (50 мМ) ацетата
натрия и 50 мл (150 мМ) триэтилортоприпионата и 5 мл (87 мМ) уксусной
кислоты. Смесь нагревалась с обратным холодильником 3 ч и выливалась в 1,5 л ледяной воды, содержащей 200 мл 2N гидроокиси
натрия. Продукт экстрагировался в этилацетате, высушивался на сульфате натрия
и десорбировался. Остаток рекристаллизовался из изопропилового спирта (эфира для выхода 7,5 г свободного основания.
Свободное основание в этаноле обрабатывалось 4,6 г циклогексан сульфаминовой кислоты
и растворитель удалялся in vacuo. Остаток перекристаллизовывался из изопропилового спирта/эфира для получения 5,8
г продукта в виде циклогексановой соли сульфаминовой кислоты, т. пл. 137-138oC.
Пример 150.
4,5-дигидро-3-этил-1-фавил-1H-2,4-бензодиазепин
(формула I: R1, R2, R4, R6=H; R3
=Et; R5Ph)
Смесь из 1,36 г (3,6 мМ) 4-бензил-4,5-дигидро-3-этил-1-фенил-1Н-2,
4-бензодиазепина, 136 мг 10% палладия на углероде и 257 мг (4,0 мМ) формата аммония в 50 мл метанола
нагревалась с обратным холодильником в азоте 3 ч, 4 порция ацетата аммония, каждая более 230 мг
добавлялись каждые два часа во время нагревания до тех пор, пока ТСХ на силикагеле с 5' диэтиламином в
этил ацетате не показывала полное преобразование. Смесь охлаждалась, фильтровалась и
десорбировалась. Остаток распределялся между водной гидроокисью натрия и эфиром. Эфирный экстракт высушивался в
сульфате натрия, обрабатывался обеспечивающим углеродом, фильтровался и десорбировался.
Остаток брался в 60:40 этил ацетата/эфире и окислялся разбавленной эфирной HCl. Полученный осадок
отфильтровывался и рекристаллизовался из изопропанол/эфира для выхода 0,61 г (610) хлористоводородной
соли продукта, т. пл. 203-204oC.
Пример 151
4,
5-дигидро-4-метил-1-фенил-1Н-2,4-бензодиазепин-3-амин моногидрохлорид
(формула I: R1, R4, R6=H; R2=Me; R3=H2, R5
=Ph)
Раствор 15 г (66 мМ) 2-[(метиламино)-метил]- α -фенилбензолметанамина в 85 мл метанола
обрабатывался 7,2 г (68 мМ) бромциана при комнатной температуре. Раствор перемешивался при
комнатной температуре в течение 18 ч и затем десорбировался. Остаток растворялся в этаноле, и этанол
десорбировался. Остаток рекристаллизовывался из метанол/изопропил ацетата для получения 4,55 г
свободного основания, т.пл. 156-159. Маточные растворы растворялись в этаноле, обрабатывались небольшим
избытком этановой HCl и перекристаллизовывались из этанола для получения 1,3 г
хлористоводородной соли, т.пл. 259-261oC.
Пример 152
1,2,4,
5-тетрагидро-4-метил-1-фенил-3H-2,4-бензодиазепин-3-тион
К суспензии 15 г (50 мМ)
2-[(метиламино)-метил]-α-фенилбензолметанамин дигидрохлорида в 100 мл изопропилового спирта
прибавлялось 10 г (100 мМ) ацетата калия с последующим добавлением 3,3 мл (55 мМ) дисульфида углерода
в 35 мл изопропилового спирта. Суспензия перемешивалась при комнатной температуре полтора часа и
затем нагревалась с обратным холодильником 30 мин. Реакция охлаждалась во льду, и внутренняя соль
карбамодитионовой кислоты, загрязненная двумя эквивалентами хлорида калия, отфильтровывалась.
Карбамодитионовая кислота суспендировалась в 125 мл 95% этанола, и туда добавлялось 1,3 мл 12N соляной
кислоты. Суспензия нагревалась с обратным холодильником в течение трех дней, охлаждалась, и было
отфильтровано 15,3 г (114%) сырого бензодиазепин-3-тиона. 6-граммовая порция сырого продукта
рекристаллизовывалась из 2-этокси этанола для получения 2,0 г (38%) продукта, т.пл. 208-209o
C.
Пример 153
3[/2-(диэтиламино)этил] амино] -4,
5-дигидро-4-метил-1-фенил-1H-2,4-бензодиазепин
(формула I: R1, R4, R6=H, R2=Me, R3=NH(CH2)2N(C2H5)2)
Шлам 11,7 г (44 мМ) 4-метил-1-фенил-1,2,4,5-тетрагидро-3H-2,4-бензодиазепин-3-тиона из
примера 152 и 146 мл этанола обрабатывался 4,2 мл (67 мМ) йодистого метила в 30 мл
этанола, добавленного по каплям при 50o. Смесь перемешивалась при температуре окружающей среды в течение
18 ч, и было собрано 13,48 г (75%) 4-метил-1фенил-3-метилтио-4,5-дигидро-IH-2,
4-бензо-диазепина, т. пл. 201 - 205, как иодистоводородной соли.
Раствор 22,7 г (55 мМ) 3-метилтиобензодиазепина в 285 мл метанола нагревался с обратным холодильником с 7,8 мл (55 мМ) N,N-диэтилэтилендиамина в течение 18 ч. Смесь фильтровалась горячей для удаления небольшого количества нерастворимых загрязнений, охлаждалась, десорбировалась и распределялась между метиленхлоридом и водным гидроксидом натрия. Органические вытяжки высушивались на сульфате магния и десорбировались. Остаток рекристаллизовывался с большой трудностью как фумаратовая соль из изопропанола. После многочисленных рекристаллизаций было получено 1,5 г продукта, как гемигидрата дифумарата, т. пл. 160 - 162oC.
Пример 154
4,5-дигидро-4-метил-1-фенил-1H-2,
4-бензодиазепин-3-сульфокислота
(формула I: R1, R4, R6=H; R2=Me; R3=SO3H; R5=Ph)
29 г (108 мМ) 1,2,4,
5-тетрагидро-4-метил-1-фенил-3H-2,4-бензодиазепин-3-тиона из примера 152 обрабатывалась 2,4 г хлорида натрия,
420 г дигидрата молиодата натрия и 35 мл 30% пероксида водорода в 50 мл воды и 10 мл
t-бутанола, в соответствии со способом Maryanoff и др., J.Org. Chem. 51, 1882 (1956). Смесь все время оставалась
суспензией, и после нагревания при 70 - 80oC в течение 2 ч продукт
отфильтровывался из охлажденной суспензии для выхода 30,6 г (90%) сульфокислоты, не требующей дальнейшей очистки, т. пл.
188 - 190oC.
Пример 155
4,
5-дигидро-4-метил-1-фенил-3/1-пирролидино/-1H-2,4- бензодиазепин
(Формула I: R1, R4, R6=H;
R2=Me; R3=C4H8N, R5=Ph)
Смесь 4,75 г (15 мМ) сульфокислоты из примера 154 и 20 мл пирролидина нагревалась с обратным холодильником 18
ч. Пирролидин десорбировался и остаток был подвергнут хроматографии
на 340 г силикагеля, с 95:5 этилацетат/диэтиламином для получения 3,12 г остатка, который рекристаллизовался из 40 мл гексана для
выхода 2,14 г (47%) продукта, т. пл. 118 - 119.
Пример
156
3-[(4,5-дигидро-4-метил-1-фенил-1H-2,4-бензодиазепин-3-ил)-тио] - N,N-диэтилпропенамин
(формула I: R1, R4, R6=H; R2=Me; R3
=(CH2)3N(C2H5)2; R5=Ph)
Раствор 12 г (45 мМ) 1,2,4,
5-тетрагидро-4-метил-1/фенил-3H-2,4-бензодиазепин-3-тиона из примера 152 в
100 мл DMF (ДМФ) обрабатывался 1,24 г (50 мМ) гидрида натрия при 70oC и 7,5 г (50 мМ) 3-диэтиламинопропил хлорида,
прибавленного по каплям при 70oC. Смесь перемешивалась при
70oC в течение 5 ч и затем при комнатной температуре - 2 дн. Смесь выливалась в 250 мл ледяной воды и дважды
экстрагировалась в этилацетат. Продукт DMF (ДМФ) экстрагировался в 150 мл 2 HCl
промывался этилацетатом, делался основным и снова экстрагировался в этилацетат. Раствор этилацетата высушивался на
сульфате магния, десорбировался, и остаток растворялся в ацетоне. Было добавлено два
эквивалента малеиновой кислоты в 40 мл ацетона с последующим добавлением небольшого количества простого эфира.
Полученный остаток рекристаллизовывался из ацетон/эфира для получения как соли малеиновой
кислоты, т. пл. 95 - 97oC.
