Способ получения бензоата и замещенных бензоатов олова (iv) - RU2660905C1
Код документа: RU2660905C1
Описание
Изобретение относится к технологии получения солей олова (IV) и может быть использовано в различных областях химической и иных практик, в аналитическом контроле и в научных исследованиях.
Известен способ получения карбоксилатов олова, в том числе и в степени окисления (IV), путем непосредственного взаимодействия раздробленного металла с карбоновой кислотой в присутствии промотора (пат. США №6303808, опубл. 10/16/2001). В качестве промотора используют 4-трет-бутилкатехол, 2,5-ди-трет-бутилгидрохинон и т.д., которые добавляют в чистом виде или в носителе типа диола, гликоля или карбоновой кислоты, в частности дипропиленгликоля и 2-этил-1-гексановой кислоты. Количество промотора оговорено от 1 до 20% от загрузки металлического олова. Этот диапазон ограничен 1-2% как наиболее распространенными вариантами. Температурный режим по ходу процесса разный. Вначале реакционную смесь нагревают до 60°С и подают воздух или иной кислородсодержащий газ. Такой режим поддерживают некоторое время, после чего реакционную смесь нагревают до 140-180°С и в таком режиме выдерживают реакционную смесь какое-то время до накопления в ней некоторого количества оловосодержащих продуктов. Далее процесс завершают или кислородсодержащий газ меняют на инертный, например азот, и еще какое-то время продолжают в указанном диапазоне температур. Выбор делается в зависимости от того, какая соль (олова (II) или олова (IV)) нужна в качестве целевого продукта. Выделение целевого продукта предлагается проводить несколькими методами. Но все они довольно сложны в исполнении.
Недостатками данного способа являются:
1. В нем нет четкого разделения на благоприятные варианты для получения карбоксилатов олова (II) и олова (IV) с перечнем характеристик, как влияют природа кислоты-реагента, промотора и других компонентов, включая и организацию контакта реакционной смеси с кислородсодержащим и инертным газом, на пооперационные составляющие благоприятных и неблагоприятных вариантов и их временные характеристики.
2. В целом цитируемый способ довольно сложен в исполнении как на этапе получения карбоксилатов олова, так и на этапах разделения реакционных смесей, выделения и очистки продуктов, а также утилизации не вошедших в продукт компонентов загрузки.
3. Недостаточная оговоренность режимных и временных характеристик фактически предлагает желающему использовать данный способ на практике дополучать их самому, т.е., по существу, выполнить определенный объем исследований, что предполагает высокую квалификацию этого человека и достаточный опыт его работы с аналогичными системами.
4. 140-180°С - далеко не малые температуры. В этом диапазоне β-форма металла может переходить в γ-форму (tпер=160°C). Возможно и прямое взаимодействие металла с кислотой с образованием Н2 (положение в ряду напряжений это позволяет). Это усложняет газовую среду (кислородсодержащий газ + водород), что свидетельствует о наличии определенных границ, выход за которые недопустим. Никакой информации по этому поводу в цитируемом способе нет.
5. Нет и определенных рекомендаций, как выделять непрореагировавшее олово, насколько оно блокировано поверхностными отложениями продукта(ов) и как его рационально использовать в дальнейшем.
6. При разделении реакционных смесей предусматривается отгонка непрореагировавшей кислоты под вакуумом, что гораздо более сложно в аппаратурном оформлении и в исполнении, чем удаление небольшого избытка растворителя жидкой фазы при фильтровании твердых продуктов с возвратом полученного фильтрата на загрузку повторного процесса.
