Способ получения (нитроксиметил)фенил эфиров производных салициловой кислоты - RU2235717C2
Код документа: RU2235717C2
Описание
Настоящее изобретение относится к способу получения (нитроксиметил)фенил эфиров производных салициловой кислоты.
Из предшествующего уровня техники известно, что (нитроксиметил)фенил эфиры производных салициловой кислоты могут быть получены различными способами синтеза. В заявке на патент WO 97/16405 описана реакция ацилхлорида ацетилсалициловой кислоты с (нитроксиметил)фенолом. (Нитроксиметил)фенол получают синтезом, включающим следующие стадии:
- реакция фенола с НВr в органическом растворителе с получением (бромметил)фенола и
- реакция (бромметил)фенола в органическом растворителе с AgNO3 с образованием (нитроксиметил)фенола.
Способ, основанный на реакции между (нитроксиметил)фенолом и ацилхлоридом ацетилсалициловой кислоты имеет следующие недостатки:
- (бромметил)фенол, полученный на первой стадии синтеза, является химически нестабильным и раздражающим соединением;
- нитрирующий агент, использованный в реакции с (бромметил)фенолом, является очень дорогим реагентом;
- (нитроксиметил)фенол является нестабильным соединением, которое может легко разлагаться неконтролируемым путем, и оно должно быть очищено перед реакцией с хлоридом ацетилсалициловой кислоты, что дополнительно увеличивает стоимость производства и требует дополнительного оборудования для производственной установки.
Следовательно, синтез упомянутых производных посредством использования интермедиата (нитроксиметил)фенола является трудным и дорогим при проведении в промышленном масштабе.
В международной заявке РСТ/ЕР 00/00353 на имя заявителя описан способ синтеза нитроксипроизводных формулы (I) (смотри здесь далее) путем нитрования посредством AgNO3(гидроксиметил)фенил эфиров ацетилсалициловой кислоты, полученной реакцией хлорида кислоты с гидроксибензальдегидом и восстановлением альдегидной группы до первичного спирта. Этот процесс так же, как и описанный выше, использует в качестве нитрирующего агента нитрат серебра и, следовательно, не является очень выгодным с промышленной точки зрения. Кроме того, общий выход такого способа не слишком высокий.
При использовании опыта предшествующего уровня техники можно получить нитроксипроизводные салициловой кислоты формулы (I) реакцией (гидроксиметил)фенил эфира ацетилсалициловой кислоты с нитрирующими реагентами, основанными на азотной кислоте. Однако в реакционных условиях предшествующего уровня техники азотная кислота вызывает нежелательные реакции, такие как, например, нитрование ароматического субстрата (см. "Nitration: Methods and Mechanism", 1984, VCH ed., стр. 269) и окисление первичных спиртов до альдегидов (см. "Industrial and Laboratory nitration", 1976, ACS publ., стр. 156).
Следовательно, указанные способы предшествующего уровня техники также неспособны решать проблему получения в промышленном масштабе нитроксипроизводных салициловой кислоты, как определено выше.
Ощущалась потребность в получении нитроксипроизводных (гидроксиметил)фенил эфиров ацетилсалициловой кислоты способом, который был бы более дешевый, чем способы предшествующего уровня техники, в отношении и использованного нитрирующего агента и выходов, и в основном без недостатков предшествующего уровня техники.
