Способ получения 2-гидрокси-4-метилтиомасляной кислоты (гидроксианалогаметионина, гам), гам - RU2130925C1
Код документа: RU2130925C1
Чертежи




Описание
Изобретение относится к способу получения 2-гидрокси-4-метилтиомасляной кислоты согласно ограничительной части пункта 1 формулы, к получаемому согласно изобретению ГАМ и к применению полученного по этому способу ГАМ.
Изобретение относится в частности к усовершенствованному способу нового типа выделения ГАМ в очень высокой концентрации и с очень высокой степенью чистоты из смеси, получаемой при осуществлении реакции.
2-Гидрокси-4-метилтиомасляная кислота (ГАМ) представляет собой гидроксианалог незаменимой аминокислоты метионин в рацемической форме и является, как и аминокислота, важной добавкой к кормам для животных. В птицеводстве ГАМ проявляет ростстимулирующие свойства, сходные с таковыми у известной этой же способностью аминокислоты. Однако и в других областях, связанных с кормлением животных, этот аддитив вызывает повышенный интерес.
В большинстве случаев ГАМ применяют в виде водных концентратов, причем эти последние наряду с мономерами содержат также некоторые количества олигомеров, главным образом ди- и тримерные линейные эфирокислоты. Содержание этих олигомеров зависит от условий получения и выбранной концентрации. Однако по причине их малой питательной ценности и неблагоприятного воздействия на текучесть, обусловленного повышением вязкости, желательно их процентную долю поддерживать на минимальном уровне. Коммерчески доступные композиции при общей концентрации 88-90 мас.% держат в общей сложности до 24 мас.%, соответственно около 27 мол.%, олигомеров, что соответствует соотношению мономеры/олигомеры 3:1.
Известно также применение в качестве добавок к кормам для животных соли кальция и смешанной кальциевоаммониевой соли ГАМ. Однако получение таких солей связано со значительным повышением их себестоимости. Кроме того, будучи порошкообразными твердыми веществами, они труднее поддаются примешиванию к композициям кормов для животных по сравнению с легко распыляемыми водными концентратами из свободной кислоты с низким содержанием олигомеров.
Синтез ГАМ включает 3 реакции.
При осуществлении общепринятого способа получения ГАМ исходят из 3-метилтиопропиональдегида, называемого также метилмеркаптопропиональдегидом или сокращенно ММПА, который взаимодействием с цианистым водородом трансформируют в 2-гидрокси-4-метилтиобутиронитрил, называемый также ММПА-циангидрином или сокращенно ММПА-ЦГ (уравнение I).
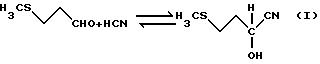
Затем образовавшийся ММПА-циангидрин обычно с помощью сильных минеральных кислот через промежуточную стадию получения 2-гидрокси-4-метилтиобутирамида, называемого также ГАМ-амид (уравнение II),

гидролизуют до гидроксианалога метионина (ГАМ) (уравнение III)

Этот гидролиз может проводиться как по одноступенчатому, так и по двухступенчатому механизму, причем под "ступенями" следует понимать то, что в реакцию гидролиза ММПА-ЦГ либо один раз, либо дважды добавляют минеральную кислоту и/или воду, т.е. число ступеней соответствует числу введения добавок.
Двухступенчатая методика, которую применяют исходя из ММПА-циангидрина, описана в патентах США 2745745, 2938053 и 3175000. Согласно этим публикациям сначала при относительно низких температурах взаимодействием с концентрированной минеральной кислотой, например, с 50-85%-ной серной кислотой, циангидрин трансформируют в ГАМ-амид и затем после добавления воды при повышенной температуре продолжают процесс гидролиза с получением ГАМ. Путем обработки омыляемой смеси гидроксидом кальция либо карбонатом кальция из нее получают кальциевую, соответственно кальциевоаммониевую соль ГАМ, а в качестве продукта реакции сочетания сульфат кальция. Для снижения неизбежного образования малоценных побочных продуктов обе первые из вышеназванных публикаций рекомендуют агент гидролиза, каковым является серная кислота, применять в меньшем стехиометрическом соотношении относительно ММПА-циангидрина, например, в соотношении 0,55-0,8: 1. Патент Великобритании 722024, в котором описывается аналогичное образование ГАМ-солей исходя из ГАМ-амида, также включает двухстадийную методику.
Двухступенчатый гидролиз применяют также в способах, описанных в европейских заявках 0142488 (взаимодействием с серной кислотой) и 0143100 (взаимодействием с минеральной кислотой), предметом изобретения в которых является получение ГАМ в жидкой форме, т.е. в виде высококонцентрированных водных растворов. Эти последние получают посредством реакции гидролиза, проводимой в указанных выше условиях концентрирования и температурного режима через стадию амида взаимодействием с избыточной минеральной кислотой, с последующей экстракцией растворителей, причем в качестве таковых применяют определенные растворители, частично смешиваемые с водой.
Согласно приведенным в этих публикациях данным отличительный признак описанного в них способа следует усматривать в получении ГАМ из экстракционного раствора, которое осуществляют таким образом, что процесс получения включает удаление органического растворителя в присутствии по крайней мере приблизительно 5 мас.% воды в пересчете на образующийся экстракт (ГАМ). ГАМ получают из экстракционного раствора путем перегонки (см. примеры), причем предпочтительна дистилляция с водяным паром. Полученный после удаления растворителя из экстракционного раствора путем перегонки с водяным паром погон представляет собой смесь ГАМ и воды. В соответствии с этим дистилляцию с водяным паром проводят таким образом, что содержание воды в погоне составляет по крайней мере 5 мас.%.
В указанных публикациях говорится далее, что при проведении дистилляции условия в колонне регулируют так, что в ней повсюду, по крайней мере, однако, в кубовом остатке, содержание воды в жидкой фазе составляет 5 мас.%.
Из этого следует, что без присутствия достаточного количества воды во время получения ГАМ из экстракционного раствора следует опасаться увеличения образования нежелательных побочных продуктов (димеров и олигомеров).
При перегонке водяной пар, кроме того, служит в качестве активного агента, способствующего полному удалению экстрагирующего агента из раствора ГАМ, например, за счет образования низкокипящей азеотропной смеси с соответствующим экстрагирующим агентом.
Концентрированные растворы ГАМ без помощи растворителя получают согласно патенту США 3773927 посредством двухстадийного гидролиза ММПА- циангидрина, осуществляемого взаимодействием с избытком водной соляной кислоты, последующим концентрированием омыляемой смеси и отделением кристаллизовавшихся хлоридов аммония. Однако полученные таким путем ГАМ-концентраты отличаются высоким содержанием олигомеров и имеют к тому же черный цвет. Выделенный хлорид аммония также сильно загрязнен примесями.
Согласно патенту США 4353924 после солянокислотного гидролиза, проводимого по двухступенчатому механизму, избыток минеральной кислоты нейтрализуют аммиаком либо другими щелочными веществами. Таким путем получают концентрированные растворы ГАМ с низкими коррозионными свойствами.
В патенте США 4310690 описывается способ, в котором после проведения гидролиза с помощью соляной кислоты в четко заданных условиях нейтрализуют едким натром и хлорид аммония преобразуют в поваренную соль и аммиак. При последующей обработке едкой известью получают кальциевую соль ГАМ в виде взвеси в практически насыщенном растворе поваренной соли. После разделения в системе твердое тело-жидкость фильтраты большей частью возвращают в процесс для повторного приготовления взвесей едкой извести. Благодаря такой методике снижают сброс сточных вод и предотвращают образование побочных продуктов, представляющих собой бесполезные или экологически небезопасные балластные вещества.
Одноступенчатые способы проведения гидролиза также описаны в патентной литературе. Так, способ, заявленный в патенте Великобритании 915193, предназначен для получения кальциевой соли ГАМ; этот способ заключается в том, что после омыления ММПА-циангидрина избытком разбавленной серной кислоты образующийся ГАМ экстрагированием с помощью высококипящих простых эфиров отделяют от омыляемого раствора и последующей обработкой экстракта гидроксидом кальция получают кальциевую соль ГАМ. Предусмотренный в этом способе, осуществляемом как непрерывный процесс, возврат водного рафината в стадию омыления приводит тем не менее к аккумулированию неорганических побочных продуктов реакции.
В европейской заявке 0330527 описан еще один одноступенчатый способ гидролиза с использованием серной кислоты в качестве агента омыления, в котором обходятся без растворителя и который приводит непосредственно к получению концентрированных водных растворов ГАМ, причем в качестве побочного продукта получают кристаллический сульфат аммония в готовом для поставок на рынок виде. Указанную цель достигают за счет того, что омыляемую смесь нейтрализуют раствором гидроксида аммония до такой степени, которая обеспечивает перевод избыточной минеральной кислоты и образовавшегося бисульфата аммония в нейтральный сульфат, причем во время этого процесса образуются две жидкие фазы, которые в свою очередь разделяют и упаривают с целью получения, во- первых, жидкого ГАМ и, во-вторых, кристаллического сульфата аммония. При этом различные процессы фильтрации и возврата комбинируют таким образом, что практически ни один продукт не идет в отходы и не образуются сточные воды, содержащие соль. Качество получаемого таким путем ГАМ сходно с таковым продукта, который получают согласно европейской заявке EP-0142488.
Однако и этот экологически безвредный способ имеет ряд недостатков. Как установил автор настоящего изобретения при работе по этому способу, для полной конверсии циангидрина требуются, что обусловлено необходимостью сравнительно более сильного разбавления серной кислоты (20-50%), гораздо большие избыточные количества кислоты, чем это указано. Кроме того, для предотвращения осаждения соли в процессе нейтрализации приходится работать с сильно разбавленной кислотой, что обусловлено необходимостью разделения обеих жидких фаз с требуемой степенью чистоты. Помимо этого, выделенный сульфат аммония имеет клейкую консистенцию и источает сильный запах, что обусловливает необходимость последующих операций по переработке, таких, например, как фильтрационная промывка или перекристаллизация, которые дополнительно удорожают способ. Более того, для проведения процесса упаривания - в отличие от указанного в заявке - требуются более высокие энергозатраты по сравнению со способом в EP-A 0142488, на который в заявке делаются ссылки. Значительное повышение материально-технических затрат связано, в частности, с работой с двумя раздельными потоками твердого материала, предусматривающей фильтрацию/центрифугирование, а также с не показанной на схеме движения материала сушкой сульфата аммония.
