Соединения для лечения заболеваний, опосредованных липазой - RU2420513C2
Код документа: RU2420513C2
Описание
Область техники, к которой относится изобретение
Изобретение относится к новым гетероциклическим соединениям бензохинонов, которые воздействуют на активность ферментов семейства генов липазы. Настоящее изобретение относится к фармацевтическим композициям, которые содержат соединения, полученные из бензохинонов, способу получения их и их фармацевтически приемлемых солей, производных, изомеров, полиморфов, их сольватов, обладающих селективной активностью в отношении заболеваний и состояний, опосредованных генами семейства липазы.
Предшествующий уровень техники
Во многих развитых и развивающихся странах продолжает нарастать тенденция к принятию рациона, содержащего большое количество жира и низкие концентрации волокон, в сочетании с низкой интенсивностью работы и сидячим образом жизни. Такое избыточное потребление жира, сопровождающееся сниженным превращением жира в энергию ввиду сидячего образа жизни, может привести к накоплению жира в организме на различных уровнях, включая биологические жидкости, клетки и ткани. Следовательно, имеется постоянный рост популяции с повышенным риском метаболических расстройств, таких как избыточная масса тела или ожирение, которое прогрессирует в ассоциированные расстройства, подобные сахарному диабету, сердечно-сосудистым расстройствам, метаболическому синдрому и гипертонии.
В целом лечение первой линии для индивидуумов, страдающих такими метаболическими расстройствами, в частности избыточной массой тела или ожирением, включает принятие рациона с низким содержанием жира и регулярную физическую нагрузку. Однако соблюдение такого режима может быть недостаточным и по мере прогрессирования заболевания становится необходимым лечение лекарственными препаратами.
Соответственно были проведены исследования в направлении разработки препаратов, которые являются безопасными и эффективными для профилактики и лечения клинических проявлений, которые возникают вследствие накопления жира в биологических жидкостях, клетках и тканях. Таким образом, продолжает оставаться необходимость в уменьшении каким-либо образом всасывания и накопления жира в организме.
Один подход к снижению накопления жира заключается в снижении или ингибировании агентов, которые содействуют перевариванию и всасыванию жира на различных уровнях в организме. Ферменты, относящиеся к семейству генов липазы, играют центральную роль в метаболизме, всасывании и транспортировке липидов.
Печеночная липаза и липопротеидлипаза представляют собой белки, которые опосредуют связывание, захват, катаболизм и перестройку липопротеидов и фосфолипидов. Липопротеидлипаза и печеночная липаза функционируют, будучи связанными с поверхностью просвета, покрытой эндотелиальными клетками, соответственно в периферических тканях и печени. Оба фермента участвуют в обратном транспорте холестерина, который представляет собой движение холестерина из периферических тканей в печень или для выделения из организма, или для завершения метаболического цикла. Известно, что генетические дефекты и в печеночной липазе, и в липопротеидлипазе вызывают семейные расстройства метаболизма липопротеидов. Дефекты в метаболизме липопротеидов приводят к тяжелым метаболическим расстройствам, включая гиперхолестеринемию, гиперлипидемию и атеросклероз.
Ферменты семейства генов липазы участвуют в широком ряду метаболических путей в диапазоне от переваривания, всасывания липидов, захвата жирных кислот, транспортировки липопротеидов, а также при воспалении (Wong Howard et al., 2002, The lipase gene family, Journal of Lipid Research, Vol. 43: 993-999).
Панкреатическая липаза представляет собой один из ключевых ферментов в липидном метаболизме. Она синтезируется панкреатическими ацинарными клетками, где она секретируется в просвет кишечника, и содействует всасыванию в кишечнике длинноцепочечных триглицеридных жирных кислот (Verger, R. 1984, Pancreatic Lipases In Lipases. B. Borgstrum and H.L. Brockman, editors. Elsevier, New York. 83-150; Lowe, M.E. 1997, Molecular mechanisms of rat and human pancreatic triglyceride lipases. J. Nutr. 127: 549-557). Считают, что действие триацилглицеринлипаз является антиатерогенным, потому что эти ферменты снижают уровни триацилглицерина и содействуют образованию HDL (липопротеидов высокой плотности) (Olivecrona, G., and Olivecrona, T. (1995) Curr. Opin. Lipid. 6:291-305). Липопротеидлипаза представляет собой основной фермент, ответственный за распределение и утилизацию триглицеридов в организме. Липопротеидлипаза гидролизует триглицириды в IDL (липопротеидах промежуточной плотности) и HDL (липопротеидах высокой плотности) и ответственна за перестройку липопротеидов. Печеночная липаза также функционирует в качестве фосфолипазы и гидролизует фосфолипиды в HDL.
Члены семейства липаз функционируют в метаболизме циркулирующих липопротеидов. Печеночная липаза играет роль в захвате холестерина HDL (Olivecrona, T. et al., 1993, Lipoprotein lipase and heratic lipase. Curr. Opin. Lipidol. 4: 187-196). Она синтезируется исключительно в печени, где она преимущественно обнаруживается (Hixenbaugh, E.A., et al., 1989, Hepatic lipase in the rat ovary. J. Biol. Chem. 264:4222-4230).
Третий член семейства генов липазы, липопротеидлипаза (LPL), распределена в разнообразных тканях при самих высоких концентрациях, встречающихся в жировой ткани и мышцах. Липаза связана с капиллярным эндотелием, где она функционирует для снабжения подлежащей ткани жирными кислотами, происходящими из богатой триглицеридами сердцевины циркулирующих хиломикронов и VLDL (липопротеидов очень низкой плотности) (Olivecrona, T. and G. Bengtsson-Olivecrona. 1993. Lipoprotein lipase and hepatic lipase. Curr. Opin. Lipidol. 4: 187-196). В этом процессе LPL трансформирует эти липопротеиды в остаток и частицы HDL. Все больше данных свидетельствуют о том, что LPL, продуцируемая макрофагами в сосудистой стенке, может стимулировать развитие атеросклероза содействием накоплению липидов внутри очага поражения. Было показано, что LPL участвует в патогенезе атеросклероза (Mead JR, et al. 1999, “Lipoprotein Lipase, a key role in atherosclerosis?” FEBS Lett., Nov 26, 462(1-2):1-6). Несколько групп также предположили, что и LPL, и печеночная липаза, как представляется, служат в качестве лигандов в метаболизме липопротеидов плазмы (Nykjaer, A., et al., 1993, The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds lipoprotein lipase and beta-migrating very low density lipoprotein associated with the lipase. J. Biol. Chem. 268: 15048-15055; Krapp, A., S. et al., 1996. Hepatic lipase mediates the uptake of chylomicrons and VLDL into cells via the LDL receptor-related protein (LRP). J. Lipid Res. 37: 926-936). У трансгенных животных, экспрессирующих человеческую липопротеидлипазу или печеночную липазу, имеются сниженные уровни триглицеридов плазмы и повышенные уровни липопротеида высокой плотности (HDL) (Shimada, M., et al (1993) J. Biol. Chem. 268:17924-17929; Liu, M.-S., et al. (1994) J. Biol. Chem. 269:11417-11424).
Позднее выявленным членом семейства генов липазы является эндотелиальная липаза. Хотя функция этой липазы в настоящее время не определена, считают, что она играет роль в метаболизме HDL (Jaye, M., et al., 1999, A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet 21:424-428).
При все более признаваемой возможной роли липаз в пути жирового метаболизма препараты, которые ингибируют или снижают активность липаз на различных уровнях в организме, формируют переднюю линию лечения заболеваний, опосредованных накоплением жира на повышенных уровнях.
Ингибитор липазы, который продается в качестве препарата против ожирения, включает Orlistat (XENICAL®) и описан в патенте США №4598089. Заявка на Европейский патент ЕР №129748 относится к Orlistat и родственным соединениям и их применению при ингибировании панкреатической липазы и лечении гиперлипидемии и ожирения. Orlistat ингибирует только кишечные липазы, такие как желудочная, панкреатическая и карбоксилэфирная липазы, в частности панкреатическую липазу, в просвете кишечника и блокирует переваривание пищевого жира предотвращением взаимодействия липазы с ее липидной мишенью. Однако, как представляется, она не оказывает эффекта на липазы, отличные от кишечных липаз, такие как печеночная липаза или эндотелиальная липаза, которые также признаны играющими роли в катализе гидролиза липидов (Drent ML, van der Veen EA. Lipase inhibition: A novel concept in the treatment of obesity. Int J. Obes. Relat. Metab. Disord. 1993; 17:241-244). Orlistat также имеет тенденцию вызывать высокую частоту возникновения неприятных (относительно безвредных) побочных эффектов, таких как диарея.
Соединения, которые ингибируют активность печеночной липазы и/или эндотелиальной липазы, были раскрыты в заявке РСТ WO №2004094393 для лечения заболеваний, опосредованных печеночной и/или эндотелиальной липазой. Эти соединения в первую очередь направлены на увеличение уровней HDL ингибированием активности печеночной и/или эндотелиальной липазы и не предназначены для нацеливания на кишечные или другие липазы.
Поэтому желательна разработка новых соединений, которые можно использовать при снижении или ингибировании метаболизма, всасывания и накопления жира на различных уровнях, включая жидкости, клетки и ткани в организме, путем ингибирования или снижения активности всех членов представляющего интерес семейства генов липазы, а не именно конкретного типа липазы.
Также сообщалось, что растительный бензохинон эмбелин (2,5-дигидрокси-3-ундецил-1,4-бензохинон), полученный из высушенного плода Embrlia ribes, известен в качестве средства против фертильности, повышает активность липогенных ферментов, малат-дегидрогеназы, глюкозо-6-фосфат-дегидрогеназы и гидроксиметилглутарил-СоА редуктазы, хотя по существу не воздействуя на активность липолитических ферментов (Gupta S. et al., Fitoterapia 60(4):331-338 (1989)). Эмбелин также используется в качестве средства против ленточных гельминтов как имеющий противоопухолевое, противовоспалительное и анальгетическое свойства (Chitra et al. Chemotherapy 40:109 (1994)) и в качестве проникающего в клетки не пептидного ингибитора Х-сцепленного ингибитора апоптоза (XIAP) (Nikolovska-Coleska et al. J. Med. Chem. 47:2430 (2004)).