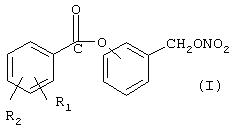
Объектом настоящего изобретения является способ получения (нитроксиметил)фенил эфиров производных салициловой кислоты, причем соединения имеют следующую формулу (I):
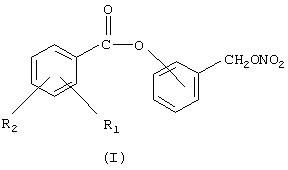
где R1 означает ОСОR3 группу, где R3 означает метил, этил или линейный или разветвленный С3 -С5 алкил, или остаток насыщенного гетероциклического кольца, имеющего 5 или 6 атомов, содержащего гетероатомы, независимо выбранные между O и N;
R2 означает водород, галоген, линейный или разветвленный, когда возможно, С1-С4алкил, линейный или разветвленный, когда возможно, C1-C4 алкоксил; линейный или разветвленный, когда возможно, C1-C4 перфторалкил, например, трифторметил; моно- или ди-(C1-C4)алкиламино;
Предпочтительно в (I) R1 является ацетокси и находится в орто-положении по отношению к карбоксильной группе, R2 означает водород; кислород сложноэфирной группы связан с ароматическим кольцом, замещенным (нитрокси)метиленовой группой, в орто-, мета- или пара-положении по отношению к (нитрокси)метиленовой группе; предпочтительно это мета-положение; упомянутый способ включает следующие стадии:
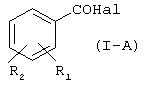
а) реакция галогенида производного салициловой кислоты формулы (I-A):
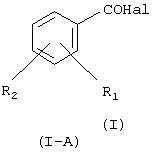
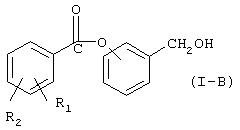
где Hal=Сl, Вr и R1 и R2 имеют вышеуказанные значения, с гидроксибензиловым спиртом в присутствии основания в органическом растворителе или в смеси воды со смешивающимся или не смешивающимся с водой органическим растворителем для получения соединения (I-B), имеющего следующую формулу (I-B):
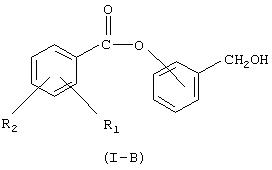
где R1 и R2 такие, как определено выше;
b) нитрование соединения (I-В) в безводных условиях в инертном органическом растворителе посредством смеси, образованной из дымящей азотной кислоты с неорганической кислотой, отличной от азотной кислоты, или с органической кислотой, или с ангидридом одной или двух органических кислот, с получением нитроксипроизводного формулы (I).
c) выделение конечного продукта путем добавления воды к органической фазе, разделения фаз, высушивания и выпаривания органической фазы.
На стадии а) основание может быть неорганическим основанием, таким как, например, гидроксиды, оксиды, карбонаты и бикарбонаты щелочных металлов (натрия, калия, лития); или органическим основанием, например, третичным амином, например, алифатическим, циклоалифатическим, гетероциклическим, гетероциклическим ароматическим, таким как триэтиламин, диизопропилэтиламин, N-метилморфолин, диазабициклооктан и т.д.
Органический растворитель, использованный на стадии а) может быть органическим растворителем, смешивающимся с водой, таким как C1 -C4 алифатические спирты, например, метанол, этанол, изопропанол, н-бутанол; или органическим растворителем, не смешивающимся с водой, например, ароматическими углеводородами, такими как толуол и ксилол, хлорированными органическими растворителями, такими как метиленхлорид, хлорбензол, другими растворителями, которые могут быть использованы, являются алифатические сложные эфиры, например, C1-C4 кислот с C1-C5 спиртами, такие как, например, этилацетат и бутилацетат и т.д.; алифатическими и циклоалифатическими кетонами, такими как C3-C12, например, ацетон, метилкетон, циклогексанон и т.д.
На стадии а) реакцию проводят при температуре в пределах от -20 до +50°С, предпочтительно 0-20°С, при использовании по отношению к молям гидроксибензилового спирта в реакции количества в молях галогенида кислоты (I-A) в соотношении между 1 и 2, предпочтительно между 1,2 и 1,5, и количества в молях основания между 0,1 и 2, предпочтительно между 0,5 и 2.
Соединение (I-В) выделяют из реакционной среды добавлением воды и необязательно, когда реакция проходит в водном растворителе или в смеси воды с водорастворимым органическим растворителем, добавлением органического растворителя, не смешивающегося с водой, такого как этилацетат или дихлорметан, фазы разделяют, органическую фазу высушивают, выпаривают и продукт выделяют. При необходимости соединение может быть очищено кристаллизацией из растворителей, таких как, например, н-гексана, н-гептана, лигроина, толуола, метанола, изопропанола, диизопропилэфира и т.п. или их смесей. Как правило, выход составляет более чем 80%.