В соответствии с указанным выше уровнем техники и с учетом недостатков известных способов в основу изобретения была положена задача разработать еще один способ получения 2-гидрокси-4-метилтиомасляной кислоты (ГАМ = гидроксианалог метионина) описанного выше типа, который был бы максимально прост по своему осуществлению, не требовал высоких материально-технических затрат на переработку реакционных продуктов и обеспечивал бы получение максимально высококонцентрированного продукта с предельно низким содержанием димеров, олигомеров и побочных продуктов. Целью изобретения является также получение более высококачественного ГАМ и его применение.
Эта, равно как и другие не указанные более подробно задачи решаются с помощью способа описанного выше типа, отличительные признаки которого представлены в пункте 1 формулы изобретения.
Предпочтительные варианты осуществления способа заявлены в пунктах формулы, зависимых от главного пункта 1. Объектом пункта 8 формулы является более высококачественный ГАМ, что представляет собой решение одной из задач согласно изобретению. Новые применения являются объектом пунктов 9 и 10 формулы изобретения.
Благодаря тому, что упаривание экстракционного раствора с целью получения ГАМ в качестве конечного продукта (целевой экстракт) проводят таким образом, что целевой экстракт содержит менее 4 мас.%, предпочтительно менее 2 мас.% воды, согласно изобретению предлагается способ, обеспечивающий получение жидкого ГАМ исключительно высокого качества и позволяющий совершенно неожиданно иметь высококонцентрированный жидкий ГАМ с незначительным содержанием олигомеров и димеров. При сравнении со способами, известными из уровня техники, прежде всего с описанными в обеих европейских заявках 0142488 и 0143100, следует признать более чем неожиданной такую возможность получения ГАМ с меньшим содержанием воды, чем это может быть очевидным для специалиста на основании вышеназванных патентов. Неожиданной согласно изобретению оказалась далее возможность получения практически 100%-ных растворов ГАМ, которые можно транспортировать без заметных потерь качества и которые затем с помощью соответствующих добавок или разбавителей можно доводить до требуемой концентрации. Эти характеристики обеспечивают огромные экономические преимущества, позволяющие применять способ в промышленном масштабе.
Таким образом, в рамках настоящего изобретения удается решить важную проблему, прежде всего касательно упаривания реакционного раствора, образующегося в результате экстракции в системе жидкость-жидкость реакционной смеси, которую получают, например, посредством гидролиза ММПА-ЦГ с помощью серной кислоты.
Применяемый для упаривания экстракционный раствор, предназначенный для получения ГАМ, может быть получен по известному специалисту способу из реакционной смеси, например, путем экстракции. Используемый при этом для экстракции органический растворитель не должен в основном обладать способностью смешиваться с водой. Допустима, правда, лишь частичная возможность смешивания органического растворителя с водой. Среди растворителей, пригодных для проведения разделения веществ путем жидкостно-жидкостной экстракции, имеется множество таковых, которые отвечают условиям химической индифферентности и малой водорастворимости. В принципе предпочтительна водорастворимость растворителя при комнатной температуре, не превышающая примерно 15 мас.%, предпочтительно не более 10 мас.%. Среди приемлемых растворителей предпочтительны такие, температура кипения которых находится в интервале от приблизительно 60oC до приблизительно 200oC, предпочтительно от приблизительно 70oC до 150oC. Коэффициент распределения растворителя, содержащегося в экстрагированном ГАМ, и водного рафината, остающегося после контактирования растворителя и ГАМ-гидролизата, должен составлять для ГАМ примерно 2 в равновесной системе. Предпочтительно этот коэффициент распределения равен по крайней мере 5. С другой стороны, коэффициент распределения для ГАМ в равновесной системе экстракционный раствор-промывочная вода не должен быть ниже примерно 1,0. Кроме того, растворитель должен обладать низкой токсичностью.
В качестве растворителей для экстракции пригоден целый ряд кетонов, альдегидов и эфиров карбоновых кислот. К особенно предпочтительным относятся кетоны с относительно малой молекулярной массой, такие, как, например, метил-н-пропилкетон, метилэтилкетон, метиламилкетон, метилизоамилкетон, метилизобутилкетон, этилбутилкетон и диизобутилкетон. Среди растворителей, отличающихся хорошими свойствами для проведения экстракции, следует назвать также альдегиды, такие, как, например, н-бутиральдегид, и сложные эфиры, как, например, этилацетат, н-бутилацетат, н-пропилацетат и изопропилацетат. Можно также использовать спирты, хотя они вследствие их взаиморастворимости с водой, медленного разделения фаз и тенденции вступать в реакцию с ГАМ, менее предпочтительны.
Собственно экстракцию можно в принципе осуществлять как непрерывный либо как полунепрерывный процесс. Для проведения полунепрерывного процесса пригодна, например, емкость с мешалкой. Предпочтительна, однако, непрерывная противоточная экстракция, проводимая в экстракционной аппаратуре, в которой для ускорения массообмена между растворителем и водной фазой предусмотрена экстракционная зона.
Так, например, целесообразно осуществлять экстракцию в каскаде, состоящем из противоточных смесителей-отстойников с непрерывным контактом фаз, загрузочной колонны, тарельчатой колонны, предпочтительно пульсационной колонны либо колонны с вибрирующим пакетом тарелок, роторно-дисковой колонны или в виде центробежного экстрактора. В особенно предпочтительном варианте выполнения экстракцию осуществляют в тарельчатой колонне в системе жидкость-жидкость. Прерывистые или импульсные, т.е. подаваемые с определенной частотой потоки, хотя и являются циклическими и, следовательно, не непрерывными, с точки зрения быстроты перемещения потока в контексте данного описания рассматриваются как "непрерывные".
Процесс экстракции предпочтительно регулируют таким образом, чтобы фаза растворителя в экстракционной зоне была непрерывной и чтобы, соответственно, сохранять ее в этом качестве.
Для снижения содержания соли в конечном продукте до минимума экстракт предпочтительно промывают водой. В непрерывной противоточной экстракционной системе экстракт можно промывать смешением с водой в зоне по ходу движения потока (относительно направления движения потока органического материала), т. е. в том месте, где гидролизат вводят в экстракционную систему жидкость-жидкость. Так, например, в вертикальной колонне при использовании растворителя, удельный вес которого предпочтительно менее 1, этот раствор подают в колонну в месте, расположенном ниже места подачи раствора гидролизата, а промывочную воду подают в колонну в месте, расположенном выше места подачи раствора гидролизата. В предпочтительном варианте выполнения раствор подают с нормой расхода из расчета приблизительно 0,5-0,6 мас.части на единицу массы гидролизата, благодаря чему получают экстракт с удельным весом порядка 0,92-0,97 и общим содержанием ГАМ (общее содержание ГАМ = сумма мономерного ГАМ + димерный ГАМ + олигомеры + необязательно ГАМ-амид) от 35 до 40 мас.%.
Продуктивность процесса экстракции повышают за счет работы при несколько повышенной температуре с тем, чтобы обеспечить для фазы растворителя в экстракционной системе относительно низкую вязкость. Проведение процесса в диапазоне температур от приблизительно 50 до приблизительно 80oC оказывает непосредственное и благоприятное воздействие на коэффициент распределения ГАМ между органической и водной фазами.
В рамках изобретения ГАМ может быть получен из экстракционного раствора, как уже указывалось выше, путем упаривания. В особенно предпочтительном варианте выполнения изобретения органический раствор удаляют во время упаривания с помощью аппарата, который обеспечивает непродолжительность пребывания экстракционного раствора на стадии упаривания. Особенно предпочтительно поэтому отделять органический растворитель при упаривании с помощью пленочного испарителя, тонкопленочного выпарного аппарата и/или глубоковакуумного испарителя или же с помощью какого-либо аппарата подобного типа.
Под понятием "аппарат подобного типа" в рамках настоящего изобретения имеется в виду, что вышеназванные аппараты с непродолжительным временем пребывания в них экстракционного раствора могут также использоваться в сочетании с другими, известными специалисту устройствами для выделения растворителя из экстракционных растворов. При этом речь должна идти не обязательно только об объединяемых в систему устройствах с непродолжительным временем пребывания в них экстракционных растворов. Можно назвать, в частности, также дистилляционные колонны, в которых при необходимости можно предусмотреть также возможность для подачи пара или каких-либо других пригодных агентов отгонки. Возможно также совместное использование нескольких из вышеназванных аппаратов с непродолжительным временем пребывания в них экстракционных растворов.
В предпочтительной модификации способа согласно изобретению целесообразно проводить упаривание экстракционного раствора таким образом, чтобы остаточное содержание растворителя было минимальным. Этого достигают, например, за счет сочетания нескольких вышеназванных аппаратов с секцией отгонки, которая может представлять собой дополнительный аппарат либо которую можно интегрировать в систему указанных выше испарителей, как это имеет место, например, при непосредственном введении отгоночной среды в один из таких испарителей.
Специфичные условия для осуществления процесса упаривания при необходимости варьируют в зависимости от особенностей применяемого для экстракции соответствующего растворителя. В принципе для упаривания с применением экстрактора с непродолжительным временем пребывания в нем экстракционного раствора предпочтительно, чтобы давление во время упаривания составляло не более 600 мбар, предпочтительно 400 мбар и особенно предпочтительно 200 мбар.
Температура упаривания, как правило, также зависит от особенностей применяемого растворителя. Однако стремятся к тому - и это согласно изобретению особенно предпочтительно, - чтобы при операции упаривания температура не превышала 150oC. При существенном превышении указанной температуры это может привести к термическому повреждению целевого продукта. При этом под температурой во время упаривания понимается не необходимая температура контакта продукта с поверхностью испарителя, оснащенного соответствующим образом для такого кратковременного контактирования с продуктом. Под температурой при операции упаривания имеется в виду скорее средняя температура в выпарном аппарате. В некоторых случаях температура на поверхности выпарного аппарата может быть значительно выше 150oC. Решающей является кратковременность контактирования в используемом выпарном аппарате. Благодаря этому предотвращается возможность термического повреждения продукта даже в том случае, если бы температура контакта заметно превысила 150oC.
Касательно распределения температуры в рамках изобретения было установлено, что продукт особенно высокого качества можно получать тогда, когда образующийся экстракт непосредственно на выходе из выпарного аппарата имеет температуру в интервале от 30 до 100oC, предпочтительно от 50 до 95oC и наиболее предпочтительно от 70 до 90oC.
Как уже упоминалось выше, длительность пребывания получаемого экстракта в аппаратуре является одним из решающих факторов, влияющих на качество и состав целевого ГАМ-продукта. В предпочтительной модификации способа согласно изобретению длительность пребывания получаемого экстракта в процессе упаривания составляет не более 1,5 ч. Это относится к продолжительности пребывания во всей выпарной системе, в которой предусмотрена по крайней мере одна операция упаривания с очень короткой продолжительностью пребывания материала. В отличие от указанной как максимальная общей продолжительности пребывания в 1, 5 ч, длительность пребывания в аппарате с кратковременной продолжительностью пребывания следует устанавливать в минутном диапазоне или менее. При всех условиях согласно изобретению в том случае, когда система упаривания состоит только из тонкопленочного выпарного аппарата и/или пленочного испарителя и/или глубоковакуумного испарителя, предпочтительно, чтобы длительность пребывания в этих аппаратах составляла не более 1 ч, предпочтительно 40 мин.