Пример 157
4,
5-дигидро-4-метил-3-метилтио-1-фенил-1H-2,4-бензодиазепин
(формула: R1, R4, R6=H;
R2=Me; R3=SMe; R5=Ph)
Раствор 8 г
(30 мМ) тиена из примера 152 и 2,7 мл (44 мМ) йодистого метила в 100 мл этанола нагревался с обратным холодильником 2 ч,
охлаждался, и гидройодид продукта отфильтровывался. Соль распределялась между
метиленхлоридом и водным бикарбонатом натрия, органический слой высушивался над сульфатом магния и десорбировался. Остаток
растворялся в этаноле, и к нему добавлялось 2,7 г метансульфокислоты с
последующим добавлением простого эфира. Полученный осадок отфильтровывался и рекристаллизовался из этанола для выхода 5,2 г
продукта как соли метансульфокислоты, т.пл. 195 - 196oC.
Пример 158
1,2,4,5-тетрагидро-4-метил-1-фенил-3H-2,4-бензодиазепин-3-он
Раствор 32,8 г (143 мМ)
2-[(метиламино)-метил]-α-фенилбензолметанамин в 215 мл хлороформа
обрабатывался 25,9 г (159 мМ) карбонилдиимидазола. Смесь перемешивалась при комнатной температуре в течение 19 ч, промывалась
четырежды водой, высушивалась над сульфатом натрия и десорбировалась in
vacuo. Камедеподобный остаток растирался в порошок и рекристаллизовывался из этилацетата для выхода 26 г (71%) продукта, т. пл.
198 - 199oC.
Пример 159
5-бутил-4,
5-дигидро-3-этил-4-метил-1-фенил-1H-2,4-бензодиазепин
(формула I: R1=nBu; R2=Me; R3=Et;
R4 и R6=H; R5=Ph)
Суспензия
14, 16 г (60 мМ) 2-метил-4-фенил-1 (2H)-фталазинона в 340 мл THF охлаждалась до -65oC в азоте и обрабатывалась 24,8 мл
(62 мМ) 2,5 Nn-бутиллития в гексане. Смесь перемешивалась в течение
20 мин при -65oC, и затем добавлялось 240 мл (240 мМ) IN комплекса боран-THF. После того, как раствор сам доходил до
комнатной температуры, в него добавлялось 340 г (9 мМ) борогидрида
натрия. Смесь нагревалась с обратным холодильником в течение 20 ч, после чего было добавлено еще 340 мг борогидрида натрия, и реакция
вновь нагревалась с обратным холодильником в течение 24 ч. Смесь
охлаждалась и остужалась 100 мл метанола, 80 мл 3,5 NHCl в метаноле добавлялось в реакционную смесь, которая затем нагревалась с
обратным холодильником 2 ч, и фильтрацией было отделено 19,9 г (93%)
дигидрохлорида 2-[1-(метиламино)пентил] - α -фенилбензометанамина. Неазолметанамин обрабатывался триэтилортопропионатом и
ацетатом натрия в изопропилацетате в соответствии с общим способом для
получения 9,48 г свободного основания продукта, т. пл. 102-114 после перекристаллизации из метил t - бутил эфир/гексана. 7 г
свободного основания переводилось в хлористоводородную соль и
рекристаллизовывалось из ацетонового эфира для выхода 5,08 г продукта в качестве соли-моногидрохлорида, т.пл. 209-211oC.
Пример 160
4,5-дигидро-1,
5-дифенил-3-этил-4-метил-1H-2,4-бензодиазепин
(Формула 1: R1; R5=Ph; R2=Me; R3+Et; R4, R6=H)
Был использован способ
из примера 159 с заменой бутиллития на фениллитий. Промежуточный продукт 2-[(метиламино)фенилметил]-α-фенилбензолметанамин кристаллизовался как
соль-дигидрохлорид, содержащая 0,6 моль воды, т.
пл. 202-216. Она циклизовалась с триэтил-ортопропионатом, как в примере 158 для получения 32% продукта в качестве хлористоводородной соли, т. пл. 275
- 276oC, из ацетона/эфира.
Пример 266
4,5-дигидро-1-(4-гидроксифенил)-4-метил-3-(2-фенил-тио/-1H-2,4-бензодиазолин
Раствор 6,78 г (17 мМ)
метокси-соединения из примера 60 в 70 мл метиленхлорида
обрабатывался 32 мл 1 М трибромида бора в метиленхлориде (32 мМ) при 0oC в азоте в течение 2 ч. Реакционная смесь выливалась в 2N
водную HCl, перемешивалась в течение 1 ч,
отфильтровывалась от солей бора в экстрагировалась в метиленхлорид со следами метанола, после того как делалась основной с помощью Na2CO3
. Органический слой высушивался,
десорбировался, и остаток помещался в метанол. Туда добавлялась метанольная HCl, и соль кристаллизовалась при добавлении эфира. Хлористый водород рекристаллизовался из
метанола, т.пл. 245-247oC выход 90%.
Пример 267
4,5-дигидро-3-[2-(4-гидроксифенил)этил] -4-метил-1-фенил-1H-2,4-бензодиазепин
(Формула; R1,
R4, R6= H;
R2=Me,

По способу, аналогичному процессу из примера 266, 1, 14 г 4, 5-дигидро-3-[2(4-гидроксифенил)этил] -4-метил-1-фенил-1H-2,4-бензодиазепин был получен в качестве хлористоводородной соли из 2,47 г (5,1 мМ) метокси-соединения из примера 35, т. пл. 160 - 162oC из метанола/эфира.
Пример 268
4,5-дигидро-1-/4-гидроксифенил/-3-[2-(4-гидроксифенил)-этил] -4-метил-1H-2,4- бензодиазепин
(формула 1; R1, R4, R6=H' R2Me;


По способу, аналогичному процессу из примера 266, 1,80 г 4,5-дигидро-1-(4-гидроксифенил)-3-[2-(4-гидроксифенил)-этил] -4-метил-1H-2, 4-бензодиазепин был получен из 4,5 г (8,7 мМ) диметокси-соединения из примера 229, используя 3,5 эквивалента трибромида бора. Свободное основание было нерастворимо в метиленхлориде. Гидрохлорид гемигидрат получался рекристаллизацией из MeO N (MeOH, т.пл. 266 - 268oC).
Разделение энантиомеров
Пример 167
(R)-(+)-4,
5-дигидро-4-метил-1-фенил-3-(2-фенилэтил)-1H-2,
4-бензодиазепин
В 1-литровую колбу Эрленмейера помещалось 100 мл метанола, 200 мл воды и 49,8 г (0,133 моль) рацемической хлористоводородной
соли из примера 25. Раствор перемешивался в
течение 10 мин, затем к гомогенному раствору добавлялось 200 мл t-бутилметилового эфира (ТБМЭ) с последующим добавлением 220 мл (0,66 моль, 5,0 экв.) 3
гидроксида натрия. Смесь перемешивалась 10 мин.
Слои разделялись и водный слой экстрагировался 100 мл насыщенного хлорида натрия. Органический слой высушивался над сульфатом магния, фильтровался и
растворитель удалялся при пониженном давлении при
30oC, чтобы получить количественный выход свободного основания. Вязкое золотисто-коричневое масло помещалось в вакуумный насос при 0,5 мм
рт. ст. на 1 ч.
Свободное основание растворялось в 40 мл метанола с небольшим подогревом. Раствор переносился в 500 мл колбу с тремя горлышками, снабженную механической мешалкой и конденсатором. Все это завершалось промыванием дополнительными 20 мл метанола. Раствор метанола подогревался по 45oC наружной водяной баней с контролируемой температурой.
В 250 мл химической стакан было добавлено 40,4 г (0,113 мМ, 0,85 экв. (D -0,0' -дибензоилвинной кислоты и 40 мл метанола. (Небольшой подогрев может быть необходим с тем, чтобы перевести хиральную кислоту в раствор). Метаноловый раствор хиральной кислоты немедленно добавлялся с постоянным помешиванием к раствору свободного основания. Полученная смесь приобретала очень светлый зеленый цвет. Дополнительные 20 мг метанола использовались для промывания, чтобы завершить перенос. После перемешивания в течение 5 мин раствор затравлялся. Продукт начинает осаждаться сразу же. Раствор перемешивался в течение ночи при 45oC. На воронке Бюхнера собирался гранулированный белый осадок, который промывался 3х25 мл холодным метанолом (5oC) и высушивался в течение ночи при 69oC при пониженном давлении.
Высушенная соль дибензоилтартрата весила 40,9 г (88%) после коррекции количества затравочного кристалла: [α] = +192oC (с=1, метанол); т.пл. 143-145oC разлож.
Соль гидрохлорида можно
получить следующей процедурой:
Было получено свободное основание из 100 г (0,143 М) из соли дибензоилтартрата, как указано выше. Свободное основание растворялось в 300 мл этилацетата. Раствор
переносился в двухлитровую
трехгорловую колбу, снабженную механической мешалкой, конденсатором и дополнительной воронкой. Перенос завершался промыванием дополнительными 300 мл этилацетата. Раствор
этилацетата подогревался до
45oC водяной баней с контролируемой температурой.