На стадии b) реакцию нитрования проводят при температуре в пределах 20 и +40°С, предпочтительно от 0 до 20°С; использованное количество в молях азотной кислоты находится в соотношении между 1 и 6, предпочтительно 1 и 3 по отношению к молям гидроксиэфира (I-B); количество в молях органической или неорганической кислоты, отличной от азотной кислоты, или ангидрида, как определено выше, находится в соотношении между 0,5 и 6, предпочтительно между 1 и 3 по отношению к молям соединения (I-B).
Неорганическая кислота, отличная от азотной кислоты, является, например, серной кислотой; органическая кислота является, например, метансульфоновой кислотой, трифторметансульфоновой кислотой, трифторуксусной кислотой, трихлоруксусной кислотой, уксусной кислотой; ангидрид органической кислоты является, например, уксусным ангидридом, трифторметансульфоновым ангидридом, трифторуксусным ангидридом, трихлоруксусным ангидридом и т.д., или смесью ангидридов, такой как, например, трифторуксусный-трифторметансульфоновый ангидрид и т.д.
Инертный органический растворитель, использованный на стадии b), является растворителем, который имеет точку кипения ниже, чем 200°С при атмосферном давлении, и может быть хлорированным растворителем, таким как, например, дихлорметан; или нитроалканом, таким как, например, нитрометан, или алифатическим или циклоалифатическим эфиром, таким как, например, метилтретбутилэфир, тетрагидрофуран и т.д.; сложным эфиром, например, этилацетатом; или алифатическим или ароматическим нитрилом, таким как, например, ацетонитрил, бензонитрил.
Объем растворителя не является критическим, как правило, объем превышает в от 1 до 20 раз количество по массе гидроксиэфира (I-B) в реакции.
Когда нитрование на стадии b) проводят в присутствии органического ангидрида, как определено выше, ангидрид предпочтительно сначала смешивают с гидроксиэфиром (I-B) и затем получившуюся смесь добавляют к раствору азотной кислоты в инертном органическом растворителе.
Предпочтительно использованный органический ангидрид является уксусным ангидридом.
На стадии с) возможна перекристаллизация полученного соединения при использовании растворителей, таких как, например, н-гексан, н-гептан, лигроин, метанол, изопропанол или их смеси.
Следующие примеры описывают изобретение без ограничения его области.
Пример 1а
Получение 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (соединение I-B) в смеси водно-органического растворителя.
3-гидроксиметилфенол (25,25 г, 0,2 моля) растворяют в 5% растворе гидроксида натрия (160 мл). К таким образом полученному раствору добавляют раствор хлорида ацетилсалициловой кислоты (40,4 г, 0,2 моля) в дихлорметане (50 мл) при комнатной температуре при перемешивании. Смесь оставляют при комнатной температуре при перемешивании в течение 2 часов и затем экстрагируют дихлорметаном (2×100 мл). Отделяют органическую фазу, обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из смеси этилацетата и гексана. Получают 3-гидроксиметилфенил эфир 2-ацетоксибензойной кислоты (45,8 г, 0,16 молей, выход 80%).
Темп. пл.: 79-81°С.
1Н-ЯМР (CDCl3)δ(м.д.): 2,29 (с, 3Н); 4,71 (с, 2Н), 7,07-8,2 (м, ароматика, 8Н).
Пример 1b
Получение 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (соединение I-B) в органическом растворителе, не смешивающемся с водой, 3-гидроксиметилфенол (10 г, 0,08 молей) растворяют в толуоле (50 мл), содержащем триэтиламин (9,8 г, 0,1 моля). К полученному таким образом раствору добавляют раствор хлорида ацетилсалициловой кислоты (16 г, 0,08 моля) в толуоле (50 мл) при температуре 5-10°С при перемешивании. Смесь поддерживают при температуре в вышеупомянутом интервале, перемешивая в течение 2 часов, затем выливают в воду и потом экстрагируют дихлорметаном (2×100 мл). Органическую фазу отделяют, промывают последовательно 25% мас./об раствором карбоната калия, водой, 3% раствором соляной кислоты и в заключение снова водой, затем обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из изопропанола. Получают 3-гидроксиметилфенил эфир 2-ацетоксибензойной кислоты (45,8 г, 0,16 моля, выход 80%).
