Новые низкомолекулярные катионные липиды для доставки олигонуклеотидов - RU2617641C2
Код документа: RU2617641C2
Чертежи


















Описание
Предпосылки к созданию изобретения
Настоящее изобретение относится к новым катионным липидам, которые могут быть использованы в комбинации с другими липидными компонентами, такими как холестерин и PEG-липиды, для формирования липидных наночастиц с олигонуклеотидами, для облегчения клеточного поглощения и эндосомального выделения и к нокдаун мРНК-мишени как in vitro, так и in vivo.
Ранее были раскрыты катионные липиды и использование катионных липидов в липидных наночастицах для доставки олигонуклеотидов, в частности, миРНК и микроРНК. Ранее были раскрыты липидные наночастицы и использование липидных наночастиц для доставки олигонуклеотидов, в частности, миРНК и микроРНК. Ранее были раскрыты олигонуклеотиды (в том числе миРНК и микроРНК) и синтез олигонуклеотидов (Смотрите патентные заявки США: US 2006/0083780, US 2006/0240554, US 2008/0020058, US 2009/0263407 и US 2009/0285881 и патентные заявки PCT: WO 2009/086558, WO 2009/127060, WO 2009/132131, WO 2010/042877, WO 2010/054384, WO 2010/054401, WO 2010/054405 и WO 2010/054406). Смотрите также Semple S.C. et al., Rational design of Cationic lipids for siRNA delivery, Nature Biotechnology, 2010, 28, 172-176.
Другие катионные липиды раскрыты в патентных заявках США: US 2009/0263407, US 2009/0285881, US 2010/0055168, US 2010/0055169, US 2010/0063135, US 2010/0076055, US 2010/0099738 и US 2010/0104629.
Общепринятые катионные липиды, такие как CLinDMA и DLinDMA, использовали для доставки миРНК в печень, но для них характерна неоптимальная эффективность доставки в пределах печени с печеночной токсичностью при более высоких дозах. Задачей данного изобретения является обеспечение каркасного катионного липида, который проявляет повышенную эффективность вместе с более низкой печеночной токсичностью в результате снижения уровней липида в печени. В настоящем изобретении используются низкомолекулярные катионные липиды с одной короткой липидной цепью для усиления эффективности и иммунности доставки in vivo миРНК.
Краткое описание изобретения
Настоящее изобретение обеспечивает новые катионные липиды, которые можно использовать в комбинации с другими липидными компонентами, такими как холестерин и PEG-липиды, для формирования липидных наночастиц с олигонуклеотидами. Задачей данного изобретения является обеспечение каркасного катионного липида, который проявляет повышенную эффективность вместе с более низкой печеночной токсичностью в результате снижения уровней липида в печени. В настоящем изобретении используются низкомолекулярные катионные липиды с одной короткой липидной цепью для усиления эффективности и иммунности доставки in vivo миРНК.
Краткое описание чертежей
Фиг.1. Эффективность LNP (соединение 1) у мышей.
Фиг.2. Эффективность LNP (соединения 32 и 33) у крыс (миРНК ApoB).
Фиг.3. Уровни катионного липида (соединения 32 и 33) в печени крыс.
Фиг.4. Эффективность LNP (соединения 32 и 33, миРНК ApoB) в NHP.
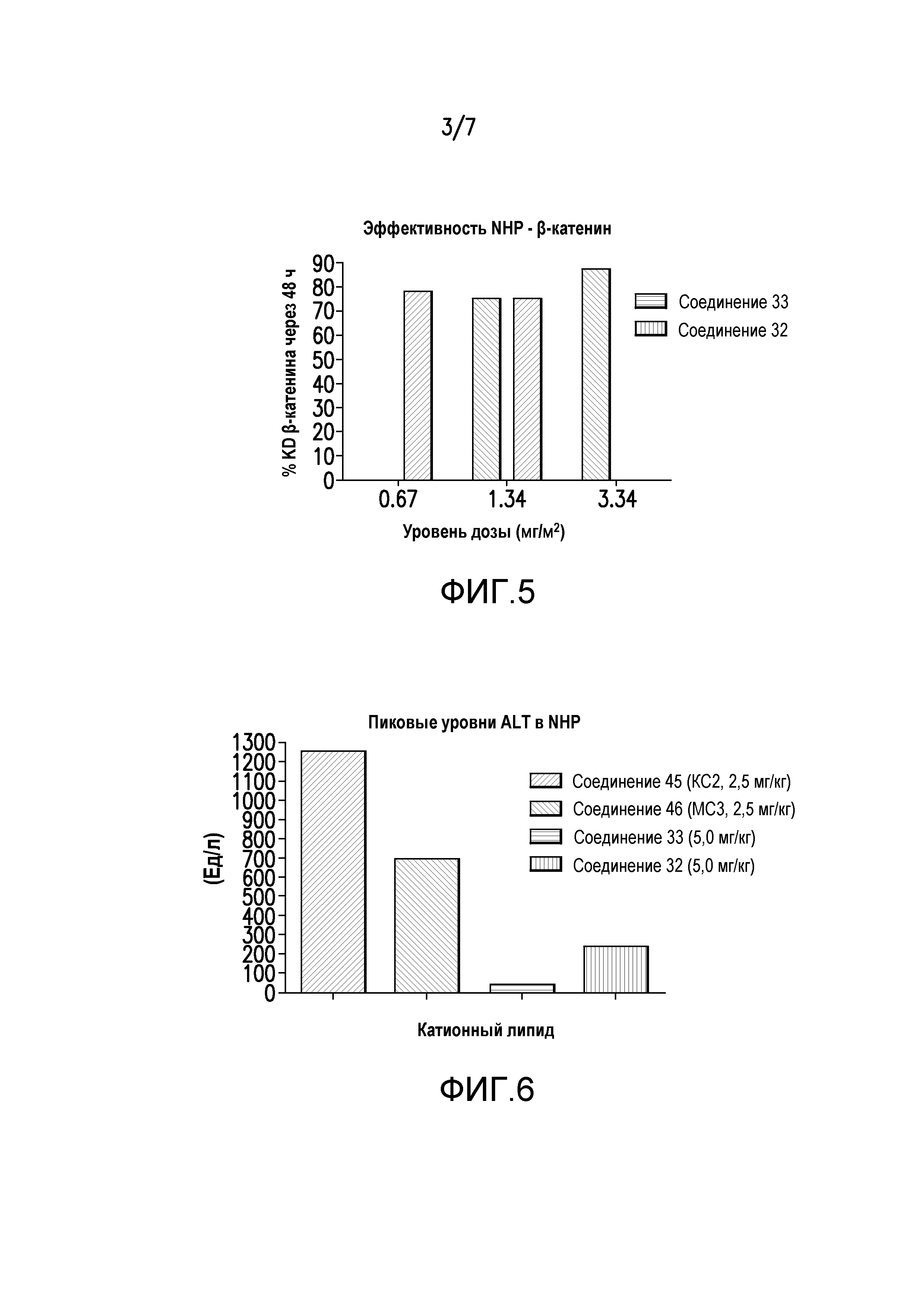
Фиг.5. Эффективность LNP (соединения 32 и 33, миРНК β-катенина) в NHP.
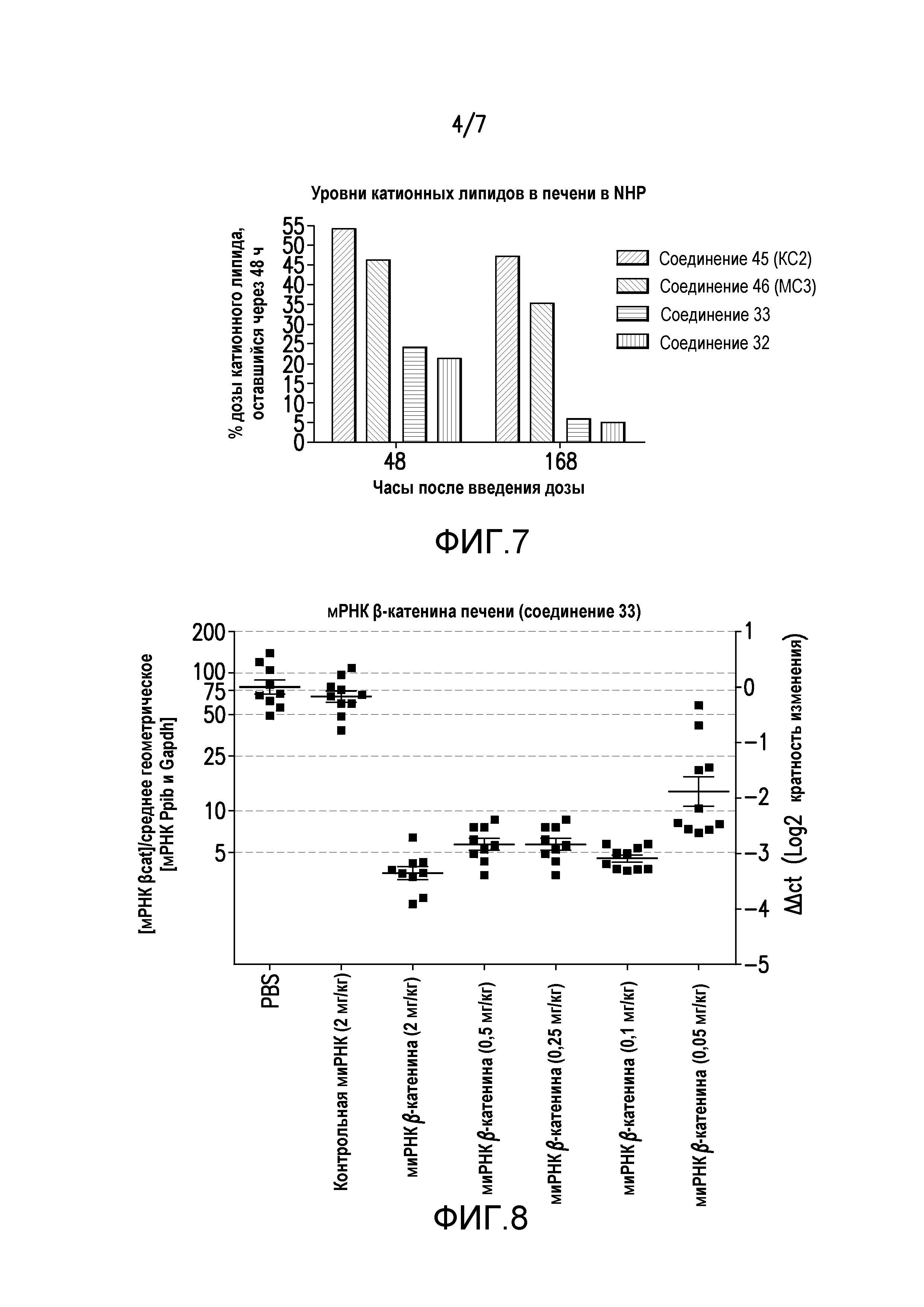
Фиг.6. Пиковые уровни ALT в NHP после дозы LNP (соединения 32 и 33).
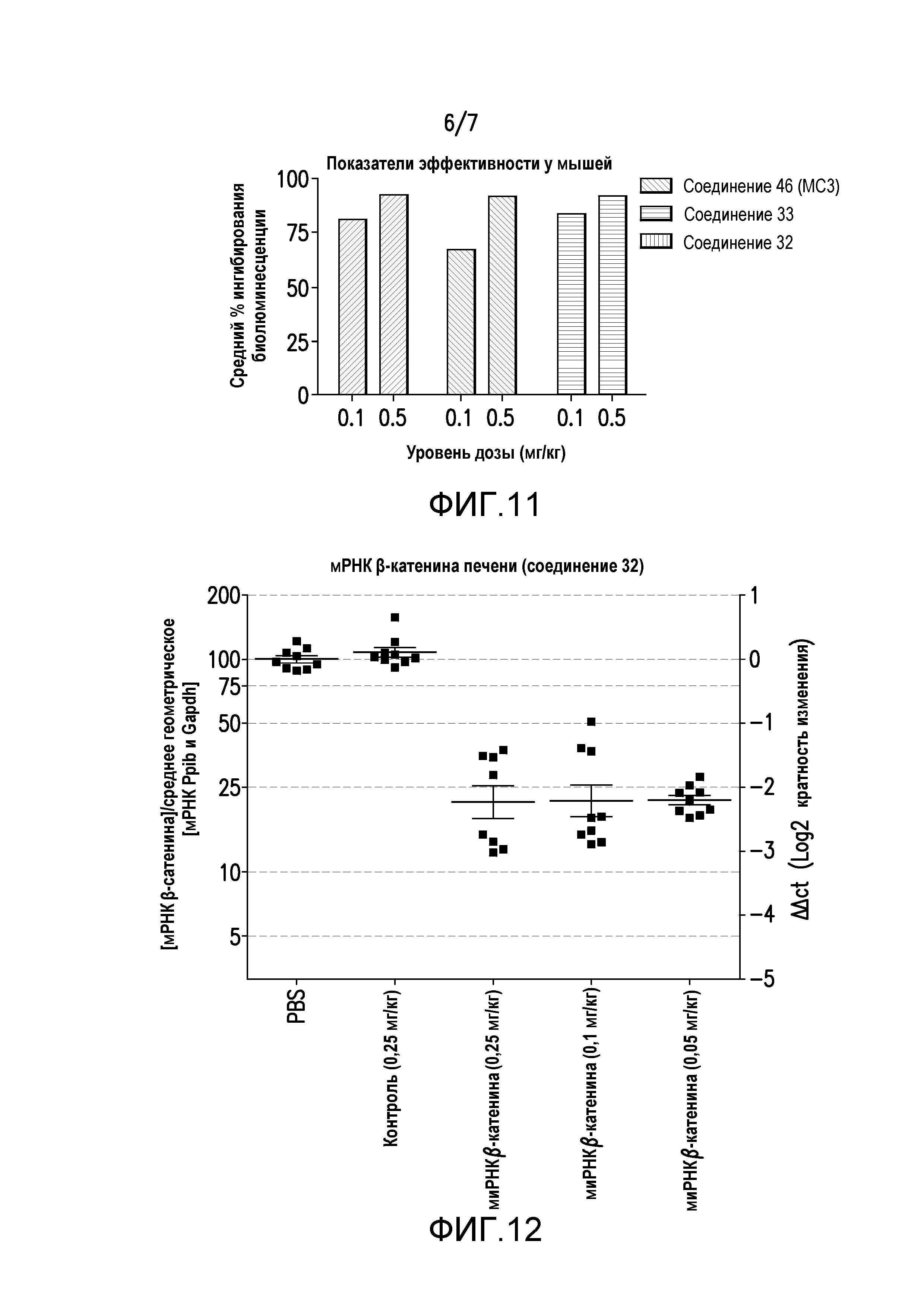
Фиг.7. Уровни катионного липида (соединения 32 и 33) в печени NHP.
Фиг.8. KD миРНК β-катенина в печени у мышей TRE-Met (соединение 33).
Фиг.9. KD миРНК β-катенина опухоли у мышей TRE-Met (соединение 33).
Фиг.10. Ингибирование роста опухоли (соединение 33) у мышей TRE-met.
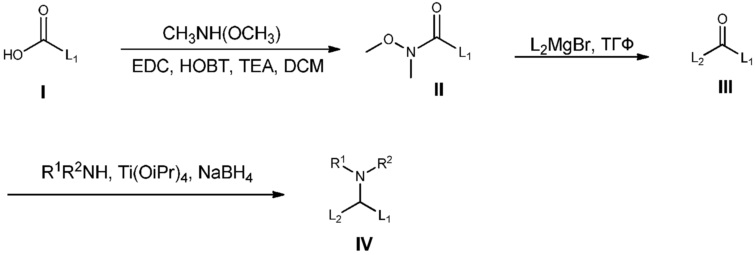
Фиг.11. Эффективность LNP (соединения 32 и 33) у мышей.
Фиг.12. KD миРНК β-катенина печени у мышей TRE-Met (соединение 32).
Фиг.13. KD миРНК β-катенина опухоли у мышей TRE-Met (соединение 32).
Фиг.14. Ингибирование роста опухоли (соединение 32) у мышей TRE-met.
Подробное описание изобретения
Различные аспекты и варианты осуществления данного изобретения направлены на практичность новых катионных липидов, эффективных в липидных наночастицах для доставки олигонуклеотидов, в частности, миРНК и микроРНК, к любому гену-мишени (Смотрите патентные заявки США: US 2006/0083780, US 2006/0240554, US 2008/0020058, US 2009/0263407 и US 2009/0285881, и патентные заявки PCT: WO 2009/086558, WO 2009/127060, WO 2009/132131, WO 2010/042877, WO 2010/054384, WO 2010/054401, WO 2010/054405 и WO 2010/054406). Также смотрите Semple S. C. et al., Rational design of Cationic lipids for siRNA delivery, Nature Biotechnology, 2010, 28, 172-176.
Катионные липиды по настоящему изобретению представляют собой эффективные компоненты в липидных наночастицах для доставки олигонуклеотидов, в особенности миРНК и микроРНК.
В первом варианте осуществления этого изобретения данные катионные липиды представлены формулой А:

в которой:
R1 и R2 независимо друг от друга выбраны из H, алкила (C1-C6), гетероциклила и полиамина, где указанный алкил, гетероциклил и полиамин, необязательно, являются замещенными одним-тремя заместителями, выбранными из R’, или R1 и R2 могут быть взяты совместно с азотом, к которому они присоединены для образования моноциклического гетероцикла с 4-7 членами, необязательно содержащего в себе, в дополнение к данному азоту, один или два дополнительных гетероатомов, выбранных из N, O и S, указанный моноциклический гетероцикл, необязательно, является замещенным с использованием от одного до трех заместителей, выбранных из R’;
R3 независимо выбран из H и алкила (C1-C6), указанный алкил, необязательно, является замещенным с использованием от одного до трех заместителей, выбранных из R’;
R’ независимо выбран из галогена, R", OR", SR", CN, CO2R" или CON(R")2;
R" независимо выбран из H и алкила (C1-C6), где указанный алкил, необязательно, является замещенным с использованием галогена и OH;
n равен 0, 1, 2, 3, 4 или 5;
L1 выбран из алкила C4-C24 и алкенила C4-C24, указанные алкил и алкенил, необязательно, являются замещенными с использованием от одного или нескольких заместителей, выбранных из R’; и
L2 выбран из алкила C3-C9 и акленила C3-C9, указанные алкил и алкенил, необязательно, являются замещенными с использованием одного или нескольких заместителей, выбранных из R’;
или любая фармацевтически приемлемая соль или его стереоизомер.
Во втором варианте осуществления данное изобретение описывает соединение, имеющее формулу А, где:
R1 и R2, каждый, представляет собой метил;
R3 представляет собой H;
n равен 0;
L1 выбран из алкила C4-C24 и алкенила C4-C24; и
L2 выбран из алкила C3-C9 и алкенила C3-C9;
или любая фармацевтически приемлемая соль или его стереоизомер.
В третьем варианте осуществления данное изобретение описывает соединение, имеющее формулу А, где:
R1 и R2, каждый, представляет собой метил;
R3 представляет собой H;
n равен 2;
L1 выбран из алкила C4-C24 и алкенила C4-C24; и
L2 выбран из алкила C3-C9 и алкенила C3-C9;
или любая фармацевтически приемлемая соль или его стереоизомер.
Специфические катионные липиды представляют собой:
(20Z,23Z)-N,N-диметилнонакоза-20,23-диен-10-амин (соединение 1);
(17Z,20Z)-N,N-диметилгексакоза-17,20-диен-9-амин (соединение 2);
(16Z,19Z)-N,N-диметилпентакоза-16,19-диен-8-амин (соединение 3);
(13Z,16Z)-N,N-диметилдокоза-13,16-диен-5-амин (соединение 4);
(12Z,15Z)-N,N-диметилгеникоза-12,15-диен-4-амин (соединение 5);
(14Z,17Z)-N,N-диметилтрикоза-14,17-диен-6-амин (соединение 6);
(15Z,18Z)-N,N-диметилтетракоза-15,18-диен-7-амин (соединение 7);
(18Z,21Z)-N,N-диметилгептакоза-18,21-диен-10-амин (соединение 8);
(15Z,18Z)-N,N-диметилтетракоза-15,18-диен-5-амин (соединение 9);
(14Z,17Z)-N,N-диметилтрикоза-14,17-диен-4-амин (соединение 10);
(19Z,22Z)-N,N-диметилоктакоза-19,22-диен-9-амин (соединение 11);
(18Z,21Z)-N,N-диметилгептакоза-18,21-диен-8-амин (соединение 12);
(17Z,20Z)-N,N-диметилгексакоза-17,20-диен-7-амин (соединение 13);
(16Z,19Z)-N,N-диметилпентакоза-16,19-диен-6-амин (соединение 14);
(22Z,25Z)-N,N-диметилгентриаконта-22,25-диен-10-амин (соединение 15);
(21Z,24Z)-N,N-диметилтриаконта-21,24-диен-9-амин (соединение 16);
(18Z)-N,N-диметилгептакоз-18-ен-10-амин (соединение 17);
(17Z)-N,N-диметилгексакоз-17-ен-9-амин (соединение 18);
(19Z,22Z)-N,N-диметилоктакоза-19,22-диен-7-амин (соединение 19); и
N,N-диметилгептакозан-10-амин (соединение 20);
(20Z,23Z)-N-этил-N-метилнонакоза-20,23-диен-10-амин (соединение 21);
1-[(11Z,14Z)-1-нониликоза-11,14-диен-1-ил]пирролидин (соединение 22);
(20Z)-N,N-диметилгептакоз-20-ен-10-амин (соединение 23);
(15Z)-N,N-диметилгептакоз-15-ен-10-амин (соединение 24);
(14Z)-N,N-диметилнонакоз-14-ен-10-амин (соединение 25);
(17Z)-N,N-диметилнонакоз-17-ен-10-амин (соединение 26);
(24Z)-N,N-диметилтритриаконт-24-ен-10-амин (соединение 27);
(20Z)-N,N-диметилнонакоз-20-ен-10-амин (соединение 28);
(22Z)-N,N-диметилгентриаконт-22-ен-10-амин (соединение 29);
(16Z)-N,N-диметилпентакоз-16-ен-8-амин (соединение 30);
(12Z,15Z)-N,N-диметил-2-нонилгеникоза-12,15-диен-1-амин (соединение 31);
(13Z,16Z)-N,N-диметил-3-нонилдокоза-13,16-диен-1-амин (соединение 32);
N,N-диметил-1-[(1S,2R)-2-октилциклопропил]гептадекан-8-амин (соединение 33);
1-[(1S,2R)-2-гексилциклопропил]-N,N-диметилнонадекан-10-амин (соединение 34);
N,N-диметил-1-[(1S,2R)-2-октилциклопропил]нонадекан-10-амин (соединение 35);
N,N-диметил-21-[(1S,2R)-2-октилциклопропил]геникозан-10-амин (соединение 36);
N,N-диметил-1-[(1S,2S)-2-{[(1R,2R)-2-пентилциклопропил]метил}циклопропил]нонадекан-10-амин (соединение 37);
N,N-диметил-1-[(1S,2R)-2-октилциклопропил]гексадекан-8-амин (соединение 38);
N,N-диметил-1-[(1R,2S)-2-ундецилциклопропил]тетрадекан-5-амин (соединение 39);
N,N-диметил-3-{7-[(1S,2R)-2-октилциклопропил]гептил}додекан-1-амин (соединение 40)
1-[(1R,2S)-2-гептилциклопропил]-N,N-диметилоктадекан-9-амин (соединение 41);
1-[(1S,2R)-2-децилциклопропил]-N,N-диметилпентадекан-6-амин (соединение 42);
N,N-диметил-1-[(1S,2R)-2-октилциклопропил]пентадекан-8-амин (соединение 43); и
(11E,20Z,23Z)-N,N-диметилнонакоза-11,20,23-триен-10-амин (соединение 44);
или любая фармацевтически приемлемая соль или его стереоизомер.
В другом варианте осуществления данные раскрытые катионные липиды являются пригодными при получении липидных наночастиц.
В другом варианте осуществления данные раскрытые катионные липиды являются пригодными компонентами в липидных наночастицах для доставки олигонуклеотидов.
В другом варианте осуществления данные раскрытые катионные липиды являются пригодными компонентами в липидных наночастицах для доставки миРНК и микроРНК.
В другом варианте осуществления данные раскрытые катионные липиды являются пригодными компонентами в липидной наночастице для доставки миРНК.
Данные катионные липиды по настоящему изобретению могут иметь ассиметричные центры, хиральные оси и хиральные плоскости (как описано у E.L. Eliel and S.H. Wilen, Stereochemistry of Carbon Compoundes, John Wiley & Sons, New York, 1994, pages 1119-1190), и встречаться в виде рацематов, рацемических смесей, и в виде отдельных диастериоизомеров, со всеми возможными изомерами и их смесями, включая оптические изомеры, входящие в данное изобретение. Кроме того, раскрытые здесь катионные липиды могут присутствовать в виде таутомеров, и обе таутомерные формы предназначены для включения в диапазон данного изобретения, несмотря на то, что описана только одна таутомерная структура.
Понятно, что заместители и схемы замещения в катионных липидах по настоящему изобретению могут быть выбраны специалистом, владеющим обычными практическими навыками в данной области, для обеспечения катионных липидов, которые являются химически стабильными и которые можно без труда синтезировать с использованием технологических приемов, известных в данной области, а также и тех методов, которые описаны ниже, из легкодоступных исходных материалов. Если сам заместитель является замещенным более чем одной группой, понятно, что эти множественные группы могут присутствовать на одном и том же углероде, или на разных углеродах, при условии получения устойчивой структуры.
Понятно, что специалист, владеющий обычными практическими навыками в данной области, может включить один или несколько атомов Si в катионные липиды по настоящему изобретению для обеспечения катионных липидов, которые являются химически стабильными и которые могут быть с легкостью синтезированы с использованием технологических приемов, известных в данной области, из легкодоступных исходных материалов.
В соединениях по формуле А атомы могут проявлять свой природный изотопный состав, или один или несколько атомов могут быть искусственно обогащенными определенным изотопом, имеющим то же самое атомное число, но атомную массу или атомное число, отличные от атомной массы или атомного числа, преимущественно встречающихся в природе. Настоящее изобретение предназначено для включения в себя всех подходящих изотопных вариантов соединений по формуле А. Например, различные изотопные формы водорода (Н), в том числе протий (1Н) и дейтерий (2Н). Протий представляет собой преобладающий изотоп водорода, встречающийся в природе. Обогащение дейтерия может обеспечить некоторые терапевтические преимущества, такие как увеличение времени полужизни in vivo, или снижение потребности в дозировках, или может обеспечить соединение, пригодное в качестве стандарта для описания характеристик биологических образцов. Изотопно-обогащенные соединения по формуле А могут быть получены без проведения излишних экспериментальных работ, используя общепринятые технологические приемы, хорошо известные специалистам в данной области, или используя процессы, аналогичные описанным в схемах и примерах, применяя соответствующие изотопно-обогащенные реагенты и/или промежуточные соединения.
Используемый здесь термин «алкил» означает насыщенный алифатический углеводород с неразветвленной цепью, циклический или разветвленный, имеющий определенное число атомов углерода.
Используемый здесь термин «алкенил» означает ненасыщенный алифатический углеводород с неразветвленной цепью, циклический или разветвленный, имеющий определенное число атомов углерода, в том числе, но не ограничиваясь этим, диеновые, триеновые и тетраеновые ненасыщенные алифатические углеводороды.
Примеры циклических «алкилов» или «алкенилов» включают в себя:

Используемый здесь термин «гетероциклил», или «гетероцикл», относится к 4-10-членному ароматическому или неароматическому гетероциклу, содержащему в себе от 1 до 4 гетероатомов, выбранных из группы, имеющей в своем составе О, N и S, и включает в себя бициклические группы. «Гетероциклил», таким образом, включает в себя следующее: бензоимидазолил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, имидазолил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, оксетанил, пиранил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридазинил, пиридил, пиримидил, пирролил, хиназолинил, хинолил, хиноксалинил, тетрагидропиранил, тетразолил, тетразолопиридил, тиадиазолил, триазолил, тиенил, триазолил, азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пирролидинил, морфолинил, тиоморфолинил, дигидробензоимидазолил, дигидробензофуранил, дигидробензотиофенил, дигидробензоксазолил, дигидрофуранил, дигидроимидазолил, дигидроиндолил, дигидроизооксазолил, дигидроизотиазолил, дигидрооксадиазолил, дигидрооксазолил, дигидропиразинил, дигидропиразолил, дигидропиридинил, дигидропиримидинил, дигидропирролил, дигидрохинолинил, дигидротетразолил, дигидротиадиазолил, дигидротриазолил, дигидротиенил, дигидротриазолил, дигидроазетидинил, метилендиоксибензоил, тетрагидрофуранил, и тетрагидротиенил, и их N-оксиды, все из которых, необязательно, являются замещенными одним-тремя заместителями, выбранными из R".
Используемый здесь термин «полиамин» означает соединения, имеющие две или более аминогрупп. Примеры включают в себя путресцин, кадаверин, спермидин и спермин.
Используемый здесь термин «галоген» означает Br, Cl, F и I.
В одном варианте осуществления формулы А, R1 и R2 независимо выбраны из H и алкила (C1-C6), где указанный алкил, необязательно, замещенный одним-тремя заместителями, выбранными из R’, или R1 и R2 могут быть взяты вместе с азотом, к которому они присоединены, для формирования моноциклического гетероцикла с 4-7 членами, необязательно содержащего в себе, в дополнение к указанному азоту, один или два гетероатома, выбранных из N, O и S, указанный моноциклический гетероцикл, необязательно, является замещенным одним-тремя заместителями, выбранными из R’.
В одном варианте осуществления формулы А, R1 и R2 независимо выбраны из H, метила, этила и пропила, где указанный метил, этил и пропил, необязательно, являются замещенными с одним-тремя заместителями, выбранными из R’, или R1 и R2 могут быть взяты вместе с азотом, к которому они присоединены, для формирования моноциклического гетероцикла с 4-7 членами, необязательно содержащего в себе, в дополнение к указанному азоту, один или два гетероатома, выбранных из N, O и S, указанный моноциклический гетероцикл, необязательно, является замещенным одним-тремя заместителями, выбранными из R’.
В одном варианте осуществления формулы А, R1 и R2 независимо выбраны из H, метила, этила и пропила.
В одном варианте осуществления формулы А, R1 и R2, каждый, представляет собой метил.
В одном варианте осуществления формулы А, R3 независимо выбран из H и метила.
В одном варианте осуществления формулы А, R3 представляет собой H.
В одном варианте осуществления формулы А, R’ представляет собой R".
В одном варианте осуществления формулы А, R" независимо выбран из H, метила, этила и пропила, где указанный метил, этил и пропил, необязательно, являются замещенными одним или несколькими галогенами и OH.
В одном варианте осуществления формулы А, R" независимо выбран из H, метила, этила и пропила.
В одном варианте осуществления формулы А, n равен 0, 1, 2 или 3.
В одном варианте осуществления формулы А, n равен 0, 1 или 2.
В одном варианте осуществления формулы А, n равен 0, 1 или 2.
В одном варианте осуществления формулы А, n равен 0.
В одном варианте осуществления формулы А, n равен 2.
В одном варианте осуществления формулы А, L1 выбран из алкила C4-C24 и алкенила C4-C24, которые, необязательно, являются замещенными галогеном и OH.
В одном варианте осуществления формулы А, L1 выбран из алкила C4-C24 и алкенила C4-C24.
В одном варианте осуществления формулы А, L1 выбран из алкенила C4-C24.
В одном варианте осуществления формулы А, L1 выбран из алкенила C12-C24.
В одном варианте осуществления формулы А, L1 представляет собой алкенил C19.
В одном варианте осуществления формулы А, L1 представляет собой:

В одном варианте осуществления формулы А, L1 представляет собой:

В одном варианте осуществления формулы А, L2 выбран из алкила C3-C9 и алкенила C3-C9, которые, необязательно, являются замещенными галогеном и OH.
В одном варианте осуществления формулы А, L2 выбран из алкила C5-C9 и алкенила C5-C9, которые, необязательно, являются замещенными галогеном и OH.
В одном варианте осуществления формулы А, L2 выбран из алкила C7-C9 и алкенила C7-C9, которые, необязательно, являются замещенными галогеном и OH.
В одном варианте осуществления формулы А, L2 выбран из алкила C3-C9 и алкенила C3-C9.
В одном варианте осуществления формулы А, L2 выбран из алкила C5-C9 и алкенила C5-C9.
В одном варианте осуществления формулы А, L2 выбран из алкила C7-C9 и алкенила C7-C9.
В одном варианте осуществления формулы А, L2 представляет собой алкил C3-C9.
В одном варианте осуществления формулы А, L2 представляет собой алкил C5-C9.
В одном варианте осуществления формулы А, L2 представляет собой алкил C7-C9.
В одном варианте осуществления формулы А, L2 представляет собой алкил C9.
В одном варианте осуществления формулы А, L1 выбран из алкила C4-C24 и акленила C4-C24, где указанные алкил и алкенил, необязательно, являются замещенными одним или несколькими заместителями, выбранными из R’; и L2 выбран из алкила C3-C9 и алкенила C3-C9, указанные алкил и алкенил, необязательно, являются замещенными одним или несколькими заместителями, выбранными из R’.
В одном варианте осуществления формулы А, L1 выбран из алкенила C12-C24, указанный алкенил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’; и L2 выбран из алкила C5-C9, указанный алкил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’.
В одном варианте осуществления формулы А, L1 выбран из алкенила C19, указанный алкенил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’; и L2 выбран из алкила C7-C9, указанный алкил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’.
В одном варианте осуществления формулы А, L1 выбран из алкенила C19, указанный алкенил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’; и L2 выбран из алкила C9, указанный алкил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’.
В одном варианте осуществления формулы А, L1 выбран из алкенила C19 с неразветвленной цепью, указанный алкенил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’; и L2 выбран из алкила C9 с неразветвленной цепью, указанный алкил, необязательно, является замещенным одним или несколькими заместителями, выбранными из R’.
В одном варианте осуществления формулы А, «гетероциклил» представляет собой пирролидин, пиперидин, морфолин, имидазол или пеперазин.
В одном варианте осуществления формулы А, «моноциклический гетероциклил» представляет собой пирролидин, пиперидин, морфолин, имидазол или пеперазин.
В одном варианте осуществления формулы А, «полиамин» представляет собой путресцин, кадаверин, спермидин или спермин.
В одном варианте осуществления «алкил» представляет собой насыщенный алифатический углеводород с неразветвленной цепью, имеющий определенное число атомов углерода.
В одном варианте осуществления «алкенил» представляет собой ненасыщенный алифатический углеводород с неразветвленной цепью, имеющий определенное число атомов углерода.
В настоящее изобретение включена свободная форма катионных липидов по формуле А, а также их фармацевтически приемлемые соли и стереоизомеры. Некоторые из приведенных здесь в качестве примеров выделенных специфических катионных липидов представляют собой протонированные соли аминовых катионных липидов. Термин «свободная форма» относится к аминовым катионным липидам в несолевой форме. Охваченные фармацевтически приемлемые соли включают в себя не только приведенные в качестве примеров выделенные соли для определенных катионных липидов, описанных здесь, но также все типичные фармацевтически приемлемые соли свободной формы катионных липидов по формуле А. Свободная форма конкретной соли описанных катионных липидов может быть выделена с использованием технологических приемов, известных в данной области. Например, свободная форма может быть восстановлена в результате обработки данной соли подходящим разбавленным водным основным раствором, таким как разбавленный водный NaOH, карбонат калия, аммиак и бикарбонат натрия. Данные свободные формы могут отличаться от их соответствующих солевых форм, в некоторой степени, некоторыми физическими свойствами, такими, как растворимость в полярных растворителях, но кислые и основные соли в остальном являются эквивалентными их соответствующим свободным формам для целей данного изобретения.
Фармацевтически приемлемые соли данных катионных липидов могут быть синтезированы из катионных липидов по этому изобретению, которые содержат в себе основные или кислотные функциональные группы, используя общепринятые химические методы. В общих чертах соли основных катионных липидов получают либо с помощью ионообменной хроматографии, или в результате реакции свободного основания со стехиометрическими количествами, или с избытком необходимой солеобразующей неорганической или органической кислоты в подходящем растворителе, или различных комбинациях растворителей. Аналогичным образом, соли кислотных соединений формируются в результате реакций с соответствующим неорганическим или органическим основанием.
Таким образом, фармацевтически приемлемые соли катионных липидов по этому изобретению включают в себя традиционные нетоксичные соли катионных липидов по этому изобретению, полученные в результате реакции основных растворимых катионных липидов с неорганической или органической кислотой. Например, традиционные нетоксичные соли включают в себя те, которые получены из неорганических кислот, таких как соляная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и тому подобное, а также соли, которые получены из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, оксиянтарная, винная, лимонная, аскорбиновая, пальмитиновая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая, трифторуксусная (TFA) и тому подобное.
В случае, когда катионные липиды по настоящему изобретению являются кислотными, подходящие «фармацевтически приемлемые соли» означают соли, полученные из фармацевтически приемлемых нетоксичных оснований, в том числе неорганических оснований и органических оснований. Соли, полученные из неорганических оснований, включают в себя соли алюминия, аммиака, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и тому подобное. Особенно предпочтительными являются соли аммиака, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают в себя соли первичных, вторичных и третичных аминов, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин кофеин, холин, N,N1-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин трипропиламин, трометамин и тому подобное.
Получение фармацевтически приемлемых солей описано выше и другие типичные фармацевтически приемлемые соли более подробно описаны у Berg et al, "Pharmaceutical Salts," J. Pharm. Sci, 1977: 66: 1-19.
Также следует отметить, что катионные липиды по настоящему изобретению представляют собой потенциально внутренние соли или цвиттерионы, поскольку в физиологических условиях депротеинированная кислотная функциональная группа в данном Соединении, например, карбоксильная группа, может быть анионной, и этот электронный заряд должен быть компенсирован внутренне относительно катионного заряда протеинированной или алкилированной основной функциональной группы, такой как четвертичный атом азота.
ПРИМЕРЫ
Представленные примеры предназначены для содействия в дальнейшем понимании данного изобретения. Конкретные используемые вещества, образцы и условия предназначены для дополнительной иллюстрации данного изобретения и не ограничивают его разумный диапазон. Реагенты, используемые при синтезе катионных липидов, являются либо коммерчески доступными, либо без труда готовятся специалистом, который обладает обычными навыками в данной области.
Синтез новых катионных липидов представляет собой линейный процесс, начинающийся с липидной кислоты (I). Связывание с N,О-диметил гидроксиламином приводит к образованию амида Вайнреба (Weinreb) II. Добавление реагента Гриньяра (Grignard) приводит к образованию кетона III. Опосредованное титаном восстановительное аминирование приводит к образованию конечных продуктов типа IV.
ОБЩАЯ СХЕМА 1
Синтез одноуглеродных допустимых катионных липидов V представляет собой линейный процесс, начинающийся с липидного кетона (III). Переход кетона в нитрил (V) осуществляется посредством обработки TOSMIC и трет-бутоксидом калия. Восстановление данного нитрила до первичного амина с последующим восстановительным аминированием обеспечивает конечные катионные липиды VI.
ОБЩАЯ СХЕМА 2

Синтез двухуглеродных допустимых катионных липидов IX представляет собой линейный процесс, начинающийся с липидного кетона (III). Переход данного кетона в α,β-ненасыщенный амид VII осуществляется в условиях реакции Петерсона. Сопряженное восстановление α,β-ненасыщенности осуществляется с использованием LS-Selectride для образования амида VIII. Восстановление данного амида с помощью алюмогидрида лития обеспечивает конечные катионные липида IX.
ОБЩАЯ СХЕМА 3

Циклопропил-содержащие липиды получены в соответствии с общей схемой 4. Ненасыщенные амиды Вайнреба II подвергают условиям циклопропанирования Симмонса-Смита для получения циклопропил-содержащих амидов Вайнреба X. Их доводят до конечных продуктов как показано в общих схемах 1-3.
ОБЩАЯ СХЕМА 4

Синтез аллиламиновых катионных липидов XVI представляет собой линейный процесс, начинающийся с альдегида XI. Добавление трет-бутилацетата приводит к образованию β-сложного гидроксиэфира XII. Преобразование данной гидроксильной функциональной группы во фтор-группу с последующей обработкой кислотой приводит к образованию β-фторзамещенной кислоты XIII. Преобразование данной кислоты в амид Вайнреба с последующим добавлением реагента Гриньяра приводит к образованию β-фтористого кетона XV. Восстановительное аминирование влечет за собой одновременное элиминирование для образования необходимого аллильного амина XVI.
ОБЩАЯ СХЕМА 5

20,23-нонакозадиен-10-амин,N,N-диметил-, (20Z,23Z) (соединение 1)

11,14-эйкозадиеновую кислоту, (11Z,14Z)- (50 г, 162 ммоль), N,О-диметилгидроксиламин гидрохлорид (31,6 г, 324 ммоль), HOAt (44,1 г, 324 ммоль), Et3N (45,2 мл, 324 ммоль) и EDC (62,1 г, 324 ммоль) смешивали в DCM (810 мл) и перемешивали в течение ночи при температуре окружающей среды. Реакционную смесь затем отмывали 5×700 мл воды, затем отмывали 1×600 мл 1 M NaOH, высушивали с помощью сульфата натрия, фильтровали через целит и конденсировали до получения 53,06 г (93%) 11,14-эйкозадиенамида, N-метокси-N-метил-, (11Z,14Z) в виде очищенного золотистого масла.1Н-ЯМР (400 МГц, CDCl3) δ 5,35 (м, 4H), 3,68 (с, 3H), 3,18 (с, 3H), 2,77 (м, 2H), 2,41 (т, J=7 Гц, 2H), 2,05 (м, 4H), 1,63 (м, 2H), 1,40-1,26 (м, 18H), 0,89 (т, J=7 Гц, 3H).

11,14-эйкозадиенамид, N-метокси-N-метил-, (11Z,14Z)- 1 (4 г, 11,38 ммоль) растворяли в сухом ТГФ (50,0 мл) в сосуде объемом 250 мл, затем 1 М нонилмагний бромид (22,76 мл, 22,76 ммоль) добавляли под азотом при температуре окружающей среды. Через 10 минут реакцию медленно подавляли с избытком насыщенного водного NH4Cl. Данную реакцию отмывали в делительной воронке с помощью гексана и воды, встряхивали, нижний водный слой удаляли, верхний слой высушивали с помощью сульфата натрия, фильтровали и конденсировали для получения неочищенного кетона в виде золотистого масла. К вышеуказанному неочищенному кетону добавляли диметиламин (2 M в ТГФ) (14,22 мл, 28,4 ммоль) с последующим добавлением Ti(O-i-Pr)4 (6,67 мл, 22,76 ммоль) и оставляли перемешиваться в течение ночи. На следующий день добавляли EtOH (50 мл), с последующим добавлением NaBH4 (0,646 г, 17,07 ммоль). После 5 мин перемешивания непосредственно вводили всю реакционную смесь в 40-г капиллярную кварцевую колонку, которая располагалась на одной линии с 330 г капиллярной кварцевой колонкой. Элюировали 10 мин 100% DCM, затем 30 мин 0-15% MeOH/DCM, собирали 20,23-нонакозадиен-10-амин, N,N-диметил-, (20Z,23Z) (1) (2,45 г, 5,47 ммоль, выход 48,1%) в виде бледно-золотистого масла.1Н-ЯМР (400 МГц, CDCl3) δ 5,35 (м, 4H), 2,78 (м, 2H), 2,23 (м, 1H), 2,21 (с, 6H), 2,05 (м, 4H), 1,45-1,16 (м, 38H), 0,89 (м, 6H). HRMS рассчитано для C31H61N 448,4877, получено 448,4872.
Соединения 2-30 представляют собой новые катионные липиды и были получены в соответствии с общей схемой 1, представленной выше.






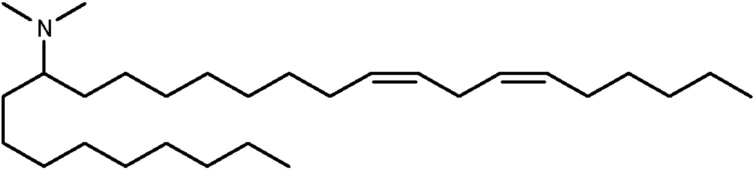

















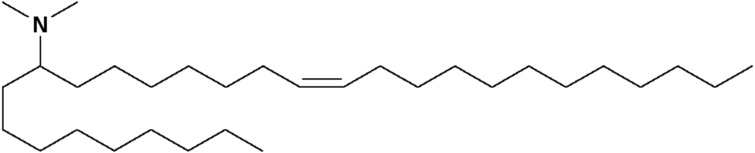


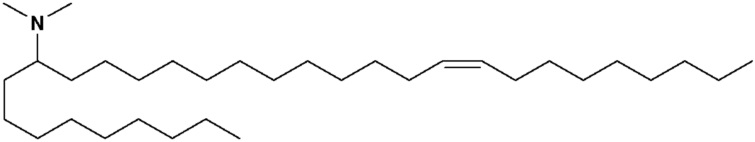

(12Z,15Z)-N,N-диметил-2-нонилгеникоза-12,15-диен-1-амин (соединение 31)

Раствор кетона iii (4,0 г, 9,55 ммоль), TOSMIC (2,4 г, 12,4 ммоль) в диметоксиэтане (45 мл) охлаждали до 0°C и обрабатывали трет-бутоксидом калия (19,1 ммоль, 19,1 мл 1 M раствора в tBuOH). Через 90 минут реакция была разделена между гексанами и водой. Органические вещества были отмыты водой, высушены сульфатом натрия, профильтрованы и конденсированы под вакуумом. Это вещество было очищено с помощью флэш-хроматографии (0-5% EtOAc/гексаны) для получения необходимого продукта (содержащего приблизительно 20% исходного продукта). Эту смесь переносили на следующий этап в виде is. ЖХ/МС (M+H)=430,6.