Краткое описание сущности изобретения
Настоящее изобретение предоставляет соединения формулы (I):

где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, С3-С13алкила, С1-С20галоидалкила, С2-С13алкенила, С2-С13алкинила, С4-С6циклоалкила, С4-С6циклоалкенила, С1-С13алкоксиалкила, С1-С5алкилциклоалкила, С1-С5алкилциклоалкенила, С1-С13алкиламина, С1-С13ариламина, С(О)С1-С6алкила, О-С(О)С1-С6алкила, гетероциклоалкила, арила, алкиларила, С(О)арила и О-С(О) арила; где каждая из указанных выше групп может необязательно иметь от 1 до 6 заместителей, независимо выбранных из водорода, галоида, нитро, амино, циано, изоциано, тио, С1-С6алкила, циклоалкила, арила, алкокси и арилокси групп. Однако в соответствии с настоящим изобретением R1 и R2 не представляют собой метил, метокси, этил, этокси, фенил и гидрокси.
Настоящее изобретение также относится к соединениям формулы (I) и их производным, включая без ограничения их полиморфы, изомеры и пролекарства, их геометрические или оптические изомеры и их фармацевтически приемлемые сложные и простые эфиры, карбаматы таких соединений, все их сольваты и гидраты и все их соли.
В другом аспекте настоящее изобретение предоставляет применение соединений формулы (I) при снижении или ингибировании метаболизма, всасывания и накопления жира на различных уровнях, включая жидкостный, клеточный и тканевой уровни в организме, путем ингибирования активности ферментов, относящихся к семейству генов липазы.
В другом варианте осуществления, изобретение предоставляет соединения формулы (II):

где каждый R1 и R2 независимо выбран из группы, состоящей из С3-С13алкила, С1-С20галоидалкила, С2-С13алкенила, С2-С13алкинила, С4-С6циклоалкила, С4-С6циклоалкенила, С1-С13алкоксиалкила, С1-С5алкилциклоалкила, С1-С5алкилциклоалкенила, С1-С13алкиламина, С1-С13ариламина, С(О)С1-С6алкила, гетероциклоалкила, арила, алкиларила и С(О)арила; где каждая из указанных выше групп может необязательно иметь от 1 до 6 заместителей, независимо выбранных из водорода, галоида, нитро, амино, циано, изоциано, тио, С1-С6алкила, циклоалкила, арила, алкокси и арилокси групп. Однако в соответствии с настоящим изобретением R1 и R2 не представляют собой метил, метокси, этил и фенил.
Настоящее изобретение также относится к соединениям формулы (II) и их производным, включая без ограничения их полиморфы, изомеры и пролекарства, их геометрические или оптические изомеры и их фармацевтически приемлемые сложные эфиры, простые эфиры, карбаматы таких соединений, все их сольваты и гидраты и все их соли.
В другом аспекте настоящее изобретение предоставляет соединения формулы (II) при снижении или ингибировании метаболизма, всасывания и накопления жира на различных уровнях, включая жидкостный, клеточный и тканевой уровни в организме, путем ингибирования активности ферментов, относящихся к семейству генов липазы.
В другом варианте осуществления изобретение предоставляет соединения формулы (III):

где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, С1-С13алкила, С1-С20галоидалкила, С2-С13алкенила, С2-С13алкинила, С4-С6циклоалкила, С4-С6циклоалкенила, С1-С13алкоксиалкила, С1-С13алкиламина, С1-С13ариламина, С1-С5алкилциклоалкила, С1-С5алкилциклоалкенила, С(О)С1-С6алкила, гетероциклоалкила, арила, алкиларила и С(О)арила; где каждая из указанных выше групп может необязательно иметь от 1 до 6 заместителей, независимо выбранных из водорода, галоида, нитро, амино, циано, изоциано, тио, С1-С6алкила, циклоалкила, арила, алкокси и арилокси групп.
Настоящее изобретение также относится к соединениям формулы (III) и их производным, включая без ограничения их полиморфы, изомеры и пролекарства, их геометрические или оптические изомеры и их фармацевтически приемлемые сложные эфиры, простые эфиры, карбаматы таких соединений, все их сольваты и гидраты и все их соли.
В другом аспекте настоящее изобретение предоставляет применение соединений формулы (III) при снижении или ингибировании метаболизма, всасывания и накопления жира на различных уровнях, включая жидкостный, клеточный и тканевой уровни в организме, путем ингибирования активности ферментов, относящихся к семейству генов липазы.
Настоящее изобретение, кроме того, предоставляет способ получения соединений формул (I), (II) и (III) и их производных.
Настоящее изобретение предоставляет фармацевтические композиции, содержащие любое из соединений по настоящему изобретению, включая их полиморфы, пролекарства, изомеры или их фармацевтически приемлемые сложные эфиры, простые эфиры и оксимы, которые можно применять при снижении или ингибировании активности ферментов семейства генов липазы, участвующих в метаболизме, всасывании и накоплении липидов в организме на различных уровнях, включая уровень биологических жидкостей, клеточный и тканевой уровни, для лечения, облегчения или предотвращения заболеваний, опосредованных ферментами семейства генов липазы, включая без ограничения избыточную массу тела и ожирение, гиперлипидемию, гиперхолестеринемию, гипертриглицеридемию, панкреатит, гипергликемию, атеросклероз, метаболические синдромы, другие сердечно-сосудистые заболевания и другие метаболические расстройства.
Настоящее изобретение в еще одном аспекте также предоставляет применение соединений формул (I), (II) и (III) и их производных для получения средств по уходу за кожей, волосами или для косметических целей.
Настоящее изобретение в еще одном аспекте также предоставляет применение соединений формул (I), (II) и (III) и их производных для предотвращения или лечения клеточного и тканевого повреждения, вызванного микробными патогенами, секретирующими липазы.
Настоящее изобретение также относится к фармацевтическим препаративным формам, содержащим любое из самих соединений формул (I), (II) и (III) и их производных или в сочетании с подходящим фармацевтически приемлемым эксципиентом. Такие препаративные формы можно применять при снижении или ингибировании активности ферментов семейства генов липазы, участвующих в метаболизме, всасывании и накоплении липидов в организме на различных уровнях, включая уровень биологических жидкостей, клеточный и тканевой уровни, для лечения, облегчения или предотвращения заболеваний, опосредованных ферментами семейства генов липазы, таких как избыточная масса тела и ожирение, гиперлипидемия, гиперхолестеринемия, гипертриглицеридемия, панкреатит, сахарный диабет, атеросклероз, другие сердечно-сосудистые заболевания, метаболические синдромы и метаболические расстройства.
Настоящее изобретение также предоставляет способ получения лекарственных препаратов, содержащих соединения формул (I), (II) и (III) и их производные в терапевтически эффективном количестве или отдельно, или в комбинации с фармацевтически приемлемым адъювантом. Соединения формул (I), (II) и (III) и их производные можно, кроме того, комбинировать с другими активными ингредиентами.
Настоящее изобретение, кроме того, относится к способу лечения заболеваний, опосредованных ферментами семейства генов липазы, введением в терапевтически эффективном количестве любого из соединений формул (I), (II) и (III) и их производных человеку или животному.
Настоящее изобретение и другие цели, признаки и преимущества настоящего изобретения станут еще более очевидными в следующем подробном описании изобретения и сопровождающих вариантов осуществления.
Подробное описание изобретения
Настоящее изобретение предоставляет новые способы и композиции для применения при снижении или ингибировании активности ферментов семейства генов липазы для лечения, облегчения или предотвращения заболеваний и состояний, опосредованных ферментами семейства генов липазы, у индивидуума.
Определения
Пока нет других уточнений, следующие определения изложены для иллюстрации и определения значения и объема различных терминов, используемых для настоящего описания изобретения.
Термин «ферменты семейства генов липазы», используемый в настоящем описании, включает без ограничения печеночную липазу, кишечные липазы, включая желудочную липазу, панкреатическую липазу и карбоксилэфирную липазу, эндотелиальную липазу, фосфолипазу и другие родственные липазы.
Используемый в настоящем описании термин «фармацевтически приемлемое» относится к веществу, включая носитель, разбавитель, носитель-эксципиент или композицию, являющиеся химически и/или токсикологически совместимыми с другими ингредиентами, составляющими препаративную форму, которые не вредны для их реципиента.
Термин «алкил», применяемый для обозначения его как такового или его как части другого заместителя, пока нет других определений, означает прямо- или разветвленно-цепочечный одновалентный углеводородный радикал, такой как, например, метил, этил, н-пропил, изопропил, н-бутил, третичный бутил, втор-бутил, н-пентил и н-гексил.
Термин «алкенил», используемый отдельно или в комбинации с другими терминами, означает прямо- или разветвленно-цепочечную одновалентную углеводородную группу, имеющую указанный диапазон количества атомов углерода, и такие группы, как винил, пропенил, кротонил, изопентенил и различные изомеры бутенила.
Термин «алкинил», используемый отдельно или в комбинации с другими терминами, означает прямую или разветвленную ациклическую углеродную цепь, которая содержит углеводородную группу с межуглеродной тройной связью, имеющую указанные диапазоны количества атомов углерода и группы.
Термин «циклоалкил» означает циклический, или моноциклический, или полициклический алкильный радикал, имеющий, по меньшей мере, 3 атома углерода, а обычно от 3 до 7 атомов углерода. Примерами являются: циклопропил, циклобутил, циклопентил, циклогексил или циклогептил.
Термин «алкокси» отдельно или в комбинации обозначает группу формулы алкил-О-, в которой термин «алкил» имеет ранее указанное значение, такое как метокси, этокси, н-пропокси, изопропокси, 2° бутокси и 3° бутокси или 2-метоксиэтокси.
Термин «С1-С5 алкилциклоалкил» означает, что любая из С1-С5 алкильная группа замещена циклоалкильной группой и составленная группа прикреплена к ядру на алкильном конце.
Термин «С1-С6 гетероциклоалкил» означает гетероциклоалкильную группу, имеющую 2-6 атомов углерода, предпочтительно 3-5 атомов углерода, и содержащую, по меньшей мере, один гетероатом, выбранный из N, O и/или S, который может быть прикреплен через гетероатом или атом углерода.
Термин «арил» означает ароматическую углеводородную группу, имеющую одиночную (например, фенильную) или конденсированную кольцевую систему (например, нафталин, антрацен, фенантрен и т.д.). Типичная арильная группа представляет собой ароматическое карбоциклическое кольцо, имеющее 6, 7, 8, 9 или 10 атомов углерода, такое как фенил, нафтил, тетрагидронафтил или индил, который может быть необязательно замещен одним или более заместителями, выбранными из гидрокси, амино, галогена, нитро, циано, С1-С4 алкила, С2-С4 алкенила, С2-С4 алкинила, С1-С4 алкокси, С1-С4 алкокси, С1-С4 диалкиламино, причем алкильные части имеют такое же значение, как определено ранее. Предпочтительной ароматической углеводородной группой является фенил.
