Днк, кодирующая фитазу aspergillus niger, рекомбинантная плазмидная днк для экспрессии фитазы (варианты), штаммы-продуценты фитазы (варианты), способ получения фитазы и рекомбинантная фитаза aspergillus niger - RU2113468C1
Код документа: RU2113468C1
Чертежи


































Описание
Изобретение относится к микробиологическому производству фитазы.
Незаменимым элементом для роста любого организма является фосфор. В животноводстве для обеспечения быстрого роста животных с простым желудком (например, свиней, рыбы, домашней птицы) корма обогащают неорганическим фосфором.
Напротив, в корма жвачных животных неорганический фосфор не добавляют, так как присутствующие в рубце микроорганизмы вырабатывают ферменты, которые катализируют превращение фитата (миоиноизитолгексакис-фосфата) в инозитол и неорганический фосфат.
Фитат представляет собой резервный источник фосфора практически во всех пищевых материалах растительного происхождения (см. обзор: фитовая кислота, химия и практическое использование (Phytie acid, chemistry and applications, E. Graf (ed.), Pilatus Press, Minneapolis, MN, USA, 1986).
Содержание фитата в орехах, зерновых, бобовых и масличных культурах, спорах и пыльце составляет 1 - 3%. Сложные соли фитовой кислоты называются фитинами. Считают, что фитовая кислота ухудшает пищеварение, поскольку она служит хелатом таких минеральных элементов, как кальций, цинк, железо и магний, и, кроме того, может вступать в реакцию с белками, уменьшая, таким образом, биологическую доступность последних и важных минеральных компонентов корма.
Содержащийся в фитате фосфор проходит через желудочно-кишечный тракт животных с однокамерным желудком и выводится с экскрементами. Хотя в толстом кишечнике и происходит частичный гидролиз фитата, освобождаемый при этом неорганический фосфор не имеет существенной питательной ценности, поскольку он может всасываться только в тонком кишечнике. В результате такие животные не используют большое количество фосфора, имеющего значительную питательную ценность, несмотря на его присутствие в корме.
Экскреция содержащегося в фитате фосфора с экскрементами также имеет известные последствия. На протяжении последних десятилетий значительно возросло поголовье сельскохозяйственных животных. Одновременно увеличилось и количество навоза, что создает угрозу чистоте окружающей среды в различных районах земного шара. Частично это обусловлено накоплением содержащегося в навозе фосфата в поверхностных водах, что в свою очередь, приводит к эутрофикации водоемов.
Ферменты, синтезируемые микроорганизмами и катализирующие превращение фитата в инозитол и неорганический фосфор, широко известны под названием фитаз. В число микроорганизмов, продуцирующих эти ферменты, входят такие бактерии, как Bacillus subtilis (V.K.Paver and V.J.Jagannathan, 1982, J. Bacteriol. , 151. 1102-1108) и Pseudomonas (D.J.Cosgrove, 1970, Austral. J. Biol. Sci., 23, 1207-1220),такие виды дрожжей, как Saccharomyces cerevisiae (N. R. Nayini and P. Markakis, 1984, Lebensmittel Wissenschft und Technologie, 17, 24-26) и такие виды грибов, как Aspergillus Terreus (K. Yamada Y. Minoda and S. Yamamoto, 1986, Aprie. Biol. Chem. 32, 1275-1282). Фитазу продуцируют и другие виды Aspergillus, среди которых Aspergillus ficuum синтезируют фермент с наиболее высокой специфической активностью и более выраженной термостабильностью по сравнению с фитазами других микроорганизмов (неопубликованные данные).
Идея добавлять бактериальную фитазу в корма животных с простым желудком высказывалась уже неоднократно (см., например, Ware J.H., Bluff L. and Shien T. R. , 1967, US Patent N 3.297.548; Nelson T.S., Shien T.R, Wodzinski R.J. and Ware J.H., 1971, J. Nutrition, 101, 1289-1294).
Однако до настоящего времени ее практическое применение представлялось экономически нерентабельным из-за высокой стоимости микробиологического производства ферментов (Y. W. Han, 1989, Animal Feed Sci & Technol., 24, 345-350). Таким образом, по причинам экономического порядка неорганический фосфор продолжают вводить в корма животных с однокамерным желудком.
Бактериальные фитазы находят и другое применение в хозяйственной деятельности. В качестве примера можно упомянуть процесс промышленного получения крахмала из зерна таких культур, как кукуруза и пшеница. Образующиеся в процессе сырого перемалывания продукты, в частности глютены, поступают на рынок в качестве кормов для сельскохозяйственных животных. В ходе вымачивания добавляют фитазу, причем технологические условия особенно благоприятны для грибных фитаз (температура порядка 50oC при pH 5,5) (см., например, заявку на Европейский патент 0321004 Alko Ltd). Важным преимуществом описанного процесса является возможность получения из отходов производства животных кормов, содержащих фосфат вместо фитата.
Предложено также использовать фитазы при переработке сои (см.Finas TM Enzymes by Alko - информационный буклет, выпущенный компанией Alko Ltd., Rajamaki, Финляндия). Соевые бобы содержат большое количество фитата - фактора, ухудшающего пищеварение, который делает белки непригодными для использования в продуктах детского питания, а также в кормах рыб, телят и нежвачных животных. Обогащение этого важного источника белков соответствующими ферментами повышает коммерческую и пищевую ценность этого материала.
Проведены исследования с целью более подробной характеристики различных фитаз и совершенствования способов их получения и практического использования. Ullah предложил метод очистки фитазы из дикого штамма Aspergillus ficuum, а также изучил ряд биохимических параметров конечного продукта такой очистки (Ullah A., 1988a, Preparative Biochem, 18, 443-438). Полученные этим автором данные приводятся в табл.1.
Аминокислотная последовательность N - концевого фрагмента фитазы A.ficuum была представлена Ullah дважды: Ullah A., 1987, Enzyme and Engineering Conference IX, October 4-8, 1987, Santa Barbara, California (poster presentation) и Ullah A., 1988b, Prep. Biochem., 18, 459-471.
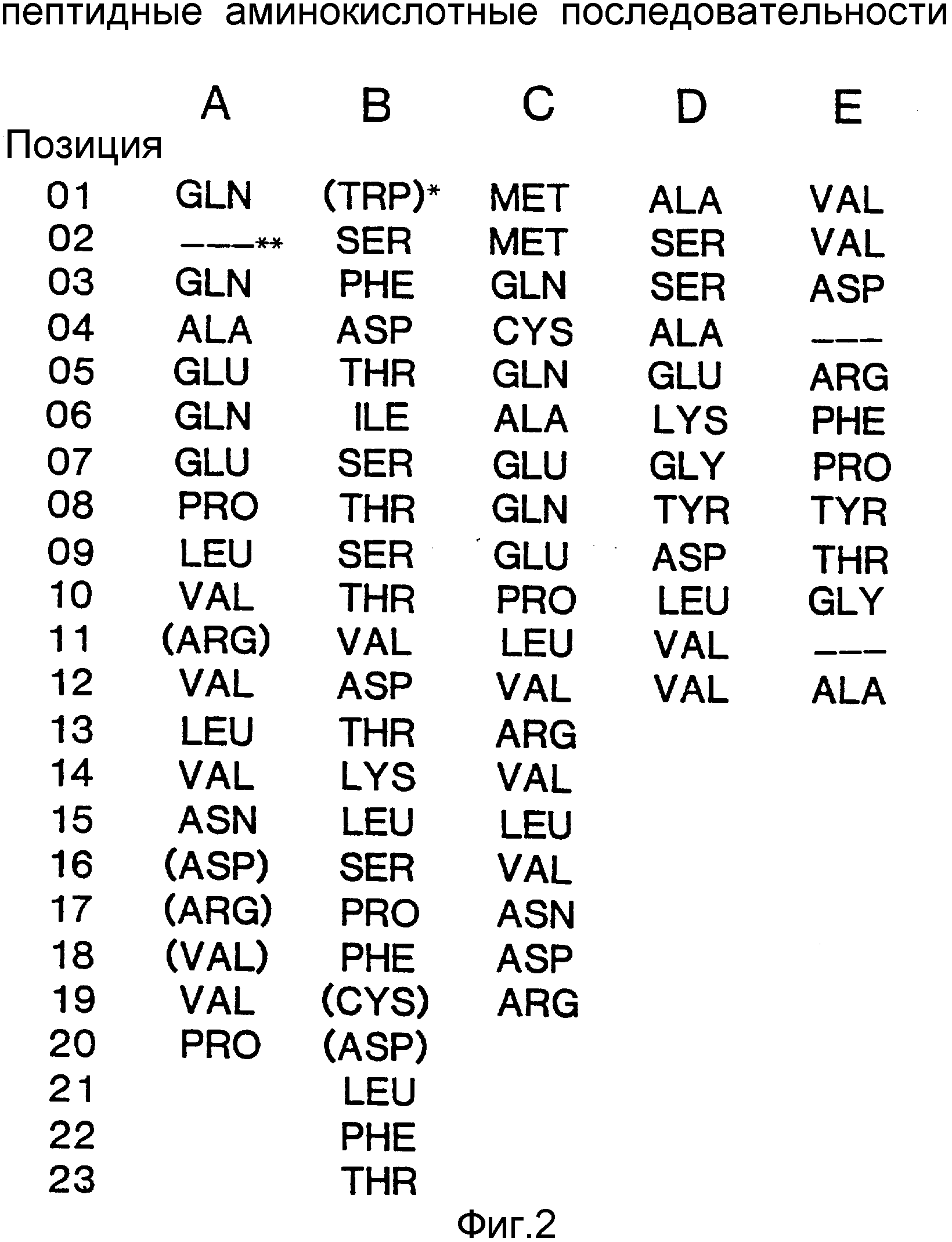
Последовательность аминокислотных остатков, расшифрованная этим автором, показана на фиг.1 (последовательность E).
Из полученных Ullah данных следуют несколько интересных выводов. Прежде всего, "очищенный" препарат, описанный им в 1988a и 1988в, при электрофорезе в полиакриламидном геле в присутствии додецил-сульфата натрия давал две белковые полосы. Нами установлено, что очищенная из A.ficuum фитаза содержит посторонний материал, который и дает при электрофорезе в полиакриламидном геле одну из полос, которую Ullah идентифицировал как фитазу.
Аналогичное заключение можно сделать на основании анализа аминокислотных последовательностей, опубликованных Ullah (1987, 1988b; сравните последовательности A и B с последовательностью С на фиг.1). Мы показали, что одна из аминокислотных последовательностей внутренних пептидов фитазы, описанных Ullah (см.рисунок 1B, последовательность E), на самом деле представляет собой посторонний белок с молекулярной массой 100 кДа (фиг.3), который присутствует в материале, получаемом по методу Ullah, и дает одну из двух полос при электрофорезе в полиакриламидном геле в присутствии додецил-сульфата натрия (Ullah, 1968a, 1968b). Ullah не признает наличия такого загрязняющего материала и считает, что указанная полоса представляет собой другую форму фитазы. Между тем этот посторонний компонент затрудняет отбор и идентификацию именно той нуклеотидной последовательности, которая кодирует биосинтез фитазы. Кроме того, его присутствие снижает удельную активность тестируемого белка.
Продолжая анализ данных Ullah, следует отметить, что аминокислотный остаток в положении 12 был идентифицирован этим автором как глицин. Используя методику секвенирования белков и ДНК, мы показали, что в этом положении на самом деле присутствует не глицин, а цистеин (см. фиг.11-17 и 19-20).
Наконец, по мнению Ullah, фитаза представляет собой белок с молекулярной массой 85 кДа, которая после гликозилирования уменьшается до 61,7 кДа (Ullah, 1988 b). Эта величина значительно ниже ранее приводившейся тем же автором - 76 кДа (Ullah A. and Gibson D., 1988, Prep. Biochem. 17 (1), 63 - 9 1) и была рассчитана на основании относительного количества углеводов, выделяющихся при гидролизе, и молекулярной массы нативного белка, которую определяли посредством электрофореза в полиакриламидном геле в присутствии додецил-сульфата натрия. Однако наши исследования показали, что гликозилированная фитаза однозначно имеет молекулярную массу 85 кДа, тогда как у дегликозилированного белка она колеблется в диапазоне от 48 до 56,5 кДа, в зависимости от степени дегликозилирования.
В сообщении Mullaney et al. (Filamentous Fungi Conference, April 1987, Pacific Grove, California (poster presentation) также приводится характеристика фитазы из A.ficuum. И в этом исследовании при электрофорезе в полиакриламидном геле с додецил-сульфатом натрия были получены две белковые полосы, одна с молекулярной массой 85 кДа, а другая с массой 100 кДа - обе с "очищенным" белковым материалом. Авторы идентифицировали обе полосы в качестве различных форм фитазы. Однако, в сообщении не описывается способ трансформации бактериальных клеток - хозяев. Описание методов клонирования и выделения последовательности ДНК, кодирующей фитазу, также отсутствует.
Очевидно, что разработка экономически рентабельного способа производства фитазы имела бы большое значение, в частности, при промышленном получении кормов для животноводства. Одним из путей достижения этой цели является использование рекомбинантной ДНК - методики, с помощью которой можно усилить экспрессию фермента в различных видах микроорганизмов, продуцирующих высоко активные пептиды и белки. Однако в настоящее время неизвестны способы выделения и клонирования последовательностей ДНК, кодирующих биосинтез фитазы.
Изобретение относится к способу получения и очистки последовательности ДНК, кодирующей фитазу. Выделение и клонирование такой последовательности осуществлялось с помощью специфических нуклеотидных зондов, которые были разработаны специально для целей настоящего изобретения. Требуемая последовательность ДНК, кодирующая фитазу, была получена из грибов, в частности из нитчатых грибов рода Aspergillus.
Другим предметом изобретения является способ получения вектора, содержащего фактор экспрессии, который в свою очередь включает по меньшей мере одну реплику по крайней мере одной (желательно гомологичной) ДНК, кодирующей фитазу. Такая последовательность ДНК функционально связана с соответствующей регуляторной областью, обеспечивающей высокий уровень экспрессии пептидов или белков с фитазной активностью в подходящих для этой цели клетках - хозяевах.
