Способ определения ингибитора, ковалентно связывающего целевой полипептид - RU2542963C2
Код документа: RU2542963C2
Чертежи





































Описание
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Соединения, ингибирующие активность полипептидов, такие как ферменты, являются важными терапевтическими средствами. Большинство ингибиторов обратимо связываются со своими целевыми полипептидами и обратимо подавляют активность последних. И хотя было разработано много обратимо действующих ингибиторов, которые стали эффективными терапевтическими средствами, им присущи определенные недостатки. Так, многие обратимо действующие ингибиторы киназ взаимодействуют с сайтом связывания АТФ (аденозинтрифосфата). Поскольку структура сайта связывания АТФ довольно консервативна среди киназ, было бы очень заманчиво создать ингибиторы, которые избирательно подавляли бы какую-либо одну или несколько определенных киназ. Кроме того, обратимо действующие ингибиторы впоследствии отъединяются от своих целевых полипептидов, поэтому продолжительность ингибирования может быть меньше, чем это необходимо. Таким образом, при использовании обратимо действующих ингибиторов в качестве терапевтических агентов, для достижения надлежащего биологического эффекта могут потребоваться повышенное количество препарата и/или более частая дозировка. А это, в свою очередь, может вызвать токсичность или другие нежелательные эффекты.
В литературе уже встречаются описания необратимо действующих ингибиторов, которые ковалентно связываются со своими целевыми полипептидами. Ковалентные необратимые ингибиторы определенных мишеней обладают многими важными преимуществами по сравнению со своими обратимыми аналогами. Пролонгированное угнетение мишеней, преследуемых лекарственными препаратами, необходимо для максимального фармакодинамического эффекта, и необратимо действующий ингибитор может помочь в достижении этой цели, так как он постоянно (стабильно) подавляет активность, которая восстанавливается, только когда синтезируется новый целевой полипептид. После введения необратимо действующего ингибитора его терапевтическую концентрацию в плазме необходимо будет поддерживать лишь до того момента, пока целевые полипептиды хотя бы слегка подвергнутся действию ингибитора, который будет необратимо подавлять деятельность мишени. Даже если после этого содержание ингибиторов в плазме будет быстро снижаться, на это можно не обращать внимания,: целевой полипептид все равно будет оставаться инактивированным. В перспективе это открывает возможность снизить минимальную концентрацию в плазме, при которой наблюдается терапевтический эффект, минимизировать введение множественных доз и избавиться он необходимости добиваться длительного времени полужизни без потери эффективности. Все это позволяет снизить токсичность препарата, обусловленную неспецифическими нецелевыми взаимодействиями, которые могут иметь место при высоких или пролонгированных концентрациях в плазме. Еще одним преимуществом необратимо действующих ингибиторов может стать возможность преодолеть резистентность к лекарственным препаратам.
В патентной заявке US 2007/0082884 описано применение способов структурной биоинформатики для идентификации цистеина (Цис) в сайте связывания нескольких киназ, которые поддаются модифицированию с помощью низкомолекулярных ингибиторов. Кроме того, в заявке описываются способы получения соединений, образующих ковалентную связь с идентифицированным Цис. В статье Pan et, al. ChemMedChem 2(1):58-61 (2007) рассмотрены идентификация остова, способного ингибировать тирозинкиназу Брутона (ВТК), путем просеивания; приготовление серий соединений на основе остова, а также идентификация ковалентных ингибиторов ВТК. В работе Wissner et al., J. Med. Chem. 48(24):7560-81 (2005) освещено приготовление серий соединений, являющихся необратимо действующими ингибиторами киназы рецептора-2 фактора роста эндотелия сосудов (VEGFR2). Такие соединения содержат квиназолиновый фибриллярный центр и высокоактивный хинон. Однако ни одна из перечисленных работ не дает ни какого-либо обобщенного/универсального способа создания необратимо действующих ингибиторов, ни способа разработки необратимо действующего ингибитора на основе его обратимого аналога. Существование таких способов позволило бы заметно сократить временные и материальные затраты на разработку необратимо действующих ингибиторов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к алгоритму и способу создания необратимо действующих ингибиторов целевых полипептидов. Необратимо действующие ингибиторы, созданные по описанным здесь алгоритму и способу, образуют ковалентную связь с аминокислотной боковой цепью в целевом полипептиде. Теперь, при использовании данного изобретения, становится возможной эффективная разработка необратимо действующего ингибитора на базе уже известного обратимого ингибитора. Такой подход позволяет сократить время и денежные средства, затрачиваемые обычно при традиционном просеивании и проработке связи активность-структура - подходах, обычно применяемых при изобретении и развитии лекарственных препаратов. Алгоритм и способ включают формирование связи между предполагаемым необратимо действующим ингибитором и целевым полипептидом.
Алгоритм и способ содержат: А) представленную структурную модель обратимого ингибитора, связанного с сайтом связывания в целевом полипептиде, где обратимый ингибитор образует не-ковалентную связь с сайтом связывания; В) идентификацию Цис-остатка в сайте связывания целевого полипептида, который является смежным с обратимым ингибитором, когда обратимый ингибитор связан с сайтом связывания; С) производство структурных моделей предполагаемых ингибиторов, которые ковалентно связаны с целевым полипептидом, где каждый предполагаемый ингибитор содержит «боеголовку», привязанную к замещаемой позиции обратимого ингибитора; «боеголовка» содержит активную химическую функциональность и - опционально - линкер, позиционирующий активную химическую функциональность в пределах длины связи Цис-остатка в сайте связывания целевого полипептида; D) определение замещаемых позиций обратимого ингибитора, что отражается на активной химической функциональности «боеголовки», находящейся в пределах длины связи Цис-остатка в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания; Е) для предполагаемого ингибитора, содержащего «боеголовку», Которая находится в пределах длины связи Цис-остатка в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания, - формирование ковалентной связи между атомом серы Цис-остатка в сайте связывания и активной химической функциональностью реакционного химического функционала «боеголовки», когда предполагаемый ингибитор связан с сайтом связывания. Длина ковалентной связи менее 2 ангстремов для связи, образовавшейся между атомом серы Цис-остатка в сайте связывания и активной химической функциональностью «боеголовки», указывает на то, что предполагаемый ингибитор является ингибитором, который ковалентно свяжется с целевым полипептидом.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
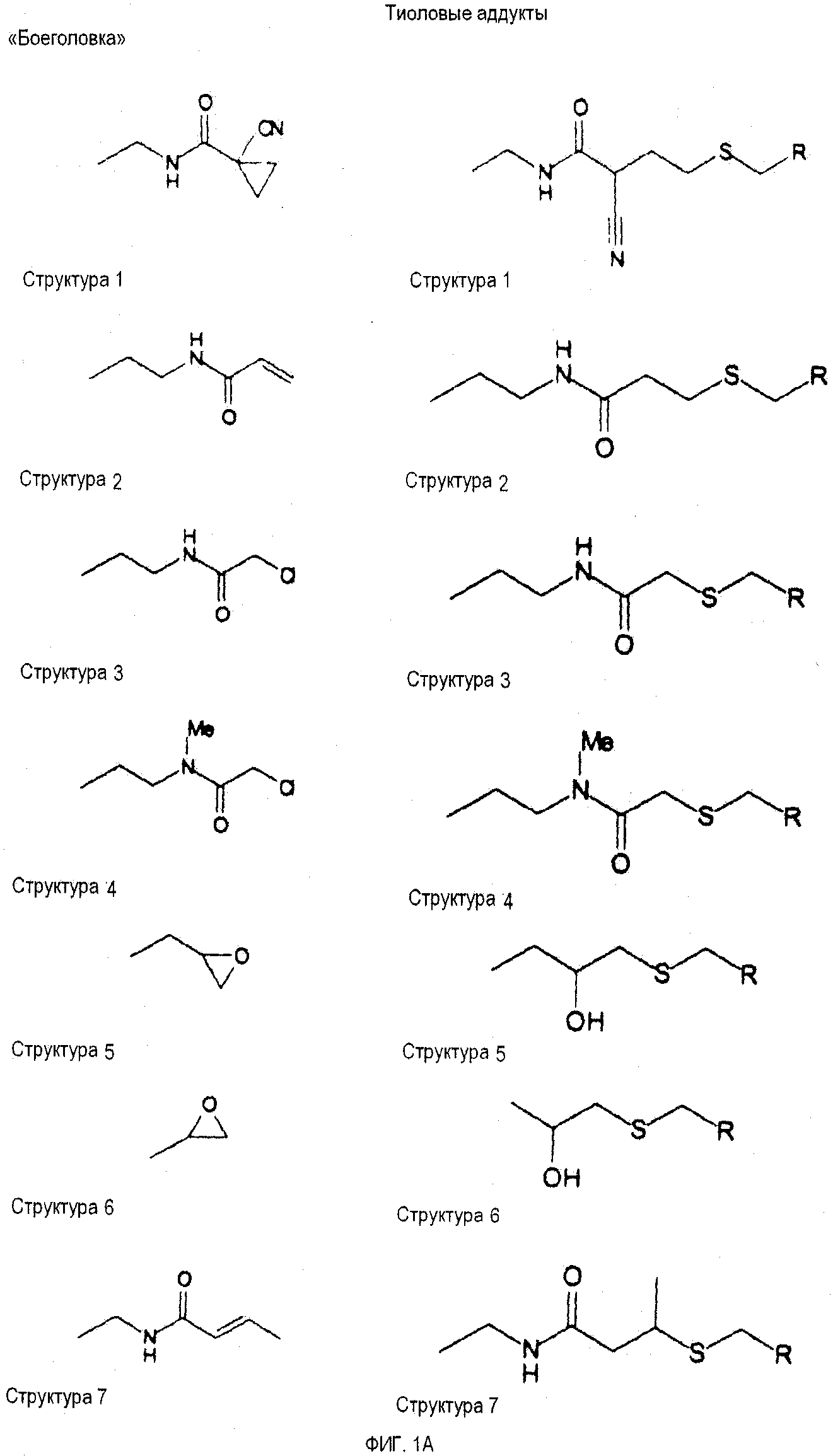
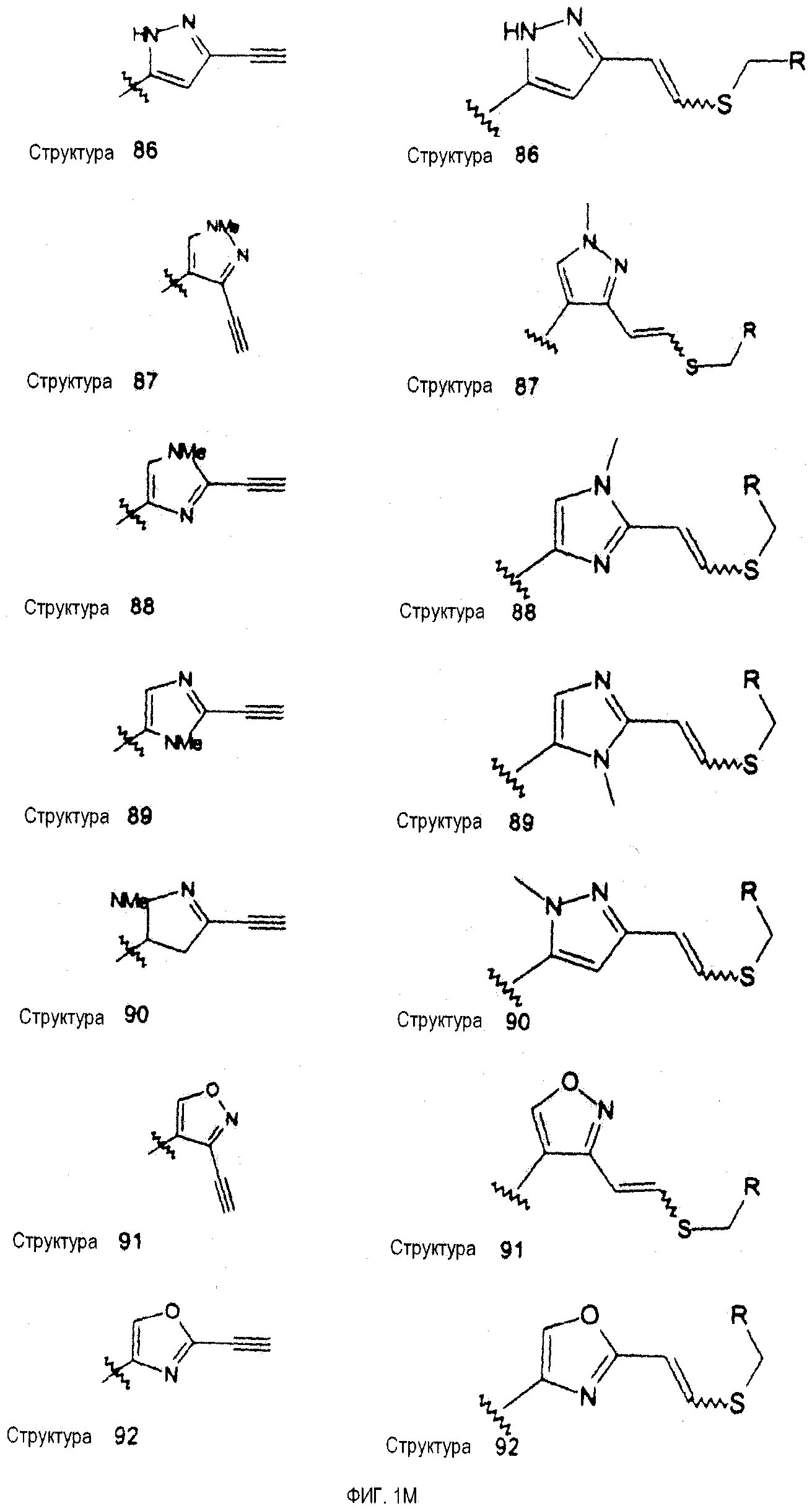
ФИГ. 1A-1Q иллюстрируют структуру 114-ти примеров «боеголовок», которые могут быть использованы в изобретении, и тиоловые аддукты, которые каждая «боеголовка» образует с Цис-остатком в целевом полипептиде. В тиоловых аддуктах атом серы Боковой цепи цистеина связан с «боеголовкой» и с β-углеродом Цис-остатка, и β-углерод Цис-остатка связан с R. R представляет собой остаток целевого полипептида.
ФИГ.2А изображает модель Соединения 1 в сайте связывания АТФ полипептида c-KIT. Также показан целевой Цис-остаток, Cys788 полипептида c-KIT.
ФИГ.2В отображает модель Соединения 1 в сайте связывания АТФ полипептида с-KIT. Согласно данному рисунку, в Соединении 1 сформирована ковалентная связь с Cys788 c-KIT.
ФИГ.3А изображает модель Соединения 4 в сайте связывания АТФ полипептида FLT3. Также показан целевой Цис-остаток, Cys828 FLT3.
ФИГ.3В изображает модель Соединения 4 в сайте связывания АТФ полипептида FLT3. Согласно данному рисунку, в Соединении 4 сформирована ковалентная связь с Cys828 FLT3.
ФИГ.4А изображает модель Соединения 5 в сайте связывания протеазы вируса гепатита С (ВГС), а именно компонент протеазы вируса NS3/4A ВГС.Также показан целевой Цис-остаток, Cysl59 протеазы ВГС.
ФИГ.4В изображает модель Соединения 5 в сайте связывания протеазы ВГС. Согласно данному рисунку, в Соединении 5 сформирована ковалентная связь с Cysl59 протеазы ВГС.
ФИГ.5 демонстрирует ингибирование в зависимости от дозы в пролиферации клеток EOL-1 по сравнению с эталонным соединением и Соединением 2.
ФИГ.6 демонстрирует результаты ингибирования PDGFR по сравнению с эталонным соединением и Соединением 2, полученные в ходе эксперимента с «вымыванием» на клетках EOL-1.
ФИГ.7 демонстрирует результаты масс-спектрального анализа триптического гидролизата PDGFR, обработанного Соединением 3. Результаты подтверждают, что Соединение 3 сформировало связь с Cys814.
ФИГ.8 демонстрирует результаты масс-спектрального анализа протеазы NS3/4A ВГС, обработанной Соединением 5. Результаты показывают, что протеаза ВГС, обработанная Соединением 5, увеличилась в массе, что обусловлено формированием аддукта между белком и Соединением 5. Формирования аддукта не происходило при использовании мутантной формы протеазы ВГС, в которой Cysl59 замещен Ser.
ФИГ.9 демонстрирует результаты масс-спектрального анализа протеазы ВГС NS3/4A, обработанной Соединением 6. Результаты показывают, что протеаза ВГС, обработанная Соединением 6, увеличилась в массе, что обусловлено формированием аддукта между белком и Соединением 6. Формирования аддукта не происходило при использовании мутантной формы протеазы ВГС, в которой Cysl59 замещен Ser.
ФИГ.10А и 10В представляют собой гистограммы, на которых показано пролонгированное ингибирование активности cKIT в результате воздействия на него необратимым ингибитором Соединения 7, по сравнению с сорафенибом, согласно результатам количественного анализа на основе фосфорилирования (10А), а также анализа клеточной сигнализации «downstream» (от первичного сигнала - через рецептор - к вторичному мессенджеру и далее к внутриклеточной мишени), при котором измеряли фосфорилирование регулируемой внеклеточными сигналами киназы (ERK) (10В).
ФИГ.11 демонстрирует результаты масс-спектрального анализа протеазы ВГС NS3/4A, обработанной Соединением 8. Результаты показывают, что протеаза ВГС, обработанная Соединением 8, увеличилась в массе, что обусловлено формированием аддукта между белком и Соединением 8.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
В данном контексте, термин "смежный" относится к аминокислотному остатку в целевом полипептиде, находящемуся рядом с обратимым ингибитором, когда обратимый ингибитор связан с целевым полипептидом. Например, аминокислотный остаток в целевом полипептиде является смежным по отношению к обратимому ингибитору, когда любой не-водородный атом аминокислотного остатка находится в пределах около 20Å, около 18 Å, около 16Å, около 14Å, около 12Å, около 10Å, около 8Å, около 6Å, около 4Å или около 2Å, любого не-водородного атома обратимого ингибитора, когда обратимый ингибитор связан с целевым полипептидом. Аминокислотный остаток в целевом полипептиде, который контактирует с обратимым ингибитором, когда обратимый ингибитор связан с целевым полипептидом, является смежным по отношению к обратимому ингибитору.
В данном контексте, "замещаемая позиция" относится к не-водородным атомам в обратимом ингибиторе, связанным с другими атомами или химическими группами (напр., водород), которые могут быть замещены и/или удалены, не нарушая связывания обратимого ингибитора с целевым полипептидом.
В данном контексте, связывание обратимого ингибитора "не нарушено", если способ связывания и время удержания обратимого ингибитора в целевом сайте связывания не претерпевает существенных изменений. Связывание обратимого ингибитора не нарушено, например, если активность ингибитора по результатам соответствующего количественного анализа (напр., IC50, Ki) изменяется на величину, меньшую, чем на три порядка, меньше чем на два порядка или меньше чем на порядок.
В данном контексте, "длина связи" относится к расстоянию, составляющему не более чем около 6Å, не более 4Å, не более 2Å.
В данном контексте, термины "ковалентная связь" и "валентная связь" относятся к химической связи между двумя атомами, образованной обобщением электронов, обычно пар электронов у связанных атомов.
В данном контексте, "не-ковалентная связь" относится к взаимодействию между атомами и/или молекулами, при котором не происходит образование ковалентной связи между ними.
В данном контексте, "необратимый ингибитор" представляет собой соединение, которое ковалентно связывается с целевым полипептидом практически постоянной ковалентной связью и ингибирует активность целевого полипептида на время, более длительное, чем функциональная жизнь белка. Необратимые ингибиторы обычно характеризуются зависимостью от времени, т.е., степень ингибирования целевого полипептида возрастает, пока его активность полностью не будет прекращена, по мере того как целевой полипептид находится в контакте с необратимо действующим ингибитором. Восстановление активности целевого полипептида после ее подавления необратимьм ингибитором зависит от начала синтеза новых белков. Активность целевого полипептида, подавленная необратимьм ингибитором, остается в значительной степени угнетенной в исследовании по "вымыванию". Профильным специалистам известны многие подходящие способы, позволяющие определить, является ли данное соединение необратимо действующим ингибитором. Например, необратимо действующее ингибирование можно идентифицировать или подтвердить при помощи кинетического анализа (напр., конкурентного, бесконкурентного, не-конкурентного) профиля ингибирования соединения с целевым полипептидом; либо масс-спектрометрии мишени лекарственного белка, модифицированного в присутствии соединения ингибитора; либо с использованием прерывистого воздействия, известного также как исследование по "вымыванию"; а также с помощью меток, например ингибитор, меченный радиоактивным изотопом, для того чтобы убедиться в наличии ковалентной модификации белка; либо воспользоваться любым другим общепринятым в данной научной области способом. В некоторых предпочтительных вариантах осуществления изобретения, целевой полипептид обладает каталитической активностью, а необратимый ингибитор формирует ковалентную связь с Цис-остатком, не являющимся каталитическим остатком.
В данном контексте, "обратимый ингибитор" - соединение, которое обратимо связывается с целевым полипептидом и угнетает активность целевого полипептида. Обратимый ингибитор может связывать свой целевой полипептид не-ковалентно или посредством механизма, включающего временную ковалентную связь. Восстановление активности целевого полипептида после ее подавления необратимым ингибитором может происходить путем отъединения обратимого ингибитора от целевого полипептида. Активность целевого полипептида можно считать восстановленной, если обратимый ингибитор "вымывается" в исследовании по вымыванию. Предпочтительные обратимые ингибиторы являются "сильными" ингибиторами активности своих целевых полипептидов. "Сильный" обратимый ингибитор подавляет активность своего целевого полипептида при значениях IC50 около 50 µМ и менее, около 1 µМ и менее, около 100 нМ и менее, или около 1 нМ и менее; и/или при Кi около 50 µМ и менее, около 1 µМ и менее, около 100 нМ, и менее, или около 1 нМ и менее.
Термины "1050" и "ингибирующая концентрация 50" - широко известные и общеупотребительные термины, обозначающие концентрацию молекул, которая подавляет на 50% активность исследуемого биологического процесса, включая, но не ограничиваясь ими, каталитическую активность, жизнеспособность клеток, активность по транслированию белков и т.п.
Термины "Кi" и "константа ингибирования" - общеупотребительные термины, обозначающие константу диссоциации комплекса полипептид (напр., белок) - ингибитор.
В данном контексте, "практически постоянная ковалентная связь" - это ковалентная связь между ингибитором и целевым полипептидом, которая сохраняется при физиологических условиях на время, более длительное, чем функциональная жизнь целевого полипептида.
В данном контексте, "временная ковалентная связь" - ковалентная связь между ингибитором и целевым полипептидом, которая сохраняется при физиологических условиях на время, меньшее, чем функциональная жизнь целевого полипептида.
В данном контексте, "боеголовкой" называется химическая группа, содержащая реакционный химический функционал или функциональную группу и - опционально-содержащая линкерную часть. Реакционноспособная химическая группа может формировать ковалентную связь с аминокислотными остатками, такими как цистеин (т.е. -SH группа в боковой цепи цистеина), или другими аминокислотными остатками, поддающимися ковалентной модификации, присутствующими в связывающем кармане целевого белка, и таким образом необратимо подавлять целевой полипептид. Следует понимать, что группа -L-Y, определение и описание которой дано ниже, дает такие группы боеголовок для ковалентного и необратимого ингибирования белка.
Термин "in silico" употребляется по отношению к компьютерным способам и процессам, например, таким как программы компьютерного моделирования, вычислительная химия, молекулярная графика, молекулярное моделирование и другие техники компьютерного моделирования.
В данном контексте, термин "программы компьютерного моделирования" относится к программному обеспечению, предназначенному для визуализации и проектирования малых молекул, включая, но не ограничиваясь ими, вычислительную химию, химическую информатику, расчеты энергии, моделирование белков и т.п. Профильным специалистам хорошо известны такие программы, некоторые из них упоминаются в данном документе.
В данном контексте, термин "выравнивание последовательностей" относится к такому расположению двух или более последовательностей белков или нуклеиновых кислот, которое позволяет сравнить их и установить их подобие (или различия). Способы и компьютерные программы для выравнивания последовательностей хорошо известны (напр., BLAST). Последовательности могут заполняться разрывами (как правило, обозначаются знаком тире) так, чтобы, где это возможно, колонки содержали идентичные или подобные знаки из последовательностей, которые подлежат выравниванию.
В данном контексте, термин "кристалл" относится к любой трехмерной упорядоченной последовательности/матрице молекул, которая преломляет рентгеновские лучи.
В данном контексте, термины "атомная система координат" и "структурная система координат" относятся к математической системе координат (представленной осями "X," "Y" и "Z"), с помощью которой описывается положение атомов в трехмерной модели/структуре model/structure или в экспериментальной структуре белка.
В данном контексте, термин "гомологичное моделирование" относится к методике производства моделей для трехмерных структур макромолекул на основе имеющихся трехмерных структур их гомологов. Гомологичные модели получают при помощи компьютерных программ, позволяющих изменять тип остатка в позиции, где последовательность исследуемой молекулы не совпадает с последовательностью молекулы с изученной / известной в науке структурой.
В данном контексте, "вычислительная химия" относится к расчетам физических и химических свойств молекул.
В данном контексте, "молекулярная графика" относится к двух- или трехмерным изображениям атомов, преимущественно на экране компьютера.
В данном контексте, "молекулярное моделирование" относится к способам и процедурам, которые могут быть воплощены на компьютере или без него, для создания одной или более моделей и, опционально, для прогнозирования связи структура-активность лигандов. Способы, применяемые при молекулярном моделировании, варьируются от молекулярной графики до вычислительной химии.
Изобретение касается алгоритмов и способов разработки необратимо действующих ингибиторов целевых полипептидов, таких как ферменты. Необратимые ингибиторы, разработанные с использованием настоящего изобретения, способны к сильному и избирательному ингибированию целевого полипептида. В целом, изобретение представляет собой рациональный алгоритм и способ разработки, в котором выбор разработки продиктован структурой целевого полипептида, структурой обратимого ингибитора целевого полипептида, а также особенностями взаимодействия обратимого ингибитора с целевым полипептидом. Необратимые ингибиторы, или предполагаемые необратимые ингибиторы, разработанные с использованием способов изобретения, включают матрицу или остов, к которому привязаны одна или более «боеголовок». Полученное соединение обладает связывающей способностью к целевому полипептиду и, однажды связавшись с ним, «боеголовка» реагирует с Цис-остатком в сайте связывания целевого полипептида и образует с ним ковалентную связь, что приводит к необратимому ингибированию целевого полипептида.
Изобретение содержит способ разработки ингибитора, который ковалентно связывает целевой полипептид. Способ содержит предоставление структурной модели обратимого ингибитора, привязанного к сайту связывания в целевом полипептиде. Обратимый ингибитор образует не-ковалентные контакты с сайтом связывания. В данном Изобретении при помощи структурной модели определен Цис-остаток в сайте связывания целевого полипептида, смежный с обратимым ингибитором, когда обратимый ингибитор связан с сайтом связывания. Также могут быть определены единичный Цис-остаток, все Цис-остатки или необходимое количество Цис-остатков, являющихся смежными с обратимым ингибитором, когда обратимый ингибитор связан с сайтом связывания.
В настоящем изобретении представлены структурные модели одного или более предполагаемых ингибиторов, разработанных для ковалентного связывания с целевым полипептидом. Предполагаемые ингибиторы содержат «боеголовку», которая связана с замещаемой позицией обратимого ингибитора. «Боеголовка» содержит реакционный химический функционал, способный или вступающий в реакцию и образующий ковалентную связь с тиоловой группой в боковой цепи Цис-остатка, и опционально -линкер, который позиционирует реакционный химический функционал в пределах длины связи одного из идентифицированных Цис-остатков в сайте связывания целевого полипептида. Определены замещаемые позиции обратимого ингибитора, результатом чего стало то, что реакционный химический функционал «боеголовки» располагается в пределах длины связи идентифицированного Цис-остатка в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания.