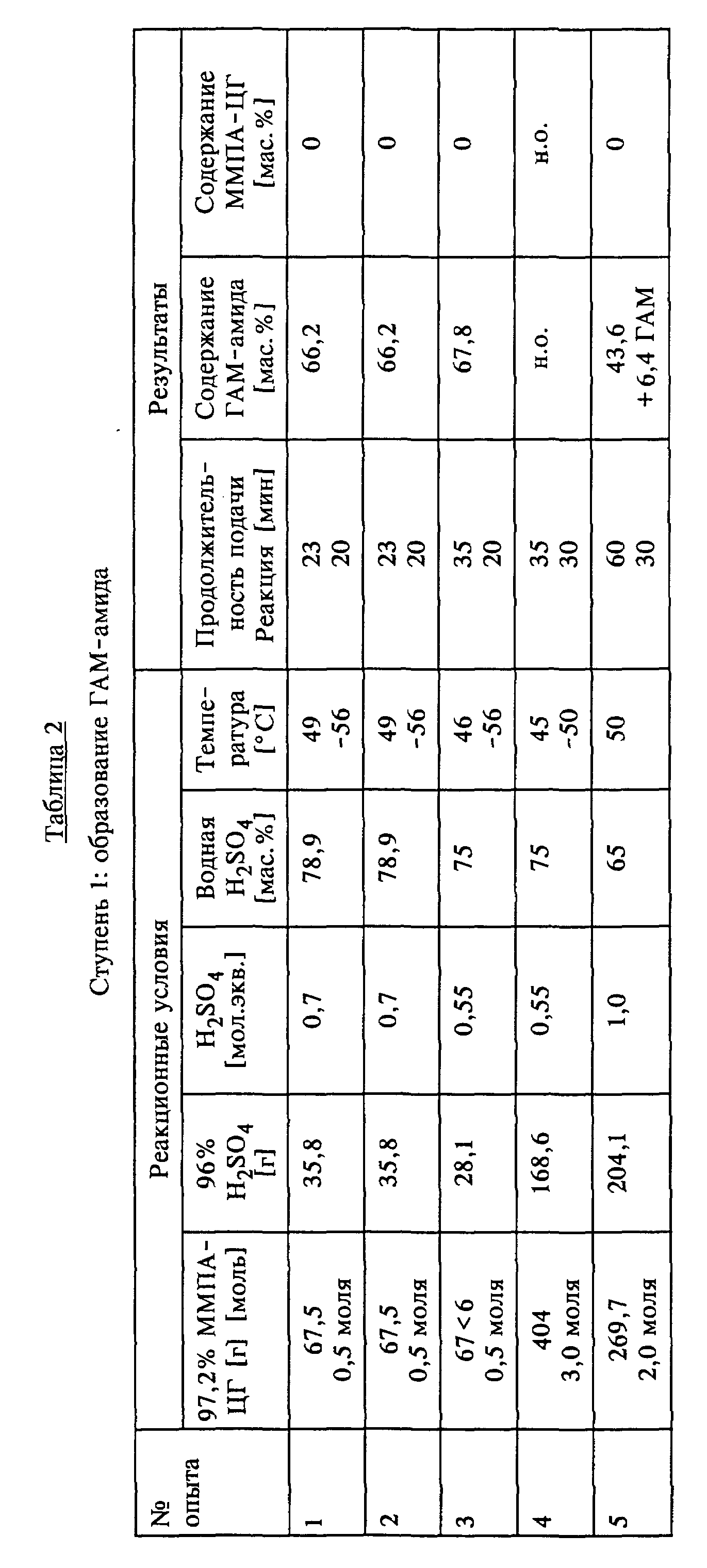
Согласно другому аспекту изобретения в дополнение к выделению ГАМ из получаемой посредством гидролиза с помощью серной кислоты реакционной смеси предлагаемый способ позволяет далее совершенствовать процесс собственно гидролиза ММПА-ЦГ. Так, в предпочтительном варианте выполнения изобретения гидролиз ММПА-ЦГ осуществляют таким образом, что на первой ступени ММПА-ЦГ гидролизуют 60-85 мас.%-ной, предпочтительно 65-80 мас.%-ной серной кислотой в молярном соотношении между ММПА-ЦГ и H2SO4 от 1,0:0,5 до 1:1, 05, предпочтительно от 1: 0,6 до 1:0,8 при температурах в диапазоне 30-90oC, предпочтительно 50-70oC с получением в основном ГАМ-амида. При этом из ММПА-циангидрина в основном образуется ГАМ-амид, причем образующаяся смесь в свою очередь в основном не содержит непрореагировавший ММПА-циангидрин. Другими словами, это означает, что имеет место практически количественный гидролиз.
Другое особое преимущество изобретения в этом аспекте состоит в том, что для дальнейшего совершенствования и полного завершения гидролиза ГАМ-амида до ГАМ гидролиз полученного на первой стадии ГАМ-амида на второй ступени осуществляют добавлением воды и минимального дополнительного количества серной кислоты до верхнего стехиометрического предела, предпочтительно, однако, без дальнейших добавок H2SO4, при температурах до 140oC, преимущественно в диапазоне от 100 до 140oC, предпочтительно в условиях дефлегмации. В принципе гидролиз на второй ступени можно проводить также при температурах ниже 100oC, например, в интервале 90-100oC. Однако в этом случае необходимы дополнительные добавки серной кислоты максимум до верхнего стехиометрического предела. Для полного завершения гидролиза ГАМ-амида до целевого ГАМ без добавления дополнительной серной кислоты, ограничиваясь лишь добавками воды, согласно изобретению была установлена целесообразность работы при температурах порядка 140oC. Таким образом, в соответствии с изобретением двухступенчатый гидролиз ММПА-циангидрина следует осуществлять с помощью более высококонцентрированной по сравнению с уровнем техники серной кислоты в соотношении от ниже стехиометрического до максимум стехиометрического, причем в случае дозировки меньше стехиометрического количества на первой стадии гидролиза (образование амида) при более низкой температуре для сокращения продолжительности реакции на второй стадии при более высокой температуре может добавляться дополнительная серная кислота, при необходимости до достижения верхнего стехиометрического предела, что необходимо для полной конверсии амида в кислоту.
Предметом изобретения является также более высококачественный ГАМ, который получают по описанному выше способу. Этот более высококачественный ГАМ отличается согласно изобретению тем, что содержит в общей сложности 95 мас.% ГАМ, причем это количество представляет собой сумму мономерного ГАМ, димерного ГАМ и ГАМ- олигомеров (= общее количество ГАМ), а также воду в количестве от более 0,1 мас.% до менее 5 мас.%. Особым преимуществом согласно изобретению оказалась возможность получения ГАМ при сохранении практически всех качественных характеристик, который отличается тем, что содержит 98 мас.% ГАМ как сумму мономерного ГАМ, димерного ГАМ и ГАМ-олигомеров, а также воду в количестве от 0,1 до менее 2 мас.% и имеет кинематическую вязкость > 100 мм2/с при 25oC. При этом неожиданным образом было установлено, что кинематическая вязкость, определяемая по Кеннону-Фенске, высококонцентрированного продукта (т.е. ГАМ с содержанием по крайней мере 98 мас.% действующего вещества) после хранения и разбавления сравнима с кинематической вязкостью 88 мас.%-ного продукта. Несмотря на относительно высокое содержание димеров и олигомеров порядка 50 мас.%, которое имел высококонцентрированный продукт после хранения в течение приблизительно 300 дней при комнатной температуре, кинематическая вязкость концентрата после разбавления водой до примерно 88 мас. % соответствует кинематической вязкости 88 мас.%-ного коммерческого продукта, имевшего в соответствующих экспериментальных условиях хранения равновесную концентрацию димеров и олигомеров, составлявшую лить около 25 мас. %. В обоих случаях, т.е. как у разбавленного концентрата, так и у коммерческого продукта, было достигнуто состояние равновесия. Этот факт очень неожидан и является существенным преимуществом, обеспечивающим получение высококонцентрированного ГАМ в одном из вариантов выполнения изобретения. С учетом того, что количество димеров и олигомеров, как правило, отрицательно проявляется в дальнейшем при практической переработке, тем более неожиданной оказалась возможность получения, несмотря на значительное исходное содержание димеров и олигомеров в так называемом высоком концентрате, легко перекачиваемой и тем самым транспортабельной смеси с приемлемой вязкостью. Это дает целый ряд преимуществ, в частности, такая вязкость и прежде всего высокое содержание действующего вещества могут обеспечить экономичную транспортировку высококонцентрированного продукта, поскольку при этом не требуется большого количества воды и, кроме того, в месте доставки, например, на мельнице по размолу кормов, его можно разбавлять водой до обычной для торговых поставок концентрации без повышения вязкости до нежелательных значений.
Далее в рамках изобретения было установлено, что при соответствующем проведении реакции гидролиза в сочетании с применяемым согласно изобретению щадящим режимом упаривания при минимально кратковременной продолжительности пребывания материала в аппаратуре, можно получать особенно высокоценный в качественном отношении ГАМ. Получаемый по особенно предпочтительной методике ГАМ отличается прежде всего суммарным количеством димеров и олигомеров, равным ≤ 10 мол.%, предпочтительно < 7 мол.% по отношению к общему количеству ГАМ. Это означает, что вопреки распространенному ошибочному мнению можно получать высококонцентрированный ГАМ, представляющий собой благодаря крайне малому содержанию димеров и олигомеров очень удобный для непродолжительных перевозок продукт. В случае увеличения продолжительности транспортировки и связанной с этим необходимостью временного складирования, вследствие чего возрастает количество образующихся димеров и олигомеров, целесообразно эти последние добавлением воды и воздействием повышенной температуры снова переводить в мономерный ГАМ.
В соответствии с изобретением возможно далее высококонцентрированный ГАМ-продукт использовать при приготовлении добавок к кормам для животных. При этом оказывается, что смешением ГАМ-концентрата с водой, метионином и/или солями ГАМ (предпочтительно аммониевый ГАМ) (при необходимости NH4 для получения NH4-ГАМ) можно получать продукт с высокими питательными свойствами при сохранении качественных показателей, удовлетворяя тем самым в принципе потребности рынка.