Алюмогидрид лития (23,9 ммоль, 23,9 мл 1 М раствора в ТГФ) добавляли непосредственно к нитрилу v (3,42 г, 8 ммоль) при температуре окружающей среды и данную реакцию перемешивали в течение 20 минут. Реакцию разбавляли с помощью 100 мл ТГФ, охлаждали до 0°C и осторожно подавляли с помощью раствора глауберовой соли. Данные твердые вещества фильтровали и отмывали с помощью ТГФ. Данный фильтрат конденсировали под вакуумом и переносили непосредственно в следующую реакционную сырьевую смесь. ЖХ/МС (M+H)=434,6.

Раствор первичного амина (3,45 г, 6,2 ммоль) в дихлорэтане (100 мл) обрабатывали формальдегидом (1,6 мл, 21,7 ммоль) с последующей обработкой триацетоксиборгидридом натрия (6,6 г, 31 ммоль). Через 5 минут данная реакция была разделена между дихлорметаном и 1 н. NaOH. Органические вещества высушивали с помощью сульфата натрия, фильтровали и конденсировали под вакуумом. Данную сырьевую смесь затем очищали посредством обращенно-фазовой препаративной хроматографии (колонка С8) для получения (12Z,15Z)-N,N-диметил-2-нонилгеникоза-12,15-диен-1-амина. HRMS рассчитано 462,5033, получено 462,5026.1Н-ЯМР (400 МГц, CDCl3) δ 5,35 (м, 4H), 2,78 (2H, т, J=5,6 Гц), 2,18 (с, 6H), 2,05 (м, 6H), 1,3 (м, 39H), 0,89 (м, 6H).
(13Z,16Z)-N,N-диметил-3-нонилдокоза-13,16-диен-1-амин (соединение 32)

Силиламидный реагент Петерсона (3,1 г, 16,7 ммоль) растворяли в ТГФ (35 мл) и охлаждали до -63°C. К этому раствору добавляли nBuLi (16,7 ммоль, 6,7 мл 2,5 M раствора). Данную реакцию нагревали до температуры окружающей среды в течение 30 минут. Кетон (5,0 г, 11,9 ммоль) растворяли в ТГФ (25 мл) во втором сосуде. Данный раствор кетона переносили к реагенту Петерсона в течение 30 минут, поддерживая температуру между -60°C и -40°C. Данную реакцию нагревали до -40°C в течение 1 часа, затем нагревали до 0°C в течение 30 минут. Данную реакцию подавляли с помощью бикарбоната натрия, разбавляли дополнительно водой и разделяли между водой/гексанами. Органические вещества отмывали солевым раствором, высушивали с помощью сульфата натрия, фильтровали и конденсировали под вакуумом. В результате очистки с помощью флэш-хроматографии получают α,β ненасыщенный амид vii.1Н-ЯМР (400 МГц, CDCl3) δ 5,75 (с, 1H), 5,36 (м, 4H), 3,01 (с, 3H), 2,99 (с, 3H), 2,78 (т, 2H), 2,28 (т, 2H), 2,05 (м, 6H), 1,35 (м, 34H), 0,89 (м, 6H).

α,β-ненасыщенный амид vii (1 г, 2,1 ммоль) и LS-Selectride (4,1 ммоль, 4,1 мл 1 M раствора) объединяли в герметически закупориваемой пробирке и нагревали до 60°C в течение 24 часов. Данную реакцию охлаждали до температуры окружающей среды и разделяли между раствором хлорида аммония и гептаном. Органические соединения высушивали с помощью сульфата натрия, фильтровали и конденсировали под вакуумом для получения амида viii. Это промежуточное соединение переносили непосредственно в следующую реакционную сырьевую смесь.
В альтернативном сопряженном восстановлению α,β-ненасыщенного амида vii используется восстановление гидрида меди:

В 5 л RB, данный медный катализатор (9,77 г, 17,13 ммоль) был растворен в толуоле (1713 мл) под азотом. К этому добавляли PMHS, от Aldrich (304 мл, 1371 ммоль) в одной дозе. Эту реакцию выдерживали в течение 5 минут. К данным растворам добавляли α,β-ненасыщенный амид vii (167,16 г, 343 ммоль). Затем к этой смеси добавляли трет-амиловый спирт (113 мл, 1028 ммоль) в течение 3 ч с помощью шприцевого насоса. После завершения добавления данный раствор был дополнен приблизительно 1700 мл 20% NH4OH небольшими порциями. Предостережение: имеет место интенсивное вспенивание и пенообразование в начале остановки реакции, и должен быть тщательный контроль, и гидроксид аммония необходимо добавлять медленно и небольшими порциями. Эту реакцию разделяли между водой и гексанами. Органические соединения фильтровали через целит и конденсировали под вакуумом. Полученное резиновое твердое вещество измельчали с помощью механической мешалки в гексанах для получения мелких частиц, которые затем фильтровали и отмывали с помощью гексанов. Органические соединения были затем конденсированы под вакуумом и очищены с помощью флэш-хроматографии (двуокись кремния, 0-15% этилацетат/гексаны) для получения требующегося амида viii. ЖХ/МС (M+H)=490,7.

К раствору амида viii (2,85 г, 5,8 ммоль) добавляли алюмогидрид лития (8,7 ммоль, 8,7 мл 1 M раствора). Данную реакционную смесь перемешивали при температуре окружающей среды в течение 10 минут, затем останавливали путем медленного добавления раствора глауберовой соли. Данные твердые соединения были профильтрованы и отмыты с помощью ТГФ, и данный фильтрат конденсировали под вакуумом. Данная сырьевая смесь была очищена с помощью обращенно-фазовой препаративной хроматографии (колонка С8) для получения (13Z,16Z)-N,N-диметил-3-нонилдокоза-13,16-диен-1-амина (соединение 32) в виде масла. HRMS (M+H) рассчитано 476,5190, получено 476,5189.1Н-ЯМР (400 МГц, CDC13) δ 5,37 (м, 4H), 2,78 (т, 2H), 2,42 (м, 8H), 2,05 (кв., 4H), 1,28 (м, 41H), 0,89 (м, 6H).
N,N-диметил-1-(2-октилциклопропил)гептадекан-8-амин (соединение 33)

К раствору олеиновой кислоты (1 г, 3,5 ммоль) в DCM (500 мл), охлажденному до 0°C, добавляли CDI (0,63 г, 3,9 ммоль). Данную реакционную смесь нагревали до температуры окружающей среды в течение 30 минут перед тем, как охладить до 0°C и обработать сначала триэтиламином (0,39 г, 3,9 ммоль), а затем диметил гидроксиламингидрохлоридом (0,38 г, 3,9 ммоль). Через 1 час данную реакцию разделяли между водой и гептаном. Органические соединения высушивали с помощью сульфата магния, фильтровали и конденсировали под вакуумом для получения неочищенного амида Вайнреба ii, который переносили непосредственно в следующую реакцию.

Раствор диэтилцинка (70,3 ммоль, 70,3 мл 1 M раствора) в дихлорметане (130 мл) охлаждали до -1°C и обрабатывали капельно с помощью TFA (8,0 г, 70,3 ммоль). Через 30 минут был добавлен дийодметан (18,8 г, 70,3 ммоль), и эту смесь выдерживали в течение 30 минут на ледяной бане. К этому раствору добавляли амид Вайнреба ii (7,6 г, 23,4 ммоль). Данную реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 1 часа. Данную реакцию останавливали путем добавления раствора хлорида аммония (100 мл), и органический слой отделяли, отмывали с помощью 10% тиосульфата натрия, высушивали сульфатом магния, фильтровали и конденсировали под вакуумом. Очистку проводили посредством флэш-хроматографии (0-30% MTBE/гептан) для получения требуемого продукта x.1Н-ЯМР (400 МГц, CDCl3) δ 3,72 (с, 3H), 3,22 (с, 3H), 2,48 (т, 2H), 1,65 (м, 2H), 1,39 (м, 22H), 1,18 (м, 2H), 0,91 (т, 3H), 0,68 (м, 2H), 0,59 (м, 1H), -0,32 (м, 1H).

Превращение амида Вайнреба x в соединение 33 осуществляли способом, аналогичным тому, который описан выше для соединения 1 (добавление нонил Гриньяра с последующим восстановительным аминированием). ЖХ/МС (M+H)=436,6.1Н-ЯМР (400 МГц, CDC13) δ 2,25 (с, 6H), 1,30 (м, 45H), 0,91 (м, 6H), 0,68 (м, 2H), 0,59 (м, 1H), -0,31 (м, 1H).
Соединения 34-43 представляют собой новые катионные липиды и были получены в соответствии с общими схемами 1-4, представленными выше.

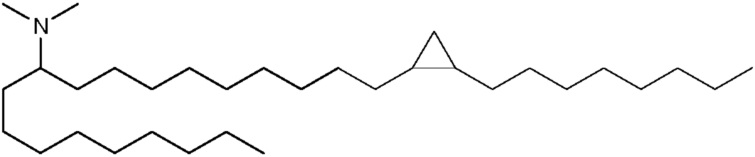








(11Е,20Z,23Z)-N,N-диметилнонакоза-11,20,23-триен-10-амин (соединение 44)

К раствору LDA (95 ммоль, 47,5 мл 2 M раствора) в ТГФ (127 мл), охлажденному до -78°C, добавляли трет-бутилацетат. Данную реакционную смесь перемешивали в течение 15 минут, с последующим добавлением альдегида xi. Данная реакция была незамедлительно остановлена путем добавления раствора хлорида аммония, нагрета до температуры окружающей среды и разделена между водой/пентаном. Органические соединения высушивали сульфатом натрия, фильтровали и конденсировали под вакуумом. ЖХ/МС (M+H-tBu)=325,4.

Кетоноспирт xii (7 г, 18,4 ммоль) растворяли в дихлорметане (150 мл) и охлаждали до 0°C и обрабатывали дезоксофтором (7,3 г, 33,1 ммоль). Данную реакционную смесь нагревали до температуры окружающей среды с перемешиванием в течение 16 часов, с последующей остановкой реакции раствором бикарбоната натрия. Данную реакционную смесь разделяли и органические соединения высушивали сульфатом натрия, фильтровали и конденсировали под вакуумом. После хроматографии на испарительной колонке (0-5% этилацетат/гексаны) получают β-фторзамещенный сложный эфир.
Промежуточный вторзамещенный сложный эфир (6 г, 15,6 ммоль) в дихлорметане был обработан хлороводородом (157 ммоль, 39,2 мл 4 M раствора в диоксане) и данную реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов. Данную реакционную смесь конденсировали под вакуумом для получения требуемой β-фторзамещенной кислоты xiii. ЖХ/МС (M+H)=327,3.

Фторкарбоновую кислоту xiii (5,1 г, 15,7 ммоль), EDC (6,0 г, 31,4 ммоль), N,О-диметилгидроксиламин гидрохлорид (3,1 г, 31,4 ммоль), триметиламин (4,0 г, 39,2 ммоль) и HOAt (4,3 г, 31,4 ммоль) смешивали в DCM (78 мл) и перемешивали при температуре окружающей среды в течение 16 часов. Эту реакционную смесь разделяли на воду/DCM и органические соединения отмывали водой (3 раза) и раствором NaOH (1 раз), высушивали сульфатом натрия, фильтровали и конденсировали под вакуумом. Сырьевое вещество очищали с помощью обратно-фазовой препаративной хроматографии для получения требуемого амида Вайнреба xiv. ЖХ/МС (M+H)=370,4.

Раствор амида Вайнреба xiv (4,3 г, 11,7 ммоль) в ТГФ (50 мл) обрабатывали нонилмагний бромидом (23,4 ммоль, 23,4 мл 1 M раствора) при температуре окружающей среды. Реакцию останавливали посредством добавления раствора хлорида аммония через 1 час и разделяли между водой и пентаном. Органические соединения высушивали сульфатом натрия, фильтровали и конденсировали под вакуумом. Это вещество переносили на следующий этап неочищенным.

Кетон xv (5,1 г, 11,7 ммоль) обрабатывали диметиламином (29,3 ммоль, 14,7 мл 2 M раствора в ТГФ) и изопропоксидом титана (IV) (6,7 г, 23,5 ммоль) и данную реакцию перемешивали при температуре окружающей среды в течение 16 часов. К данной реакционной смеси добавляли этанол (50 мл) с последующим добавлением боргидрида натрия (0,67 г, 17,6 ммоль). Данную реакцию нагружали непосредственно на кварцевую колонку и очищали посредством флэш-хроматографии (0-15% MeOH/DCM). Данному веществу необходима вторая очистка посредством препаративной обратно-фазовой хроматографии для получения (11E,20Z,23Z)-N,N-диметилнонакоза-11,20,23-триен-10-амина. HRMS рассчитано 446,4720, получено 446,4724.1Н-ЯМР (400 МГц, CDCl3) δ 5,48 (м, 1H), 5,37 (м, 4H), 5,23 (м, 1H), 2,78 (т, 2H), 2,58 (м, 1H), 2,22 (с, 6H), 2,04 (м, 6H), 1,56 (м, 1H), 1,30 (м, 31H), 0,89 (м, 6H).
Соединение 45 представляет собой DLinKC2DMA, как описано в Nature Biotechnology, 2010, 28, 172-176, WO 2010/042877 A1, WO 2010/048536 A2, WO 2010/088537 A2 и WO 2009/127060 A1.

