Производные карбоновой кислоты - RU2175315C2
Код документа: RU2175315C2
Чертежи
Описание
Изобретение относится к органическим кислотам, более конкретно к новым производным карбоновой кислоты, обладающим биологической активностью, в частности активностью, ингибирующей рецептор эндотелина.
Эндотелин представляет собой пептид, состоящий из 21 аминокислоты, который синтезируется и выделяется сосудистым эндотелом. Эндотелин существует в трех изомерных формах, ЕТ-1, ЕТ- 2 и ЕТ-3. В дальнейшем под названиями "эндотелин" или "ЕТ" понимают одну или все изомерные формы эндотелина. Эндотелин представляет собой потенциальный вазоконстриктор и оказывает сильный эффект на сосудистый тонус. Известно, что вазоконстрикция вызывается связыванием эндотелина с рецептором (см. "Nature", N 332, стр. 411 - 415, 1988 г. ; "FEBS Letters", N 231, стр. 440 - 444, 1988 г., "Biochem. Biophys. Res. Commun., N 154, стр. 868 - 875, 1988 г.).
Повышенное или ненормальное выделение эндотелина приводит к постоянной контрактуре в периферийных, почечных и мозговых кровеносных сосудах, которая может вызвать заболевания. Как описано в литературе, повышенное содержание эндотелина обнаруживается в плазме пациентов, страдающих от гипертонии, острого инфаркта миокарда, легочной гипертонии, синдрома Рейно, атеросклероза, а также в дыхательных путях пациентов, страдающих от астмы ("Japan J. Hypertension", N 12, стр. 79, 1989 г., "J. Vascular Med. Biology", N 2, стр. 207, 1990 г., "J. Am, Med. Association", N 264, стр. 2868, 1990 г.).
Следовательно, вещества, которые специфично ингибируют связывание эндотелина с рецептором, должны были бы также антагонизировать вышеуказанные различные физиологические эффекты эндотелина и поэтому могут представлять собой ценные фармакологические препараты.
Из международной заявки N WO 94/02474 известно, что определенные производные карбоновой
кислоты общей формулы Q
представляют собой хорошие ингибиторы рецепторов эндотелина
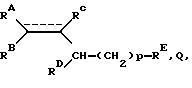
Rc = COOH и др.
При этом изучаются преимущественно соединения с двойной связью. Наряду с заместителями RA и RB у β-центра допускается максимально один атом водорода.
Неожиданно было найдено, что этот атом водорода может заменяться алкильными остатками. При этом образуется четвертичный β-центр, что приводит к повышению действия на рецепторы эндотелина.
Поэтому объектом предлагаемого изобретения являются производные карбоновой кислоты формулы I
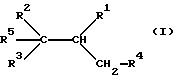
где R1 - карбоксил или гидролизируемый в группу COOH остаток,
R2 и R3 могут быть одинаковыми или различными и означают фенил, который может быть необязательно замещен одним или несколькими остатками из группы, включающей алкил с 1-4 атомами углерода и алкоксил с 1-4 атомами углерода, которые связаны в орто-положении через непосредственную связь, метилен, этилен или
R4 - фенил, который может быть замещен одним или несколькими остатками из группы, включающей алкил с 1 - 4 атомами углерода, гидрокси, алкоксил с 1-4 атомами углерода, бензилоксигруппу; нафтил, метилендиоксифенил, который может быть замещен алкоксилом с 1-4 атомами углерода, этилендиоксифенил;
R5 - алкил с 1-8 атомами углерода.
Соединения, а также промежуточные продукты, образующиеся при их получении, как, например, соединения нижепредставленной формулы Va, могут содержать один или несколько асимметрически замещенных атомов углерода. Такие соединения могут иметься в виде чистых энатиомеров или диастереомеров, или в виде их смесей. Предпочитают применение энантиомерно чистого соединения в качестве активного начала.
Соединения формулы I получают за счет того, что кетон формулы II подвергают взаимодействию со сложным фосфоновым эфиром формулы III в присутствии основания с получением соединений формулы IV.
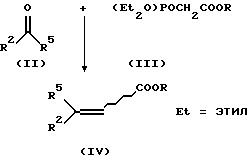
В качестве растворителя используют апротонные полярные растворители, как, например, диметилформамид или тетрагидрофуран.
В качестве основания можно использовать гидрид щелочного или щелочноземельного металла, как гидрид натрия, гидрид калия или гидрид кальция, карбонат, как карбонат щелочного металла, как, например, карбонат натрия или калия, гидроокись щелочного или щелочноземельного металла, как гидроокись натрия или калия, металлорганическое соединение, как бутиллитий, алкоголят щелочного металла, как метанолат натрия или трет.-бутанолат калия, или амид щелочного метала, как диизопропиламид лития.
При этом реакция осуществляется предпочтительно при температурах от 0oC до точки кипения растворителя или смеси растворителей.
Соединения формулы IV
можно подвергать взаимодействию с ароматическими соединениями в присутствии катализатора с получением производных карбоновой кислоты общей формулы Va
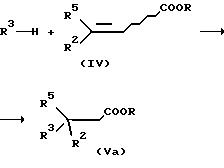
При этом в качестве катализаторов используют сильные неорганические кислоты, а также кислоты Льюиса. Примерами являются, в том числе, серная кислота, трихлорид алюминия, хлорид цинка или трихлорид железа. При применении серной кислоты можно непосредственно получать свободную кислоту.
Альтернативно можно получать симметричные производные карбоновой кислоты формулы Vb из β-дикарбонилсоединения формулы VI и ароматического соединения в присутствии катализатора.
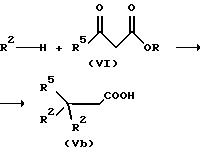
При этом в качестве катализаторов пригодны сильные неорганические кислоты, а также кислоты Льюиса. Примерами являются, например, серная кислоты, трихлорид алюминия, хлорид цинка или трихлорид железа (см. также Gogte G.R. и др., J. Univ. Bombay, Sect. Al, N 27, стр. 41, 1958 г.).
Другой возможностью
получения соединений формулы Va является взаимодействие кетона формулы VII
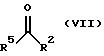
со сложным изопропилиденовым эфиром малоновой кислоты в присутствии основания, как пиридина или гидрида натрия, с получением соединений формулы VIII

Если подвергать соединения формулы VIII взаимодействию с реагентом Гриньяра общей формулы IX
R3 - MgY,
где Y означает бром, хлор или йод,
в среде диэтилового эфира получают соединения формулы X
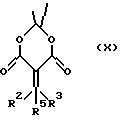
причем выгодным может быть дополнительное использование солей меди, как хлорида меди, бромида меди, йодида меди или цианида меди, а также присутствие кислоты Льюиса, как триметилсилилхлорида или бортрифторидэтерата.
Гидролизом соединений формулы X с минеральными кислотами, как соляной кислотой или серной кислотой, можно получать соединения формулы Va, где R означает водород.
Дальнейшие возможности получения соединений формулы Va описаны в литературе: Zimmermann Н.Е. и др., "J. Am. Chem. Soc.", N 83, стр. 1196, 1961 г. , Yu A.J. и др. "J. Org. Chem.", N 23, стр. 1004, 1958 г.