К подогретому раствору этилацетата со свободным основанием медленно добавлялись 69 мл 2,3 N хлороводорода/этилацетата. Добавление кислоты завершалось в течение получаса с последующим перемешиванием при 45oC 1 ч. Этилацетатная суспензия полученной соли гидрохлорида нагревалась с обратным холодильником 1 ч, чтобы избавиться от избытка хлороводорода, присутствующего в растворителе, и охлаждалась до комнатной температуры. Хлопьевидный белый осадок собирался на воронке Бюхнера и промывался 3х150 мл этилацетатом. Продукт высушивался в течение ночи при 80oC при пониженном давлении.
Высушенная соль гидрохлорида весила 50,9 г [α] = +234oC (с = 1, метанол); т. пл. 197 - 199oC.
Пример 168
(S)-(-)-4,
5-дигидро-4-метил-1-фенил-3-(2-фенилэтил)-1H-2,4-бензодиазепин
Маточные жидкости из кристаллизации в примере 167 десорбировались и свободное основание высвобождалось как раньше с
использованием tBuOMe и водного NaOH. Свободное основание растворялось в 100 мл метаноле, обрабатывалось 34,3 г дибензоил-L-винной кислоты и затравлялся. Было получено 37,2 г диастереометрической
соли
(-) изомера, т.пл. 160 - 170, [α] = -198oC (с = 1, MeOH). Свободное
основание
было образовано как выше, и соль HCl была образована и рекристаллизована из ацетонитрила/эфира, т. пл. 198 - 199 [α] = -249oC (с =1, CHCl3).
Пример 169
(+)-4,5-дигидро-1-фенил-1,3,4-триметил-1H-2,4-бензодиазепин
Способом,
аналогичным способу примера 167, включающему множественную рекристаллизацию, 1,4 г (+)-4,5-дигидро-1-фенил-1, 2,4-триметил-1H-2,4-бензодиазепина было получено из 8,9 г (33,7 мМ) гидрата
дибензоил-L-винной кислоты. Свободное основание было получено без рекристаллизации десорбцией t Bu OMe, т.пл. 115-116, [α] = + 101oC (с=1, MeOH).
Пример 170
(-)-4,5-дигидро-1-фенил-1,3,4-триметил-1H-2,4-бензодиазепин
Способом аналогичным
способу
примера 168,19 г (-)-4,5-дигидро-1-фенил-1,3,4-триметил-1H-2,4-бензодиазепина было получено из маточных жидкостей примере 169, т.пл. 116-117. [α] = -93oC (с = 1, MeOH).
Пример 171 (R)-(+)-4,5-дигидро-3-этил-4-метил-1-фенил-1H-2,
4- бензодиазепин
Способом
аналогичным примеру 167, 7,5 г (R(+)-4,5-дигидро-3-этил-4-метил-1-фенил-1H-2,4-бензодиазепина было получено из 92 г свободного основания рацемического продукта
примера 8 после перекристаллизации. Соль
гидрохлорида была получена из зтанол/эфира, т.пл. 244-247, [α] = +347oC (с =1,
CHCl3).
Соль d-10-камфорсульфокислоты была получена из ацетонитрила, т. пл. 215-218, [α] = 203oC (с =1,MeOH, [α] =242oC (с =1, CHCl).
Пример 172
(S)(-)-4,5-дигидро-3-этил-4-метил-1-фенил-1H-2,4-бензодиазепин
Способом аналогичным способу
примера 168, из маточных жидкостей примера 171 было
получено 15 г левого энантиомера. Продукт кристаллизовался как гидрохлорид из этанол/эфира, т.пл. 247-249oC, [α] =-343oC (с= 1, CHCl3).
Пример 173
(S)-(-) 4,
5-дигидро-3-этил-4-метил-1-фенил-1H-2,4-бензодиазепин
Следующая
процедура описывает альтернативный синтез соединения примера 172.
2 г (6,3 мМ) моногидрохлорида (S)N-[[[2-2/метиламино/метил]фенил]пропанамид (пример 181 перемешивались в 15 мл толуола в азоте и было добавлено при 0oC 3,45 мл 2М триметил алюминия в толуоле. Смесь перемешивалась 2 ч при комнатной температуре, затем 1,5 ч при нагревании с обратным холодильником. Далее она охлаждалась и остужалась 0,31 мл воды, а затем 0,93 мл 30 водного NaOH. Добавлялись метиленхлорид, небольшое количество метанола и немного сульфата натрия, смесь фильтровалась, десорбировалась и остаток рекристаллизовался из MeOH/эфира как гидрохлорид, т.пл. 247-248oC.
Пример
174
2-[[(1,1-диметилэтил/амино]метил]-α- фенилбензолметанамин
(Формула IV: R1a, R6=H, R2a=Bu; R5a H=pH)
Раствор из 15.91
г (90 мМ) N -t-бутилбензамида в 390 мл ТГФ охлаждался по - 15oC в азоте и
было добавлено 77 мл (193 мМ) -2,5 Mn-бутиллития в гексане. Смесь перемешивалась при - 5±3oC 1 ч
и в течение 10 мин при - 10oC было добавлено 17,5 г (99 мМ)
триметилсилилимина бензальдегида, полученного в соответствии с процедурой Hart et al.,J. Ord. Chem 48, 289-294 (1983). Реакция
перемешивалась при 0oC 1 ч, затем 5oC - 45 мин. Она
выливалась в 400 мл ледяной воды, содержащей 225 мл 2 NHCl и дважды промывалась эфиром. Водный слой делался основным
гидроокисью натрия и экстрагировался в эфир. Эфирные экстракты высушивались над
сульфатом натрия и десорбировались для выхода 25,4 г 2[(амино) (фенио/метил]-N-(1,1-диметил-этил/бензамида.
Вся часть аминоамида в 50 мл ТГФ объединялась с 450 мл (450 мМ) комплекса 1 боран-ТГФ и смесь перемешивалась при нагревании с обратным холодильником 18ч. Реакция охлаждалась, добавлялось 225 мл метанола и раствор нагревался с обратным холодильником 1 ч. Он был вновь охлажден и добавлено 200 мл полунасыщенной метаноловой HCl. Раствор вновь нагревался с обратным холодильником 1 ч, выпаривался в вакууме и остаток рекристаллизовался из хлороформ/эфира для выхода 21,3 г (70%) продукта в виде гидрохлорида, т.пл. 222-231oC.
Пример 175
2-(аминометил)-N-метил-α-фенилбензолметанамин
(Формула IV: R1a=R, R2a=Me, R5a, R6=H)
75 г (0,36 М) 2-бензоилбензальдегида
растворялось в 70 мл ТГФ и в течение 30 мин при 0oC добавлялись 18,5 г метилгидразина. Суспензия
стала однородным раствором, который выстаивался 4 дн. Первая порция, содержащая 8,5 г
продукта, была получена добавлением гексана и фильтрацией. Вторая порция 27,5 г продукта была получена
хроматографией на силикагеле с 85:15 метилен хлорид/этилацетатом. Т. пл. гидразона после
рекристаллизации из метилен хлорид-гексана была 164-165.
Раствор из 44,5 г метилгидразона в 80 мл ТГФ обрабатывался 374 мл 1М борана. После 6 дн реакция дорабатывалась, как описано в примере 164, и дигидрохлорид рекристаллизировался из метанол эфира, т. пл. 224-226oC.
Пример 176
2-(аминометил)-α-N-(фенилметил)бензолметанамин
(Формула IV: R1a=Ph; R2a=RZl; R5a = Ph)
62 г (0,30 М)
2-бензоилбензальдегида в 140 мл ТГФ реагировало с 81,5 г (0,59 М) карбоната калия и 64 г (0,32 М)
дигидрохлорида бензиглидразина. Смесь перемешивалась 30 мин при 0oC и было добавлено 40 мл
метилен хлорида. Смесь перемешивалась при комнатной температуре 1 день, фильтровалась и
десорбировалась в вакууме для получения 131 г 2-бензил-1-фенил фталазиний хлорида.
Сырой фталазин хлорид в 175 мл ТГФ обрабатывался 1,325 л 1 М боран-ТГФ при нагревании с обратным холодильником в азоте. После 24 ч реакция дорабатывалось, как описано в примере 175, и соль дигидрохлорида была получена осаждением на эфире эфирной HCl, чтобы получить 36,8 г дигидрохлорида продукта, т.пл. 175-178oC.
Пример 270
N-метил-α′-фенил-2,
3-тиофендиметанамин
(формула XXXVI: A = тиофен, R1a=H; R2a=ME; R5a=Ph)
Следуя процедуре общего способа T, 19 г N-метил-α′-фенил-2,
3-тиофендиметанамина было получено из 32,6 т (0,135 М) 6,7-дигидро-6-метил-7-оксо-4-фенилтиено[2,
3-d]пирадазина. Продукт был рекристаллизирован в виде его дигидрохлорида на этанол-воды, т.пл.
290-292oC.