Крайне неожиданным при осуществлении изобретения оказалось прежде всего то, что начиная с момента выхода целевого ГАМ-продукта из стадии упаривания доступно получение смесей не только за счет добавок соответствующих компонентов, таких, как вода, метионин и/или аммониевый ГАМ, но также и то особое преимущество, что в случае смеси с аммониевым ГАМ аммиак можно непосредственно вводить в ГАМ-продукт после упаривания. При этом в зависимости от добавляемого количества аммиака требуемое количество ГАМ трансформируют в аммониевый ГАМ.
Особенно предпочтительным оказалось далее то, что существует возможность получения практически не содержащего димеров и олигомеров продукта на основе экстракционного раствора в том случае, когда в этот экстракционный раствор добавляют аммиак и обе образовавшиеся фазы разделяют на органическую и водную и водную фазу подают в выпарной аппарат, предусмотренный предпочтительно для кратковременной продолжительности пребывания в нем материала. Благодаря такой методике на выходе из стадии упаривания получают смесь из ГАМ и аммониевого ГАМ, практически полностью свободную от димеров и олигомеров. Допустима далее непосредственная подача аммиака в испаритель. Кроме того, аммиак можно использовать в качестве среды для перегонки.
В одном из наиболее предпочтительных вариантов выполнения изобретения получаемый по описанному способу ГАМ, содержащий свыше 98 мас.% ГАМ, определенных как сумма мономерного ГАМ, димерного ГАМ и ГАМ-олигомеров, а также более 0,1 и менее 2 мас.% воды и имеющий кинематическую вязкость > 100 мм2/с при 25oC, может использоваться для приготовления смесей добавочных компонентов к кормам для животных, причем такое применение продукта по изобретению отличается тем, что приготавливают смесь с метионином, содержание ГАМ в которой, включая мономеры, димеры и олигомеры, составляет < 80 мас.%, а содержание олигомеров в которой, включая димерный ГАМ и олигомеры, после хранения в течение 300 дней при комнатной температуре составляет < 25 мол.% в пересчете на общее количество действующих веществ ГАМ, димерного ГАМ и олигомеров, а также метионина.
Смеси из ГАМ и метионина представляют собой добавляемые в корма для животных компоненты с великолепными свойствами. О самом метионине известно, что он практически во всех органических растворителях плохо либо совсем не растворяется (даже в воде). Согласно изобретению особенно неожиданным оказался тот факт, что метионин даже при нормальной температуре хорошо растворяется в ГАМ. Далее, о коммерчески доступном 88 мас.%-ном ГАМ известно, что, как было установлено экспериментальным путем, после хранения в течение 300 дней при комнатной температуре в нем образуется примерно 25 мол.%-ная равновесная концентрация димеров и олигомеров. Как уже указывалось выше, содержание димеров и олигомеров в высококонцентрированном ГАМ при 300-дневном хранении заметно выше и составляет около 50 мол.%. При исследовании смесей из метионина и ГАМ в опыте по хранению неожиданно было установлено, что по истечении 300 дней хранения доля олигомеров и димеров значительно ниже (что следует рассматривать как положительный фактор) и составляет, например, 20 мол.%, причем следует учесть еще и возможное дальнейшее снижение этой доли за счет замещения метионина в ГАМ'е, что приведет к изменению этого показателя до примерно 21,5 мол.%. Несмотря на такое незначительное изменение оно тем не менее существенно, поскольку в принципе, чем ниже содержание олигомеров, тем соответственно выше биологическая ценность мономера. Даже такая незначительная корректировка, составляющая "всего лишь" 3,5 мол.%, может способствовать заметному улучшению эффективности продукта как компонента, добавляемого в корма для животных. Решающей, таким образом, является прежде всего тенденция, направленная на повышение эффективности. Кроме того, следует отметить неожиданное существенное повышение устойчивости смесей из ГАМ и метионина при хранении в условиях низких температур. Смесь из 78 мас.% ГАМ и 10 мас.% метионина при -20oC по истечении 4 недель не обнаруживает никакой тенденции к кристаллизации (т.е. ни одна субстанция не выпадает в осадок), каковой фактор является неожиданным, так как в других растворителях метионин при прочих равных условиях сразу же выпадает в осадок. Ходовой коммерческий товар с содержанием ГАМ порядка 88 мас.% уже при -20oC через 3-21 день начинает выпадать в осадок. Тем самым при длительных перевозках вышеуказанных смесей из метионина и ГАМ в условиях низких температур предпочтительно можно отказаться в определенной степени от дополнительных мер по обогреву. Хранение в соответствующих низкотемпературных условиях тем самым также значительно упрощается, поскольку отпадает необходимость в дорогостоящих дополнительных обогревательных устройствах. Кроме того, следует отметить, что благодаря добавкам метионина повышается биологическая ценность ГАМ. Это является новым и неожиданным, так как до настоящего времени метионинсодержащие жидкие композиции ГАМ не были известны и возможность приготовления таких смесей для специалиста отнюдь не была очевидной, поскольку она базируется на неожиданной способности метионина растворяться в ГАМ. Смесь из ГАМ и метионина представляет собой прежде всего первую жидкостную добавку к кормам, превосходящую по своей биологической ценности известные жидкостные кормовые аддитивы на основе ГАМ.