Фактор экспрессии согласно настоящему изобретению может быть введен в вектор, предпочтительно плазмиду, способный трансформировать бактериальные клетки - хозяева и внедряться в геном.
Еще один предмет настоящего изобретения - способ получения трансформированной клетки, желательно бактериальной, которая претерпевает изменения под воздействием вектора, описанного в предшествующем параграфе. Согласно изобретению такими трансформированными клетками могут служить нитчатые грибы родов Aspergillus, Trichoderma, Mucor и Penicillium дрожжи родов Kluyveromyces и Saccaromyces или бактерии рода Bacillus. Наиболее предпочтительно использование в качестве клеток - хозяев нитчатых грибов рода Aspergillus. Трансформированные клетки способны синтезировать большие количества рекомбинантной фитазы в промышленном масштабе и при достаточном уровне экономической рентабельности.
Другие аспекты изобретения относятся к рекомбинантным белкам и пептидам, которые обладают фитазной активностью как в гликозилированной, так и в негликозилированной форме; к способу получения таких негликозилированных белков и пептидов; к белкам и пептидам с фитазной активностью, свободным от посторонних примесей; к моноклональным антителам, реагирующим с этими рекомбинантными или очищенными белками.
В табл. 1 приведены сравнительные биохимические характеристики фитазы, полученной Ullah из дикого штамма A.ficuum, и дополнительно очищенной фитазы из A.ficuum дикого типа, полученной согласно настоящему изобретению. Следует обратить особое внимание на данные об удельной активности, которые свидетельствуют о том, что у полученного нами фермента она в два раза выше, чем у фитазы, описанной Ullah.
Настоящее изобретение относится также к нуклеотидной последовательности, кодирующей белки с фитазной активностью, а также к аминокислотным последовательностям таких белков. Полученные последовательности можно использовать для разработки олигонуклеотидных зондов, которые в свою очередь применяются в гибридизационных скрининговых исследованиях с целью идентификации генов фитазы из других источников, особенно из различных видов микроорганизмов, которые в последующем могут быть выделены и клонированы.
Последовательности, получаемые согласно настоящему изобретению, можно использовать в качестве исходных материалов для конструирования фитаз "второго поколения". Фитазы "второго поколения" - это фитазы, измененные под воздействием мутагенных факторов (в частности, методом направленного мутагенеза) и обладающие свойствами, которые отличаются от свойств фитаз дикого типа или рекомбинантных фитаз, например, получаемых согласно настоящему изобретению. Для оптимизации того или иного процесса изменяют такие его параметры, как температура и оптимальная величина pH, удельная активность белков или сродство субстратов.
Применительно к данному изобретению понятие фитаза охватывает семейство ферментов, которые катализируют реакции, направленные на удаление неорганического фосфора из разнообразных миоинозитолфосфатов.
Для определения активности фитазы существует много способов, выбор которых не лимитируется настоящим изобретением. В порядке иллюстрации можно отметить способ количественного определения активности фермента по его расходу на освобождение неорганического фосфора из 1,5 мМ фитата натрия со скоростью 1 мкмоль/мин при 37oC и pH 5,50.
Следует также иметь в виду, что термин "фитаза", употребляемый в тексте настоящей спецификации, относится ко всем белкам и пептидам, обладающим фитазной активностью. Это положение иллюстрируется фиг.1, на котором сопоставляются последовательности A и B (последовательности, полученные в ходе настоящего исследования) с последовательностью C, полученной Ullah (1988в). Фиг.1 показывает, что с помощью нашего метода можно получить белки, в которых отсутствуют первые 4 аминокислотные остатка нативной фитазы A.ficuum (белок с последовательностью А не имеет первых 7 аминокислотных остатков). Указанные белки сохраняют тем не менее фитазную активность. Полная аминокислотная последовательность белка фитазы, полученная на основании результатов анализа соответствующей нуклеотидной последовательности, показана на фиг.19-20.
Фитазы, получаемые по предлагаемому способу, могут использоваться в самых разнообразных процессах, требующих превращения фитата в инозитол и неорганический фосфор.
В частности, получение фитазы согласно настоящему изобретению позволяет снизить стоимость ее промышленного микробиологического производства и обеспечить экономическую рентабельность последнего при изготовлении кормов для животных, а в конечном счете добиться такого же соотношения затрат и эффективности использования кормов, как в случае применения добавок неорганического фосфата. Кроме того, значительно снизится содержание фосфора в навозе.
Получение фитаз по ценам, сопоставимым с ценами неорганического фосфата, расширит возможности промышленности кормоматериалов в смысле увеличения ассортимента высококачественных кормов. Например, обогащение кормов фитазой позволяет отказаться от добавок неорганического фосфата и повысить в них долю фитатсодержащего материала.
Помимо использования получаемой согласно настоящему изобретению фитазы в качестве добавки к кормам животных и при переработке сои (см. выше), она может
найти применение в следующих отраслях промышленности:
- производство жидких кормов для свиней и домашней птицы. В настоящее время широкое распространение получила практика замачивания кормов
за несколько часов до скармливания животным. На протяжении этого периода фитаза превращает фитат в инозитол и неорганический фосфат;
- промышленное производство инозитола или
инозитолфосфатов
из фитата;
- другие промышленные процессы с использованием фитатсодержащих субстратов, такие как производство крахмала и ферментация, включая пивоварение. Хелирование
металлических ионов
фитатом может сделать их недоступными для микроорганизмов. Ферментативный гидролиз фитата устраняет эту проблему.
Эти и другие предметы и преимущества настоящего изобретения еще более очевидны из следующего ниже подробного его описания.
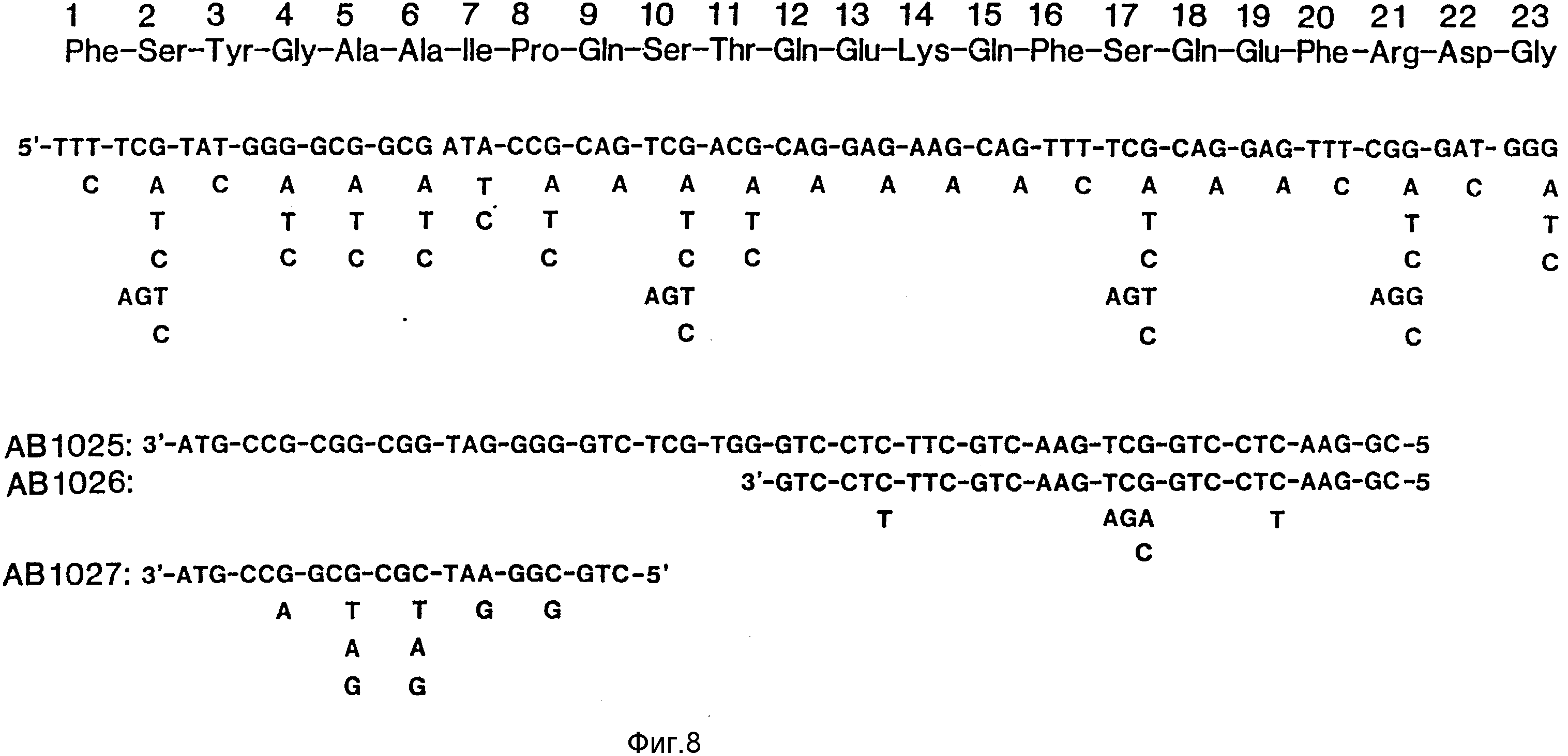
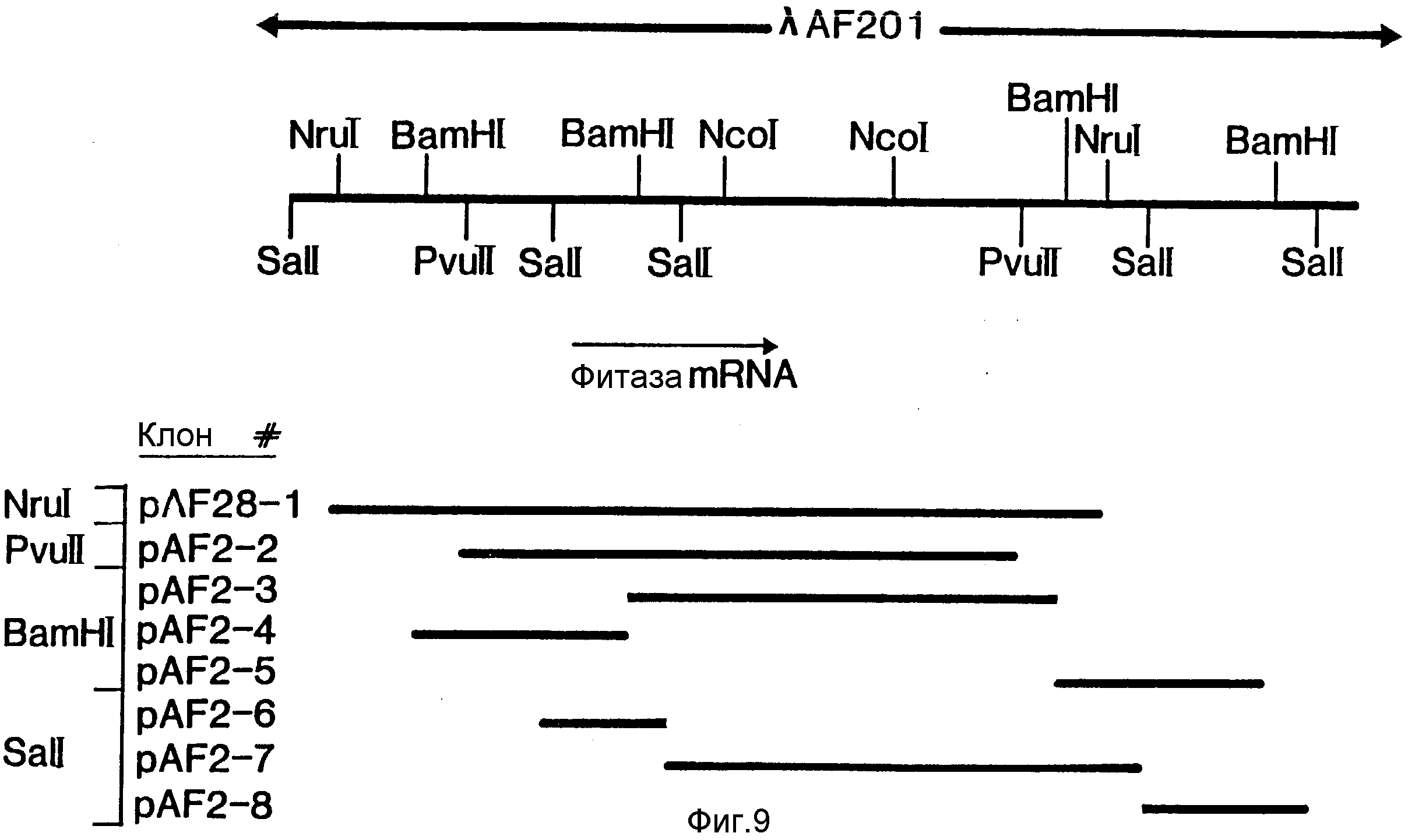
На фиг. 1 представлена N-концевая аминокислотная последовательность очищенной фитазы. Аминокислотные последовательности, обозначенные буквами A и B, получены в ходе настоящего исследования с использованием подформ фитазы с изоэлектрическими точками соответственно 5,2 и 5,4. Последовательность C приводится по публикациям Ullah (1987, 1938в, supra). Согласно нашим данным аминокислотный остаток в положении 12 последовательностей A и B не является глицином (знак * означает сомнительную идентификацию, а знак ** отсутствие определяемого остатка); на фиг.2 представлена N-концевая последовательность внутренних фрагментов фитазы после расщепления CNBr. Аминокислотные последовательности, обозначенные буквами A и B, с приблизительными молекулярными массами соответственно 2,5 и 36 кДа получены в ходе настоящего исследования. Последовательности C и E даны по Ullah (1988 b, supra); на фиг.3 - N-концевая аминокислотная последовательность белка с молекулярной массой 100 кДа, который по данным настоящего исследования присутствует в неочищенных препаратах фитазы; на фиг. 4,5 - олигонуклеотидные зонды, сконструированные на основании данных фиг.1 (пептиды A - E); на фиг.6,7 - олигонуклеотидные зонды, сконструированные на основании данных фиг.2 (пептиды A и B); на фиг.8 - олигонуклеотидные зоны, использовавшиеся для выделения гена, кодирующего кислую фосфатазу; на фиг. 9 - рестрикционная карта бактериофага лямбда AF 201, содержащего локус фитазы A.ficuum. Стрелка показывает положение гена фитазы и направление транскрипции. Клон # указывает субклоны, полученные с помощью указанных рестрикционных ферментов из фага AF201 в pAN 8-1 (для pAF 28-1) и в pUC 19 (для всех других субклонов); на фиг.10 - физическая карта pAF 1-1. Фрагмент Bam HI длиной в 10 килооснований, внедренный в pUC19, содержит полный ген, кодирующий кислую фосфатазу A.ficuum; на фиг.11-17 представлены нуклеотидные последовательности плазмид pAF 2-3, pAF 2-6 и pAF 2-7, включающие хромосомные генные локусы фитазы. Кодирующая фитазу область локализуется между нуклеотидами в положениях 210 и 1713. Интрон размещается в хромосомном гене между нуклеотидами 254 и 355. Показаны также другие существенные признаки: начальный и конечный кодоны фитазы, рестрикционные участки и положение интрона; на фиг. 18 представлена подробная физическая карта секвенированного хромосомного локуса фитазы. Стрелки показывают локализацию двух экзонов области, кодирующей фитазу; на фиг.19,20 - нуклеотидная последовательность трансляционной зоны фрагмента кДНК фитазы и выведенная из нее последовательность аминокислотных остатков в молекуле фитазы. Начало зрелой молекулы белка обозначено как положение +1. N-конец внутреннего белка с молекулярной массой 36 кДа находится в положении 241, тогда как белок с молекулярной массой 2,5 кДа начинается в положении 390; на фиг.21 - физическая карта блока экспрессии фитазы pAF 2-2S. Стрелки указывают направление транскрипции генов; на фиг.22,23 - доказательство сверхэкспрессии фитазы в трансформированных клетках A.ficuum NRRL 3135 методами изоэлектрофокусировки и электрофореза в полиакриламидном геле. Равные объемы культуральной жидкости A. ficuum (полоса ) и трансформированных клеток pAF 2-2S SP7 (полоса 2), выращивавшихся в одинаковых условиях, анализировали в системе Phast (Framacia) методами электроизофокусировки и электрофореза в полиакриламидном геле при pH от 4,5 до 6.0. Для сравнения анализировали образец фитазы a.ficuum, очищенной до гомогенного состояния, либо отдельно (полоса 4), либо в смеси с культуральной жидкостью (полоса 3). Гели окрашивали или на фосфатазу, как описано в тексте (A), или на общий белок (кумасси бриллиантовый синий, B). Полосы, соответствующие фитазе, обозначены звездочками; на фиг. 24,25 представлено доказательство сверхэкспрессии фитазы в трансформированных клетках A.niger CBS 513.88 методами изоэлектрофокусировки и электрофореза в полиакриламидном геле. Равные объемы культуральной жидкости родительского штамма A.niger (полоса 1) или трансформированных клеток pAF - 2-2S # 8 (полоса 2) pFYT3 # 205 (полоса 3) и # 282 (полоса 4) анализировали методами изоэлектрофокусировки и электрофореза в полиакриламидном геле, как описано в подписи к фиг.22,23. Гели окрашивали либо на общую фосфатазную активность (A), либо на общий белок (B). Полосы, соответствующие фитазе, обозначены звездочками; на фиг. 26 представлена физическая карта pAB 6-1. Вставка ДНК Hind III длиной 14,5 килооснований в pUC19 содержит полный локус глюкоамилазы из A.niger (AG); на фиг.27 дано схематическое изображение полимеразной цепной реакции для слияния промотора AG и гена фитазы. Последовательности всех олигонуклеотидных затравок приведены в тексте; на фиг.28 представлена физическая карта блока экспрессии фитазы PAF 2-2S H; на фиг.29-31 - физическая карта промежуточных блоков экспрессии фитазы pXXFYT1 и PXXFYT2 и блока pXXFYT3, в которых XX - лидирующая последовательность (L). В p18FYT# и p 24FYT# включены соответственно лидирующие последовательности AG 18 aa и 24 aa, тогда как в pFYT# в качестве лидера использована фитаза; на фиг.32 - физическая карта плазмиды pFYT3 Qmds; на фиг.33 - физическая карта плазмиды pFYT3INT; на фиг.34 - физическая карта замещающего вектора pREPFYT3 фитазы /AG; на фиг.35,36 - ауторадиограммы хромосомной ДНК, после переваривания pVU II (A) и Bam HI (B) и гибридизации с меченной32P кДНК фитазы A.ficuum, в качестве зонда следующих микроорганизмов: S.cerevisiae (полоса 2), B.subtilis (полоса 3), K.lactis (полоса 4), P.crysogenus (полоса 5), P.aeruginosa (полоса 6), S.lividans (полоса 7), A.niger, 1 мкг (полоса 8), A.niger, 5 мкг (полоса 9), контроль (полоса 10), C.thermocellum (полоса 11). Полоса 1 - маркерная ДНК.