Соединения формулы Va, b можно переводить в анион (или дианион, если R = Н) при температурах от -78oC до комнатной температуры в
присутствии
сильного основания, как, например, бутиллития или литийдиизопропиламида, в среде инертного растворителя, как, например, диэтилового эфира или тетрагидрофурана, в атмосфере инертного газа,
как азота
или аргона. Данный анион реагирует с алкилирующими средствами формулы VII при температурах от -78oC до комнатной температуры. После резкого охлаждения концентрированным хлоридом
аммония или
разбавленной минеральной кислотой, как HCl, получают соединения формулы I
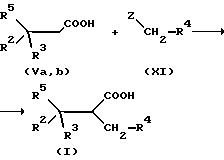
Z = галоген, триалкиламин
Соединения формулы I можно получать и тем, что соответствующие карбоновые кислоты, т. е. соединения формулы I, где R1 означает COOH, переводят обычным образом в активированную форму такую, как галогенид кислоты, ангидрид или имидазолид, и затем подвергают взаимодействию с соответствующим гидроксисоединением формулы HOR. Реакция может осуществляться в среде обычных растворителей, и часто следует добавлять основание, причем можно использовать вышеуказанные основания. Оба шага можно упрощать, например, тем, что гидроксисоединение подвергают воздействию карбоновой кислоты в присутствии водоотщепляющего средства, как карбодиимида.
Кроме того, соединения формулы I можно получать и за счет того, что используют соли соответствующих карбокислот, т.е. соединений формулы I, где R1 означает остаток COR и R - ОМ, причем М представляет собой катион щелочного металла или эквивалент катиона щелочноземельного металла. Эти соли можно подвергать взаимодействию с многими соединениями формулы R-A, причем A означает обычную нуклеофугную удаляемую группу, например, галоген, как, хлор, бром, йод, или незамещенный или замещенный галогеном, алкилом или галоидалкилом арил- или алкилсульфонил, как, например, толуолсульфонил и метилсульфонил, или другую подобную удаляемую группу. Соединения формулы R-A, где A представляет собой реакционноспособный заместитель, известны или легко получаются специалистом. Реакция осуществляется в среде обычных растворителей и выгодно происходит при добавки основания, причем используют указанные основания.
Энантиомерно чистые соединения формулы I можно получать за счет того, что осуществляют классическое отщепление рацемата с использованием рацемических или диастереомерных соединений формулы VI в присутствии пригодных энантиомерно чистых оснований, таких как, например, бруцин, стрихнин, хинин, хинидин, цинхонидин, цинхонин, иохимбин, морфин, дегидроабиэтиламин, эфедрин (-), (+), деоксиэфедрин (-), (+), трео-2-амино-1-(п-нитрофенил)-1.3- пропандиол (-), (+), трео-2-(N,N-диметиламино)-1-(п-нитрофенил)- 1, 3-пропандиол (+), (-) трео-2-амино-1-фенил-1,3-пропандиол (+), (-) α метилбензиламин (+), (-), α (1-нафтил)этиламин (+), (-), α -(2-нафтил)этиламин (+), (-), аминометилпинан, N, N-диметил-1- фенилэтиламин, N-метил-1-фенилэтиламин, 4-нитрофенилэтиламин, псевдоэфедрин, норэфедрин, норпсевдоэфедрин, производные аминокислот, производные пептидов.
В качестве
гидролизируемого в группу COOH остатка R1 можно назвать, например, остаток формулы
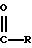
где R означает:
а) сукцинилимидоксигруппу,
б) связанный через атом азота 5-членный гетероаром, как пирролил, пиразолил, имидазолил и триазолил, который может нести 1 - 2 атома галогена, в частности фтор и хлор, и/или один или два остатка из группы, включающей: алкил с 1-4 атомами углерода, как метил, этил, 1-пропил, 2-пропил, 2-метил-2-пропил, 2-метил-1-пропил, 1-бутил, 2-бутил;
галоидалкил с 1-4 атомами углерода, в частности галоидалкил с 1-2 атомами углерода, как, например, фторметил, дифторметил, трифторметил, хлордифторметил, дихлорфторметил, трихлорметил, 1-фторэтил, 2-фторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил, 2-хлор-2,2-дифторэтил, 2,2-дихлор-2-фторэтил, 2,2,2-трихлорэтил и пентафторэтил;
галоидалкоксил с 1 - 4 атомами углерода, в частности галоидалкоксил с 1 - 2 атомами углерода, как дифторметоксил, трифторметоксил, хлордифторметоксил, 1-фторэтоксил, 2-фторэтоксил, 2,2-дифторэтоксил, 1,1,2,2-тетрафторэтоксил, 2,2, 2-трифторэтоксил, 2-хлор-1,1,2-трифторэтоксил и пентафторэтоксил, в частности трифторметоксил;
алкоксил с 1-4 атомами углерода, как метоксил, этоксил, пропоксил, 1-метилэтоксил, бутоксил, 1-метилпропоксил, 2-метилпропоксил, 1,1-диметилэтоксил, в частности метоксил, этоксил, 1-метилэтоксил;
алкилтиогруппу с 1-4 атомами углерода, как метилтиогруппу, этилтиогруппу, пропилтиогруппу, 1-метилэтилтиогруппу, бутилтиогруппу, 1-метилпропилтиогруппу, 2-метилпропилтиогруппу, 1.1-диметилэтилтиогруппу, в частности метилтиогруппу и этилтиогруппу;
в) группу формулы
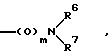
где m означает 0 или 1,
R6 и R7 могут быть одинаковыми или различными и означают водород,
алкил с 1 - 8 атомами углерода, в частности вышеуказанный алкил с 1-4 атомами углерода,
алкенил с 3-6 атомами углерода, как, 2-пропенил, 2-бутенил, 3-бутенил, 1-метил-2-пропенил, 2-метил-2-пропенил, 2-пентенил, 3-пентенил, 4-пентенил, 1-метил-2-бутенил, 2-метил-2-бутенил, 3-метил-2-бутенил, 1-метил-3-бутенил, 2-метил-3-бутенил, 3-метил-3-бутенил, 1,1-диметил-2-пропенил, 1,2-диметил-2-пропенил, 1-этил-2-пропенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 1-метил-2-пентенил, 2-метил-2-пентенил, 3-метил-2-пентенил, 4-метил-2-пентенил, 3-метил-3-пентенил, 4-метил-3-пентенил, 1-метил-4-пентенил, 2-метил-4-пентенил, 3-метил-4-пентенил, 4-метил-4-пентенил, 1,1-диметил-2-бутенил, 1, 1-диметил-3-бутенил, 1,2-диметил-2-бутенил, 1, 2-диметил-3-бутенил, 1,3-диметил-2-бутенил, 1,3-диметил-3-бутенил, 2,2-диметил-3-бутенил, 2,3-диметил-2-бутенил, 2,3-диметил-3-бутенил, 1-этил-2-бутенил, 1-этил-3-бутенил, 2-этил-2-бутенил, 2-этил-3-бутенил, 1,1,2-триметил-2-пропенил, 1-этил-1-метил-2-пропенил и 1-этил-2-метил-2-пропенил, в частности, 2-пропенил, 2-бутенил, 3-метил-2-бутенил и 