Согласно другому предпочтительному варианту выполнения предметом изобретения является применение ГАМ, получаемого по описанному способу, для приготовления смесей в качестве добавляемых к кормам для животных компонентов, причем это применение отличается тем, что смесь получают взаимодействием с газообразным аммиаком, водным аммиаком и/или аммониевым ГАМ и содержание ГАМ в смеси, включая мономеры, димеры и олигомеры, составляет < 80 мас.%, а содержание олигомеров, включая димерный ГАМ и ГАМ-олигомеры, после хранения в течение 30 дней при 40oC составляет < 25 мол.% в пересчете на общее количество действующих веществ, включающих ГАМ, димерный ГАМ и ГАМ-олигомеры. Касательно устойчивости при хранении при комнатной температуре, соответственно при низких температурах смеси из ГАМ и аммониевого ГАМ проявляют уникальные преимущества по сравнению с чистым ГАМ, причем эти преимущества названных смесей, равно как и смесей из ГАМ и метионина невозможно было предвидеть. Аммиак оказывает прежде всего положительное воздействие на образование равновесия между мономерами, димерами и олигомерами.
Ниже изобретение подробнее поясняется на примерах со ссылками на прилагаемые чертежи, на которых
показано:
на
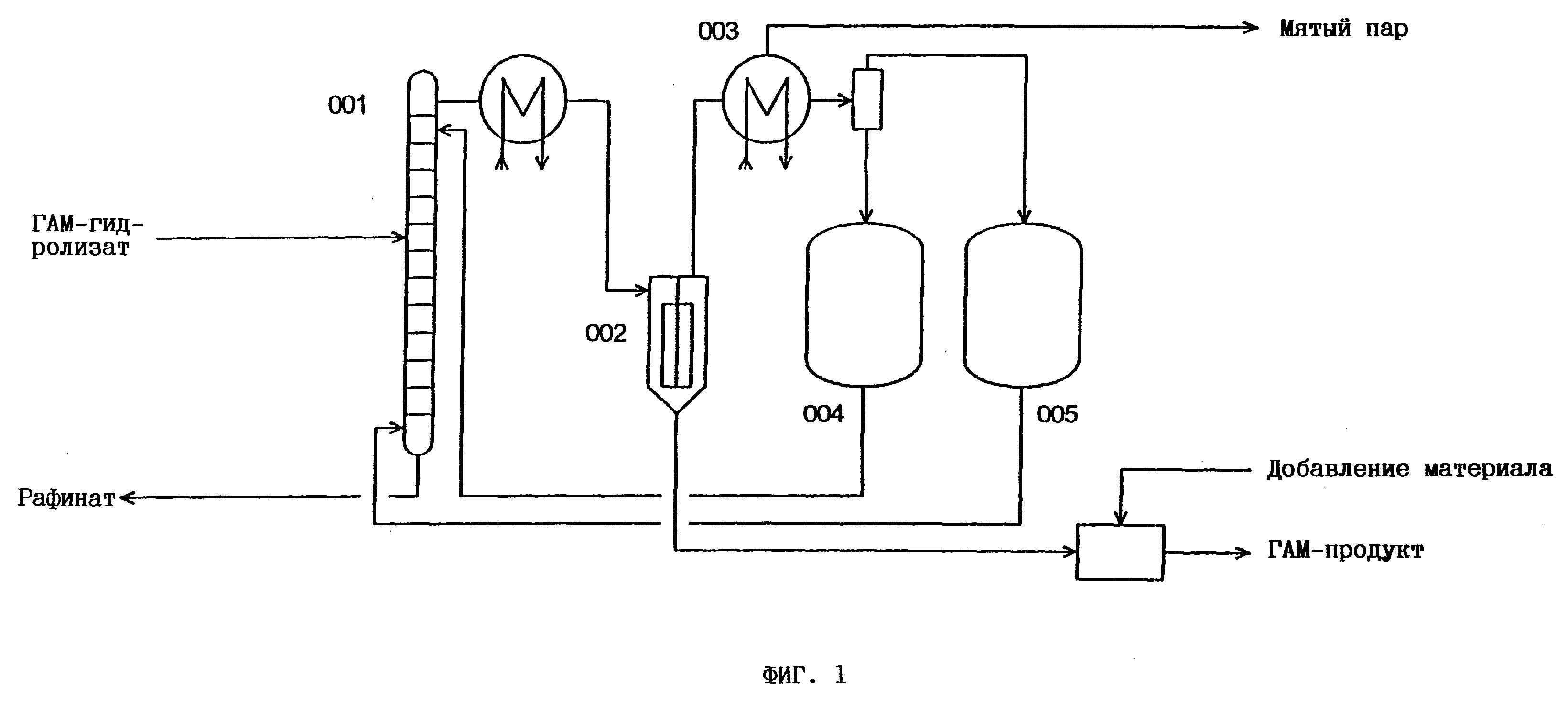
фиг. 1 - схематическое изображение части промышленной установки по получению ГАМ, причем в целях упрощения показана только секция по переработке;
на фиг. 2 - диаграмма
изменения
кинематической вязкости по Кеннону-Фенске высококонцентрированного ГАМ (приблизительно 98 мас.%-ного) по истечении 300 дней хранения в сравнении с 88 мас.%-ным имеющимся в продаже продуктом
и с
раствором ГАМ, приготовленным разбавлением водой находившегося на хранении концентрата до содержания 88 мас.% ГАМ, при различных температурах;
на фиг. 3 - диаграмма зависимости содержания
ГАМ и димеров + олигомеров в мол. % при хранении ГАМ-концентрата (приблизительно 98 мас.%-ного) в течение 300 дней при комнатной температуре;
на фиг. 4 - диаграмма зависимости содержания ГАМ
и димеров + олигомеров в мол. % при хранении ГАМ (88 мас.%-ного) в течение 300 дней при комнатной температуре;
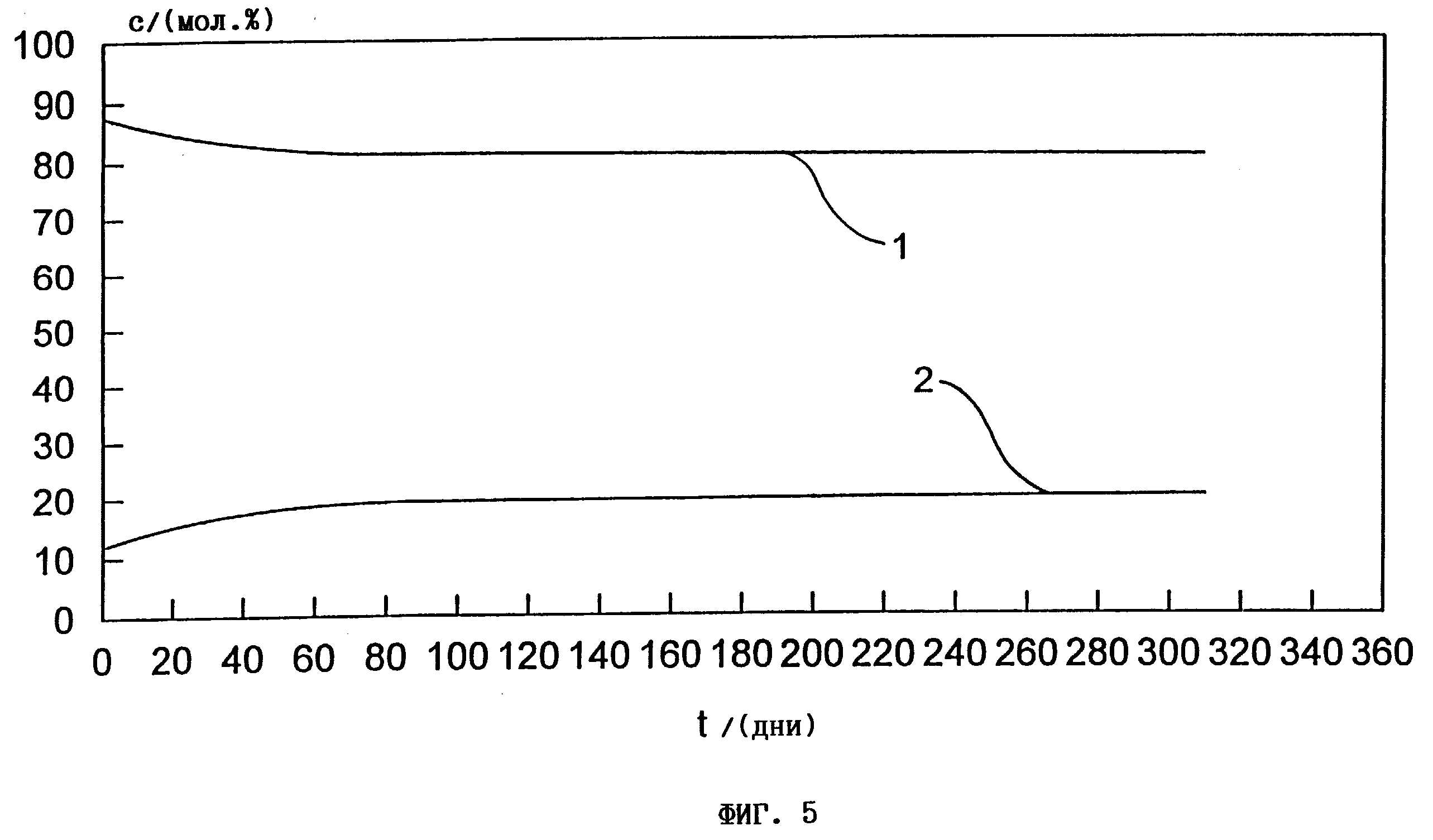
на фиг. 5 - диаграмма зависимости содержания ГАМ + метионина, димеров +
олигомеров, а также метионина в мол.% при хранении смесей ГАМ из 78 мас.% ГАМ и 10 мас.% метионина в течение 300 дней при комнатной температуре; и на
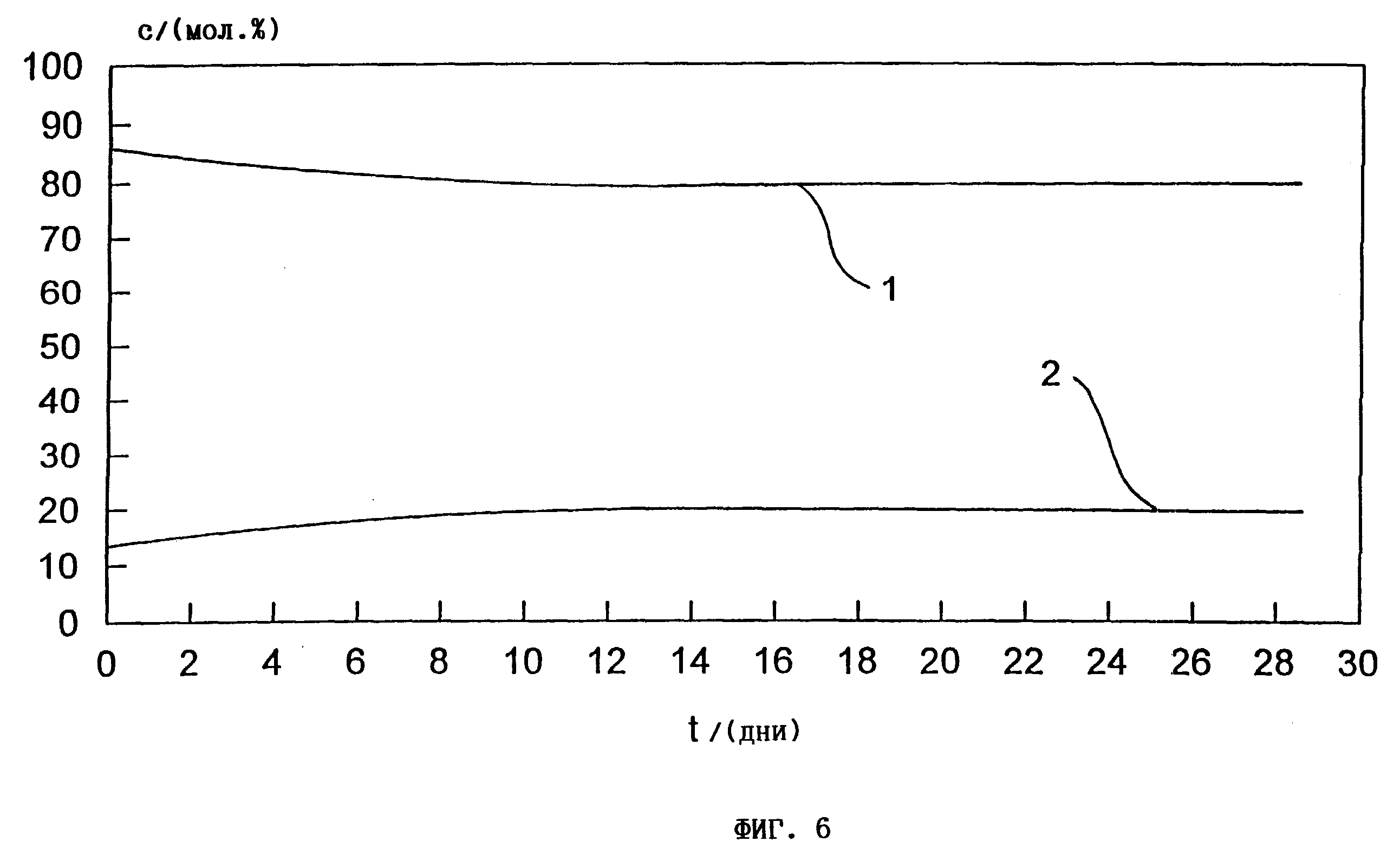
фиг. 6 - диаграмма зависимости
содержания
ГАМ + аммониевого ГАМ, димеров + олигомеров, аммониевого ГАМ в мол.% при хранении смесей ГАМ из 78 мас.% ГАМ + 10 мас.% аммониевого ГАМ в течение 30 дней при 40oC.
Примеры.
Аналитические методы определения и пояснения.
Количества ММПА-циангидрина, ГАМ-амида, мономерного ГАМ, соответственно метионина определяли в обрабатывающих растворах посредством ЖХВД путем сравнения с внешним эталоном (чистая субстанция).
Общее количество ГАМ = ГАМ-амид (необязательно) + мономерный ГАМ + ГАМ - (димеры +
олигомеры) +
метионин (необязательно)
определяли посредством титриметрии тиоэфирной функции с помощью стандартного раствора KBr/KBrO3 и выражали в виде суммы соответствующих
ГАМ-мономерных
эквивалентов в [мас.%], соответственно в [г], соответственно в [молях], соответственно в [мол.%].
Содержание ГАМ-димеров + ГАМ-олигомеров (ДИМ + ОЛИ) определяли по разнице между общим количеством ГАМ и мономерного ГАМ + (необязательно ГАМ-амид и/или метионин) и выражали в виде суммы соответствующих ГАМ-мономерных эквивалентов в [мас.%], соответственно в [г], соответственно в [молях], соответственно в [мол.%].
С помощью стандартных методов определяли: содержание воды - титрованием по Карлу-Фишеру, содержание МИБК (метилизобутилкетона) - посредством ГХ или по разнице, содержание сульфата, соответственно аммония - посредством ионной хроматографии.
Пример 1.
В серии опытов исследовали степень конверсии ММПА-циангидрина при контакте с водной серной кислотой в различных условиях в ГАМ-амид.
Проведение опыта 1.
В трехгорлую колбу вместимостью 1 л, снабженную обратным холодильником, капельной воронкой, внутренним термометром и магнитной мешалкой, загружали 141 г (1,03 моля) 65%-ной H2SO4 при 50oC и при перемешивании добавляли по каплям в течение 30 мин 138 г (1,0 моль) 95%-ного ММПА-циангидрина. По истечении еще 15 мин, в течение которых реакция продолжалась при этой температуре, наличие ММПА-циангидрина в реакционном растворе обнаружено не было.
Проведение опыта 2.