Клонирование генов, кодирующих определенные белки, которые продуцируются тем или иным микроорганизмом, может осуществляться различными способами. Один из них состоит в очистке требуемого белка, последующего определения его N-концевой аминокислотной последовательности и скрининга геномной библиотеки данного микроорганизма с использованием олигонуклеотидного зонда ДНК, полученного исходя из расшифрованной аминокислотной последовательности N-концевого фрагмента. В качестве примера успешного применения этой методики можно указать клонирование гена изопенициллин-N-синтетазы Cephalosporium acremonium (S.M.Samson et al., 1985, Nature, 318, 191-194) и выделение гена, кодирующего ТАКА амилазу Aspergillus oryzae (Boel et al., 1986, европейская патентная заявка 0238023).
Используя этот подход, мы попытались изолировать ген, кодирующий фитазу из Aspergillus ficuum. Белок тщательно очищали и определяли ряд его биохимических параметров. Полученные данные сравнивали с результатами исследования Ullah (1968a). Материалы обоих исследований представлены в табл.1.
Для выделения кодирующего фитазу гена сначала получали набор олигонуклеотидных зондов, конструировавшихся, как описано выше (фиг.4,5), на основании данных расшифровки аминокислотной последовательности. В качестве контроля всего процесса, аналогичным образом выделяли ген, кодирующий кислую фосфатазу, используя для этой цели характеристики белка, полученные Ullah и Cummins (Prep. Biochem., 17, 397-422, 1987). Выделение гена кислой фосфатазы не представляло существенных трудностей. Совсем иной была ситуация с выделением гена фитазы. Несмотря на многочисленные попытки с использованием зондов, полученных исходя из последовательности N-концевых аминокислотных остатков, не удалось выделить геномные фрагменты ДНК или клоны геномной библиотеки, в которых можно было бы однозначно показать присутствие гена, кодирующего фитазу.
Для решения этой задачи очищенную фитазу расщепляли CNBr и выделяли образовавшиеся белковые фрагменты, определяя их N-концевые аминокислотные последовательности (фиг.2).
Используя полученные данные, конструировали новые олигонуклеотидные зонды (фиг.6,7). Эти олигонуклеотидные зонды оказались способными идентифицировать специфические фрагменты ДНК и пригодными для выявления требуемых клонов геномной библиотеки. Перекрестная гибридизация между полученными таким образом новыми клонами или фрагментами ДНК и первым набором олигонуклеотидных зондов или клонами, выделенными с использованием первого набора зондов, отсутствовала.
В связи с этим второй набор зондов также можно использовать при идентификации кодирующих последовательностей для соответствующих фитаз.
Вновь выделенные клоны были использованы для гибридизации (Northern blot hybridization). Дискретные мРНК удавалось определять только в тех случаях, когда их выделяли из мицелия, вырабатывающего фитазу. Гибридизационный сигнал отсутствовал, если мРНК получали из мицелия, в котором отсутствовала продукция фитазы. мРНК имела в длину примерно 1800 оснований и теоретически давала белок с максимальным молекулярным весом около 60 кДа. Эта величина соответствует молекулярной массе негликозилированного белка и молекулярной массе белка, рассчитанной на основании данных о нуклеотидной последовательности ДНК.
Более того, после внедрения такой мРНК в грибную клетку посредством трансформации наблюдали увеличение фитазной активности. Это неопровержимо свидетельствует о том, что была получена нуклеотидная последовательность, кодирующая фитазу. Последовательность аминокислотных остатков в очищенной фитазе и в белковых фрагментах, полученных после переваривания CNBr, была такой же, как аминокислотная последовательность, рассчитанная на основании нуклеотидной последовательности клонированного гена. Нуклеотидная последовательность и выведенная из нее последовательность аминокислотных остатков представлены на фиг.11-17, 19, 20, которые служат дополнительной иллюстрацией структуры клонированного гена, кодирующего фитазу.
Выделение нуклеотидной последовательности, кодирующей фитазу, является предпосылкой получения последней в промышленных масштабах на основании современной технологии рекомбинантных ДНК, включающей такие операции, как амплификация гена, замена регуляторных элементов (промоторов, сигналов секреции) или комбинация различных методов.
В связи с этим предметом настоящего изобретения являются также трансформированные клетки - хозяева, в которых происходит эффективная экспрессия пептидов и белков, обладающих фитазной активностью, при высоких концентрациях этих соединений, а также (в случае необходимости) - эффективная экспрессия кислых фосфатаз. Такими клетками могут служить нитчатые грибы родов Mucor, Aspergillus, Trichoderma и Penicillium, дрожжи родов Kluyveromyces и Saccharomyces и бактерии, принадлежащие к роду Bacillus. Рекомендуется в качестве организмов - хозяев отбирать формы, способные к интенсивной секреции собственных эндогенных белков.
Особый интерес представляют промышленные штаммы Aspergillus, в частности, A. niger, A. ficuum, A.awamori, A.oryzae. В качестве альтернативы, могут использоваться Trichoderma reesei, Mucor, miehei, Kluyveromyces lactis, Saccharomyces cerevisiae, Bacillus subtilis, Bacillus licheniformis.
Трансформированные клетки содержат нуклеотидные последовательности, которые кодируют требуемый белковый продукт, и обычно включают сигнальную последовательность секреции, способную функционировать в данном хозяине и необходимую для освобождения синтезируемого пептидного или белкового продукта.
Настоящее изобретение допускает применение различных сигнальных последовательностей. Прежде всего, можно использовать сигнальные последовательности, гомологичные клонированной нуклеотидной последовательности, которая подлежит экспрессии. С другой стороны, для оптимизации гомологичной рекомбинации можно использовать сигнальную последовательность, которая гомологична или в значительной степени гомологична сигнальной последовательности в соответствующем локусе гена клетки-хозяина. Кроме того, допускается применение сигнальной последовательности, сконструированной специально, с целью улучшения секреции требуемого продукта, продуцируемого данным организмом, например, описанной в работах Von Heyne (1983), Eur. J.Biochem., 133, 17-21, Perlman & Halverson (1983) J.Med.Biochem., 167. Последовательность ДНК, кодирующая сигнальную последовательность, может присоединяться непосредственно к последовательности, кодирующей требуемый белок, через последовательность, кодирующую сигнал процессинга (участок узнавания места расщепления), либо через короткий мостик, длиной обычно не более десятка кодонов.
Согласно настоящему изобретению предпочтительно использовать сигнальные последовательности, гомологичные клонированной нуклеотидной последовательности, подлежащей экспрессии, сигнальную последовательности в форме 18-членной глюкоамилазы (AG) или глюкоамилазы (AG) из 24 аминокислотных остатков. Две последние последовательности могут быть либо гомологичны, либо гетерологичны подлежащей экспрессии нуклеотидной последовательности.
Продуктом экспрессии или требуемой нуклеотидной последовательностью могут служить ДНК, гомологичные или гетерологичные для данного организма-хозяина.
Для целей настоящего изобретения под термином "гомологичная" ДНК понимается ДНК из клеток, относящихся к тому же биологическому роду, что и организм-хозяин. Например, клетки грибов Aspergillus трансформируются ДНК из грибов рода Aspergillus. Такой подход позволяет улучшить свойства клеток гриба данного рода, избежав придания им тех свойств, которые не свойственны данному организму в отсутствие трансформации.
"Гетерологичная" ДНК - это ДНК, полученная из клеток более чем одного рода микроорганизмов (как следует из примера, приведенного в предыдущем параграфе, гетерологичной ДНК может служить ДНК из иных, нежели Aspergillus, микроорганизмов, которую используют для экспрессии в клетках Aspergillus).
Нуклеотидные последовательности, кодирующие белки с фитазной активностью, желательно получать из грибов. Наиболее предпочтительны кодирующие фитазу нуклеотидные последовательности из грибов Asperhillus ficuum или Aspergillus niger.
Область 5' открытой считываемой последовательности требуемой нуклеотидной последовательности должна включать регуляторную зону инициирования транскрипции (или промотор). Можно использовать любую функционально активную область нуклеотидной последовательности клетки-хозяина, в том числе промотор, гомологичный кодирующей фитазную активность нуклеотидной последовательности, которая подлежит экспрессии. Однако в большинстве случаев используют область, гомологичную области локуса-мишени. Это позволяет заменить продукт экспрессии локуса-мишени требуемым продуктом. В этом случае регуляторная зона инициирования транскрипции обычно удовлетворяет предъявляемым требованиям в том смысле, что достигаемый уровень экспрессии и секреции белка, кодируемого локусом-мишенью, обеспечивает достаточно эффективную продукцию последнего. Тем не менее, в отдельных случаях может требоваться более высокий уровень транскрипции, нежели тот, что обеспечивается локусом-мишенью, либо возникает необходимость получать продукт экспрессии, используя специфический индуцирующий фактор. В таких случаях используют регуляторную зону инициирования транскрипции, которая отличается от зоны, присутствующей в локусе-мишени данного гена. Известно большое число функционально активных регуляторных зон инициирования транскрипции из различных видов нитчатых грибов. К их числу относятся зоны, кодирующие гликоамилазу (AG), грибную амилазу, кислую фосфатазу, GAPDH, TrpC, AmdS, AlcA, AbdA, гистон H2A, Pyr G, Pyr4, изопенициллин-N-синтетазу, ФГК, кислую протеазу, ацилтрансферазу и т.д..