3-метил-2-пентенил; алкинил с 3-6 атомами углерода, как 2-пропинил, 2-бутинил, 3-бутинил, 1-метил-2-пропинил, 2-пентинил, 3-пентинил, 4-пентинил, 1-метил-3-бутинил, 2-метил-3-бутинил, 1-метил-2-бутинил, 1,1-диметил-2-пропинил, 1-этил-2-пропинил, 2-гексинил, 3-гексинил, 4-гексинил, 5-гексинил, 1-метил-2-пентинил, 1-метил-3-пентинил, 1-метил-4-пентинил, 2-метил-3-пентинил, 2-метил-4-пентинил, 3-метил-4-пентинил, 4-метил-2-пентинил, 1,1-диметил-2-бутинил, 1,1-диметил-3-бутинил, 1,2-диметил-3-бутинил, 2,2-диметил-3-бутинил, 1-этил-2-бутинил, 1-этил-3-бутинил, 2-этил-3-бутинил и 1-этил-1-метил-2-пропинил, предпочтительно 2-пропинил, 2-бутинил, 1-метил-2-пропинил и 1-метил-2-бутинил, в частности 2-пропинил; циклоалкил с 3-8 атомами углерода, как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, циклооктил, причем данные алкильные, циклоалкильные, алкенильные и алкинильные группы могут нести по 1-5 атомов галогена, в частности фтор или хлор, и/или 1 - 2 остатка из группы, включающей вышеуказанные алкил с 1-4 атомами углерода, алкоксил с 1 - 4 атомами углерода, алкилтиогруппу с 1 - 4 атомами углерода и галоидалкоксил с 1 - 4 атомами углерода, алкенилоксигруппу с 3-6 атомами углерода, алкенилтиогруппу с 3-6 атомами углерода, алкинилоксигруппу с 3-6 атомами углерода, алкинилтиогруппу с 3 - 6 атомами углерода, причем содержащиеся в этих остатках алкенильные и алкинильные части предпочтительно имеют вышеуказанные значения: алкилкарбонил с 1 - 4 атомами углерода в алкильной части, в частности метилкарбонил, этилкарбонил, пропилкарбонил, 1-метилэтилкарбонил, бутилкарбонил, 1-метилпропилкарбонил, 2-метилпропилкарбонил, 1,1- диметилэтилкарбонил; алкоксикарбонил с 1-4 атомами углерода в алкоксильной части, как метоксикарбонил, этоксикарбонил, пропилоксикарбонил, 1-метилэтоксикарбонил, бутилоксикарбонил, 1-метилпропилоксикарбонил, 2-метилпропилоксикарбонил, 1,1-диметилэтоксикарбонил; алкенилкарбонил с 3-6 атомами углерода в алкенильной части, алкинилкарбонил с 3-6 атомами углерода в алкинильной части, алкенилоксикарбонил с 3-6 атомами углерода в алкенильной части и алкинилоксикарбонил с 3-6 атомами углерода в алкинильной части, причем алкенильные или алкинильные части предпочтительно имеют вышеуказанное значение;
фенил, который незамещен или замещен одно- или многократно, например, одно-трехкратно галогеном, нитрогруппой, цианогруппой, алкилом с 1 - 4 атомами углерода, галоидалкилом с 1 - 4 атомами углерода, алкоксилом с 1-4 атомами углерода, галоидалкоксилом с 1-4 атомами углерода или алкилтиогруппой с 1 - 4 атомами углерода, как, например, 2-фторфенил, 3-хлорфенил, 4-бромфенил, 2-метилфенил, 3-нитрофенил, 4-цианофенил, 2-трифторметилфенил, 3-метоксифенил, 4-трифторэтоксифенил, 2-метилтиофенил, 2, 4-дихлорфенил, 2-метокси-3-метилфенил, 2,4-диметоксифенил, 2-нитро-5-цианофенил, 2,6-дифторфенил; диалкиламиногруппу с 1 - 4 атомами углерода в каждой алкильной части, в частности диметиламиногруппу, дипропиламиногруппу, N-пропил-N-метиламиногруппу, N-пропил-N-этиламиногруппу, диизопропиламиногруппу, N-изопропил-N-метиламиногруппу, N-изопропил-N-этиламиногруппу, N-изопропил-N-пропиламиногруппу;
R6 и R7 означают фенил, который может быть замещен одним или несколькими, например, 1 - 3 остатками из группы, включающей галоген, нитрогруппу, цианогруппу, алкил с 1 - 4 атомами углерода, галоидалкил с 1 - 4 атомами углерода, алкоксил с 1 - 4 атомами углерода, галоидалкоксил с 1 - 4 атомами углерода или алкилтиогруппу с 1 - 4 атомами углерода, имеющие, в частности, вышеуказанное значение;
R6 и R7 вместе образуют алкиленовую цепь с 4 - 7 атомами углерода, которая представляет собой кольцо и, при необходимости, замещена, например, алкилом с 1 - 4 атомами углерода, и которая может содержать гетероатом из группы, включающей кислород, серу или азот, как, например, -(CH2)4-, -(CH2)5-, -(CH2)6-, -(CH2)7-, -(CH2)2-O-(CH2)2-, -CH2-S- (CH2)3-,
-(CH2)2-O-(CH2)3-, -NH-(CH2)3-, -CH2-NH-(CH2)2-, -CH2-CH=CH-CH2-,
-CH=CH-(CH2)3-;
г) группу формулы
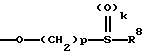
где k - 0, 1 и 2,
p - 1, 2, 3 и 4,
R8 означает алкил с 1-4 атомами углерода, галоидалкил с 1 - 4 атомами углерода, алкенил с 3-6 атомами углерода, алкинил с 3-6 атомами углерода или незамещенный или замещенный фенил, в частности, вышеуказанный фенил;
д) группу формулы OR9,
где R9 означает водород, катион щелочного металла, такого как литий, натрий, калий, или катион щелочноземельного металла, такого как кальций, магний и барий, или экологически безвредный органический ион аммония, как третичный алкиламмоний с 1-4 атомами углерода, или ион аммония; вышеуказанный циклоалкил с 3-8 атомами углерода, который может нести 1 - 3 алкильные группы с 1 - 4 атомами углерода;
алкил с 1 - 8 атомами углерода, в частности метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1, 2-диметилпропил, 1,1-диметилпропил, 2,2-диметилпропил, 1-этилпропил, гексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 1, 1-диметилбутил, 2,2-диметилбутил, 3,3-диметилбутил, 1,1,2-триметилпропил, 1,2, 2-триметилпропил, 1-этил-бутил, 2-этилбутил, 1-этил-2-метилпропил, который может нести 1 - 5 атомов галогена, в частности фтор и хлор, и/или один из следующих остатков:
алкоксил с 1 - 4 атомами углерода, алкилтиогруппу с 1 - 4 атомами углерода, цианогруппу, алкилкарбонил с 1-4 атомами углерода в алкильной части, циклоалкил с 3-8 атомами углерода, алкоксикарбонил с 1 - 4 атомами углерода, фенил, феноксил или фенилкарбонил, причем ароматические остатки в свою очередь могут нести по 1-5 атомов галогена и/или 1 - 3 остатка из группы, включающей нитрогруппу, цианогруппу, алкил с 1-4 атомами углерода, галоидалкил с 1-4 атомами углерода, алкоксил с 1 - 4 атомами углерода, галоидалкоксил с 1 - 4 атомами углерода и/или алкилтиогруппу с 1-4 атомами углерода, имеющие, в частности, вышеуказанное значение;