Определение/установление, является ли предполагаемый необратимый ингибитор,содержащий «боеголовку», которая прикреплена к идентифицированной замещаемой позиции и находится в пределах длины связи идентифицированного Цис-остатка в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания, вероятно, является ингибитором, который ковалентно связывает целевой полипептид, и предпочтительно является необратимым ингибитором целевого полипептида, производится путем формирования ковалентной связи между атомом серы Цис-остатка в сайте связывания и реакционным химическим функционалом «боеголовки», когда предполагаемый ингибитор связан с сайтом связывания. Длина ковалентной связи в пределах от приблизительно 2,1 ангстремов до около 1,5 ангстремов, или менее 2 ангстремов для связи, образованной между атомом серы Цис-остатка в сайте связывания и реакционным химическим функционалом «боеголовки», указывает на то, что предполагаемый ингибитор является ингибитором, который ковалентно связывает целевой полипептид.
Способ изобретения может применяться с использованием любых подходящих структурных моделей, таких как физические модели или, предпочтительнее, модели, полученные с помощью компьютерной графики. Способ можно применять с использованием ручного труда или автоматизированных процессов. Желательно, чтобы способ применялся in silico.
Как будет видно из более подробного описания, приведенного ниже, концептуально алгоритм и способ изобретения включают: А) предоставление мишени и обратимого ингибитора. В) идентификацию целевого цистеина. С) производство структурных моделей предполагаемых ингибиторов, содержащих «боеголовку», D) определение достижимости боеголовкой целевого цистеина и Е) формирование ковалентной связи.
А) Предоставление мишени и обратимого ингибитора
Изобретение включает предоставление структурной модели обратимого ингибитора, привязанного к сайту связывания в целевом полипептиде, в котором обратимый ингибитор образует не-ковалентные контакты с сайтом связывания. Может быть представлена и использована любая подходящая структурная модель обратимого ингибитора, привязанного к сайту связывания в целевом полипептиде. В общем, известный или уже существующий сильный обратимый ингибитор целевого полипептида используется для получения отправной точки (напр., матрицу или остов) для разработки ингибитора, который ковалентно связывает целевой полипептид с использованием настоящего изобретения. Например, если обратимый ингибитор целевого белка ранее был идентифицирован {напр., на основе научной литературы или при помощи любого общепринятого способа), известный обратимый ингибитор может использоваться для создания структурной модели целевого полипептида, образующего комплекс с ингибитором. Тем не менее, если потребуется, новый или ранее неизвестный обратимый ингибитор может использоваться для создания структурной модели целевого полипептида, образующего комплекс с ингибитором.
Алгоритм и способ настоящего изобретения может использоваться для разработки необратимых ингибиторов на основе любых подходящих обратимых ингибиторов, таких как сильные обратимые ингибиторы, слабые обратимые ингибиторы или обратимые ингибиторы умеренной силы. Например, как описано в Примере 8, алгоритм и способ изобретения может использоваться для увеличения силы обратимых ингибиторов путем разработки способности ковалентно связываться с целевым белком. В некоторых вариантах осуществления изобретения, алгоритм и способ задействуют структуру сильного обратимого ингибитора. В других вариантах осуществления изобретения, алгоритм и способ используются для улучшения силы путем разработки ковалентного связывания, и задействуют структуру ингибитора низкой или умеренной силы, такого как ингибитор с показателями IC50 или Кi на уровне ≥10 нМ, ≥100 нМ, между 1 мМ и около 10 нМ, между около 1 мМ и около 100 нМ, между около 100 мМ и 1 мМ, или между около 1 мМ и около 1 мМ.
Трехмерные структуры многих подходящих целевых полипептидов хорошо изучены и всегда доступны из открытых источников, таких как База данных белковых структур в Исследовательской коллаборатории структурной биоинформатики (RCSB PDB), доступная в режиме онлайн: www.pdb.org; см. также Н.М.Berman et al.,: Nucleic Acids Research, 28 pp.235-242 (2000) и www.rcsb.org), а также всемирная База данных белковых структур (wwPDB; Berman et al, Nature Structural Biology 10(12):980 (2003)). Неполный список подходящих целевых полипептидов, структуры которых имеются в Базе данных белковых структур, представлены в Табл. 1. Если необходимо, трехмерная структура целевого белка может быть получена при помощи любого приемлемого способа. Приемлемые способы определения структуры хорошо известны профильным специалистам, например спектроскопия ядерно-магнитного резонанса (ЯМР) жидкой фазы, спектроскопия ЯМР твердой фазы, рентгенокристаллография и т.п. (См., напр., Blow, D, Outline of Crystallography for Biologists. Oxford: Oxford University Press. ISBN 0-19-851051-9(2002)).
Структурные модели целевых полипептидов также могут быть получены при помощи общепринятых способов компьютерного моделирования, таких как гомологичное моделирование, или исследование фолдинга, основанное, например, на первичной и вторичной структурах белка. Приемлемые способы получения гомологичных моделей хорошо известны специалистам. (См., напр., John, В. and Sali, A. Nucleic Acids Res 31(14):3982-92 (2003)). Приемлемые программы для гомологичного моделирования включают, например, Modeler (компания Accelrys, Inc., Сан-Диего, США) и Prime (компания Schrodinger Inc., Нью-Йорк). Например, как описано в настоящем документе в Примере 3, гомологичная модель FLT3-киназы была получена на основе уже известной структуры киназы Авроры. Неполный список подходящих целевых полипептидов, информация о последовательностях которых общедоступна и которые могут быть использованы для получения гомологичных моделей, приведен в Табл. 2. Предпочтительные структурные модели производятся с использованием атомной системы координат для целевого полипептида, или, по крайней мере, сайта связывания целевого полипептида, в комплексе с обратимым ингибитором. Такие атомные координаты доступны в Базе данных белковых структур для многих целевых полипептидов, образующих комплекс с обратимыми ингибиторами, и могут определяться при помощи рентгенокристаллографии, спектроскопии ЯМР, с использованием гомологичного моделирования и т.п.
Таким же образом структурные модели обратимых ингибиторов - отдельно взятых или в комплексе с целевыми полипептидами, могут быть созданы на основе уже известных атомных координат или с использованием других приемлемых способов. Приемлемые способы и программы для «стыковки» (докинга) ингибиторов к целевым белкам хорошо известны специалистам. (См., напр., Perola et al., Proteins: Structure, Function и Bioinformatics 56:235-249 (2004).) В общем, если структура обратимого ингибитора, образующего комплекс с целевым полипептидом, неизвестна, то модель этого комплекса можно составить на основе возможного или предположительного типа связывания обратимого ингибитора. Профильный специалист без труда сможет назвать возможные или предположительные типы связывания для обратимых ингибиторов, например, основываясь на подобии структур обратимого ингибитора и другого ингибитора в уже известных типах связывания. Например, как описано в Примере 5, известна структура комплексов протеазы ВГС с боле чем 10-ю различными ингибиторами, кроме того, было обнаружено, что все ингибиторы имеют структурное сходство в своем типе связывания с протеазой. Основываясь на этих данных о вероятном типе связывания обратимого ингибитора V-1, авторы изобретения создали структурную модель комплекса ингибитора V-1 и протеазы ВГС, которая была успешно использована для разработки необратимо действующего ингибитора, который ковалентно связывается с Cysl 59 протеазой ВГС.
Структурная модель обратимого ингибитора, привязанного к сайту связывания в целевом полипептиде, является преимущественно компьютерной моделью. Компьютерные модели могут быть получены и визуализированы с помощью любого приемлемого программного обеспечения (ПО), например, VIDA™, ПО для визуализации, (компания OpenEye Scientific Software, Нью-Мексико, США), Insight II® или Discovery Studio®, программа для графического молекулярного моделирования (Accelrys Software Inc., Сан-Диего, Калифорния, США).
В) Идентификация целевого цистеина
Данное изобретение включает идентификацию Цис-остатка в сайте связывания целевого полипептида, смежного с обратимьм ингибитором, когда обратимый ингибитор связан с сайтом связывания. При помощи структурной модели целевого полипептида, образующего комплекс с обратимым ингибитором, идентифицируются Цис-остатки целевого полипептида, подходящие в качестве мишени для формирования ковалентной связи с «боеголовкой». Цис-остатки, подходящие в качестве мишени для формирования ковалентной связи с «боеголовкой», являются смежными по отношению к обратимому ингибитору в структурной модели. Цис-остатки, смежные с обратимым ингибитором в структурной модели, могут быть идентифицированы любым приемлемым способом для определения межмолекулярных расстояний. Профильным специалистам хорошо известны программные продукты для измерения межмолекулярных расстояний в компьютерных моделях, например, VIDA™, ПО для визуализации (OpenEye Scientific Software, Нью-Мексико, США), Discovery Studio, программа для визуализации (Accelrys, Inc., Сан-Диего, Калифорния, США) и т.п.
В одном примере определяется межмолекулярное расстояние {напр., в ангстремах) между всеми не-водородными атомами Цис-остатков в сайте связывания целевого полипептида и всеми не-водородными атомами обратимого ингибитора. Цис-остатки, смежные с обратимым ингибитором, хорошо поддаются идентификации по этим межмолекулярным расстояниям. Желательно, чтобы смежный Цис-остаток находился на расстоянии в пределах около 10 ангстремов, около 8 ангстремов или около 6 ангстремов от обратимого ингибитора.
При необходимости, Цис-остатки, смежные с обратимым ингибитором, можно идентифицировать, проведя анализ изменений на доступной поверхности Цис-остатков в целевых полипептидах. Сделать это можно, например, рассчитав площадь доступной поверхности Цис-остатков в целевых полипептидах (напр., сайт связывания ингибитора целевого полипептида), когда целевой полипептид образует комплекс с обратимым ингибитором, или когда целевой полипептид не образует комплекса с обратимым ингибитором. Цис-остатки, в которых наблюдаются изменения в площади доступной поверхности, когда обратимый ингибитор образует комплекс с целевым полипептидом, вероятно, являются смежными с обратимым ингибитором. См., напр., Lee, В. and Richared, F.M., J. Mol. Biol. 55:379-400 (1971) относительно вопроса о доступной площади. При желании можно дополнительно подтвердить этот вывод, проведя измерение межмолекулярных расстояний.
С) Производство структурных моделей предполагаемых ингибиторов, содержащих «боеголовку»
Изобретение включает производство структурных моделей предполагаемых ингибиторов, разработанных для ковалентного связывания целевого полипептида, где каждый предполагаемый ингибитор содержит «боеголовку», которая связана с замещаемой позицией обратимо действующего ингибитора. Предполагаемые ингибиторы, способные сформировать ковалентную связь со смежным Цис-остатком, разработаны путем добавления группы «боеголовок» к замещаемой позиции на обратимом ингибиторе. Например, «боеголовка» может быть привязана к ненасыщенному атому углерода, смежному с Цис-остатком в целевом полипептиде, в другом примере, в комплексе, образованном обратимым ингибитором и целевым полипептидом, Цис-остаток поглощен или частично поглощен областью обратимого ингибитора. В таком случае область обратимого ингибитора может быть удалена и заменена на подходящую «боеголовку» с целью получить ингибитор, который ковалентно связывается с Цис-остатком, полностью или частично поглощенным обратимым ингибитором. Такой подход приемлем, если область обратимого ингибитора, удаленная и замененная на «боеголовку», может быть удалена без ущерба для связующей способности обратимого ингибитора. Области обратимого ингибитора, которые могут быть удалены без ущерба для связующей способности, без труда можно идентифицировать; в их число входят, например, области, не участвующие в связывании водорода, ван-дер-ваальсовых взаимодействиях и/или гидрофобных взаимодействиях с целевым полипептидом.
«Боеголовка» содержит реакционный химический функционал, способный реагировать с боковой цепью цистеина и образовывать ковалентную связь между реакционным химическим функционалом и атомом серы боковой цепи цистеина. «Боеголовка» опционально содержит линкер, который позиционирует реакционный химический функционал в пределах длины связи боковой цепи Цис в сайте связывания целевого полипептида. Выбор «боеголовки» зависит от желаемой степени реакционной способности боковой цепи Цис. Если присутствует линкер, он служит для позиционирования реакционного химического функционала в пределах длины связи целевого Цис-остатка. Например, когда смежный Цис-остаток находится слишком далеко от обратимого ингибитора, что не дает реакционному химическому функционалу непосредственно связаться с замещаемой позицией обратимого ингибитора, реакционный химический функционал может быть привязан к замещаемой позиции обратимого ингибитора через посредство подходящего линкера, например бивалентной, от С1 до С18, насыщенной или ненасыщенной, прямой или разветвленной углеводородной цепи.
Примеры «боеголовок» включают те, что описаны здесь, например, на ФИГ.1. Некоторые приемлемые «боеголовки» имеют формулу *-X-L-Y, где * указывает на место присоединения к замещаемой позиции обратимого ингибитора.
Х представляет собой связь или бивалентную C1-С6 насыщенную или ненасыщенную, прямую или разветвленную углеводородную цепь, где опционально одна, две или три метиленовых единицы углеводородной цепи независимо замещены на: -NR-, -O-, -С(O)-, -ОС(O)-, -С(O)O-, -S-, -SO-, -SO2-, -C(=S)-, -C(=NR)-, -N=N- или -C(=N2)-;
L - ковалентная связь или бивалентная C1-8 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, где одна, две или три метиленовых единицы L опционально и независимо замещены на: циклопропилен, -NR-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2-, -SO2N(R)-, -O-, -C(O)-, -OC(O)-, -C(O)O-, -S-, -SO-, -SO2-, -C(=S)-, -C(=NR)-, -N=N- или -C(=N2)-;
Y - водород, С1-6 алифатик, опционально замещенный на оксо, галоген, NO2, или CN, или 3-10-звенное моноциклическое или бициклическое, насыщенное, частично ненасыщенное, или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и где указанное кольцо заменяется на 1-4 Re группы; и
каждая Re независимо выбрана из -Q-Z, оксо, NO2, галоген, CN, приемлемой уходящей группы, или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN, где:
Q - ковалентная связь или бивалентная C1-6 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, где одна или две метиленовые единицы Q опционально и независимо заменены на -N(R)-, -S-, -O-, -С(O)-, -ОС(O)-, -С(O)O-, -SO- или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2- или -SO2N(R)-; и
Z - водород или С1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN.
В некоторых вариантах осуществления изобретения Х представляет собой связь, -O-, -NH-, -S-, -O-СН2-С≡С-, -NH-CH2-C≡C-, -S-CH2-C≡C-, -O-CH2-CH2-O-, -O-(СН2)3-, или -O-(СН2)2-С(СН3)2-.
В определенных вариантах осуществления изобретения, L является ковалентной связью.
В определенных вариантах осуществления изобретения, L - бивалентная C1-8 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь. В определенных вариантах осуществления изобретения, L является -CH2-.
В определенных вариантах осуществления изобретения, L является ковалентной связью, -СН2-, -NH-, -CH2NH-, -NHCH2-, -NHC(O)-, -NHC(O)CH2OC(O)-, -CH2NHC(O)-, -NHSO2-, -NHSO2CH2-, -NHC(O)CH2OC(O)- или -SO2NH-.
В некоторых вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и одна или две дополнительные метиленовые единицы L опционально и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, -C(O)O-, циклопропилен, -O-, -N(R)- или -C(O)-.
В определенных вариантах осуществления изобретения, L - бивалентная С2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -С(O)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -C(O)O-, и одна или две дополнительные метиленовые единицы L опционально и независимо заменены на циклопропилен, -O-, -N(R)- или -С(O)-.
В некоторых вариантах осуществления изобретения, L - бивалентная С2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, по меньшей мере одна метиленовая единица L замещена на -С(O)-, и одна или две дополнительные метиленовые единицы L опционально и независимо заменены на циклопропилен, -O-, -N(R)- или -С(O)-.
Как упоминалось выше, в определенных вариантах осуществления изобретения L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь. Для профильных специалистов очевидно, что подобные двойные связи могут существовать внутри остова углеводородной цепи, либо могут быть вне пределов ("экзо") по отношению к остову цепи и таким образом формировать алкилиденовую группу. Например, такая L группа, имеющая алкилиденовую разветвленную цепь, включает -СН2С(=CH2)СН2-. Так, в некоторых вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну алкилиденовую двойную связь.
Среди примеров L групп можно назвать -NHC(O)С(=СН2)СН2-.
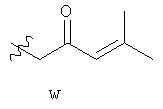
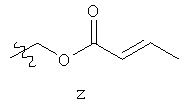
В определенных вариантах осуществления изобретения, L - бивалентная С2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -С(O)-. В определенных вариантах осуществления изобретения, L является -С(O)СН=СН(СН3)-, -С(O)СН=СНСН2NH(СН3)-, -С(O)СН=СН(СН3)-, -С(O)СН=СН-, -СН2С(O)СН=СН-, -СН2С(O)СН=СН(СН3)-, -СН2СН2С(O)СН=СН-, -СН2СН2С(O)СН=СНСН2-, -СН2СН2С(O)СН=CНСН2NH(СН3)-, или -СН2СН2С(O)СН=СН(СН3)-, или -СН(СН3)ОС(O)СН=СН-.
В определенных вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -ОС(O)-.
В некоторых вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -С(O)O-, и одна или две дополнительные метиленовые единицы L опционально и независимо заменены на циклопропилен, -O-, -N(R)- или -С(O)-. В некоторых вариантах осуществления изобретения, L является -СН2OС(O)СН=СНСН2-, -СН2-ОС(O)СН=СН- или -CH(CH=CH2)OC(O)CH=CH-.
В определенных вариантах осуществления изобретения, L является -NRC(O)CH=CH-, -NRC(O)CH=CHCH2N(CH3)-, -NRC(O)CH=CHCH2O-, -CH2NRC(O)CH=CH-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, -NRC(O)(C=N2)C(O)-, -NRC(O)CH=CHCH2N(CH3)-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, -NRC(O)CH=CHCH2O-, -NRC(O)C(=CH2)CH2-, -CH2NRC(O)-, -CH2NRC(O)CH=CH-, -CH2CH2NRC(O)- или -СН2NRC(O)циклопропилен-, где каждая R независимо является водородом или опционально замещенным C1-6алифатиком.
В определенных вариантах осуществления изобретения, L является -NHC(O)CH=CH-, -NHC(O)СН=СНСН2N(СН3)-, -NHC(O)CH=CHCH2O-, -CH2NHC(O)CH=CH-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, -NHC(O)(C=N2)C(O)-, -NHC(O)СН=СНСН2N(СН3)-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, -NHC(O)CH=CHCH2O-, -NHC(O)C(=CH2)CH2-, -CH2NHC(O)-, -CH2NHC(O)CH=CH-, -CH2CH2NHC(O)- или -СН2NHC(O)циклопропилен-.
В некоторых вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну тройную связь. В определенных вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну тройную связь, и одна или две дополнительные метиленовые единицы L опционально и независимо заменены на -NRC(O)-, -C(O)NR-, -S-, -S(O)-, -SO2-, -C(=S)-, -C(=NR)-, -O-, -N(R)- или -С(O)-. В некоторых вариантах осуществления изобретения, L имеет по меньшей мере одну тройную связь, и по меньшей мере одна метиленовая единица L замещена на -N(R)-, -N(R)C(O)-, -С(O)-, -С(O)O- или -ОС(O)- или -O-.
Среди примеров L групп можно назвать -С≡С-, -С≡ССН2N(изопропил)-, -NHC(O)C≡CCH2CH2-, -CH2-C≡C-CH2-, -С≡ССН2O-, -СН2С(O)С≡С-, -С(O)С≡С- или -CH2OC(=O)C≡C-.
В определенных вариантах осуществления изобретения, L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где одна метиленовая единица L замещена на циклопропилен, и одна или две дополнительные метиленовые единицы L независимо заменены на -С(O)-, -NRC(O)-, -C(O)NR-, -N(R)SO2- или -SO2N(R)-. Примерами L групп могут служить -NHC(O)-циклопропилен-SO2- и -NHC(O)-циклопропилен-.
Как упоминалось выше, Y представляет собой водород, C1-6 алифатик, опционально замещенный на оксо, галоген, NO2, или CN, или 3-10-звенное моноциклическое или бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и где упомянутое кольцо замещено на 1-4 Re группы; каждая Re независимо выбрана из -Q-Z, оксо, NO2, галоген, CN или C1-6 алифатика, где Q - ковалентная связь или бивалентная С 1-6 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, где одна или две метиленовые единицы Q опционально и независимо заменены на -N(R)-, -S-, -O-, -С(O)-, -ОС(O)-, -С(O)O-, -SO- или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2- или -SO2N(R)-; и Z - водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN.
В определенных вариантах осуществления изобретения, Y является водородом. В определенных вариантах осуществления изобретения, Y - C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN. В некоторых вариантах осуществления изобретения, Y - С2-6 алкенил, опционально замещенный на оксо, галоген, NO2 или CN. В других вариантах осуществления изобретения, Y - С2-6 алкинил, опционально замещенный на оксо, галоген, NO2 или CN. В некоторых вариантах осуществления изобретения, Y является С2-6 алкенилом. В других вариантах осуществления изобретения, Y является C2-4 алкинилом.
В других вариантах осуществления изобретения, Y - C1-6 алкил, замещенный на оксо, галоген, NO2 или CN. Такие Y группы включают -CH2F, -СН2Сl, -CH2CN и -CH2NO2.
В определенных вариантах осуществления изобретения, Y - насыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где Y замещен на 1-4 Re группы, где каждая Re соответствует данному выше определению и описан в данном документе.
В некоторых вариантах осуществления изобретения, Y - насыщенное 3-4-звенное гетероциклическое кольцо, имеющее 1 гетероатом, выбранный из кислорода или азота, где указанное кольцо замещено на 1-2 Re группы, где каждая Re соответствует данному выше определению. Примерами таких колец могут служить эпоксидные и окситановые кольца, где каждое кольцо замещено на 1-2 Re группы, где каждая Re соответствует данному выше определению.
В других вариантах осуществления изобретения, Y - насыщенное 5-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранных из кислорода или азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. Такие кольца включают пиперидин и пирролидин, где каждое кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y -