Примеры 177-184, 208, 209, 273
Аминоамиды с формулой
III
был получены как побочные продукты синтеза соответствующих бензодиазепинов и обычно выделялись через хроматографию на силикагеле, с использованием основных растворителей для анализа. Они также получались гидролизом соответствующих бензодиазепинов, как описано в общем способе U примеры даны в табл. H (см. в конце текста).
Общий способ U
Раствор подходящего
бензодиазепина в 3-5 мл метанола на каждый миллимоль диазелина перемешивался при комнатной
температуре от 1 до 4 дн с 3-5 эквивалентами гидроксида калия в 1-2 мл воды на каждый миллимоль диазепина.
Добавлялся водный хлорид натрия, и аминоамид экстрагировался в эфир. Эфирный слой
высушивался над MgSO4 фильтровался, десорбировался, и остаток обрабатывался так, как показано в табл. H.
Предполагается, что с помощью этой процедуры все виды, описанные в табл. A-E,
должны превращаться в соответствующий аминоамид.
Общий способ V
Подводящее нитросоединение, в
виде его соли, обычно фумарата, растворялось примерно в 10 мл сухого метанола
на каждый миллимоль нитросоединения, и добавлялось от 0,1 до 0,15 г 10% Pd на углероде на каждый миллимоль
нитросоединения. Реакционная смесь перемешивалась при 18-24oC и туда добавлялось
от 7,5 до 8,0 эквивалентов формата аммония. По прохождении 1-2 ч реакционная смесь фильтровалась,
десорбировалась, распределялась между метиленхлоридом и 2N NaOH, разделялась, высушивалась и
десорбировалась. Остаток кристаллизовался, как показано в табл. Z (см. в конце текста).
Общий способ W
Подходящее нитросоединение в виде его хлористоводородной соли или
свободное основание плюс один эквивалент метанольной HCl, растворялось в примерно 20 мл метанола или этанола
на каждый миллимоль нитросоединения, и добавлялось 0.05-0,1 Pd на углероде на каждый
миллимоль нитросоединения. Смесь гидрогенизировалась при 3,5-1,4 атм на шейкер Парра. Когда было потреблено
расчетное количество водорода, реакционная смесь фильтровалась, добавлялся избыток эфирной
HCl и раствор десорбировался. Остаток кристаллизовался, как показано в табл. Z.
Пример
274
4-[2-/4,5-дигидро-3-этил-1H-2,4-бензодиазепин-4-ил/этил] бензоламин
(формула I: R1, R4, R6 = U,

По общему способу W, 4,50 г (10,3 мМ) 4,5-дигидро-3-этил-4-[2-/4-нитрофенил/этил] -1H-2, 4-бензодиазепин гидрохлорида из примера 276 было восстановлено до 3,33 г 4[2-/4,5-дигидро-3-этил-1H-2, 4-бензодиазепин-4-ил/этил] бензоламина в виде его моногидрохлорида, т.пл. 149-151 из KtOH-эфира.
Общий способ X
Раствор подходящего амина в виде его дигидрохлорида и от 3
эквивалентов пиридина до 30 эквивалентов пиридина перемешивались при 0oC в
примерно 10 мл метиленхлорида на каждый миллимоль амино в азотной атмосфере, в то время как 1.1-1.5 эквивалентов
метансульфонилхлорида или ацетилхлорида добавлялось по каплям. Реакционная смесь
перемешивалась при 0oC в течение 1-2 ч, и был прибавлен один объем насыщенного водного Na2CO3. В нескольких случаях, когда TCX показывала незавершенную реакцию, пере
раствором Na2CO3 дополнительно добавлялось 1-3 эквивалентов хлорида. Слои разделялись, и
органический слой десорбировался. Остаток был подвержен мгновенной хроматографии, если
это необходимо, на силикагеле с MeOH/MeOtBu изопропиламином 49:49:2.
Продукт
рекристаллизовывался, как показано в табл. M (см. в конце текста)
Пример 275
N-[4-[2-/4-4,5-дигидро-3-этил-1H-2,4-бензодиазепин-4-ил/этил] фенил] метансульфонамид
(формула
I: R1, R4, R6=H;

По общему способу X, 3,25 г 4-[2-/4,5-дигидро-3-этил-1H-2,4-бензодиазепин-4-ил/этил] бензоламин из примера 274 было превращено в 3,49 г N-[4-[2-/4-дигидро-3-этил-1H-2,4-бензодиазепин-4-ил/этил]фенил] метансульфонамид, т. пл. 129-142, в виде свободного основания из EtOH-эфир-метиленхлорида.
Исходные материалы
Фиалазиноны, являющиеся исходными материалами для синтеза
диаминов, описанного в таблице G, обычно получаются способами, известными в литературе. Наиболее часто
они синтезируются конденсацией соответствующих y - кетокислот с подходящим гидразином.
Пример 85
2-метил-4-фенил-(2H)-фталазином
Резервуар из нержавеющей стали
емкостью в 100 галлонов загружался 40,0 кг 2-бензоилбензойной кислоты и 87,5 кг толуола. Спустя
около 45 мин добавлялся метилгидразин, внутренняя температура при этом поднималась до 34o
C.
Полученная тонкая суспензия нагревалась с обратным холодильником (95-118oC) в течение 4,5 ч с одновременным сбором около 7,5 л воды.
Реакционная смесь медленно охлаждалась с заметным началом выпадения осадка при 88oC. Полученная суспензия охлаждалась до 0 - -5oC и затем собирались кристаллы бежевого света. Слежавшийся кусок промывали 2х20 л холодного толуола и высушивали in vacno при 45-40oC в течение ночи, чтобы получить 38,0 кг (выход 91,0%) 2-метил-4-фенил-1(2H)-фталазинона, т. пл. 166-168oC.
Пример 271
6,7-дигидро-6-метил-7-оксо-4-фенилтиено[2,
3-d]пиридазин
Раствор 31,2 г (0,134 моль) 3-бензоил-2-тиофенилкарбоновой кислоты в 400 мл этанола обрабатывался 9,3
(0,2 моль) метилгидразина при комнатной температуре в течение 18 ч,
нагревался с обратным холодильником 3 ч, охлаждался и 30,7 продукта отфильтровывалось, т. пл. 174-175oC.
3-бензоил-2-тиофен карбоновая кислота была получена из 3-бромтиофена методом Mac Dowell (J. Org. chem. 42, 3717 (1977).
В случае, когда подходящий алкилгидразин для конденсации фталазинона не всегда имеется в распоряжении, y-кетокислота конденсируется с гидразином, и полученный 2-незамещенный 1-фталазинон алкилируется.
Пример 186
4-фенил-2-/2-фенилэтил/-1(2H)-фталазинон
85 г (0,38 моль)
4-фенил-1(2H)-фталазинона добавлялось к 18,4 г (0,47 моль) гидрида натрия в 1 л DMSO (DMCO) четырьмя порциями. Смесь перемешивалась в
течение 2 ч при комнатной температуре до тех пор, пока не
прекратилось выделение водорода, и туда добавлялось 95,5 г (0,52 моль) 2-бромэтилбензола. Смесь перемешивалась 1,5 ч при комнатной температуре,
затем добавлялся 1 л 2N NaOH, и суспензия выливалась в
1 л воды. Продукт отфильтровывался и высушивался для выхода 118 г (95%) 4-фенил-2-(2-фенилэтил)-1(2H)-фталазинона, т.пл. 135-138o
C.
Пример 278
2-[2-/4-нитрофенил/этил] -4-фенил-1(2H)фталазинон (формула VIII:

Способом, аналогичным описанному в примере 186, 11,8 г 2-[2-/4-нитрофенил/этил] -4-фенил-1(2H)-фталазинон готовился из 10,0 г (45 мМ) 4-фенил-1(2H)фталазинона и 11,6 г (50 мМ) 4-нитрофенил бромида. Продукт рекристаллизовывался из EtOAc-эфиргексана, т. пл. 152-155oC.
Синтез сложных эфиров с формулами R8CH=CH-COOEt и R8CH2CH2 COOEt, где R8 является гетероарилом.
В этих случаях, когда подходящие эфиры-пропаноаты и эфиры-пропаноаты не являлись коммерчески доступными пропеноат синтезировался конденсацией этилацетата с подходящим альдегидом в присутствии 1 эквивалента металлического натрия. Ненасыщенные сложные эфиры восстанавливались большим избытком металлического магния в метаноле, чтобы получить пропаноаты.
Смешанные синтезы диаминов, фталазинов и предшествующих соединений показаны ниже:
Пример 187
4-бензил-2-метил-1(2H)-фталазинон
Раствор 30 г гидроксида калия и 31,1 г (140 мМ) бензилиденфталида в 100 мл воды нагревался до состояния гомогенности и выливался в
раствор 40 мл H2SO4 в 250 мл воды. После
охлаждения полученное твердое вещество собиралось и растворялось в водном бикарбонате натрия. Добавлялась 2N HCl до первых признаков
осаждения, водный раствор промывали четыре раза хлороформом, и
затем подкислялся избытком 2N HCl. Белый осадок 20,3 г 2-/1-оксо-2-фенил-этил/бензойной кислоты отфильтровывался и сушился, т. пл.