Опыт 2 проводили аналогично опыту 1, с той разницей, что использовали 72 г (0,55 моля) 75%-ной H2SO4 и 138 г (1,0 моль) 95%-ного ММПА-циангидрина. По истечении 150 мин, в течение которых продолжалась реакция, была обнаружена еще 0,1% от теории ММПА-циангидрина.
Проведение опыта 3.
В четырехгорлую колбу вместимостью 1 л, снабженную обратным холодильником, двумя капельными воронками, внутренним термометром и магнитной мешалкой, в течение 15 мин одновременно загружали 155 г (1,03 моля) 65%-ной H2SO4 и 138 г (1,0 моль) 95%-ного ММПА-циангидрина при температуре 50oC. По истечении последующих 15 мин, в течение которых реакция протекала при этой температуре, наличие ММПА-циангидрина обнаружено не было.
Проведение опытов 4-11.
Опыты 4-11 проводили аналогично опыту 3. Параметры реакции и полученные результаты представлены в таблице 1 (см. в конце описания).
Как видно из таблицы 1, в опытах 1, 3, 6, 7, 10 и 11 в указанное реакционное время имела место полная конверсия ММПА-ЦГ.
Реакционное время, необходимое для полной конверсии ММПА-ЦГ, осуществляемой взаимодействием с H2SO4, возрастает с увеличением молярного соотношения между ММПА-ЦГ и H2SO4 (ср. опыты 8 и 11). Практиковавшаяся в опытах различная последовательность подачи применяемых материалов (соответственно2)) не оказывает решающего влияния на результат реакции (ср. опыты 1 и 3, соответственно 2 и 4).
Пример 2.
Проведение опыта 1.
В трехгорлой колбе вместимостью
500 мл, снабженной обратным холодильником, капельной воронкой,
внутренним термометром и магнитной мешалкой, разбавляли водой 35,8 г (0,35 моля, 0,7 мол.экв.) 96%-ной серной кислоты до концентрации
серной кислоты 78,9 мас.% H2SO4. Затем в
течение 23 мин при перемешивании по каплям добавляли при температуре 49-56oC 67,5 г (0,5 моля) 97,2%-ного ММПА-ЦГ. По
истечении последующих 20 мин, в течение которых при температуре порядка
50oC протекала реакция, анализ посредством ЖХВД показал полную конверсию циангидрина (0 мас.%) в ГАМ-амид (66,2
мас.%). Затем содержимое колбы с помощью 42,3 г воды перемещали в стальной
автоклав объемом 200 мл со стеклянной вставкой, термометром для измерения внутренней температуры, манометром и магнитной
мешалкой, где концентрация имевшейся в реакционной смеси водной серной кислоты
составляла 44 мас.%
После того, как автоклав закрывали, реакционную смесь в течение 45 мин нагревали до 140oC и в течение последующих 120 мин перемешивали при этой температуре.
После охлаждения и открывания автоклава получали 150,4 г ГАМ-гидролизата. Титрованием сернокислотных эквивалентов раствором
бромид-бромат получали в общей сложности 71,5 г ГАМ (общий выход 0,48 моля,
95,2% от теории). Анализ посредством ЖХВД показал наличие 57,5 г (76,6% от теории) мономерного ГАМ. Это дает разницу в 14,0
г (18,6% от теории), приходящуюся на долю ГАМ-(димеров+олигомеров), что
выражено в виде ГАМ-мономера. ЖХВД наличия ГАМ-амида не показала.
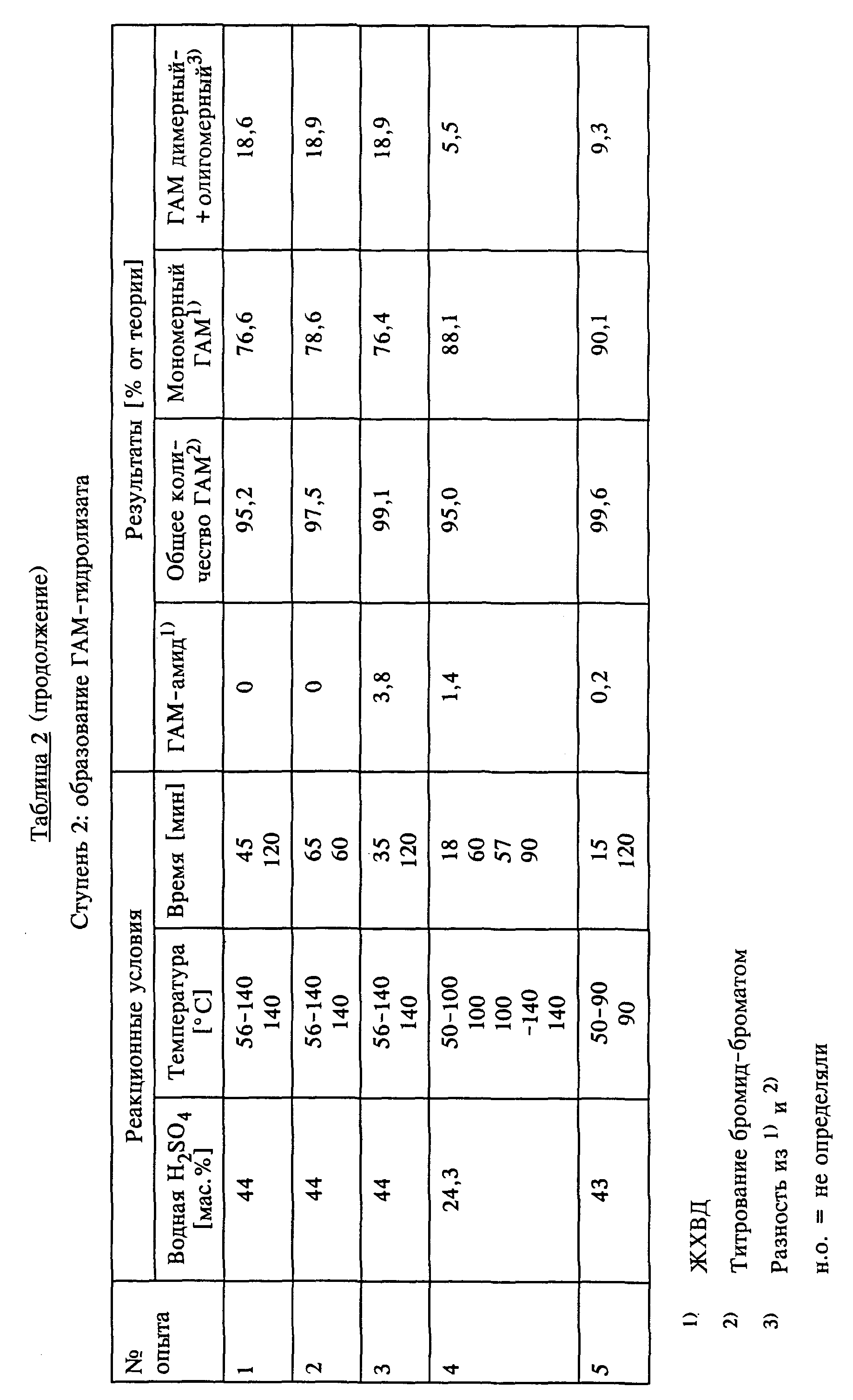
Проведение опытов 2-5.
Опыты 2-5 проводили аналогично опыту 1. Параметры реакций полученные результаты представлены в таблице 2 (см. в конце описания). В опыте 4 на стадии 1 использовали 2-литровую стеклянную колбу, на стадии 2 - 2-литровый автоклав, в опыте 5 на обеих стадиях использовали 2-литровую стеклянную колбу.
Пример 3.
Выделение ГАМ-концентрата из ГАМ-гидролизата жидкостно-жидкостной экстракцией и упариванием экстракционного раствора. Описание способа со ссылками на фиг. 1.
На фиг. 1 схематически изображена применявшаяся в примере 3 аппаратура. Эта система состоит
в основном из следующих аппаратов:
экстракционная колонна (001)
= пульсационная тарельчатая колонна длиной 3 м, с внутренним диаметром 2,1 см, пакетом из 60 тарелок, двойной рубашкой с
обогревом;
тонкопленочный испаритель (002) = испаритель Sambay с
поверхностью обмена 0,08 м2, двойной рубашкой с обогревом;
конденсационная секция (003) = стеклянный
холодильник с водяным охлаждением;
сборник (004/005) для возвращаемых
в процесс воды, соответственно растворителя.
Поступающий из ступени гидролиза ГАМ ГАМ-гидролизат, в состав которого входят в основном ГАМ (мономеры + димеры + олигомеры + необязательно амид), (NH4)SO4 и/или NH4HSO4, а также вода, после предварительного нагревания до экстракционной температуры подают над сороковой тарелкой в экстракционную колонну 001. Растворитель (в данном случае метилизобутилкетон (МИБК)) также после предварительного нагревания закачивают в нижнюю часть колонны (принцип противотока). В головную часть колонны дополнительно подают промывочную воду. Содержащий в основном (NH4)2SO4 и/или NH4HSO4 и воду рафинат удаляют через низ колонны. Содержащий в основном ГАМ, растворитель и воду экстракционный раствор отводят через головную часть колонны, после чего подают в испаритель Sambay 002. Там под вакуумом и при дополнительной подаче под давлением водяного пара и потока N2 незадолго до спуска из испарителя экстракционного раствора из последнего удаляют совместно МИБК и H2О. Операцию упаривания осуществляли таким образом, что в поступающем из испарителя Sambay высококонцентрированном ГАМ содержалось < 2 мас.% H2О (титрование по Карлу Фишеру) и в нем практически отсутствовал растворитель.
Поступающую из испарителя 002 смесь растворитель-вода сначала конденсировали в конденсационной секции 003 и затем для разделения направляли в сепаратор. Растворитель, соответственно воду собирали в сборниках 004, соответственно 005, откуда их возвращали в экстракционную систему. Продукт из испарителя Sambay после охлаждения до комнатной температуры направляли в предусмотренный для этой цели приемник.
Условия и результаты отдельных опытов 1-4 представлены ниже.
Состав экстракционного раствора анализировали непосредственно после выхода из головной части экстракционной колонны 001, а состав раствора рафината определяли непосредственно после выхода из нижней части этой колонны.
Состав высококонцентрированного ГАМ-продукта анализировали в поступавшем из испарителя Sambay материале непосредственно на выходе.