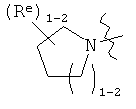
В некоторых вариантах осуществления изобретения, Y - насыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y представляет собой циклопропил, циклобутил, циклопентил или циклогексил, где каждое кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y -

В определенных вариантах осуществления изобретения, Y - частично ненасыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению.
В некоторых вариантах осуществления изобретения, Y - частично ненасыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. В некоторых вариантах осуществления изобретения, Y - циклопропенил, циклобутенил, циклопентенил или циклогексенил, где каждое кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y -

В определенных вариантах осуществления изобретения, Y - частично ненасыщенное 4-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y выбран из:




где каждая R и Re соответствуют данному выше определению.
В определенных вариантах осуществления изобретения, Y - 6-звенное ароматическое кольцо, имеющее 0-2 атома азота, где указанное кольцо замещено на 1-4 Reгруппы, где каждая Re группа соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y - фенил, пиридил или пиримидинил, где каждое кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению.
В некоторых вариантах осуществления изобретения, Y выбран из:





где каждая Re соответствует данному выше определению.
В других вариантах осуществления изобретения, Y - 5-звенное гетероарильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению. В некоторых вариантах осуществления изобретения, Y - 5-звенное частично ненасыщенное или арильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода и серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re группа соответствует данному выше определению. Примерами таких колец могут служить изоксазолил, оксазолил, тиазолил, имидазолил, пиразолил, пирролил, фуранил, тиенил, триазол, тиадиазол и оксадиазол, где каждое кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению. В определенных вариантах осуществления изобретения, Y выбран из:

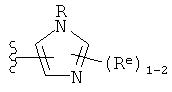





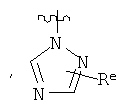







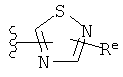
где каждые R и R6 соответствуют данному выше определению.
В определенных вариантах осуществления изобретения, Y - 8-10-звенное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению. Согласно другому аспекту, Y - 9-10-звенное бициклическое, частично ненасыщенное или арильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению. Примеры таких бициклических колец включают 2,3-дигидробензо[d]изотиазол, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению.
Как упоминалось выше, каждая Re группа независимо выбрана из -Q-Z, оксо, NO2, галоген, CN, приемлемой уходящей группы или C1-6 алифатика, опционально замещенных на оксо, галоген, NO2 или CN, где Q - ковалентная связь или бивалентная С1-6 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, где одна или две метиленовые единицы Q опционально и независимо заменены на -N(R)-, -S-, -O-, -С(O)-, -ОС(O)-, -С(O)O-, -SO- или -SO2-, -N(R)C(O)-, -C(O)N(R)-, -N(R)SO2- или -SO2N(R)-; и Z является водородом или C1-6 алифатиком, опционально замещенным на оксо, галоген, NO2 или CN.
В определенных вариантах осуществления изобретения, Re - C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN. В других вариантах осуществления изобретения, Re - оксо, NO2, галоген или CN.
В некоторых вариантах осуществления изобретения, Re является -Q-Z, где Q представляет собой ковалентную связь и Z является водородом (т.е. Re является водородом). В других вариантах осуществления изобретения, Re представляет собой -Q-Z, где Q - бивалентная C1-6 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, где одна или две метиленовые единицы Q опционально и независимо заменены на -NR-, -NRC(O)-, -C(O)NR-, -S-, -O-, -С(O)-, -SO- или -SO2-. В других вариантах осуществления изобретения, Q - бивалентная С2-6 прямая или разветвленная углеводородная цепь, имеющая по меньшей мере одну двойную связь, где одна или две метиленовые единицы Q опционально и независимо заменены на -NR-, -NRC(O)-, -C(O)NR-, -S-, -O-, -С(O)-, -SO- или -SO2-. В определенных вариантах осуществления изобретения, Z - часть группы Re является водородом. В некоторых вариантах осуществления изобретения, -Q-Z является -NHC(O)CH=CH2 или -С(O)СН=СН2.
В определенных вариантах осуществления изобретения, каждая Re независимо выбрана из оксо, NO2, CN, фтор-, хлор-, -NHC(O)CH=CH2, -С(O)СН=СН2, -СН2СН=СН2, -С≡СН, -С(O)ОСН2Сl, -C(O)OCH2F, -C(O)OCHCN, -C(O)CH2Cl, -C(O)CH2F, -C(O)CH2CN или -СН2С(O)СН3.
В определенных вариантах осуществления изобретения, Re является приемлемой уходящей группой, т.е. группой, подлежащей нуклеофильному замещению. Под "приемлемой уходящей группой" понимается химическая группа, которая легко можно заменить на желаемую входящую химическую часть, например тиоловую часть исследуемого цистеина. Примеры приемлемых уходящих групп широко известны в данной научной области, напр., см., "Advanced Organic Chemistry," Jerry March, 5th Ed., pp.351-357, John Wiley and Sons, N.Y. Такие уходящие группы включают, но не ограничиваясь ими, галоген, алкокси, сульфонилокси, опционально замещенную алкилсульфонилокси, опционально замещенную алкенилсульфонилокси, опционально замещенную арилсульфонилокси, ацил и диазониевые части. Примеры приемлемых уходящих групп включают хлор, йод, бром, фтор, ацетил, метансульфонилокси (месилокси), тосилокси, трифлулокси, нитро-фенилсульфонилокси (носилокси) и бром-фенилсульфонилокси (бросилокси).
В определенных вариантах осуществления изобретения применяются следующие воплощения и комбинации -L-Y:
(a) L - бивалентная С2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и одна или две дополнительные метиленовые единицы в L опционально и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)-, -C(O)O-, циклопропилен, -O-, -N(R) или -C(O)-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(b) L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -С(O)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-. -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -С(O)O- и одна или две дополнительные метиленовые единицы в L опционально и независимо заменены на циклопропилен, -O-, -N(R)- или -С(O)-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(c) L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица в L замещена на -С(O)-, и одна или две дополнительные метиленовые единицы в L опционально и независимо заменены на циклопропилен, -O-, -N(R)- или -С(O)-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(d) L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -С(O)-; и Y представляет собой водород или С1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(e) L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну двойную связь, и по меньшей мере одна метиленовая единица L замещена на -ОС(O)-; а Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(f) L является -NRC(O)CH=CH-, -NRC(O)CH=CHCH2N(CH3)-, -NRC(O)CH=CHCH2O-, -CH2NRC(O)CH=CH-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, -NRC(O)(C=N2)-, -NRC(O)(C=N2)C(O)-, -NRC(O)CH=CHCH2N(CH3)-, -NRSO2CH=CH-, -NRSO2CH=CHCH2-, -NRC(O)CH=CHCH2O-, -NRC(O)C(=CH2)CH2-, -СН2NRC(O)-, -CH2NRC(O)CH=CH-, -CH2CH2NRC(O)- или -СH2NRC(O)циклопропилен-; где R является H или опционально замещенным С1-6алифатиком; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
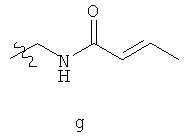
(g) L является -NHC(O)CH=CH-, -NHC(O)CH=CHCH2N(CH3)-, -NHC(O)CH=CHCH2O-, -CH2NHC(O)CH=CH-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, -NHC(O)(C=N2)-, -NHC(O)(C=N2)C(O)-, -NHC(O)CH=CHCH2N(CH3)-, -NHSO2CH=CH-, -NHSO2CH=CHCH2-, -NHC(O)CH=CHCH2O-, -NHC(O)C(=CH2)CH2-, -CH2NHC(O)-, -CH2NHC(O)CH=CH-, -CH2CH2NHC(O)- или -СН2NHC(O)циклопропилен-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(h) L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну алкилидениловую двойную связь, и по меньшей мере одна метиленовая единица в L замещена на -С(O)-, -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -С(O)O-, и одна или две дополнительные метиленовые единицы в L опционально и независимо заменены на циклопропилен, -O-, -N(R)- или -С(O)-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(i) L - бивалентная С2-8 прямая или разветвленная углеводородная цепь, где L имеет по меньшей мере одну тройную связь, и одна или две дополнительные метиленовые единицы в L опционально и независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -С(O)O-, и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(j) L is -C≡C-, -С≡ССН2N(изопропил)-, -NHC(O)C≡CCH2CH2-, -CH2-С≡C-CH2-, -С≡ССН2O-, -СН2С(O)С≡С-, -С(O)С≡С- или -СН2OС(=O)С≡С-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(k) L - бивалентная C2-8 прямая или разветвленная углеводородная цепь, где одна метиленовая единица в L замещена на циклопропилен, и одна или две дополнительные метиленовые единицы в L независимо заменены на -NRC(O)-, -C(O)NR-, -N(R)SO2-, -SO2N(R)-, -S-, -S(O)-, -SO2-, -OC(O)- или -С(O)O-; и Y представляет собой водород или C1-6 алифатик, опционально замещенный на оксо, галоген, NO2 или CN; или
(1) L является ковалентной связью, а Y выбран из таких элементов:
(i) C1-6 алкил, замещенный на оксо, галоген, NO2 или CN;
(ii) C2-6 алкенил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iii) С2-6 алкинил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iv) насыщенное 3-4-звенное гетероциклическое кольцо, имеющее 1 гетероатом, выбранный из кислорода или азота, где указанное кольцо замещено на 1-2 Re группы, где каждая Re соответствует данному выше определению; или
(v) насыщенное 5-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома выбранных из кислорода или азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(vi)
(vii) насыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(viii) частично ненасыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1 -4 Re группы, где каждая Re соответствует данному выше определению; или
(iх) частично ненасыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(х)
(хi) частично ненасыщенное 4-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
где каждая R и Re соответствуют данному выше определению; или
(хiii) 6-звенное ароматическое кольцо, имеющее 0-2 атома азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re группа соответствует данному выше определению; или
(xiv)
где каждая Re соответствует данному выше определению; или
(xv) 5-звенное гетероарильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению; или
где каждая R и Re соответствуют данному выше определению; или
(xvii) 8-10-звенное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению;
(m) L является -С(O)-, и Y выбран из следующих элементов:
(i) С1-6 алкил, замещенный на оксо, галоген, NO2 или CN; или
(ii) С2-6 алкенил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iii) C2-6 алкинил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iv) насыщенное 3-4-звенное гетероциклическое кольцо, имеющее 1 гетероатом, выбранный из кислорода или азота, где указанное кольцо замещено на 1-2 Reгруппы, где каждая Re соответствует данному выше определению; или
(v) насыщенное 5-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранных из кислорода или азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(vi)
(vii) насыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(viii) частично ненасыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(ix) частично ненасыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению;
(х)
(xi) частично ненасыщенное 4-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(xii)
где каждая R и Re соответствуют данному выше определению; или
(xiii) 6-звенное ароматическое кольцо, имеющее 0-2 атома азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re группа соответствует данному выше определению; или
(xiv)
где каждая Re соответствует данному выше определению; или
(xv) 5-звенное гетероарильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению; или
где каждая R и Re соответствуют данному выше определению; или
(xvii) 8-10-звенное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению;
(n) L является -N(R)C(O)-, a Y выбран из таких элементов:
(i) С1-6 алкил, замещенный на оксо, галоген, NO2 или CN; или
(ii) С2-6 алкенил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iii) С2-6 алкинил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iv) насыщенное 3-4-звенное гетероциклическое кольцо, имеющее 1 гетероатом, выбранный из кислорода или азота, где указанное кольцо замещено на 1-2 Re группы, где каждая Re соответствует данному выше определению; или
(v) насыщенное 5-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранных из кислорода или азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(vi)
(vii) насыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(viii) частично ненасыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(ix) частично ненасыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению;
(х)
(xi) частично ненасыщенное 4-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1 -4 Re группы, где каждая Re соответствует данному выше определению; или
(xii)
где каждая R и Re соответствуют данному выше определению; или
(xiii) 6-звенное ароматическое кольцо, имеющее 0-2 атома азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re группа соответствует данному выше определению; или
(xiv)
где каждая Re соответствует данному выше определению; или
(xv) 5-звенное гетероарильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению; или
где каждая R и Re соответствуют данному выше определению; или
(xvii) 8-10-звенное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению;
(о) L - бивалентная C1-8 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь; a Y выбран из таких элементов:
(i) C1-6 алкил, замещенный на оксо, галоген, NO2 или CN;
(ii) С2-6 алкенил, опционально замещенный на оксо, галоген, NO2 или CN;
или:
(iii) С2-6 алкинил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iv) насыщенное 3-4-звенное гетероциклическое кольцо, имеющее 1 гетероатом, выбранный из кислорода или азота, где указанное кольцо замещено на 1-2 Re группы, где каждая Re соответствует данному выше определению; или
(v) насыщенное 5-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранных из кислорода или азота, где указанное кольцо замещено на 1-4 Reгруппы, где каждая Re соответствует данному выше определению; или
(vi)
(vii) насыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(viii) частично ненасыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(ix) частично ненасыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению;
(х)
(хi) частично ненасыщенное 4-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(xii)
где каждая R и Re соответствуют данному выше определению; или
(xiii) 6-звенное ароматическое кольцо, имеющее 0-2 атома азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re группа соответствует данному выше определению; или
(xiv)
где каждая Re соответствует данному выше определению; или
(xv) 5-звенное гетероарильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению; или
где каждая R и Re соответствуют данному выше определению; или
(xvii) 8-10-звенное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Re соответствует данному выше определению;
(р) L представляет собой ковалентную связь, -СН2-, -NH-, -С(O)-, -CH2NH-, -NHCH2-, -NHC(O)-, -NHC(O)CH2OC(O)-, -CH2NHC(O)-, -NHSO2-, -NHSO2CH2-, -NHC(O)CH2OC(O)- или -SO2NH-; a Y выбран из таких элементов:
(i) C1-6 алкил, замещенный на оксо, галоген, NO2 или CN; или
(ii) С2-6 алкенил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iii) C2-6 алкинил, опционально замещенный на оксо, галоген, NO2 или CN;
или
(iv) насыщенное 3-4-звенное гетероциклическое кольцо, имеющее 1 гетероатом, выбранный из кислорода или азота, где указанное кольцо замещено на 1-2 Reгруппы, где каждая Re соответствует данному выше определению; или
(v) насыщенное 5-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, выбранных из кислорода или азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(vi)
(vii) насыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(viii) частично ненасыщенное 3-6-звенное моноциклическое кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(ix) частично ненасыщенное 3-6-звенное карбоциклическое кольцо, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению;
(x)
(хi) частично ненасыщенное 4-6-звенное гетероциклическое кольцо, имеющее 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где каждая Re соответствует данному выше определению; или
(xii)
где каждая R и Re соответствуют данному выше определению; или
(xiii) 6-звенное ароматическое кольцо, имеющее 0-2 атома азота, где указанное кольцо замещено на 1-4 Re группы, где каждая Re группа соответствует данному выше определению; или
(xiv)
где каждая Re соответствует данному выше определению; или
(xv) 5-звенное гетероарильное кольцо, имеющее 1-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-3 Re группы, где каждая Re группа соответствует данному выше определению; или
где каждая R и Re соответствуют данному выше определению; или
(xvii) 8-10-звенное бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, где указанное кольцо замещено на 1-4 Re группы, где Reсоответствует данному выше определению.
В определенных вариантах осуществления изобретения, Y-группа Формулы I выбрана из элементов, указанных в Табл. 3, где каждая волнистая линия обозначает точку присоединения к остальной части молекулы. Каждая Re группа, изображенная в Табл. 2, независимо выбрана из галогенов.










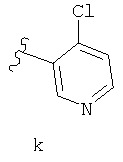




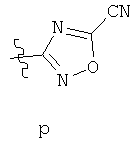










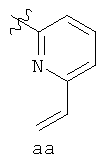

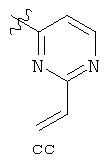
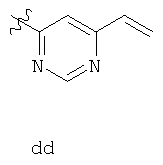


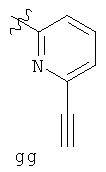












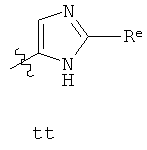








































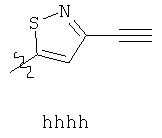



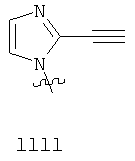


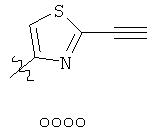













В Табл. 4 каждая волнистая линия обозначает точку присоединения к остальной части молекулы. В определенных вариантах осуществления изобретения, R1 представляет собой -С≡СН, -С≡ССН2NH(изопропил), -NHC(O)C≡CCH2CH3, -СН2-С≡С-СН3, -C≡CCH2OH, -СН2С(O)С≡СН, -С(O)С≡СН или -СН2OС(=O)С≡СН. В некоторых вариантах осуществления изобретения, R1 выбрана из -NHC(O)CH=CH2, -NHC(O)СН=СНСН2N(СН3)2 или -CH2NHC(O)CH=CH2.
В определенных вариантах осуществления изобретения, R1 выбрана из соединений, перечисленных части молекулы.


























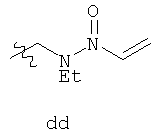
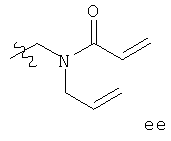





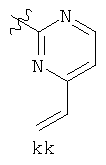













































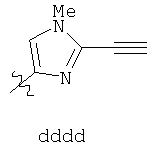



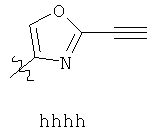






















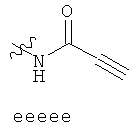






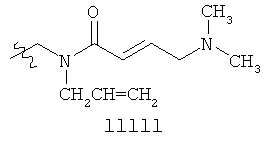
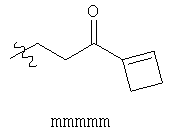