74-75oC, после перекристаллизации из этаноловой воды. Эта
y-кетокислота обрабатывалась метилгидразином в соответствии со способом из примера 185, для получения 17,3 г фталазинонового
продукта, т. пл. 144-146oC.
Пример 188
2-метил-4-тиенил-1(2H)-фталазинон
Раствор 74,1 г (0,5 моль) фталового ангидрида в 300 мл нитробензола обрабатывался
174 г (1,1 моль) хлорида алюминия. Раствор перемешивался в течение двух
часов, и при 40-45oC за 80 мин по каплям добавлялось 42,1 г (0,5 моль) тиофена. Реакционная смесь перемешивалась при
50-55oC в течение 2 ч и затем была оставлена при комнатной
температуре на ночь. После этого реакционная смесь выливалась в 2,8 л холодной воды, перемешивалась, разделялась, и нитробензол
удалялся из нитробензолового слоя перегонкой с водяным паром. Остаток
рекристаллизовывался из толуола, чтобы получить 31,1 г (27%) 2-тиеноилбензойной кислоты, т. пл. 141-143oC.
Тиеноилбензойная кислота обрабатывалась метилгидразином, как описано в примере
185, чтобы получить 24,4 г (75%) фталезинонового продукта, т. пл. 143-144oC, после перекристаллизации из
этилацетата.
Пример 189
5-фтор-3,
4-дигидро-3-метил-1-фенилфтазалин
К раствору 20 г (0,14 моль 2-фтор-6-хлоро-толуола в 125 мл ТГФ добавлялось 6,9 г (0,28 моль)
магниевых стружек. Смесь нагревалась с обратным холодильником с
одновременным прибавлением по каплям на протяжении 3 ч 12 мл 1,2-диброметана в 50 мл бензола. Далее реакционная смесь нагревалась с
обратным холодильником еще 1 ч, и добавлялось 15 мл бензонитрила.
Далее смесь нагревалась с обратным холодильником еще 2 ч, охлаждалась, и остужалась 50 мл воды, прибавленной по каплям. Смесь
экстрагировалась в этилацетат, высушивалась над сульфатом натрия, и
десорбировалась. Остаток растворялся в 50 мл этанола, добавлялось 25 мл 1N HCl и смесь нагревалась с обратным холодильником 3 ч.
Этанол десорбировался, продукт экстрагировался в этилацетат,
этилацетат высушивался над сульфатом натрия, и десорбировался. Остаток 3-фторо-2-метил-бензофенона подвергался хроматографии на силикагеле
с 20% эфиром в гексане.
Раствор 1,69 г (7, 89 мМ) бензофенона в 40 мл 4-хлористого углерода обрабатывался 100 мг пероксида бензоила и 1,5 г (8,43 мМ) N-бросукцинимида. Реакционная смесь перемешивалась при комнатной температуре 2 ч и затем дополнительно нагревалась с обратным холодильником 4 ч, в течение которых была добавлена вторая порция 50 мг пероксида бензола и 400 мг N-бром-сукцинимида. Реакционная смесь охлаждалась, отфильтровывалось небольшое количество примесей, и фильтрат концентрировался in vacuo для получения 2,5 г 2-бромометилфторобензофенона, предположительно содержащего связанный 4-хлористый углерод.
Остаток α - бромметилкетона растворялся в 40 мл хлороформа, и по каплям прибавлялась смесь 1,5 мл триэтиламина и одного эквивалента метилгидразина. Реакционная смесь перемешивалась 1 ч, промывалась 20 мл водного бикарбоната натрия и фильтровалась непосредственно через силикагель с 25% этилацетатом в гексане. Концентрирование in vacuo дало 1,86 г (98%) продукта, который немедленно восстанавливался как показано в таблице. G (см. в конце текста).
Пример 190
2-бензоил-5-фторобензойная кислота
Следуя процессу из примера 189, 4,
0 г (21,2 мМ) 2-бромо-5-фторотолуола обрабатывалось 2,4 мл (23,5 мМ) бензонитрила. Имин, полученный из конденсации не
гидролизовался. Вместо этого 28,6 г (0,13 моль) 4-фторо-2-метил-бензофенонимина в
200 мл воды и 100 мл пиридина нагревалось с обратным холодильником в течение 8 ч и обрабатывалось четырьмя порциями
перманганата калия через строго 2-часовые интервалы. Эти порции были по 53, 28, 20
и 10 г. Реакционная смесь охлаждалась, фильтровалась через диатомовую землю и концентрировалась in vacuo. Остаток
распределялся между водной уксусной кислотой и этилацетатом; этилацетат сушился над
сульфатом магния и этилацетат удался in vacuo, чтобы получить 16,2 г (51%) желтой смолы, которая используется по
назначению.
Пример 191
2-бензоил-4-фторбензойная кислота
Процесс из примера 190 использовался для получения 14,2 г (53%) продукта из 21,3 г 2-бром-4-фторотолуола.
Пример 192
3,4-дигидро-3-метил-1-фенилбензо [f] фталазин
Раствор 10 г (45 мМ) 1-бром-2-метилнафталина в 75 мл ТГФ нагревался с обратным холодильником с 1,2 г (50 мМ)
магниевых стружек в течение 3 ч. Реакционная смесь охлаждалась на льду, и в нее
добавлялось 4,8 мл (43 мМ) бензальдегида. Лед удалялся, реакционная смесь перемешивалась 45 мин и охлаждалась 5 мл IN
HCl с последующим добавлением 50 мл воды. Смесью экстрагировалась в этилацетат,
сушилась над сульфатом натрия и подвергалась мгновенной хроматографии через силикагель с 5 - 10% этилацетата в гексане
для получения 8,4 г (75%) 2-метил-а/фенил-1-нафталин-метанола в виде
бледно-желтой смолы.
Раствор 10,0 г (40 мМ) вторичного спирта в дихлорметане обрабатывался 12,4 г (58 мМ) хлорохромата пиридина, быстро нагревался с обратным холодильником, перемешивался при комнатной температуре 1 ч. Смесь разбавлялась 150 мл эфира и фильтровалась через флоризил. Концентрирование фильтрата in vacuo давало 7,92 г (80%) 1-бензоил-2-метилнафталина в виде ярко-оранжевой смолы, которая медленно кристаллизовалась.
Способом, описанным в примере 189, 4,4 г (18 мМ) бензоилиафталина было превращено в 1,73 г фталазинового продукта, который немедленно восстанавливался как показано в табл. G.
Пример 193
3,4-дигидро-3,
8-диметил-1-фенилфталазин
Способом, полностью аналогичным способу из примера 192,
фталазин синтезировался из 2-бромо-m-ксилена
Пример 194
2-аминометил-α
-фенилбензолметанамин
К суспензии 3,8 г (100 мМ) литий алюминийгидрида в 120 мл ТГФ добавлялось
11,1 г (50 мМ) 4-фенил-1-(2H)фталазинона.
Смесь нагревалась с обратным холодильником 1 ч, охлаждалась, разбавлялась 100 мл эфира и последовательно обрабатывалась 3,8 мл воды, 3,8 мл 15% водного гидроксида натрия и 11,4 мл воды. Смесь перемешивалась 30 мин, и гранулярный осадок отфильтровывался. Фильтрат разбавлялся небольшим количеством толуола, сушился над сульфатом натрия и десорбировался для получения 14,1 г масла, которое растворялось в 180 мл этанола и гидрогенизировалось при 50 ф/дюйм2 в присутствии 20 мл этанольной HCl и 1,5 г 10% палладия на углероде. По прошествии 24 ч образовывался осадок. Смесь фильтровалась, осадок суспендировался в 250 мл горячего метанола и снова фильтровался. Объединенные фильтраты десорбировались по 100 мл и разбавлялись эфиром. При охлаждении 7,3 г 1,2,3,4-тетрагидро-1-фенилфталазина в виде соли-моногидрохлорида отфильтровалось. Он рекристаллизовался из метанол/эфира для выхода 6,93 г (56%) продукта, т. пл. 251 - 253oC.
Тетрагидрофталазин повторно растворялся в 200 мл метанола нагреванием и гидрогенизировался при давлении 50 ф/дюйм2 при 66oC 20 ч в присутствии 3,5 г никелевого катализатора Ранея. Катализатор отфильтровывался, и фильтрат десорбировался. Остаток рекристаллизовался из метано/эфира для получения 99% выхода соли диаминадигидрохлорида, т. пл. 270 - 273oC.