Наиболее близким к заявленному является способ трибохимического получения металлсодержащих мыл - компонентов жирующих смесей (пат. РФ №2092533, опубл. 10.10.1997; БИ №28), в соответствии с которым соли переходных и иных металлов получают путем прямого взаимодействия оксидов, гидроксидов и карбонатов металлов с С7 и выше карбоновыми кислотами индивидуально и в виде смесей в присутствии 1-2% воды и 0,08-0,10% по массе трибохимического катализатора, лучшими из которых являются нитраты аммония, первичные, вторичные, третичные амины и амиды кислот. При этом в качестве растворителя используют парафиновые и ароматические углеводороды, их технические смеси и масло ПОД, в который вводят масляную составляющую (рыбий жир и подсолнечное масло, сложноэфирные и неомыляемые составляющие талового и кислых масел, индустриальные масла, отходы СЖК). Сначала готовят раствор при интенсивном перемешивании и нагревании всех компонентов, кроме металлсодержащего, в бисерной мельнице с высокооборотной мешалкой лопастного типа, в который дозируют металлсодержащий реагент и ведут процесс получения целевого продукта, остающегося в реакционной смеси, используемой как единое целое.
Недостатками данного способа являются:
1. Цитируемый способ не предполагает получение с выделением индивидуального карбоксилата, подтвержденного соответствующей идентификацией. Поэтому нет никаких сведений о том, представляет ли получаемый карбоксилат смесь основной и средней соли или же является одной из них.
2. В реакционной смеси рассматриваемого способа есть восстановители, в частности непредельные соединения, которые могут окисляться солями металлов в более высокой степени окисления, как это имеет место при получении плавленых сиккативов (пиролюзит → соли марганца (II), оксид кобальта → в соли Со (II)). Правда, температуры здесь значимо более низкие, но исключать получение соли двухвалентного металла из его оксида в степени окисления 4 без экспериментального подтверждения никак нельзя.
3. Присутствие масляной составляющей в реакционной смеси на получение и выделение целевого карбоксилата просто недопустимо. Мало того, что эта составляющая содержит восстановители, но она существенно влияет на растворимость солей в сторону увеличения, а также затрудняет отделение перешедшей в твердую фазу целевой соли, требует дополнительной отмывки и очистки продукта от себя. А это дополнительные сложности и затраты при выделении и очистке продукта, что выразится в значимом снижении выхода целевого продукта.
4. Цитируемый способ предполагает использование довольно большого ассортимента многокомпонентного, переменного по составу и примесям от партии к партии технического сырья, удовлетворяющего требованиям кожевенного производства, использующего и получаемые растворы мыл как единое целое. При получении конкретного карбоксилата металла как целевого продукта в этом нет никакой необходимости, и использование такого подхода может оказаться крайне вредным.
5. В цитируемом решении нет четкого качественного и количественного соотнесения в системе оксид металла:карбоновая кислота:трибохимический катализатор. Полученные данные последних лет при работе с чистыми моделями таких систем показывают, что каждая из таких систем имеет оптимальный вариант, который предстоит найти. Причем совсем не обязательно, что он будет находиться в диапазонах, обозначенных в цитируемом решении.
Задачей предлагаемого решения является подобрать такие условия проведения процесса непосредственного взаимодействия оксида олова (IV) с бензойной и рядом замещенных бензойных кислот, которые бы обеспечивали высокие выход и избирательность получения карбоксилата олова (IV) при практически количественном расходовании диоксида олова и небольших стехиометрических избытках кислоты в отсутствие трибохимического катализатора и при соизмеримых с комнатной температурах, а также при накоплении основной массы целевого продукта в твердой фазе, легко отделяемой путем простого фильтрования и освобождаемой от непрореагировавшей кислоты отмывкой растворителем.