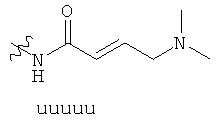












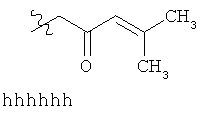







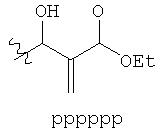





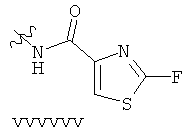


где каждая Re независимо является приемлемой уходящей группой, NO2, CN или оксо.
Структурные модели предполагаемых ингибиторов, содержащих «боеголовку», могут быть получены любым подходящим способом. Например, как описано в настоящем документе, трехмерное изображение «боеголовки», насаженной на матрицу обратимого ингибитора, можно получить, использовав приемлемую программу компьютерного моделирования. К приемлемым программам компьютерного моделирования относятся: Discovery Studio® и Pipeline Pilot™ (ПО для молекулярного моделирования, Accelrys Inc., Сан-Диего, Калифорния), Combibuild, Combilibmaker 3D, (ПО для создания библиотек соединений, Tripos L.P., Сент-Луис, Миссури), SMOG (программа для компьютерного дизайна (разработки) малых молекул; DeWitte and Shakhnovich, J. Am. Chem. Soc. 118:11733-11744 (1996); DeWitte et al., J. Am. Chem. Soc. 119:4608-4617 (1997); Shimada et al., Protein Sci. 9:765-775 (2000); Maestro™, CombiGlide™, Glide™ и Jaguar™ (программные пакеты для моделирования, Schrödinger, LLC. 120, 45-я Западная улица, Нью-Йорк, штат Нью-Йорк, 10036-4041)). «Боеголовку» можно присоединять к каждой замещаемой позиции, являющейся смежной по отношению к Цис-остатку в целевом полипептиде, или к выбранной замещаемой позиции, либо к единичной замещаемой позиции, если это необходимо. Присоединение «боеголовки» к соединению может осуществляться с использованием любого приемлемого способа или программы, например, FROG (генератор 3D-конформаций молекул; Bohme et al., Nucleic Acids Res. 35(электронная версия): W568-W572 (2007).), Discovery Studio® или Pipline Pilot™ (Accelrys, Inc., Сан-Диего), Combilibmaker 3D (Tripos, Сент-Луис), SMOG (DeWitte and Shakhnovich, J. Am. Chem. Soc. 118:11733-11744 (1996); DeWitte et al., J. Am. Chem. Soc. 119:4608-4617 (1997); Shimada er al., Protein Sci. 9:765-775 (2000)) и т.п.
«Боеголовки» могут присоединяться вручную, как, например, в Discovery Studio®, или в автоматическом режиме, как, например, в Pipline Pilot™ (Accelrys, Inc., Сан-Диего).
В некоторых предпочтительных вариантах осуществления изобретения, производятся структурные модели множества предполагаемых ингибиторов. Структурные модели включают соединения, в которых «боеголовка» присоединена к разным замещаемым позициям, и присоединение к каждой возможной замещаемой позиции представлено по меньшей мере одним соединением.
D) Определение достижимости «боеголовкой» целевого цистеина
Изобретение содержит определение замещаемых позиций обратимого ингибитора, что влияет на реакционный химический функционал «боеголовки», находящейся в пределах длины связи Цис-остатка в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания. Структурные модели предполагаемых ингибиторов подвергаются анализу с целью определить, какие именно замещаемые позиции в обратимом ингибиторе позволят достичь реакционного химического функционала «боеголовки», находящегося в пределах длины связи Цис-остатка в сайте связывания целевого полипептида. Комбинации Цис-остаток - замещаемая позиция, способствующие размещению реакционного химического функционала в пределах длины связи Цис-остатка в структурной модели, идентифицируются при помощи любого приемлемого способа определения межмолекулярных расстояний, с или без ограничений. Например, комбинации Цис-остаток - замещаемая позиция, способствующие размещению реакционного химического функционала в пределах длины связи Цис-остатка в структурной модели, могут быть идентифицированы с использованием приемлемого компьютерного способа, в котором 1) целевой полипептид удерживается в зафиксированном положении, кроме случаев, когда боковой цепи цистеина позволяют изгибаться, и предполагаемый ингибитор удерживается в зафиксированном положении, кроме случаев, когда «боеголовке» позволяют изгибаться, 2) целевому полипептиду позволяют изгибаться, и предполагаемому ингибитору позволяют изгибаться, 3) целевому полипептиду позволяют изгибаться, а предполагаемый ингибитор удерживается в зафиксированном положении, кроме случаев, когда «боеголовке» позволяют изгибаться или 4) целевой полипептид удерживается в зафиксированном положении, кроме случаев, когда боковой цепи цистеина позволяют изгибаться, и предполагаемому ингибитору позволяют изгибаться. Предпочтительно, чтобы целевой полипептид удерживался в зафиксированном положении, кроме случаев, когда боковой цепи цистеина позволяют изгибаться, и предполагаемый ингибитор удерживался в зафиксированном положении, кроме случаев, когда «боеголовке» позволяют изгибаться.
Многие компьютерные способы, приемлемые для идентификации комбинаций Цис-остаток - замещаемая позиция, способствующие размещению реакционного химического функционала в пределах длины связи Цис-остатка, хорошо известны профильным специалистам. Это могут быть программы для расчета межмолекулярных расстояний, молекулярной динамики, минимизации энергии, системного конформационного поиска и мануального моделирования. Для этих целей подойдут, например, программы Discovery Studio® и Charmm (Accelrys, Inc. Сан-Диего), Amber (Amber Software Administrator, USSF, 600 16-я улица, комната 552, Сан-Франциско, Калифорния 94158 и сайт ambermd.org/) и т.п. Программы для оценки энергии деформации соединений и электростатических взаимодействий также известны и доступны и включают, например, Gaussian 92, редакция С (М.J.Frisch, Gaussian, Inc., Питтсбург, Пенсильвания); AMBER, версия 4.0 (Р.A.Kollman, Калифорнийский университет в Сан-Франциско, Калифорния); QUANTA/CHARMM (Accelrys, Inc., Берлингтон, Массачусетс). Такие программы можно применять, например, на компьютерных рабочих станциях. Кроме них, можно использовать и другое аппаратное обеспечение и пакеты программ, известные профильным специалистам. «Стыковка» предполагаемых ингибиторов может осуществляться с помощью специальных программ, таких как Flexx (Tripos, Сент-Луис, Миссури), Glide (Schrodinger, Нью-Йорк), ICM-Pro (Molsoft, Калифорния) и т.п., с последующими программами по минимизацией энергии и молекулярной динамике со стандартными для молекулярной механики полями сил, например, OPLS-AA, CHARMM или AMBER.
Е) Формирование ковалентной связи
Изобретение включает формирование ковалентной связи между атомом серы Цис-остатка в сайте связывания и реакционным химическим функционалом «боеголовки». Идентификация комбинаций Цис-остаток - замещаемая позиция, способствующие размещению реакционного химического функционала в пределах длины связи Цис-остатка, позволяет выявить предполагаемые ингибиторы, которые будут с высокой вероятностью ковалентно модифицировать Цис-остаток. Так, сферическое сближение реакционного химического функционала и боковой цепи цистеина в модели само по себе не является достаточным индикатором того, что между реакционным химическим функционалом и боковой цепью цистеина будет сформирована ковалентная связь. Соответственно, в алгоритме и способе изобретения сформирована связь между реакционным химическим функционалом и боковой цепью цистеина, и длина этой связи подвергнута анализу. Длина ковалентной связи приблизительно от 2,1 ангстремов до 1,5 ангстремов или, предпочтительнее, менее 2 ангстремов, характеризующая связь между атомом серы Цис-остатка в сайте связывания и реакционного химического функционала «боеголовки», указывает, что предполагаемый ингибитор является ингибитором, который ковалентно свяжется с целевым полипептидом. Предпочтительно, чтобы длина связи, сформированной между реакционным химическим функционалом и боковой цепью цистеина, составляла около 2 ангстремов, около 1,9 ангстремов, около 1,8 ангстремов, около 1,7 ангстремов, около 1,6 ангстремов или около 1,5 ангстремов. Приемлемые способы и программы для формирования связи и анализа ее длины широко известны, например, можно упомянуть Discovery Studio® и Charmm (Accelrys, Inc. Сан-Диего), Amber (Amber Software Administrator, USSF, 600 16-я улица, комната 552, Сан-Франциско, Калифорния 94158 и сайт http://ambermd.org/), Guassian (340 ул. Квиннипиак, здание 40, Уоллингфорд, Коннектикут 06292 США, веб-сайт www.gaussian.com/), Qsite (Schrodinger Inc., Нью-Йорк) и программы ковалентной «стыковки» (BioSolvIT Гмбх, Германия www.biosolveit.de). Maestro™, MacroModel™ и Jaguar™ (Modeling softwar packages, Schrödinger, LLC. 120 Западная 45-я улица, Нью-Йорк, штат Нью-Йорк 10036-4041).
Если необходимо, соединения, разработанные по способу изобретения, в дальнейшем могут быть подвергнуты анализу и/или структурно очищены. Например, изобретение может включать следующий этап, позволяющий определить, заблокирован ли сайт связывания целевого полипептида (т.е. когда лиганд, субстрат или кофактор не может связываться с сайтом связывания), когда сформирована ковалентная связь между атомом серы Цис-остатка в сайте связывания и реакционным химическим функционалом «боеголовки». Воплощение этого этапа возможно с использованием структурной модели целевой полипептид - необратимый ингибитор, который ковалентно связывается с комплексом Цис-остатка. Возможно изменение в характере связывания ингибитора с целевым полипептидом в результате формирования ковалентной связи между реакционным химическим функционалом и Цис-остатком. Тем не менее, в большинстве случаев соединение все-таки блокирует сайт связывания целевого полипептида и не позволяет лигандам, субстратам или кофакторам связываться с сайтом связывания.
Изменения в режиме связывания ингибитора под воздействием формирования ковалентной связи; остается ли при этом сайт связывания заблокированным, можно определить посредством анализа структурной модели ингибитора, образовавшего комплекс с целевым полипептидом после формирования ковалентной связи, используя специальные способы и программы, упомянутые в данном документе.
В другом примере соединения, разработанные при помощи данного изобретения, подвергаются анализу на предмет благоприятных или предпочтительных характеристик, таких как конформация образованной ковалентной связи. Как описано в Примерах 1 и 6, ковалентные связи, сформированные между Цис и акриламидной «боеголовкой», могут иметь Цис-конформацию или транс-конформацию амида, причем транс-конформация является предпочтительной. Еще в одном примере, предпочтительные соединения выбраны из соединений, имеющих подобную структуру, на основе энергии продукта, полученного после реакции «боеголовки» с Цис-остатком, при этом предпочтительными являются продукты с более низкой энергией. Энергия продуктов измеряется любым приемлемым способом квантовой или молекулярной механики.
Изобретение может использоваться для разработки ингибиторов, которые ковалентно связываются с любым желаемым целевым полипептидом, образуя ковалентную связь с Цис-остатком в сайте связывания целевого полипептида. Предпочтительно, чтобы Цис-остаток, образующий ковалентную связь с ингибитором и разработанный в соответствии с настоящим изобретением, не был консервативным в семействе белков, содержащем целевой полипептид. В силу того, что Цис-остаток не был консервативным, можно конвертировать разнородные обратимые ингибиторы, которые угнетают несколько представителей семейства белков, в более селективно действующие необратимые ингибиторы, которые угнетают меньшее количество представителей этого семейства или даже только один из них.
В некоторых вариантах осуществления изобретения, целевой полипептид обладает Каталитической активностью. Например, целевой полипептид может быть киназой, протеазой, например, вирусной протеазой, фосфатазой или другим ферментом. Если целевой полипептид обладает каталитической активностью, предпочтительно, чтобы Цис-остаток, образующий ковалентную связь с ингибитором, разработанным согласно настоящему изобретению, не был каталитическим остатком. В определенных предпочтительных вариантах осуществления изобретения, необратимый ингибитор, разработанный с использованием данного изобретения, не является суицидным или механизм-обусловленным ингибитором - ингибиторами, являющимися результатом процесса, когда фермент преобразует субстрат в ковалентный инактиватор во время каталитического процесса.
Предпочтительно, чтобы обратимый ингибитор связывался с таким сайтом на целевом полипептиде, который является сайтом связывания лиганда, кофактора или субстрата. Если целевой полипептид является киназой, предпочтительно, чтобы обратимый ингибитор связывался или взаимодействовал с АТР-сайтом связывания киназы. Например, обратимый ингибитор может взаимодействовать с шарнирной областью АТР-сайта связывания.
Алгоритм и способ, описываемые здесь, могут быть воплощены с использованием полной структуры сайта связывания целевого полипептида и структуры обратимого ингибитора. Опционально, при воплощении алгоритма рассматриваются только цистеин сайта связывания целевого полипептида и структура обратимого ингибитора. В этом случае, трехмерная ориентация Цис-остатка и обратимого ингибитора остаются такими же, как в остальной структуре сайта связывания целевого полипептида. Если необратимый ингибитор или предполагаемый необратимый ингибитор разрабатываются только на основе цистеина сайта связывания, в дальнейшем, если необходимо, может быть рассмотрена полная модель сайта связывания, что поможет получить дополнительную информацию о структуре и конструкциях, которая помогает идентифицировать стерические столкновения, которые сокращают количество замещаемых позиций, что приводит к размещению «боеголовки» в пределах длины связи цистеина в сайте связывания. В описанных в данном документе примерах алгоритм применяется в отношении структур обратимого ингибитора и цистеина сайта связывания целевого полипептида. Такой подход позволил получить необратимые ингибиторы нескольких целевых полипептидов. Количество примеров замещаемых позиций на обратимых ингибиторах, идентифицированных и описанных в данном документе, достаточно невелико, поэтому в дополнительных конструкциях, которые могут быть продиктованы полной моделью сайта связывания, нет необходимости, однако при желании они могут быть использованы.
Для удобства этапы алгоритма и способа описаны здесь в порядке, способствующем более понятному и краткому описанию изобретения. И хотя предпочтительнее, чтобы этапы по применению способа предпринимались последовательно, в описанном здесь порядке, при желании этот порядок может быть изменен на любой другой. Например, возможно такое воплощение способа: формирование связи между «боеголовкой» и Цис-остатком для создания аддукта, и затем привязывание «боеголовки» к замещаемой позиции на обратимом ингибиторе, опционально посредством линкера.
Енон-содержащие «боеголовки», необратимые ингибиторы и конъюгаты
Изобретение также относится к необратимым ингибиторам с «боеголовкой», которая содержит сопряженный енон, α, β ненасыщенный карбонил. Изобретение также относится к полипептидным конъюгатам, образованным в результате реакции сопряженного енона «боеголовки» с -SH цистеинового остатка полипептида. Еноны представляют собой класс реакционных функционалов, содержащих структуру -С(O)-СН=СН-. Такая структура может быть частью линейной, разветвленной или циклической химической части. Преимущество енонов состоит в том, что они, как правило, отличаются низкой реакционной способностью и не вступают в реакцию с -SH цистеина в растворе. Однако, если енон расположен в пределах длины связи Цис в полипептиде, он может избирательно реагировать с -SH цистеинового остатка. Таким образом, конъюгированные еноны можно использовать для получения «боеголовок» и необратимых ингибиторов высокоизбирательного действия.
В одном аспекте «боеголовка», содержащая конъюгированный енон, имеет такую формулу

где R1, R2 и R3 независимо являются водородом, C1-С6 алкилом, или С1-С6 алкилом, замещенным на -NRxRy; Rx и Ry независимо являются водородом или С1-С6 алкилом.
Примеры «боеголовок», содержащих конъюгированный енон, включают соединения 1-а - I-h.







Изобретение относится к необратимым ингибиторам, содержащим «боеголовку» конъюгированного енона, которая образует ковалентную связь с цистеиновым остатком целевого полипептида, в частности к необратимым ингибиторам, разработанным по алгоритму настоящего изобретения. В некоторых вариантах осуществления изобретения, «боеголовка» конъюгированного енона описывается Формулой I. В отдельных вариантах осуществления изобретения, «боеголовка» конъюгированного енона выбрана из I-a, I-b, I-с, I-d, 1-е, I-f и I-g.
Изобретение также относится к способу необратимого угнетения целевого полипептида, достигаемого путем контактирования с полипептидом, содержащим сайт связывания, который имеет цистеиновый остаток с необратимым ингибитором, который содержит «боеголовку» конъюгированного енона, образующую ковалентную связь с цистеиновым остатком целевого полипептида, в частности необратимого ингибитора, разработанного при помощи алгоритма настоящего изобретения.
Изобретение также относится к полипептидным конъюгатам, образованным в результате реакции конъюгированной енон-содержащей «боеголовки» с -SH группой Цис-остатка. Такие конъюгаты имеют множество применений. Например, количество конъюгированного целевого полипептида по сравнению с неконъюгированным целевым полипептидом в биологическом образце, полученном у пациента, который получал лечение необратимым ингибитором, содержащим «боеголовку» конъюгированного енона, может использоваться для адаптации дозировки (напр., изменение вводимого количества препарата и/или временных интервалов между приемами). В одном из аспектов конъюгат имеет формулу

Где Х - химическая часть, которая связывается с сайтом связывания целевого полипептида, где сайт связывания содержит цистеиновый остаток;
М - модификатор, образованный ковалентным связыванием группы конъюгированной енон-содержащей «боеголовки» с атомом серы указанного цистеинового остатка;
S-CH2 - боковая цепь серы-метилена указанного цистеинового остатка;
R - остаток целевого полипептида.
В некоторых вариантах осуществления изобретения, конъюгированная енон-содержащая «боеголовка» описывается формулой I, а конъюгат имеет формулу II

где Х - химическая часть, которая связывается с сайтом связывания целевого полипептида, где сайт связывания содержит цистеиновый остаток;
S-СН2 - боковая цепь указанного цистеинового остатка;
R - остаток целевого полипептида;
R1, R2 и R3 являются независимо водородом, C1-С6 алкилом или C1-С6 алкилом, замещенным на -NRxRy;
a Rx и Ry являются независимо водородом или C1-С6 алкилом.
В отдельных вариантах осуществления изобретения, конъюгат имеет формулу, выбранную из II-а, II-b, II-с, II-d, II-e, II-f, II-g и II-h, где X и R описываются Формулой II.








ПРИМЕРЫ
Пример 1. Необратимо действующий иматиниб
Иматиниб - сильный обратимый ингибитор киназ cKIT, PDGFR, ABL и CSF1R. При помощи алгоритма разработки, описанного в этом документе, данный обратимый ингибитор был быстро и эффективно преобразован в необратимый ингибитор киназ cKit, PDGFR и CSF1R. Кроме того, будет показано, что описываемый способ позволяет определить, в каких случаях невозможно легко конвертировать обратимый ингибитор мишени в необратимый ингибитор этой мишени, как было в следующем примере, касающемся иматиниба и мишени ABL.

A. cKIT
Способ разработки
Координаты рентгеновского комплекса cKIT, связанного с иматинибом (код PDB: 1Т46) были получены в базе данных белковых структур (www.rcsb.org). Затем были извлечены координаты иматиниба, и все белковые Цис-остатки иматиниба в пределах 20 ангстремов, после их связывания с cKIT, идентифицировали при помощи Discovery Studio (v2.0,1.7347; Accelrys Inc., Калифорния). Всего было идентифицировано семь остатков: Cys660, Cys673, Cys674, Cys788, Cys809, Cys884 и Cys906. После этого 15 замещаемых позиций исследовали в трех измерениях на матрице иматиниба (Формула I-1), чтобы выяснить, какая из них может быть замещена на «боеголовку» так, чтобы «боеголовка» образовала ковалентную связь с одним Цис-остатком (Cys660, Cys673, Cys674, Cys788,Cys809, Cys884 или Cys906) в сайте связывания cKIT.
Способ разработки 1.1
Согласно данному способу, «боеголовки» вручную насадили на матрицу иматиниба, а затем с помощью молекулярной динамики оценивали их способность образовывать связи с Цис в сайте связывания cKit. Акриламидные «боеголовки» были надстроены в трехмерном пространстве на матрицу иматиниба в программе Discovery Studio. Матрица иматиниба изображена на Формуле I-1. Затем проверяли структуру конечного соединения с целью определить позицию «боеголовки» и выяснить, способна ли «боеголовка» достичь любого из идентифицированных Цис-остатков в сайте связывания.

Для того чтобы проверить гибкость «боеголовки» и позиций боковой цепи, на основе молекулярной динамики была создана и проанализирована симуляция «боеголовок» и позиций боковых цепей с целью выяснить, находится ли «боеголовка» в пределах 6 ангстремов от любого Цис-остатка в сайте связывания и не происходят ли стерические столкновения между «боеголовками» и остатками. Для молекулярно-динамической были использованы стандартные настройки протокола «Каскадные симуляции стандартной динамики» приложения Discovery Studio. Применялась силовое поле «Ракета многорежимного наведения, запускаемая по принципу выстрелил/забыл» (MMFF) с 4ps симуляцией в приложении Discovery Studio. Во время молекулярно-динамической симуляции координаты позиций не-«боеголовок» атомов Цис главной цепи удерживались в зафиксированном положении.
Данная симуляция позволила выявить три матричных позиции, находившихся возле Cys788, и две - возле Cys809 cKIT (Табл. 5).
Затем эти пять матричных позиций были подвергнуты финальной фильтрации, критерием которой было формирование продукта акриламидной реакции между предполагаемым ингибитором и Цис-остатком (Cys788 или Cys809), в том числе формирование связи длиной менее 2, ангстремов в стандартной молекулярно-динамической симуляции. В результате этого отсеивания осталось три матричных позиции: R1, R2 и R4, и только один Цис-остаток, Cys788. Из этих матричных позиций связи, возникающие у «боеголовок» в позициях R2 и R4, затрагивали цис-конформацию «боеголовки» амидной группы, что было не очень желательно. Связь, возникшая у «боеголовки» в позиции R1, затрагивала транс-конформацию «боеголовки» амидной группы, - этот вариант является наиболее предпочтительным.
Способ разработки 1.2
В данном способе «боеголовки» в автоматическом режиме моделирования «накладывали» на матрицу иматиниба и затем с помощью способа молекулярной стыковки оценивали их способность к формированию связи с Цис в сайте связывания IcKit.
«Боеголовки» надстраивали на матрицу иматиниба при помощи протокола Accelryes SciTegic Pipeline enumeration, в результате чего получили 13 виртуальных соединений из 15 возможных. Это обусловлено тем, что R2 и R4, так же как R3 и R5 (Формула I-1) являются эквивалентными в силу своей симметрии, а потому в дальнейших исследованиях рассматривались только R2 и R3. Данные соединения затем конвертировали в SD-формат при помощи протокола приготовления лигандов в приложении Discovery Studio. После этого трехмерные виртуальные соединения стыковали с рентгеновской структурой cKit при помощи протокола CDOCKER программы Discovery Studio. Применялся алгоритм стыковки с ограничениями, в котором ядро иматиниба в рентгеновской структуре (Формула I-1) использовалось в качестве ограничителя в процессе стыковки. Произведено десять конформаций каждого виртуального соединения, и высшая конформация каждого соединения оценивалась на предмет близости по расстоянию до Цис в сайте связывания cKit. После применения фильтра дальности (<6 ангстремов), осталось только два соединения, с «боеголовками» на R1 и R2, находящихся достаточно близко к Цис в сайте связывания. Оба этих соединения расположены близко к Cys788, что дало основания произвести анализ их способности образовывать химические связи. Белок и соединения зафиксировали в пространстве, а к боковой цепи Cys788 и «боеголовке» не применяли никаких ограничений. После завершения процесса минимизации, исследовали новообразовавшуюся ковалентную связь и потенциальную энергию. Виртуальное соединение с «боеголовкой» в позиции R1 было признано наиболее предпочтительным.
Как будет подробно описано ниже, синтезированы два соединения с акриламидом в позиции R1 - Соединение 1 и Соединение 2 - которые способны ингибировать cKit.
Синтез Соединения
Синтез промежуточного соединения А
Этап 1: 3-диметиламино-1-пиридин-3-ид-пропенон: 3-Ацетилпиридин (2,5 г, 20,64 ммоль) и N, N-диметил-формамид диметилацетал (3,20 мл, 24 ммоль) кипятили с обратным конденсированием в этаноле (10 мл) в течение ночи. Реакционную смесь охлаждали до комнатной температуры и выпаривали при пониженном давлении. Диэтиловый эфир (20 мл) добавляли в остаток и смесь охлаждали до 0°С. Отфильтровав смесь, в результате получали 3-диметиламино-1-пиридин-3-ил-пропенон (1,9 г, 10,78 ммоль) в виде желтых кристаллов. (Выход: 52%). Этот материал был использован в последующих этапах без дополнительной очистки.
Этап 2: N-(2-метил-5-нитро-фенил)-гуанидиния нитрат: 2-метил-5-нитро-анилин (10 г, 65 ммоль) растворяли в этаноле (25 мл), затем концентрированную HNO3 (4,6 мл) добавляли в раствор по каплям, а за ним - 50%-ный водный раствор цианамида (99 ммоль). Реакционную смесь кипятили с обратным конденсированием в течение ночи и затем охлаждали до 0°С. Полученную смесь фильтровали и остаток промывали этилацетатом и диэтиловым эфиром и сушили, получая N-(2-метил-5-нитро-фенил)-гуанидиний селитры (4,25 г, выход: 34%).
Этап 3: 2-метил-5-нитрофенил-(4-пиридин-3-ил-пиримидин-2-ил)-амин: К суспензии 3-диметиламино-1-пиридин-3-ил-пропенона (1,70 г, 9,6 ммоль) и N-(2-метил-5-нитро-фенил)-гуанидиния селитры (2,47 г, 9,6 ммоль) в 2-пропаноле (20 мл) добавили NaOH (430 мг, 10,75 ммоль) и полученную смесь кипятили с обратным конденсированием в течение 24 ч. Реакционную смесь охлаждали до 0°С и полученный осадок отфильтровали. Твердый остаток суспендировали в воде и фильтровали, а затем промывали в 2-пропаноле и диэтиловом эфире и сушили. После этого отделили 0,87 г (2,83 ммоль) 2-метил-5-нитрофенил-(4-пиридин-3-ил-пиримидин-2-ил)-амина (выход: 30%).
Этап 4: 4-метил-N-3-(4-пиридин-3-ил-пиримидин-2-ил)-бензол-1,3-диамин (Промежуточное соединение А): раствор SnCl2·2 Н2О (2,14 г, 9,48 ммоль) в концентрированной соляной кислоте (8 мл) был добавлен к 2-метил-5-нитро-фенил-(4-пиридин-3-ил-пиримидин-2-ил)-амину (0,61 г, 1,98 ммоль) при интенсивном перемешивании. После 30 минут перемешивания смесь вылили на колотый лед, подщелачивали при помощи К2СО3 и три раза экстрагировали этилацетатом (50 мл). Органические фазы объединили, просушили над MgSO4 и выпаривали досуха. После перекристаллизации из дихлорметана получили 252,6 мг (0,91 ммоль) 4-метил-N-3-(4-пиридин-3-ил-пиримидин-2-ил)-бензол-1,3-диамина (выход: 46%) в виде беловатого твердого вещества.
Синтез Соединения 1

Соединение 1
Этап 1: 4-(акриламидо)бензойная кислота А Раствор 4-аминобензойной кислоты (1,40 г, 10 ммоль) в ДМФ (10 мл) и пиридине (0,5 мл) охлаждали до 0°С. К этому раствору добавили акрилоил хлорид (0,94 г, 10 ммоль) и полученную смесь перемешивали в течение 3 ч. Смесь добавили к 200 мл воды. Полученное белое твердое вещество фильтровали, промывали водой и эфиром. После сушки в высоком вакууме получили 1,8 г желаемого продукта, который был использован в последующих этапах без очистки.
Этап 2: 4-(акриламидо)бензойную кислоту (82 мг, 0,43 ммоль) и Промежуточное соединение А (100 мг, 0,36 ммоль) растворили в пиридине (4 мл) в присутствии азота и перемешали. К этому раствору добавили 1-пропан циклический ангидрид фосфоновой кислоты (0,28 г, 0,43 ммоль) и полученный раствор перемешивали в течение ночи при комнатной температуре. Растворитель упаривали до небольшого объема, а затем выливали в 50 мл холодной воды. Твердый осадок фильтровали и получили желтый порошок. Очистка сырого продукта с помощью колоночной хроматографии (95:5 СНСl3:МеОН) позволила получить 30 мг 4-акриламидо-N-(4-метил-3-(4-(пиридин-3-ил)пиримидин-2-ил амино)фенил)бензамид (Соединение 1) в виде белого порошка. МС (масс-спектрометрия) (М+Н+): 251,2;1Н ЯМР (ДМСО-D6, 300 МГц) δ (ppm): 10,42 (s, 1H). 10,11 (s, 1H), 9,26 (d, 1H, J=2,2 Гц), 8,99 (s, 1H), 8,68 (dd, 1H, J=3,0 and 1,7 Гц), 8,51 (d, 1H, J=5,2 Гц), 8,48 (m, 1H), 8,07 (d, 1H, J=1,7 Гц), 7,95 (d, 2H, J=8,8 Гц), 7,79 (d, 2H, J=8,8 Гц), 7,45 (m, 3Н), 7,19 (d, 1H, J=8,5 Гц), 6,47 (dd, 1H, J=16,7 and 9,6 Гц), 6,30 (dd, Н, J=16,7 и 1,9 Гц), 5,81 (dd, 1H, J=9,9 и 2,2 Гц), 2,22 (s, 3Н).
Синтез Соединения 2
4-акриламидо-N-(4-метил-3-(4-пиридин-3-ил)пиримидин-2-ил амино)фенил-3-(трифторметил)бензамид

1) метил-4-акриламидо-3-нитробензоат
Метилиодид (1,4 г, 9,86 ммоль) добавляли по каплям к перемешанному раствору 4-нитро-3-(трифторметил) бензойной кислоты (1,0 г, 4,25 ммоль) и карбоната калия (1,5 г, 10,85 ммоль) в 30 мл ДМФ при комнатной температуре. Смесь перемешивали при комнатной температуре в течение ночи. Добавили диэтиловый эфир (120 мл) и смесь промыли водой, просушили над Na2SO4, отфильтровали и концентрировали при пониженном давлении, получая 1,0 г сырого метил-4-нитро-3-(трифторметил) бензоата. Раствор из 0,87 г (3,49 ммоль) метил-4-нитро-3-(трифторметил) бензоата и 0,2 г 10%-ного палладиевого катализатора на углеродном носителе (Pd/C) в 30 мл метанола, перемешивали в присутствии водорода (40 фунтов на квадратный дюйм) при комнатной температуре в течение ночи. Полученную смесь фильтровали и концентрировали при пониженном давлении, получая 0,8 г сырого метил-4-амино-3-(трифторметил) бензоата в виде белого твердого вещества. Акрилоил хлорид (0,35 мл, 3,65 ммоль) добавляли в раствор 0,8 г метил-4-амино-3-(трифторметил) бензоата и триэтиламина (0,9 г, 8,9 ммоль) в 40 мл дихлорметана при 0°С. После перемешивания в течение 3 ч при комнатной температуре этот раствор промывали последовательно насыщенным водным NaHCO3 и насыщенным водным NaCl. Раствор дихлорметана сушили над Na2SO4 и концентрировали в вакууме, получая сырой продукт, который затем очищали с помощью хроматографии на силикагеле с использованием 1% CH3OH-CH2Cl2, в результате получая 0,818 мг титульного соединения в виде белого твердого вещества.
2) 4-Акриламидо-3-нитробензойная кислота