вышеуказанный алкил с 1-8 атомами углерода, который может нести 1 - 5 атомов галогена, в частности фтор и/или хлор, и остаток из группы, включающей 5-членный гетероаромат, содержащий 1 - 3 атома азота, или 5-членный гетероаромат, содержащий атом азота и атом кислорода или серы, который может содержать 1 - 4 атома галогена и/или 1 - 2 остатка из группы, включающей нитрогруппу, цианогруппу, алкил с 1-4 атомами углерода, галоидалкил с 1 - 4 атомами углерода, алкоксил с 1 - 4 атомами углерода, фенил галоидалкоксил с 1-4 атомами углерода и/или алкилтиогруппу с 1-4 атомами углерода. В частности можно назвать 1-пиразолил, 3-метил-1-пиразолил, 4-метил-1-пиразолил, 3, 5-диметил-1-пиразолил, 3-фенил-1-пиразолил, 4-фенил-1-пиразолил, 4-хлор-1-пиразолил, 4-бром-1-пиразолил, 1- имидазолил, 1-бензимидазолил, 1,2,4-триазол-1-ил, 3-метил-1,2,4-триазол-1-ил, 5-метил-1,2, 4-триазол-1-ил, 1- бензтриазолил, 3-изопропилизоксазол-5-ил, 3-метилизоксазол-5-ил, оксазол-2-ил, тиазол-2-ил, имидазол-2-ил, 3-этилизоксазол-5-ил, 3-фенилизоксазол-5-ил, 3-трет.-бутилизоксазол-5-ил;
алкил с 2-6 атомами углерода, который в положении 2 несет остаток из группы, включающей алкоксииминогруппу с 1 - 4 атомами углерода, алкинилоксииминогруппу с 3-6 атомами углерода, галоидалкенилоксииминогруппу с 3-6 атомами углерода или бензилоксииминогруппу;
алкенил с 3-6 атомами углерода или алкинил с 3-6 атомами углерода, причем данные группы в свою очередь могут нести 1 - 5 атомов галогена;
R9 означает дальше фенил, который может нести 1 - 5 атомов галогена и/или 1 - 3 остатка из группы, включающей нитрогруппу, цианогруппу, алкил с 1 - 4 атомами углерода, галоидалкил с 1 - 4 атомами углерода, алкоксил с 1 - 4 атомами углерода, галоидалкоксил с 1-4 атомами углерода и/или алкилтиогруппу с 1-4 атомами углерода, которые имеют, в частности, вышеуказанное значение;
связанный через атом азота 5-членный гетероаромат, содержащий 1 - 3 атома азота, который может нести 1 - 2 атома галогена и/или 1 - 2 остатка из группы, включающей алкил с 1 - 4 атомами углерода, галоидалкил с 1-4 атомами углерода, алкоксил с 1 - 4 атомами углерода, фенил, галоидалкоксил с 1 - 4 атомами углерода и/или алкилтиогруппу с 1 - 4 атомами углерода. В частности можно назвать 1-пиразолил, 3-метил-1-пиразолил, 4-метил-1-пиразолил, 3,5-диметил-1-пиразолил, 3-фенил-1-пиразолил, 4-фенил-1-пиразолил, 4-хлор-1-пиразолил, 4-бром-1-пиразолил, 1-имидазолил, 1-бензимидазолил, 1,2,4-триазол-1-ил, 3-метил-1,2,4-триазол-1-ил, 5-метил-1,2,4-триазол-1-ил, 1-бензтриазолил, 3,4-дихлоримидазол-1-ил;
R9 означает дальше остаток формулы
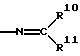
где R19 и R11 могут быть одинаковыми или различными и означают алкил с 1-8 атомами углерода, алкенил с 3-6 атомами углерода, алкинил с 3-6 атомами углерода, циклоалкил с 3-8 атомами углерода, причем данные остатки могут нести алкоксил с 1 - 4 атомами углерода, алкилтиогруппу с 1 - 4 атомами углерода и/или незамещенный или замещенный фенил, которые имеют, в частности, вышеуказанное значение;
фенил, который может быть замещен одним или несколькими, например, 1 - 3 остатками из группы, включающей галоген, нитрогруппу, цианогруппу, алкил с 1-4 атомами углерода, галоидалкил с 1-4 атомами углерода, алкоксил с 1-4 атомами углерода, галоидалкоксил с 1-4 атомами углерода или алкилтиогруппу с 1 - 4 атомами углерода, причем данные остатки имеют, в частности, значение выше указанных остатков;
или R10 и R11 вместе образуют алкиленовую цепь с 3-12 атомами углерода, которая может нести 1 - 3 алкильные группы с 1 - 4 атомами углерода и гетероатом из группы, включающей кислород, серу и азот, и имеет, в частности, указанное для заместителей R6 и R7 значение;
е) группу формулы
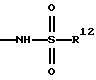
где R12 означает алкил с 1-4 атомами углерода, алкенил с 3-6 атомами углерода, алкинил с 3-6 атомами углерода, циклоалкил с 3- 8 атомами углерода, имеющие, в частности, вышеуказанное значение, причем данные остатки могут нести вышеуказанные алкоксил с 1-4 атомами углерода, алкилтиогруппу с 1-4 атомами углерода и/или фенил; незамещенный или замещенный фенил, имеющий, в частности, вышеуказанное значение;
ж) группу формулы
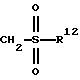
где R12 имеет вышеуказанное значение.
В отношении
биологической активности предпочитают производные карбоновой кислоты общей формулы I как в виде чистых энантиомеров или чистых диастереомеров, так и в виде их
смеси, в которых заместители имеют
следующее значение:
R1 - карбоксил или гидролизуемый в карбоксил остаток;
R2 и R3 могут быть одинаковыми или
различными и означают фенил, который
может быть необязательно замещен одним или несколькими остатками из группы, включающей метил, этил, пропил, изопропил, метокси, этокси, пропокси, изопропокси,
R4 - фенил, который
может быть замещен одним или несколькими остатками из группы, включающей метил, этил, пропил, изопропил, метокси, этокси, пропокси, изопропокси, бутилокси;
метилендиоксифенил, который может быть
замещен метокси, этокси, пропокси, изопропокси или бутилокси,
R5 метил, этил, пропил, изопропил, бутил, 2-метилпропил, трет.-бутил, пентил,
3-метилбутил, гексил, пент-3-ил,
4-метилпентил, 2-этилбутил.
Соединения настоящего изобретения, которые относятся к категории малотоксичных веществ, предлагают новые возможности для лечения гипертонии, легочной гипертонии, инфаркта миокарда, стенокардии, почечной недостаточности, острой почечной недостаточности, вазоспазмов мозга, ишемии мозга, субарахноидальных кровоизлияний, мигрени, астмы, атеросклероза, эндотоксического шока, вызванной эндотоксином недостаточности органов, внутрисосудистой коагуляции, рестеноза после ангиопластии, доброкачественной гипеплазии простаты, почечной недостаточности или гипертониии в результате ишемии и интоксикации.
Активность соединений показывается на следующих опытах.
Изучение связывания рецептора
Для изучения связывания
применяют клонированные клетки яичников китайского хомячка (CHO), экспримирующие ЕТA-рецептор, и мембраны мозжечка морской свинки с > 60% ЕТB
-рецепторов по сравнению с
ЕТA-рецепторами.