Используемые для экстракции растворы ГАМ-гидролизата приготавливали в 400-литровой емкости с мешалкой под постоянным давлением из 114,7 кг (874 моля) ММПА-циангидрина и 85,7 кг (874 моля, 1 мол.экв.) H2 SO4 в условиях, указанных в примере 2, опыт 5, соответственно из 142, 9 кг (1089 молей) ММПА-циангидрина и 110,0 кг (1122 моля, 1,03 мол.экв.) H2SO4 согласно примеру 2, опыт 5, соответственно из 115,3 кг (879 молей) ММПА-циангидрина и 47,5 кг (484 моля, 0,55 мол.экв.) H2SO4 согласно примеру 2, опыт 4. Из растворов неочищенного гидролизата после окончания реакции соответственно под вакуумом удаляли необязательно имевшиеся легколетучие побочные продукты и затем анализировали. Полученные при этом составы ГАМ-гидролизата, который применяли для проведения экстракции, представлены соответственно в описании отдельных опытов в примере 3.
Получение высококонцентрированного ГАМ-продукта.
Опыт 1.
Применение ГАМ-гидролизата из ММПА-циангидрина и 1,0 мол.экв. H2SO4.
Применение при экстракции:
Расход
МИБК - 5 кг/ч
ГАМ-гидролизат - 8,4 кг/ч
общее количество ГАМ - 3,4 кг/ч
промывочная H2
O - 0,5 кг/ч
МИБК/гидролизат - 0,6 [-]
Состав ГАМ-гидролизата (получение
аналогично примеру 2, опыт 5)
общее количество ГАМ - 42 мас.%; ГАМ 90 мол.%; димеры+олигомеры 10
мол.%
H2O - 28,9 мас.%
-SO: - 25,9 мас.%
Экстракция
(001):
температура - 60oC (средняя)
состав:
экстракционного раствора - МИБК 47 мас.%; общее количество ГАМ 40 мас.%; H2O 13 мас.%
рафината
- общее количество ГАМ 0,1 мас.%
Упаривание (002):
давление: - 100
бар
Sambay - температура:
в обогреваемой рубашке - 150oC
в головной части
- 62oC
в нижней части - не опр.
- пар для отгонки 0,
5 кг/ч
- газ для отгонки N2 100 л/ч
Состав ГАМ-концентрата в материале, поступающем
из испарителя:
общее кол-во ГАМ: - 98 мас.%; ГАМ 86 мол.%, димеры + олигомеры
14 мол.%
H2O - 2 мас.%
МИБК - 40 част./млн
Из материала, поступающего из
испарителя Sambay, получали приблизительно 3,5 кг/ч высококонцентрированного
ГАМ-продукта указанного выше состава.
Опыт 2.
Применение ГАМ-гидролизата из ММПА-циангидрина и 1,03 мол.экв. H2SO4.
Применение
при экстракции:
Расход
МИБК - 3,6 кг/ч
ГАМ-гидролизат - 5,38 кг/ч
общее
количество ГАМ - 2,10 кг/ч
промывочная H2O - 0,5 кг/ч
МИБК/гидролизат - 0,67 [-]
Состав ГАМ-гидролизата (получение аналогично примеру 2, опыт 5):
общее
количество ГАМ - 39 мас.%; ГАМ 86,9 мол.%; димеры+олигомеры 13,1 мол.%
H2O - приблизительно 30 мас.% (остаток)
-SO - 25,4 мас.%
NH4HSO4 - 30,4 мас.%
Экстракция (001):
температура - 54oC (средняя)
состав:
экстракционного раствора - МИБК ≤ 54 мас.%; общее
количество ГАМ 34,1 мас.%; H2O 11,9 мас.%
рафината - общее количество ГАМ 0,3 мас.%; SO 30,4 мас.%; МИБК 0,17 мас.%
Упаривание (002):
давление - 100 мбар
Sambay
- температура:
в обогреваемой рубашке - 150oC
в головной
части - 65oC
в нижней части - 92oC
- пар для отгонки 0,7 кг/ч
- газ для отгонки N2 100 л/ч
Состав ГАМ-концентрата в материале,
поступающем из испарителя:
общее кол-во ГАМ: - 99,2 мас.%; ГАМ 86,9 мол.%; димеры+олигомеры 13,1 мол.%
H2O: - 0,8 мас.%
SO: - 0,43 мас.%
МИБК: - 28 част./млн
Из материала, поступающего из испарителя Sambay, получали
приблизительно 2,1 кг/ч высококонцентрированного ГАМ-продукта указанного выше состава.
Опыт 3.
Применение ГАМ-гидролизата из ММПА-циангидрина и 0,55 мол.экв. H2SO4.
Применение при экстракции:
Расход
МИБК - 3,6 кг/ч
ГАМ-гидролизат - 6,55
кг/ч
общее количество ГАМ - 2,62 кг/ч
промывочная H2O - 0,5 кг/ч
МИБК/гидролизат - 0,55 [-]
Состав ГАМ-гидролизата (получение аналогично примеру 2, опыт
4):
общее количество ГАМ - 40 мас.%; ГАМ 94,7
мол.%; димеры+олигомеры 5,3 мол.%
H2O - приблизительно 40,2 мас.%
-SO
: - 13,9 мас.%
-NH: - 4,85
мас.%
Экстракция (001):
температура - 52oC (средняя)
состав:
экстракционного раствора: - МИБК ~49 мас.%; общее количество ГАМ 35,8 мас.%; H2O
14,9 мас.%
рафината - общее количество ГАМ 0,13
мас.%
Упаривание (002):
давление: - 100 мбар
Sambay - температура:
в обогреваемой рубашке - 150o
C
в головной части - 59oC
в
нижней части - 92oC.
- пар для отгонки 0,6 кг/ч
- газ для отгонки N2 100 л/ч
Состав
ГАМ-концентрата в материале, поступающем из испарителя:
общее кол-во ГАМ - 98,8 мас.%; ГАМ 86 мол.%, димеры + олигомеры 4,9 мол. %;
H2O - 0,5 мас.%
SO: - 0,18 мас.%
МИБК - 477 част./млн
Из материала, поступающего из испарителя Sambay,
получали приблизительно 2,7 кг/ч высококонцентрированного
ГАМ-продукта указанного выше состава.
Опыт 4.
Применение ГАМ-гидролизата из ММПА-циангидрина и 0,55 мол.экв. H2SO4.
Применение
при экстракции:
Расход
МИБК - 3,6 кг/ч
ГАМ-гидролизат - 6,89 кг/ч
общее количество ГАМ - 2,76 кг/ч
H2O - 0,5 кг/ч
МИБК/гидролизат - 0,
52 [-]
Состав ГАМ-гидролизата (получение аналогично примеру 2, опыт 4):
общее количество ГАМ - 40 мас.%; ГАМ 94,7 мол.%;
димеры+олигомеры 5,3 мол.%
H2O - 40,2
мас.%
-SO: - 13,9 мас.%
-NH: - 4,85 мас.%
Экстракция (001):
температура - 52oC (средняя)
состав:
экстракционного раствора - МИБК ~49,
3 мас.%; общее количество ГАМ 35,8 мас.%; H2O 14,9 мас.%
рафината - общее количество ГАМ 0,1 мас.%
Упаривание
(002):
давление: - 100 мбар
Sambay
- температура:
в обогреваемой рубашке - 150oC
в головной части - 59oC
в нижней части - 95oC
- пар для отгонки 0,6 кг/ч
- газ для отгонки N2 100 л/ч
Состав ГАМ-концентрата в материале, поступающем из испарителя материале:
общее кол-во
ГАМ - 99,0 мас.%; ГАМ 94,9 мол.%; димеры+олигомеры 5,1
мол. %
H2O - 0,2 мас.%
SO: - 0,16 мас.%
МИБК - 482
част./млн
Из материала, поступающего из испарителя Sambay, получали приблизительно 2,8 кг/ч высококонцентрированного ГАМ-продукта указанного
выше состава.
Как показывает сравнение опытов 1-4, можно получать высококонцентрированный ГАМ-продукт с общим содержанием ГАМ в количестве ≥ 98 мас.% и содержанием воды от 0,2 до 2 мас.% без повреждения продукта [увеличенная доля ГАМ - (димеров + олигомеров)]. Этот неожиданный эффект проявляется особенно в ГАМ-концентрате, получаемом из более бедных сульфатом растворов ГАМ-гидролизата (опыты 3 и 4). В этих случаях доля ГАМ - (димеров + олигомеров) составляет лишь 4,9-5,1 мол.%, тогда как этот показатель в опытах 1 и 2 равен 13,1-14 мол. %. Соотношение весовых частей между мономерным ГАМ и суммой димеры + олигомеры [= ГАМ/ГАМ-(ДИМ + ОЛИ)] составляет приблизительно 19, соответственно 6,1-6,6. Таким образом, во всех приведенных случаях доля ГАМ - (димеров + олигомеров) значительно ниже тех же показателей у коммерчески доступного ГАМ-продукта, в котором доля ГАМ-(димеров + олигомеров) составляет обычно 20-25 мол.%.
Кроме того, удалось решающим образом улучшить соотношение весовых частей ГАМ/ГАМ-(ДИМ + ОЛИ) по сравнению с теми же показателями, указанными в EP 0142488 в примерах 1-6: 3,3; 3,2; 3,7 (пример 1), 1,8 (пример 2), 3,0 (пример 3), 5,4 (пример 5) и 5,2 (пример 6).
Пример 4.
Получение смешанного продукта состава ГАМ-метионин-H2О из ГАМ-концентрата.
Опыт 1.
Опыт 1 проводили
аналогично примеру 3, опыт 2, с тем отличием, что материал, поступавший из испарителя Sambay, смешивали в емкости с мешалкой с метионином и водой и при
перемешивании получали гомогенный раствор,
содержащий 89 мас. % общего количества ГАМ (указанное общее количество ГАМ = ГАМ-(мономеры + димеры + олигомеры) + мет.) и около 10 мас.% воды. В этот
раствор добавляли в общей сложности 25,7 кг
ГАМ-концентрата указанного выше состава совместно с 3,2 кг D,L-метионина (= мет) и 3,2 кг воды и при перемешивании гомогенизировали. Полученный в результате
продукт (32,1 кг) имел следующий состав,
который определяли аналитическим путем:
общее количество ГАМ (указанное общее количество ГАМ = ГАМ-(мономеры + димеры + олигомеры) + мет.) - 89,2
мас.% ~100,0 мол.%
мономерный ГАМ
- 64,8 мас.% ~ 72,6 мол.%
ГАМ-(димеры+олигомеры) - 15,1 мас.% ~16,7 мол.%
мет - 9,2 мас.% ~10,7 мол.%
SO - 0,8мас.%
МИБК - 14 част./млн
Опыт 2.