К перемешанному раствору метил 4-акриламидо-3-нитробензоата (0,8 г, 2,93 ммоль) в 20 мл ТГФ добавили 20 мл раствора 1 N LiOH. Полученный раствор подкисляли до рН 1 при помощи 10%-ного водного раствора НСl и затем экстрагировали тремя порциями этилацетата по 40 мл. Комбинированный этилацетат экстракт промывали насыщенным водным NaCl, сушили над Na2SO4, фильтровали и концентрировали до сухого состояния в вакууме, получая 0,75 г титульного соединения в виде белого твердого вещества.
3) 4-Акриламидо-N-(4-метил-3-(4-пиридин-3-ил)пиримидин-2-ил амино)фенил-3-(трифторметил)бензамид

К перемешанному раствору N-(4-метил-3-(4-пиридин-3-ил)пиримидин-2-ил амино)фениламина (87 мг, 0,31 ммоль) и 4-акриламидо-3-(трифторметил) бензойной кислоты (95 мг, 0,37 ммоль) в 10 мл пиридина добавили 250 мг (0,39 ммоль) пропилфосфонового ангидрида. Полученный раствор перемешивали при комнатной температуре в течение 72 ч. Растворитель удаляли в вакууме, а остаток перемешивали с 50 мл воды, получая твердое вещество желтого цвета, которое было выделено путем фильтрации. Очистка сырого продукта хроматографией на силикагеле с использованием 5% СН3ОН-СН2Сl2 позволила получить 101 мг титульного соединения.1H ЯМР (ДМСО-D6, 300 МГц) δ (ppm): 10,41 (s, 1Н), 9,90 (s, 1H), 9,28 (d, 1H), 8,98 (s, 1H), 8,69 (d, 1H), 8,68 (dd, 1H), 8,49 (m, 1Н), 8,29 (s, 1H), 8,24 (d, 1H), 8,08 (d, 1H), 7,79, (m, 3Н), 7,23 (d, 1H), 6,59 (dd, 1H), 6,28 (dd, 1H),5,81 (dd, 1H), 2,24 (s, 3H).
Оценка ингибирующей способности по отношению к cKIT
Оценка соединений как ингибиторов тирозинкиназного рецептора c-KIT производилась с использованием человеческого рекомбинантного c-KIT (полученного в Millipore, номер по каталогу 14-559) с применением фосфорилирования флуоресцеин-меченого пептидного субстрата (1,5 мМ). Реакции проводили в 100 мМ HEPES, т.е. N-2-гидроксиэтилпиперазин-N-2-этансульфоновой кислоты (рН 7,5), 10 мМ MnCl2, 1 мМ DDT, т.е. дихлородифенилтрихлорэтана, 0,015% Бридж-35 и 300 мМ АТФ, т.е. аденозинтрифосфата, с исследуемым соединением и без него. Реакция началась с добавления АТФ и инкубирования в течение 1 ч при комнатной температуре. Реакция была остановлена путем добавления останавливающего буфера, содержащего 100 мМ HEPES (рН 7,5), 30 мМ ЭДТА, 0,015% Бридж-35 и 5% ДМСО. Фосфорилированные и нефосфорилированные субстраты были разделены по зарядам с помощью смещения электрофоретической подвижности. Полученный продукт сравнивали с контрольными лунками, чтобы определить наличие ингибирования или усиления активности ферментов.
Данные об ингибировании c-KIT для Соединения 1 и Соединения 2 представлены в Табл. 6.
В. PDGFR (рецептор к фактору роста, выделенному из тромбоцитов)
Способ разработки
Используя систему координат для рентгеновского комплекса cKIT, связанного с иматинибом (код PDB: 1T46), как описано выше, авторы создали гомологичную модель PDGFR-альфа-киназ (код UniProt (универсального ресурса белковых последовательностей): Р 16234). Гомологичная модель была построена с использованием модуля Build Homology в программе Discovery Studio с использованием показанного cKIT-PDGFRα выравнивания. Затем 15 замещаемых позиций на матрице иматиниба были исследованы в трех измерениях с целью определить, какая из них может быть замещена на «боеголовку» так, чтобы «боеголовка» сформировала ковалентную связь с Цис в сайте связывания.
Использованный способ позволил выделить три матричных позиции - R1, R2 и R4 - и Cys814, способные образовывать ковалентную связь с акриламидной «боеголовкой». Из этих матричных позиций связи, которые возникают в «боеголовке» на позициях R2 и R4, затрагивают цис-конформации амидной группы «боеголовки», что является наименее предпочтительным вариантом. Связи, возникающие в «боеголовке» в положении R1, затрагивают транс-конформации амидной группы «боеголовки», - такой вариант наиболее предпочтителен.

Оценка ингибирующей способности по отношению к PDGFR
Способ А:
Оценка ингибирующей способности соединений по отношению к PDGFR производилась способом, подобным тому, что описан корпорацией Invitrogen Corp (Invitrogen Corporation, 1600 Фарадэй авеню, Карлсбад, Калифорния, Калифорния;
www.invitrogen.com/downloads/Z-LYTE_Brochure_1205.pdf) с использованием процедуры биохимического анализа Z′-LYTE™ или другого подобного биохимического анализа. При анализе Z′-LYTE™ используется основанный на флуоресценции формат сцепленных ферментов, который основан на разной чувствительности фосфорилированных и нефосфорилированных пептидов к протеолитическому расщеплению.
Соединение 1 исследовали при 0,1 мМ и 1 мМ в двух повторностях. Соединение 1 продемонстрировало средний показатель ингибирования PDGFR-α на уровне 76% при 1 мМ, и 29% - при 0,1 мМ.
Способ В
Вкратце, сток 10Х фермента PDGFRα (PV3811), субстраты 1.13Х АТФ (AS001A) и Y12-Sox пептида (KCZ1001) были приготовлены в IX киназа реакционном буфере, состоящем из 20 мМ трис, рН 7,5, 5 мМ MgCl2, 1 мМ ЭГТК, 5 мМ β-глицерофосфата, 5% глицерина (10Х сток, КВ002А) и 0,2 мМ ДТТ (DS001A). 5 мкл предварительно инкубировали в Coming (# 3574) 384-луночных, белых титрационных микропланшетах с несвязывающей поверхностью (Coming, Нью-Йорк) в течение 30 мин при 27°С с 0,5 мкл объема 50%-ного ДМСО и серийно разбавленного соединения, приготовленного в 50% ДМСО. Киназные реакции были начаты с добавлением 45 мкл ATP/Y9 или Y12-Sox пептидной субстратной смеси и контролировались каждые 30-90 секунд в течение 60 мин при λех360/λem485 на планшет-ридере Synergy4 компании BioTek (Винуски, Вермонт). В конце каждого оценочного анализа прогрессирующие кривые для каждой лунки рассматривались относительно линейной кинетики реакции и статистики пригодности (R2, 95% ДИ (доверительный интервал), абсолютная сумма квадратов). Начальная скорость (от 0 минут до 20+ минут) из каждой реакции определялась по наклону зависимости относительных единиц флуоресценции vs время (в минутах), а затем строили кривую против концентрации ингибитора с целью оценить IC50 от log [Ингибитор] vs Ответ, модель Переменного уклона (Variable Slope) в приложении GraphPad Prism компании GraphPad Software (Сан-Диего, Калифорния). [PDGFRα]=2-5 нМ, [АТР]=60 мМ и [Y9-Sox пептид]=10 мМ (АТФ КМмарр=61 мМ).
Данные об ингибировании PDGFR Соединением 1 и Соединением 2 приведены ниже в Табл. 7.
PDGFR: Масс-спектральный анализ Соединения 1
Произведен масс-спектральный анализ PDGFR-a в присутствии Соединения 1. Белок PDGFR-α (получен в компании In vitro gen: PV3811) инкубировали с 1 мМ, 10 мМ и 100 мМ Соединения 1 в течение 60 мин. В частности, 1 мкл 0,4 мкг/мкл PDGFR-α (Invitrogen PV3811) стоковый раствор (50 мМ Трис НСl ph 7,5, 150 мМ NaCl, 0,5 мМ ЭДТА, 0,02% Тритон Х-100, 2 мМ ДТТ, 50% глицерин) добавили к 9 мкл Соединения 1 в 10% ДМСО (финальная концентрация 1 мМ, 10 мМ и 100 мМ). Через 60 мин добавили 9 мкл 50 мМ бикарбоната аммония, 3,3 мкл 6 мМ йодацетамида в 50 мМ бикарбоната аммония и 1 мкл 35 нг/мкл трипсина для остановки реакции.
Триптический гидролизат исследовали на масс-спектрометре (MALDI-TOF) при 10 мМ. Из пяти цистеиновых остатков, обнаруженных в белке PDGFR-α, четыре были идентифицированы как модифицированные йодацетамидом, тогда как пятый цистеиновый остаток был модифицирован соединением 1. Масс-спектральный анализ триптического гидролизата показал соответствие с Соединением 1, ковалентно связанным с белком PDGFR-α в Cys814. Тандемная масс-спектрометрия триптического гидролизата подтвердила присутствие Соединения 1 в Cys814.
Оценка пролиферации клеток EOL-1
Клетки EOL-1, приобретенные в компании DSMZ (АСС 386) хранили в RPMI-растворе (Invitrogen #21870)+10% FBS (эмбриональная бычья сыворотка)+1% пенициллина/стрептомицина (Invitrogen # 15140-122). Для оценки пролиферации клетки в полной среде разместили в 96-луночных планшетах при плотности 2×104 клеток/лунку и инкубировали в двух повторностях с соединением в диапазоне от 500 нМ до 10 пикометров в течение 72 ч. Пролиферацию клеток оценивали путем измерения метаболической активности при помощи реагента Alamar Blue (Invitrogen cat # DAL1100). Через 8 ч инкубации при помощи Alamar Blue при 37°С, поглощение считывалось при 590 нм, и концентрацию IC50 клеточной пролиферации рассчитывали в программе GraphPad. Ингибирование дозовой зависимости пролиферации клеток EOL-1 эталонным соединением и Соединением 2 изображено на ФИГ.5.
Анализ по «вымыванию» клеток EOL-1
Клетки EOL-1 выращивали в суспензии в полной среде, затем добавляли соединение к 2×106 клеткам на образец в течение 1 ч. Через 1 ч клетки осаждали центрифугированием, среду удаляли и заменяли на другую среду, не содержащую Соединение. Клетки промывали каждые 2 ч и ресуспендировали в свежей среде, не содержащей Соединение, Клетки собирали в определенные временные точки, лизировали в экстракционном буфере и загружали 15 мг лизата суммарного белка в каждой вертикальной дорожке. Оценку фосфорилирования PDGFR проводили путем вестерн-блоттинга с использованием антител компании Santa Cruz sc-12910. Результаты данного эксперимента показаны на ФИГ.6, на которой видно, что по сравнению с контрольным ДМСО и обратимо действующим эталонным соединением, Соединение 2 сохраняло способность ингибировать фермент PDGFR в клетках EOL-1 после теста на "вымывание" через 0 ч и 4 ч.
С.CSF1R
Способ разработки
Используя систему координат для рентгеновского комплекса cKIT, связанного с иматинибом (код PDB: 1T46), как описано выше, авторы создали гомологичную модель CSF1R киназы (код Uniprot: P07333). Гомологичную модель выстраивали с использованием модуля Build Homology программы Discovery Studio, применяя выравнивание cKIT-CSF1R. Затем 15 замещаемых позиций на матрице иматиниба исследовали в трех измерениях с целью определить, какая из них может быть замещена на «боеголовку» так, чтобы «боеголовка» сформировала ковалентную связь с Цис в сайте связывания. Данный способ позволил идентифицировать две матричные позиции (R1 и R2) и Cys774, которые могут сформировать связь с акриламидной «боеголовкой».

Оценка ингибирования CSF1R
Оценка Соединения как ингибитора PDGFR производилась по способу, подобному тому, который описан корпорацией Invitrogen Corp (Invitrogen Corporation, 1600 Фарадэй авеню, Карлсбад, Калифорния, СА) с использованием процедуры биохимического анализа Z′-LYTE™ или другого биохимического анализа. Пептидную смесь 2Х CSF1R (FMS) / Tyr 01 приготовили в 50 мМ HEPES рН 7,5, 0,01% Бридж-35, 10 мМ MgCl2, 1 мМ ЭГТА. Финальная 10 мкл Киназная реакция состояла из 0,2-67,3 нг CSF1R (FMS) и 2 мМ Tyr 01 пептида в 50 мМ HEPES рН 7,5, 0.01% Бридж -35, 10 мМ MgCl2, 1 мМ ЭГТА. Через 1 ч после инкубирования киназной реакции, добавили 5 мкл 1:128 разбавленного раствора Реагента Развития В.
Соединение 1 продемонстрировало 72% ингибирования против CSF1R при 10 мМ, а Соединение 2 показало 89% ингибирования против CSF1R при 10 мМ.
Данные масс-спектрального анализа
Масс-спектральный анализ применялся с целью выяснить, является ли Соединение 2 ковалентным модификатором CSF1R. CSF1R (0.09 мкг/мкл) инкубировали с Соединением 2 (усредненная молекулярная масса 518,17) в течение 3 ч при 10Х избыток перед триптическим гидролизатом. Йодацетамид использовали в качестве алкилирующего агента после инкубирования соединения. Для триптического гидролизата аликвоту 2 мкл (0,09 мкг/мкл) разбавили с помощью 10 мкл of 0,1% ТФК перед микро картридж-колонкой С 18 Zip Tipping непосредственно на мишень MALDI (лазерная ионизация и десорбция из матрицы) с использованием альфа-циано-4-гидрокси коричной кислоты в качестве матрицы (5 мг/мл в 0,1% ТФК: ацетонитрил 50:50).
Для триптического гидролиза оборудование настроили на режим Reflectron с пульсационным выводом 1800. Калибровку производили с использованием стандарта Laser Biolabs Pep Mix (1046,54, 1296,69, 1672,92, 2093,09, 2465,20). Для анализа CID/PSD (индуцированная столкновениями диссоциация/ спектральная плотность мощности) пептиды выбирались с использованием курсоров для назначения ионного стробирования и фрагментации, происходившей при мощности лазерного излучения около 20% выше. Гелий использовался в качестве газа для соударений в CID. Калибрование фрагментов производили с использованием P14R фрагментационной калибровки для Рефлектрона искривленного поля. Поиск по базе данных триптического гидролизата CSF1R идентифицирует его корректно. Включение модификации Соединения 2 (518,17) также идентифицировало исследуемый целевой пептид NCIHR (SEQ ID NO: 8) (МН+642,31+518,17=1160,48) как единственный модифицированный пептид, имеющийся в наличии. Анализ PSD данного пептидного сигнала (1160,50) позволил получить достаточно фрагментов для ионного поиска по базе тандемной масс-спектрометрии, которая подтвердила последовательность данного пептида.
D. ABL
Способ разработки
При помощи координат для рентгеновского комплекса cKIT и связанного с ним иматиниба (код PDB: 1T46), как описано выше, была построена гомологичная модель ABL-киназы (код Uniprot: P00519). Построение гомологичной модели производилось при помощи модуля Build Homology программы Discovery Studio с использованием выравнивания cKIT-ABL, показанного ниже. Затем 15 замещаемых позиций исследовали в трех измерениях на матрице иматиниба с целью найти положение акриламидной «боеголовки», способной сформировать ковалентную связь с Цис в сайте связывания. Однако данный способ не выявил матричных позиций или подходящего Цис, поддающихся модификации.

Пример 2. Необратимо действующий нилотиниб
Нилотиниб - сильный обратимый ингибитор ABL, cKIT, PDGFR и CSF1R киназ. Описанный в данной работе алгоритм, основанный на структурных особенностях, позволяет быстро и эффективно преобразовать нилотиниб в необратимо действующий ингибитор, способный подавлять cKIT и PDGFR.


A. ABL
Координаты рентгеновского комплекса нилотиниба и связанного с ним ABL (код PDB: 3CS9) были получены в базе данных белковых структур (www.rcsb.org). Координаты ниотиниба извлекли, и все белковые Цис-остатки в пределах 20 ангстремов от нилотиниба, связанные с ABL, идентифицировали. Затем 14 замещаемых позиций на матрице нилотиниба (11-1) исследовали в трех измерениях, чтобы определить, какая из них может быть замещена на хлорацетамидную «боеголовку» так, чтобы образовалась ковалентная связь с Цис в сайте связывания. Однако данный способ не выявил матричных позиций или подходящего Цис, поддающихся модификации.
В. PDGFRa
Гомологичная модель PDGFR альфа киназы (код Uniprot: PI 6234) разрабатывалась с использованием рентгеновских структур нилотиниба, связанного с ABL-матрицей (код PDB 3CS9). Гомологичная модель строилась с использованием модуля Build Homology в программе Discovery Studio, с ABL-PDGFRa выравниванием, изображенным ниже. Затем 14 замещаемых позиций на матрице нилотиниба (II-1) исследовали в трех измерениях с целью определить, которая из них может быть замещена на «боеголовку» так, чтобы образовалась ковалентная связь с Цис в сайте связывания. Использованный способ позволил выявить одну позицию в матрице (R11) и один Цис (Cys814), между которым может образоваться ковалентная связь с хлорацетамидной «боеголовкой». Далее было синтезировано Соединение 3, содержащее хлорацетамид в R11.

С.CSF1R
Гомологичная модель CSF1R киназы (код Uniprot: P07333) разрабатывалась на основе рентгеновских структур нилотиниба, связанного с ABL-матрицей (код PDB 3CS9). Гомологичная модель строилась с применением модуля Build Homology в приложении Discovery Studio с ABL-CSF1R выравниванием, показанным ниже. Затем 14 замещаемых позиций на матрице нилотиниба (II-1) исследовали в трех измерениях с целью определить, какая из них может быть замещена на «боеголовку» так, чтобы сформировалась ковалентная связь с Цис в сайте связывания. Использованный способ позволил выявить одну позицию в матрице (R11) и один Цис (Cys774), которые могут создать связь с хлорацетамидной «боеголовкой».

D. cKIT
Гомологичная модель cKIT киназы (код Uniprot: PI 0721) строилась на основе рентгеновских структур нилотиниба, связанных с матрицей ABL (код PDB 3CS9). Гомологичная модель разрабатывалась в модуле Build Homology программы Discovery Studio с выравниванием ABL-cKIT, изображенным ниже. Затем 14 замещаемых позиций на матрице нилотиниба (11-1) исследовали в трех измерениях, чтобы определить, какая из них может быть замещена на хлорацетамидную «боеголовку» так, чтобы сформировалась ковалентная связь с Цис в сайте связывания. Таким ограничительным условиям соответствовали одна позиция в матрице (R11) и один Цис (Cys788).

Синтез Соединения 3

Схема 3-А

a) NH2CN, EtOH/HCl, 90°С, 15 ч, b) ДМФ-ДМА, этанол, обратный конденсат, 16 ч, с) NaOH/EtOH, обратный конденсат, 16 ч
Этап-1: К перемешанному раствору эфира анилина (5 г, 30,27 ммоль) в этаноле (12,5 мл) добавили концентрированную HNO3 (3 мл), а затем 50% водного раствора цианамида (1,9 г, 46,0 ммоль) при комнатной температуре. Реакционную смесь нагревали при 90°С в течение 16 ч, затем охлаждали до 0°С. Выпавшее в осадок твердое вещество фильтровали, промыли в этилацетате (10 мл), диэтиловом эфире (10 мл) и просушили, в результате получали соответствующий гуанидин (4,8 г, 76,5%) в виде светло-розового твердого вещества, которое использовалось без дальнейшей очистки.
Этап-2: Перемешанный раствор 3-ацетил пиридина (10,0 г, 82,56 ммоль) и N, N-диметилформамид диметил ацеталя (12,8 г, 96,00 ммоль) в этаноле (40 мл) кипятили с обратным конденсированием в течение 16 ч. Затем состав охлаждали до комнатной температуры и концентрировали при пониженном давлении, получая сырую массу. Остаток помещали в эфир (10 мл), охлаждали до 0°С и фильтровали, чтобы получить соответствующий енамид (7,4 г, 50,8%) в виде желтого кристаллического твердого вещества.
Этап-3: Перемешанную смесь производного гуанидина (2 г, 9,6 ммоль), производного енамида (1,88 г, 10,7 ммоль) и NaOH (0,44 г, 11,0 ммоль) в этаноле (27 мл) кипятили с обратным конденсированием при 90°С в течение 48 ч. Затем реакционную смесь охлаждали и концентрировали при пониженном давлении, чтобы получить остаток. Остаток помещали в этилацетат (20 мл) и промывали водой (5 мл). Органические и водные слои были отделены и обрабатывались отдельно, чтобы получить соответствующий эфир и Промежуточное соединение С соответственно. Водный слой охлаждали и подкисляли 1,5 N НСl (рН ~3-4), когда выпадал белый твердый осадок. Осадок отфильтровывали, сушили, а избыток воды удаляли азеотропной дистилляцией над толуолом (2х10 мл), чтобы получить Промежуточное соединение С (0,5 г) в виде бледно-желтого твердого вещества. 1Н ЯМР (ДМСО-d6, 400 МГц) (ppm): 2,32 (s, 3Н), 7,36 (d, J=10,44 Гц, 1Н), 7,53 (d, J=6.84 Гц, 1Н), 7,60-7,72 (m, 2Н), 8,26 (s, 1Н), 8,57 (d, J=6,84 Гц, 1Н), 8,64 (d, J=10,28 Гц, 1Н), 8,70-8,78 (bs, 1Н), 9,15 (s, 1Н), 9,35 (s, 1Н). Органический экстракт промывали солевым раствором (3 мл), сушили (Na2SO4) и концентрировали при пониженном давлении, чтобы получить эфир Промежуточного соединения С в виде сырого твердого вещества. В дальнейшем его очищали с помощью колоночной хроматографии (SiO2, меш 60-120 сетка, МеОН/СНСl3: 10/90), чтобы получить эфир Промежуточного соединения С (0,54 г) в виде твердого вещества желтого цвета.
Схема 3-В