Темп. пл.: 79-81°С.
1Н-ЯМР (CDCl3)δ(м.д.): 2,29 (с, 3Н); 4,71 (с, 2Н), 7,07-8,2 (м, ароматика, 8Н).
Пример 1с
Получение 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (соединение I-B) в органическом растворителе, смешивающемся с водой, 3-гидроксиметилфенол (10 г, 0,08 моля) растворяют в ацетоне (50 мл). В полученный раствор суспендируют порошок карбоната калия (22,2 г, 0,16 моля). К суспензии добавляют раствор хлорида ацетилсалициловой кислоты (16 г, 0,08 моля) в ацетоне (50 мл) при температуре 5-10°С при перемешивании. Смесь поддерживают при температуре в вышеуказанном интервале при перемешивании в течение 2 часов, затем фильтруют и растворитель выпаривают под вакуумом. Остаток кристаллизуют из изопропанола. Получают 3-гидроксиметилфенил эфир 2-ацетоксибензойной кислоты (21,0 г, 0,07 моля, выход 91%).
Темп. пл.: 79-81°С.
1Н-ЯМР (CDCl3)δ(м.д.): 2,29 (с, 3Н); 4,71 (с, 2Н), 7,07-8,2 (м, ароматика, 8Н).
Пример 2
Получение 3-нитроксиметилфенил эфира 2-ацетоксибензойной кислоты нитрованием дымящей азотной кислотой в присутствии серной кислоты 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты.
Раствор дымящей азотной кислоты (3,92 г, 62,2 ммоля, 3 моля по отношению к молям гидроксиэфира I-B) и 96% серной кислоты (6,10 г, 62,2 ммоля, 3 моля по отношению к молям гидроксиэфира I-B) в дихлорметане (25 мл) охлаждают до 0°С и добавляют в течение 1 часа при перемешивании и в атмосфере азота в раствор 3-гидроксиметилфенил эфира 2 ацетоксибензойной кислоты (6 г, 20,7 моля) в 25 мл дихлорметана. Смесь затем разбавляют дихлорметаном (50 мл) и выливают в воду и лед (100 г). Органическую фазу отделяют, промывают водой, обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из изопропанола, получая 3-нитроксиметилфенил эфир 2-ацетоксибензойной кислоты (5,6 г, 17 ммолей, выход 82%).
Темп. пл.:61-62°С.
1Н-ЯМР (CDCl3)δ(м.д.): 2,31 (с, 3Н); 5,44 (с, 2Н), 7,16-8,22 (м, ароматика, 8Н).