Пример 195
2-/4-метоксибензоил/бензойная кислота
Смесь 57 г (0,4 моль) фталевого ангидрида и 43 мл (0,4 моль)
анизола в 400 мл бензола обрабатывалась 105 г (0,8 моль) хлорида алюминия при 5o
C. Реакционная смесь держалась в течение 5 дн при 5oC, выливалась в 600 мл 2N водной HCl и льда,
и фильтровалась. Остаток растирался в порошок в водном карбонате натрия и фильтровался
несколько раз до тех пор, пока твердое вещество больше не содержало продукта. Вытяжки карбоната натрия
соединялись, промывались эфиром и подкислялись 2N водной HCl. Продукт экстрагировался в эфир,
высушивался над сульфатом натрия и десорбировался. Он рекристаллизовался из толуола для получения 80%
продукта, т. пл. 145 - 147oC.
Пример 196
2-(2-метоксиэтид)-4-фенил-1(2H)-фталазинон
85 г (0,38 моль) 2-бензоилбензойной кислоты и 28,6 г (0,38 моль)
гидроксиэтилгидразина реагировали согласно процессу из примера 185. Полученный
гидроксиэтилфталазинон суспендировался в 300 мл DMF и 200 мл ТГФ, и в течение 40 мин в азоте добавлялся по порциям 12,3
г 60% гидрида натрия в масле. Реакционная смесь перемешивалась дополнительно 45
мин при комнатной температуре, и выделение водорода прекращалось. На протяжении 1,5 ч добавлялся 31 мл йодистого метила,
и реакция перемешивалась при мягком нагревании с обратным холодильником в
течение 16 ч. Затем смесь выливалась в воду и экстрагировалась в эфир. Эфирные слои высушивались над сульфатом натрия и
десорбировались. Остаток подвергался хроматографии на силикагеле с 5% этиламином
в этилацетате для выхода 39 г (47%) продукта, т. пл. 115 - 118oC после перекристаллизации из
циклогексана.
Пример 197
2-бензоил-4,5-диметоксибензойная кислота
500 миллилитров 37% раствора формалина насыщалось хлористоводородным газом при 15 - 20oC,
и было добавлено 70 г (0,38 моль) вератровой кислоты одной порцией. Смесь нагревалась при 60
- 70oC 7 ч и оставлялась при комнатной температуре на 14 ч. Раствор концентрировался in vacuo,
растворялся примерно 300 мл воды, охлаждался и делался основным с помощью гидроксида аммония.
Полученное твердое вещество собиралось фильтрацией и высушивалось для выхода 65% диметоксифталида.
108 г (0,56 моль) фталида окислялось 258 (1,64 моль) перманганата калия в соответствии с процессом из примера 190.
81 г диметоксифталевой кислоты превращалось в 72 г соответствующего диметоксифталевого ангидрида быстрым нагреванием в 200 мл уксусного ангидрида.
30 г (0,14 моль) 4,5-диметоксифталевого ангидрида суспендировалось в 300 мл ТГФ, и 87 мл (0,17 моль) фенилмагний хлорида добавлялось на протяжении 2 ч. Реакционная смесь перемешивалась при комнатной температуре 14 ч, нагревалась с обратным холодильником 2 ч, охлаждалась и выливалась в насыщенный хлорид аммония. Смесь делалась кислотой 6NHCl, экстрагировалась в хлороформ, сушилась над сульфатом магния, концентрировалась для выхода 30 г 2-бензоил-4,5-диметоксибензойной кислоты.
Пример 272
Метил-4-/диэтиламиносульфонил/бензопропаноат
К 14,5 г N, N диэтил-4-бромбензолсульфонамида (50 мМ) (готовился реакцией 4-бромбензол сульфонилхлорида с
диэтиламином) прибавлялось 13,8 г
тетрабутиламмоний хлорида (50 мМ), 10,3 г NaHCO3 (124 мМ), 266 мг ацетата палладия (II) (1,07 мМ) и 100 мл DMF в указанном порядке. К этой суспензии
добавлялось 8,8 мл метилакрилата (98 мМ),
и реакционная смесь перемешивалась 1 ч при 80oC. Реакционная смесь охлаждалась, в нее добавлялись 500 мл воды и 900 мл эфира, слои разделялись, и
эфирный слой фильтровался для удаления Pd
(0) и соединялся с 2 последующими эфирными промывками водной фазы. Объединенные эфирные растворы сушились над MgSO4, фильтровались,
десорбировались и рекристаллизовались из метанол/эфира
для выхода 9,4 г метил 4-диэтиламинсульфонил/бензол-2-пропеоната, 6 г пропеоната восстанавливалось в этаноле при 3,5 атм 10% Pd на углероде в
шейкере Парра для получения продукта в виде желтого масла.
В этой форме он использовался в примере 269.
Пример 198
1[4-/диэтиламино/фенил]-3-этил-4-метил-1H-2,
4-банзодиазепин
(Формула) R1, R4, R6=U; R2=Me; R3=Et; R5=Et2;

С помощью процесса, аналогичного процессу из примера 41, видно, что 1-[4-/диэтиламинофенил] -3-этил-4-1H-2,4-бензодиазепин может быть синтезирован из 2-[4-/диэтиламино/бензоил] бензойной кислоты (смотри патент США N 4106174), метилгидразина, и триэтилортопропионата.
Пример 199
3-метил-1-[2-[/1-оксопропил/амино] фенил] -4-/3-фенилпропил/-1H-2,4-бензодиазепин
(Формула I:
R1, R4, R6=U; R2=(CU2)3Ph;
R3=Ml;

С помощью процесса, аналогичного процессу из примера 41, видно, что 1-/2-аминофенил/4-/3-фенилпропил/-3-метил-1H-2,4-бензодиазепин можно синтезировать из 2-/2-аминобензоил/-бензойной кислоты, гидразина, бромбензопропана и триэтил-ортоацетата. Далее заключается, что этот продукт может ацилироваться обработкой пропионовым ангидридом при комнатной температуре для получения 3-метил-1-/2-/ (1-оксопропил/амино/-фенил/-4-/3-фенилпропил/1H-2,4-бензодиазепина.
Иминоэфиры
(алкоксиимины)
R3COR12
• HCl
NH
Этокси и метокси имины, используемые для конденсации с диаминами, были получены
из соответствующих нитрилов способами, хорошо известными в литературе. В общем, нитрил
растворялся в эфире, добавлялся 1,1 эквивалентов алканола, впускалось 1,1 эквивалентов сухого HCl газа, и смесь
выдерживалась при 5oC в течение 24-48 ч; хлористоводородная соль иминоэфира
была регененирована простой фильтрацией.
Триалкилортоэстеры получались обработкой соответствующего иминоэфира с подходящим алканолом в условиях, известных в литературе.
Соединения этого изобретения, имеющие формулы XXXVI, XXX, III, II и XXXVII, обладают антиаритмической активностью, как показали результаты стандартных фармакологических испытаний, проведенных на показательных примерах, описанных ниже.
Антиаритмическая активность демонстрировалась с помощью процедуры, которая является вариантом стандартной запрограммированной техникой проведения электрофизиологических испытаний, используемой для крупных животных и для клинических экспериментов. Мужские особи крыс Duman-Hartlly (600-800 г) обезболивали пентобарбиталом натрия (30 мг/кг) и искусственно вентилировали респиратором Harvard. Производилась торакотомия слева и через переднюю стенку левого желудочка вводился катетер, наполненный жидкостью и датчик (Millar Micro-tip, Model 4F, Millfr Jnst, Jnc., Nousson, texas), чтобы контролировать левое вентикулярное давление (ЛВД). Первые показания ЛВД (dp/dt) были получены от дифференциатора (Grass) Model 7P20B) и они использовались как индекс функции сокращения. ЭКГ вместе с ЛВД и dp/dt непрерывно записывалось на полиграфе Grass/model 7B). Индекс работы сердца рассчитывался с использованием пикового систолического ЛВД и сердечных сокращений (СС). Периоды эффективной невосприимчивости (ПЭН) определялись в левом вентрикулярном ритме. Подкожные электроды Grass вживлялись подкожно как диплолюсные вентрикулярные электроды для получения стимула от стимулятора (Bloom Electriniss, Jne., Reoding, Pennsylvania). Сердца стимулировались с наименьшей частотой, позволяющей постоянный ритм (S), 240-300 с/мин с использованием 2-мс импульсов при двойном диастолическом пороге. Порог определялся увеличение напряжения стимуляции до того, пока не наблюдалась вентикулярная реакция на стимул 1:1. Подавался шлейф из 8 обычных импульсов с последующим преждевременным (S2) импульсом. Интервал между последующим S1 и преждевременным S2 импульсом снижался в 10-мс увеличениях до тех пор, пока не появляется вентрикулярный ответ (реакция). Самый длинный интервал S1 - S2, который не давал вентрикулярной реакции, определялся как ПЭН. Ритм и ЭКГ показался с частотой 92 Гц на App микрокомпьютере с использованием двухканального преобразователя 8-bit (R.C. Electronics, Comp-Scope APL-D2, Santa Barbara).