а) (ВОС)2O, DMAP, Et3N, ТГФ, b) H2, Pd/C, CH3OH
Этап-1: К перемешиваемому раствору нитроанилина (0,15 г, 0,7 ммоль) в ТГФ (0,3 мл) добавили Et3N (0,11 мл, 0,73 ммоль) и ДМАП (0,05 г, 0,44 ммоль). К ним добавили трет-бутоксикарбонил (ВОС) ангидрид (0,33 мл, 1,52 ммоль) и реакционную смесь кипятили с обратным конденсированием в течение 5 ч. Затем реакционную смесь охлаждали, разбавляли ТГФ (15 мл) и промывали солевым раствором (5 мл). Органическую фазу отделили, сушили над Na2S04, фильтровали и концентрировали при пониженном давлении, чтобы получить сырую массу. Сырой продукт далее очищали с помощью колоночной хроматографии (SiO2, меш 230-400, гексан/EtOAc: 8/2), чтобы получить соответствующий ди-Вос защищенный анилин (0,25 г, 88%) в виде белого кристаллического твердого вещества, которое использовалось в следующем Этапе без дополнительной очистки.
Этап-2: Раствор Вос-защищенного анилина (0,25 г, 0,62 ммоль) в метаноле (5 мл) гидрогенизировали (H2, 3 кг) более 10% Pd/С (0,14 г, 0,13 ммоль) при 20°С в течение 12 ч. Реакционную смесь пропускали через короткий планшет Celite®, концентрировали при пониженном давлении, чтобы получить соответствующий анилин в виде беловатого твердого вещества (0,18 г, 77,6%).
1Н ЯМР (CD3OD, 400 МГц) 5 (ppm): 1,36 (s, 18 H), 6.84-6.87 (m, 1H), 6.95-6.97 (m, 2H).
Схема 3-С
a) HATU, DIEA, CH3CN, 85°С, 16 ч, b) TFA/CH2Cl2, 0°C до комнатной температуры, 3 ч
Этап 1: Взаимодействие Промежуточного соединения С с di-BOC защищенным анилином в присутствии HATU, DIEA в ацетонитриле позволяет получить соответствующий амид.
Этап 2: Снятие защиты с ВОС-группы для получения Промежуточного соединения D может быть достигнуто путем обработки амида с ТФК в метиленхлориде при 0°С, а затем нагревания до комнатной температуры.
К перемешанному раствору Промежуточного соединения D (0,1 г, 0,22 ммоль) в ТГФ (10 мл) при 0°С добавили Et3N (0,033 г, 0,32 ммоль) в присутствии N2. Хлорацетилхлорид (0,029 г, 0,26 ммоль) добавили по каплям при перемешивании; реакционной смеси дали нагреться до комнатной температуры и перемешивали ее в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении, получая остаток, который помещали в EtOAc (10 мл). Этот раствор промывали водой (2 мл) и водный слой снова экстрагировали этилацетатом (2×10 мл). Фракции EtOAc объединяли и промывали солевым раствором (2 мл). После сушки над Na2SO4 и фильтрации раствор EtOAc концентрировали при пониженном давлении, получая сырой продукт, который затем очищали с помощью колоночной хроматографии (SiO2, меш 60-120, СНCl3/МеОН: 9/1), получая III-14 (50 мг, 43%) в виде бледно-желтого твердого вещества. 1H ЯМР (ДМСО-d6) δ ppm: 2,34 (s, 3Н), 4,30 (s, 2H), 7,42-7,48 (m, 4H), 7,73-7,75 (m, 1H), 8,05-8,10 (m, 1H), 8,24 (d, J=2,2 Гц, 1H), 8,29 (s, 1H), 8,43 (d, J=8,04 Гц, 1H), 8,54 (d, J=5,16 Гц, 1Н), 8,67 (dd, J=1,6 & 4,76 Гц, 1Н), 9,16 (s, 1H), 9,26 (d, J=2,2 Гц, 1H), 9,89 (s, 1H), 10,50 (s, 1H); ЖХМС: m/e 541,2 (M+1)
Ингибирование мишени
Способность Соединения 3 подавлять сКIT или PDGFR исследовалась по способу оценки ингибирования cKIT или PDGFR, описанному в Примере 1. Полученные данные подтверждают, что Соединение 3 является сильным ингибитором cKIT (IC50=0,7 нМ) и PDGFR (IC50=9 нМ) (Табл. 8).
Масс-спектральный анализ PDGFR
Производился масс-спектральный анализ PDGFR-a в присутствии Соединения 3. PDGFR-a (43 пикомоль) инкубировали с Соединением 3 (434 пмоль) в течение 3 ч при 10Х доступе перед триптическим гидролизатом, Йодацетамид использовался в качестве алкилирующего агента после инкубирования соединения. Для триптического гидролизата аликвоту 5 мкл (7 пмоль) разбавляли при помощи 10 мкл 0,1% ТФК перед микро картридж-колонкой С 18 Zip Tipping непосредственно на мишень MALDI с использованием альфа-циано-4-гидрокси коричной кислоты в качестве матрицы (5 мг/мл в 0,1% ТФК:Ацетонитрил 50:50).
Триптический гидролизат анализировали на масс-спектрометре (MALDI-TOF). Результаты масс-спектрального анализа триптических гидролизатов были идентичны таковым для Соединения 3, ковалентно связанного с белком PDGFR-α в Cys814. (ФИГ.7) Тандемная масс-спектрометрия триптических гидролизатов подтвердила наличие Соединения 3 в Cys814.
Масс-спектральный анализ с-КIТ
Произведен масс-спектральный анализ с-КIТ в присутствии Соединения 3. В частности, киназу с-КIТ (86 пмоль) инкубировали с Соединением 3 (863 пмоль) в течение 3 ч при 10Х доступе перед триптической гидролизацией. Йодацетамид использовался в качестве алкилирующего агента после инкубирования соединения. Для триптического гидролизата аликвота 5 мкл (14 пмоль) разбавляли при помощи 10 мкл 0,1% ТФК перед микро картридж-колонкой С 18 Zip Tipping непосредственно на мишень MALDI с использованием альфа-циано-4-гидрокси коричной кислоты в качестве матрицы (5 мг/мл в 0,1% ТФК:Ацетонитрил 50:50).
Триптический гидролизат анализировали на масс-спектрометре (MALDI-TOF). Результаты масс-спектрального анализа триптических гидролизатов были идентичны таковым для Соединения 3, ковалентно связанного с белком с-К1Тв двух целевых цистеиновых остатках Cys788 (в большей степени) и: Cys808 (в меньшей степени).
Оценка «вымывания» cKIT
Клетки GIST430 (См. Bauer et al., Cancer Research, 66(18):9153-9161 (2006)) высевали в 6-луночный планшет при плотности 8×105 клеток/лунка и обрабатывали 1 мМ Соединения 3, разбавленного в полной среде в течение 90 мин на следующий день. По прошествии 90 мин среду удаляли и клетки промывали средой, не имеющей в своем составе исследуемого Соединения. Клетки промывали каждые 2 ч и ресуспендировали в свежей среде, не содержащей Соединения. Сбор клеток производили в определенные временные точки, клетки лизировали в экстракционном буфере Cell Extraction Buffer (Invitrogen FNN0011), в который были добавлены Roche - цельные ингибирующие протеазу таблетки (Roche 11697498001) и ингибиторы фосфатазы (Roche 04906837001). Далее 10 мкг общего белкового лизата загружали в каждую вертикальную дорожку. Фосфорилирование с-КIТ оценивали путем вестерн-блоттинга с использованием антител pTyr (4G10) и общих антител kit производства компании Cell Signaling Technology. Результаты отображены в Табл. 9, из которой видно, что Соединение 3 сохраняет ингибирующую способность по отношению к ферментам с-КIТ в клетках GIST430 после "вымывания" в точках 0 ч и 6 ч.
Пример 3. Необратимо действующий VX-680
VX-680 - сильный обратимый ингибитор FLT3 киназ. При помощи алгоритма разработки, описанного в этом документе, VX-680 был быстро и эффективно Преобразован в необратимый ингибитор FLT-3.
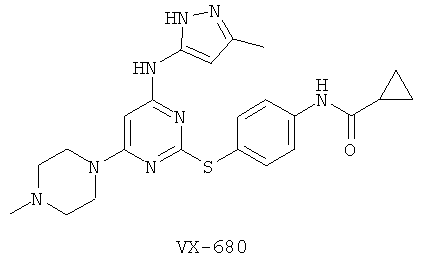
Характер связывания VX-680 с Flt3 определялся путем логических умозаключений на основе характера связывания VX-680 с родственной киназой Аврора, поскольку кристаллическая структура Авроры, образующей комплекс с VX-680, известна. Гомологичная модель FLT3 строилась с использованием рентгеновских структур киназы Аврора (код PDB: 2F4J) на основе компонента для моделирования белков в приложении Accelrys Discovery Studio (Discovery Studio v2.0-1.7347, Accelrys Inc). Выравнивание при построении модели основывалось на структурном выравнивании рентгеновских комплексов FLT3 и киназы Аврора. Высокая степень структурного подобия между этими двумя белками и значительное подобие позиций сайта связывания в дальнейшем были подтвердили правильность выбранной стратегии гомологичного моделирования.
Выравнивание структур FLT3 (1RJB) и киназы Аврора /VX-680 комплекса (2FB4) с 256 структурно эквивалентными позициями, со среднеквадратичным отклонением (RMSD) 3Å.
Выравнивание структур FLT3 (1RJB) и киназы Аврора /VX-680 комплекса (2FB4) с 256 структурно эквивалентными позициями, со среднеквадратичным отклонением (RMSD) 3Å.

Гомологичная модель Flt3 с VX680 позволила выявить шесть Цис-остатков в Flt3, находящихся в пределах 20 ангстремов от связи VX680 (Cys694, Cys695, Cys681, Cys828, Cys807 и Cys790). Затем 7 замещаемых позиций на матрице VX-680 (Формула III-1) исследовали в трех измерениях с целью определить, какая из них может быть замещена на «боеголовку», чтобы образовалась ковалентная связь с одним из идентифицированных Цис-остатков в сайте связывания FLT3. «Боеголовки» надстраивали в трехмерном пространстве на матрицу VX-680 в программе Discovery Studio. Структуру полученного соединения исследовали, чтобы определить, может ли «боеголовка» достичь Цис в сайте связывания.

Чтобы получить проверить гибкость «боеголовок» и позиций боковой цепи, была построена стандартная молекулярно-динамическая симуляция «боеголовок» и позиций боковой цепи. По этой симуляции смотрели, находится ли «боеголовка» в пределах 6 ангстремов от любого из идентифицированных Цис-остатков в сайте связывания. Использовались стандартные настройки в протоколе Standard Dynamics Cascade Simulations программы Accelrys Discovery Studio v2.0.1.7347 (Accelrys Inc) для молекулярно-динамических симуляций. Координаты позиций не-«боеголовок» и атомов главной цепи Цис удерживались при этом в зафиксированном положении.
В результате было выявлено 3 матричных позиции (R4, R6 и R7), расположенных поблизости от Cys828 (Табл. 10). Эти позиции затем отсеивали по критерию, чтобы мог образоваться продукт акриламидной реакции между предполагаемым ингибитором и Cys828, для чего должна была образоваться связь менее 2 ангстремов (отсеивание производилось при помощи стандартной молекулярно-динамической симуляции. Этим условиям успешно удовлетворяли все три позиции. Далее было синтезировано Соединение 4, которое содержит акриламид в позиции R7.
Синтез Соединения 4

Этап 1. 4,6-дихлор-2-метилсульфонил пиримидин
4,6-дихлор-2-(метилтио) пиридин (24 г, 0,123 моль) растворяли в 500 мл CH2Cl2, при перемешивании и на ледяной бане. Мета-хлорпероксибензойную кислоту (около 0, 29 моль) медленно добавляли в течение 40 мин. Реакционную смесь перемешивали в течение 4 ч, разбавляли с помощью СН2Cl2 и затем обрабатывали 50% раствором Na2S2O3/NaHCO3. Органическую фазу промывали насыщенным водным NaCl, сушили над MgSO4 и затем фильтровали. После удаления растворителя под вакуумом получили выход 24 г титульного соединения в виде светло-красного твердого вещества.
Этап 2. Трет-бутил N-(4-меркаптофенил)карбамат

4-аминотиофенол (25 г, 0,2 моль) растворяли в 250 мл этилацетата. Раствор охлаждали на ледяной бане и добавляли по каплям ди-трет-бутилдикарбонат (48 г, 0,22 моль), перемешивая. После перемешивания в течение 1 ч был добавлен насыщенный NaHCO3 в воде (200 мл). Реакционную смесь перемешивали в течение ночи. Органическую фазу промывали водой, насыщенным водным раствором NaCl, сушили над MgSO4 и затем фильтровали. После удаления органического растворителя под вакуумом получили выход 68 г желтого масла, которые обработали гексаном, получая выход около 50 г титульного соединения в виде желтого твердого вещества.
Этап 3. Трет-бутил 4-(4,6-дихлорпиримидин-2-ил тио)фенилкарбамат

Смесь трет-бутил N-(4-меркаптофенил) карбамата (5 г, 0,022 моль) и 4,6-дихлор-2-метилсульфонилпиримидина (5 г, 0,026 моль) в 150 мл t-BuOH нагревали с обратным конденсированием в течение 1 ч и затем добавляли NaOAc (0,5 г). Реакционную смесь нагревали с обратным конденсированием дополнительно в течение 14 ч. Растворитель удаляли в вакууме и остаток растворяли в этилацетате. Органическую фазу промывали последовательно раствором К3СО3 и насыщенным водным NaCl, сушили над MgSO4 и затем фильтровали. После удаления растворителя получили выход около 5 г титульного соединения в виде как желтого твердого вещества.
Этап 4. Трет-бутил 4-(4-хлор-6-(3-метил-1H-пиразол-5-ил амино)пиримидин-2-ил тио)фенилкарбамат

Раствор трет-бутил-4-(4,6-дихлорпиримидин-2-ил тио)фенилкарбамата (100 мг, 0,27 ммоль), 3-метил-5-амино-1Н-пиразола (28,7 мг, 0,3 ммоль), диизопропилэтиламина (41,87 мг) и NaI (48,6 мг, 0,324 ммоль) в 1 мл DMF нагревали при 85°С в течение 4 ч. После охлаждения и разбавления с 20 мл этилацетата, органическую фазу последовательно промывали водой и насыщенным водным NaCl, сушили над MgSO4 и затем фильтровали. После удаления растворителя в вакууме получили выход около 120 мг сырого продукта, который очищали хроматографией на силикагеле (30% этилацетат / гексаны) с получением 64 мг титульного соединения.
Этап 5. Трет-бутил 4-(4-(3-метил-1H-пиразол-5-ил амино)-6-(4-метилпиперазин-1-ил)пиримидин-2-ил тио)фенилкарбамат

Смесь трет-бутил-4-(4-хлор-6-(3-метил-1Н-пиразол-5-ил амино) пиримидин-2-ил тио) фенилкарбамата (61 мг, 0,14 ммоль) и 1 мл метилпиперазина нагревали при 110°С в течение 2 ч. Реакционную смесь разбавляли 20 мл этилацетата. Органическую фазу промывали водой, сушили над MgSO4 и затем фильтровали. После удаления растворителя в вакууме получали выход около 68,2 мг сырого продукта в виде светло-коричневого твердого вещества, которое очищали на силикагеле (30% EtOAc/гексаны), получая 49,5 мг титульного соединения. МС (М+Н+):497,36.
Этап 6. 2-(4-Аминфенилтио)-N-(3-метил-1H-пиразол-5-ил амино)-6-(4-метилпиперазин-1-ил)пиримидин-4-амин

Раствор трет-бутил-4-(4-(3-метил-1Н-пиразол-5-ил амино)-6-(4-метилпиперазин-1-ил) пиримидин-2-ил тио) фенилкарбамата (44,5 мг) в 5 мл МеОН обрабатывали 2 мл 5N НСl. Когда тонкослойная хроматография показала, что исходный материал полностью отсутствует, реакционную смесь разбавили этилацетатом. Органическую фазу промывали NaHCO3 и насыщенным водным NaCl, сушили над MgSO4 и затем фильтровали. После удаления растворителя в вакууме получили около 32,1 мг титульного соединения.1Н ЯМР (300 МГц, ДМСО-d6): δ 11,68 (s, 1H), 9,65 (s, 1H), 9,25 (s, 1H), 7,60 (d, 2H), 7,45 (d, 2H), 6,00 (s, 1H), 5,43 (s, 1H), 2,38 (m, 4H), 2,20 (m, 2H), 2,05 (m, 2H), 1,52 (s, 6H), MC (M+H+):397,18.
Этап 7. N-(4-(4-(3-Метил-1H-пиразол-5-ил амино)-6-(4-метилпиперазин-1-ил)пиримидин-2ил тио)фенил)акриламид

Акрилоил хлорид (6,85 мл, 7,33 мг, 0,081 ммоль) добавляли к раствору из 2-(4-аминфенилтио)-N-(3-метил-1Н-пиразол-5-ил)-6-(4-метилпиперазин-1-ил)-пиримидин-4-амин (32,1 мг, 0,081 ммоль) в 3 мл CH2Cl2 при 0°С. Через 30 мин реакционную смесь разбавляли этилацетатом. Органическую фазу промывали раствором NaHCO3, насыщенным водным раствором NaCl, сушили над MgSO4 и затем фильтровали. После удаления растворителя получали сырой продукт, который очищали на силикагеле, получая выход 20 мг продукта. MC(М+Н+):451,36.
Биохимические исследования
У Соединения 4 показатель IC50 составил 2,2 нМ в отношении ингибирования фосфорилирования FLT3 в биохимическом анализе FLT3. У соединения VX-680 показатель IC50 был равен 10,7 нМ, согласно этому же анализу.
Биохимический анализ FLT3
Для измерения активности Соединения в отношении активного FLT-3 фермента использовался анализ киназы с постоянным считыванием. Анализ проводился по способу, подобному тому, который описан поставщиком (компанией Invitrogen, Карлсбад, Калифорния, www.invitrogen.com/content.cfm?pageid=11338). Вкратце, 10Х стоков KDR, приобретенных у Invitrogen или BPS Bioscience (PV3660 или 40301) или FLT-3 (PV3182) ферменты, 1.13Х АТР (AS001A) и Y9-Sox или Y12-Sox пептидных субстратов (KCZ1001) были приготовлены в IX киназном реакционном буфере, состоящем из 20 мМ Трис, рН 7,5, 5 мМ MgCl2, 1 мМ ЭГТА, 5 мМ β-глицерофосфата, 5% глицерина (10Х сток, КВ002А) и 0,2 мМ ДТТ (DS001A). Далее 5 мкл фермента предварительно инкубировали в 384-луночных, белых, титрационных микропланшетах с несвязывающей поверхностью Coming (#3574) (компания Coming, Нью-Йорк) в течение 30 мин при 27°С с добавлением 0,5 мкл объема 50% ДМСО и последовательно разбавляли соединения, приготовленные в 50% ДМСО. Киназные реакции начинались с добавления 45 мкл ATP/Y9 или Y5-Sox пептидной субстратной смеси и наблюдались каждые 30-90 с в течение 60 мин при λех360/λem485 в планшетном ридере Synergy4 производства BioTek (Винуски, Вермонт). В конце каждого оценочного анализа, прогрессирующие кривые из каждой лунки рассматривались относительно линейной кинетики реакции и статистики пригодности (R, 95% ДИ, абсолютная сумма квадратов). Начальная скорость (от 0 минут до 20+ минут) из каждой реакции определялась по наклону зависимости относительных единиц флуоресценции vs время (в минутах), а затем строили кривую против концентрации ингибитора с целью оценить IC50 от log [Ингибитор] vs Ответ, модель Переменного уклона (Variable Slope) GraphPad Prism приложения GraphPad Software (Сан-Диего, Калифорния). [Реагент], использованный в оптимизированных протоколах:
[PDGFRα]=2-5 нМ, [АТР]=60 µМ и [Y9-Sox пептид]=10 мМ (АТР КМарр=61 мМ)
[FLT-3]=15 нМ, [АТР]=500 мМ и [Y5-Sox пептид]=10 мМ (АТР КМарр=470 мМ)
Масс-спектральный анализ
Flt3 инкубировали с Соединением 4 в течение 3 ч при 100Х избытке перед триптическим гидролизом. Йодацетамид использовался в качестве алкилирующего агента после инкубирования соединения. Для триптического гидролиза аликвоту 5 мкл (7 пмоль) разбавляли при помощи 10 мкл 0,1% ТФК перед микро картридж-колонкой С18 Zip Tipping непосредственно на мишень MALDI с использованием альфа-циано-4-гидрокси коричной кислоты в качестве матрицы (5 мг/мл в 0,1% ТФК: ацетонитрил 50:50).
Аппарат для масс-спектрометрии настроили на режим Reflectron с пульсационным выводом 1800. Калибровку производили с использованием стандарта Laser Biolabs Pep Mix (1046,54, 1296,69, 1672,92, 2093,09, 2465,20). Для анализа CID/PSD пептиды выбирались с использованием курсоров для назначения ионного стробирования и фрагментации, происходившей при мощности лазерного излучения около 20% выше; Гелий использовался в качестве газа для соударений в CID. Калибрование фрагментов производили с использованием P14R фрагментационной калибровки для Рефлектрона искривленного поля.
Модифицированная форма триптического пептида с последовательностью ICDFGLAR (SEQ ID NO: 9) с присоединенным Соединением 4 достигла пика при 1344,73. Контрольный гидролизат не показал свидетельств пика 1344, представляющего модифицированный Соединением 4 пептид.
Пример 4. Необратимо действующий боцепревир
Боцепревир - сильный обратимый ингибитор протеазы вируса гепатита С (ВГС). При помощи основанного на структуре алгоритма разработки, описанного в данном документе, боцепревир был быстро и эффективно преобразован из обратимо действующего в необратимо действующий ингибитор протеазы ВГС.
Координаты рентгеновского комплекса боцепревира и связанной с ним протеазы ВГС (код PDB 20C8) были получены из базы данных белковых структур. Координаты боцепревира извлекались, и все белковые Цис-остатки в пределах 20 ангстремов от боцепревира идентифицировались. В результате было выявлено пять остатков: Cys16, Cys47, Cys52, Cys145 и Cys159. Затем 4 замещаемые позиции на матрице боцепревира (Формула IV-1) исследовали в трех измерениях с целью определить, которая из них может быть замещена на «боеголовку» так, чтобы «боеголовка» образовала ковалентную связь с Цис в сайте связывания боцепревира. Акриламидная «боеголовка» была надстроена в трехмерном пространстве на матрицу боцепревира (Формула IV-1) с использованием программы Accelrys Discovery Studio v2.0.1.7347 (Accelrys Inc. Калифорния). Структура полученных соединений исследовалась, чтобы выяснить, может ли «боеголовка» достичь одного из идентифицированных Цис-остатков в сайте связывания протеазы ВГС.