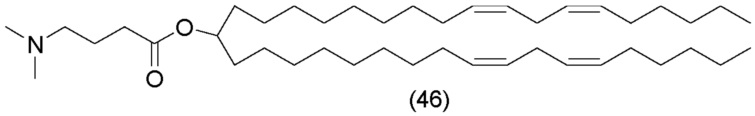
Соединение 46 представляет собой MC3, как описано в WO 2010/054401 и WO 2010/144740 A1.
Композиции LNP
Следующие композиции липидных наночастиц (LNP) по настоящему изобретению являются пригодными для доставки олигонуклеотидов, в частности, миРНК и микроРНК:
катионный липид/холестерин/PEG-DMG 56,6/38/5,4;
катионный липид/холестерин/PEG-DMG 60/38/2;
катионный липид/холестерин/PEG-DMG 67,3/29/3,7;
катионный липид/холестерин/PEG-DMG 49,3/47/3,7;
катионный липид/холестерин/PEG-DMG 50,3/44,3/5,4;
катионный липид/холестерин/PEG-C-DMA/DSPC 40/48/2/10;
катионный липид/холестерин/PEG-DMG/DSPC 40/48/2/10; и
катионный липид/холестерин/PEG-DMG/DSPC 58/30/2/10.
Описание процесса LNP
Данные липидные наночастицы (LNP) получены с использованием процесса сталкивающихся струй. Данные частицы сформированы посредством смешивания липидов, растворенных в спирте, с миРНК, растворенной в цитратном буфере. Коэффициент смешения липидов и миРНК задан как 45-55% липидов и 65-45% миРНК. Данный липидный раствор содержит в себе новый катионный липид по настоящему изобретению, вспомогательный липид (холестерин), липид PEG (например, PEG-C-DMA, PEG-DMG), и DSPC в концентрации 5-15 мг/мл с расчетной 9-12 мг/мл в спирте (например, этиловом). Соотношение данных липидов имеет диапазон молярных процентов 25-98 для данного катионного липида, с расчетным 35-65, данный вспомогательный липид имеет диапазон молярных процентов от 0-75 с расчетным 30-50, данный липид PEG имеет диапазон молярных процентов от 1-15 с расчетным 1-6, и DSPC имеет диапазон молярных процентов 0-15 с расчетным 0-12. Данный раствор миРНК содержит в себе одну или несколько последовательностей миРНК в интервале концентраций от 0,3 до 1,0 мг/мл, с расчетной 0,3-0,9 мг/мл в солевом растворе забуференном цитратом натрия, со значением рН в диапазоне 3,5-5. Данные две жидкости нагревали до температуры в диапазоне 15-40°C, рассчитано 30-40°C, и затем смешивали в смесительном аппарате со сталкивающимися струями, сразу же с образованием LNP. Данный teeID имеет диапазон от 0,25 до 1,0 мм и общую скорость потока 10-600 мл/мин. Комбинация скорости потока и внутреннего диаметра трубы (tubing ID) обладает эффектом контроля размера частиц данных LNP между 30 и 200 нм. Данный раствор затем смешивают с забуференным раствором при более высоком значении рН с коэффициентом смешивания в диапазоне от 1:1 до 1:3 об./об., но рассчитано 1:2 об./об. Температура данного забуференного раствора находится в диапазоне 15-40°C, расчетная 30-40°C. Данные смешанные LNP выдерживают от 30 минут до 2 часов до этапа анионообменной фильтрации. Температура во время инкубирования находится в диапазоне 15-40°C, расчетная 30-40°C. После инкубирования данный раствор фильтруют через 0,8-мкм фильтр, включающий в себя анионообменную ступень. В этом процессе используются ID труб в диапазоне от 1 мм ID до 5 мм ID, и скорость потока от 10 до 2000 мл/мин. Данные LNP являются концентрированными посредством процесса ультрафильтрации, в котором спирт удаляется и цитратный буфер замещен на конечный буферный раствор, такой как забуференный фосфатом физиологический раствор. В данном процессе ультрафильтрации используется формат проточной фильтрации вдоль потока (TFF). В этом процессе используется мембрана с отсечкой номинальной молекулярной массы в диапазоне 30-500 кДа. Данная мембрана может быть в виде пористого волокна или кассеты с плоской поверхностью. Процесс TFF с соответствующей точкой отсечки молекулярной массы сохраняет LNP в ультраконцентрате и данный фильтрат или ультрафильтрат содержит в себе спирт; цитратный буфер; излишки конечного буфера. Данный процесс TFF представляет собой многоступенчатый процесс, с первичной концентрацией к концентрации миРНК 1-3 мг/мл. После концентрирования, данный раствор LNP диафильтруют против 10-20 объемов конечного буфера для удаления спирта и осуществления обмена буферов. Данное вещество затем концентрируют дополнительно 1-3-кратно. Конечными этапами процесса LNP являются стерилизирующая фильтрация концентрированного раствора LNP и помещение данного продукта в ампулы.
Аналитическая процедура
1) Концентрация миРНК
Концентрации дуплекса миРНК были определены посредством высокоемкой анионообменной высокоэффективной жидкостной хроматографии (SAX-HPLC) с использованием системы Waters 2695 Alliance (Water Corporation, Milford MA) с детектором 2996 PDA. Данные LNP, иначе называемые Системы доставки RNAi (RDV), обработаны с помощью 0,5% Triton X-100 для высвобождения общей миРНК и проанализированы посредством сепарирования SAX с использованием колонки Dionex BioLC DNAPac PA 200 (4×250 мм) с УФ-детектором при 254 нм. Мобильная фаза состоит из A: 25 мМ NaClO4, 10 мМ Tris, 20% EtOH, pH 7,0 и B: 250 мМ NaClO4,10 мМ Tris, 20% EtOH, pH 7,0 с линейным градиентом от 0-15 мин и скоростью потока 1 мл/мин. Количество миРНК определено посредством сравнения с калибровочной кривой миРНК.
2) Степень инкапсулирования
Флуоресцентный реагент SYBR Gold использовали для количественного анализа РНК для контроля степени инкапсулирования RDV. RDV с или без Triton X-100 использовали для определения количества свободной миРНК и общей миРНК. Данное исследование осуществлено с использованием микропланшетного спектрофотометра SpectraMax M5e компании Molecular Devices (Sunnyvale, CA). Образцы стимулировали при 485 нм и измеряли флуоресценцию при 530 нм. Количество миРНК определяли путем сравнения с калибровочной кривой миРНК.
Степень инкапсулирования=(1-свободная миРНК/общая миРНК)×100%
3) Размер частиц и полидисперсные свойства
RDV, содержащие в себе 1 мкг миРНК, разводили до конечного объема 3 мл с помощью 1×PBS. Размер частиц и полидисперсные свойства данных образцов оценивали методом динамического рассеяния света с использованием прибора ZetaPALS (Brookhaven Instruments Corporation, Holtsville, NY). Интенсивность рассеянного излучения оценивали посредством He-Ne лазера при 25°C с углом рассеивания 90°.
4) Анализ дзета-потенциала
RDV, содержащие в себе 1 мкг миРНК, разводили до конечного объема 2 мл с помощью 1 М Tris-буфера (pH 7,4). Электрофоретическую подвижность образцов оценивали с использованием прибора ZetaPALS instrument (Brookhaven Instruments Corporation, Holtsville, NY) с электродом и He-Ne лазером в качестве источника света. Предел Смолуховского был принят в расчете дзета-потенциалов.
5) Липидный анализ
Индивидуальные липидные концентрации определяют посредством обращенно-фазовой высокоэффективной жидкостной хроматографии (RP-HPLC), используя систему Waters 2695 Alliance (Water Corporation, Milford MA) с детектором заряженного аэрозоля Corona (CAD) (ESA Biosciences, Inc, Chelmsford, MA). Индивидуальные липиды в RDV анализируют, используя колонку Agilent Zorbax SB-C18 (50×4,6 мм, размер частиц 1,8 мкм) с CAD при 60°C. Мобильная фаза состоит из A: 0,1% TFA в H2O и B: 0,1% TFA в IPA. Градиент изменяется от 60% мобильной фазы A и 40% мобильной фазы B с момента времени 0 до 40% мобильной фазы A и 60% мобильной фазы B в момент времени 1,00 мин; 40% мобильной фазы A и 60% мобильной фазы B с 1,00 по 5,00 мин; 40% мобильной фазы A и 60% мобильной фазы B с 5,00 мин до 25% мобильной фазы A и 75% мобильной фазы B на 10,00 мин; 25% мобильной фазы A и 75% мобильной фазы B с 10,00 мин до 5% мобильной фазы A и 95% мобильной фазы B на 15,00 мин; и 5% мобильной фазы A и 95% мобильной фазы B с 15,00 до 60% мобильной фазы A и 40% мобильной фазы B на 20,00 мин со скоростью потока 1 мл/мин. Индивидуальную липидную концентрацию определяют путем сравнения с градуировочной кривой со всеми липидными компонентами в данных RDV с подбором квадратической кривой. Молярную концентрацию каждого липида рассчитывали, исходя из его молекулярной массы.
Описание общего процесса LNP для формирования соединения 32
Данные липидные наночастицы получены с использованием процесса сталкивающихся струй. Данные частицы сформированы посредством смешивания липидов, растворенных в спирте, с миРНК, растворенной в цитратном буфере. Данный липидный раствор содержал в себе катионный липид, вспомогательный липид (холестерин), липид PEG (например, PEG-C-DMA, PEG-DMG) и DSPC в концентрации 5-15 мг/мл, с заданной 9-12 мг/мл, в спирте (например, этиловом спирте). Соотношение данных липидов имеет диапазон молярных процентов 25-98 для данного катионного липида, с расчетным 35-65, данный вспомогательный липид имеет диапазон молярных процентов от 0-75 с расчетным 30-50, данный липид PEG имеет диапазон молярных процентов от 1-15 с расчетным 1-6, и DSPC имеет диапазон молярных процентов 0-15 с расчетным 0-12. Данный раствор миРНК содержит в себе одну или несколько последовательностей миРНК в интервале концентраций от 0,3 до 1,0 мг/мл, с расчетной 0,3-0,9 мг/мл в солевом растворе забуференном цитратом натрия, со значением рН в диапазоне 3,5-5. Данные две жидкости нагревали до температуры в диапазоне 15-40°C, расчетная 30-40°C, и затем смешивали в смесительном аппарате со сталкивающимися струями, сразу же с образованием LNP. Данный teeID имеет диапазон от 0,25 до 1,0 мм и общую скорость потока 10-600 мл/мин. Комбинация скорости потока и внутреннего диаметра трубы (tubing ID) обладает эффектом контроля размера частиц данных LNP между 30 и 200 нм. Данную суспензию LNP затем смешивают с забуференным раствором при более высоком значении рН с коэффициентом смешивания в диапазоне от 1:1 до 1:3 об./об., но рассчитано 1:2 об./об. Температура данного забуференного раствора находится в диапазоне 15-40°C, расчетная 30-40°C. Данные смешанные LNP выдерживают от 30 минут до 2 часов до этапа анионообменной фильтрации. Температура во время инкубации находится в диапазоне 15-40°C, расчетная 30-40°C. После инкубирования данный раствор фильтруют через 0,8 мкм фильтр, включающий в себя анионообменную ступень. В этом процессе используются ID труб в диапазоне от 1 мм ID до 5 мм ID и скорость потока от 10 до 2000 мл/мин. Данные LNP являются концентрированными посредством процесса ультрафильтрации, в котором спирт удаляется и цитратный буфер замещен на конечный буферный раствор, такой как забуференный фосфатом физиологический раствор. В данном процессе ультрафильтрации используется формат проточной фильтрации вдоль потока (TFF). В этом процессе используется мембрана с отсечкой номинальной молекулярной массы в диапазоне 30-500 кДа. Данная мембрана может быть в виде пористого волокна или кассеты с плоской поверхностью. Процесс TFF с соответствующей точкой отсечки молекулярной массы сохраняет LNP в ультраконцентрате и данный фильтрат или ультрафильтрат содержит в себе спирт; цитратный буфер; излишки конечного буфера. Данный процесс TFF представляет собой многоступенчатый процесс, с первичной концентрацией к концентрации миРНК 1-3 мг/мл. После концентрирования, данный раствор LNP диафильтруют против 10-20 объемов конечного буфера для удаления спирта и осуществления обмена буферов. Данное вещество затем концентрируют дополнительно 1-3-кратно. Конечными этапами процесса LNP являются стерилизирующая фильтрация концентрированного раствора LNP и помещение данного продукта в ампулы.
Аналитическая процедура
Концентрация миРНК
Концентрации дуплекса миРНК были определены посредством высокоемкой анионообменной высокоэффективной жидкостной хроматографии (SAX-HPLC) с использованием системы Waters 2695 Alliance (Water Corporation, Milford MA) с детектором 2996 PDA. Данные LNP, иначе называемые Системы доставки RNAi (RDV), обработаны с помощью 0,5% Triton X-100 для высвобождения общей миРНК, и проанализированы посредством сепарирования SAX с использованием колонки Dionex BioLC DNAPac PA 200 (4×250 мм) с УФ-детектором при 254 нм. Мобильная фаза состоит из A: 25 мМ NaClO4, 10 мМ Tris, 20% EtOH, pH 7,0 и B: 250 мМ NaClO4, 10 мМ Tris, 20% EtOH, pH 7,0 с линейным градиентом от 0-15 мин и скоростью потока 1 мл/мин. Количество миРНК определено посредством сравнения с калибровочной кривой миРНК.
Степень инкапсулирования
Флуоресцентный реагент SYBR Gold использовали для количественного анализа РНК для контроля степени инкапсулирования RDV. RDV с или без Triton X-100 использовали для определения количества свободной миРНК и общей миРНК. Данное исследование осуществлено с использованием микропланшетного спектрофотометра SpectraMax M5e компании Molecular Devices (Sunnyvale, CA). Образцы стимулировали при 485 нм и измеряли флуоресценцию при 530 нм. Количество миРНК определяли путем сравнения с калибровочной кривой миРНК.
Степень капсулирования=(1-свободная миРНК/общая миРНК)×100%
Размер частиц и полидисперсные свойства
RDV, содержащие в себе 1 мкг миРНК, разводили до конечного объема 3 мл с помощью 1×PBS. Размер частиц и полидисперсные свойства данных образцов оценивали методом динамического рассеяния света с использованием прибора ZetaPALS (Brookhaven Instruments Corporation, Holtsville, NY). Интенсивность рассеянного излучения оценивали посредством He-Ne лазера при 25°C с углом рассеивания 90°.
Анализ дзета-потенциала
RDV, содержащие в себе 1 мкг миРНК, разводили до конечного объема 2 мл с помощью 1 М Tris-буфера (pH 7,4). Электрофоретическую подвижность образцов оценивали с использованием прибора ZetaPALS instrument (Brookhaven Instruments Corporation, Holtsville, NY) с электродом и He-Ne лазером в качестве источника света. Предел Смолуховского был принят в расчете дзета-потенциалов.