Термин «С3-С9 гетероарил» означает замещенную или незамещенную ароматическую группу, имеющую 3, 4, 5, 6, 7, 8 или 9 атомов углерода, по меньшей мере, включая один гетероатом, выбранный из N, O и/или S, подобную имидазолилу, тиадиазолилиу, пиридилу, (бензо)тиенилу, (бензо)фурилу, хинолилу, тетрагидрохинолилу, хиноксалилу или индолилу. Заместители на гетероарильной группе могут быть выбраны из группы заместителей, перечисленных для арильной группы. Гетероарильная группа может быть прикреплена через атом углерода или, если возможно, гетероатом.
Термин «С6-С10 арилокси» означает арильную группу, содержащую 6, 7, 8, 9 или 10 атомов углерода, как определено выше, прикрепленную к атому кислорода. С3-С9 гетероарилокси группы представляют собой аналоги С6-С10 арилокси групп, по меньшей мере, содержащих один гетероатом, выбранный из N, O или S.
Термин «галоид» означает фтор-, хлор-, бром- или йод-.
Термин «амино», отдельно или в комбинации, обозначает первичную, вторичную или третичную аминогруппу, связанную через атом азота со вторичной аминогруппой, несущей алкильный или циклоалкильный заместитель, и третичную аминогруппу, несущую 2 одинаковых или различных алкильных или циклоалкильных заместителя, или 2 заместителя азота вместе образуют кольцо, такое как, например, -NH2, метиламино, этиламино, диметиламино, диэтиламино, метилэтиламино, пирролидин-1-ил или пиперидино и т.д., предпочтительно амино, диметиламино и диэтиламино, особенно предпочтительно первичную амино.
Термин «циано», отдельно или в комбинации, обозначает -CN группу.
Термин «нитро», отдельно или в комбинации, обозначает -NO2 группу.
Термин «гетероциклическая группа» относится к радикалам или группам, полученным из моноциклических или полициклических насыщенных или ненасыщенных, замещенных или незамещенных гетероциклических ядер, имеющих 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14 кольцевых атомов и содержащих 1, 2-3 гетероатома, выбранных из группы, состоящей из азота, кислорода или серы.
Термин «заместитель» представляет собой «не вносящие помехи» заместители. Под термином «не вносящая помехи» подразумевается, что группа является подходящей химически и достаточно устойчивой для занятия предназначенного положения и выполнения предназначенной или предполагаемой роли. Таким образом, неподходящие группы исключены из определения «не вносящие помехи».
Кроме того, соединения формулы (I) и их производные могут быть меченными изотопом (например,3H,14C,35S,125I и т.д.).
«Пролекарство» относится к соединениям, способным превращаться в соединения по настоящему изобретению взаимодействиями фермента, желудочного сока или им подобного в физиологических условиях in vivo, в частности соединения, способные превращаться в соединения по настоящему изобретению после ферментативного окисления, восстановления, гидролиза или им подобного, или соединения, способные превращаться в соединения по настоящему изобретению после гидролиза или ему подобного желудочным соком или ему подобным.
«Полиморф» относится к соединению, которое встречается в двух или более формах.
Фраза «терапевтически эффективное количество» означает количество соединения по настоящему изобретению, которое лечит или предотвращает определенное заболевание, состояние; или ослабляет, облегчает или устраняет один или более симптомов конкретного заболевания, состояния или расстройства; или предотвращает или задерживает начало одного или более симптомов описанного в настоящем описании заболевания, состояния или расстройства.
Использование терминов C1-Cn используется для обозначения каждого из C1, C2, C3 … Cn. Таким образом, C1-C20включает C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11, C12, C13, C14, C15, C16, C17, C18 C19 и C20. C1-C13включает каждый из C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11, C12, C13. C2-C13включает каждый из C2, C3, C4, C5, C6, C7, C8, C9, C10, C11, C12, C13 и т.д.
Для специалистов в данной области будет очевидно, что любые модификации и материалов, и способов могут осуществляться без отхода от назначения и цели настоящего изобретения. Следующие общие термины, встречающиеся в примерах, объясняются ниже:
а) все операции, проводимые при комнатной или окружающей температуре, были в интервале от 18 до 25°С;
b) течение реакции контролировали тонкослойной хроматографией (TLC) и величины времени взаимодействия приведены только для иллюстрации;
с) точки плавления не корректированы, точки плавления представлены для материалов, полученных, как описано, полиморфизм может привести к выделению материалов с другими точками плавления у тех же препаратов;
d) структура и чистота всех конечных продуктов подтверждалась, по меньшей мере, одной из следующих методик: TLC, NMR (ЯМР, ядерным магнитным резонансом), спектроскопией, масс-спектроскопией или микроаналитическими данными;
е) выходы представлены только для иллюстрации;
f) данные ЯМР, если они приводятся, представлены в виде величин дельта (δ) для основных диагностических протонов, приведенных в частях на миллион (м.д.) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, определенного при 300 МГц или 400 МГц с использованием указанного растворителя;
g) химические символы имеют их обычные значения: также использовались следующие аббревиатуры: об. (объем), масс. (масса), т.к. (точка кипения), т.п. (точка плавления), л (литры), мл (миллилитры), г (граммы), мг (миллиграммы), моль (моли), ммоль (миллимоли), экв. (эквиваленты), °С (градусы Цельсия), конц. (концентрированный), SiO2 (диоксид кремния), Na2SO4 (сульфат натрия);
h) промежуточные соединения, использованные в следующих примерах, были получены от компании Sigma.
Липазы
Были описаны 3 члена семейства человеческих триацилглицеринлипаз: панкретическая липаза, липопротеидлипаза и печеночная липаза (Goldberg, I.J., Le, N.-A., Ginsberg, H.N., Krauss, R.M., and Lindgren, F.T. (1988) J. Clin. Invest. 81, 561-568; Goldberg, I.J., Le, N., Paterniti J.R., Ginsberg, H.N., Lindgren, F.T., и Brown, W.V. (1982) J. Clin. Invest. 70, 1184-1192; Hide, W.A., Chan, L., и Li, W.-H. (1992) J. Lipid. Res. 33, 167-178). Панкреатическая липаза в первую очередь ответственна за гидролиз пищевых липидов. Были описаны варианты панкреатической липазы, но их физиологическая роль не была определена (Giller, T., Buchwald, P., Blum-Kaelin, D., и Hunziker, W. (1992) J. Biol. Chem. 267,16509-16516).
Полипептиды липазы, кодируемые генами этой липазы, имеют длину приблизительно 450 аминокислот с лидерными сигнальными пептидами для содействия секреции. Белки липазы состоят из двух основных доменов (Winkler, K., D'Arcy, A., and Hunziker, W. (1990) Nature 343, 771-774). Аминоконцевой домен содержит каталитический сайт, в то время как карбоксильный домен считается ответственным за связывания субстратов, ассоциацию кофакторов и взаимодействие с клеточными рецепторами (Wong, H., Davis, R.C, Nikazy, J., Seebart, K.E., and Schotz, M.C. (1991) Proc. Natl. Acad. Sci. USA 88, 11290-11294; van Tilbeurgh, H., Roussel, A., Lalouel, J.-M., and Cambillau, C. (1994) J. Biol. Chem. 269, 4626-4633; Wong, H., Davis, R.C, Thuren, T., Goers, J.W., Nikazy, J., Waite, M., and Schotz, M.C. (1994) J. Biol. Chem. 269, 10319-10323; Chappell, D.A., Inoue, L, Fiy, G.L., Pladet, M.W., Bowen, S.L., Iverius, P.-H., Lalouel, J.-M., and Strickland, D.K. (1994) J. Biol. Chem. 269, 18001-18006). Общий уровень аминокислотной гомологии между членами семейства составляет 22-65%, причем локальные области высокой гомологии соответствуют структурным гомологиям, которые связаны с ферментативной функцией.
Члены семейства триацилглицеринлипазы разделяют ряд сохраняемых структурных признаков. Один такой признак представляет собой мотив “GXSXG”, в котором центральный сериновый остаток представляет собой один из трех остатков, составляющих «каталитическую триаду» (Winkler, K., D'Arcy, A., и Hunziker, W. (1990) Nature 343, 771-774; Faustinella, F., Smith, L.C, и Chan, L. (1992) Biochemistry 31, 7219-7223). Сохраненные аспартатный и гистидиновый остатки составляют остальную часть каталитической триады. Короткий участок 19-23 аминокислот («крышечная область») образует амфипатическую спиральную структуру и покрывает каталитический карман фермента (Winkler, K., D'Arcy, A., и Hunziker, W. (1990) Nature 343, 771-774). Сравнения печеночной и липопротеидлипазы продемонстрировали, что различия активности ферментов триацилглицеринлипазы и фосфолипазы частично опосредованы этой крышечной областью (Dugi, K.A., Dichek H.L., и Santamarina-Fojo, S. (1995) J. Biol. Chem. 270, 25396-25401). Триацилглицеринлипазы обладают варьирующимися степенями активности связывания гепарина. Липопротеидлипаза имеет самый высокий аффинитет к гепарину, и эта активность связывания была картирована к фрагментам секвенирования положительно заряженных остатков в аминоконцевом домене (Ma, Y., Henderson, H.E., Liu, M.-S., Zhang, H., Forsythe, I.J., Clarke-Lewis, L, Hayden, M.R., и Brunzell, J. D. J. Lipid Res. 35, 2049-2059).
Сообщалось о генетических последовательностях, кодирующих человеческую панкреатическую липазу, печеночную липазу и липопротеидлипазу (номера доступа в генетическом банке соответственно #M93285, #J03540 и #M15856). Матричные РНК человеческой печеночной липазы и панкреатической липазы имеют длину приблизительно 1,7 и 1,8 тысяч гетероциклических оснований нуклеиновой кислоты соответственно. 2 транскрипта мРНК 3,6 и 3,2 тысячи пар оснований продуцируются из гена человеческой липопротеидлипазы (Ranganathan, G., Ong, J.M., Yukht, A., Saghizadeh, M., Simsolo, R.B., Pauer, A., и Kern, P.A. (1995) J. Biol. Chem. 270, 7149-7155).