Получение препарата мембран
Клетки CHO, экспримирующие ЕТA-рецептор, культивируют в среде F12 с
10% телячьей эмбриональной
сывороткой, 1% глутамином, 100 Е/мл пенициллина и 0,2% стрептомицином (Гибко БРЛ, Гайтерсбург, МД, США). Спустя 48 ч клетки промывают забуференным фосфатом физиологическим
раствором (3ФР) и инкубируют
5 мин в 3ФР, содержащем 0,05% трипсина. После этого нейтрализуют средой Fi12 и клетки центрифугируют при ускорении 300 g. Для лизиса клеток осадок быстро
промывают буфером (5 мМ Трис-HCL,
pH=7,4 с 10% глицерином) и после этого инкубируют при концентрации клеток 10-7/мл лизисного буфера в течение 30 мин при 4oC. Мембраны
центрифугируют при ускорении 20 000 g в
течение 10 мин и осадок помещают в жидкий азот. Мозжечки морских свинок гомогенизируют в гомогенизаторе Поттера-Эльвейхема и препарат выделяют с помощью
дифференциального центрифугирования в течение
10 мин при ускорении 1000 g и повторного центрифугирования надосадочной жидкости в течение 10 мин при ускорении 20 000 g.
Испытание на
связывание
Для испытания на связывание
ЕТA - и ЕТB-рецепторов суспендируют мембраны в инкубационном буфере (50 мМ Трис-HCL, pH 7,4 с 5 мМ MnCL2, 40 мкг/мл
бацитрацина и 0,2% бычьего сыворотного альбумина)
при концентрации 50 мкг протеина на испытуемую загрузку и инкубируют при 25oC с 25 пМ [125J]-ET1
(испытание на ЕТA
-рецептор) или с 25 пМ [125 J]-RZ3
(испытание на ЕТB-рецептор) в присутствии или в отсутствие испытуемого вещества. Неспецифическое связывание определяют с 10-7 М
ET1. Спустя 30 мин свободный и
связанный радиолиганд разделяют фильтрацией через фильтр со стекловолокном (фирмы Ватман, Англия) в сборнике клеток (фирмы Скатрон, Лиер, Норвегия) и фильтр
промывают охлажденным льдом буфером
Трис-HCL с pH 7,4, содержащим 0,2% бычьего сывороточного альбумина. Радиоактивность собранного с фильтра остатка определяют количественно с помощью жидкостного
сцинтилляционного счетчика Пакард 2200
CА.
Функциональная система испытания in vitro для поиска антагонистов рецептора эндотелина (подтип А)
Эта система является
функциональным, базирующимся на клетках испытанием
для рецептора эндотелина. Определенные клетки, если они стимулированы эндотелином 1 (ET1), показывают повышение внутриклеточной
концентрации кальция. Это повышение может быть измерено в
интактных клетках, которые нагружены красителями с повышенной чувствительностью к кальцию.
Выделенные у крыс 1-фибробласты, у которых был определен эндогенный рецептор эндотелина (подтип А), нагружают следующим образом флюоресцентным красителем фура-2-аном.
После обработки трипсином клетки повторно суспендируют в буфере A (120 мМ NaCl, 5 мМ KCl, 1,5 мМ MgCl2, 1 мМ CaCl2, 25 мМ HEPES, 10 мМ глюкозы, pH 7,4) до плотности 2 • 106/мл и инкубируют 30 мин при 37oC в темноте с фура-2-аном (2 мкм), плуроником F-127 (0,04%) и 0,2%-ным диметилсульфоксидом. После этого клетки дважды промывают буфером A и повторно суспендируют до плотности 2 • 106/мл. Постоянно регистрируют при 30oC сигнал флюоресценции клеток с плотностью 2 • 106/мл при соотношении экстинции к эмиссии, равном 380: 510. К клеткам добавляют испытуемое соединение и после инкубирования в течение 3 мин добавляют ET1. В течение 30 мин определяют максимальное изменение флюоресценции. Ответ клеток на ET1 без предварительного добавления испытуемого соединения служит контролем и означает 100%.
Испытание ЕТ-антагонистов in vivo
Самцов крыс SD весом 250-300 г наркотизируют
амобарбиталом, вводят искусственное дыхание, ваготомизируют и извлекают спинной
мозг. Вставляют катетеры в Arteria carotis и Vena jugularis. У контрольных животных внутривенное введение 1 мкг/кг
ET1 приводит к значительному повышению кровяного давления, которое
держится длительное время. За 5 мин или 30 мин до введения ET1 испытуемым животным внутривенно вводят
исследуемое соединение (1 мл/кг). Для определения ЕТ-антагонистических свойств
сравнивают повышение давления у испытуемых животных и у контрольных животных.
Гибель мышей вследствие
внезапной остановки сердца, наведенной эндотелином-1
Принцип испытания
состоит в ингибировании вызванной эндотелином внезапной остановки сердца у мышей, вероятно обусловленной сужением
сосудов сердца, путем предварительной обработки антагонистами рецептора эндотелина.
После внутривенной инъекции 10 нмоль/кг эндотелина в объеме 5 мл/кг веса тела животные погибают через несколько
минут.
Летальную дозу эндотелина-1 определяют на небольшой группе животных. Если испытуемое соединение вводят внутривенно, то обычно спустя 5 мин следует летальная инъекция эндотелина-1 в сравнительной группе животных. При других способах введения это время предварительного введения соединения удлиняется иногда до нескольких часов.
Регистрируют степень выживания и эффективные дозы, которые защищают 50% животных в течение 24 ч и более от внезапной остановки сердца, вызванной эндотелином (ED 50).
Функциональное испытание сосудов на
антагонисты рецепторов эндотелина
На сегментах аорты кролика после
предварительного напряжения в 2 g и релаксации в течение одного часа в растворе Кребса-Хенселайта при 37oC и pH=
7,3-7,5 сначала снимают K+-контрактуру. После промывки строят
кривую доз активности эндотелина до максимального значения. Потенциальные антагонисты эндотелина наносят на другие препараты
из той же аорты за 15 мин до начального значения на кривой доз активности
эндотелина. Эффекты эндотелина рассчитывают в % K+-контрактуры. В случае активных антагонистов эндотелина
происходит сдвиг вправо кривой доз активности эндотелина.
Предлагаемые соединения могут вводиться обычным образом орально или парентерально (подкожно, внутривенно, внутримышечно, внутрибрюшинно). Можно вводить их также через носовую полость с парами или аэрозолями.
Дозировка зависит от возраста, состояния и веса пациента, а также от вида введения. Обычно дневная доза активного начала составляет от 0,5 до 50 мг/кг веса тела при оральном введении и от 0,1 до 10 мг/кг веса тела при парентеральном введении.
Новые соединения могут применяться в виде обычных галеновых твердых или жидких форм, например в виде таблеток, покрытых оболочкой таблеток, капсул, порошков, гранул, драже, свечей, растворов, мазей, кремов или аэрозолей. Эти формы изготавливают обычным способом. Активные вещества могут перерабатываться вместе с обычными галеновыми вспомогательными средствами, например связующими, наполнителями, консервантами, разрыхлителями, средствами для регулирования текучести, пластификаторами, смачивающими средствами, диспергаторами, эмульгаторами, растворителями, средствами, замедляющими выделение активного начала, антиокислителями и/или рабочими газами (см. Н. Sucker и др.: Pharmazeutische Technologie, изд. Тиме, Штуттгарт, 1991 г.).
Полученные таким образом лекарственные формы содержат активное начала обычно в количестве от 0,1 до 90 вес.%.