Желательно, чтобы локус-мишень кодировал ген белка с высоким уровнем экспрессии, т.е. ген, продукт экспрессии которого синтезируется в концентрации не менее 0,1 г/л (в конце процесса ферментации). Продолжительность такого процесса, помимо прочих факторов, может зависеть от характера требуемого белка. В качестве примера такого гена можно указать на ген, кодирующий гликоамилазу (AG). Среди других представляющих интерес генов: гены грибной альфа-амилазы, кислой фосфатазы, протеазы, кислой протеазы, липазы, фитазы и целлобиодеградазы. Особенно ценны локусы гена гликоамилазы A.niger, гена амилазы A.oryzae, гена целлобиогидролазы T.reesei, гена кислой протеазы Mucor michei, гена лактазы Kluyveromyces lactis и гена инвертазы Saccharomyces cerevisiae.
Регуляторная зона окончания транскрипции может быть получена из используемого гена, локуса-мишени или любой другой подходящей последовательности. При наличии в клетке других последовательностей, расположенных ниже используемого гена (по направлению транскрипции), зона окончания транскрипции, в случае ее гомологичности локусу-мишени, должна быть значительно короче, чем гомологичная фланкирующая область.
Обычно используют маркер селективности, являющийся частью трансформированного генома, подлежащего экспрессии, или отделенный от трансформированного участка, в результате чего он может быть внедрен за пределами используемого гена. Поскольку рекомбинантные молекулы согласно настоящему изобретению трансформируются преимущественно в клетки промышленных штаммов микроорганизмов, маркеры селективности для контроля трансформации должны быть по преимуществу доминантными маркерами. Иными словами, их введение в клетки-хозяева не должно сопровождаться мутациями. Примером таких маркеров, обеспечивающих рост трансформированных клеток на контролируемой питательной среде, может служить ген A. amdnudulansS, который поддерживает рост трансформированных клеток A.niger на ацетамиде в качестве единственного источника питания, а примером маркеров, обеспечивающих резистентность к антибиотикам, - ген ble, ответственный за устойчивость к флеомицину, или ген hph, от которого зависит устойчивость к гигромицину В.
Ген селективности должен иметь собственные регуляторные зоны инициирования и окончания транскрипции и трансляции, так как это необходимо дли независимой экспрессии маркера. Как указывалось выше, известно большое число регуляторных зон инициирования транскрипции, которые можно использовать в сочетании с маркерным геном. В тех случаях, когда используется феномен резистентности к антибиотикам, концентрация последних может варьироваться в зависимости от типа последних от 30 до 300 мкг/мл.
Отдельные последовательности можно присоединять с помощью одного из существующих методов, например, рестрикции, соединения комплементарных рестрикционных участков и их лигирования, получения тупых концов посредством заполнения выступов с последующим лигированием, Bal 31 резекции, репарации праймера, мутагенеза in vitro и т.п.. В случае необходимости можно использовать множественные связки (полилинкеры) и адаптеры. Их внедряют и удаляют с помощью общепринятых методов с целью облегчения сборки конструируемых единиц экспрессии. На каждой стадии синтеза таких блоков применяются клонирование фрагментов, анализ рестрикционных ферментов, секвенирование, гибридизация и другие операции. Имеется большое число векторов клонирования, однако выбор того или иного из них не является лимитирующим фактором в данном изобретении. Обычно клонирование производится в E.coli.
Фланкирующие области могут включать по крайней мере часть открытой считываемой зоны локуса-мишени, особенно его сигнальную последовательность, регуляторные области 5' и 3' генного локуса-мишени или могут простираться за пределы регуляторных областей. Обычно длина фланкирующей области не менее 100 пар оснований, предпочтительно не менее 200 пар оснований, но может достигать 500 пар оснований или более. Фланкирующие области выбирают таким образом, чтобы обеспечить разрыв гена-мишени и воспрепятствовать его экспрессии. Эта цель может быть достигнута посредством внедрения блока экспрессии (включающего подлежащую экспрессии нуклеотидную последовательность и (при необходимости) такие дополнительные элементы, как сигнальная последовательность, регуляторная зона инициирования транскрипции и/или регуляторная зона окончания транскрипции) в считываемую область рядом с 5'-областью путем замещения всего или части гена-мишени сконструированной единицей экспрессии, или внедрением последней между регуляторной зоной инициирования транскрипции локуса-мишени и открытой считываемой последовательностью. Как уже указывалось, в случае гомологичности регуляторной зоны окончания транскрипции и зоны локуса-мишени, длина 3' - фланкирующей области должна быть значительно больше чем длина регуляторной зоны окончания транскрипции, имеющейся в трансформированном геноме.
Предметом настоящего изобретения является также исходный материал для конструирования фитаз "второго поколения", т.е. ферментов с фитазной активностью, которые по своим свойствам отличаются от выделенных до сих пор ферментов. Фитазы "второго поколения" могут характеризоваться измененными оптимумами pH, температуры, специфической активности или сродства к субстратам, а также другими отличиями, которые повышают пригодность ферментов для использования в тех или иных процессах. E.coli является наилучшим объектом для получения таких мутаций, в том числе направленных. E.coli не имеет механизма, обеспечивающего удаление интронов, которые могут присутствовать в гене фитазы. Для экспрессии в этих организмах предпочтительно использовать клонированную кДНК фитазы. Клонированная последовательность кДНК легко поддается мутациям хорошо известными способами после чего мутантный ген может быть введен в желаемый геном для экспрессии.
Вновь сконструированный блок можно трансформировать в качестве клонирующего вектора в клетку-хозяин либо линейно, либо в форме циклической структуры, а при необходимости он может быть удален из клонирующего вектора. Предпочтительным клонирующим вектором является плазмида, которую обычно переводят в линейную форму в пределах примерно 1 килооснования используемого гена. Желательно интегрировать блок экспрессии фитазы, получаемый согласно настоящему изобретению, в геном строго определенного вида микроорганизмов.
Существует множество методов трансформирования нитчатых грибов. Среди этих методов следует отметить слияние или трансформацию протопластов, электроперфорацию или микробомбардировку клеток. Особенно хорошие результаты дает трансформация протопластов, применение которой имеет ряд преимуществ.
Сначала мицелий гриба требуемого штамма переводят в протопласты посредством ферментативного переваривания клеточной стенки в присутствии таких стабилизаторов осмотического давления, как KCl или сорбитол. Добавление CaCl2 способствует более интенсивному включению ДНК в протопласты. Аналогичным эффектом обладает концентрированный раствор полиэтиленгликоля, вызывающий к тому же агрегацию протопластов, в процессе которой происходит включение ДНК в агрегаты с последующим захватом трансформирующего фактора протопластами. Затем протопласты регенерируют на твердой среде, содержащей стабилизатор осмотического давления, и, в случае необходимости, фактор селективности, резистентность к которому кодируется трансформирующей ДНК.
После отбора трансформированных клеток присутствие требуемого гена можно установить различными способами. При синтезе продукта, гетерологичного хозяину, наличие экспрессии гена устанавливается с помощью антител. Другим способом является применение одной из разновидностей гибридизации, которая позволяет обнаруживать присутствие встроенного гена или продукта его транскрипции.
Амплификация нуклеотидной последовательности или экспрессия трансформированного генома достигаются с помощью стандартных методов, таких как введение в трансформирующий вектор множественных копий трансформанта или использование в качестве маркера избирательности гена amdS (см., например, Weinans et al., 1985, Current Genetics, 9, 361-368). Подлежащая амплификации последовательность ДНК может представлять собой, как указано выше, либо гомологичную, либо гетерологичную клетке-хозяину ДНК.
После завершения всех этих операций клетки можно выращивать в обычной питательной среде. При этом используют невысокие концентрации протеазных ингибиторов, таких как фторофенилметилсульфонил, альфа-2- макроглобулины, пепстатин и другие. Обычно это концентрации порядка 1 мкг/мл - 1 мг/мл. Ген(ы) протеазы можно инактивировать, чтобы избежать разрушения белкового продукта или уменьшить его.
Трансформированные клетки можно выращивать в ферментерах для непрерывного или периодического культивирования, с последующей изоляцией питательной среды и экстракцией конечного продукта.
При необходимости очистки конечного продукта можно применять самые разнообразные методы, включая хроматографию (в частности, ВЭЖХ), экстракцию растворителя, электрофорез, а также другие методы и их сочетание.
Предметом настоящего изобретения является также способ
нисходящего процессинга, при котором фильтрация ферментационного бульона (при необходимости
предварительно очищенного) сопровождается повторной фильтрацией в стерильных условиях, после которой
отфильтрованный раствор подвергают концентрированию. Полученный таким образом жидкий концентрат
можно использовать для следующих целей:
а) осаждения из него фитазы и других белков путем
добавления ацетона до конечной концентрации 60% (в объемном соотношении) при непрерывном
перемешивании. Осадок можно высушивать под вакуумом при температуре 35oC. После измельчения сухого
порошка ферментативный продукт используют как таковой в опытах по его практическому
применению. Выход конечного продукта составляет при этом 90%.
б) высушивания в форме аэрозоля с
помощью обычно применяемых для этой цели методов с выходом 80-99%;
в)
смешивания с носителями, например, с пшеничными отрубями. Получаемые при этом смеси можно высушивать в оросительных
колоннах или над слоем жидкости.
г) для осмотической стабилизации соответствующими агентами, например, сорбитолом. Для предотвращения загрязнения микроорганизмами можно добавлять консерванты, в частности, бензойную кислоту.
Все четыре описанные формы конечного продукта могут поступать на рынок для продажи изготовителям полуфабрикатов, предприятиям по изготовлению комбикормов, другим потребителям и фермерам.
Нижеследующие примеры служат для иллюстрации изобретения и не ограничивают сферу его применения. Специалистам в данной области не составляет труда понять, что ген фитазы согласно настоящему изобретению можно использовать в экспериментах по гетерологичной гибридизации, проводимых с целью выделения кодирующего фитазу гена из других видов микроорганизмов.
Пример 1. Ферментация A.ficuum NRRL 3155.
Штамм NRRL 3133 Aspergillus ficuum получали из лаборатории Northern Region Research Lab. USDA, 1815 Nortern Univ.str. Peoria, США. Препараты грибных спор готовили с помощью стандартной методики.
Споры, а затем и клетки многократно пассировали в колбах Эрленмейера и переносили в ферментер на 10 л. После циклического культивирования содержимое ферментера использовали в качестве инокулята для конечной ферментации в объеме 500 л.
Состав использовавшейся среды: 91 г/л кукурузного крахмала (BDH Chemicals, Ltd. ), 38 г/л глюкозы, H2O, 0,6 г/л MgSO4•7H2O, 0,6 г/л KCl, 0,2 г/л FeSO4•4H2O и 12 г/л KNO3. Величина pH поддерживалась на уровне 4,6 ± 0,3 системой автоматического титрования с использованием 4H NaOH или 4H H2SO4.
Клетки выращивали при 28oC и автоматически регулируемой концентрации растворенного кислорода, эквивалентной 25%-ному насыщению воздуха. Максимальную продукцию фитазы от 5 до 10 ед/мл регистрировали через 10 дней после начала ферментации.
Пример 2. Очистка и свойства фитазы A.ficuum.
А. Определение активности фитазы A.ficuum.
100 мкл
ферментационного бульона после фильтрации (в случае необходимости - разведения)
или супернатанта, или разбавителя (в качестве контроля) добавляли в инкубационную смесь следующего состава:
0,
25 М натрий - ацетатный буфер pH 5,5 или
глициновый HCl - буфер pH 2,
5
1 мМ фитовая кислота, натриевая соль
разбавитель до конечного объема 900 мкл
Полученную смесь
инкубировали при 37oC в течение 30 мин. Реакцию останавливали
добавлением 1 мл 10% ТХК (трихлоруксусной кислоты). После прекращения реакции добавляли 2 мл реактива (3,66 г FeSO4
•7H2O в 50 мл раствора молибдата аммония /2,5 г (NH4)6Mo7O24•4H2O и 8 мл H2SO4, разведенных в 250
мл разбавителя/). Интенсивность синей окраски измеряли
спектрофотометрически при длине волны 750 нм. Результаты измерения свидетельствуют о том, что количество фосфата, выделяемого в результате
реакции, составляет 0 - 1 ммоль/л (в сопоставлении с
калибровочной кривой фосфата).
Окрашивание на фосфатазу.
Компоненты, обладающие фосфатазной активностью, определяли методом изоэлектрофокусировки с использованием обычного красителя. Гель инкубировали в присутствии раствора альфа-нафтил-фосфата и соли Fast Garnet GBC, Sigma (соответственно 0,1 и 0,2% (вес/объем)) в 0,6 М натрийацетатном буфере pH 5,5. Реакцию, в результате которой появлялся осадок черного цвета, останавливали смесью метанола с уксусной кислотой в объемном соотношении 30:10% или, при необходимости получения в дальнейшем белка с фитазной активностью, промывкой дистиллированной водой.
Б. Очистка фитазы A.ficuum.
Фитазу очищали до гомогенного состояния из культуральной среды A.ficuum NRRL 3135. Предварительно бульон стерилизовали фильтрованием. Затем полученный фильтрат концентрировали в ультрафильтровальной установке Filtron с фильтрами 30 кД. Ионную силу и pH проб доводили до необходимой для очистки величины, промывая их 10 мМ натрий-ацетатный буфером pH 4,5. Процедура очистки обеспечивала 20-кратную концентрацию проб.
После концентрации пробы наносили на катионообменник (колонка HR 16/10, 20 мл, из S-сефарозы Fast-Flow фирмы Pharmacia) и хроматографировали в системе для ускоренной очистки белков (Waters Preparative 650 Advanced Protein Purification System). Связанные белки элюировали в градиенте 0-1 М натрий - ацетатного буфера, а фитазу в 250 мМ NaCl. Фракции, содержавшие фитазную активность, объединяли, концентрировали и обессоливали с помощью ультрацентрифугирования. Полученный раствор наносили на колонку ионообменника (катионообменная Q - сефароза Fast-Flow, HR 16/10, 20 мл, фирмы Pharmacia), после чего белки повторно элюировали в градиенте 0 - 1 М хлористого натрия в ацетатном буфере, как описано выше. С этой колонки фитазу элюировали в 200 мМ растворе хлористого натрия.