Липидный анализ
Индивидуальные липидные концентрации определяют посредством обращенно-фазовой высокоэффективной жидкостной хроматографии (RP-HPLC), используя систему Waters 2695 Alliance (Water Corporation, Milford MA) с детектором заряженного аэрозоля Corona (CAD) (ESA Biosciences, Inc, Chelmsford, MA). Индивидуальные липиды в RDV анализируют, используя колонку Agilent Zorbax SB-C18 (50×4,6 мм, размер частиц 1,8 мкм) с CAD при 60°C. Мобильная фаза состоит из A: 0,1% TFA в H2O и B: 0,1% TFA в IPA. Градиент изменяется от 60% мобильной фазы A и 40% мобильной фазы B с момента времени 0 до 40% мобильной фазы A и 60% мобильной фазы B в момент времени 1,00 мин; 40% мобильной фазы A и 60% мобильной фазы B с 1,00 по 5,00 мин; 40% мобильной фазы A и 60% мобильной фазы B с 5,00 мин до 25% мобильной фазы A и 75% мобильной фазы B на 10,00 мин; 25% мобильной фазы A и 75% мобильной фазы B с 10,00 мин до 5% мобильной фазы A и 95% мобильной фазы B на 15,00 мин; и 5% мобильной фазы A и 95% мобильной фазы B с 15,00 до 60% мобильной фазы A и 40% мобильной фазы B на 20,00 мин со скоростью потока 1 мл/мин. Индивидуальную липидную концентрацию определяют путем сравнения с калибровочной кривой со всеми липидными компонентами в данных RDV с подбором квадратической кривой. Молярную концентрацию каждого липида рассчитывали, исходя из его молекулярной массы.
Общее получение LNP для различных композиций в таблице 1
Суспензии наночастиц миРНК, представленных в таблице 1, были получены путем растворения миРНК и/или молекул носителя в 20 мМ натрий цитратном буфере (рН 5,0) в концентрации приблизительно 0,40 мг/мл. Липидные растворы получают путем растворения смеси катионного липида (например, 32, смотрите структуру в таблице 2), DSPC, холестерина, и PEG-DMG (соотношения представлены в таблице 1) в абсолютном этаноле в концентрации приблизительно 8 мг/мл. Соотношение азота к фосфату составляет приблизительно 6:1.
Практически одинаковые объемы растворов миРНК/носитель и липидных растворов доставляются с помощью двух насосов FPLC при одинаковой скорости потока в смесительный Т-образный коннектор. Возвратный клапан используется для регулирования необходимого размера частиц. Полученную млечную смесь собирали в стерильные стеклянные бутыли. Эту смесь затем разбавляли равным объемом цитратного буфера, с последующим добавлением равного объема PBS (pH 7,4), фильтровали через ионообменный мембранный фильтр для удаления любых свободных миРНК/носитель из данной смеси. Ультрафильтрацию против PBS (7,4) используют для удаления этанола и для замены буфера. Конечный LNP получали путем концентрирования до требуемого объема и фильтрации в стерильных условиях через 0,2 мкм фильтр. Полученные LNP описываются с точки зрения размера частиц, дзета-потенциала, общего содержания липида, инкапсулированной нуклеиновой кислоты, и общей концентрации нуклеиновой кислоты.
Процесс изготовления LNP
В неограничивающем примере LNP получают в большом объеме следующим образом. Данный процесс состоит из (1) получения липидного раствора; (2) получения раствора миРНК/носитель; (3) смешивания/формирования частиц; (4) инкубации; (5) разведения; (6) ультрафильтрования и концентрирования.
Получение липидного раствора
Стеклянные флаконы для реактивов объемом 2 л и мерные цилиндры были апирогенированы. Липиды нагревали до комнатной температуры. В данный стеклянный флакон для реагентов переносили 0,8 г соединения 32 с помощью пипетки и добавляли 1,2 г DSPC, 3,5 г холестерина, 0,9 г PEG-DMG. К данной смеси добавляли 1 л этилового спирта. Данный флакон для реагентов помещали в нагретую водяную баню, в которой температура не превышает 50°C. Данную липидную суспензию перемешивают с помощью магнитной мешалки. Датчик термопары помещают в данную суспензию через одно горлышко флакона с круглым дном с герметичным переходником. Данную суспензию нагревают при 30-40°C до тех пор, пока она не станет прозрачной. Данный раствор оставляют остывать до комнатной температуры.
Получение раствора миРНК/носитель
В стерильный контейнер, такой как бутыль для хранения Corning, взвешивают 0,4 г с поправкой на фактор стабилизации воды (приблизительно 1,2) порошка миРНК-1. Данную миРНК переносят в апирогенный стеклянный флакон для реагентов объемом 2 л. Данный контейнер для взвешивания споласкивают 3 раза цитратным буфером (20 мМ, pH 5,0), и данные смывы помещают в данный стеклянный флакон объемом 2 л, и доводят объем цитратным буфером до 1 л. Концентрацию данного раствора миРНК определяют с помощью УФ спектрофотометра, применяя следующую процедуру. Из данного раствора забирают 20 мкл, разводят в 50 раз до 1000 мкл и считывают показание УФ прибора, записанные при А269 нм после выставления бланка по цитратному буферу. Этот процесс повторяют. Если показатели для двух образцов сопоставимы, берется среднее значение и проводится расчет концентрации, принимая во внимание коэффициенты экстинции данных миРНК. Если конечная концентрация выпадает из диапазона 0,40±0,01 мг/мл, данную концентрацию корректируют путем добавления большего количества порошка миРНК/носитель, или добавления большего количества цитратного буфера. Этот процесс повторяют для второй миРНК, если применяется.
Альтернативно, если данный раствор миРНК/носитель включает в себя дуплекс единственной миРНК и/или носитель, вместо коктейля двух или более дуплексов миРНК и/или носителя, тогда это раствор миРНК/носитель растворяют в 20 мМ цитратном буфере (pH 5,0) для получения конечной концентрации 0,4 мг/мл.
Растворы липида и этанола затем фильтруют в стерильных условиях через Pall Acropak 20 0,8/0,2 мкм стерильный фильтр PN 12203 в апирогенный стеклянный сосуд, используя перистальтический насос Master Flex модель 7520-40, для получения стерильного исходного материала для процесса инкапсулирования. Процесс фильтрации осуществляют в 80 мл с площадью мембраны 20 см2. Скорость потока составляет 280 мл/минуту. Этот процесс является шкалируемым посредством увеличения диаметра трубы и площади фильтрации.
Этап формирование частиц - смешивание
Используя двухцилиндровый шприцевый насос с приводом (Harvard 33 Twin Syringe), данный стерильный раствор липид/этанол и данный стерильный раствор миРНК/носитель или миРНК/коктейль носителей/цитратный буфер (20 мМ цитратный буфер, pH 5,0) смешивали в Т-смесителе с внутренним диаметром (ID) 0,5 мм (стадия смешивания I) при равных, или практически равных, скоростях потока. Полученная на выходе суспензия LNP содержала в себе 40-50 об.% этанола. При необходимости иметь на выходе суспензию с 45 об.% этанола, данные стерильные растворы липид/этанол, миРНК/носитель или миРНК/коктейль носителей/цитратный буфер, смешивали при скорости потока 54 мл/мин и 66 мл/мин, так чтобы получить общую скорость потока данной смеси на выходе 120 мл/мин.
Разведение
Выходящий поток, получено на стадии смешивания I подавали непосредственно в Т-смеситель с ID 4 мм (стадия смешивания II), где он разводится забуференным раствором при более высоком значении рН (20 мМ цитрат натрия, 300 мМ хлорид натрия, pH 6,0) в соотношении 1:1 об./об.%. Этот забуференный раствор находится при температуре в диапазоне 30-40°C и доставляется в 4 мм Т-смеситель посредством перистальтического насоса (Cole Parmer MasterFlex L/S 600 RPM) при скорости потока 120 мл/мин.
Выходящий поток, получено на стадии смешивания II, подавали непосредственно в Т-смеситель с ID 6 мм (стадия смешивания III), где он разводится забуференным раствором при более высоком значении рН (PBS, pH 7,4) в соотношении 1:1 об./об.%. Этот забуференный раствор находится при температуре в диапазоне 15-25°C и доставляется в 6 мм Т-смеситель посредством перистальтического насоса (Cole Parmer MasterFlex L/S 600 RPM) при скорости потока 240 мл/мин.
Инкубирование и удаление свободной миРНК
Выходящий поток, получено на стадии смешивания III, после смешивания инкубируют в течение 30 минут. Данное инкубирование проводится при температуре 35-40°C и данную обрабатываемую суспензию защищают от воздействия света. После инкубирования свободную (неинкапсулированную) миРНК удаляют посредством анионного обмена с помощью фильтров для хроматографии Mustang Q (капсулы). Перед применением данные фильтры для хроматографии предварительно обрабатывают, последовательно промывая струей 1 н. NaOH, 1 M NaCl, и конечным раствором 12,5 об.% этанола в PBS. Значение рН данной конечной промывочной жидкости проверяют для обеспечения значения рН<8. Данную инкубированную фракцию LNP затем фильтруют через фильтры Mustang Q посредством перистальтического насоса (Cole Parmer MasterFlex L/S 600 RPM) при скорости потока приблизительно 100 мл/мин. Данную профильтрованную фракцию собирают в стерильный стеклянный контейнер для ультрафильтрования и концентрирования, как указано далее.
Ультрафильтрование, концентрирование и стерилизующее фильтрование
Процесс фильтрования представляет собой регулируемый во времени процесс и скорости потока должны контролироваться тщательным образом. Это двухэтапный процесс; первый этап представляет собой этап концентрирования, на котором берется данное разведенное вещество и концентрируется приблизительно 8-кратно, для получения концентрации миРНК приблизительно 0,3-0,6 мг/мл.
На данном первом этапе кольцевой штатив со встроенной ультрафильтрационной мембраной 100 кДа PES (Spectrum Labs), присоединен к перистальтическому насосу (Spectrum KrosFloII System). В данный резервуар добавляют 9,2 л стерильной дистиллированной воды; 3 л сливают и выбрасывают, и оставшееся количество фильтруют через пермеат и выбрасывают. 5,3 л 0,25 н. гидроксида натрия добавляют в данный резервуар, 1,5 л сливают и выбрасывают, и 3,1 л фильтруют через пермеат и выбрасывают. Оставшееся количество гидроксида натрия оставляют в данной системе для очистки (по меньшей мере, 10 минут), и затем данный насос осушают. 9,2 л 70% (об./об.) изопропилового спирта добавляют в данный резервуар, 1,5 л сливают и выбрасывают, и оставшееся количество фильтруют через пермеат и выбрасывают. Добавляют 6 л кондиционирующего буфера (12% этанол в забуференном фосфатом физиологическом растворе), 1,5 л сливают и выбрасывают, а оставшееся количество фильтруют через пермеат до тех пор, пока стоковая жидкость не будет иметь нейтральное значение рН (7-8). Значение потока сквозь мембрану записывают, а затем насос осушают.
Данный разведенный раствор LNP помещают в данный резервуар до метки 1,1 л. Насос включают на 2,3 л/мин. После 5 минут циркуляции в замкнутой системе, данный насос переключают на 62,5 мл/мин и уровень жидкости в данном резервуаре находится неизменным приблизительно на 950 мл. Данный разведенный раствор LNP концентрируют с 9,8 л до 1,1 л в течение 140 минут и насос останавливают, когда весь разбавленный раствор LNP перемещен в резервуар.
Второй этап представляет собой этап диафильтрации, на котором происходит обмен этанол/водяного буфера на забуференный фосфатом физиологический раствор. На этом этапе используют приблизительно 10-20 диафильтрующих объемов забуференного фосфатом физиологического раствора. После диафильтрования предпринято второе концентрирование для концентрирования данной суспензии LNP 3-кратно до приблизительно 1-1,5 мг/мл миРНК. Данную концентрированную суспензию собирали в стерильные пластиковые PETG сосуды. Конечную суспензию затем фильтровали последовательно через фильтры Pall 0,45 мкм PES и Pall 0,2 мкм PES для конечной стерилизации перед наполнением флаконов.
В одном варианте осуществления композиция LNP по настоящему изобретению содержит в себе катионный липид по формуле А, холестерин, DSPC и PEG-DMG.
В другом варианте осуществления композиция LNP по настоящему изобретению дополнительно содержит в себе криопротектор.
В другом варианте осуществления данный криопротектор представляет собой сахарозу, трегалозу, рафинозу, стахиозу, вербаскозу, маннит, глюкозу, лактозу, мальтозу, мальтотриоз-гептаозу, декстран, оксиэтилированный крахмал, инсулин, сорбит, глицерин, аргинин, гистидин, лизин, пролин, диметилсульфоксид или любую их комбинацию.
В другом варианте осуществления данный криопротектор представляет собой сахарозу.
В другом варианте осуществления данный криопротектор представляет собой трегалозу.
В другом варианте осуществления данный криопротектор представляет собой комбинацию сахарозы и трегалозы.
В другом варианте осуществления данная композиция LNP содержит в себе катионный липид (13Z,16Z)-N,N-диметил-3-нонилдокоза-13,16-диен-1-амин (соединение 32), холестерин, DSPC и PEG-DMG.
Полученные LNP охарактеризованы на основании размера частиц, дзета-потенциала, содержания спирта, общего содержания липида, инкапсулированных нуклеиновых кислот и общей концентрации нуклеиновых кислот.
Специалисту в данной области будет полностью понятно, что настоящее изобретение хорошо адаптируется для осуществления целей и получения результатов и упомянутых преимуществ, а также тех, которые связаны с этим. Способы и композиции, описанные здесь, в качестве в настоящее время типичных вариантов осуществления, являются иллюстративными и не предназначены для ограничения диапазона данного изобретения. Специалисту в данной области будут очевидны изменения и другие применения, которые охвачены сущностью данного изобретения и определены диапазоном формулы изобретения.