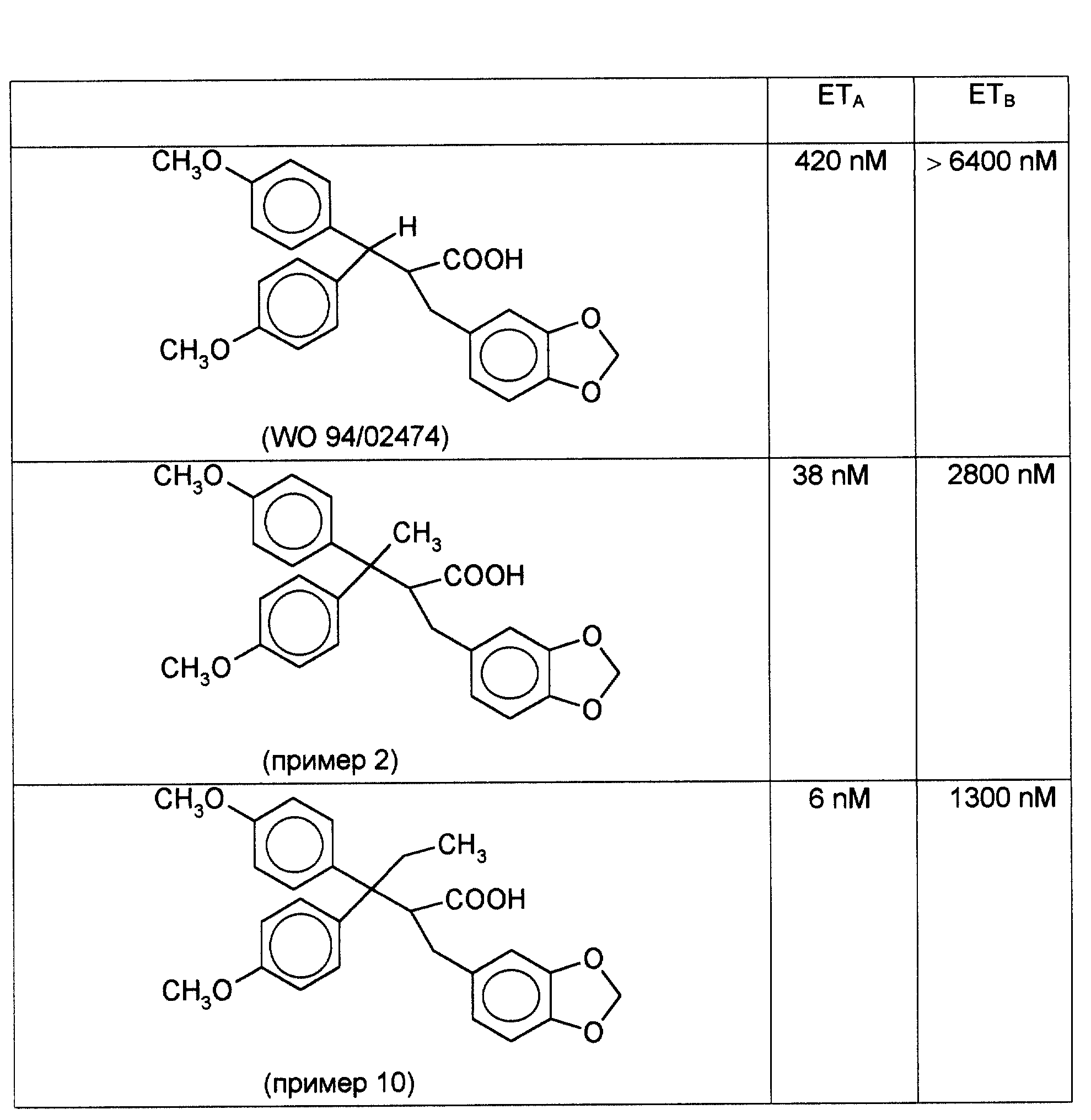
Вышеописанным образом определяли активность соединения примеров 2 и 10 и описанного в вышеуказанной заявке WO 94/02474 соединения аналогичной структуры. Результаты исследований приведены в таблице.
Соединения остальных примеров данной заявки проявляют аналогичную активность, что и соединения примеров 2, 10.
Получение соединений формулы (I) иллюстрируется следующими примерами.
Пример 1
Получение 3,3-бис(4-метоксифенил)бутановой кислоты
а) 6,6 г (30
ммоль) сложного этилового эфира (2E,Z)-3-(4-метоксифенил)бут-2-еновой
кислоты растворяют в 4,9 г (45 ммоль) анизола при 0oC, и затем в смесь осторожно подают 50 мл 80%-ной серной кислоты.
Двухфазную смесь сильно смешивают в течение 20 ч при комнатной
температуре, затем ее подают на лед и продукт экстрагируют сложным этиловым эфиром уксусной кислоты. Органическую фазу сушат над
сульфатом натрия, фильтруют, сгущают, остаток подают в простой эфир,
экстрагируют с использованием 2 н. натрового щелока и эфирную фазу удаляют. Щелочную фазу доводят до pH = 2 с использованием 2 н.
HCl и продукт экстрагируют сложным этиловым эфиром уксусной кислоты.
Затем органическую фазу сушат над сульфатом натрия, фильтруют, сгущают и твердый остаток размешивают в среде диизопропилового
эфира. Продукт отсасывают и сушат. Получают 5,1 г белого порошка (56%),
точка плавления: 161-164oC.
Маточный раствор обрабатывают дополнительно, причем получают 1,1 г (12%) кислоты.
Альтернативно кислоту можно получать следующим
образом:
б) при 0oC 32 мл (294 ммоль) анизола смешивают с 33 мл (258 ммоль) сложного этилового эфира
ацетатуксусной кислоты, в смесь осторожно подают 150 мл 70%-ной серной кислоты,
а получаемую таким образом двухфазную смесь сильно смешивают при комнатной температуре в течение 72 ч. Затем смесь
подают на лед и обрабатывают по способу, описанному под пунктом 1а). Остаток
перекристаллизуют из диизопропилового эфира. Получают 15,3 г (35%) белого твердого вещества.
в) Аналогично способу получения 3,3-дифенилбутановой кислоты (примеры 3, 4).
Пример 2
Получение (2R, S)-3,3-бис-(4-метоксифенил)-2- (3',4'-метилендиоксибензил)-бутановой кислоты
В
раствор 3,1 мл (22 ммоль) диизопропиламина в среде 50 мл сухого
тетрагидрофурана в атмосфере азота при -10oC подают 13,8 мл (22 ммоль) 1,6 М бутиллития в среде гексана, смешивают в течение
5 мин при -10oC, и затем при 0oC капают 3,
0 г (10 ммоль) 3,3-бис-(4-метоксифенил)бутановой кислоты в среде 15 мл абсолютного тетрагидрофурана. После этого смешивают при
комнатной температуре в течение часа и охлаждают до -20oC,
прибавляют 2,6 г (12 ммоль) пиперонилбромида в среде 10 мл тетрагидрофурана и затем смешивают при комнатной температуре в течение
72 ч. После этого смесь резко охлаждают насыщенным раствором NH4Cl, органическую фазу отделяют и водную фазу экстрагируют сложным этиловым эфиром уксусной кислоты. Объединенные
органические экстракты сушат над сульфатом натрия, фильтруют и сгущают в
ротационном испарителе. Коричневый остаток хроматографируют на силикагеле с применением в качестве элюента смеси метанола и
дихлорметана в соотношении, равном 1:19, причем получают 1,3 г (30%)
продукта в виде белой пены, точка плавления: 137-140oC (из диизопропилового эфира).