Продуктом этих стадий очистки был очищенный препарат фитазы с удельной активностью порядка 40 - 50 ед/мг белка, что свидетельствует о 25-кратной очистке.
Анализ чистоты частично очищенной фитазы выявил присутствие одного основного загрязняющего компонента с молекулярной массой примерно 100 кДа (фиг. 2, последовательность Е). С помощью методов изоэлектрофокусировки показано присутствие нескольких ферментов, обладающих фосфатазной активностью, в том числе 3-4 форм фитазы, изоэлектрические точки которых колебались от 5,0 до 5,4 (фиг.1, последовательности А и В).
Чтобы получить гомогенный препарат фитазы, осуществляли дальнейшую двукратную очистку посредством разделения компонентов частично очищенного фермента методом изоэлектрофокусировки в системе LKB Multiphor на пластинах Ampholine PAG (в диапазоне pH от 4,0 до 6,5). Белки с фосфатазной активностью (в том числе фитаза) определяли окрашиванием на общую фосфатазу, как описано выше. Соответствующие полосы вырезали из геля и элюировали активный белок посредством инкубации срезов в 10 мМ натрий - ацетатном буфере pH 5,5 в течение 16 ч. Белковые фракции анализировали с целью определения удельной активности фитазы, как описано в примере 2, тем самым дифференцируя содержащие фитазу фракции от остальных кислых фосфатаз. Конечный коэффициент очистки фитазы достигал 60-кратной величины (удельная активность очищенного белка равнялась 100 ед/мг). На этом заключительном этапе очистки удавалось также выделять различные подформы фитазы (фиг.1, последовательности A и B).
Эффективность процедуры очистки обеспечивалась использованием моноклональных антител к A. ficuum. Антитела иммобилизировали на активированной цианоген - бромидом сефарозе 4B (5 мг/мл геля) и использовали полученный матрикс в колонках для иммуноаффинной хроматографии. Связывающая способность матрикса составляла примерно 1 мг фитазы/мл. Фитазу элюировали с колонки буфером с pH 2,5 (100 мМ HCl -глицина, 500 мМ NaCl) без потери активности. Эту процедуру можно использовать для выделения гомогенной фитазы из неочищенных фильтратов культуральной среды в одну стадию с выходом 80% при 60-кратной степени очистки.
В. Дегликозилирование фитазы.
Фитазу A. ficuum (70 мкг белка) инкубировали с 2,5 ед N- гликаназы (Genzyme) в 0,2 М натрий-фосфатной буфере pH 6,8 и 10 мМ 1, 10-фенантролине, в общем объеме 30 мкл.
Через 16 ч после начала инкубации при 37oC степень дегликозилирования оценивали с помощью электрофореза в системе Phast System (Pharmacia). Молекулярная масса фитазы уменьшалась с 85 кДа до примерно 56,5 кДа. Окраска на сахаре реактивом Шиффа, применяемая для идентификации гликопротеиновой природы нативной фитазы, не выявила остаточных углеводных компонентов в белковом материале. Полное отсутствие углеводов было дополнительно подтверждено с помощью высоко чувствительной методики блоттинга в лектине. Нативную и дегликозилированную фитазы (по 1,5 мкг) подвергали электрофорезу в стандартном полиакриламидном геле с додецилсульфатом натрия для переноса на мембрану PVDF (Immobilon Millipore)в 25 мМ трис-глициновом буфере pH 8,3 и 20% метаноле, в течение 16 ч., при 30В.
После этого мембрану инкубировали с 1% сывороточным альбумином крупного рогатого скота в фосфатном буферном растворе, а затем с конканавалин-A-пероксидазой (Sigma) в том же буфере (10 мкг/мл). После этого пероксидазу окрашивали 4-хлоро-1-нафтолом фирмы Sigma.
Этот чувствительный метод также не выявил остаточных углеводов в дегликозилированной фитазе.
После дегликозилирования фермент полностью утрачивал активность, вероятно вследствие агрегации его молекул.
Пример 3. Определение аминокислотной последовательности фитазы и конструирование олигонуклеотидных зондов.
А. Определение N-концевой аминокислотной последовательности.
Фитазу электрофоретическим методом переносили из полиакриламидного геля с додецилсульфатом натрия или такого же геля для изоэлектрофокусировки на мембрану. Электроблоттинг осуществляли в 10 мМ ЦАРК (3-циклогексиламино-пропансульфоновой кислоте) в форме буферного раствора с pH 11,0 в присутствии метанола (10% в объемном соотношении) на протяжении 16 ч. при 30 В и 4oC.
Локализацию белка определяли с помощью окраски кумасси бриллиантовым синим. Соответствующую полосу вырезали из геля, обесцвечивали в метаноле и подвергали газофазовому секвенированию. Эту процедуру повторяли несколько раз, используя несколько отдельных образцов. Полученные в этом исследовании результаты приведены на рисунке 1А (последовательности A и B).
Одновременно определяли аминокислотную последовательность белка с молекулярной массой 100 кДа, который присутствовал в неочищенных препаратах. Эти данные представлены на фиг.22, 23. Расшифрованная таким образом последовательность аминокислотных остатков характеризуется значительной степенью гомологичности последовательности кислой фосфатазы, которая была выделена из Aspergillus niger (MacRae et al., 1988, Gene, 71, 339-648).
Б. Определение внутренних аминокислотных последовательностей.
Фрагментация белка цианоген-бромидом.
Очищенную до гомогенного состояния фитазу с помощью ультрацентрифугирования переносили в 100 мМ NaHCO3, используя микроконцентратор Centricon 30 (Amicon). После этого белок лиофилизировали, разводили в 70%-ной трифторуксусной кислоте (объемное соотношение) и инкубировали с 300-кратным молярным избыточным количеством CNBr в течение 6 ч. Реакцию останавливали, разбавляя инкубационную систему водой. Полученные фрагменты снова лиофилизировали. После этого пробу разводили в буфере для электрофореза в полиакриламидном геле с додецилсульфатом натрия, содержавшем ДТТ (дитиотреитол), и с помощью указанной электрофоретической методики оценивали степень фрагментации. Для аналитического электрофореза в полиакриламидном геле использовали аппарат Phast System (Pharmacia) и 20%-гель с додецилсульфатом натрия. С целью повышения эффективности разделения малых пептидов предварительно проводили электрофорез без анализируемых проб, получая непрерывную буферную систему для данного геля (в соответствии с инструкцией). Пептиды определяли методом окрашивания серебром, который широко используется в аналогичных случаях, поскольку окраска кумасси бриллиантовым синим не позволяла выявлять самые мелкие пептиды. Результатом описанной процедуры была полная деградация фитазы, распадавшейся на пептиды с молекулярной массой менее 2,5, 36, 57 и 80 кДа.
Эти пептиды выделяли методом газофазового секвенирования в условиях электрофореза в полиакриламидном геле с трицином и додецилсульфатом натрия, как описано Schagger & Jagow (1987, Anal. Biochem. 166, 368-379), после чего проводили электроблоттинг в соответствии с ранее описанной процедурой.
N-концевая последовательность фрагмента с молекулярной массой 57 кДа оказалась идентичной такой же последовательности фитазы, как она была описана Ullah (1968b, Supra), с тем исключением, что в указанном фрагменте отсутствовали первые четыре аминокислотных остатка (фиг.1, последовательность В). N-концевые последовательности пептидов с молекулярными массами 2,5 кДа и 36 кДа показаны на фиг.2 (последовательности А и В).
B. Олигонуклеотидные зонды.
Конструирование олигонуклеотидных зондов осуществляли на основании данных о последовательности аминокислотных остатков (фиг.1,2). Их получали в синтезаторе ДНК Applied Biosystems ABI 380 В. Эти олигонуклеотиды показаны на фиг.4-7.
Пример 4. Гибридизация геномных комплексов и геномных библиотек с первым набором олигонуклеотидных зондов.
Геномные ДНК A.ficuum выделяли после измельчения мицелия в жидком озоне, используя стандартные методики (в частности, методику Yelton et al., 1984, Proc. Natl. Acad. США, 1470-1474). Геномную библиотеку конструировали в векторе-бактериофаге лямбда EMBL 3, используя частично переваренную Sau3A хромосомную ДНК штамма NRRL 3135 A.ficuum (в соответствии со стандартной процедурой, описанной в Руководстве по лабораторному клонированию молекул /Maniatis et al., Molecular Cloning: A Laboratory Manual. Gold Spring Harbor Lab. N4. , 1982/). Полученная таким образом геномная библиотека содержала 60-70-кратное количество генома A. ficuum. Контроль библиотеки на наличие бляшек без вставки осуществляли посредством гибридизации с более закрытым фрагментом фага лямбда EMBL 3A. Гибридизация бляшек с зондом лямбда EMBL 3A не превышала 1%. Размер вставки составлял 13 - 17 килооснований.
Для выяснения условий и свойств зондов, необходимых для скрининга геномной библиотеки, геномную ДНК переваривали несколькими рестрикционными ферментами, разделяли на агарозе и подвергали блоттингу на Genescreen в соответствии с указаниями предприятия - изготовителя. Полученные препараты гибридизировались со всеми олигонуклеотидными зондами. Гибридизацию проводили в условиях, отличавшихся степенью лимитированности (6 х SSC, от 40 до 60oC для гибридизации и до 0,2 х SSC при 65oC для промывания). Для скрининга геномной библиотеки были выбраны зонды 1068 и 1024 (фиг.4,5), хотя общие фрагменты ДНК, которые специфически гибридизировались бы с обоими зондами, не обнаружены. Зонд 1025 для кислой фосфатазы (фиг.8) давал специфический дискретный сигнал гибридизации, в связи с чем он был выбран для скрининга геномной библиотеки кислой фосфатазы.
Применение всех трех зондов позволило выявить в геномной библиотеке гибридизирующиеся бляшки. Гибридизационный сигнал, соответствующий зонду 1025 (кислой фосфатазе), характеризовался высокими интенсивностью и воспроизводимостью. Зонды 1024 и 1068 давали гибридизационные сигналы переменной интенсивности, характеризовавшие свойства фитазы. Перекрестные реакции между обеими сериями не наблюдались. Был проведен повторный скрининг всех трех серий бляшек, из восьми одиночных, способных к гибридизации бляшек, были выделены ДНК (Maniatis et al., supra). В каждой серии были идентифицированы клоны, содержавшие идентичные гибридизирующиеся фрагменты. Это свидетельствует о том, что вставки, имеющиеся в этих клонах, могут частично перекрываться в пределах одной и той же области геномной ДНК. Однако и в этом случае перекрестная гибридизация отсутствовала (при использовании двух серий специфических зондов фитазы 1024 и 1068). Таким образом, несмотря на то, что оба зонда, использовавшиеся для выделения двух серий клонов, были получены из N-концевой последовательности белка, идентифицировались и клонировались разные фрагменты геномной ДНК.
Все три серии клонов гибридизировались с бляшками, содержавшими мРНК, выделенными из активированного и не активированного мицелия (Northern blots, пример 6). Клоны, специфичные для кислой фосфатазы, а также выделенный из этих клонов внутренний фрагмент Sal 1 длиной 3,1 килооснований, гибридизировался исключительно с образцами индуцированной мРНК. Идентифицированная с помощью специфичных для кислой фосфатазы зондов мРНК имела длину приблизительно 1800 оснований, что согласуется с известными размерами белка (68 кДа по данным Ullah & Cummins 1987, Prep. Biochem., 17,397-422). Гибридизация специфичных для фитазы клонов со специфическими мРНК отсутствовала. В связи с этим мы пришли к выводу о том, что вышеописанная методика не позволяла клонировать ген, кодирующий фитазу. Другой вывод состоял в том, что невозможность использования этого метода для этой цели не была связана с его несостоятельностью, поскольку его с успехом использовали для клонирования гена, кодирующего кислую фосфатазу. Клон лямбда, содержащий ген кислой фосфатазы, был передан на хранение в Центральное бюро грибковых культур в Барне, Нидерланды (Centraal Bureaen voor Schimmelcultures, Baarn, Netherlands) 24 апреля 1989 г. , где ему был присвоен входящий номер CBS 214.89. Из бляшки Z1, был выделен фрагмент Bam H1 длиной 10 килооснований, который клонировали в pUC19. Этот подклон включает полный ген, кодирующий кислую фосфатазу. Подклон, закодированный как 1-1 (фиг. ), был передан на хранение 24 апреля 1989г. и получил номер CBS 213.89.
Пример 5. Выделение гена, кодирующего фитазу, с использованием второго набора олигонуклеотидных зондов.
Зонды конструировали на основании данных о последовательности N-концевых аминокислотных остатков в пептидных фрагментах, полученных посредством переваривания CNBr (фиг.6,7, зонды 1295, 1296 и 1297), и гибридизировали с геномной ДНК, как описано выше. Приемлемость этих зондов для выделения кодирующего фитазу гена снова оценивали посредством их гибридизации с геномными комплексами (Southern hybridization). На этот раз, при использовании всех трех зондов были получены гибридизационные фрагменты соответствующей длины, несмотря на то, что зонды были сконструированы на основании последовательностей неперекрывающихся областей. Гибридизация между новым набором зондов и клонами, выделенными с помощью первого набора, отсутствовала (пример 4). Поэтому были проведены специальные эксперименты с целью повторного скрининга геномной библиотеки с использованием всех трех зондов. Установлена также гибридизация подгруппы клонов (лямбда AF 201, 219, 241 и 243), выделенных с помощью одного из зондов, с каждым из двух других зондов. Это говорит о том, что при использовании трех различных зондов были выделены клоны из одной и той же области генома. Была предпринята попытка провести гибридизацию вновь выделенных клонов с зондами 1024 и 1083. В обоих случаях гибридизация с новыми клонами отсутствовала в условиях, при которых оба зонда давали гибриды с клонами, выделенными с их помощью (см. пример 4). Это показывает, что вновь изолированные клоны были негомологичны зондам из N-концевой последовательности фитазы.
Клон лямбда - EMBL 3, который гибридизовался со всеми тремя зондами (1295, 1298 и 1297), получил наименование лямбда - AF201 (фиг.9). Он был передан на хранение 9 марта 1969 г. и получил номер CBS 155.89.