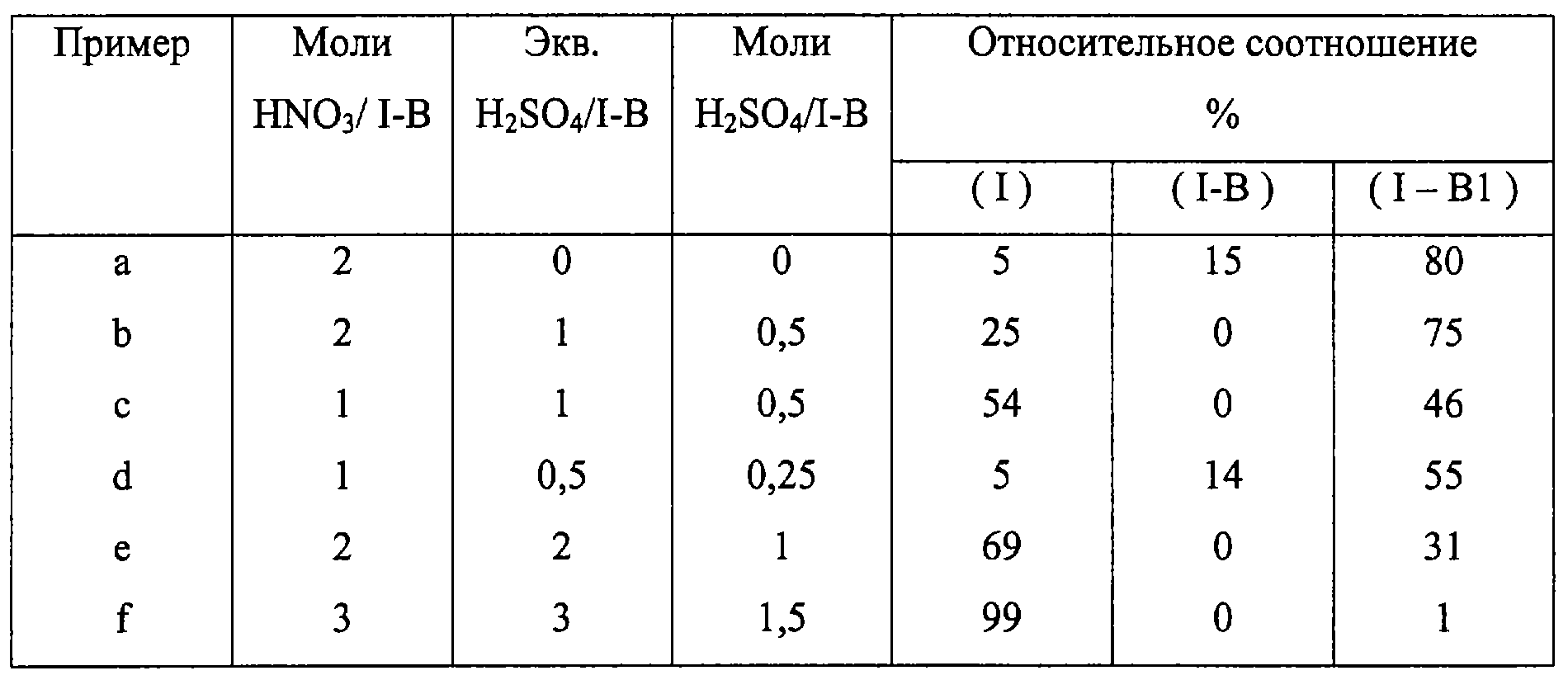
Примеры 2a-2f
Пример 2 повторяют, изменяя количество молей азотной кислоты и серной кислоты по отношению к количеству молей интермедиата 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (I-B). В таблице представлены молярные соотношения использованных реагентов по отношению к соединению I-B и относительные процентные соотношения между 3-нитроксиметилфенил эфиром 2-ацетоксибензойной кислоты (I), 3-(формил)фенил эфиром 2-ацетоксибензойной кислоты (I-B1), учитывая, когда присутствует, также исходное соединение (I-B).
Таблица показывает, что наибольший выход получают при использовании молярного соотношения азотная кислота/соединение (I-B), равного 3, и соотношения серная кислота/соединение (I-B), равного 1, 5.
Пример 3
Получение 3-нитроксиметилфенил эфира 2-ацетоксибензойной кислоты нитрованием дымящей азотной кислотой, в присутствии уксусного ангидрида, 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты.
Раствор дымящей азотной кислоты (1,44 г, 22,8 ммолей), уксусного ангидрида (2,33 г, 22,8 ммолей) в дихлорметане (25 мл) охлаждают до 0°С и при перемешивании добавляют в течение 1 часа в атмосфере азота к раствору 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (6 г, 20,7 ммолей) в 25 мл дихлорметана. Смесь нагревают до 20°С в течение часа и затем разбавляют дихлорметаном (50 мл) и выливают в воду и лед (100 г). Отделяют органическую фазу, промывают водой, обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из изопропанола и получают 3-нитроксиметилфенил эфир 2-ацетоксибензойной кислоты (5,6 г, 17 ммолей, выход 82%).