Соединения, которые воздействуют на активность липазы
Настоящее изобретение относится к соединениям формулы (I)
и к их производным, включая без ограничения их полиморфы, изомеры и пролекарства, их геометрические или оптические изомеры и их фармацевтически приемлемые сложные эфиры, простые эфиры, карбаматы таких соединений, все их сольваты и гидраты и все их соли, где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, С3-С13алкила, С1-С20галоидалкила, С2-С13алкенила, С2-С13алкинила, С4-С6циклоалкила, С4-С6циклоалкенила, С1-С13алкоксиалкила, С1-С5алкилциклоалкила, С1-С5алкилциклоалкенила, С1-С13алкиламина, С1-С13ариламина, С(О)С1-С6алкила, О-С(О)С1-С6алкила, гетероциклоалкила, арила, алкиларила, С(О)арила и О-С(О) арила; где каждая из указанных выше групп может необязательно иметь от 1 до 6 заместителей, независимо выбранных из водорода, галоида, нитро, амино, циано, изоциано, тио, С1-С6алкила, циклоалкила, арила, алкокси и арилокси групп. Однако в соответствии с настоящим изобретением R1 и R2 не представляют собой метил, метокси, этил, этокси, фенил и гидрокси.
В другом варианте осуществления изобретение относится к соединениям формулы (II):
и к их производным, включая без ограничения их полиморфы, изомеры и пролекарства, их геометрические или оптические изомеры и их фармацевтически приемлемые сложные эфиры, простые эфиры, карбаматы таких соединений, все их сольваты и гидраты и все их соли, где каждый R1 и R2 независимо выбран из группы, состоящей из С3-С13алкила, С1-С20галоидалкила, С2-С13алкенила, С2-С13алкинила, С4-С6циклоалкила, С4-С6циклоалкенила, С1-С13алкоксиалкила, С1-С5алкилциклоалкила, С1-С5алкилциклоалкенила, С1-С13алкиламина, С1-С13ариламина, С(О)С1-С6алкила, гетероциклоалкила, арила, алкиларила и С(О)арила; где каждая из указанных выше групп может необязательно иметь от 1 до 6 заместителей, независимо выбранных из водорода, галоида, нитро, амино, циано, изоциано, тио, С1-С6алкила, циклоалкила, арила, алкокси и арилокси групп. Однако в соответствии с настоящим изобретением R1 и R2 не представляют собой метил, метокси, этил и фенил.
В другом варианте осуществления изобретение относится к соединениям формулы (III):
и к их производным, включая без ограничения их полиморфы, изомеры и пролекарства, их геометрические или оптические изомеры и их фармацевтически приемлемые сложные эфиры, простые эфиры, карбаматы таких соединений, все их сольваты и гидраты и все их соли, где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, С1-С13алкила, С1-С20галоидалкила, С2-С13алкенила, С2-С13алкинила, С4-С6циклоалкила, С4-С6циклоалкенила, С1-С13алкоксиалкила, С1-С13алкиламина, С1-С13ариламина, С1-С5алкилциклоалкила, С1-С5алкилциклоалкенила, С(О)С1-С6алкила, гетероциклоалкила, арила, алкиларила и С(О)арила; где каждая из указанных выше групп может необязательно иметь от 1 до 6 заместителей, независимо выбранных из водорода, галоида, нитро, амино, циано, изоциано, тио, С1-С6алкила, циклоалкила, арила, алкокси- и арилокси групп.
В одном варианте осуществления соединения формулы (III) имеют R1 и R2, независимо выбранные из С(О)арила, С(О)алкиларила, С(О)галоидарила, С(О)нитроарила или С(О)алкоксиарила.
Другой вариант осуществления относится к соединениям формулы (III), где R1 и R2 независимо выбраны из метилфенилкарбонила, этилфенилкарбонила, пропилфенилкарбонила, бутилфенилкарбонила, хлорфенилкарбонила, бромфенилкарбонила, йодфенилкарбонила, фторфенилкарбонила, нитрофенилкарбонила, метоксифенилкарбонила или этоксифенилкарбонила.
Другие варианты осуществления относятся к соединениям формулы (III) где R1 и R2 независимо выбраны из 2-метилфенилкарбонила, 3-метилфенилкарбонила, 4-метилфенилкарбонила, 4-трет-бутилфенилкарбонила, 2-хлорфенилкарбонила, 3-хлорфенилкарбонила, 4-хлорфенилкарбонила, 2-бромфенилкарбонила, 3-бромфенилкарбонила, 4-бромфенилкарбонила, 2-йодфенилкарбонила, 3-йодфенилкарбонила, 4-йодфенилкарбонила, 2-фторфенилкарбонила, 3-фторфенилкарбонила, 4-фторфенилкарбонила, 2-нитрофенилкарбонила, 3-нитрофенилкарбонила, 4-нитрофенилкарбонила, 2-метоксифенилкарбонила, 3-метоксифенилкарбонила и 4-метоксифенилкарбонила.
Соответственно настоящее изобретение также охватывает пролекарства соединений по настоящему изобретению. Термин «пролекарство» включает соединение, которое трансформируется in vivo для выхода соединения формул (I), (II) или (III). Информацию о применении пролекарств можно найти в публикациях "Pro-drugs as Novel Delivery Systems", Vol. 14 of the A.C.S. Symposium Series, by T. Higuchi and W. Stella, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
В настоящее изобретение также включены подходящие активные метаболиты соединений в пределах объема формул (I), (II) или (III).
Соединения по настоящему изобретению могут содержать асимметричные или хиральные центры и поэтому могут существовать в различных стереоизомерных формах. Все подходящие оптические изомеры и стереоизомерные формы соединений по настоящему изобретению, а также их смеси, включая рацемические смеси, формируют часть настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение по настоящему изобретению включает двойную связь или конденсированное кольцо, цис- и транс-формы, а также смеси входят в объем изобретения. В отношении таких соединений, настоящее изобретение включает в зависимости от подходящей формы применение рацемата, одиночной энантиомерной формы, одиночной диастереомерной формы или их смесей. Кроме того, такие соединения могут также существовать в виде таутомеров. Соответственно настоящее изобретение относится к применению всех таких подходящих таутомеров и их смесей. Диастереомерные смеси можно разделить на их отдельные диастереоизомеры на основе их физико-химических различий способами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры могут быть отделены превращением энантиомерной смеси в диастереомерную смесь взаимодействием с соответствующим оптически активным соединением (например, хиральным вспомогательным агентом, таким как хиральный спирт или хлорид кислоты Мошера), разделением диастереоизомеров и превращением (например, гидролизом) отдельных дистереоизомеров в соответствующие чистые энантиомеры или разделением рацемической формы методиками перекристаллизации, синтезом из оптически активных исходных материалов, хиральным синтезом или хроматографическим разделением с использованием хиральной стационарной фазы. Также некоторые из соединений по настоящему изобретению могут представлять собой атропизомеры (например, замещенные биарилы) и считаются частью настоящего изобретения. Энантиомеры можно также отделить использованием хиральной колонки ВЭЖХ.
Кроме того, соединения по настоящему изобретению могут проявлять полиморфизм. Объем настоящего изобретения включает все полиморфные формы соединений в соответствии с изобретением, которые составляют еще один аспект изобретения. Следует понимать, что настоящее изобретение охватывает любую и все рацемические, оптически активные, полиморфные и стереоизомерные формы или их смеси, причем эта форма или формы обладают свойствами, которые можно использовать при лечении указанных в настоящем описании состояний.
Кроме того, настоящее изобретение также включает меченные изотопами соединения по настоящему изобретению, которые идентичны приведенным здесь соединениям, но в которых один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно обнаруживаемого в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как соответственно2H,3H,11C,13C,14C,13N,15N,15O,17O,18O, 31P, 32P,35S,18F,123I,125I и36Cl. Некоторые меченные изотопами соединения по настоящему изобретению (например, соединения, меченные3H и14C) можно использовать в анализах распределения в тканях соединений и/или субстратов. Особенно предпочтительны изотопы, меченные тритием (т.е.3H) и углеродом-14 (т.е.14C) в виду легкости их получения и возможности выявления. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е.2H), может предоставить некоторые терапевтические преимущества, возникающие в результате большей метаболической стабильности (например, увеличенного периода полувыведения или потребностей сниженной дозировки), и, следовательно, в некоторых обстоятельствах может быть предпочтительно. Испускающие позитроны изотопы, такие как15O,13N,11C и18F, можно использовать для исследований позитронной эмиссионной томографией (РЕЕТ) для исследования занятости рецепторов субстратов. Меченные изотопами соединения по настоящему изобретению можно в целом получить процедурами, аналогичными процедурам, раскрытым ниже в «Примерах», путем замещения меченного изотопом реагента на не меченного изотопом реагента.
Одним аспектом изобретения является предоставление способа получения соединений формул (I), (II) или (III). Специалистам в данной области будет понятно из настоящего описания, как получить наиболее предпочтительные соединения по настоящему изобретению с использованием любого подходящего известного способа. Соединения формул (I), (II) или (III) и, пока нет других указаний, R1, R2, как описано выше, можно подходящим образом получить в соответствии со схемой 1.
Кроме того, представленные в настоящем описании примеры, кроме того, иллюстрируют получение соединений по настоящему изобретению. Кроме того, специалистам в данной области будет понятно из настоящего описания, как модифицировать схему 1 и детали описанных ниже в настоящем описании примеров для получения по желанию любого определенного соединения формул (I), (II) или (III) по настоящему изобретению. Следует понимать, что схема 1 представлена исключительно в целях иллюстрации и изображает возможный путь синтеза соединений формул (I), (II) или (III) и не ограничивает изобретение. Специалистам в данной области будет понятно, что для синтеза соединений по настоящему изобретению можно использовать другие пути синтеза. Хотя в схеме 1, проиллюстрированной ниже, также изображены определенные исходные материалы и реагенты, подходящее замещение может быть легко произведено для предоставления разнообразных производных и условий взаимодействия. Кроме того, многие из соединений, полученных описанным ниже способом, могут быть дополнительно модифицированы в свете описания с использованием обычной химии, известной специалистам в данной области.
Схема I

На схеме I изображен общий протокол получения соединения формул (I), (II) или (III) из 2,5-дигидрокси-3-ундецил-1,4-бензохинона или его оксима, или замещенного оксима, или его подходящей соли или аналогов. Исходный материал - 2,5-дигидрокси-3-ундецил-1,4-бензохинон - взаимодействует с алкилхлоридом или ацилхлоридом, или арилхлоридом или ароилхлоридом, или замещенным арилхлоридом или замещенным ароилхлоридом в подходящем инертном галогенизированном растворителе (например, дихлорметане) в присутствии подходящего ароматического основания (например, пиридина) в контролируемых условиях, таких как температура от примерно 10°С до примерно 40°С, в течение периода от примерно 1 ч до примерно 24 ч для выхода соединения формул (I), (II) или (III) в неочищенной форме. Для выделения и очистки соединений по настоящему изобретению можно использовать обычные способы и/или методики разделения и очистки, известные среднему специалисту в данной области. Такие методики будут хорошо известны среднему специалисту в данной области и могут включать, например, все типы хроматографии (высокоэффективную жидкостную хроматографию (ВЭЖХ), колоночную хроматографию с использованием обычных адсорбентов, таких как силикагель, и тонкослойную томографию), методики перекристаллизации и дифференциальной (т.е. жидкостно-жидкостной) экстракции.