Для того чтобы проверить гибкость «боеголовки» и позиций боковой цепи, была создана стандартная молекулярно-динамическая симуляция «боеголовки» и позиций боковой цепи с целью выяснить, находится ли «боеголовка» в пределах 6 ангстремов от любого идентифицированного Цис-остатка в сайте связывания. Данная симуляция позволила выявить 2 матричных позиции (R1 и R3), находившихся возле Cys159 (Табл. 11). Затем эти две матричные позиции были подвергнуты финальной фильтрации, критерием которой было формирование продукта реакции между предполагаемым необратимым ингибитором и Cys159, в том числе формирование связи длиной менее 2 ангстремов, с применением стандартной молекулярно-динамической симуляции. В результате этого отсеивания осталась одна матричная позиция: R3. После этого было синтезировано Соединение 5. Биохимический анализ показал IC50_АPP данного Соединения на уровне 1,3 мМ (Оценка Протеазы ВГС FRET); оценка клеточной репликации подтвердила, что Соединение ингибирует репликацию ВГС, при этом значение ЕС50 составило 230 нМ.
Синтез Соединения 5

Соединение 5 было получено с использованием этапов и промежуточных соединений, описанных ниже.
Схема 4-А

Этап 1: Промежуточное соединение 4а

К раствору (1R, 2S, 5S)-3-трет-бутил-2-метил 6,6-диметил-3-азабицикло [3.1.0] гексан-2,3-дикарбоксилата (0,30 г, 1,1 ммоль) в 4 мл ТГФ/ МеОН (1:1) был добавлен 1 N водный раствор LiOH (2,0 мл). После перемешивания при комнатной температуре в течение 10 ч, реакционную смесь нейтрализовали 1,0 N НСl. Органические растворители выпаривали в вакууме и оставшуюся водную фазу подкисляли до рН ~ 3 с помощью 1,0 N НСl и экстрагировали этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. После удаления растворителя было получено 0,28 г Промежуточного соединения 1a: MC m/z (масса к заряду): 254,2 (ES-).
Этап 2: Промежуточное соединение 4b

К раствору продукта Этапа 1 (0,28 г, 1,0 ммоль) и 3-амино-4-циклобутил-2-гидроксибутанамида (0,27 г, 1,3 ммоль) в 10,0 мл безводного ацетонитрила добавили HATU (0,45 г, 1,2 ммоль) и DIEA (0,5 мл, 3,0 ммоль) при комнатной температуре при перемешивании. Тонкослойная хроматография показала завершение реакции сочетания через 10 ч. Далее добавили 50 мл EtOAc и смесь промывали водным раствором NaHCO3 и солевым раствором. Органический слой отделяли и сушили над Na2SO4. После удаления растворителя сырой продукт подвергли хроматографии на силикагеле (элюенты: EtOAc/гексан). Всего было получено 0,4 г титульного соединения (88%). MC m/z: 432,2 (ES+, M+Na).
Этап 3: Промежуточное соединение 4с

Продукт Этапа 2 (0,40 г, 1,0 ммоль) растворяли в 5 мл 4 N НСl в диоксане. Смесь перемешивали при комнатной температуре в течение 1 ч. После удаления растворителей в смесь долили 10 мл DCM с последующим выпариванием досуха. Этот процесс добавления DCM и последующего выпаривания повторяли четыре раза, чтобы получить остаток в виде твердого вещества, которое использовалось непосредственно для следующего Этапа.
MC m/z: 310,1 (М+H+).
Этап 4: Промежуточное соединение 4d

К раствору продукта Этапа 3 (0,10 г, 0,28 ммоль) и (S)-3-(трет-бутоксикарбониламино)-2-(3-трет-бутилуреидо)пропановой кислоты (0,10 г, 0,33 ммоль) в 3,0 мл безводного ацетонитрила добавляли HATU (125 мг, 0,33 ммоль) и DIEA (0,17 мл, 1,0 ммоль) при комнатной температуре при перемешивании. Через 1 ч добавили 15 мл этилацетата и смесь промывали водным раствором NaHCO3 и солевым раствором. Органический слой отделяли и сушили над Na2SO4. После удаления растворителя сырой продукт подвергали хроматографии на силикагеле (элюенты: EtOAc/гексан) с получением 103 мг титульного соединения (60%). МС m/z: 595,2 (М+Н+).
Этап 5: Промежуточное соединение 4е

Продукт Этапа 4 (75 мг, 0,12 ммоль) растворяли в 3 мл 4 N НСl в диоксане и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. После удаления растворителей долили 3 мл DCM и выпаривали досуха. Этот процесс добавления DCM с последующим выпариванием повторяли три раза, чтобы получить светло-коричневое твердое вещество, которое непосредственно использовалось для следующего Этапа.
МС m/z: 495,2 (М+Н+).
Этап 6: Промежуточное соединение 4f

Акриловую кислоту (13,6 мг, 0,19 ммоль) соединяли с продуктом Этапа 5 и с HATU (65 мг, 0,17 ммоль) по методике, описанной в Этапе 2, получая при этом титульное соединение (60 мг сырого продукта). МС m/z: 549,3 (M+H+).
Этап 7: Сырой продукт Этапа 6 (60 мг, 0,11 ммоль) растворяли в 5 мл дихлорметана с последующим добавлением реагента Десс-Мартина (Dess-Martin periodinane) (60 мг, 0,15 ммоль). Полученный раствор перемешивали в течение 1 ч при комнатной температуре. Растворитель удаляли и остаток подвергали хроматографии на силикагеле (элюенты: EtOAc/гептаны), чтобы получить 13 мг Соединения 5. МС m/z: 547,2 (М+H+).
Масс-спектральный анализ
Масс-спектральный анализ ВГС в присутствии Соединения 5 проводился по следующему протоколу: ВГС NS3/4A дикого типа (дт) инкубировали в течение 1 ч при 10Х-кратном избытке Соединения 5 по отношению к белку. Аликвоты образцов 2 мкл разбавляли с помощью 10 мкл 0,1% ТФК перед микрокартридж-колонкой С4 Zip Tipping непосредственно на мишень MALDI с использованием синапиновой кислоты в качестве десорбирующей матрицы (10 мг/мл в 0,1% ТФК:Ацетонитрил 50:50). Аппарат был настроен на линейный режим с пульсационным выводом 24,500 и апомиоглобином в качестве эталона для калибровки оборудования. По сравнению с белком без Соединения 5, белок, инкубированный с Соединением 5, прореагировал в значительной степени, в результате чего образовался новый вид, существенно более тяжелый (547 Да), чем протеаза ВГС, и соответствующий массе Соединения 5, равной 547 Да.
Дополнительный анализ Соединения 5 с использованием мутированной формы протеазы ВГС, в котором Cys159 мутировал до Ser, показал, что Соединение 5 не модифицирует мутантную протеазу ВГС.
Экспрессирование и очистка единичной цепи белка протеазы ВГС (дт)
Одноцепочечный протеолитический домен (NS4A21-32-GSGS-NS33-631) ("GSGS" раскрыт в последовательности SEQ ID NO: 10) был клонирован в pET-14b (Novagen, Мадисон, Висконсин) и трансформирован в клетки DH10B (Invitrogen). Полученная плазмида была перенесена в Escherichia coli BL21 (Novagen) для белкового экспрессирования и очистки способом, описанным выше (1, 2). Вкратце, культуры выращивали при 37°С в среде Лурия-Бертани, содержащей 100 мкг/мл ампициллина, пока оптическая плотность при 600 нм (OD600) не достигла 1.0. Затем культуры подвергали индуцированию, добавляя изопропил-β-D-тиогалактопиранозид (IPTG) до 1 мМ. После дополнительного инкубирования при 18°С в течение 20 ч бактерии собирали центрифугированием при 6,000 × г в течение 10 мин и ресуспендировали в лизирующем буфере, содержащем 50 мМ Na3PO4, рН 8,0, 300 мМ NaCl, 5 мМ 2-меркаптоэтанола, 10% глицерина, 0,5% реагента Igepal CA630 и ингибирующий протеазу коктейль из 1 мМ фенилметилсульфонил фторида, 0,5 мкг/мл лейпептина, пепстатина А и 2 мМ бензамидина. Клетки лизировали путем замораживания и оттаивания, с последующим разрушением ультразвуком. Клеточный детрит удаляли центрифугированием при 12,000 × г в течение 30 мин. Надосадочную жидкость очищали, пропуская через фильтр размером 0,45 мкм (Coming), после чего ее помещали в HiTrap хелатообразующую колонку, обработанную NiSO4 (Amersham Pharmacia Biotech). Связанный белок элюировали с помощью раствора имидазола в линейном градиенте 100-к-500 мМ. Выбранные фракции пропускали через Ni2+ колоночную хроматографию и анализировали на 10% додецилсульфата натрия (SDS)-полиакриламидном геле. Очищенный белок разлагали электрофорезом в 12% SDS-PAGE геле и затем переносили на нитроцеллюлозную мембрану. Белок анализировали способом вестерн-блоттинга с использованием моноклональных антител к NS3. Белки визуализировали с помощью набора для хемилюминесценции (Roche), с козьими антимышиными антителами, конъюгированными с пероксидазой хрена (Pierce) в роли вторичных антител. Белок разделяли на аликвоты и хранили при -80°С.
Клонирование и экспрессирование протеазы ВГС и варианта C159S
Мутантные фрагменты ДНК NS4A/NS3 создавались с помощью полимеразной цепной реакции (ПЦР) и клонировались в экспрессионный вектор рЕТ. После трансформации в BL21 компетентные клетки индуцировалась экспрессия с изопропилтиогалактозидом (IPTG) в течение 2 ч. Гистидин-меченые белки слияния очищали с использованием колонки для аффинной хроматографии, с последующей эксклюзионной хроматографией размеров.
Биохимический анализ
Применялся анализ ВГС Протеазы FRET с использованием ВГС NS3/4A 1b белка (IС50_АРР). Для создания "кажущихся" значений IC50 (IС50_АРР) применялся следующий протокол. Не желая привязываться к какой-либо одной теории, авторы изобретения склонны полагать, что IС50_АРР, сопоставленная с значениями IC50, может служить более целесообразным указателем ингибирования, зависимого от времени, и таким образом является более показательным в отношении связывающей способности. Протокол представляет собой модифицированный основанный на FRET анализ (v_03), разработанный для оценки силы (эффективности) соединения, порядка ранжирования и профиля резистентности по отношению к дикому типу и мутантам C159S, A156S, А156Т, D168A, D168V, R155K протеазных ферментов вируса гепатита С NS3/4A 1b. Протокол состоит в следующем: 10Х стоков NS3/4A протеазных ферментов, приобретенных у Bioenza (Маунтин-Вью, Калифорния), и 1,13Х 5-FAM/QXL™520 FRET пептидного субстрата от Anaspec (Сан-Хосе, Калифорния) приготовили в 50 мМ Трис-HCl, рН 7,5, 5 мМ ДТТ, 2% CHAPS и 20% глицерина. 5 мкл каждого фермента добавляли в Corning (#3575) 384-луночные, черные планшеты для микротитрования (Corning, Нью-Йорк) после нанесения 0,5 мкл объема 50% ДМСО и последовательно разбавляли соединения, приготовленные в 50% ДМСО. Протеазные реакции начинались сразу после добавления фермента, с добавлением 45 мкл FRET субстрата, и контролировались в течение 60-90 мин при λех485/λem520 в Synergy4 планшет-ридере BioTek (Винуски, Вермонт). В конце каждого оценочного анализа прогрессирующие кривые из каждой лунки рассматривались относительно линейной кинетики реакции и статистики пригодности (R2, 95% ДИ, абсолютная сумма квадратов). Начальная скорость (от 0 минут до 15+ минут) из каждой реакции определялась по наклону зависимости относительных единиц флуоресценции vs время (в минутах), а затем строили кривую против концентрации ингибитора в виде процента отсутствия контрольных данных ингибитора и отсутствия контрольных данных фермента с целью оценить кажущийся IC50 от log [Ингибитор] vs Ответ, модель Variable Slope в программе GraphPad Prism производства GraphPad Software (Сан-Диего, СА).
Соединение 5 ингибировало протеазу ВГС с показателем IC50, равным 1,3 мМ, согласно проведенному анализу.
Анализ репликона
Соединения исследовали с целью оценить их противовирусную активность и цитотоксичность соединения, используя репликон-производную активность люциферазы.
В данном анализе использовались клеточные линии ЕТ (luc-ubi-neo/ET), представляющие собой человеческие Huh7 клеточные линии гепатомы, которые содержат РНК-репликон ВГС со стабильным люциферазным (Luc) репортером и три адаптивные мутации культуры клеток.
Клеточная линия ЕТ выращивалась в модифицированной по способу Дульбекко среде Игла (DMEM), 10% фетальной бычьей сыворотки (FBS), 1% пенициллин-стрептомицин (пен-стреп), 1% глутамина, 1% заменимой аминокислоты, 400 мкг/мл G418 в 5% СO2 инкубаторе при 37°С. Все реагенты клеточных культур были получены в Компании Invitrogen (Карлсбад). Клетки трипсинизировали (1% трипсин:ЭДТА) и высаживали по 5×103 клеток/лунку в белые 96-луночные планшеты (Costar), в зависимости от планируемого исследования: анализ количества клеток (цитотоксичность) или антивирусной активности. Соединения добавляли в шести 3-кратных концентрациях каждая и проводили анализ в DMEM, 5% FBS, 1% пен-стреп, 1% глутамина, 1% заменимой аминокислоты. Человеческий интерферон альфа-2b (PBL Biolabs, Нью Брансуик, Нью-Джерси) использовали на каждой стадии в качестве позитивной регуляции соединения. Клетки выдерживали 72 ч после добавления соединения, когда они все еще оставались субконфлюэнтными. Противовирусную активность оценивали, анализируя репликон-производную активность люциферазы при помощи системы Steady-Glo Luciferase Assay System (Promega, Мадисон, Висконсин), соблюдая инструкции производителя. Количество клеток в каждой лунке определяли с помощью Cell Titer Blue Assay (Promega). Профиль соединения получали, рассчитывая соответствующие значения ЕС50 (эффективная концентрация, ингибирующая репликацию вируса на 50%), ЕС90 (эффективная концентрация, ингибирующая репликацию вируса на 90%), IС50 (концентрация, снижающая жизнеспособность клеток на 50%) и SI50 (селективный индекс: EC50/IC50).
Согласно данному анализу. Соединение 5 ингибирует активность вируса при ЕС50_АPP=230 нМ.
Пример 5. Необратимый ингибитор протеазы вируса гепатита С
Соединение V-1 является сильным обратимьм ингибитором протеазы ВГС (IС50_APP 0,4 нМ, согласно биохимическому анализу, описанному в Примере 4).

Координаты рентгеновского комплекса боцепревира, связанного с протеазой ВГС (код PDB 20C8), были получены из базы данных белковых структур (www.rcsb.org). Определялась кристаллическая структура протеазы ВГС в комплексе с более 10 малыми молекулами ингибиторов с пептидной основой, и в способе их связывания наблюдалось существенное сходство. Структура боцепревира использовалась для построения модели структуры V-1 в протеазе ВГС, в приложении Discovery Studio.
Идентифицировались все белковые Цис-остатки в пределах 20 ангстремов от V-1 в созданной модели. Было выявлено пять остатков: Cys16, Cys47, Cys52, Cys145 и Cys159. Затем 4 замещаемые позиции на V-1 (с использованием матрицы V-2) исследовали в трех измерениях с целью определить, которая из них может быть замещена на «боеголовку» так, чтобы «боеголовка» образовала ковалентную связь с идентифицированным Цис-остатком в сайте связывания протеазы ВГС. «Боеголовки» надстраивали в трехмерном пространстве на матрицу (Формула V-2) в приложении Discovery Studio (Accelrys Inc, Калифорния). Структуру полученного соединения анализировали с целью выяснить, сможет ли «боеголовка» достичь одного из идентифицированных Цис-остатков в сайте связывания.
Формула V-2

Для того чтобы проверить гибкость «боеголовки» и позиций боковой цепи, была создана стандартная молекулярно-динамическая симуляция «боеголовок» и позиций боковых цепей с целью выяснить, находится ли «боеголовка» в пределах 6 ангстремов от любого из идентифицированных Цис-остатков в сайте связывания. Данная симуляция позволила выявить две матричные позиции (R1 и R3), находившихся возле Cys159. Затем эти две матричные позиции были подвергнуты финальной фильтрации, критерием которой было формирование продукта акриламидной реакции между предполагаемым ингибитором и Cys159, в том числе формирование связи длиной менее 2 ангстремов, в стандартной молекулярно-динамической симуляции. В результате этого отсеивания осталась одна матричная позиция, R3.
Было синтезировано Соединение 6, которое, как было доказано, является сильным ингибитором протеазы ВГС (IC50 0,4 нМ) и способно модифицировать протеазу ВГС в Cys159 (Фиг.4).
Синтез Соединения 6

N-[(1,1-диметилетокси)карбонил]-3-[(2-пропеноил)амино]-L-аланил-(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R, 2S)-циклопропанкарбоксамид: Титульное соединение было подготовлено в соответствии с этапами и промежуточными соединениями, описанными ниже.
Промежуточное соединение 5-1

Этил-1-[[[(2S, 4R)-1-[(1,1-диметилетокси)карбонил]-4-[(7-метокси-2-фенил-4-хинолинил)окси]-2-пирролидинил]карбонил]амино]-2-этенил-(1R, 2S)-циклопропанкарбоксилат: К раствору (1R, 2S)-1-амино-2-винилциклопропан карбоновой кислоты этиловый эфир толуолсульфокислоты (2,29 г, 7,0 ммоль) и N- Вос (2S, 4R)-(2-фенил-7-метокси хинолин-4-оксо) пролина (3,4 г, 7,3 ммоль) в 100 мл дихлорметана были добавлены HATU (3,44 г, 9,05 ммоль), а затем DIEA (3,81 мл, 21,9 ммоль) при перемешивании.
Смесь перемешивали при комнатной температуре в течение двух часов. После полного израсходования исходных материалов реакционную смесь промывали солевым раствором в два раза и сушили над MgSO4. После удаления растворителя сырой продукт подвергали хроматографии на силикагеле (гексан:этилацетат = 2:1). Было получено 3,45 г титульного соединения: Rf0,3 (этилацетат:гексан = 2:1); МС m/z: 602,36 (M+H+).
Промежуточное соединение 5-2
1-[[[(2S, 4R)-1-[(1,1-диметилетокси)карбонил]-4-[(7-метокси-2-фенил-4-хинолинил)окси]-2-пирролидинил]карбонил]амино]-2-этенил-(1R, 2S)-циклопропанкарбоновая кислота: К раствору продукта Промежуточного соединения 5-1 (1,70 г, 2,83 ммоль) в 140 мл ТГФ/ Н2O /МеОН (9:5:1.5) был добавлен лития гидрокисид моногидрат (0,95 г, 22,6 ммоль). После перемешивания при комнатной температуре в течение 24 ч реакционную смесь нейтрализовали 1,0 N НСl. Органические растворители выпаривали в вакууме, а оставшуюся водную фазу подкисляли до рН~3 с помощью 1,0 N НСl и экстрагировали этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. После удаления растворителя было получено 1,6 г титульного соединения: Rf0,2 (EtOAc:MeOH = 10:1); MC m/z: 574,36 (M+H+).
Промежуточное соединение 5-3

N-(1,1-диметилетокси)карбонил)-(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R, 2S)-пиклопропанкарбоксамид: К раствору продукта Промежуточного соединения 5-2 (1.24g, 2,16 ммоль) в 20 мл ДМФ добавили HATU (0,98 г, 2,58 ммоль) и DIEA (1,43 мл, 8,24 ммоль), смесь перемешивали в течение одного часа, прежде чем добавить раствор из бензолсульфонамида (1,30 г, 8,24 ммоль), DMAP (1,0 г, 8,24 ммоль) и DBU (1,29 г, 8,4 ммоль) в 15 мл ДМФ. Перемешивание продолжали дополнительно в течение четырех часов. Реакционную смесь разбавляли этилацетатом и промывали водным буфером NaOAc (pH ~ 5, 2×10 мл), раствором NaHCO3 и солевым раствором. После сушки над MgSO4 и удаления растворителя чистый продукт осаждали, добавляя одну часть DCM. Фильтрат концентрировали и остаток подвергали хроматографии на силикагеле с использованием гексан/EtOAc (1:1 ~ 1:2). В общей сложности было получено 0,76 г титульного соединения: Rf 0,3 (EtOAc:гексан = 3:1), MC m/z: 713,45 (M+H+), 735,36 (M+Na+).
Промежуточное соединение 5-4

(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R, 2S)-циклопропанкарбоксамид: К раствору продукта Промежуточного соединения 5-3 в 30 мл дихлорметана добавляли по каплям 15 мл ТФК. Смесь перемешивали при комнатной температуре в течение двух часов. После удаления растворителей доливали часть объемом 20 мл DCM, с последующим выпариванием досуха. Этот процесс добавления DCM с последующим выпариванием повторяли четыре раза. Толуол (20 мл) добавляли, а затем удаляли выпариванием досуха. После двукратного повторения этого цикла получили затвердевший остаток массой 0,9 г в виде белого порошка, как ТФК-соль титульного соединения. Маленький образец ТФК-соли был нейтрализован с помощью NaHCO3, в результате чего получили титульное соединение: Rf 0,4 (DCM:MeOH = 10:1); MC m/z: 613,65 (M+H+).
Промежуточное соединение 5-5

N-[(1,1-диметилетокси)карбонил]-3-[[(9Н-флуорен-9-ил метокси)карбонил]амино]-L-аланил-(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R, 2S)-циклопропанкарбоксамид: К раствору продукта Промежуточного соединения 5-4 (0,15 г, 0,178 ммоль) и N-Boc-3-(Fmoc)амино-L-аланина (0,107 г, 0,25 ммоль) в 3,0 мл ДМФ добавили HATU (85,1 мг, 0,224 ммоль) и NMM (90,5 мг, 0,895 ммоль) при комнатной температуре при перемешивании.
ТСХ (тонкослойная хроматография) показала завершение реакции связывания через 1 ч. Затем добавили 20 мл EtOAc и смесь промывали буфером (рН ~ 4, AcONa/АсОН), NaHCO3 и солевым раствором и сушили над MgSO4. После удаления растворителя сырой масляный продукт подвергали хроматографии на силикагеле (элюенты: EtOAc/гексан). В общей сложности было получено 0,12 г титульного соединения: Rf0,4 (EtOAc:гексан = 1:1); МС m/z: 1021,56 (М+Н+).
Промежуточное соединение 5-6

N-[(1,1-диметилетокси)карбонил]-3-амино-L-аланил-(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R, 2S)-циклопропанкарбоксамид: Раствор из 110 мг продукта Промежуточного соединения 5-5 (0,108 ммоль) в 1 мл ДМФ с 12% пиперидина перемешивали в течение 1,5 ч при комнатной температуре, а затем выпаривали досуха при высоком вакууме. Остаток тритурировали с гексаном / эфиром (4:1), получая 70 мг титульного соединения: Rf 0,25 (EtOAc:MeOH = 10:1); МС m/z: 798,9 (M+H+).
Соединение 6

N-[(1,1-диметилетокси)карбонил]-3-[(2-пропеноил)амино]-L-аланил-(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R, 2S)-циклопропанкарбоксамид: Акрилоил хлорид (11 мкл, 0,132 ммоль) добавили по каплям при 0°С к перемешанному раствору из 69 мг продукта Промежуточного соединения 5-6 в 3 мл DCM, содержащего 3 экв. триэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч, а затем разбавляли 10 мл DCM. Полученный раствор промывали дважды солевым раствором и сушили над сульфатом магния. После удаления растворителя получили сырой продукт, который очищали с помощью хроматографии на силикагеле, элюирование производили сначала составом гексан/EtOAc (1:3 ~ 1:5), а затем с DCM-метанол (50:1 ~ 25:1)). В общей сложности было получено 36 мг титульного соединения: Rf 0,25 (DCM:MeOH = 25:1); МС m/z: 892,55 (M+H+).
Масс-спектральный анализ
Масс-спектральный анализ показал наличие модификации протеазы WT, однако не мутанта C159S, который поддерживает специфическую модификацию целевого Cys Соединением 6.
Проводили масс-спектральный анализ ВГС дикого типа или ВГС варианта C159S в присутствии исследуемого соединения. 100 пмоль ВГС дикого типа (Bioenza, Калифорния) инкубировали с Соединением в течение 1 ч и 3 ч при 10-кратном доступе Соединения 6 к белку. Аликвоты 1 мкл образцов (общим объемом 4,24 мкл) разбавляли с помощью 10 мкл 0,1% ТФК перед микрокартридж-колонкой С4 Zip Tipping непосредственно на мишень MALDI с использованием синапиновой кислоты в качестве десорбирующей матрицы (10 мг/мл в 0,1% ТФК:Ацетонитрил 50:50). Анализ производили на Shimadzu Biotech Axima TOF2 (Shimadzu Instruments) времяпролетном масс-спектрометре с лазерной ионизацией и десорбцией из жидкой матрицы (MALDI-TOF). Такую же процедуру произвели для 100 пмоль ВГС C159S мутанта протеазы ВГС в течение 3 ч при 10-кратном избытке Соединения 6 к белку.
Интактный белок ВГС встречался при МН+ 24465 с соответствующими синапинными (матрикс) аддуктами, встречавшимися примерно на 200 Да выше. Встречалось стехиометрическое включение Соединения 6 (MW 852 Да), вызывавшее новый пик массы на 850-860 Да выше (МН+ 25320-25329). (ФИГ.9) Это соответствует инкорпорированию единичной молекулы Соединения 6. Значительная реакция происходила даже через 1 ч при 10х концентрации соединения практически с полной конверсией после 3 ч при 10х концентрации. В вариантной форме C159S фермента не было обнаружено никаких свидетельств модификации, а это подтверждает, что соединение модифицирует Cys159.
Данные масс-спектрального анализа подтвердили, что добавление Соединения 6 к протеазе ВГС приводит к сдвигу по массе 853 Дальтона, что доказывает, что сформирован аддукт протеазы ВГС с соединением. Кроме того, Соединение 6 не образовало аддукт с мутированной формой протеазы ВГС, в которой Cys159 был изменен на серин, как ожидалось, учитывая разную химическую активность Cys и Ser с акриламидной «боеголовкой». Полученные данные показали, что способы, описанные в данном документе, были использованы для разработки специфического необратимого ингибитора протеазы ВГС.
Данные биохимического и клеточного анализа
Соединение 6 испытывали при проведении анализа репликона и биохимического анализа, описанных в Примере 4. Соединение 6 имеет IС50_АPP 2,8 нМ (по результатам биохимического анализа) и ЕС50 174 нМ (по результатам анализа репликона).
Пример 6. Необратимо действующий Сорафениб
Сорафениб - сильный обратимый ингибитор сКIT-киназного домена. При помощи алгоритма разработки, описанного в этом документе, сорафениб был быстро и эффективно преобразован в необратимо действующий ингибитор cKIT.
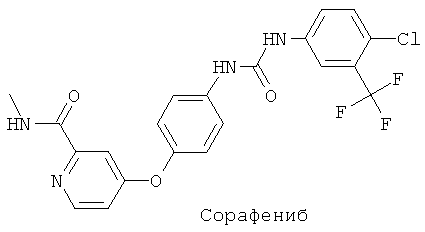
Гомологичная модель cKIT киназы (код Uniprot: P10721) была создана с использованием рентгеновских структур сорафениба, связанного с B-Raf в качестве матрицы (код PDB 1UWH). Гомологичную модель выстраивали с использованием модуля Build Homology программы Discovery Studio, применяя выравнивание cKIT - B-RAF, результаты которого изображены ниже. Затем 10 замещаемых позиций на матрице сорафениба (Формула VI-1) исследовали в трех измерениях с целью определить, которая из них может быть замещена на «боеголовку» так, чтобы «боеголовка» образовала ковалентную связь с Цис в сайте связывания. Данный способ позволил идентифицировать две матричные позиции (R9 и R10) и один Cys (Cys788), которые могут сформировать ковалентную связь с акриламидной «боеголовкой». Связи, возникающие в позиции R9, затрагивали внедрение цис-амида, что было наименее предпочтительно, тогда как связи, возникающие в позиции R10, были способны образовать транс-амид, что является наиболее предпочтительным. Было синтезировано Соединение 7, которое подтвердило важность наличия «боеголовки» в позиции R10.