Пример 4
Получение 3-нитроксиметилфенил эфира 2-ацетоксибензойной кислоты нитрованием дымящей азотной кислотой, в присутствии уксусного ангидрида, 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (уксусный ангидрид смешан с гидроксиэфиром).
Раствор дымящей азотной кислоты (1,44 г, 22,8 ммолей) в дихлорметане (25 мл) охлаждают до 0°С и добавляют в течение 1 часа при перемешивании и в атмосфере азота в раствор 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (6 г, 20,7 ммолей) и уксусного ангидрида (2,33 г, 22,8 ммолей) в 25 мл дихлорметана. Смесь нагревают до 20°С в течение 1 часа и затем разбавляют дихлорметаном (50 мл) и выливают в воду и лед (100 г). Отделяют органическую фазу, промывают водой, обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из изопропанола и получают 3-нитроксиметилфенил эфир 2-ацетоксибензойной кислоты (6, 42 г, 19,5 ммолей, выход 94%).
Пример 5
Получение 3-нитроксиметилфенил эфира 2-ацетоксибензойной кислоты нитрованием дымящей азотной кислотой в присутствии метансульфоновой кислоты 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты.
Раствор дымящей азотной кислоты (1,44 г, 22,8 ммолей) и метансульфоновой кислоты (2,55 г, 22,8 ммолей) в дихлорметане (25 мл) охлаждают до 0°С и добавляют в течение 1 часа при перемешивании в атмосфере азота в раствор 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (6 г, 20,7 ммолей) в 25 мл дихлорметана. Смесь разбавляют дихлорметаном (50 мл) и выливают в воду и лед (100 г). Отделяют органическую фазу, промывают водой, обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из изопропанола и получают 3-нитроксиметилфенил эфир 2-ацетоксибензойной кислоты (2,73 г, 8,29 ммолей, выход 40%).
Пример 6
Получение 3-нитроксиметилфенил эфира 2-ацетоксибензойной кислоты нитрованием дымящей азотной кислотой, в присутствии уксусного ангидрида, 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (уксусный ангидрид смешан с гидроксиэфиром).
Раствор дымящей азотной кислоты (990 мг, 15,2 ммолей), уксусного ангидрида (1,55 г, 15,2 ммоля) в дихлорметане (25 мл) охлаждают до 0°С и добавляют в течение 1 часа при перемешивании в атмосфере азота в раствор 3-гидроксиметилфенил эфира 2-ацетоксибензойной кислоты (4 г, 13,8 ммолей) в 25 мл дихлорметана. Смесь нагревают до 20°С в течение 1 часа и затем разбавляют дихлорметаном (50 мл) и выливают в воду и лед (100 г). Отделяют органическую фазу, промывают водой, обезвоживают сульфатом натрия и выпаривают растворитель под вакуумом. Остаток кристаллизуют из изопропанола и получают 3-нитроксиметилфенил эфир 2-ацетоксибензойной кислоты (4,1 г, 12,28 ммолей, выход 89%).
Реферат
Изобретение относится к способу получения (нитроксиметил)фенил эфиров производных салициловой кислоты формулы (I)
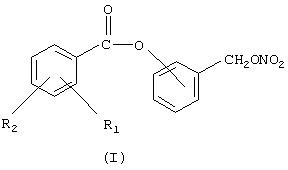
где R1 означает OCOR3 группу, где R3 означает метил, этил или линейный или разветвленный С3-С5 алкил;R2 означает водород. Способ включает следующие стадии: а) реакция галогенида производного салициловой кислоты с гидроксибензиловым спиртом в присутствии основания в органическом растворителе; b) нитрование полученного продукта в безводных условиях в инертном органическом растворителе, имеющего точку кипения ниже, чем 200°С при атмосферном давлении, смесью азотной кислоты с отличной неорганической кислотой, или органической кислотой, или ангидридом одной или двух органических кислот; с) выделение конечного продукта. Технический результат – удешевление процесса, уменьшение побочных реакций, увеличение выхода продукта. 5 з.п. ф-лы, 1 табл.
Формула
Комментарии