Связанные с липазой заболевания и состояния
Аспектом настоящего изобретения является предоставление соединений формул (I), (II) или (III) и их производных для применения в качестве терапевтически активных веществ.
Описанные выше соединения по настоящему изобретению можно применять для снижения или ингибирования активности ферментов семейства генов липазы для лечения, облегчения или предотвращения заболеваний, опосредованных ферментами семейства генов липазы. Эти соединения можно применять для снижения или ингибирования метаболизма, всасывания и накопления жира на различных уровнях, включая уровни биологических жидкостей, клеточный и тканевой уровни в организме, путем ингибирования или снижения активности ферментов, относящихся к семейству генов липазы. Таким образом, соединения по настоящему изобретению и их производные, включая их композиции, можно применять при снижении или ингибировании активности ферментов, относящихся к семейству генов липазы, участвующих в метаболизме, всасывании и накоплении липидов в организме на различных уровнях, включая уровень биологических жидкостей, клеточный и тканевой уровни, для лечения, облегчения или предотвращения заболеваний, опосредованных ферментами семейства генов липазы, включая без ограничения избыточную массу тела и ожирение, гиперлипидемию, гиперхолестеринемию, гипертриглицеридемию, панкреатит, гипергликемию, атеросклероз, метаболические синдромы, другие сердечно-сосудистые заболевания и другие метаболические расстройства.
В другом аспекте соединения по настоящему изобретению и их производные, включая их композиции, можно применять при предотвращении или лечении клеточного и тканевого повреждения, вызванного микробными патогенами, секретирующими липазы.
В другом аспекте соединения по настоящему изобретению и их производные, включая их композиции, можно также применять для средств ухода за кожей и волосами или для косметических целей.
Вариантом осуществления настоящего изобретения является предоставление способа лечения состояний, заболеваний и/или расстройств, опосредованных ферментами, относящимися к семейству генов липазы, включая без ограничения ожирение, гиперлипидемию, гиперхолестеринемию, гипертриглицеридемию, панкреатит, гипергликемию, атеросклероз, метаболические синдромы, другие сердечно-сосудистые заболевания и другие метаболические расстройства, снижением или ингибированием метаболизма, всасывания и накопления жира на различных уровнях, включая уровень биологических жидкостей, клеточный и тканевой уровни в организме, путем ингибирования или снижения активности ферментов, относящихся к семейству генов липазы, у млекопитающего, включая человека, который включает введение указанному млекопитающему эффективного лечащего количества соединения формул (I), (II) или (III) или его производных.
В другом варианте осуществления настоящее изобретение предоставляет способ лечения или предотвращения клеточного и тканевого повреждения, вызванного микробными патогенами, секретирующими липазы, ингибированием или снижением активности ферментов, относящихся к семейству генов липазы, у млекопитающего, включая человека, который включает введение указанному млекопитающему эффективного лечащего количества соединения формул (I), (II) или (III) или его производных.
Соответственно одним вариантом осуществления настоящего изобретения является предоставление фармацевтической композиции, содержащей терапевтически эффективное количество соединения формул (I), (II) или (III) или его производного и фармацевтически приемлемый инертный адъювант, разбавитель или носитель. Альтернативно, фармацевтическая композиция может включать, по меньшей мере, одно дополнительное фармацевтически активное средство. Дополнительное фармацевтически активное средство может быть выбрано из химически синтезированных соединений или соединений естественного происхождения, имеющих желаемую фармакологическую активность.
Препаративные формы
Соединение формул (I), (II) или (III) или его производное можно вводить любым обычным путем перорально, буккально, интраназально, ингаляционным распылением в стандартной лекарственной форме, парентерально (например, внутривенной, внутримышечной, подкожной, внутригрудинной и инфузионной методиками), местно (например, порошок, мазь или капли), трансдермально, в цистерны, во влагалище, внутрибрюшинно, в мочевой пузырь или ректально. В другом аспекте изобретения соединение по настоящему изобретению и, по меньшей мере, одно другое фармацевтически активное средство можно вводить или отдельно, или в фармацевтической композиции, содержащей их оба. В целом предпочтительно, чтобы такое введение было пероральным. Однако если получающий лечение индивидуум не способен глотать или пероральное введение затруднено иным образом или нежелательно, может быть целесообразным парентеральное или трансдермальное введение.
Соединение формул (I), (II) или (III) или его производное можно вводить в форме любых препаративных форм модифицированного высвобождения, контролируемого высвобождения или отмеренного по времени высвобождения (см., например, Langer, Science 249:1527-1533 (1990)). В одном варианте осуществления можно использовать насос (Langer, Science 249:1527-1533 (1990); Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al., Surgery 88:507 (1980); и Saudek et al, N. Engl. J. Med. 321:574 (1989)) В другом варианте осуществления можно использовать полимерные материалы (см. Medical Applications of Controlled Release (Langer и Wise eds., 1974); Controlled Drug Bioavailability, Drug Product Design и Performance (Smolen and Ball eds., 1984); Ranger and Peppas, J. Macromol. Sci. Rev. Macromol. Chem. 23:61 (1983); Levy et al., Science 228:190 (1985); During et al., Ann. Neurol. 25:351 (1989); и Howard et al., J. Neurosurg. 71:105 (1989)).
Доза соединения формул (I), (II) или (III) или его производного, подлежащая введению млекопитающему, включая человека или животного, по указанному выше назначению, определенно не ограничена. Она скорее достаточно вариабельна и зависит от патологий, состояний, симптомов или возраста субъекта и суждения лечащего врача или ветеринара. Общий диапазон эффективных частот введения соединений по настоящему изобретению составляет от примерно 0,001 мг/кг массы тела до примерно 100 мг/кг массы тела субъекта в день. Предпочтительный диапазон эффективных частот введения соединений по настоящему изобретению составляет от примерно 0,01 мг/кг массы тела до примерно 50 мг/кг массы тела субъекта в день. Количества выбирают на основании различных факторов, включая среду, в которую вводится композиция, участок клеток, подлежащих лечению, возраст, состояние здоровья, пол и масса тела пациента или животного, подлежащего лечению, и т.д. Полезные количества включают 1, 5, 15, 20, 25, 30, 40, 60, 150, 200 мг, 1 г, 2 г, 3 г и находятся в диапазоне от 10 мг до 100 мг, от 50 мг до 5 г, от 100 мг до 10 г, от 250 мг до 2,5 г, от 500 мг до 1,25 г и т.д. на дозировку. Хотя может быть целесообразным введение суточной дозы соединения по настоящему изобретению порциями в различные часы суток в каждом данном случае, количество соединения по настоящему изобретению будет зависеть от таких факторов, как растворимость соединения, пролекарства, изомера или фармацевтически приемлемой соли по настоящему изобретению, используемой препаративной формы и путь введения (например, перорально, трансдермально, парентерально или местно).
Дозировки соединений по настоящему изобретению можно вводить людям любым подходящим путем, причем пероральное введение является предпочтительным. Отдельная пероральная лекарственная форма, например таблетки или капсулы, должна в целом содержать от примерно 0,1 мг до примерно 100 мг соединения по настоящему изобретению в подходящем фармацевтически приемлемом наполнителе, разбавителе или носителе. В зависимости от потребности дозировки для внутривенного введения находятся в целом в диапазоне от примерно 0,1 мг до примерно 10 мг на одну дозу. Для интраназального или ингаляционного введения дозировка в целом составляется в виде раствора с концентрацией от примерно 0,1% до примерно 1% (масс./об.). На практике врач определит действительную дозировку, которая будет наиболее подходящей для отдельного пациента, и она будет варьироваться, например, в зависимости от возраста, массы тела и реакции конкретного пациента. Указанные выше дозировки являются иллюстративными для среднего случая, но, конечно, могут быть отдельные случаи, когда возможны диапазоны более высоких или более низких дозировок. И такие дозировки соединений по настоящему изобретению находятся в пределах объема настоящего изобретения.
Другим предпочтительным вариантом осуществления настоящего изобретения является предоставление соединений формул (I), (II) или (III) или их производных для изготовления фармацевтических препаративных форм для профилактики и лечения состояний, заболеваний и/или расстройств, опосредованных ферментами, относящимися к семейству генов липазы, включая без ограничения избыточную массу тела и ожирение, гиперлипидемию, гиперхолестеринемию, гипертриглицеридемию, панкреатит, гипергликемию, атеросклероз, метаболические синдромы, другие сердечно-сосудистые заболевания и другие метаболические расстройства. Фармацевтическую препаративную форму, содержащую соединение формул (I), (II) или (III) или его производные, можно составить обычным образом, известным специалистам в данной области, с использованием одного или нескольких фармацевтически приемлемых разбавителей, носителей или наполнителей.
Для перорального введения фармацевтические препаративные формы, которые можно применять в настоящем изобретении, включают таблетки, жевательные таблетки, таблетки контролируемого высвобождения, капсулы, пастилки, гранулы, порошки, пилюли, микрокапсулы, эликсиры, сиропы и суспензии.
В целом таблетки можно получить способами, известными в фармацевтической науке, прямым прессованием, влажным гранулированием или сухим гранулированием. Их препаративные формы обычно включают разбавители, связывающие агенты, смазывающие вещества и разрыхлители, а также соединение по настоящему изобретению. Обычные разбавители включают, например, различные типы крахмала, лактозу, манит, каолин, фосфат или сульфат кальция, неорганические соли, такие как хлорид натрия и порошкообразный сахар. Можно также использовать порошкообразные производные целлюлозы. Обычные таблеточные связывающие агенты включают такие вещества, как крахмал, желатин и сахара, такие как лактоза, фруктоза, глюкоза и им подобные. Также подходят естественные и синтетические смолы, включая акацию, альгинаты, метилцеллюлозу, поливинилпирролидон и им подобные. В качестве связывающих агентов могут также служить полиэтиленгликоль и воски.