Поставленная задача достигается тем, что оксид олова (IV) в количестве 0,1-0,68 моль/кг вводят в контакт с раствором-суспензией кислоты в уайт-спирите, предварительно приготовленным в том же реакторе после загрузки стеклянного бисера, подогретого до 49-60°С растворителя и взятой с 5%-ным избытком в отношении стехиометрического на среднюю соль кислоты включением и проведением перемешивания в отсутствие подвода внешнего тепла в течение 10-20 мин, после чего проводят процесс при контроле за расходованием кислоты до момента практически полного расходования диоксида, затем перемешивание прекращают, суспензию твердой соли отделяют от перетирающего агента и фильтруют, остатки реакционной смеси в бисерной мельнице и на перетирающем агенте удаляют промывным растворителем, который в дальнейшем используют для промывки осадка продукта на фильтре, осадок хорошо отжимают, снимают с фильтра, сушат на воздухе до постоянной массы и определяют содержание в нем основного вещества. При этом в качестве кислоты используют бензойную, салициловую, п-оксибензойную, анисовую, антраниловую, п-аминобензойную, п-нитробензойную, м-нитробензойную, фенилантраниловую, м-хлорбензойную, ацетилсалициловую, 5-аминосалициловую, галловую, а фильтрат и промывной растворитель возвращают в повторный процесс.
Характеристика используемого сырья:
Оксид олова (IV) ГОСТ 22516-77
Кислоты:
Бензойная ГОСТ 6413-77
Салициловая ГОСТ 624-70
п-Оксибензойная ТУ 6-09-3646-74
Анисовая ТУ 6-09-2796-73
Антраниловая ТУ 6-09-3821-74
п-Аминобензойная ТУ 6-09-3395-78
п-Нитробензойная ТУ 6-09-1935-77
м-Нитробензойная ТУ 6-09-19-73
Фенилантраниловая ТУ 6-09-3592-74
м-Хлорбензойная ТУ 6-09-08-1128-76
Ацетилсалициловая ФС 42-0220-07
5-Аминосалициловая ТУ 6-09-07-691-76
Галловая ТУ 6-09-3591-74
Уайт-спирит ГОСТ 3134-78
Проведение процесса заявляемым способом следующее. В бисерную мельницу вертикального типа с высокооборотной лопастной мешалкой, стеклянным корпусом с небольшими различиями в диаметрах входного отверстия и корпуса вводят расчетные количества стеклянного бисера как перетирающего агента, предварительно подогретого до 49-60°С уайт-спирита и кислоты. Корпус с загруженными компонентами помещают в предназначенное для него гнездо каркасной рамы, соединяют с крышкой бисерной мельницы, снабженной сальниковой коробкой с механической мешалкой с лопастью из нержавеющей стали, отверстиями для помещения датчика температуры и пробоотборника, и должным образом крепят. Включают механическое перемешивание и в течение определенного времени готовят раствор-суспензию кислоты. По истечении его в реактор без прекращения перемешивания загружают расчетное количество диоксида олова (IV) и этот момент принимают за начало процесса получения карбоксилата. Контроль ведут методом отбора проб и определения в них содержания непрореагировавшей кислоты. Как только последнее приближается к выбранному значению избыточного в загрузке, перемешивание прекращают, снимают крепление корпуса в каркасной раме, отсоединяют крышку и опускают корпус таким образом, чтобы остатки реакционной смеси с вала и лопасти мешалки и других элементов стекали в корпус. Далее содержимое корпуса мельницы выливают в воронку узла отделения перетирающего агента с сеткой с ячейками 0,3×0,3 мм. Оставшийся на сетке перетирающий агент аккуратно возвращают в корпус бисерной мельницы. Проводят повторную сборку и крепление бисерной мельницы, прокручивают вручную вал мешалки, вводят некоторое количество растворителя жидкой фазы, включают механическое перемешивание и проводят отмывку корпуса реактора, мешалки и перетирающего агента от остатков реакционной смеси (PC). По истечении заданного времени перемешивание прекращают и проводят повторное отделение перетирающего агента от промывного растворителя, который собирают в отдельную емкость и в дальнейшем используют для промывки осадка на фильтре. Параллельно отделенную от стеклянного бисера суспензию PC фильтруют, осадок на фильтре промывают промывным растворителем, отжимают, вместе с фильтром снимают и отправляют на сушку, а при необходимости на очистку путем перекристаллизации. Фильтрат и промывной растворитель взвешивают, анализируют на содержание карбоксилата и кислоты и используют в загрузках повторных процессов.