Пример 3
Получение сложного этилового эфира 3,3-дифенилбутановой кислоты
При 0oC 65 г (487 ммоль) AlCl3 суспендируют в 500 мл бензола и в смесь медленно подают 61,7 г сложного
этилового эфира (2E,Z)-3-фенил-бут-2-еновой кислоты. Темно-красный раствор
смешивают при комнатной температуре в течение 20 часов и затем подают на смесь льда и концентрированной соляной кислоты.
Органическую фазу отделяют, водную фазу экстрагируют сложным этиловым эфиром
уксусной кислоты. Объединенные органические фазы экстрагируют натровым щелоком, сушат над сульфатом натрия, фильтруют и
сгущают. Получают 66,8 г темно-коричнего масла. 56,5 г этого масла подвергают
перегонке, причем получают 46,3 г продукта в виде бесцветного масла.
Пример 4
Получение 3,
3-дифенилбутановой кислоты
В среде 30 мл диоксана растворяют 4,9 г (18,3
ммоль) сложного этилового эфира 3,3-дифенилбутановой кислоты, в смесь подают 36 мл 1 М гидроокиси калия и смешивают
при 60 - 70oC в течение 6 ч. Затем диоксан удаляют в ротационном
испарителе, водный остаток разбавляют водой и экстрагируют диэтиловым эфиром. Затем водную фазу доводят до pH 1 и
экстрагируют сложным этиловым эфиром уксусной кислоты. Органическую фазу сушат над
сульфатом натрия, фильтруют и сгущают. Твердый остаток размешивают в среде гептана, и получают 2,35 г белого порошка
(55%). Маточный раствор не подвергают дополнительной очистке.
Пример 5
Получение (2R,S)-3,3-дифенил-2-(3',4'-метилендиоксибензил)бутановой кислоты
В раствор 2,4 г
(10 ммоль) 3,3-дифенилбутановой кислоты в среде 40 мл абсолютного
тетрагидрофурана при -20oC капают 15 мл (24 ммоль) 1,6 М бутиллития в среде гексана, и затем смешивают при температуре от
-10 до - 20oC в течение часа. Затем прибавляют 2,2 г (13
ммоль) пиперонилхлорида в среде 10 мл тетрагидрофурана, смешивают при комнатной температуре в течение 16 ч и резко охлаждают с
использованием насыщенного раствора NH4Cl. Органическую фазу
отделяют, водную фазу экстрагируют сложным этиловым эфиром уксусной кислоты, объединенные органические экстракты сушат над
сульфатом натрия, фильтруют и сгущают. Остаток хроматографируют на силикагеле
с применением в качестве элюента смеси дихлорметана и метанола в соотношении, равном 19:1, причем получают 2,4 г
желаемого продукта (65%).
Кислоту растворяют в среде дихлорметана и встряхивают вместе с насыщенным раствором карбоната натрия. Органическую (!) фазу отделяют, сушат над сульфатом натрия, фильтруют и сгущают. Получают 2,5 г соли натрия с кислотой, точка плавления: 308-310oC (разл.).
Пример 6
Получение 3,
3-бис-(4-метокси-3-метилфенил)бутановой кислоты
Получение осуществляется аналогично примеру 16, с той разницей, что
главным образом выделяют соответствующий сложный этиловый эфир, так что
необходимо дополнительное омылование (аналогично примеру 4), точка плавления: 121-124oC.
Пример 7
Получение (2R, S)-3,3-бис-(4-метокси-3-метилфенил)-2- (3',
4'-метилендиоксибензил)бутановой кислоты
Получение осуществляется аналогично примеру 2. Из 3,25 мл (23 ммоль) диизопропиламина,
15,6 мл (23 ммоль) 1,5 М бутиллития в среде гексана, 3,28 г
(10 ммоль) 3,3-бис-(4-метокси-3-метилфенил)бутановой кислоты, 2,19 г (13 ммоль) пиперонилхлорида получают 4,1 г сырого продукта.
Хроматографией на силикагеле с применением в качестве элюента смеси
дихлорметана и метанола в соотношении, равном 19: 1, получают 1,6 г продукта (35%), точка плавления: 152-153oC.
Пример 8
Получение (2R,S)-3,3-дифенил-2-(3',
4'-диметоксибензил)бутановой кислоты
Получение осуществляется аналогично примеру 5. Из 2,4 г (10 ммоль) 3,3-дифенилбутановой
кислоты, 15,6 мл (23 ммоль) 1,5 М бутиллития в среде гексана, 2,2
г (13 ммоль) 3,4-диметоксибензилхлорида получают 3,8 г сырого продукта. Очищают на силикагеле с применением в качестве элюента смеси
гептана и сложного этилового эфира уксусной кислоты в соотношении,
равном 1:1. Получают 2,1 г продукта (54%), точка плавления: 141-143oC.
Пример 9
Получение 3,
3-бис-(4-метоксифенил)пентановой кислоты 7,0 г (30 ммоль) сложного
этилового эфира (2E, Z)-3-(4- метоксифенил)пент-2-еновой кислоты растворяют при 0oC в среде 4,9 г (45 ммоль) анизола и в
смесь осторожно подают 50 мл 80%-ной серной кислоты. Двухфазную
смесь сильно смешивают при комнатной температуре в течение 30 ч, затем ее подают на лед и продукт экстрагируют дихлорметаном.
Органическую фазу сушат над сульфатом натрия, фильтруют, сгущают, остаток
подают в простой эфир, экстрагируют с использованием 2 н. натрового щелока и эфирную фазу удаляют. Щелочную фазу доводят до pH
2 с использованием 2 н. HCl и продукт экстрагируют сложным этиловым
эфиром уксусной кислоты. Органическую фазу сушат над сульфатом натрия, фильтруют, сгущают и твердый остаток размещают в среде
гептана. Продукт отсасывают и сушат. Получают 6,8 г белого порошка (72%),
точка плавления: 136-139oC.
Пример 10
Получение (2R, S)-3,3-бис-(4-метоксифенил)-2- (3',
4'-диметилендиоксибензил)-пентановой кислоты
В раствор 6,2 г (20
ммоль) 3,3-бис-(4-метоксифенил)пентановой кислоты в среде 100 мл абсолютного тетрагидрофурана при -20oC капают 29
мл (46 мммоль) 1,6 М бутиллития в среде гексана и затем смешивают при
комнатной температуре в течение часа. После этого в смесь при -10oC прибавляют 4,4 г (24 ммоль) пиперонилхлорида в
среде 10 мл тетрагидрофурана, смешивают при комнатной температуре в
течение 72 ч и смесь резко охлаждают с использованием насыщенного раствора NH4Cl. Органическую фазу отделяют, водную фазу
экстрагируют сложным этиловым эфиром уксусной кислоты,
объединенные органические экстракты сушат над сульфатом натрия, фильтруют и сгущают. 11,2 г остатка хроматографируют на силикагеле с применением
в качестве элюента смеси дихлорметана и метанола в
соотношении, равном 24:1, причем получают 3,1 г желаемого продукта (34%) (размещенного в среде гептана), точка плавления: 84-86oC.