Смазывающее вещество в целом необходимо в таблетированной препаративной форме для предотвращения прилипания таблетки и штампов к головке экструдера. Смазывающее вещество выбирают из таких скользящих твердых веществ, как тальк, стеарат магния и кальция, стеариновая кислота и гидрированные растительные масла. Таблеточные разрыхлители включают вещества, которые набухают при смачивании для разрушения таблетки и высвобождения соединения, пролекарства, изомера или фармацевтически приемлемой соли по настоящему изобретению. Они включают крахмалы, глины, целлюлозы, альгины и смолы. Конкретнее, можно использовать, например, кукурузный и картофельный крахмалы, метилцеллюлозу, агар, бентонит, целлюлозу древесины, порошкообразную натуральную губку, катионно-обменные смолы, альгиновую кислоту, кизельгур, цитрусовую пульпу и карбоксиметилцеллюлозу, а также лаурилсульфат натрия. Таблетки часто покрыты сахаром в качестве отдушки и герметика или образующими пленку защитными агентами для модификации свойств растворения таблетки. Соединения по изобретению можно также составлять в виде жевательных таблеток путем использования больших количеств веществ с приятным вкусом, таких как манит в препаративной форме, что в настоящее время достаточно принято в настоящей области.
Как обсуждено выше, действие соединения по настоящему изобретению можно контролировать, то есть задерживать, или пролонгировать, или ограничивать по времени соответствующей препаративной формой. Например, можно получить медленно растворимую гранулу соединения по настоящему изобретению и включить в таблетку или капсулу. Методику можно усовершенствовать изготовлением гранул с несколькими различными скоростями растворения с последующим заполнением капсул смесью этих гранул. Таблетки или капсулы могут быть покрыты пленкой, которая противодействует растворению в течение прогнозируемого периода времени.
Капсулы можно получить смешиванием соединения по изобретению с подходящим разбавителем и заполнением капсул соответствующим количеством смеси. Обычные разбавители включают инертные порошкообразные вещества, такие как крахмал многих различных видов, порошкообразная целлюлоза, особенно кристаллическая и микрокристаллическая целлюлоза, сахара, такие как фруктоза, манит и сахароза, мука злаков и аналогичные пищевые порошки.
Жидкие препараты для перорального введения могут, например, принимать форму растворов, сиропов или суспензий или могут быть представлены в виде сухого продукта для растворения водой или другим подходящим носителем перед применением. Такие жидкие препараты можно получить обычными средствами с фармацевтически приемлемыми добавками, такими как суспендирующие/увеличивающие вязкость агенты (например, сироп сорбита, метилцеллюлоза или гидрированные пищевые жиры); эмульгирующие агенты (например, лецитин или акация); неводные носители (например, миндальное масло, маслянистые сложные эфиры или этиловый спирт, среднецепочечные триглицериды) и консерванты (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота).
Для парентерального введения соединения по изобретению можно составлять в инъекционной форме, включая использование обычных методик катетеризации или вливания. Препаративные формы для инъекций могут быть представлены в стандартной лекарственной форме, например в ампулах или в многодозовых контейнерах с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать такие составляющие агенты, как суспендирующие, стабилизирующие и/или диспергирующие агенты. Инъецируемые растворы или суспензии можно составлять в соответствии с известными способами, используя подходящие нетоксичные, приемлемые для парентерального введения разбавители или растворители, такие как манит, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия, или подходящие диспергирующие или смачивающие и суспендирующие агенты, такие как стерильные, мягкие, фиксированные масла, включая синтетические моно- или диглицериды, и жирные кислоты, включая олеиновую кислоту, и поверхностно-активные вещества, такие как, например, гидроксипропилцеллюлоза, причем при необходимости рН раствора также подходящим образом доводится и забуферивается. В целом масляные растворы подходят для целей внутрисуставных, внутримышечных и подкожных инъекций. Такие водные растворы подходят для целей внутривенных инъекций. Альтернативно, активный ингредиент может быть представлен в порошкообразной форме для растворения подходящим носителем, например стерильной апирогенной водой, перед применением. Парентеральные препараты можно также изготавливать длительно действующими растворением или суспендированием соединения, пролекарства или фармацевтически приемлемой соли по настоящему изобретению, как возможно, например, в масляных или эмульгированных носителях, которые дают ему возможность лишь медленно диспергироваться в сыворотке.
Для интраназального введения или введения путем ингаляции соединения по настоящему изобретению приемлемо доставляются в форме раствора или суспензии из снабженного насосом контейнера с аэрозолем, который выдавливается или выкачивается пациентом, или в виде подачи распыленного аэрозоля из находящегося под избыточным давлением контейнера или распылителя с использованием подходящего газа-вытеснителя, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае сжатого аэрозоля стандартную дозировку можно определить предоставлением клапана для подачи отмеренного количества. Находящийся под избыточным давлением контейнер или распылитель может содержать раствор или суспензию активного соединения. При введении интраназальным аэрозолем или ингаляцией эти композиции получают в соответствии с методиками, хорошо известными в области фармацевтических препаративных форм, и их можно получить в виде растворов в солевом растворе с использованием бензилового спирта или других подходящих консервантов, стимуляторов всасывания для усиления биологической доступности, фторуглеродов и и/или других солюбилизирующих или диспергирующих агентов, известных в данной области. Капсулы и кассеты (изготовленные, например, из желатина) для использования в ингаляторе или инсуффляторе могут составляться с содержанием порошковой смеси соединения по изобретению и подходящего порошка для ингаляционной основы, такого как лактоза или крахмал.
Соединения по настоящему изобретению можно также вводить местно, и это можно осуществлять, например, с помощью кремов, желе, смазок, лосьонов, гелей, паст, мазей и им подобных в соответствии со стандартной фармацевтической практикой. Соединения по настоящему изобретению можно также вводить трансдермально (например, путем применения трансдермальной системы).
Можно использовать любую подходящую препаративную форму для трансдермального применения, содержащую соединение по настоящему изобретению, и такие препаративные формы должны в целом также содержать подходящий трансдермальный носитель, например всасываемый фармакологически приемлемый растворитель, для стимуляции и содействия прохождению соединения через кожу субъекта. Например, подходящие трансдермальные устройства могут иметь вид повязки, имеющей элемент подложки и резервуар, содержащий обсуждаемое соединение. Такие трансдермальные устройства типа повязки могут, кроме того, включать подходящие носители, регулирующие скорость высвобождения барьеры и средства для прикрепления трансдермального устройства к коже субъекта.
Когда желательно введение соединения по настоящему изобретению в виде суппозитория, то можно использовать любую подходящую основу. Масло какао представляет собой традиционную основу суппозиториев, которую можно модифицировать добавлением восков для подъема точки плавления. Широко используются смешиваемые с водой основы суппозиториев, содержащие, в частности, полиэтиленгликоли различной молекулярной массы.
В других вариантах осуществления соединение по настоящему изобретении может быть включено в пищу или напитки.
Соединения по настоящему изобретению можно также вводить млекопитающему, кроме человека. Способ введения и дозировка, подлежащая введению такому млекопитающему, будет зависеть, например, от вида животного и заболевания или расстройства, подлежащего лечению. Соединения по настоящему изобретению можно вводить животным любым подходящим способом, например перорально, парентерально или трансдермально, в любой подходящей форме, такой как, например, капсула, болюс, таблетка, пилюля, например, полученные смешиванием соединения, пролекарства, изомера или фармацевтически приемлемой соли по настоящему изобретению с подходящим разбавителем, таким как карбовоск или воск карнаубы, вместе со смазывающим веществом, жидким смачивающим агентом или пастой, например, полученной диспергированием соединения по настоящему изобретению в фармацевтически приемлемом масле, таком как арахисовое масло, кунжутное масло или кукурузное масло. Соединения, пролекарства, изомеры или фармацевтически приемлемые соли по настоящему изобретению можно также вводить животным в виде имплантата. Такие препаративные формы получают обычным путем в соответствии со стандартной ветеринарной практикой. В качестве альтернативы, соединения по настоящему изобретению можно вводить с подачей воды, например, в форме жидкости или растворимого в воде концентрата. Кроме того, соединения по настоящему изобретению, например, внутри фармацевтических композиций по настоящему изобретению, можно вводить в корм животных, например, можно получить концентрированную кормовую добавку или премикс для смешивания с обычным кормом для животных обычно наряду с подходящим для этого носителем. Носитель содействует равномерному распределению соединения, пролекарства, изомера или фармацевтически приемлемой соли по настоящему изобретению, например, в готовом корме, с которым смешивается премикс. Походящие носители включают без ограничения жидкости, например воду, масла, такие как соевое, кукурузное, семян хлопка, или летучие органические растворители и твердые вещества, например небольшую часть корма или различные подходящие пищевые продукты, содержащие люцерну, соевые бобы, масло семян хлопка, масло семян льна, сердцевину кукурузных початков, кукурузу, мелассу, мочевину и костную муку и минеральные смеси.
В другом аспекте настоящего изобретения биологические анализы, проведенные с использованием соединений формул (I), (II) и (III), демонстрируют сильное ингибирование липаз.
Следующие примеры иллюстрируют варианты осуществления настоящего изобретения. Однако следует понимать, что варианты осуществления изобретения не ограничиваются определенными деталями этих примеров, так как среднему специалисту в данной области будут известны или очевидны его различные вариации в свете настоящего описания.
ПРИМЕРЫ
Пример 1
Экстракция и выделение исходного материала: 2,5-дигидрокси-3-ундецил-1,4-бензохинона

Структура № 1: 2,5-дигидрокси-3-ундецил-п-бензохинон (эмбелин)
Порошкообразные ягоды Embella ribes экстрагируют последовательно простым петролейным эфиром, хлороформом, этилацетатом, метанолом и водой. Хлороформный экстракт подвергают повторной кристаллизации с использованием простого петролейного эфира в качестве кристаллизующего агента. Стадию кристаллизации повторяют для увеличения выхода. Маточный раствор фракционируют хроматографией на колонке силикагеля (100-200 меш) с использованием простого петролейного эфира-хлороформа в качестве элюирующих растворителей (градиентное элюирование) для дополнительного улучшения выхода. Все фракции контролируют на пластине для TLC (Силикагель 60 F254, Merck) н-пропанол:н-бутанол:жидким аммиаком (6:1:3) в качестве системы растворителя TLC. На основании TLC аналогичные фракции объединяют вместе и концентрируют под пониженным давлением. Соединение очищают повторной кристаллизацией в простом петролейном эфире для выхода 140 г (3,5%) экстракта. Примерно 50 г экстракта кипятят в сосуде с обратным холодильником в простом петролейном эфире (500 мл), до тех пор пока экстракт не растворяется в растворителе. Раствор охлаждают до комнатной температуры и фильтруют. Указанную выше процедуру повторяют до получения соединения желаемой чистоты. Полученное соединение проявляет1Н ЯМР (300 МГц, CDCl3) δ: 0,87 (м, 3H), 1,0-1,75 (м, 18H), 2,43 (м, 2H), 5,99 (с, 1H), 7,66 (уш.с, 2H). TOF MS ES: 295 (M+H). Тпл. 142-143°C.