Опыт 2 проводили аналогично примеру 3, опыт
3, с тем отличием, что материал,
поступавший из испарителя Sambay, смешивали в емкости с мешалкой с метионином и водой и при перемешивании получали гомогенный раствор, содержащий 88 мас. % общего
количества ГАМ (указанное общее
количество ГАМ = ГАМ-(мономеры + димеры + олигомеры) + мет.) и около 11 мас.% воды. В этот раствор добавляли в общей сложности 27,0 кг ГАМ-концентрата указанного выше
состава совместно с 3,5 кг D,
L-метионина и 4,0 кг воды и при перемешивании растворяли. Полученный в результате продукт (34,5 кг) имел следующий состав, который определяли аналитическим путем:
общее количество ГАМ - 88,1
мас.% ~100,0 мол.%
мономерный ГАМ - 73,8 мас.% ~ 83,8 мол.%
ГАМ-(димеры+олигомеры) - 4,3 мас.% ~ 4,9 мол.%
мет - 10,0 мас.% ~ 11,4 мол.%
SO - 0,16 мас.%
МИБК - 482 част./млн
Пример 5.
Получение ГАМ-NH4-солевых растворов из ГАМ-концентрата.
98,3 г экстракционного раствора ГАМ, содержащего общее количество ГАМ 40 мас. % (0,26 моля), 13 мас.% воды и приблизительно 47
мас.% МИБК и полученного
аналогично примеру 3, опыт 1, упаривали в водоструйном вакууме при 55-70oC в течение 2,4 ч. Остаток (общее количество ГАМ 99,9 мас.%) при перемешивании смешивали с
17,7 г (0,26 моля)
25%-ного водного раствора аммиака. При этом температура повышалась до 53oC. После охлаждения до комнатной температуры получали 57,6 г коричневой прозрачной жидкости
следующего состава:
общее количество ГАМ - 68,3 мас.%
мономерный ГАМ - 59,5 мас.%
ГАМ-амид - 0,0 мас.%
ГАМ-(димеры+олигомеры) - 8,8 мас.%
NH - 8,1 мас.%
H2O - 22,4 мас.%
Пример 6.
Получение ГАМ-NH4 -солевого раствора ГАМ из экстракционного раствора.
ГАМ 3502 г экстракционного раствора ГАМ, содержавшего общее количество ГАМ 35,8 мас.%, 14,9 мас.% воды и приблизительно 49 мас.%
МИБК и
полученного аналогично примеру 3, опыт 4, охлаждали в ледяной бане до 5oC. В этот раствор в течение 3,5 ч при температуре в интервале от 5 до 21oC вводили 300 г (17,6
молей)
газообразного аммиака. При этом образовывались две жидкие фазы. Более тяжелую фазу (2202 г) отделяли и из нее с помощью водяного пара, вводимого при 60oC и давлении 100 мбар, удаляли
остаточный МИБК. Остаток продолжали концентрировать в водоструйном вакууме при 50oC в течение 2 ч, в результате чего получали маслянистую коричнево-желтую жидкость (1583 г) следующего
состава:
общее кол-во ГАМ - 81,2 мас.% ~103% от теории)
включая: - мономерный ГАМ - 79,3 мас.%
ГАМ-амид - 1,9 мас.%
ГАМ-(димеры + олигомеры) - 0,0 мас.%
NH - 9,0%
вода - 9,8 мас.%
После разбавления 230 г воды и 20 г 25%-ного
раствора
аммиака получали светло-коричневую масловязкую жидкость (1743 г) следующего состава:
общее количество ГАМ - 70,0 мас.%
включая: - мономерный ГАМ - 68,3 мас.%
ГАМ-амид - 1,7
мас.%
ГАМ-(димеры + олигомеры) - 0,0 мас.%
NH - 8,1 мас.%
Н2
О - 20,0 мас.%
Неожиданно после такой обработки в
растворе продукта не было больше обнаружено наличия нежелательных ГАМ-(димеров + олигомеров). Единственным побочным
компонентом было лишь
совсем незначительное количество ГАМ-амида.
Пример 7.
Получение смешанного продукта состава ГАМ-ГАМNH4-H2O из экстракционного раствора ГАМ.
Опыт 1.
В условиях, в основном аналогичных указанным в примере 3, опыт 2, 10 кг экстракционного раствора ГАМ, содержавшего общее
количество ГАМ 39,7 мас.%,
приблизительно 12 мас.% H2О и приблизительно 48 мас.% МИБК, упаривали с помощью испарителя Sambay. Выходящий из испарителя раствор разбавляли в общей сложности 0,
636 кг (3,06 моля) 8,
2%-ного водного раствора NH3 и равномерно перемешивали. Таким путем получали маслянистую коричнево-желтую жидкость (4,60 кг; pH ~2,3) следующего состава:
общее количество ГАМ - 86,
2 мас.% (соответствует 100% от теории)
включая: - мономерный ГАМ - 74,3 мас.%
ГАМ-амид - 0,0 мас.%
ГАМ-(димеры + олигомеры) - 11,9 мас.%
NH - 1,2 мас.%
SO - 0,22 мас.%
H2O - 13,5 мас.%
ГАМ-NH4 (расчетный показатель количества ГАМ-NH4 (соответствует 11,2 мол.% мономеров,
димеров и олигомеров))
- 10,75 мас.%
Опыт 2.
В условиях, в основном аналогичных описанным в примере 3, опыт 2, 10 кг экстракционного раствора ГАМ, содержавшего общее
количество ГАМ 35,1 мас.%,
приблизительно 12,7 мас. % H2О и приблизительно 52 мас.% МИБК, упаривали с помощью испарителя Sambay. Выходящий из испарителя раствор разбавляли в общей
сложности 0,45кг (4,23 моля)
16%-ного водного раствора NH3 и равномерно перемешивали. Таким путем получали маслянистую коричнево-желтую жидкость (3,96 кг; pH приблизительно 2,7) следующего
состава:
общее
количество ГАМ - 88,6 мас.% (соответствует 100% от теории)
включая: - мономерный ГАМ - 76,9 мас.%
ГАМ-амид - 0,0 мас.%
ГАМ-(димеры + олигомеры) - 11,
7 мас.%
NH - 11,94 мас.%
SO - 0,15 мас.%
H2O - 9,8 мас.%
FAM-NH4 (расчетный показатель количества ГАМ-NH4
(соответствует 17,7 мол.% мономеров,
димеров и олигомеров)) - 17,74 мас.%
Пример 8.
Определение вязкости по Кеннону-Фенске.
Как это лучше всего видно на
фиг. 2, кинематическую вязкость
определяли с помощью вискозиметра типа Cannon Fenske opaque по методу ISO 3105-1976 в зависимости от температуры для следующих характеристик ГАМ:
ГАМ 98%,
получение согласно примеру 3, опыт 2,
и хранение при комнатной температуре в течение > 300 дней, соответствует кривой со ссылочной цифрой 1 по фиг. 2;
ГАМ 88% из ГАМ 98%, получение
последнего согласно примеру 3, опыт 2,
хранение при комнатной температуре в течение > 300 дней и последующее разбавление до 88%, соответствует кривой со ссылочной цифрой 2 по фиг. 2; и
ГАМ 88% обычный коммерчески доступный
продукт, соответствует кривой со ссылочной цифрой 3 по фиг. 2.
Несмотря на относительно высокое содержание димеров и олигомеров непосредственно после разбавления водой показатели вязкости полученного таким путем ГАМ 88 из ГАМ 98 (2) соответствуют этим показателям коммерчески доступного продукта ГАМ 88 (3).
Пример 9.
Опыты по хранению ГАМ-продуктов.
Представленные на фиг. З-6 продукты хранились соответственно в закрытой стеклянной емкости без перемешивания при указанной температуре в течение периода времени до 310 дней. Через регулярные промежутки времени брали пробы и определяли содержание общего количества ГАМ, ГАМ-мономеров, ГАМ-(димеров+олигомеров) и необязательно мет (ср. указанные выше методы).
Фиг. 3.
ГАМ-концентрат, полученный согласно примеру 3, опыт 2, по истечении приблизительно 3 месяцев хранения при комнатной температуре обнаруживает установившуюся равновесную систему из 47 мол.% ГАМ-мономеров (1) и 53 мол.% ГАМ-(димеров + олигомеров) (2).
Фиг. 4.
ГАМ 88, полученный аналогично примеру 3, опыт 2, и последующим разбавлением водой до содержания 88% ГАМ, по истечении приблизительно 3 месяцев хранения при комнатной температуре обнаруживает установившуюся равновесную систему из 47 мол.% ГАМ-мономеров (1) и 26 мол.% ГАМ-(димеров+олигомеров) (2).
Это соотношение было обнаружено также в коммерчески доступном продукте.
Фиг. 5.
ГАМ 78 + мет 10, полученный согласно примеру 4, опыт 1, по истечении 3 месяцев хранения при комнатной температуре обнаруживает установившуюся равновесную систему из 80 мол. % мономерного ГАМ + мет (1) и 20 мол.% ГАМ-(димеров + олигомеров) (2).
Фиг. 6.
ГАМ 78 +ГАМNH4 10, полученный согласно примеру 7, по истечении уже 14 дней хранения при температуре 40oC обнаруживает установившуюся равновесную систему из 80 мол.% ГАМ-мономеров (1) и 20 мол.% ГАМ-(димеров + олигомеров) (2).
Сравнение фигур 3, 4, 5 и 6 показывает, что содержание димеров+олигомеров в смешанных продуктах, таких, как ГАМ 78 + мет 10, соответственно ГАМ 78 + FAMNH4 10, после длительного хранения заметно благоприятнее по сравнению с коммерчески доступным ГАМ 88. В ГАМ 78 + FAMNH4 10 равновесное состояние (обусловленное повышенной температурой) удавалось достичь уже значительно быстрее (фиг. 6), чем в других описанных случаях.
Реферат
Описывается способ получения 2-гидрокси-4-метилтиомасляной кислоты (ГАМ), в котором ГАМ выделяют из реакционной смеси, получаемой путем присоединения синильной кислоты (HCN) к метилмеркаптопропиональдегиду (ММПА) и гидролиза образующегося при этом метилмеркаптопропиональдегидциангидрина (ММПА-ЦГ), осуществляемого взаимодействием с серной кислотой, причем реакционную смесь вводят в контакт с в основном не смешиваемым с водой органическим растворителем в экстракционной системе жидкость - жидкость для получения экстракционного раствора, содержащего в своем составе растворитель и извлеченный из реакционной смеси ГАМ, и ГАМ получают в виде экстракта из этого экстракционного раствора упариванием, отличающийся тем, что упаривание проводят таким образом, что содержание воды в получаемом экстракте составляет менее 4 мас.%. Описывается также ГАМ - гидроксианалог аминокислоты метионина в рацемической форме, которая является важной добавкой к кормам для животных. Технический результат - увеличение степени чистоты целевого продукта и получение его с высокой концентрацией. 2 с. и 6 з.п.ф-лы, 6 ил., 2 табл.
Комментарии