Пример 11
Получение сложного
метилового эфира 3,3-бис-(4-метоксифенил)гексановой кислоты 6,6 г (61 ммоль) анизола растворяют в среде 200 мл дихлорэтана, при 0oC порциями
прибавляют 12,3 г (92 ммоль) трихлорида
алюминия и затем при смешивании капают 18 г (61 ммоль) сложного метилового эфира (2E,Z)-3-(4-метоксифенил)гекс-2-еновой кислоты. Реакционную смесь смешивают при
5oC в течение 2 ч и затем
при комнатной температуре в течение 2 суток. Для обработки смесь подают на ледяную воду, экстрагируют дихлорметаном, объединенные органические фазы промывают
насыщенным раствором хлорида натрия и
сушат над сульфатом магния. Оставшийся после сгущения остаток очищают хроматографией на силикагеле с применением в качестве элюента н-гептана в виде смеси с 7,5%
сложного этилового эфира уксусной
кислоты. Таким образом получают 5,2 г (25%) бесцветного масла.
1H-ЯМР (CDCl3), δ: 0,9 (м, 3H), 1,1 и 2,2 (м, 2H), 3,08 (с, 2H), 3,4 (с, 3H) (с, 6H), 6,8 и 7,1 (м, 4H) м.д.
Пример 12
Получение 3,3-бис-(4-метоксифенил)гексановой кислоты 5,2 г (15,2 ммол) сложного метилового эфира 3,
3-бис-(4-метоксифенил)гексановой кислоты подают
в 20 мл диоксана, прибавляют 1,05 г (18,2 ммоль) гидроокиси калия и кипятят примерно час. Затем смесь разбавляют водой, промывают сложным этиловым
эфиром уксусной кислоты, водную фазу доводят до pH 3
с использованием разбавленной соляной кислоты и экстрагируют сложным этиловым эфиром уксусной кислоты. Органическую фазу промывают насыщенным
раствором хлорида натрия, сушат над сульфатом магния и
сгущают. Хроматографией на силикагеле с применением в качестве элюента дихлорметана в виде смеси с 3% метанола получают 4,1 г тонкожелтоватого
масла (84%).
1H-ЯМР (CDCl3), δ: 0,9 (м, 3H), 1,1 и 2,2 (м, 2H), 3,1 (с, 3H), 3,8 (с, 6H); 6,8 и 7,1 (м, 4H) м.д.
Пример 13
Следующие соединения получают аналогично примеру 5.
(2R, S)-3,3-дифенил-2-(метилнафт-2'-ил)бутановая кислота, точка плавления: 163-166oC, масс-спектроскопия (бомбардировка
быстрыми атомами) (далее: бба/мс): 380 (М+),
(2R,S)-3,3-дифенил-2-(3',5'-диметилбензил)бутановая кислота, точка плавления: 141-143oC, бба/мс: (М+),
(2R,S)-3,
3-дифенил-2-(4'-бензилокси-3'- метоксибензил)бутановая кислота, точка плавления: 163-166oC, бба/мс: 466 (М+),
(2R, S)-3,
3-бис-(4-метоксифенил)-2-(4'-бензилокси-3'-метоксибензил)бутановая кислота, точка плавления: 137-140oC, бба/мс: 526 (М+),
(2R, S)-3,
3-дифенил-2-(4'-гидрокси-3'-метоксибензил)бутановая кислота, точка плавления: 153-155oC, бба/мс: 376 (М+),
(2R, S)-3,
3-бис-(4-метоксифенил)-2-(4'-гидрокси-3- метоксибензил)-бутановая кислота, точка плавления: 157-160oC, бба/мс: 436 (М+),
(2R,S)-3,3-бис-(4-метокси-3-метилфенил)-2-(3',
5'- диметилбензил)бутановая кислота, точка плавления: 150-152oC, бба/мс: 446 (М+),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(метилнафт-2'-ил)бутановая кислота, точка
плавления: 162-164oC, бба/мс: 440 (М+),
(2R,S)-3,3-бис-(4-метоксифенил)-2-(3',5'- диметилбензил)бутановая кислота, точка плавления: 125-128oC, бба/мс: 418
(М+),
(2R, S)-3,3-бис-(4-метокси-3-метилфенил)-2-(3', 4'- диметоксибензил)бутановая кислота, точка плавления: 155-157oC, бба/мс: 478 (М+),
(2R, S)-3,
3-бис-(4-метоксифенил)-2-(3',4'- диметоксибензил)бутановая кислота, точка плавления: 148-150oC, бба/мс: 450 (М+),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(5'-метокси-3',
4'- метилендиоксибензил)пентановая кислота, точка плавления: 138-141oC, бба/мс: 478 (М+),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(5'-метокси-3',
4'- метилендиоксибензил)бутановая кислота, точка плавления: 134-136oC, бба/мс: 464 (М+),
(2R, S)-3,3-дифенил-2-(5'-метокси-3', 4'- метилендиоксибензил)бутановая кислота,
точка плавления: 135-138oC, бба/мс: 464 (М+),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(3', 4'-этилендиоксибензил)пентановая кислота, точка плавления: 168-170oC,
бба/мс: 462 (М+),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(3', 4'-этилендиоксибензил)бутановая кислота, точка плавления: 161-163oC, бба/мс: 448 (М+),
(2R,
S)-3,3-бис-(4-метоксифенил)-2-(3',4'-метилендиоксибензил)гексановая кислота, точка плавления: 142-145oC (из н-гептана),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(3',
4'-этилендиоксибензил)гексановая кислота, точка плавления: 163-165oC (из смеси н-гептана и диэтилового эфира),
(2R, S)-3,3-бис-(4-метоксифенил)-2-(3',
4'-метилендиокси-5'-метоксибензил)гексановая кислота, точка плавления: 180-182oC (из смеси н-гептана и диэтилового эфира).
Реферат
Изобретение относится к новым производным карбоновой кислоты формулы (I), где R1 - карбоксил или гидролизируемый в группу СООН остаток; R2 и R3 могут быть одинаковыми или различными и означать фенил, который может быть необязательно замещен одним или несколькими остатками из группы, включающей алкил с 1-4 атомами углерода и алкоксил с 1-4 атомами углерода, которые связаны в орто-положении через непосредственную связь, метилен, этилен; R4 - фенил, который может быть замещен одним или несколькими остатками из группы, включающей алкил с 1-4 атомами углерода, гидроксил, алкоксил с 1-4 атомами углерода, бензилоксигруппу; или нафтил, метилендилксифенил, который может быть замещен алкоксилом с 1-4 атомами углерода, этилендиоксифенил; R5 алкил с 1-8 атомами углерода. Технический результат - получение новых производных карбоновой кислоты. 1 табл.
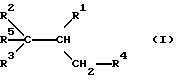
Формула
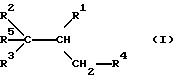
где R1 - карбоксил или гидролизуемый в группу COOH остаток;
R2 и R3 могут быть одинаковыми или различными и означают фенил, который может быть необязательно замещен одним или несколькими остатками из группы, включающей алкил с 1 - 4 атомами углерода и алкоксил с 1 - 4 атомами углерода, которые связаны в орто-положении через непосредственную связь, метилен, этилен или
R4 - фенил, который может быть замещен одним или несколькими остатками из группы, включающей алкил с 1 - 4 атомами углерода, гидрокси, алкоксил с 1 - 4 атомами углерода, бензилоксигруппу; нафтил, метилендиоксифенил, который может быть замещен алкоксилом с 1 - 4 атомами углерода, этилендиоксифенил;
R5 - алкил с 1 - 8 атомами углерода.
Комментарии