Пример 2
2,5-Ди-О-(3-фторфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 2: 2,5-бис-(3-фторфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (10 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 3-фторбензоилхлорид (1,35 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,96 г, 52,7%).
1H ЯМР (300 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,9 Гц), 1,0-1,6 (м, 18H), 2,5 (т, 2H5 J = 7,6 Гц), 6,8 (с, 1H), 7,3-8,0 (м, 8H). TOF MS ES: 539 (M+H). Тпл. 73,2-75,6°C.
Пример 3
2,5-ди-О-(4-трет-бутилфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 3: 2,5-бис-(4-трет-бутилфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 4-трет-бутилбензоилхлорид (1,67 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,47 г, 28,3%).
1H ЯМР (300 МГц, CDCl3) δ: 0,86 (т, 3H, J = 7,0 Гц), 1,0-1,6 (м, 36H), 2,5 (т, 2H, J = 7,5 Гц), 6,75 (с, 1H), 7,4-8,2 (м, 8H). TOF MS ES: 615 (M+H). Вязкая масса.
Пример 4
2,5-Ди-О-(2-фторфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 4: 2,5-бис-(2-фторфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 2-фторбензоилхлорид (1,35 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,96 г, 52,7%).
1H ЯМР (300 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,9 Гц), 1,1-1,7 (м, 18H), 2,5 (т, 2H, J = 8,0 Гц), 6,8 (с, 1H), 7,2-8,2 (м, 8H). TOF MS ES: 539 (M+H). Тпл. 66,2-68°C.
Пример 5
2,5-Ди-О-(2-бромфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 5: 2,5-бис-(2-бромфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 2-бромбензоилхлорид (1,9 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,56 г, 69,6%).
1H ЯМР (400 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,8 Гц), 1,0-1,8 (м, 18H), 2,5 (т, 2H, J = 6,8 Гц), 6,8 (с, 1H), 7,4-8,2 (м, 8H). TOF MS ES: 680 (25, M++Na), 682 (100, M++2+Na), 684 (25, M++4+Na). Тпл. 77,1-78,6°C.
Пример 6
2,5-Ди-О-(3-бромфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 6: 2,5-бис-(3-бромфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 3-бромбензоилхлорид (1,9 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,4 г, 62%).
1Н ЯМР (400 МГц, CDCl3) δ: 0,87 (т, 3H, J = 7,2 Гц), 1,0-1,8 (м, 18H), 2,51 (т, 2H, J = 7,2 Гц), 6,8 (с, 1H), 7,4-8,4 (м, 8H). TOF MS ES: 680 (5, M++Na), 682 (20, M++2+Na), 684 (5, M++4+Na). Тпл. 98,2-99,6°C.
Пример 7
2,5-Ди-О-(3-хлорфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 7: 2,5-бис-(3-хлорфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 3-хлорбензоилхлорид (1,5 г, 8,50 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,25 г, 64,4%).
1H ЯМР (300 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,9 Гц), 1,1-1,7 (м, 18H), 2,5 (т, 2H, J = 7,6 Гц), 6,8 (с, 1H), 7,4-8,2 (м, 8H). TOF MS ES: 593 (9, M++Na), 595 (3, M++2+Na). Тпл. 102,8-104,6°C.
Пример 8
2,5-Ди-О-(2-хлорфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 8: 2,5-бис-(2-хлорфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 2-хлорбензоилхлорид (1,5 г, 8,50 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,14 г, 58,7%).
1H ЯМР (300 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,9 Гц), 1,0-1,6 (м, 18H), 2,5 (т, 2H, J = 8,0 Гц), 6,8 (с, 1H), 7,2-8,2 (м, 8H). TOF MS ES: 593 (100, M++Na), 595 (35, M++2+Na). Тпл. 56,2-57,8°C.
Пример 9
2,5-Ди-О-(4-хлорфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 9: 2,5-бис-(4-хлорфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 4-хлорбензоилхлорид (1,5 г, 8,50 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,21 г, 62,4%).
1H ЯМР (400 МГц, CDCl3) δ: 0,87 (т, 3H, J = 7,0 Гц), 1,1-1,7 (м, 18H), 2,5 (т, 2H, J = 7,6 Гц), 6,8 (с, 1H), 7,3-8,1 (м, 8H). TOF MS ES: 593 (10, M++Na), 595 (3, M++2+Na). Тпл. 110,2-112°C.
Пример 10
2,5-Ди-О-(4-бромфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 10: 2,5-бис-(4-бромфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 4-бромбензоилхлорид (1,9 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,49 г, 66,5%).
1Н ЯМР (400 МГц, CDCl3) δ: 0,8 (т, 3H, J = 6,8 Гц), 1,0-1,8 (м, 18H), 2,5 (т, 2H, J = 7,6 Гц), 6,7 (с, 1H), 7,6-8,2 (м, 8H). TOF MS ES: 680 (5, M++Na), 682 (20, M++2+Na), 684 (5, M++4+Na). Тпл. 124,4-126,7°C.
Пример 11
2,5-Ди-О-(3-нитрофенилкарбонил)-3-ундецил-1,4-бензохинон
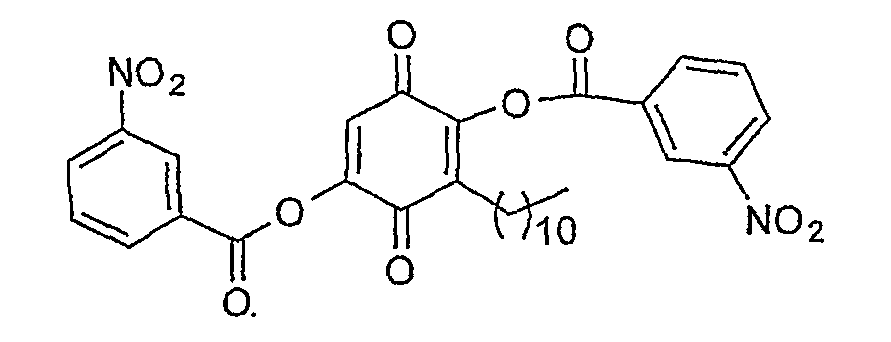
Синтез структуры № 11: 2,5-бис-(3-нитрофенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 3-нитробензоилхлорид (1,6 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,95 г, 47%).
1Н ЯМР (300 МГц, CDCl3) δ: 0,86 (т, 3H, J = 6,9 Гц), 1,1-1,6 (м, 18H), 2,5 (т, 2H, J = 8,0 Гц), 6,8 (с, 1H), 7,6-8,6 (м, 8H). TOF MS ES: 593 (M+H). Тпл. 112,6-114,4°C.
Пример 12
2,5-Ди-О-(4-метилфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 12: 2,5-бис-(4-метилфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (0,15 г, 0,5 ммоль) в дихлорметане (20 мл) добавляют пиридин (0,41 мл, 5,1 ммоль). К нему добавляют 4-толуоилхлорид (0,23 г, 1,53 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (84 мг, 32%).
1H ЯМР (300 МГц, CDCl3) δ: 0,85 (м, 3H), 1,0-1,8 (м, 18H), 2,44 (м, 8H), 6,74 (с, 1H), 7,25-8,25 (м, 8H). TOF MS ES: 553 (100, M++Na). Тпл. 94,8-96,2°C.
Пример 13
2,5-Ди-О-(3-метилфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 13: 2,5-бис-(3-метилфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (0,5 г, 1,7 ммоль) в дихлорметане (20 мл) добавляют пиридин (0,55 мл, 6,8 ммоль). К нему добавляют 3-толуоилхлорид (0,654 г, 4,25 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,64 г, 71%).
1H ЯМР (300 МГц, CDCl3) δ: 0,85 (т, 3H, J = 7,5 Гц), 1,0-1,5 (м, 18H), 2,3-2,5 (м, 8H), 6,75 (с, 1H), 7,30-7,61 (м, 5H), 7,9-8,10 (м, 3H). TOF MS ES: 553 (100, M++Na). Тпл. 80,4-81,7°C.
Пример 14
2,5-Ди-О-(2-нитрофенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 14: 2,5-бис-(2-нитрофенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 2-нитробензоилхлорид (1,6 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,95 г, 47%).
1H ЯМР (400 МГц, CDCl3) δ: 0,86 (т, 3H, J = 6,5 Гц), 1,1-1,6 (м, 18H), 2,6 (т, 2H, J = 8,0 Гц), 6,9 (с, 1H), 7,7-8,1 (м, 8H). TOF MS ES: 615 (100, M++Na). Тпл. 72,2-73,8°C.
Пример 15
2,5-Ди-О-(фенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 15: 2,5-бис-(фенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (0,3 г, 1,02 ммоль) в дихлорметане (20 мл) добавляют пиридин (0,3 мл, 4,08 ммоль). К нему добавляют бензоилхлорид (0,26 г, 2,55 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,285 г, 56,80%).
1H ЯМР (400 МГц, CDCl3) δ: 0,85 (м, 3H), 1,10-1,68 (м, 18H), 2,50 (м, 2H), 6,77 (с, 1H), 7,42-7,64 (м, 6H), 8,11-8,22 (м, 4H). TOF MS ES: 525 (100, M++Na). Тпл. 98,2-99,6°C.
Пример 16
2,5-Ди-О-(4-фторфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 16: 2,5-бис-(4-фторфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 4-фторбензоилхлорид (1,35 г, 8,5 ммоль) при 15-20°С и перемешивают, дают возможность достичь температуры 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,85 г, 46,4%).
1H ЯМР (400 МГц, CDCl3) δ: 0,86 (т, 3H, J = 6,7 Гц), 1,0-1,7 (м, 18H), 2,5 (т, 2H, J = 7,3 Гц), 6,7 (с, 1H), 7,1-7,2 (м, 4H), 8,1-8,2 (м, 4H). Тпл. 95,8-97,7°C.
Пример 17
2,5-Ди-О-(3-метоксифенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 17: 2,5-бис-(3-метоксифенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 3-метоксибензоилхлорид (1,45 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,32 г, 69%).
1H ЯМР (400 МГц, CDCl3) δ: 0,87 (т, 3H, J = 7,0 Гц), 1,1-1,7 (м, 18H), 2,5 (т, 2H, J = 7,9 Гц), 3,9 (с, 6H), 6,8 (с, 1H), 7,2-7,7 (М, 8H). TOF MS ES: 585 (100, M++Na). Тпл. 77,1-78,6°C.
Пример 18
2,5-Ди-О-(4-метоксифенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 18: 2,5-бис-(4-метоксифенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 4-метоксибензоилхлорид (1,45 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (1,37 г, 71,7%).