Используя вышеописанные процессы LNP, были установлены специфические LNP в следующих соотношениях:
Номинальная композиция
Катионный липид/холестерин/PEG-DMG 60/38/2
Катионный липид/холестерин/PEG-DMG/DSPC 58/30/2/10
Luc миРНК
5’-iB-AUAAGGCUAUGAAGAGAUATT-iB 3’ (SEQ ID NO:1)
3’-UUUAUUCCGAUACUUCUCUAU-5’ (SEQ ID NO:2)
AUGC - рибоза
iB - инвертированный дезокси с удаленными азотистыми основаниями
UC - 2’-фтор
AGT - 2’-дезокси
AGU - 2’-OCH3
Номинальная композиция
Катионный липид/холестерин/PEG-DMG 60/38/2
Катионный липид/холестерин/PEG-DMG/DSPC 40/48/2/10
Катионный липид/холестерин/PEG-DMG/DSPC 58/30/2/10
миРНК ApoB
5’-iB-CUUUAACAAUUCCUGAAAUTsT-iB-3’ (SEQ ID NO:3)
3’-UsUGAAAUUGUUAAGGACUsUsUsA-5’ (SEQ ID NO:4)
AUGC-рибоза
iB - инвертированный дезокси с удаленными азотистыми основаниями
UC - 2’-фтор
AGT - 2’-дезокси
AGU - 2’-OCH3
UsA - фосфоро-серная связь
миРНК бета-катенина
5’-iB-CUGUUGGAUUGAUUCGAAAUsU-iB-3’ (SEQ ID NO:5)
3’-UsUGACAACCUAACUAAGCUUU-5’ (SEQ ID NO:6)
AUGC - рибоза
iB - инвертированный дезокси с удаленными азотистыми основаниями
UC - 2’-фтор
AGT - 2’-дезокси
AGU - 2’-OCH3
UsA - фосфоро-серная связь
5’-iB-ACGACUAGUUCAGUUGCUUUsU-iB-3’ (SEQ ID NO:7)
3’-UsUUGCUGAUCAAGUCAACGAA-5’ (SEQ ID NO:8)
AUGC - рибоза
iB - инвертированный дезокси с удаленными азотистыми основаниями
UC - 2’-фтор
AGT - 2’-дезокси
AGU - 2’-OCH3
UsA - фосфоро-серная связь
5’-iB-ACGACUAGUUCAGUUGCUUUU-iB-3’ (SEQ ID NO:9)
3’-UUUGCUGAUCAAGUCAACGAA-5’ (SEQ ID NO:10)
AUGC - рибоза
iB - инвертированный дезокси с удаленными азотистыми основаниями
UC - 2’-фтор
AGT - 2’-дезокси
AGU - 2’-OCH3
UsA - фосфоро-серная связь
Синтез олигонуклеотидов хорошо известен в данной области (Смотрите патентные заявки США: US 2006/0083780, US 2006/0240554, US 2008/0020058, US 2009/0263407 и US 2009/0285881, и патентные заявки PCT: WO 2009/086558, WO 2009/127060, WO 2009/132131, WO 2010/042877, WO 2010/054384, WO 2010/054401, WO 2010/054405 и WO 2010/054406). Раскрытые и используемые в примерах миРНК были синтезированы с использованием стандартных твердофазных процедур.
Пример 1
Оценка эффективности in vivo на мышах
Была проведена оценка эффективности in vivo LNP, применяемых в соединениях 1-44, в номинальных композициях, описанных непосредственно выше. Данные миРНК нацелены на транскрипт мРНК гена люциферазы жука-светляка (Photinus pyralis) (№ доступа M15077). Первичная последовательность и характер модификации химической структуры данной миРНК люциферазы представлен выше. В модели in vivo люциферазы используется трансгенная мышь, у которой кодирующая последовательность люциферазы жука-светляка присутствует во всех клетках. Трансгенные мыши ROSA26-LoxP-Stop-LoxP-Luc (LSL-Luc), запатентованные Институтом Dana Farber Cancer Institute, индуцированы для экспрессии гена люциферазы сначала посредством удаления данной последовательности LSL с помощью рекомбинантного вируса Ad-Cre (Vector Biolabs). Благодаря органотропной природе данного вируса, экспрессия ограничена в печени, при доставке посредством инъекции в хвостовую вену. Уровни экспрессии люциферазы в печени вычисляли количественно, оценивая световой выход, используя систему для формирования изображений IVIS (Xenogen), после введения субстрата люциферина (Caliper Life Sciences). Уровни люминисценции до введения препарата оценивали перед введением данных RDV. Люциферин в PBS (15 мг/мл) вводили внутрибрюшинно (в/б) в объеме 150 мкл. После инкубационного периода длительностью четыре минуты мышам проводили анестезию с помощью изофлюрана и помещали их в систему для формирования изображения IVIS. Данные RDV (содержащие в себе миРНК) в носителе PBS, инъекционно вводили в хвостовую вену в объеме 0,2 мл. Уровни конечной дозы находятся в диапазоне от 0,1 до 0,5 мг/кг миРНК. В качестве контроля вводился носитель PBS. Через 48 часов после введения дозы, используя метод, описанный выше, получали сформированные изображение от данных мышей. Изменения светового выхода люциферина напрямую коррелируют с уровнями мРНК люциферазы и представляют собой непрямое измерение активности миРНК люциферазы. Результаты эффективности in vivo выражаются как % ингибирования люминесценции относительно уровней люминесценции до введения дозы. Системное введение RDV, содержащих в себе миРНК люциферазы, снижает экспрессию люциферазы в дозозависимой манере. Более высокую эффективность наблюдали у мышей, которым вводили RDV, содержащие в себе соединение 1, по сравнению с RDV, содержащим в себе катионный липид октил-CLinDMA (OCD) (фиг.1). OCD является известным и описан в WO 2010/021865. Схожую эффективность наблюдали у мышей, которым вводили дозы RDV, содержащих в себе соединения 32 и 33, по сравнению с RDV, содержащим в себе катионный липид MC3 (соединение 46) (фиг.11).
Пример 2
In vitro анализ связывания ApoE
LNP инкубировали при 37°C в 90% резус-сыворотке при конечной концентрации LNP 4 мкг/мл. Инкубирование продолжается в течение 20 минут с орбитальным вращением. После инкубирования образцы разводили 1:20 с помощью PBS и 100 мкл аликвот каждого разведенного образца помещали в лунки 96-луночного планшета с покрытием анти-PEG антитела (Life Diagnostics по каталогу № P-0001PL). После инкубирования при комнатной температуре в течение 1 часа, планшет отмывали 5× с помощью 300 мкл PBS. После отмывки в каждую лунку добавляли 50 мкл 0,2% Triton X-100 и планшет инкубировали при 37°C в течение 10 минут с последующим встряхиванием на планшетном шейкере в течение 1 минуты при 750 об/мин. Образцы замораживали до осуществления ApoE ELISA и ПЦР-анализа структуры «стебель-петля» образцов.
Анализ ApoE ELISA осуществляют для количественного определения связывания ApoE с данными LNP после инкубирования в резус-сыворотке. Анти-ApoE антитело (Milipore, по каталогу № AB947) разводят 1:1000 в PBS и 100 мкл разведенного антитела добавляют в каждую лунку полистирольного планшета с высокой степенью связывания. Данный планшет с антителом инкубируют в течение ночи при 4°C, после чего планшет отмывают 2× с помощью 200 мкл PBS. Далее, в каждую лунку добавляют 200 мкл буфера, содержащего в себе 1% BSA и 0,05% Tween-20 в PBS (инкубационный буфер), с последующим инкубированием при комнатной температуре в течение 1 часа. Планшеты отмывают 5× с помощью PBS, содержащего в себе 0,05% Tween-20. Замороженные образцы для лизиса тритоном размораживали и разводили 1:6 буфером для инкубирования и аликвоту по 100 мкл пробного образца помещали в лунки планшета с ApoE антителом. Инкубирование длится в течение 1 часа при комнатной температуре, с последующими отмывками 5× с помощью PBS, содержащем в себе 0,05% Tween-20. После отмывки 100 мкл биотинилированного анти-ApoE антитела (Mabtech, по каталогу № E887-biotin), разведенного 1:500 в буфере для инкубирования, добавляли в каждую лунку и инкубировали в течение 1 часа при комнатной температуре, с последующими отмывками 5× с помощью 0,05% Tween-20 в PBS. Затем добавляли стрептавидин-HPR (Thermo, по каталогу № TS-125-HR) 100 мкл на лунку и инкубировали в течение 1 часа при комнатной температуре. После отмывки 5× с помощью 0,05% Tween-20 в PBS, в каждую лунку добавляли 100 мкл субстрата TMB (Thermo, по каталогу № 34028), с последующим инкубированием при комнатной температуре в течение 20 минут в темноте. Данную колориметрическую реакцию останавливали, добавляя 100 мкл стоп-раствора TMB (KPL, по каталогу № 50-85-04), и определяли поглощение при 450 нм. Калибровочную кривую ApoE получали посредством разведения резус рекомбинантного АроЕ в буфере для разведения с 0,03% Triton X-100, с концентрациями в диапазоне от 100 нг/мл до 0,78 нг/мл. Стандарты ApoE определяли в анализе ELISA в параллели с данными опытными образцами. Контрольную резус-сыворотку (не содержащую в себе LNP) использовали для получения фоновых субстратов для сигнала АроЕ, независимого от LNP, в данном анализе ELISA.
Протокол RT-ПЦР для структуры «стебель-петля»
Для нормализации связывания АроЕ с количеством LNP, связанного на анти-PEG антитело планшете, данное количество миРНК, оставшееся в лунке с анти-PEG антителом, определяли количественно с помощью ПЦР «стебель-петля» и определяли соотношение с числом инкапсулированных миРНК на LNP, для получения приблизительной оценки общего количества связанных частиц LNP на лунку.
Получение маркировочных калибровочных кривых образцов
Калибровочную кривую получали с использованием молекулярной массы данной миРНК (13693 г/моль для ApoB 17063) для подсчета числа копий. Высокий стандарт должен содержать в себе 1011копий в 3 мкл. 10-кратные серийные разведения были осуществлены сквозь ряд аналитического планшета до тех пор, пока низший стандарт содержит 102 копий в 3 мкл. Разводят 1:80 0,2% Triton X-100 в воде и с помощью пипетки вносят 20 мкл данного разведенного Triton X-100 в 10 лунок 96-луночного планшета. В каждую лунку данного планшета добавляют 30 мкл серийных разведений из калибровочной кривой и смесь. 10 мкл из маркировочной калибровочной кривой используют в реакции обратной транскрипции.
RT-ПЦР «стебель петля» - опытные образцы и калибровочная кривая
Лизаты Triton из планшета с иммобилизованным антителом PEG разводили 1 к 2000 в воде, не содержащей нуклеазы. В каждую лунку 96-луночного планшета Micro-Amp QPCR (ABI, по каталогу № N801-0560) добавляли 10 мкл смеси праймеров «RT-Primer Mix» (Applied Biosystem’s TaqMan MicroRNA Reverse Transcription Kit, по каталогу № 4366596).
Последовательность праймера ApoB RT: 5’ GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCTTTAACA3’ (SEQ ID NO:11)
Аликвоты по 10 мкл каждого опытного образца (разведенного 1 к 2000) или маркировочной калибровочной кривой (выше) были внесены в 96-луночный планшет. Данный планшет накрывали матрасиком (ABI, по каталогу № N801-0550), для сведения к минимуму испарения. Данный планшет центрифугировали при 800 об/мин в течение 1 минуты. Затем данный планшет помещали в термоциклер и осуществляли реакцию, используя следующие циклические параметры:
Далее, в каждую лунку добавляют 10 мкл смеси «RT Mix» (Applied Biosystem’s TaqMan MicroRNA Reverse Transcription Kit, по каталогу № 4366596).
Данная циклическая реакция RT содержит в себе 10 мкл опытного образца, 10 мкл смеси праймеров RT и 10 мкл компонентов смеси RT, с общим объемом реакционной смеси 30 мкл. Конечная концентрация RT-праймера в общем объеме RT-смеси 30 мкл составляет 200 нМ. Затем данный планшет герметически закрывают тем же матрасиком для планшета, центрифугируют при 800 об/мин в течение 1 минуты, затем помещают в термоциклер и осуществляют реакцию, используя следующие циклические параметры:
Далее, в каждую лунку нового 96-луночного планшета Fast (Applied Biosystem’s TaqMan Fast Universal PCR Master Mix, по каталогу № 4352042) добавляют 15 мкл смеси Fast Enzyme/праймер-зонд.
Последовательности праймеров ApoB и зонда:
17063DC F3 GGCGCGAAATTTCAGGAATTGT (SEQ ID NO:12)
17063DC Pr2 CACTGGATACGACCTTTAACA (SEQ ID NO:13)
Universal R2 AGTGCAGGGTCCGAG (SEQ ID NO:14)
В планшет Fast Enzyme Mix вносили по 5 мкл каждой RT-реакции. Данный планшет центрифугировали при 1000 об/мин в течение 1 минуты и осуществляли анализ QPCR на AB17900 с Fast Block. Циклические параметры следующие: 1 цикл - 95°C в течение 20 секунд, с последующими 40 циклами - 95°C в течение 1 секунды, 60°C в течение 20 секунд.
Результат, получено в QPCR используют для расчета концентрации миРНК в лизатах Triton в планшете с иммобилизованным антителом PEG. Исходя из 500 миРНК на частицу LNP, может быть подсчитано число LNP, оставшихся в каждой лунке данного планшета с анти-PEG антителом. Используя концентрацию АроЕ на лунку, как определено в анализе ApoE ELISA, и число частиц LNP на лунку, можно рассчитать приблизительное количество связанных молекул АроЕ на частицу LNP.
#ApoE связанных молекул на LNP
Пример 3
HI-TRAP™ анализ связывания на гепарин-сефарозе
Липидные наночастицы (LNP) с нейтральным зарядом поверхности не задерживаются после ввода пробы на гепарин-сефарозе с 1× физиологическим раствором, забуференным фосфатом Дульбекко (DPBS), в качестве рабочего буфера, но элюируются в свободный объем колонки. Аполипопротеин Е сыворотки (АроЕ) проявляет высокоаффинное связывание с гепарин сульфатом и было показано, что LNP связываются с гепарин-сефарозой в степени, зависящей от их внутренней способности связываться с АроЕ (в зависимости и от состава липидной наночастицы, и от концентрации АроЕ) после инкубирования с очищенными и/или рекомбинантными образцами АроЕ человека или образцами сыворотки. Липидные наночастицы с поверхностно связанным АроЕ связываются с гепарин-сефарозой с высокой аффинностью и элюируются только при высоком содержании соли (1 M NaCl).
Анализ связывания гепарин-сефарозы был осуществлен для определения связывания АроЕ сыворотки с липидными наночастицами, исходя из высокоаффинного взаимодействия, которое комплексы АроЕ-LNPA демонстрируют по отношению к гепарин-сефарозе.
Инкубирование
Липидные наночастицы инкубировали при 37°C в течение 20 мин в конечной концентрации миРНК 50 мкг/мл с различными концентрациями либо очищенного, либо рекомбинантного АроЕ человека, или 0,1-50% сывороткой крысы/мыши/макаки-резус/человека в 1× физиологическом растворе забуференном фосфатом Дульбекко (DPBS). После инкубирования с АроЕ или сывороткой образцы LNP разводили 10-кратно, используя 1×DPBS, и проводили анализ с помощью хроматографии на гепарин-сефарозе. Площадь пика удержанного LNP (после образования субстрата соответствующих сигналов гашения) сравнивали с общей площадью пика контрольного LNP без инкубирования с ApoE и/или сывороткой, для определения процентного содержания LNP, который претерпевает сдвиг в сторону высокоаффинного взаимодействия с гепарином после инкубирования с ApoE/сывороткой.
Условия для хроматографии на гепарин-сефарозе HI-TRAP™
Колонку для хроматографии на гепарин-сефарозе HI-TRAP™ (GE Healthcare; объем слоя 1 мл) уравновешивают либо с помощью 1×, либо 2×PBS Дульбекко; более высокая 2× концентрация соли используется для LNP с более высокой природной способностью к задержанию на гепарин-сефарозе (предположительно вследствие более высокого положительного поверхностного заряда).
Мобильная фаза A: 1× или 2×DPBS
Мобильная фаза B: 1 M NaCl в 10 мМ натрий-фосфатном буфере, pH 7,0
100% A доставляли изократически в течение 10 мин, с последующим ступенчатым градиентом до 100% В; выдерживали в течение дополнительных 10 мин; осуществляли ступенчатый градиент обратно к 100% А и повторно уравновешивали в течение дополнительных 10 мин перед введением следующего образца.
Скорость потока: 1 мл/мин
Объем вводимого образца: 50 мкл
Регистрация: УФ при 260 нм
HI-TRAP™ результаты связывания после инкубирования с сывороткой макаки-резус (2×DPBS условия)
Пример 4
Оценка in vivo эффективности и токсичности у крыс