Фрагмент Bam HI клона AF201 длиной 5,1 килооснований, субклонированный в pUC19 и обозначенный как pAF 2-3 (см. фиг.9), давал гибриды со всеми тремя олигонуклеотидными зондами и был использован для блоттинга. В этих экспериментах была выделена дискретная мРНК длиной 1800 килооснований. Эта мРНК присутствовала только в активированном мицелии. Сходные результаты были получены при использовании в качестве зондов олигонуклеотидов. Поэтому, используя новый набор зондов, идентифицировали общий фрагмент ДНК, который специфически гибридизируется с индуцированной мРНК. Длина этой РНК (1800 оснований) достаточна для кодирования белка с молекулярной массой порядка 60 кДа, т. е. примерно того же размера, что и негликозилированный белок. Очевидно, что выделенные фрагменты содержат по крайней мере часть гена, кодирующего фитазу.
Пример 6. Выделение "индуцированной" и "неиндуцированной" мРНК.
Из литературы известно, что биосинтез фитазы в клетках A.ficuum строго контролируется зависимым от фосфата механизмом (Han and Callagher, 1987, J. Indust. Mecrobiol., 1, 295-301). В связи с этим мы считали, что, показав наличие сходного механизма регуляции выделенного гена, мы получим доказательство клонирования этого гена.
Для того, чтобы выделить мРНК, синтезированную в продуктивных и непродуктивных условиях, клетки A. ficuum (штамм NRRL 3135) выращивали следующим образом. Сначала в течение ночи споры проращивали в неиндуцирующей среде. На следующий день собирали мицелий, отмывали его стерильной водой и инокулировали в активирующую или неактивирующую среды следующего состава: 20 г/л кукурузного крахмала; 7,5 г/л глюкозы; 0,5 г/л MgSO4•7H2O; 0,2 г/л FeSO4•7H2O и 7,2 г/л KHO3. Для индукции фитазы в среду добавляли до 2 г/л кукурузной вытяжки, тогда как неиндуцированная среда содержала 2 г/л K2 HPO4. Мицелий выращивали на протяжении по меньшей мере еще 100 ч., отбирая пробы через определенные промежутки времени. Продукцию фитазы контролировали с помощью методики ее определения, описанной в примере 2A. Денатурированную мРНК отделяли посредством электрофореза и использовали для блоттинга на Genescreen plus. Полученные пятна гибридизировали с меченным32P pAF 2-3 или с изолированным из pAF 1-1 фрагментом SalI длиной 3,1 килооснований из примера 4 (для кислой фосфатазы). Результаты этого эксперимента представлены в табл.2.
Положительную гибридизацию специфичного для фитазы фрагмента BamHI длиной 5,1 килооснований и специфичного для кислой фосфатазы фрагмента SalI длиной 3,1 килооснований с изолированной мРНК наблюдали только в тех случаях, когда клетки выращивались в условиях, индуцирующих биосинтез фитазы и кислой фосфатазы. На основании этих результатов был сделан вывод о том, что выделенные гены регулируются постулированными для фитазы и кислой фосфатазы механизмами.
Гибридизация пятен со специфичным для фитазы фрагментом BamHI длиной 5,1 килооснований (A) или специфичным для кислой фосфатазы фрагментом SalI длиной 3,1 килооснований (B), использовавшимися в качестве зондов. Знак +а указывает на присутствие мРНК фитазы длиной 1800 оснований или мРНК кислой фосфатазы длиной 1800 оснований. Относительную фитазную активность определяли в пробах, полученных через 24 ч. после начала культивирования. В активированных культурах фитазная активность была в 10 раз выше, чем в неактивированных.
Пример 7. Доказательство клонирования гена фитазы.
Для получения бесспорного доказательства успешного выделения кодирующего фитазу гена и изучения возможности повышения уровня его экспрессии, этот ген субклонировали в соответствующем векторе и трансформировали в A.niger 402 (АТСС 9092). С этой целью выделяли ген фитазы из клона лямбда AF201 в форме фрагмента NruI длиной 10 килооснований и проводили его клонирование в сайте StuI вектора pAN 1-8, который в качестве маркера селективности содержит ген ble, обеспечивающий устойчивость к флеомицину (Mattern J.E. and Punt P.J., 1988, Fungal Genetics Newsletter, 35, 25). Полученный материал получил наименование pAF 28-1 (фиг. 9) и был трансформирован в A.niger 402 с помощью методики, описанной в примере 9, с тем исключением, что протопласты наслаивали на минимальную питательную среду для Aspergillus, в которую добавляли 30 мкг флеомицина/мл, и отверждали примесью 0,75%-ного агара. Отдельные трансформанты после выделения подвергали очистке и оценивали их продуктивность во встряхиваемых колбах, как описано в примерах 1 и 2. В качестве контроля использовали трансформанты, содержавшие только вектор, или нетрансформированные клетки-хозяева (табл.3). Было установлено, что фитаза, реагировавшая со специфическими моноклональными антителами к фитазе A.ficuum, синтезировалась только A. niger 402, содержащими pAF 28-1. Фермент, дававший реакцию с указанными антителами, элюировали с колонки для иммунно-аффинной хроматографии при pH 2,5. По молекулярной массе, степени гликозилирования, изоэлектрической точке и удельной активности он был неотличим от фитазы из A.ficuum. Эти данные со всей очевидностью подтверждают, что в клетках A.niger 402, трансформированных pAF 28-1, имеет место экспрессия фитазы, практически идентичной ферменту из A.ficuum. Аналогичная экспрессия в обоих типах клеток, служивших контролем, не зарегистрирована.
Штаммы грибов выращивали в активирующей среде (пример 6).
Пробы отбирали через 96 ч после начала культивирования.
Пример 8. Характеристика гена фитазы.
Клоны лямбда, содержавшие ген фитазы, анализировали посредством переваривания различными рестрикционными ферментами.
Карта геномной области, включающей ген фитазы, представлена на фиг.9.
Ранее (пример 5) было показано, что присутствующий в pAF 2-3 фрагмент Bam HI длиной 5,1 килооснований, содержит по крайней мере часть гена фитазы. Более того, олигонуклеотидные зонды 1295 и 1297 оказались способными гибридизироваться со вставкой SalI pAF 2-7 (см. фиг.6,7; положения клонов pAF 2 показаны на фиг.9), тогда как зонд 1296, по-видимому, соединяется с сайтом SalI между фрагментами pAF 2-6 и pAF 2-7. Результаты этих экспериментов показывают, что кодирующая фитазу последовательность локализована в левосторонней части вставки BamHI в pAF 2-3.
В ходе последующих исследований была полностью расшифрована нуклеотидная последовательность вставок плазмид pAF 2-3, pAF 2-6 и pAF 2-7. Для этой цели использовали методику прерывания дидезокси-цепей (Sanger et al., 1977, Proc. Natl. Acad. Sci. USA, 74, 5463-5467) и методику бомбардировки, описанную Messing et al. (Nucl. Acids Res. 1981, 9, 309-321). Специфические олигонуклеотиды синтезировали на основании данных о нуклеотидной последовательности, полученных в процессе секвенирования.
Полная нуклеотидная последовательность клонов pAF 2-3, pAF 2-8 и pAF 2-7, включающая хромосомный локус гена фитазы, представлена на фиг.11-17, а схематическое изображение локуса - на фиг.18.
Анализ способности полной последовательности кодировать белок показал, что N-концевая последовательность аминокислотных остатков в сформировавшейся молекуле белка кодируется нуклеотидным участком, начинающимся в положении 381 (по данным Ullah N-конец локализован в положении 369). Установлено также, что N-концевые последовательности внутренних пептидных фрагментов с молекулярными массами 38 кДа и 2,5 кДа (см.фиг.2, последовательности В и А) кодируются нуклеотидными последовательностями, которые начинаются в положениях соответственно 1101 и 1548. Открытая считываемая последовательность заканчивается нуклеотидом в положении 1713.
Эти результаты со всей очевидностью подтверждают наличие кодирующей фитазу последовательности ДНК в описанном выше хромосомном локусе.
В считываемой последовательности, примыкающей к считываемой последовательности зрелого белка, в восходящем направлении от хромосомной нуклеотидной последовательности, которая кодирует полную молекулу фитазы, не обнаружен стартовый ATG кодон. Тем не менее, на основании свойств пограничной между интроном и экзоном зоны можно постулировать присутствие интрона между нуклеотидами в положениях 254 и 355. Это позволяет предположить наличие ATG кодона в положении 210 последовательности, включающей последовательность для кодирования зрелой фитазы. Расчетная последовательность аминокислотных остатков в N-концевом участке полностью соответствует закономерностям строения последовательности, ответственной за секреторный сигнал, которая была установлена von Heyne (Eur. J. Biochem., 1963, 133, 17-21).
Для подтверждения высказанных гипотез была предпринята попытка выделить кДНК фитазы с помощью амплификации специфической затравкой, с использованием в качестве матрицы общей популяции мРНК/ДНК. Процедура выделения описана ниже.
Выделение поли-A+ РНК из Aspergillus ficuum.
Общую РНК выделяли из штамма NHHL 3135 A.ficuum, культивировавшегося в активирующей среде, как описано в примере 6. Сухой мицелий замораживали в жидком азоте и измельчали. После этого порошок гомогенизировали в гомогенизаторе uetra-Furrax в течение 1 мин при максимальной скорости в 3 M ZiCl и 6 М мочевине при 0oC. Гомогенат оставляли на ночь при 4oC, как описано Auffrey and Rougeon (Eur. J.Biochem., 107, 303-314, 1980). Общую клеточную РНК получали после центрифугирования при 16000 g в течение 30 мин и двух последовательных экстракций смесью фенола, хлороформа и изоамилового спирта (50:48: 2), РНК осаждали этанолом и разводили в 1 мл 10 мМ трис-HCl буфера (pH 7,4) с 0,5% додецилсульфатом натрия. Для отбора поли-A+ РНК общую РНК прогревали при 60oC в течение 5 мин, доводили до 0,5 М раствором NaCl и наносили на колонку из олиго (dT)-целлюлозы. После нескольких промывок раствором, содержащим 10 мМ трис-HCl буфер pH 7,4 с 0,5% додецилсульфата натрия и 0,1 М NaCl, поли-A+ РНК элюировали 10 мМ трис-HCl буфером pH 7,4 и 0,5%-ным додецилсульфатом натрия.
Получение комплекса мРНК/кДНК.
Для синтеза первой нити кДНК 5 мкг поли-A+ РНК разводили в 16,5 мкл H2O и добавляли в полученный раствор следующие компоненты: 2,5 мкл PHазина (30 ед/мкл), 10 мкл буферного раствора с 50 мМ трис-HCl pH 7,6, 6 мМ MgCl2 и 40 мМ KCl; 2 мкл 1 М KCl, 5 мкл 0,1 М ДТТ, 0,5 мкл олиго (dT)12-18 (2,5 мг/мл), 5 мкл 8 мМ dNTP-mix, 5 мкл сывороточного альбумина крупного рогатого скота (1 мг/мл) и 2,5 мкл ревертивной транскриптазы Moloney MLV (200 ед/мл). Смесь инкубировали в течение 30 мин при 37oC, реакцию останавливали добавлением 10 мкл 0,2 М ЭДТА и 50 мкл H2O. Для экстракции использовали хлороформ, а после центрифугирования к супернатанту последовательно добавляли 110 мкл 5 М NH4Ac и 440 мкл этанола. Комплекс мРНК/кДНК осаждали смесью этанола с сухим льдом в течение 30 мин. Осажденный комплекс собирали посредством центрифугирования, отмывали 70%-ным этанолом, охлажденным до 0oC, и разбавляли 20 мкл H2O.
Клонирование фрагментов кДНК фитазы.
Последовательности кДНК, кодирующие фитазу, выделяли с помощью цепной полимеразной реакции (ЦПР) в составе двух фрагментов. Исходя из кодирующей фитазу геномной последовательности,
показанной на
фиг.11-17, конструировали олигонуклеотидные затравки:
Oliga 1:5' - GGG.TAG.AAT.TCA.AAA.ATG.GGC.GTC.TCT.GCT.GTT.CTA-3'
Oligo 2:5'
- AGT.GAC.GAA.TTC.GTG.CTG.GTG.GAG.ATG.GTG.TCG-3'
Oligo 3:5' - GAG.CAC.CAA.GCT.GAA.GGA.TCC-3'
Oligo 4:5' - AAA.CTG.CAG.GCG.TTG.AGT.GTG.ATT.GTT.TAA.AGG.G-3'
Олиго 1 содержит
нуклеотидную последовательность, расположенную в
нисходящем направлении от стартового кодона ATG (в положениях 210-231) и фланкированную по 5'-границе сайтом EcoRI. Олиго 2 содержит нуклеотидную
последовательность, непосредственно примыкающую к
восходящему концу сайта SalI (положения 1229-1109) и также фланкированную дополнительным сайтом EcoR1. Олиго 3 содержит нуклеотидную
последовательность, локализованную рядом с сайтом Bam HI (положения
845-866). Олиго 4 содержит нуклеотидную последовательность, расположенную в нисходящем от стоп-кодона фитазы направлении (в
положениях 1890 - 1867) и фланкированную дополнительным сайтом Pst 1.
Цепную полимеразную реакцию осуществляли в соответствии с рекомендациями фирмы - изготовителя Tag-полимеразы (Cetus). В качестве матрицы использовали раствор (1,5 мкл), содержащий вышеописанные гибриды мРНК/кДНК, а в качестве затравок - олиго 1 и 2 (по 0,3 мкг каждой) в реакции амплификации N-концевой части кДНК фитазы и олиго 3 и 4 в реакции амплификации C-концевой части той же кДНК (см.фиг.19,20). После денатурации в течение 7 мин при 100oC и добавления 2 ед Tag-полимеразы реакционную смесь подвергали 25 циклам амплификации в ДНК - амплификаторе модели Perkin-Elmer/Cetus (каждый цикл по 2' при 55oC, 3 при 72oC, 1' при 94oC. В последнем цикле стадию денатурации опускали. После переваривания (Eco R1 для N-концевой части кДНК и Bam HI и Pst I для C-концевой части кДНК) оба фрагмента кДНК клонировали в соответствующие сайты pTZ18R (Promega).