Пример 1
В бисерную мельницу вертикального типа с высокооборотной (3000 об/мин) мешалкой с размерами лопасти 41×21×2,5 мм из нержавеющей стали на целесообразную загрузку 100-110 г вводят 100 г стеклянного бисера, 83,41 г подогретого до 55°С уайт-спирита и 12,81 г (0,105 моль) бензойной кислоты. Стеклянный корпус реактора помещают в предназначенное для него гнездо каркасной рамы, соединяют с крышкой, должным образом крепят, проверяют вручную прокручивание вала мешалки, включают механическое перемешивание и этот момент принимают за начало приготовления раствора кислоты. Последнее длится 15 мин. За это время вся введенная в реактор кислота перешла в раствор. Не прекращая перемешивания, в реактор через отверстие для отбора проб вводят 3,77 г (0,025 моль) диоксида олова и этот момент принимают за начало получения карбоксилата. Процесс ведут при естественно складывающейся температуре и текущем контроле за расходованием кислоты на образование продукта. Результаты такого контроля приведены в таблице 1.
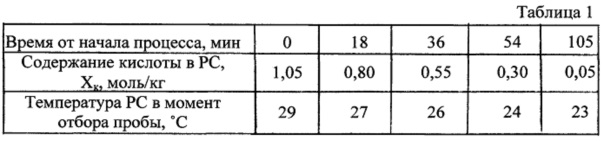
Когда величина остаточной кислоты достигает расчетного значения (0,05 моль/кг), перемешивание прекращают, корпус реактора отсоединяют от крышки с механической мешалкой и опускают вниз таким образом, чтобы остатки PC могли стечь в корпус с вала, лопасти механической мешалки и поверхностей других закрепленных на крышке элементов. На это отводится около 5 мин. Далее корпус перемещают к узлу отделения суспензии PC от стеклянного бисера и выливают его содержимое в приемлемую воронку для выполнения этой операции. В качестве фильтровальной перегородки в ней использована сетка с размерами ячеек 0,3×0,3 мм, которая легко пропускает суспензию PC и на которой полностью задерживается находившийся в зоне реакции стеклянный бисер. Отделенный бисер тщательно снимают с сетки и переносят в корпус реактора. Бисерную мельницу как реактор собирают вновь, вводят 35 г уайт-спирита, включают механическое перемешивание и в течение 12 мин проводят отмывку стенок и содержимого реактора от остатков PC. По истечении указанного времени перемешивание прекращают, проводят повторное отделение стеклянного бисера, а промывной растворитель собирают и используют в промывке осадка продукта на фильтре, который получают фильтрованием отделенной ранее от стеклянного бисера PC. Промытый осадок тщательно отжимают, аккуратно снимают вместе с фильтром и отправляют на воздушную сушку. Во время сушки его периодически несколько раз измельчают, добиваясь превращения в порошкообразное состояние. Такой порошок периодически взвешивают, пока получаемые значения не достигнут минимальной стабильной величины. После этого определяют эквивалентную массу и выход выделенного и высушенного продукта. Он оказался равным 96,1%. Оценка потерь при выделении в выбранных условиях составила 3,8%.
Фильтрат и промывной растворитель взвешивают, объединяют и определяют содержание соли олова (IV) и остаточной кислоты. В фильтрате они оказались равными 0,054 и 0,006 моль/кг. После этого их направляют на загрузку повторных процессов. Прямым экспериментом подтверждено, что такой вариант не только возможен, но и целесообразен.