LNP, в которых используются соединения в номинальных композициях, описанных выше, оценивали на in vivo эффективность и повышение аланинаминотрансферазы и аспартатаминотрансферазы на самках крыс Sprague-Dawley (Crl:CD(SD)) (Charles River Labs). Данная миРНК нацелена на транскрипт мРНК для гена АроВ (№ доступа NM 019287). Первичная последовательность и характер модификации химической структуры миРНК АроВ представлены выше. Данные RDV (содержащие в себе миРНК) в носителе PBS были инъекционно введены в хвостовую вену в объеме от 1 до 1,5 мл. Скорость введения составляла приблизительно 3 мл/мин. В каждой группе использовали по пять крыс. После введения LNP крыс помещали в клетки и содержали с нормальным рационом и наличием воды. Через шесть часов после введения дозы из клеток убирали еду. Вскрытие животных осуществляли через 24 часа после введения дозы LNP. Крысам проводили анестезию изофлураном в течение 5 минут, затем поддерживали под анестезией, помещая их в носовые конусы и продолжая подачу изофлурана до тех пор, пока не закончится обескровливание. Кровь собирают из полой вены, используя набор для венепункции типа «бабочка» 23 калибра, и аликвоты помещали в вакуумный контейнер сепаратора сыворотки для химического анализы сыворотки. Брали пункции удаленных хвостатых долей печени и помещали их в RNALater (Ambion) для анализа мРНК. Фиксированную ткань печени гомогенизировали и выделяли общую РНК, используя бисерную мельницу Qiagen bead mill и набор для выделения РНК Qiagen miRNA-Easy RNA isolation kit, следуя инструкциям фирмы-изготовителя. Уровни мРНК АроВ печени определяли с помощью количественной RT-ПЦР. Информационную РНК амплифицировали из очищенной РНК, используя набор коммерческих зондов АроВ крысы (Applied Biosystems, по каталогу № RN01499054_m1). Реакцию ПЦР осуществляли на оборудовании ABI 7500 с 96-луночным Fast Block. Уровень мРНК ApoB нормализовали к мРНК PPIB «домашнего хозяйства» (NM 011149). Уровни мРНК PPIB были определены с помощью RT-ПЦР, используя коммерческий набор зондов (Applied Biosytems, по каталогу № Mm00478295 m1). Результаты представлены в виде соотношения мРНК ApoB/мРНК PPIB. Все данные мРНК выражены по отношению к контрольной дозе PBS. Анализы ALT и AST сыворотки были осуществлены на оборудовании Siemens Advia 1800 Clinical Chemistry Analyzer, с использованием реагентов Siemens аланинаминотрансферазы (по каталогу № 03039631) и аспартатаминотрансферазы (по каталогу № 03039631). Схожую эффективность и повышенную сопротивляемость наблюдали у крыс, которым вводили дозы RDV, содержащих в себе соединение 32 или 33, в сравнении с RDV, содержащим в себе катионный липид DLinKC2DMA (соединение 45) или MC3 (соединение 46, фиг.2).
Пример 5
Определение уровней катионных липидов в печени крыс/обезьян
Ткань печени взвешивали в пробирках объемом 20 мл и гомогенизировали в воде 9 об/вес, используя GenoGrinder 2000 (OPS Diagnostics, 1600 ударов/мин, 5 мин). Аликвоту 50 мкл каждого гомогената ткани смешивали с 300 мкл сольвента для экстракции/преципитации белка (50/50 ацетонитрил/метанол содержащий 500 нМ внутреннего стандарта) и данный планшет центрифугировали до осаждения белкового осадка. 200 мкл каждого супернатанта затем переносили в изолированные лунки 96-луночного планшета и проводили непосредственный анализ 10 мкл образцов с помощью ЖХ/МС-MC.
Стандарты готовили путем внесения известных количеств метилового исходного раствора соединения в необработанный гомогенат печени крысы (9 об. воды/вес печени). Аликвоты (50 мкл) каждого стандарт/гомогенат печени смешивали с 300 мкл сольвента для экстракции/преципитации белка (50/50 ацетонитрил/метанол содержащий 500 нМ внутреннего стандарта) и данный планшет центрифугировали до осаждения белкового осадка. 200 мкл каждого супернатанта затем переносили в изолированные лунки 96-луночного планшета и проводили непосредственный анализ 10 мкл образцов с помощью ЖХ/МС-MC.
Абсолютное количественное определение в сопоставлении со стандартом, приготовленным и выделенным из гомогенатов печени, было осуществлено с использованием системы Aria LX-2 HPLC (Thermo Scientific), соединенной с масс-спектрометром с тремя квадрупольными линзами API 4000 (Applied Biosystems). Для каждой серии измерений, общий объем 10 мкл образца вносили в колонку BDS Hypersil C8 HPLC (Thermo, 50×2 мм, 3 мкм) при температуре окружающей среды.
Мобильная фаза A: 95% H2O/5% метанол/10 мМ муравьинокислый аммоний/0,1% муравьиная кислота. Мобильная фаза B: 40% метанол/60% n-пропанол/10 мМ муравьинокислый аммоний/0,1% муравьиная кислота. Скорость потока составляла 0,5 мл/мин и профиль градиентного элюирования был следующим: выдержать при 80% A в течение 0,25 мин, линейно изменить до 100% B за 1,6 мин, выдержать при 100% B в течение 2,5 мин, затем возвратить и выдержать при 80% A в течение 1,75 мин. Общее время выполнения составляло 5,8 мин. Параметры источника API 4000 были следующие CAD: 4, CUR: 15, GS1: 65, GS2: 35, IS: 4000, TEM: 550, CXP: 15, DP: 60, EP: 10.
У крыс, получавших дозы RDV, содержащего в себе соединения 32 или 33, уровни в печени были либо аналогичными, либо более низкими по сравнению с RDV, содержащим в себе катионный липид DLinKC2DMA (соединение 45) или MC3 (соединение 46, фиг.3). У макак, получавших дозы RDV, содержащего в себе соединения 32 или 33, уровни в печени были более низкими по сравнению с RDV, содержащим в себе катионный липид DLinKC2DMA (соединение 45) или MC3 (соединение 46, фиг.7).
Пример 6
Оценка in vivo эффективности ApoB у макак-резус
LNP, в которых используются соединения в номинальных композициях, описанных выше, оценивали на in vivo эффективность на самцах или самках макак-резус Macaca mullata. Данная миРНК нацелена на транскрипт мРНК для гена АроВ (№ доступа XM 001097404). Первичная последовательность и характер модификации химической структуры миРНК АроВ представлены выше. Данные RDV (содержащие в себе миРНК) в носителе PBS были введены посредством внутривенной инъекции в подкожную вену со скоростью введения 20 мл/мин до уровня дозы миРНК 0,25 мг/килограмм. Вводимые объемы были от 1,9 до 2,1 мл/килограмм, и вес макак находился в диапазоне от 2,5 до 4,5 килограмма. RDV или контрольный PBS вводили трем обезьянам. Через несколько дней после введения дозы, из бедренной артерии был произведен забор крови объемом 1 мл для проведения химического анализа сыворотки. Перед забором крови обезьяны голодали в течение ночи. В качестве меры эффективности, осуществляли мониторинг LDL-C как суррогатного маркера в 5’-3’ направлении уменьшения мРНК АроВ. На 4 день после системного введения RDV, содержащих в себе соединения 32 и 33 (0,25 мг/кг), сывороточные урони LDL-C были снижены до менее чем 30% от уровней до введения дозы (фиг.4).
Пример 7
Оценка in vivo эффективности β-катенина у макак-резус
В день исследования -7 перед введением дозы были собраны образцы биопсии печени (приблизительно 0,5-1 грам/образец), полученные от самцов макак-резус в результате лапароскопической хирургической резекции (резекция одного образца биопсии из наружного края одной, выбранной случайным образом, доли печени на обезьяну). Пункцию ткани размером 5 мм использовали для проведения анализа трех несмежных образцов приблизительно 50 мг из каждой биопсии, взятой перед введением дозы. Образцы сохраняли в RNAlater™ (Ambion) для дальнейшего анализа мРНК CTNNB1.
В день исследования 0 обезьянам вводили суспензии испытательных образцов липидных наночастиц (LNP) в физиологическом растворе, забуференном фосфатом (0,05-0,1 мг миРНК/мл) посредством внутривенной болюсной инъекции однократной дозы с расчетными дозами 0,67, 1,34 или 3,34 мг миРНК/мл. Для целей дозирования, площадь поверхности тела (BSA) (м2) оценивалась по массе тела (BW) в соответствии с установленной аллометрической шкалой отношений, представленной ниже (1):
BSA (м2)=0,11×BW(в кг)0,65
В дни исследования 2 и 7, через 48 часов и 168 часов после введения LNP, образцы биопсии печени (приблизительно 0,5-1 грамм/образец) собирали от обезьян посредством лапароскопической хирургической резекции (были проведены резекции 2 раздельных, выбранных случайным образом, долей печени на обезьяну). Пункцию ткани размером 5 мм использовали для проведения анализа трех несмежных образцов приблизительно 50 мг из каждой биопсии, взятой через 48 часов и 168 часов. Образцы сохраняли в RNAlater™ (Ambion) для дальнейшего анализа мРНК CTNNB1.
Уровни мРНК CTNNB1 измеряли посредством сравнительной количественной RT-ПЦР, с использованием набора праймер/зонд, утвержденного для CTNNB1, и нормализованы относительно уровней мРНК пептидилпролилизомеразы В (также известной как PPIB или циклофилин В) и уровней РНК 18S рибосомальной РНК (18S рРНК). Изменение экспрессии мРНК CTNNB 1 в печени определяли как разницу порогового количества циклов ПЦР (AACt) между образцами после введения дозы и каждым соответствующим образцом ткани печени обезьяны, взятым до введения дозы.
Расчет нокдаун мРНК CTNNB1 (относительно уровней перед введением дозы) проводился исходя из AACt, с использованием следующего соотношения:
мРНК (% нокдаун)-100-(100/2-δδCt)
обезьяны, получавшие дозы RDV, содержащих в себе соединения 32 и 33 и миРНК бета-катенина, проявляли устойчивый KD при дозах в диапазоне 0,67-3,34 мг/м2 (фиг.5).
(1) Руководящий документ Управления по контролю за продуктами и лекарствами США (FDA Guidance Document): «Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers» July 2005, US Department of Health and Human Services, Food and Drug Administration - Center for Drug Evaluation and Research (CDER).
Пример 8
Оценка in vivo повышения ALT у макак-резус
Аланинаминотрансферазу (ALT) измеряют в сыворотке, полученной из коагулированной цельной крови обезьяны после центрифугирования. Автоматический химический анализатор Roche Modular System измеряет ферментативную активность ALT в сыворотке с применением стандартизированных Международной Федерацией по Клинической Химии (International Federation of Clinical Chemistry) процедур и реагентов. Вычислительное устройство данного анализатора использует измерения оптической плотности для расчета активности ALT в данном образце по сравнению с калибровочной кривой. Активность ALT представлена в Международных Единицах на литр (МЕ/л).
У обезьян, получавших дозы RDV, содержащих в себе соединения 32 и 33, наблюдали более низкие подъемы пика ALT, чем у тех, которые получали дозы RDV, содержащего в себе катионный липид DLinKC2DMA (соединение 45) или MC3 (соединение 46, фиг.6).
Пример 9
Оценка на модели гепатоцеллюлярной гарциномы на мышах
Активность LNP в доставке миРНК β-катенина (миРНКβ-cat) в гепатоцеллюлярную карциному оценивали на модели гепатоцеллюлярной карциномы (НСС) у мыши, называемой TRB-MET. Мыши TRE-MET представляют собой трансгенных мышей с генетическим фоном FVB/N, где трансген МЕТ человека экспрессируется с промотороа hCMV с гептамерными трет-оперонами в направлении 3’-5’. Если мышей TRE-MET скрестить с линией LAP-tTA, то двойные трансгенные мыши (TRE-MET/LAP-tTA) экспрессируют МЕТ в печеночноспецифической манере, что может быть подавлено посредством введения диксоциклина. У этих мышей развивается НСС в возрасте приблизительно 3 месяцев с визуально определяемыми опухолевыми узелками на поверхности печени и данные опухоли проявляют диффузный трабекулярный характер роста, типичный для НСС, и экспрессируют опухолевый маркер НСС альфа-фетопротеин (AFP). Кроме того, мутационный анализ данной опухоли мышей TRE-MET также выявил активирующие мутации в гене бета-катенина приблизительно в 95% опухолей. Эти особенности делают модель НСС на мышах TRE-MET подходящей для оценки LNP-опосредованной доставки миРНК β-катенина и результирующей эффективности на рост опухоли.
Эффект LNP, содержащих в себе β-катенин, на молчащих мРНК β-катенина как в печени, так и в опухолевых тканях, сначала был оценен в исследовании фармакодинамики (PD) на мышах TRE-MET, несущих опухоли. Различные дозы LNP, или высокие дозы LNP с контрольной миРНК, были введены внутривенно и через 72 часа было осуществлено вскрытие для забора печени и опухолевых тканей для определения уровней мРНК β-катенина посредством Taqman. Как представлено на фиг.8 и 9, соединение 33 индуцирует устойчивое и дозозависимое выключение мРНК β-катенина как в печени, так и в опухолевых тканях, в то время как у животных, получавших контрольную миРНК или PBS не наблюдали выключение β-катенина. 0,1 мг/кг и 0,05 мг/кг LNP индуцировали 88% и 69% KD в нормальной печени, соответственно. KD в опухолях колеблется в пределах от 70% (2 мг/кг) до приблизительно 40% (0,1 или 0,05 мг/кг). Как представлено на фиг.12 и 13, соединение 32 индуцирует устойчивое и дозозависимое выключение мРНК β-катенина как в печени, так и в опухолевых тканях, в то время как у животных, получавших контрольную миРНК или PBS не наблюдали выключение β-катенина. 0,1 мг/кг и 0,05 мг/кг LNP индуцировали 76% и 78% KD в нормальной печени, соответственно. KD в опухолях колеблется в пределах от 47% (0,25 мг/кг) до приблизительно 20-30% (0,1 или 0,05 мг/кг).
Эффект LNP на опухолевый рост оценивали в исследовании с использованием многократных доз. Мышам TRE-MET HCC вводили дозы соединения 33/миРНКβ-cat, соединения 32/миРНКβ-cat, контрольной миРНК или PBS, еженедельно в течение 3 недель (3 дозы) и объем опухоли у каждого животного определяли посредством микроКТ сканирования за 7 дней до 1ой дозы и через 3 дня после последней дозы (фиг.10 и 14). Кроме того, через 7 дней после финальной дозы производили забор печени и опухолевых тканей для оценки уровней мРНК β-катенина. В то время как у мышей, получавших PBS или контрольную миРНК, наблюдался рост опухолевой нагрузки 360-470%, мыши, получавшие соединение 33/миРНКβ-cat демонстрировали основательное ингибирование роста опухоли или регресс в дозозависимой манере (фиг.10). Дозы 2 мг/кг и 0,5 мг/кг соединения 33/миРНКβ-cat индуцировали 60% и 40% регресс опухоли, соответственно, и доза 0,05 мг/кг вызывала остановку роста опухоли. В то время как у мышей, получавших PBS или контрольную миРНК, наблюдался рост опухолевой нагрузки приблизительно 350%, мыши, получавшие соединение 32/миРНКβ-cat демонстрировали основательное ингибирование роста опухоли или регресс в дозозависимой манере (фиг.14). Дозы 0,5, 0,25 и 0,1 мг/кг соединения 32/миРНКβ-cat индуцировали 37, 58 и 37% регресс опухоли, соответственно, и доза 0,05 мг/кг вызывала остановку роста опухоли.
Реферат
Настоящее изобретение относится к новому катионному липиду по формуле А, или его любой фармацевтически приемлемой соли, которые используются для формирования липидных наночастиц (LNP) для доставки олигонуклеотидов, к композиции липидных наночастиц (LNP), содержащей предлагаемый катионный липид, и к применению катионного липида. В формуле А значения R, R, R, R', Rʺ, n, Lи Lтакие, как указаны в формуле изобретения. 7 н. и 3 з.п. ф-лы, 14 ил., 2 табл., 9 пр.
Формула

Документы, цитированные в отчёте о поиске
Система доставки нуклеиновых кислот
Комментарии