Нуклеотидные последовательности обоих полученных фрагментов определяли методом прерывания дидезокси-цепей (Sanger, supra), используя синтетические олигонуклеотиды, сконструированные на основании данных о последовательности хромосомного гена фитазы. Эти олигонуклеотиды служили затравками, а в качестве матрицы применяли общую амплифицированную ДНК, а также клонированные фрагменты кДНК. Последовательность области кДНК, кодирующей фитазу, и расчетная аминокислотная последовательность этого фермента показаны на фиг.19,20.
Анализ последовательности кДНК подтвердил постулированную ранее локализацию интрона и показал отсутствие в пределах хромосомной генной последовательности других интронов.
Ген фитазы кодирует первичный продукт трансляции, состоящий из 467 аминокислотных остатков, с молекулярной массой 51091. Процессинг первичного продукта с отщеплением сигнального пептида, приводит к образованию зрелого белка с фитазной активностью, который построен из 444 аминокислотных остатков при молекулярной массе 48851 или из 448 остатков (включая 4 первые остатка, о которых сообщал Ullah) при молекулярной массе порядка 49232.
Пример 9. Сверхэкспрессия фитазы в грибах рода Aspergillus после введения дополнительных геномных последовательностей ДНК фитазы.
Конструирование вектора экспрессии.
Все векторы конструировали с помощью стандартных методов молекулярной биологии, как описано в Руководстве по лабораторному клонированию молекул (Maniatis et al., Molecular cloning: A Laboratory manual. Celd spring Harbor Laboratory, NY, 1982).
Вектор экспрессии pAF 2-2S получали посредством субклонирования фрагмента ДНК pVUII длиной 6 килооснований из геномного клона фитазы лямбда AF201 в сайт Sma I pUC19. Полученная таким образом плазмида была обозначена шифром pAF. 2-2 (фиг.9). В качестве маркера селекции для трансформирования Aspergillus использовали фрагмент ДНК EcoR1/. Kpn1 плазмиды pGW 325 (Wernarsk. Thesis, Agr. Univ. Wageningen, Netherlands, 1986), содержащий гомологичный ген amdS aspergillus nidulans. Этот фрагмент вставляли в сайты EcoR1/Kpn1 pAF 2-2. Синтезированный таким образом вектор экспрессии получил обозначение pAF2-2S. Он показан на фиг.21.
А. Сверхэкспрессия фитазы в A.ficuum NRRL 3135.
Плазмиду pAF2-2S интродуцировали в клетки штамма NRRL 3135 A.ficuum
с помощью методики трансформации, описанной Tilburn J. et al., (Gene, 1983, 26, 205-221) и Kelly J. and
Hynes M. (EMBO, J., 1985, 4, 457-479) со следующими изменениями:
- мицелий выращивали
на минимальной питательной среде для грибов Aspergillus (Cove D., Biochem. Biophys. Acta., 1966, 113,
51-56), обогащенной 10 мМ аргинина и 10 мМ пролина, при 30oC на протяжении 16 ч в колбе,
встряхиваемой с частотой 300 оборотов/мин,
- для формирования протопластов вместо хеликазы
применяли Novozym 234 (NOVO Industri)
- через 90 мин после образования протопластов в
суспензию добавляли 1 объем буфера STC (1,2 М сорбитол, 10 мМ трис-HCl pH 7,5, 50 мМ CaCl2) и
центрифугировали ее при 2500g и 4oC в течение 10 мин во вращающемся роторе; затем
протопласты отмывали и ресуспендировали в буфере STC до получения концентрации 108 клеток/мл,
- к 100 мкл суспензии протопластов добавляли плазмидную ДНК в 10 мкл буфера TE (10 мМ
трис-HCl pH 7,5 и 0,1 мМ ЭДТА);
- после инкубации суспензии протопластов и ДНК при 0oC в течение 25 мин в нее по каплям добавляли 200 мкл раствора полиэтиленгликоля (25%
полиэтиленгликоль 4000 фирмы Merck) и 10 мМ трис-HCl pH 7,5 с 50 мМ CaCl2. Затем доливали еще 1
мл раствора полиэтиленгликоля (60% полиэтиленгликоля 4000 в 10 мМ трис-HCl pH 7,5 с 50 мМ
CaCl2), медленно и при непрерывном перемешивании. После инкубации при комнатной температуре
суспензию разбавляли буфером STC и перемешивали посредством взбалтывания и последующего
центрифугирования при 2000g и 4oC в течение 10 мин. Протопласты осторожно ресуспендировали в 200 мкл
буфера STC и наслаивали на минимальную питательную среду для грибов Aspergillus,
содержавшую 20 мМ ацетамида в качестве единственного источника азота, а также 15 мМ CSCl и 1 М сахарозы, и
отверждавшуюся 0,75%-ным бактериологическим агаром # 1 (Oxoid). Рост продолжался при 35oC в течение 6 - 10 дней.
Выделяли отдельные трансформанты, обозначавшиеся как SP4, SP7 и SP8, очищали их и анализировали на способность продуцировать фитазу во встряхиваемых колбах, используя процедуру, описанную в примерах 1 и 2. Аналогичные операции проводили с трансформантами, содержавшими только вектор (ген amdS в pUC 19), или с нетрансформированными хозяевами (контрольные эксперименты).
Штамм грибов выращивали в условиях активации (см. пример 6), пробы для анализа отбирали спустя 96 ч после начала культивирования. Анализы включали определение фитазной активности (табл.4) и изоэлектрическую фокусировку в полиакриламидном геле.
Пробы, полученные из ферментированных культур A. ficuum и A.ficuum pAF2-2S SP7, выращивавшихся в идентичных условиях, в равных объемах наносили на полиакриламидный гель для изоэлектрофокусировки (Phast System, Pharmacia) в диапазоне pH от 4,5 до 6,0. Электрофорез проводили в соответствии с указаниями предприятия - изготовителя системы. Затем гель окрашивали кумасси бриллиантовым синим на общие белки (фиг.23) или красителем, выявлявшим общую фосфатазную активность, как описано в примере 2 (фиг.22).
Кроме того, использовали образец фитазы A.ficuum, очищенной до гомогенного состояния посредством иммунноаффинной хроматографии, как описано в примере 7, либо как таковой, либо в смеси с культуральной жидкостью.
Как отмечалось ранее, фитаза присутствует в отдельных пробах в разных изоформах (указаны звездочками). Очищенная фитаза при обоих способах окраски дает хорошо видимые полосы 3 и 4, соответствующие двум основным изоферментам (A и B). Аналогичные полосы едва заметны при окраске фитазы из родительского штамма A. ficuum. В трансформированном штамме pAF 2-2S SP7 они значительно более интенсивны.
Б. Сверхэкспрессия
фитазы в грибах A.niger CBS 513.88
Вектор экспрессии pAF 2-2 интродуцировали в
клетки A.niger CBS 513.88 в процессе трансформации, как описано для A.ficuum. Отдельные трансформаты выделяли,
очищали и определяли их способность продуцировать фитазу во встряхиваемых колбах в
условиях индуцированного роста, как описано в примере 6.
Уровень экспрессии фитазы в отдельных трансформантах, обозначенных как pAF 2-2S ≠ 8, ≠ 20 и ≠ 33, а также в контрольных штаммах определяли, как описано в примере 9A и показано в табл.5.
Уровень экспрессии фитазы в трансформированных клетках.
A.niger был сопоставим с ее уровнем в трансформированных A.ficuum. Кроме того, полученные результаты показывают, что промотор фитазы A.ficuum активен в A.niger.
Дальнейшие анализ проводили с использованием культуральной среды трансформированных клеток pAF 2-2S ≠ 8, которую подвергали электрофорезу в полиакриламидном геле с pH 4,5-6,0 для изоэлектрофокусировки (Phast System, Pharmacia) в соответствии с ранее описанной процедурой. Равные объемы супернатанта культуральной жидкости родительского штамма и трансформированных клеток pAF 2-2S ≠ 8, выращивавшихся в идентичных условиях, наносили на гель, который окрашивали по завершении электрофореза, как описано выше.
Родительский штамм A.niger синтезировал очень небольшие количества фитазы и ее не удавалось выявлять методом гелевого электрофореза. Штамм pAF 2-2S ≠ 8 продуцировал примерно в 90 раз больше фермента, чем родительский штамм. Это различие хорошо видно на фиг.24, 25.
Выявлено несколько изоформ фермента (обозначены звездочками). Окрашивание на общие белки показало, что интенсивность соответствующих полос резко увеличивается, тогда как полосы других белков отсутствуют.
Пример 10. Экспрессия фитазы в A.niger, трансформированных векторами экспрессии, содержащими ген фитазы A.ficuum, слитый с промотором и/или сигнальной последовательностью гена амилоглюкозидазы (AG) A.niger.
Конструирование векторов экспрессии.
Для достижения сверхэкспрессии фитазы в A.niger были получены дополнительные блоки экспрессии, в которых ген фитазы A.ficuum контролируется промотором амилоглюкозилазы (AG) A. niger в сочетании с различными сигнальными последовательностями. В p18FYT3 и p24FYT3 соответствующие 18 и 24 - членные лидирующие аминокислотные последовательности (aa) гена AG из A.niger слиты с фрагментом гена фитазы, который кодирует зрелый белок. В векторе экспрессии pFYT3 последовательность промотора AG слита с последовательностью, кодирующей фитазу, которая включает лидирующую последовательность для биосинтеза этого белка.
Конструирование p18FYT3.
Слияние промотора AG и 18
aa AG - лидирующей последовательности с последовательностью, кодирующей полностью сформированную
фитазу, осуществляли с помощью цепной полимеразной реакции. При этом использовали две различные
матрицы: pAF 2-2S, содержащую, как указано выше, полный ген фитазы, и pAB6-1 - плазмиду, включающую
полный локус AG из A.niger, которая была выделена из плазмидной библиотеки A. niger, содержащей
фрагменты HindIII длиной 13-15 килооснований в pUC19. Для выделения использовали AG - специфичные
олигонуклеотиды:
AG-1: 5'-GACAATGGCTACACCAGCACCGCAACGGACATTGRRRGGCCC-3'
AG-2:
5'-AAGCAGCCATTGCCCGAAGCCGAT-3'
Оба олигонуклеотида получены на основании нуклеотидной
последовательности для A. niger (Boel et. al., 1984. EMBOJ., 3., 1097-1102; Boel et al., 1984, Mol.
& Cell. Biol., 4, 2306-15). Олигонуклеотидные зонды конструировались, исходя из строения
последовательностей, окружающих интрон 2: олиго AG-I локализован в положении 3' по отношению к интрону и
имеет такую же полярность, как AG мРНК, а олиго AG-2 расположен вверх от интрона 2 и
антипараллелен AG мРНК. Плазмида pAB6-I содержит ген AG во фрагменте Hind III длиной 14,5 килооснований.
В качестве затравок для амплификации цепной полимеразной реакции применяли
четыре синтетические олигонуклеотида, имеющих показанное ниже строение:
Oligo 1:
5'-CTCTGCAGGAATTCAAGCTAG-3' (AG-специфичная последовательность вокруг EcoR1 - сайта, примерно на 250
килооснований вверх от ATG - стартового кодона
Oligo 18-2:
5'-CGAGGCGGGCACTGCCAGTGCCAACCCTGTGCAGAC-3' зрелая фитаза ←_→ 18 AA AG-лидирующая последовательность
Oligo 18-3: 5'-GTCTGCACAGGGTTGGCACTGGCAGTCCCCGCCTCG-3' 18 aa
AG-лидирующая последовательность - зрелая фитаза
Oligo 4: 5'-GGCACGAGGATCCTTCAGCTT-3' (специфичная для фитазы последовательность,
локализованная на сайте BamHI в положении 861).
Цепную полимеразную реакцию осуществляли в соответствии с методикой Saiki et al., 1988, Science 239, 487-491), с небольшими изменениями (см. пример 8).
Для слияния последовательностей AG с последовательностями, кодирующими фитазу, ставили две отдельные цепные полимеразные реакции. В первой из них для амплификации фрагмента ДНК длиной 300 пар оснований, содержащего 3' - участок AG - промотора и 18 aa AG-лидирующую последовательность, фланкированную по 3'-границе нуклеотидами гена фитазы, использовали в качестве затравок олигонуклеотиды 1 и 18-2, а в качестве матрицы - pAB 6-1. Во второй реакции использовали матрицу pAF 2-2 и олигонуклеотиды 18-3 и 5 в качестве затравок для амплификации фрагмента ДНК длиной 600 пар оснований, содержащего 5'-участок гена фитазы, фланкированный по 5'-границе 18 нуклеотидами сигнального пептида AG. Процедура амплификации в схематическом виде показана на фиг.27.
Полученные таким образом два фрагмента ДНК очищали посредством электрофореза в полиакриламидном геле и преципитации этанолом, а затем использовали в качестве матриц в третьей цепной полимеразной реакции с олигонуклеотидами 1 и 4 для затравки при слиянии AG-фитазы. Образовавшийся при этом фрагмент ДНК переваривали EcoR1 и BamHI и субклонировали в pTZ18R. Полученный комплекс секвенировали и обозначали как p18FYT1.
Остальная, восходящая часть промотора AG длиной 3,5 килооснований была получена посредством переваривания pAB 6-1 Kpn1 и частично EcoRI, а затем присоединена к фрагменту EcoRI/BamHI p18FYT1 длиной 1,1 килооснования и клонирована в сайты KpnI/MamHI pTZ18R. Полученная описанным способом плазмида показана на фиг.29-31.
Дополнительный рестрикционный участок Hind III
индродуцировали, используя вставку синтетического фрагмента

в сайт EcoR1, фланкирующий ген amdS pAF2-2S. Полученной таким образом плазмиде присваивали обозначение pAF2-2S (фиг.28) и использовали ее в качестве стартовой плазмиды для замены последовательностей промотора фитазы фрагментами ДНК, полученными в результате слияния AG-фитазы в цепной полимеразной реакции.
На заключительном этапе конструирования плазмиды p18FYT2 и pAF2-2S переваривали KpnI и частично BamHI. Фрагмент ДНК p18FYT2 длиной 4,6 килооснований и фрагмент ДНК pAF 2-2S длиной 11 килооснований после выделения подвергали очистке посредством гелевого электрофореза, а затем лигировали и переносили в E. coli. Полученный этим способом вектор экспрессии получил обозначение p18FYT3 (фиг.29-31).