Функция гемодинамики определялась с последующим определением ПЭН. До дачи лекарства ритм был прерывистым и возобновлялся через определенные интервалы для определения ПЭН. Лекарства вводились (1 мл/кг) через левый вентрикулярный катетер через 15-секундный интервал для дозировок менее 10 мг/кг. Более увеличенные дозы (> 10 мг/кг) медленно вводились через 1-минутный интервал. Дозы кумулятивно увеличивались каждые 25 мин до тех пор, пока не были доведены до максимально допустимой дозы, при которой отмечалось сниженное dp/dt на 50%. Через 10 мин после каждой дозы определялась вновь гемодинамика и ПЭН.
Данные анализировались с использованием анализа варианта, для повторных мер необработанных данных и выражались в средних величинах. Эффективная доза, которая увеличивала ПЭН минимум на 20 мс (ЭД20), которая постоянно была статистически существенным увеличением, была получена для каждого животного из линейной регрессии данных и выражена как средняя величина для лечения. Биологическое значение было определено с вероятностью ошибок менее 0,05. Результаты представлены в табл. N, приведенной в конце текста.
Соединения изобретения можно получить для использования в обычных фармацевтических процедурах, т.е. растворением или суспендированием их или их фармацевтически приемлемых солей в фармацевтически приемлемой среде, например воде, водном спирте, гликоле, масляном растворе или водо-масляной эмульсии для парентерального или орального назначения; или введением их в форме дозировки в виде капсул или таблеток для орального назначения либо в чистом виде, либо с обычными наполнителями, например карбонатом кальция, крахмалом, лактозой, тальком, стеаратом магния, гуммиакацией и прочее.
Процентный состав активного компонента в композиции и метол лечения или профилактики аритмии можно варьировать с тем, чтобы получить приемлемую дозу. Дозировка, назначаемая конкретному пациенту меняется в зависимости от клинической оценки, используемой как критерии: вид назначения, длительность лечения, вес и состояние пациента, потенцию активного компонента и ответную реакцию пациента. Таким образом, эффективную дозировку активного компонента может определить врач с учетом всех критериев и оценки здоровья пациента.
Составы, примеры которых приведены в конце текста, лиофилизируются при отсутствии воды. Указанный объем воды используется для восстановления объема лиофилизированного состава перед его введением пациенту.
Сахароза и хлорид натрия в составах выполняют роль стабилизаторов, в то время как молочная кислота и гидроксид натрия выполняют роль буферов.
Процесс лиофилизации вышеуказанных и им подобных составов заключаются в следующем.
Ампулы необходимых размеров для заполнения составом выбираются, исходя из требований дозировки. При выборе ампул, соответствующих дозе, заполняемый объем не должен превышать определенную часть объема ампулы. Например, 5 мл не должны содержаться в ампуле объемом менее 10 л. После наполнения ампулы помещаются в сушильный шкаф прямо на рефрижераторные полки, предварительно охлажденные до 4oC. Внутрь некоторых ампул помещаются термопары для наблюдения над температурой состава во время лиофилизации. Затем ампулам дается возможность уравновеситься до температуры полок (4oC), прежде чем температура полок будет снижена до -40oC для сахарозы состава и -30oC для маннитозного состава. При достижении -40 и -30oC, соответственно, для сахарозных и маннитозных составов, ампулы держат при этой температуре, в течение приблизительно 2 ч, чтобы обеспечить полное замерзание составов. (Для маннитозных составов на этом этапе проводится стадия отжига). После этого спирали конденсатора охлаждаются до -60oC и включается вакуумный насос для откачивания воздуха из конденсаторной камеры, а затем проводятся процессы первичного и вторичного высушивания. Во время первичного процесса высушивания главный клапан между конденсатором и сушильной камерой открыт и из последней откачивается воздух до тех пор, пока давление не составит приблизительно 100 мкм с выводом азота. Этот период цикла лиофилизации (первичная сушка) занимает 40 - 50 ч. Первичный процесс сушки считается завершенным, когда с замершей матрицы исчезнет весь лед. Во время второго процесса сушки, температуру повышают от -20 или -30 до +25oC с целью удаления всей остаточной влаги, которая не была удалена во время первичного процесса сушки. Этот вторичный период сушки занимает приблизительно 15 ч.
После завершения вторичного процесса сушки, главный клапан закрывается и сушильный шкаф заполняется азотом с тем, чтобы поддерживать в нем небольшой вакуум. Затем включается поршень и ампулы (флаконы) закрываются. Затем сушильный шкаф уравновешивается до атмосферного давления и дверца шкафа открывается для того, чтобы внутрь флаконы и запечатать их гофрированным герметиком. Флаконы затем хранятся при указанной температуре до тех пор, пока их содержимое не будет разведено водой для инъекций.
Твердые лекарственные формы для орального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивается с по крайней мере одним обычным инертным наполнителем (или носителем), таким как цитрат натрия или дифосфат кальция или (а) наполнителями или удлинителями, такими, например, как крахмалы, лактоза, глюкоза, маннит и силициловая кислота, (б) связующими, такими как, например, карбоксиметил целлюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и акация, (в) увлажнителями, такими как, например, глицерин, (г) веществами, вызывающими распад, такими как, например, агар-агар, карбонат кальция, (д) заместителями растворения, такими как, например, парафин, (е) ускорителями абсорбции, такими как, например, четвертичные соединения аммония, (ж) увлажнителями, такими как, например, цетиловый спирт и моностеарат глицерина, (з) абсорбенты, такие как, например, каолин и бентонит, и (и) смазывающие вещества, такие как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленовые гликоли, сульфат лаурила натрия, или их смеси. Лекарственные формы, представляющие собой капсулы, таблетки и пилюли, лекарственные формы могут также включать буферные вещества.
Пример F. Мягкие или
твердые капсулы
Компоненты - Количества, мг
Активные соединения или их соли - 1,00
Крахмал - 51,00
Моногидрат лактозы - 203,33
Поливинилпирролидон - 4,
30
Лаурилсульфат натрия - 0,17
Карбоксиметил целлюлоза - 8,50
Достаточное количества очищенной воды для влажного
гранулирования
Стеарат магния - 1,70
Вышеуказанный состав помещался в капсулу, содержащую 170 мг соединения.
Пример G
Компоненты - Количества, мг
Активное соединение - 10,00
Крахмал - 51,00
Моногидрат лактозы - 94,33
Поливинилпирролидон - 4,30
Лаурилсульфат натрия - 0,17
Карбоксиметил целлюлоза - 8,
50
Достаточное количество очищенной воды для
влажного гранулирования
Стеарат магния, г - 1,70
170 мг вышеуказанного состава помещались в капсулу.
Пример
H
Компоненты - Количества, мг
Активное
соединение - 30,00
Крахмал - 51,00
Моногидрат лактозы - 74,33
Поливинилпирролидон - 4,30
Лаурилсульфат
натрия - 0,17
Карбоксиметил целлюлоза - 8,50
Достаточное количество очищенной воды для влажного гранулирования
Стеарат магния, г - 1,70
170 мг вышеуказанного
состава помещались в капсулу.
Жидкие лекарственные формы для орального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и элексиры. Как основа для жидких составов могут приготавливаться составы натрия. Помимо активных соединений жидкие лекарственные формы могут содержать обычно используемые инертные разбавители, такие как вода или другие растворители, солюбилизирующие вещества и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1, 3-бутиленгликоль, диметилформамидные масла, особенно хлопковое масло, арахисовое масло, продезинфицированное кукурузное масло, оливковое масло, касторовое масло и кунжутное масло, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и жирные кислотные эфиры сорбитана или смеси этих веществ и т.п. Помимо таких инертных разбавителей, эти составы могут также включать присадки, такие как увлажняющие, эмульгирующие и суспендирующие вещества, подслащающие, ароматизирующие и отдушивающие вещества.
Помимо активного соединения суспензии могут содержать суспендирующие вещества, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленовый сорбит, полиэтиленгликоли, имеющие различный молекулярный вес, и эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, или смеси этих веществ и т.п.
Составы для аэрозольного использования приготавливаются путем растворения активного вещества в воде или подходящем растворителе, например, простой спиртовой эфир, или другие инертные растворители, смешивания с летучим пропеллентом, расфасовки в герметичные емкости, снабженные дозирующим клапаном разбрызгивания состава в виде мелких капель.
Используемый сжиженный пропеллент обычно представляет собой пропеллент, имеющий точки кипения ниже температуры окружающей среды при атмосферном давлении. Сжиженный пропеллент, используемый в составах для изготовления медицинских аэрозолей, должен быть нетоксичным. Подходящими сжиженными пропеллентами являются низшие алканы, содержащие до 5 атомов углерода, такие как бутан и пентан, или алкилхлорид, такой как хлориды метила, этила или пропила. Подходящими сжиженными пропеллентами могут также быть фторированные и фторхлорированные алканы, продающиеся под торговыми марками "Фреон" и "Генетрон". Можно также использовать смеси вышеуказанных пропеллентов.