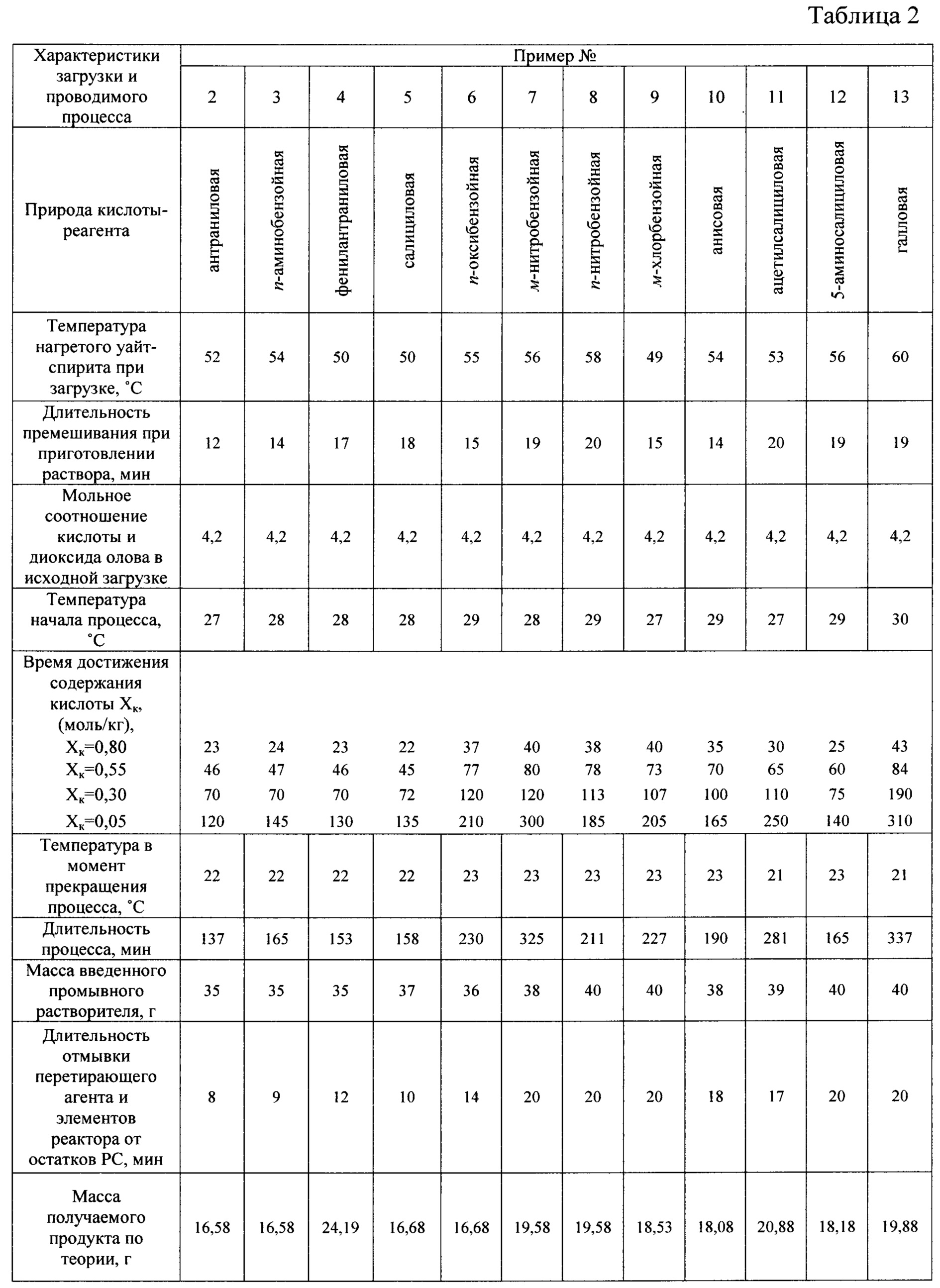
Примеры 2-13
Реактор, исходный диоксид олова, растворитель жидкой фазы, загрузка диоксида, мольное соотношение диоксид:кислота, соотношение масс загрузки и стеклянного бисера, последовательности операций при приготовлении раствора-суспензии, проведении процесса, его контроле, выборе момента завершения, завершении, отделении перетирающего агента, отмывке его, внутренней поверхности корпуса и поверхностей других элементов реактора от остатков PC, выделении из последней твердого продукта, его промывке, сушке и измельчении, а также при возврате фильтрата и промывного растворителя на загрузку повторного процесса аналогичны описанным в примере 1. Отличаются природой используемой в качестве реагента кислоты, в качестве которой выбраны некоторые замещенные бензойные кислоты, и связанными с ней растворимостью кислоты и продукта, а также временными характеристиками операций. Указанные различия и другие характеристики процесса сведены в таблицу 2.