Конструирование p24FYT3.
Слияние - промотора и 24 aa AG - лидирующей
последовательности с последовательностью, кодирующей зрелую фитазу, осуществляли в процессе амплификации цепной
полимеразной реакции по методике, описанной выше для конструирования p18FYT3, с тем
отличием, что в качестве затравок использовали другие олигонуклеотиды:
Oligo 24-2:
5'-CGAGCCGGGGACTGCCAGGCGCTTGGAAATCACATT-3' зрелая фитаза ←_→ 24 AA AG-лидирующая
последовательность
Oligo 24-3: 5'-AATGTGARRRCCAAGCGCCTGGCAGTCCCCGCCTCG-3' 24 aa
AG-лидирующая последовательность - зрелая фитаза
Проводили две разные цепные полимеразные реакции. В
первой из них в качестве матрицы использовали pAB 6-1, а олигонуклеотиды 1 и 24-2
применяли в качестве затравок для амплификации фрагмента ДНК длиной 318 пар оснований, который содержал 3' - участок
промотора AG и 24 aa AG-лидирующую последовательность, фланкированную по 3'
- границе 18 нуклеотидами гена фитазы. Во второй реакции в качестве матрицы применяли pAF 2-2S, а олигонуклеотиды 24-3 и 4
использовали для затравки при амплификации фрагмента ДНК, содержащего
5'-участок гена фитазы, фланкированный по 5'-границе 18 нуклеотидами 24 aa AG - лидирующей последовательности. Процедура
амплификации в схематическом виде представлена на фиг.29.
На заключительном этапе конструирования вектора экспрессии p24FYT3 с использованием плазмид p25FYT1 и p24FYT2, применяли ту же методику клонирования, которая описана для случая получения вектора p18FYT3 через промежуточные стадии конструирования p18FYT1 и p18FYT2 (фиг.29-31).
Конструирование pFYT3.
Слияние AG - промотора с последовательностью гена фитазы, включавшего
лидирующую последовательность, осуществляли в процессе амплификации цепной полимеразной реакции, как описано выше при
конструировании p18FYT3, с тем отличием, что применяли следующие
олигонуклеотиды:
Oligo fyt-2: 5'-AACAGCAGAGACGCCCATTGCTGAGGTGTAATGATG-3' лидирующая последовательность ←_→
AG-промотор
Oligo fyt-3:
5'-CATCATTACACCTCAGCAATGGGCGTCTCTGCTGTT-3' AG - промотор ←_→ лидирующая последовательность
Ставили две разные цепные полимеразные
реакции. В первой из них в качестве
матрицы использовали pAB 6-1, а олигонуклеотиды 1 и fyt-2 служили в качестве затравок при амплификации фрагмента ДНК длиной 282 пары оснований, содержащего 3'
- участок AG-промотора, фланкированного
по 3'-границе 18 нуклеотидами лидирующей последовательности фитазы. Во второй реакции в качестве матрицы использовали pAF 2-2S, а олигонуклеотиды fyt-2 и 4
служили для затравки при амплификации
фрагмента ДНК, содержащего 5'-участок гена фитазы (включая лидирующую последовательность фитазы) и фланкированного по 5'-границе 18 нуклеотидами AG-промотора.
Процедура амплификации в схематическом
виде представлена на фиг.29.
На заключительном этапе конструирования вектора экспрессии pFYT3 через промежуточные плазмиды pFYT1 и pFYT2, применяли методику клонирования, описанную при конструировании p18FYT1 и p18FYT2 для получения блока экспрессии p18FYT3 (фиг.29-31).
Экспрессия гена фитазы под контролем промотора AG
B
A.niger
Из описанных
выше блоков экспрессии фитазы посредством переваривания Hind III удаляли последовательности E.coli. После этого штамм CBS 513.88 A.niger (передан на хранение
10 октября 1988 г.) трансформировали
фрагментом ДНК в количестве 10 мкг, как описано в примере 9. Из каждого блока экспрессии выделяли индивидуальные трансформаты A.niger. Споры высевали на
селективные ацетамид-агаровые пластины. Споры
каждого трансформанта собирали после выращивания клеток на пластинах, покрытых агаром с 0,4% картофельной декстрозой (Oxoid, England), при 37oC
в течение 3 дней. Продукцию фитазы оценивали
во встряхиваемых колбах при следующих условиях культивирования.
Примерно 108 спор вносили в 100 мл исходной среды, содержавшей 1 г/л KH2PO4, 30 г/л мальтозы, 5 г/л дрожжевого экстракта, 10 г/л казеинового гидролизата, 0,5 г/л MgSO4•7H2O и 3 г/л твина 80, pH полученной смеси доводили до 5,5.
После культивирования во вращающемся сосуде при 34oC в течение ночи 1 мл растущей культуры переносили в 100 мл основной среды: 2 г/л KH2PO4, 70 г/л мальтодекстрина (Maldex MDO3, Amylum), 12,5 г/л дрожжевого экстракта, 25 г/л казеинового гидролизата, 2 г/л K2SO4; 0,5 г/л MgSO4• 7H2O; 0,03 г/л ZnCl2; 0,02 г/л CaCl2, 0,05 г/л MnSO4•4H2O и FeSO4, pH полученной смеси доводили до 5,6.
Мицелий выращивали на протяжении по меньшей мере 140 ч. Продукцию фитазы оценивали, как описано в примере 2. Результаты этой оценки с использованием нескольких произвольно выбранных трансформантов из каждого блока экспрессии, показаны в табл.6.
Продукция фитазы несколькими штаммами A.niger CBS 513.88, трансформированными плазмидами, которые содержат ген фитазы A.ficuum, контролируемый промотором AG A. niger в сочетании с различными лидирующими последовательностями.
Эти данные убедительно свидетельствуют о высоком уровне экспрессии фитазы в трансформированных клетках A.niger, в которых ген фитазы контролируется AG - промотором A.niger. Они показывают также, что максимальная продукция фитазы достигается при использовании вектора экспрессии pFYT3, который включает лидирующую последовательность фитазы. При использовании сходных векторов экспрессии, в которых имеется безинтронный ген фитазы (после трансформации в A.niger), экспрессия фитазы была сопоставима с таковой в A.niger, трансформированных.
Дополнительно супернатанты культур клеток, трансформированных pFYT3 # 205 и # 282, подвергали электрофорезу в полиакриламидном геле для изоэлектрофокусировки в диапазоне pH от 4,5 до 6,0. Равные объемы супернатантов из культур родительского штамма A.niger и обоих трансформантов, выращивавшихся в идентичных условиях, наносили на гель и по окончании электрофореза окрашивали, как описано в примере 9. Родительский штамм A.niger синтезировал очень небольшие количества фитазы, которую не удавалось определить использовавшимся методом. Штаммы pFYT3 # 205 и # 282 синтезировали соответственно в 250 и 1400 раз больше фитазы по сравнению с родительским штаммом (сравните продукцию фитазы по сравнению с родительским штаммом и сравните продукцию фитазы по данным табл.4 и 5). Это различие особенно хорошо иллюстрируется фиг. 24, 25. Идентифицированы несколько изоформ фитазы (обозначены звездочками). Окраска на общие белки сопровождалась появлением чрезвычайно интенсивных полос фитазы, тогда как полосы, соответствующие другим основным белкам, отсутствовали.
Пример 11. Сверхэкспрессия фитазы в A.ficuum и A.niger при культивировании в промышленных масштабах.
А. A.ficuum
Штаммы pAR 2-2S # 4 и NRRL 3135 A.ficuum культивировали, как описано в
примере 1. Оба типа трансформированных клеток
синтезировали примерно в 50 раз больше фитазы чем клетки дикого штамма того же гриба.
Сверхэкспрессия фитазы трансформированными клетками A.ficuum, содержащими множественный ген фитазы. Клетки культивировали, как описано в примере 1.
Б. A.niger
Штамм A.niger pAF2-2S #8, трансформированный штамм A.niger CBS
513.88 и родительский штамм A.niger
культивировали, как описано в примере 1. Трансформированные клетки синтезировали примерно в 1000 раз больше фитазы по сравнению с исходным родительским штаммом
A.niger (табл.8).
Сверхэкспрессия фитазы трансформированными клетками A. niger (CBS 513.88), содержащими множественный ген фитазы. Клетки культивировали, как описано в примере 1.
Пример 12. Для конструирования вектора pREPFYT3, с помощью которого достигались одновременно экспрессия фитазы и замена гена AG, плазмиду pFYT3 переваривали Kpn1. Полученный линейный фрагмент ДНК Kpn1 использовали для лигирования в двух разных случаях.
Лигирование 1 с использованием адаптера Kpn1-HindIII:

Лигирование 2 с использованием адаптера Kpn1-HindIII*, в котором рестрикционный участок Hind III не сохраняется после
лигирования:

Последовательность, полученную при лигировании 1, частично переваривали HindIII. После удаления фрагмента, содержащего ген amdS, посредством гелевого электрофореза, остаточный фрагмент ДНК переводили посредством лигирования в кольцеобразную форму и переносили в E.coli. Полученную таким образом плазмиду обозначали как pFYT3 amdS (см. фиг.32).
Последовательность, полученную в результате лигирования 2, также переваривали HindIII, фрагмент ДНК Hind III/Hind III* длиной 4 килооснования, содержащий ген amdS, выделяли с помощью электрофореза и лигировали с частично переваренной Hind III плазмидой HindIII/HindIIi*, после чего ее переносили в E.coli. Плазмида, у которой на 3'-конце гена фитазы имелся ген amdS, получила обозначение pFYT3INT (см. фиг.33).
Для интродуцирования фрагмента SalI/HindIII ДНК
плазмиды pAB6-1 длиной 6
килооснований, содержащего 3'-фланкирующую AG - область, плазмиду pFYT3INT частично переваривали Hind III и присоединяли сначала к адаптеру

(в котором рестрикционный участок Hind III* не сохраняется после лигирования), а затем к фрагменту SalI/HindIII плазмиды pAB6-1. После трансформации в E. coli получали искомую плазмиду pREPFYT3, содержащую 3'-фланкирующую последовательность AG в требуемом положении.
Экспрессия фитазы в A.niger посредством замещения гена AG.
Перед трансформаций A. niger плазмидой pREPFYT3 из последней удаляли последовательности E.coli (путем переваривания Hind III и гелевого электрофореза). Штамм CBS 513.88 A.niger трансформировали 10 мкг фрагмента ДНК, используя процедуру, описанную в примере 9. Селекцию и культивирование трансформированных клеток осуществляли, как описано в примере 9. Лишь незначительное количество отобранных трансформантов (приблизительно 20%) утрачивало AG-активность. Гибридационный анализ хромосомной ДНК в AG-отрицательных и положительных по фитазе трансформантах использовали для контроля замены гена AG геном фитазы.
Пример 13. Консервация гена фитазы в разных видах микроорганизмов.
Для выяснения степени консервативности гена фитазы в микроорганизмах проводили гибридизационный анализ хромосомных ДНК десяти различных видов, используя в качестве зонда кДНК фитазы A.ficuum.
Хромосомные ДНК для анализа получали из нитчатых грибов, дрожжей и бактерий. Из каждой группы организмов отбирали ограниченное число видов, в частности: нитчатые грибы Penicillium chrysogenium и Aspergillus niger, дрожжи Saccharomyces cerevisiae и Kluyveromyces betis, такие прокариотные организмы, как грам-положительные формы, Bacillus subtilis, Clostridium Thermocellum, Streptomyces lividans, а также грам-отрицательные бактерии Pseudomonas aeruginosa.
Высокомолекулярные хромосомные ДНК этих видов переваривали pVU II и Bam HI (раздельно) и подвергали электрофорезу в 0,7%-ном агарозном геле.
После переноса на нитроцеллюлозные фильтры проводили гибридизацию в нелимитирующих условиях (6 x SSC при 50oC в течение ночи) с меченным32P фрагментом 5' кДНК фитазы (как описано в примере 8). Комплексы отмывали в 6 x SSC при комнатной температуре и на протяжении 18 ч подвергали воздействию рентгеновского облучения.
Как показано на фиг. 35, 36, практически на каждой электрофореграмме имеются дискретные полосы, свидетельствующие о высокой степени гомологичности генов фитазы из различных видов микроорганизмов.
Реферат
Изобретение может быть использовано в микробиологическом производстве фитазы. Разработан экономически рентабельный способ производства фитазы. Получен фрагмент геномной ДНК и к ДНК, кодирующих фитазу Aspergillus niger и имеющих определенные аминокислотные последовательности. Рекомбинантная плазмидная ДНК p FYT3 для эспрессии фитазы в Aspergillus имеет размер 15,3 т.п. о. и состоит из вектора РТZ 18 и фрагмента ДНК, оперативно связанного с промотором глюкоамилазы Aspergillus niger, содержит ген amds Aspergillus nidulans в качестве селективного маркера. Рекомбинантная плазмидная дНК pAF - 2 - 2 S для экспрессии фитазы в Aspergillus имеет размер 13, 4 т.п.о., состоит из вектора pUC 19 и pVU II фрагмента, размером 6,1 т.п.о. Последний содержит ДНК фрагмент. Плазмида включает ген amds Aspergillus nidulons в качестве селективного маркера. Штамм Aspergillus niger CBS 513.88 трансформирован рекомбинантной плазмидой pFYT3 или pAF 2 - 2S. Штамм Aspergillus ficuum NRRL 3135 трансформирован рекомбинатной плазмидой pFYT3. Штаммы являются продуцентами фитазы Aspergillus. Фитазу получают культивированием клеток-продуцентов фитазы Aspergillus niger или Aspergillus ficuum. Рекомбинантную фитазу Aspergillus niger получают в клетках Aspergillus ficuum NRRL 3135 или Aspergillus niger СВ 513.88 кодированием фрагментом ДНК. Фитаза имеет N - концевую последовательность Leu Ala Val Pro Ala Ser Arg Asn Gln Ser Gly Asp Thr Val Asp, удельную активность 100 ед/мг, оптимум pH 5,5, тампературу 50oC, молекулярную массу 85 кД. 9 с.п. ф-лы, 36 ил., 8 табл.

Комментарии