1H ЯМР (400 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,7 Гц), 1,1-1,7 (м, 18H), 2,5 (т, 2H, J = 7,6 Гц), 3,9 (с, 6H), 6,9 (с, 1H), 7,0-7,2 (м, 4H), 8,10-8,13 (м, 4H). TOF MS ES: 585 (100, M++Na). Тпл. 99,2-100,5°C.
Пример 19
2,5-Ди-О-(2-йодфенилкарбонил)-3-ундецил-1,4-бензохинон

Синтез структуры № 19: 2,5-бис-(2-йодфенилкарбонилокси)-3-ундецил-1,4-бензохинона
К перемешанному раствору 2,5-дигидрокси-3-ундецил-1,4-бензохинона (1,0 г, 3,4 ммоль) в дихлорметане (20 мл) добавляют пиридин (1,1 мл, 13,6 ммоль). К нему добавляют 2-йодбензоилхлорид (2,26 г, 8,5 ммоль) при 15-20°С и перемешивают, доводят температуру до 30°С и перемешивание продолжают в течение 3 ч (TLC). Органический слой экстрагируют дихлорметаном, промывают (вода, рассол), сушат (Na2SO4), концентрируют до неочищенного продукта, который очищают хроматографией на колонке SiO2 (10-20% EtOAc в гексане) до чистой массы (0,89 г, 35%).
1H ЯМР (300 МГц, CDCl3) δ: 0,87 (т, 3H, J = 6,9 Гц), 1,2-1,7 (м, 18H), 2,5 (т, 2H, J = 7,6 Гц), 6,8 (с, 1H), 7,2-8,2 (м, 8H). TOF MS ES: 777 (100, M++Na). Тпл. 64,2-67,6°C.
Пример 20
Анализ ингибирующей липазу активности
Получение испытуемых образцов: структуры №№ 1-19 растворяли в 1 мл DMSO (диметилсульфоксида) для получения 10 мМ маточного раствора, перемешивали вихревой мешалкой до растворения и хранили при 4°С. Различные концентрации рабочих образцов готовили в 0,2 М фосфатном буфере при рН 8,0. Этот образец использовали в качестве испытуемого образца для всех дальнейших анализов.
Анализ липазы: анализ липазы выполняли способом, описанным Winkler и Stuckmann, 1979, с модификацией (Winkler, U.K. & Stuckmann, M. Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia marcescens. J. Bacteriol. 138: 663-670 (1979)). Анализ структурировали, используя 96-луночный формат. Субстрат, используемый в данном анализе, представлял собой п-нитрофенолпальмитат (Sigma, каталожный № N-2752). 4,5 мг п-нитрофенолпальмитата растворяли в 200 мкл N,N-диметилформамида (Sigma, каталожный № D-4551) и объем доводили до 10 мл 0,1 М фосфатным буфером при рН 8,0. Образец панкреатической липазы (Sigma, каталожный № L-3126) готовили растворением фермента в 0,1М фосфатном буфере в концентрации 5 мг/мл. Реакционная смесь состояла из 150 мкл раствора субстрата; 40 мкл фосфатного буфера (рН 8,0, 0,2 М) и 10 мкл раствора липазы. Реакционную смесь инкубировали при 37°С и оптическую плотность измеряли при 405 нм после инкубации. Активность фермента представляли в форме международной единицы (МЕ).
Активность липазы: одна ферментная единица липазы определяется как то количество, которое высвобождает 1 нм свободного фенола из субстрата (п-нитрофенолпальмитат) в 1 мин на 1 мл в стандартных условиях анализа (Winkler and Stuckmann, 1979; Yadav RP, Saxena RK, Gupta R, Davidson WS. Purification and characterization of a region-specific lipase from Aspergillus terreus. Biotechnol. Appl. Biochem. (1998) 28, (243-249)). Ее получают из стандартной графы п-нитрофенола (Sigma, 104-8).
Анализ ингибирования липазы: анализ ингибирования фермента выполняли зависимым от дозы образом. Проверенные концентрации синтетических аналогов были 100 мкМ и 200 мкМ. Анализ был аналогичен анализу, описному выше, за исключением того, что использовали 40 мкл испытуемого образца вместо фосфатного буфера в контроле. Оптическую плотность измеряли через 0 ч и после инкубации при 37°С.
Ингибирование фермента: ингибирование фермента было представлено в виде относительной активности и процентной доли ингибирования на основании изменения в международных единицах (МЕ).
Пример 21
Определение IC50
Приготовление испытуемых образцов: синтетические аналоги растворяли в 1 мл DMSO для получения 10 мМ маточного раствора. Образцы перемешивали вихревой мешалкой до растворения и хранили при 4°С. Различные концентрации рабочих образцов получали в 0,2 М фосфатном буфере, рН 8,0. Этот образец использовали в качестве испытуемого образца для всех дальнейших анализов.
Анализ ингибирования липазы: анализ ингибирования фермента выполняли зависимым от дозы образом. Проверенные концентрации синтетических аналогов были от 6,25 мкМ до 200 мкМ. Анализ был аналогичен анализу, описному выше, за исключением того, что использовали 40 мкл испытуемого образца, вместо фосфатного буфера в контроле. Оптическую плотность измеряли через 0 ч и после инкубации при 37°С. Активность фермента измеряли в международных единицах (МЕ).
Активность липазы: одна ферментная единица липазы определяется как то количество, которое высвобождает 1 нм свободного фенола из субстрата (п-нитрофенолпальмитат) в 1 мин на 1 мл в стандартных условиях анализа. Ее получают из стандартной графы п-нитрофенола (Sigma, 104-8).
Ингибирование в процентах: ингибирование фермента было представлено в виде процентной доли ингибирования на основании изменения в международных единицах (МЕ).
Расчет IC50: IC50 каждого аналога рассчитывали вручную по графе зависимости от дозы (от 6,25 мкМ до 200 мкМ) каждого аналога в концентрации, при которой % ингибирования липазы измерялся как 50% в двух близких прямых точках (ингибирование более и менее 50%). Величину IC50 получали по линейной регрессии. Полученная величина основана на интерполированных данных.
Пример 22
Ингибирование всасывания липазы
Голодавших в течение ночи 4-недельных самцов крыс Wistar (диапазон массы тела: 150-200 г) использовали для исследования всасывания липидов. Крыс делили на 2 группы по 6 крыс каждая и 100 мкл крови брали из орбитальной пазухи для оценки липидного профиля плазмы через 0 ч. 1 мл богатой жиром жидкости вводили РО (перорально) через желудочный зонд. Кроме того, экспериментальной группе перорально вводили 100 мкл испытуемого соединения (структуры №№ 1-19 производных 2,5-ди-О-ароил-3-ундецил-1,4-бензохинона), растворенного в 500 мкл носителя; контрольная группа получала такой же объем носителя.
100 мкл крови брали через 1 ч после кормления для оценки общего содержания триглицеридов. Результаты показаны в следующей таблице. У экспериментальной группы был значимо более низкий уровень триглицеридов.
Пример 23
Ингибирование клеточного захвата липидов
Клеточную линию мышиных макрофагов (J774A.1) высевали в 6-луночные культуральные планшеты (1×105 клеток на лунку) в модифицированную Дульбекко среду Игла (DMEM), содержащую 10% фетальную телячью сыворотку. В каждую лунку добавляли 100 мкг окисленных липопротеидов низкой плотности (LDL). В каждую планшету в лунки в трех повторениях добавляли одно из соединений (структуры №№ 1-19) (10 мкМ), в то время как 3 лунки сохраняли в качестве контроля. Эксперимент заканчивали через 48 ч. Клетки окрашивали красным нейтральным маслом, окрашивали в противотоке гематоксилином и осматривали под микроскопом. По сравнению с контролем, обработка соединениями структур №№ 2-19 значительно ингибировала захват LDL макрофагами.
Все публикации и патентные заявки, приведенные в настоящем описании, включены в него в качестве ссылок, как если бы каждая публикация и патентная заявка была бы конкретно и отдельно указана как включенная в качестве ссылки.
Хотя предшествующее изобретение было описано в некоторых деталях путем иллюстрации и примера в интересах ясности и понимания, для средних специалистов в данной области в свете положений настоящего изобретения будет совершенно очевидно, что в него можно внести определенные изменения и модификации без отхода от сущности или объема прилагаемой формулы изобретения.
Реферат
Изобретение относится к новым соединениям - производным бензохинонов формулы (I): ! ! где каждый R1 и R2 представляет собой O-С(O)фенил; где фенил замещен 1 заместителем, выбранным из галоида, нитро, C1-С6алкила или С1-С6алкокси, и к их фармацевтически приемлемым солям. Изобретение также относится к фармацевтической композиции, ингибирующей или снижающей активность панкреатической липазы на основе соединений формулы (I). 4 н. и 2 з.п. ф-лы, 3 табл.
Формула
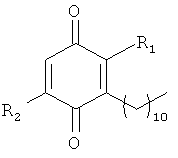
где каждый R1 и R2 представляет собой O-С(O)фенил; где фенил замещен 1 заместителем, выбранным из галоида, нитро, C1-С6алкила или С1-С6алкокси;
и его фармацевтически приемлемые соли.

где каждый R1 и R2 представляет собой С(O)фенил; где фенил замещен 1 заместителем, независимо выбранным из галоида, нитро, С1-С6алкила или С1-С6алкокси;
и его фармацевтически приемлемые соли.

где каждый R1 и R2 представляют собой фенил; где фенил замещен 1 заместителем, выбранным из галоида, нитро, С1-С6алкила и С1-С6алкокси;
и его фармацевтически приемлемые соли.
i) 2,5-бис-(3-фторфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
ii) 2,5-бис-(4-трет-бутилфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
iii) 2,5-бис-(2-фторфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
iv) 2,5-бис-(2-бромфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
v) 2,5-бис-(3-бромфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
vi) 2,5-бис-(3-хлорфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
vii) 2,5-бис-(2-хлорфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
viii) 2,5-бис-(4-хлорфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
ix) 2,5-бис-(4-бромфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
x) 2,5-бис-(3-нитрофенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xi) 2,5-бис-(4-метилфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xii) 2,5-бис-(3-метилфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xiii) 2,5-бис-(2-нитрофенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xv) 2,5-бис-(4-фторфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xvi) 2,5-бис-(3-метоксифенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xvii) 2,5-бис-(4-метоксифенилкарбонилокси)-3-ундецил-1,4-бензохинона;
xviii) 2,5-бис-(2-йодфенилкарбонилокси)-3-ундецил-1,4-бензохинона;
и его фармацевтически приемлемые соли.
Комментарии