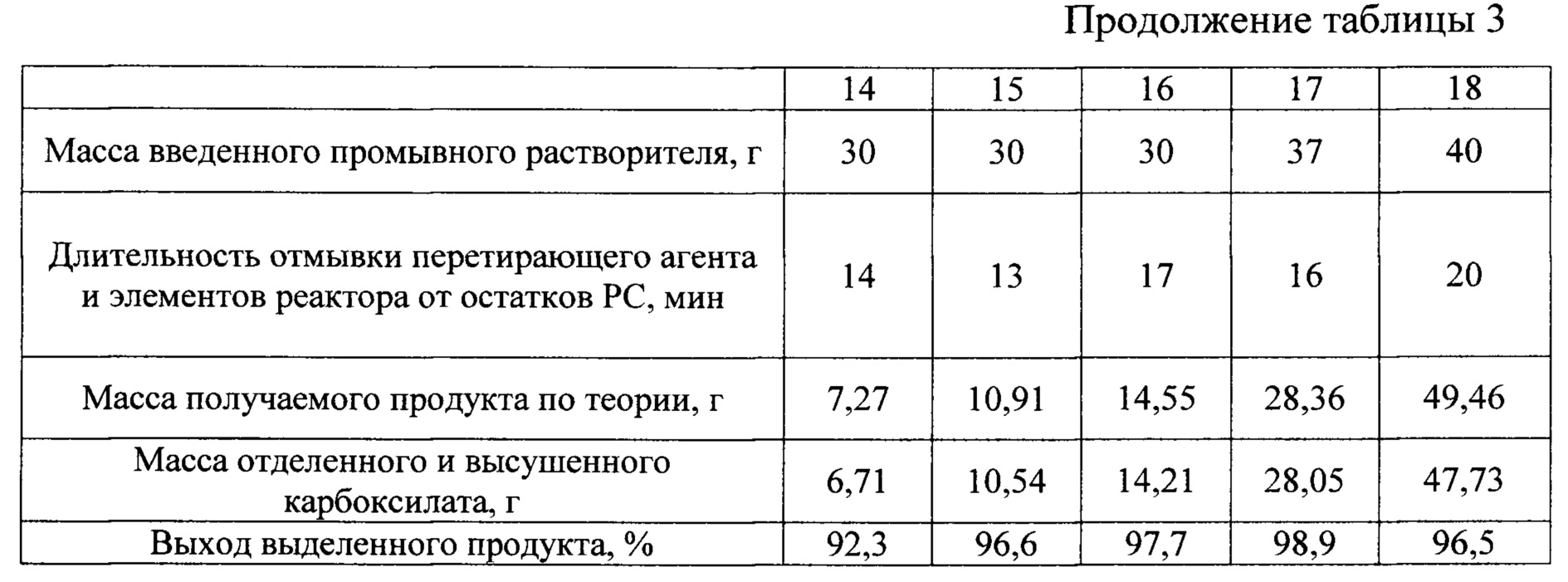
Примеры 14-18
Реактор, исходные реагенты, растворитель жидкой фазы, перетирающий агент, последовательности операций при загрузке, приготовлении раствора-суспензии кислоты, проведении процесса, его завершении, отделении перетирающего агента, отмывке его и элементов реактора от остатков PC, выделении из последней твердого целевого продукта и использовании фильтрата и промывного растворителя в загрузке повторных процессов аналогичны описанным в примере 12. Отличаются величиной начальной загрузки диоксида олова при сохранении постоянным мольного соотношения кислоты и диоксида в загрузке. Все это вместе с другими характеристиками сведено в таблицу 3.