Реферат
Новые арил-конденсированные 2,4-диазениновые соединения общей формулы 1

где А - нафтил, фенил, незамещенный или замещенный галоидом, нитро, низшим алкилом или алкокси; R1 - Н, низший алкил или фенил; R2 - Н, низший алкил, циклопропил (низший) алкил, бензил, фенил, незамещенный или замещенный низшим алкокси, или группа - CH2CH2R7, где R7 - низший алкокси, ди(низший) алкиламино или морфолино; R3 - радикал Yp - (СН2)m - Xn - R8, где J - NН-, -S-, -0- или -СН(СH3 ), р = 0 или 1, m = 0 - 7, n = 0 - 1 Х- -S-, -0-, - SO2- или -СН = СН; R8 - Н, низший алкил, фенил, пиридинил, возможно замещенный низшим алкилом; фенил с 1 или 2 заместителями, выбранными из галоида, низшего алкила, низшего алкокси, низшего алкилсульфонамида ди(низшего)алкиламиносульфонила; R4 - Н, низший алкил или низший алкоксиалкил; R5 - Н, низший алкил, бензил, фенил, возможно замещенный галоидом или низшим алкокси, или нафтил, обладающие антиаритмической активностью. Используются как антиаритмический агент и в композиции для лечения сердечной аритмии совместно с фармацевтически приемлемым растворителем или наполнителем. Описываются новые промежуточные соединения

где R1a - Н, низший алкил, фенил; R5a - Н, фенил, возможно замещенный галоидом или низшим алкокси, нафтил; R6 - галоид или сконденсированное бензольное кольцо; R9 - Н, низший алкил, бензил или ди(низший)алкиламино (низший) алкил; R10 - Н, низший алкил, фенил, возможно замещенный галоидом, низшим алкилом, низшим алкокси, низшим алкилсульфонамидом, или пиридинил, незамещенный или замещенный низшим алкилом; w - 0 или 1. R2a - низший алкил, бензил, фенил, возможно замещенный низшим алкокси; или группа - CH2CH2R7, где R7 - низший алкокси, ди(низший) алкиламино или морфолино, R5a - Н, бензил, нафтил, фенил, возможно замещенный галоидом, низшим алкокси, используемые в синтезе соединений общей формулы I. 4 с. и 7 з.п.ф-лы, 11 табл.
Формула
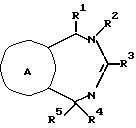
где A - нафтил, фенил, незамещенный или замещенный галоидом, нитро, низшим алкилом или низшим алкоксирадикалом;
R1 - водород, низший алкил или фенил;
R2 - водород, низший алкил, циклопропил (низший) алкил, бензил, фенил, незамещенный или замещенный низшим алкокси, или группа -CH2CH2R7, где R7 - низший алкокси, ди(низший) алкиламино или морфолино;
R3 представляет радикал формулы:
Yp - (CH2)m - Xn - R8,
где Y означает - NH-, -S-, -O- или

p = 0 или 1;
m - целое число 0 - 7;
n = 0 или 1;
X означает -S-, -O-, -SO2- или -CH = CH-;
R8 - водород, низший алкил, фенил, пиридинил, незамещенный или замещенный низшим алкилом; фенил с одним или двумя заместителями, выбранными независимо друг от друга из группы, включающей галоген, низший алкил, низший алкоксил, низший алкилсульфонамидо, ди(низший)алкиламиносулфонил;
R4 - водород, низший алкил или низший алкоксиалкил;
R5 означает водород, низший алкил, бензил, фенил, незамещенный или замещенный галоидом или низшим алкокси, или нафтил,
при условии, что общее число атомов углерода в R1, R2, R4 и R5 должно быть не менее 5, или их кислотно-аддитивные соли.

где R1 - водород, низший алкил или фенил;
R2 - водород, низший алкил, бензил, фенил, незамещенный или замещенный низшим алкокси, или группа формулы -CH2CH2R7, где R7 - низший алкокси, ди(низший)алкиламино или морфолино;
R3 - группа формулы
Yp - (CH2)m - Xn - R8,
где Y означает - NH-, -O-, -S- или
p = 0 или 1;
m - целое число 0 - 7;
n = 0 или 1;
X означает -S-, -O-, -SO2- или -CH = CH-;
R8 - водород, низший алкил, фенил, пиридинил, незамещенный или замещенный низшим алкилом, фенил, фенил с одним или двумя заместителями, выбранными независимо друг от друга из группы, включающей галоид, низший алкил, низший алкокси, низший алкилсульфонамидо или ди(низший)алкиламиносульфонил;
R4 - водород, низший алкил или низший алкокси-алкил;
R5 - водород, низший алкил, бензил, фенил, незамещенный или замещенный галоидом или низшим алкокси, или нафтил;
R6* - водород, галоид или нитро, или сконденсированное бензольное кольцо,
при условии, что общее число атомов углерода в R1, R2, R4 и R5 должно быть не менее 5.
4,5-дигидро-4-метил-1-фенил-3-(2-фенилэтил)-1H-2,4-бензодиазеин, R-(+)-4, 5-дигидро-4-метил-1-фенил-3-(2-фенилэтил)-1H-2,4-бензодиазепин,
N-[4-[2-(4,5-дигидро-4-метил-1-фенил-1H-2,4-бензодиазепин-3-ил)этил] - фенил]метансульфонамид,
N-[4-[2-(4, 5-дигидро-3-этил-1-фенил-1H-2,4-бензодиазепин-4-ил)этил] - фенил]метансульфонамид,
N-[4,5-дигидро-4-метил-1-фенил-3-(2-фенилэтил)-1H-2,4-бензодиазепин-3-ил] метансульфонамид,
1-(4-хлорфенил)-4,5-дигидро-4-метил-3-(2-фенилэтил)-1H-2, 4-бензодиазепин,
3-[2-(4-хлорфенил)этил] -4,5-дигидро-4-метил-1-фенил-1H-2,4-бензодиазепин,
3-[2-(4-хлорфенил)этил] -4, 5-дигидро-1,4-диметил-1-фенил-1H-2,4-бензодиазепин,
N, N-диэтил-4-(4,5-дигидро-5-метил-1-фенил-1H-2,4-бензодиазепин-4-ил)бензолсульфонамид,
4, 5-дигидро-1-(4-гидроксифенил)-4-метил-3-(2-фенилэтил)-1H-2,4-бензоди- азепин,
4, 5-дигидро-3-[2-(4-гидроксифенил)этил] -4-метил-1-фенил-1H-2,4-бензод- иазепин
или его кислотно-аддитивная соль по п.2.

где R1a - водород, низший алкил или фенил;
R5a - водород, фенил, незамещенный или замещенный галоидом или низшим алкокси, или нафтил;
R6 - галоид или сконденсированное бензольное кольцо;
R9 - водород, низший алкил, бензил или ди(низший)-алкиламино-низший алкил;
R10 - водород, низший алкил, фенил, незамещенный или замещенный галоидом, низшим алкилом, низшим алкокси, низшим алкилсульфонамидом, или пиридил, незамещенный или замещенный низшим алкилом;
n означает 0 или 1.
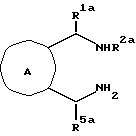
где A - нафтил, фенил, незамещенный или замещенный галоидом, нитро, низшим алкилом или низшим алкоксирадикалом;
R1a - водород, низший алкил или фенил;
R2a - низший алкил, бензил, фенил, незамещенный или замещенный низшим алкокси, или группа формулы - CH2CH2R7, в которой R7 представляет низший алкокси, ди(низший)алкиламино или морфолино;
R5а - водород, бензил, нафтил, фенил, незамещенный или замещенный галоидом, низшим алкокси, при условии, что по меньшей мере один из R1а и R5а не означает водород.
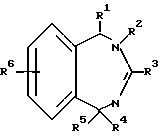
где значения R1, R2, R4 и R5 указаны в п.2.
R6 - водород, галоген или сконденсированное бензольное кольцо, при условии, что общее число атомов углерода в R1, R2, R4 и R5 должно быть не менее 5.
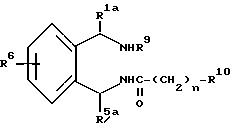
где R1а, R5а, R6 и R9 имеют значения, указанные в п.4;
R10 - водород, низший алкил, фенил, незамещенный или замещенный галогеном, низшим алкилом или низшим алкокси, или пиридил.

где R1а, R2а и R5а имеют значения, определенные в п.5;
R6 - водород, галоид и нитро, или сконденсированное бензольное кольцо, при условии, что по меньшей мере один из R1а и R5а не означает водород.
10.09.90. - по пп.2 и 4, за исключением значений R8 - фуранил, тиенил, пиридинил, фенил, замещенный, нитро, гидрокси, низшим алкиламино, низшим алкилсульфонамидо, ди(низшим) алкиламиносульфонилом и амино; или 5-членный гетероцикл, содержащий 1 или несколько атомов кислорода; или X и R8, взятые вместе, означают циклогексилидин; по п.3 в отношении соединений 1,2,6 - 8 и пп. 6, 9 - 13;
13.06.91 - по пп.1 и 5, и п.3 в отношении соединений 3 - 5, 9 - 11.
Комментарии