Положительный эффект предлагаемого решения состоит в том, что:
1. Предлагаемое решение может быть с успехом реализовано для многих замещенных бензойных кислот без какой-либо существенной корректировки на ее природу, растворимость в выбранной жидкой фазе, различия в молекулярных массах как кислоты, так и продукта.
2. Способ прост в исполнении и в аппаратурном оформлении. Он не нуждается в организации ни подвода внешнего тепла, ни отвода реакционного. В нем нет необходимости в разделении жидких фаз реакционной смеси - их просто как единое целое возвращают на загрузку повторных процессов. В результате этого нет потерь и растворимых в них целевых продуктов. Сказанное в полной мере относится и к промывному растворителю.
3. Предлагаемый способ не требует значимых избытков кислоты-реагента, что исключает, с одной стороны, необходимость решения вопросов возврата непрореагировавшей кислоты в повторный процесс, а с другой - возможное загрязнение целевого продукта плохо растворимой в выбранной жидкой фазе кислотой.
4. Целевые продукты в выбранном растворителе являются очень плохо растворимыми компонентами, что предопределяет их накопление в твердой фазе и отделение путем простого фильтрования, т.е. одним из наиболее простых и дешевых способов.
Реферат
Изобретение относится к способу получения бензоата и замещенных бензоатов олова (IV) прямым взаимодействием диоксида олова с бензойной, салициловой, п-оксибензойной, анисовой, антраниловой, п-аминобензойной, п- и м-нитробензойными, фенилантраниловой, м-хлорбензойной, ацетилсалициловой, 5-аминосалициловой и галловой кислотами в уайт-спирите в бисерной мельнице с высокооборотной (3000 об/мин) мешалкой в присутствии стеклянного бисера как перетирающего агента. Сначала загружают расчетные количества бисера, подогретого до 49-60°C уайт-спирита и кислоты 5%-ный избыток по стехиометрии, включают механическое перемешивание и ведут приготовление раствора-суспензии кислоты при естественно складывающейся температуре. Затем, не прекращая перемешивания, вводят расчетное количество диоксида олова и ведут процесс получения карбоксилата при текущем контроле за содержанием непрореагировавшей кислоты до достижения величины избыточно загруженного количества этого реагента. После достижения этого перемешивание прекращают, отсоединяют реактор со своего гнезда в каркасной раме, снимают крышку, дают возможность остаткам реакционной смеси стечь в корпус, последний освобождают от содержимого переносом его в воронку с сеткой, где отделяют перетирающий агент от суспензии реакционной смеси. Перетирающий агент возвращают в корпус реактора, собирают бисерную мельницу, добавляют растворитель и ведут отмывку корпуса, элементов реактора и бисера от остатков реакционной смеси, после чего бисер отделяют повторно. Суспензию реакционной смеси фильтруют, осадок на фильтре промывают промывным растворителем, отжимают, снимают с фильтра и сушат до постоянной массы. По отношению этой массы к теоретическому значению определяют выход выделенного продукта. Он оказался равным 95±2,5%, что меньше степени превращения диоксида (приближается к 100%) за счет потерь при разгрузке реактора и в процессе выделения продукта. Фильтраты с промывным растворителем возвращают в повторные процессы. Способ позволяет получать продукты с высокими выходом и избирательностью. 2 з.п. ф-лы, 3 табл., 18 пр.
Комментарии