Синтез Соединения 7
4-(4-(3-(4-Акриламидо-3-(трифторметил)фенил)уреидо)фенокси)-N-метилпиколинамид

Этап 1. C,C′-Bis-трет-бутил N-4-амино-2-трифторметилфенил)иминодикарбонат

К перемешанному раствору 4-нитро-2-трифторметиланилина (4,12 г, 20 ммоль) в 1,4-диоксане (50 мл) добавили 4-DMAP (1,22 г, 10 ммоль) и Вос ангидрид (13,13 г, 50 ммоль) при комнатной температуре. Реакционную смесь нагревали при 110°С в течение 2 ч. Затем реакционную смесь охлаждали, концентрировали при пониженном давлении и остаток растворяли в этилацетате (25 мл). После его промывали 10%-ным раствором лимонной кислоты (5 мл), водой (5 мл) и насыщенным водным NaCl (2 мл). Высушивание над Na2SO4 и концентрирование при пониженном давлении позволили получить остаток, который очищали с помощью колоночной хроматографии (SiO2, 60-120, ПЭТ эфир/этилацетат, 6/4), чтобы получить 5,3 г (13 ммоль) bis-Boc промежуточного соединения в виде бледно-желтого твердого вещества. Этот материал растворяли в 50 мл метанола. К этому раствору в присутствии азотной атмосферы добавили уксусную кислоту (3 мл), затем порошок железа (1,71 г, 19,4 г-атом). Реакционную смесь нагревали при 70°С в течение 2 ч, охлаждали до комнатной температуры и фильтровали через целит®. Фильтрат концентрировали при пониженном давлении и остаток разбавляли этилацетатом (30 мл). Далее его промывали водой (2 мл) и насыщенным водным NaCl (2 мл) и сушили над Na2SO4. После концентрации при пониженном давлении получили остаток, который затем очищали колоночной хроматографией (SiO2, 60-120, ПЭТ эфир/этилацетат, 6/4), получая 3,19 г титульного соединения в виде беловатого твердого вещества.
Этап 2. 4-(4-(3-(4-амино-3-(трифторметил)фенил)уреидо)фенокси)-N-метилпиколинамид

К перемешанному раствору С,С′-bis-трет-бутил N-4-амино-2-трифторметилфенил)иминодикарбоната (0,5 г, 1,32 ммоль) и Et3N (0,6 мл, 5,97 ммоль) в толуоле (5 мл) добавили фосген (20% раствор в толуоле, 0,91 мл, 1,85 ммоль). Реакционную смесь нагревали с обратным конденсированном в течение 16 ч, затем охлаждали до комнатной температуры. Добавили 4-(4-аминофенокси)-N-метил-2-пиридинкарбоксамид (0,32 г, 1,32 ммоль) и нагревали реакционную смесь с обратным конденсированием в течение 2 ч. После этого реакционную смесь гасили водой (5 мл) в вытяжном шкафу, экстрагировали этилацетатом (2×20 мл). Этилацетатный экстракт промывали насыщенным водным NaCl (15 мл), сушили над NO2SO4 и концентрировали при пониженном давлении, получая 0,62 г титульного соединения.
Этап 3. 4-(4-(3-(4-Акриламидо-3-(трифторметил)фенил)уреидо)фенокси)-N-метилпиколинамид

К перемешанному раствору 4-(4-(3-(4-амино-3-(трифторметил)фенил)уреидо)фенокси)-N-метилпиколинамида (0,1 г, 0,22 ммоль) и пиридина (0,035 г, 0,45 ммоль) в ДМФ (5 мл) добавили акрилоил хлорид (0,03 г, 0,33 ммоль) при 0°С. Реакционной смеси дали нагреться до комнатной температуры и далее перемешивали в течение 12 ч, после чего гасили ледяной водой (10 мл) и экстрагировали этилацетатом (2×20 мл). Этилацетатный экстракт промывали насыщенным водным раствором NaCl (5 мл), сушили над Na2SO4 и концентрировали при пониженном давлении, чтобы получить сырой CNX-43. Сырой продукт очищали сначала с помощью колоночной хроматографии с нейтральным оксидом алюминия, а затем подготовительной ВЭЖХ, получая 18 мг титульного соединения в виде белого твердого вещества.1Н ЯМР (MeOD) δ ppm: 2,94 (s, 3Н), 5,82 (d, J=10,0 Гц, 1H), 6,37 (dd, J=1,76 & 17,16 Гц, 1Н). 6,50 (dd, J=10,28 & 17,16 Гц, 1H), 7,06 (dd, J=2,6 & 5,94 Гц, 1H), 7,11-7,15 (m, 2H), 7,45 (d, J=8,64 Гц, 1H), 7,56-7.61 (m, 3Н), 7,67 (dd, J=2,24 & 8,48 Гц, 1H), 8,0 (s, 1H), 8,45-8,55 (m, 1H); ЖХМС: m/е 501 (М+2)
БИОХИМИЧЕСКОЕ ТЕСТИРОВАНИЕ
Сорафениб обладает IС50 50,5 нМ против ингибирования фосфорилирования cKIT, тогда как Соединение 7 имеет IС50 31 нМ против ингибирования фосфорилирования cKIT. Биохимическое тестирование проводилось по способу, описанному в Примере 1 для cKIT.
Клеточный анализ GIST882
Клетки GIST882 высевали в 6-луночный планшет при плотности 8×105 клеток/лунка в полной среде. На следующий день производили обработку клеток 1 мМ соединения, разбавленного в полной среде, в течение 90 мин. Через 90 мин среду удаляли, а клетки промывали средой, не содержащей Соединения. Клетки промывали каждые 2 ч и ресуспендировали в свежей среде, не содержащей Соединения. Сбор клеток осуществлялся в определенные временные точки, клетки лизировали в экстракционном буфере Cell Extraction Buffer (Invitrogen FNN0011), в который были добавлены Roche - цельные ингибирующие протеазу таблетки (Roche 11697498001) и ингибиторы фосфатазы (Roche 04906837001). Лизаты разделяли, пропуская их через шприц на 28,5 gauge, по 10 раз каждый. Далее измеряли концентрацию белка и загружали по 10 мкг общего белкового лизата в каждую дорожку. Фосфорилирование cKIT оценивали путем вестерн-блоттинга с антителами pTyr (4G10) и общих антител kit производства Cell Signaling Technology.
Сорафениб и Соединение 7 исследовали в отношении клеточной активности на клеточных линиях GIST882 при 1 мкмоль. Оба соединения ингибировали аутофосфорилирование cKIT, а также клеточную сигнализацию «downstream» регулируемой внеклеточными сигналами киназы. Чтобы выявить, оказывает ли необратимый ингибитор пролонгированное угнетение, клетки промывали без соединения. В случае с Сорафенибом ингибирующее действие ckit и downstream-сигнализация было истощено, тогда как необратимое ингибирование, оказываемое Соединением 7, сохранялось по меньшей мере 8 ч. Эти данные подтверждают превосходство необратимо действующего ингибитора Соединения 7 по продолжительности действия, по сравнению с обратимым ингибитором Сорафенибом.
Масс-спектральный анализ
с-КIТ (15 пмоль) инкубировали с Соединением 7 (150 пмоль) в течение 3 ч при 10Х доступе перед триптическим гидролизом. Йодацетамид использовали в качестве алкилирующего агента после инкубации соединения. Также был подготовлен контрольный образец, в который не было добавлено Соединение 7. Для триптического гидролиза аликвоту 2 мкл (3,3 пмоль) разбавляли 10 мкл 0,1% ТФК перед микрокартридж-колонкой С18 Zip Tipping непосредственно на мишень MALDI с использованием альфа-циано-4-гидрокси коричной кислоты в качестве матрицы (5 мг/мл в 0,1% ТФК:Ацетонитрил 50:50).
Оборудование:
Для триптического гидролиза оборудование настроили на режим Reflectron с пульсационным выводом 2200. Калибровку производили с использованием стандарта (1046,4, 1296,9, 1672,2, 2093,9, 2465,0). Для анализа CID/PSD пептиды выбирались с использованием курсоров для назначения ионного стробирования и фрагментации, происходившей при мощности лазерного излучения около 20% выше; Гелий использовался в качестве газа для соударений в CID. Калибрование фрагментов производили с использованием P14R фрагментационной калибровки для Рефлектрона искривленного поля.
Пептид, который должен был быть модифицирован Соединением 7, имеет последовательность NCIHR (SEQ ID NO: 8) и наблюдался при МН+ 1141,5. (Моноизотопная масса Соединения 7 составляла 499,15.) Для сравнения, контрольный гидролизат cKIT, не содержавшего Соединение 7, продемонстрировал полное отсутствие этого пика массы. Полученные данные также позволяют предположить существование модификации пептида пептида, имеющего последовательность ICDFGLAR (SEQ ID NO: 9).
Пример 7. Необратимо действующий ингибитор протеазы вируса гепатита С
Как описано в Примере 5, Соединение V-1 является сильным обратимо действующим ингибитором протеазы ВГС. Используя смоделированную структуру V-1 в протеазе ВГС (См. Пример 5), были идентифицированы все белковые Цис-остатки в пределах 20 ангстремов от V-1 в данной модели. Таких остатков было пять: Cys16, Cys47, Cys52, Cys145 и Cys159. Затем 4 замещаемые позиции на V-1, которые могли быть замещены на еноновую «боеголовку» так, чтобы «боеголовка» образовала ковалентную связь с идентифицированным Цис-остатком в сайте связывания протеазы ВГС, исследовали в трехмерном пространстве. «Боеголовки» надстраивали в трехмерном пространстве на матрицу (Формула V-2) в программе Discovery Studio (Accelrys Inc, Калифорния), и структуру полученного соединения исследовали с целью выяснить, может ли «боеголовка» достичь одного из идентифицированных Цис-остатков в сайте связывания.
Для того чтобы проверить гибкость «боеголовки» и позиций боковой цепи, была создана стандартная молекулярно-динамическая симуляция «боеголовок» позиций боковых цепей с целью выяснить, находится ли «боеголовка» в пределах 6 ангстремов от любого из идентифицированных Цис-остатков в сайте связывания. Данная симуляция позволила выявить две матричные позиции (R1 и R3), находившихся возле Cys159. Затем эти две матричные позиции были подвергнуты финальной фильтрации, критерием которой было формирование продукта еноновой реакции между предполагаемым ингибитором и Cys159, а именно формирование связи длиной менее 2 ангстремов (в стандартной молекулярно-динамической симуляции). В результате этого отсеивания осталась только одна матричная позиция, R3.
Было синтезировано Соединение 8. Было показано, что оно является сильным ингибитором протеазы ВГС (IС50_АPP<0,5 нМ) и что оно модифицирует протеазу ВГС на Cys159.

Синтез Соединения 8
Соединение 8 трет-бутил-(S)-1-((2S,4R)-2-((1R,2S)-1-(циклопропилсульфонилкарбамоил)-2-винилциклопропилкарбамоил)-4-(7-метокси-2-фенилхинолин-4-ил окси)пирролидин-1-ил)-7-метил-1,5-диоксоокт-6-ен-2-илкарбамат:
Титульное соединение было подготовлено в соответствии с этапами промежуточными соединениями, описанными ниже:

Промежуточное соединение 8-1:
К раствору Промежуточного соединения 5-2 (0,9 г, 1,57 ммоль) в 6 мл ДМФ добавили CDI (0,28 г, 1,7 ммоль). Смесь перемешивали в течение одного часа, прежде чем добавить раствор из циклопропилсульфонамида (0,25 г, 2,0 ммоль), DBU (0,26 мл, 1,8 ммоль) и DIEA (0,9 мл, 5 ммоль) в 2 мл ДМФ. Полученную смесь перемешивали при 60°С в течение 10 ч. Реакционную смесь разбавляли этилацетатом и промывали водным буфером NaOAc (pH ~ 5, 2×10 мл), раствором NaHCO3 и солевым раствором. После сушки над Na2SO4 и удаления растворителя остаток подвергали хроматографии на силикагеле с использованием гексана/EtOAc (1:1 ~ 1:2). Всего было получено 0,8 г Промежуточного соединения 8-1: Rf0,3 (EtOAc гексан = 3:1), МС m/z: 677,2 (М+Н+).
Промежуточное соединение 8-2:
Промежуточное соединение 8-1 (0,8 г, 1,18 ммоль) растворяли в 5 мл 4 N НСl в диоксане, реакционную смесь перемешивали в течение 1 ч при комнатной температуре. После удаления растворителей доливали порцию 20 мл DCM, с последующим выпариванием досуха. Этот процесс добавления DCM с последующим выпариванием повторяли три раза, чтобы получить Промежуточное соединение 8-2 в виде его HCl-соли. МС m/z: 577,2 (М+Н+).
Промежуточное соединение 8-3:
К раствору N-Boc-пироглутаминовой кислоты (0,23 г 1,0 ммоль) в 10,0 мл безводного ТГФ медленно добавляли 2-метилпроп-1-енил) магний бромид (0,5 М в ТГФ, 5 мл, 2,5 ммоль) при -78°С. Реакционную смесь перемешивали в течение 1 ч при -78°С. Затем добавили водный раствор 1 N НСl (2,5 мл) и смесь медленно нагревали до комнатной температуры, РН доводили до ~ 3 при помощи 1 N НСl. ТГФ удаляли в вакууме, а оставшуюся водную фазу экстрагировали с помощью DCM (3X 20 мл). Органический слой сушили над Na2SO4, фильтровали и растворитель удаляли, получая сырой продукт.
Соединение 8:
трет-бутил-(S)-1-((2S,4R)-2-((1R,2S)-1-(циклопропилсульфонилкарбамоил)-2-винилциклопропилкарбамоил)-4-(7-метокси-2-фенилхинолин-4-ил окси)пирролидин-1-ил)-7-метил-1,5-диоксоокт-6-ен-2-ил карбамат:
Титульное соединение было получено путем связывания Промежуточного соединения 8-2 с Промежуточным соединением 8-3, с использованием HATU после реакции связывания, описанной для Промежуточного соединения 5-5 в разделе «Синтез Соединения 6».
Всего было получено 70 мг титульного соединения (65%): Rf0,5 (EtOAc);
МС m/z: 844,2 (M+H+).
Биохимические данные
Соединение 8 исследовали в биохимическом анализе, описанном в Примере 4. Было показано, что данное соединение является сильным ингибитором протеазы ВГС
(IС50_АРР<0,5 нМ).
Масс-спектральный анализ
Масс-спектральный анализ производили способом, описанным в Примере 5. Результаты подтверждают, что добавление Соединения 8 к протеазе ВГС приводит к сдвигу по массе приблизительно на 844 Дальтона, что доказывает, что сформирован аддукт протеазы ВГС с соединением (ФИГ.11).
Пример 8. Усиление активности при помощи ковалентной связи.
На данном Примере показано практическое применение алгоритма и способа разработки сильных необратимо действующих ингибиторов на основе обратимо действующих ингибиторов с умеренной или слабой активностью.
8.А. Ингибитор Btk-киназы
: Соединение 9 является слабым обратимым ингибитором Btk киназы (IС50 8,6 мМ, согласно биохимическому анализу). При помощи основанного на структуре алгоритма разработки, описанного в этом документе, Соединение 9 было быстро и эффективно преобразовано в необратимо действующий ингибитор Btk.

Соединение 9
Данные о режиме связывания Соединения 9 в Btk были получены способом стыковки, с использованием апо-структуры Btk (код PDB: 1K2P) и ко-кристаллической структуры ингибитора EGFR (код PDB: 2RGP) с белок0моделирующим компонентом в приложении Discovery Studio (Discovery Studio v2.0-1.7347, Accelrys Inc).
Модель связывания Соединения 9 с Btk позволила идентифицировать пять Цис-остатков, находившихся в пределах 20 ангстремов (Cys464, Cys481, Cys502, Cys506 и Cys527) от Соединения 9. Однако анализ в трехмерном пространстве показал, что четыре (Cys464, Cys502, Cys506 и Cys527) из пяти цистеинов блокируются боковыми цепями или остовом белка. Однако доступ к этим цистеинам затруднен из-за стерических столкновений. Таким образом, в пределах досягаемости был только один цистеин (Cys481), находящийся на приемлемом расстоянии. Одну замещаемую позицию на матрице Соединения 9 исследовали в трехмерном пространстве (R1) в Формуле VIII-1). «Боеголовку» (акриламид) надстраивали на матрицу Соединения 9 в программе Discovery Studio. Структуру полученного соединения стыковали с Btk при помощи Accelrys Discovery Studio v2.0.1.7347 (Accelrys Inc). Финальная трехмерная структура исследовалась с целью выяснить, способна ли «боеголовка» достичь Цис в сайте связывания (т.е. отстояла бы не далее чем 6 ангстремов от Цис).
Формула VIIIA-1

Такой подход подтвердил, что выбранная позиция R1 на матрице Соединения 9 расположена возле Cys481, на расстоянии менее 6 ангстремов. Между предполагаемым ингибитором и Cys481 был сформирован продукт акриламидной реакции, включая образование связи длиной менее 2 ангстремов, в стандартной молекулярно-динамической симуляции. Выбранная позиция успешно удовлетворяла данному ограничительному условию.
Применяя способы и оценочный анализ, описанные ниже, авторы изобретения синтезировали Соединение 10, которое содержит акриламид в позиции R1. Было показано, что оно является сильным ингибитором Btk-киназы, с показателем IС50 1,8 нМ, по данным биохимического анализа. Таким образом, очевидно значительное улучшение активности по сравнению с Соединением 9 (IС50 8,6 мМ). Активность Соединения 10 также оценивалась с использованием Ramos-клеток. Поскольку Соединение 9 оказалось достаточно слабым ингибитором Btk, согласно биохимическому анализу, авторы не ожидали получить данные о его ингибирующей активности в данном анализе клеток. Тем не менее, при концентрации 1 мМ, Соединение 10 продемонстрировало 85% ингибирования сигнализации Btk в Ramos-клетках, Полученные данные подтверждают, что алгоритм и способ изобретения позволили усилить активность слабого обратимо действующего ингибитора (Соединение 9), и что сильный необратимый ингибитор (Соединение 10) обладает активностью в клетках.
Синтез Соединения 10
N-{3-[6-(4-феноксифениламино)-пиримидин-4-ил амино]фенил}-2 пропенамид

Соединение 10
Раствор 3-[6-(4-феноксифениламино)-пиримидин-4-ил амино] фениламина (250 мг, 0,7 ммоль) и триэтиламина (180 мг, 1,75 ммоль) в 5 мл ТГФ перемешивали при комнатной температуре. Акрилоил хлорид (80 мг, 0,9 ммоль) добавили к реакционной смеси и перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляют вакуумным испарением, сырой продукт очищали с помощью флэш-хроматографии на силикагеле с EtOAc/DCM системой растворителей, с получением 115 мг (выход 40%) Титульного соединения в виде светлого твердого тела. МС (m/z): МН+ = 424.1Н ЯМР (ДМСО): 9,14 (s, 1Н), 9,10 (s, 1H), 8,22 (s, 1H), 7,89 (s, 1H), 7,52 (d, 2H, J=9,0 Гц), 7,35 - 6.92 (m, 11H), 6,42 (dd, 1H, J1=10,1 Гц, J2=16,9 Гц), 6,22 (dd, 1H, J1=1,9 Гц, J2=16,9 Гц), 6,12 (s, 1H), 5,70 (dd, 1H, J1=1,9 Гц, J2=10,1 Гц) ppm.
Протокол общего анализа для оценки активности против Btk
Нижеследующий протокол описывает непрерывно считываемый анализ киназ, проведенный для измерения специфической активности соединения против активных форм Btk-ферментов. Механизм проведения анализа лучше всего описан производителями (Invitrogen, Карлсбад, Калифорния) и приведен на его веб-сайте:
www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/Drug-Discovery/Target-and-Lead-Identification-and-Validation/KinaseBiology/KB-Misc/Biochemical-Assays/Omnia-Киназ-Assays.html. Вкратце 10Х сток 5 нМ Btk от Invitrogen, 1.13Х 40 мМ АТФ (AS001A) и 10 мМ (АФТ КМарр ~ 36 мМ) Tyr-Sox конъюгированный пептидный субстрат (KCZ1001) приготовили в 1X киназном реакционном буфере, который состоит из 20 мМ Трис, рН 7,5, 5 мМ MgCl2, 1 мМ ЭГТА, 5 мМ β-глицерофосфата, 5% глицерина (10Х сток, КВ002А) и 0,2 мМ ДТТ (DS001A). 5 мкл фермента предварительно инкубировали в лунках Coming (#3574) 384-луночных, белых титрационных микропланшетах с несвязывающей поверхностью (Corning, Нью-Йорк) в течение 30 мин при 27°С с 0,5 мкл объема 50%-ного ДМСО и серийно разбавленного соединения, приготовленного в 50% ДМСО. Киназные реакции были начаты с добавлением 45 мкл ATP/Tyr-Sox пептидной субстратной смеси и контролировались каждые 30-90 секунд в течение 60 мин при λех360/λem485 на планшет-ридере Synergy4 компании BioTek (Винуски, Вермонт). В конце каждого оценочного анализа прогрессирующие кривые для каждой лунки рассматривались относительно линейной кинетики реакции и статистики пригодности (R2, 95% ДИ, абсолютная сумма квадратов). Начальная скорость (от 0 минут до ~30 минут) из каждой реакции определялась по наклону зависимости относительных единиц флуоресценции vs время (в минутах), а затем строили кривую против концентрации ингибитора с целью оценить IC50 от log[Ингибитор] vs Ответ, модель Variable Slope в приложении GraphPad Prism производства GraphPad Software (Сан-Диего, СА).
Btk анализ Ramos-клеток
Соединение CNX-85 испытывали на Ramos человеческих клетках лимфомы Беркитта. Ramos-клетки выращивали в суспензии в колбах Т225, направляли вниз, ресуспендировали в 50 мл бессывороточной среды и инкубировали в течение 1 ч. Исследуемое Соединение добавили в Ramos-клетки cells в бессывороточной среде до итоговой концентрации 1, 0,1, 0,01 или 0,001 мМ. Ramos-клетки инкубировали с Соединением в течение 1 ч, снова промывали и ресуспендировали в 100 мкл бессывороточной среды. Затем клетки стимулировали с помощью 1 мкг козьего F(ab′)2 Анти-человеческого IgM и инкубировали на льду в течение 10 мин, чтобы активировать сигнальный путь клеточных рецепторов В. Через 10 мин клетки промывали сначала PBS, а затем лизировали на льду с помощью клеточного экстракционного буфера Invitrogen Cell Extraction buffer. Далее 16 мкг общего белка из лизата помещали на гель и пятна тестировали на фосфорилирование субстрата Btk, препаративная тонкослойная хроматография PLCγ2.
8.В. Ингибитор Протеазы ВГС
Соединение 11 - слабый обратимый ингибитор протеазы ВГС (IC50 165 нМ, по данным биохимического анализа). При помощи основанного на структуре алгоритма разработки, описанного в данном документе, Соединение 11 было быстро и эффективно преобразовано в необратимый ингибитор протеазы ВГС.
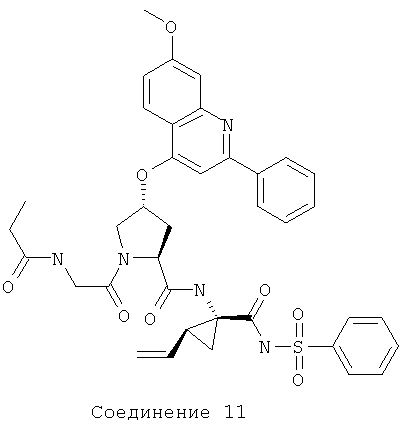
Определялась кристаллическая структура протеазы ВГС в комплексе с более 10 малыми молекулами ингибиторов с пепетидной основой, и в способе связывания ингибиторов наблюдалось существенное сходство структуры. Рентгеновские структуры комплекса с боцепревиром (код PDB 20C8) были получены в базе данных белковых структур (www.rcsb.org) и использованы для построения моделей структуры Соединения 11 в протеазе ВГС с использованием приложения Discovery Studio.
Были идентифицированы все Цис-остатки протеазы ВГС в пределах 20 ангстремов в пристыкованном соединении модели. Таких остатков было пять: Cys16, Cys47, Cys52, Cys145 и Cys159. После этого одну замещаемую позицию на матрице Соединения 11 (R1 в Формуле VIIIB-1) исследовали в трехмерном пространстве, чтобы выяснить, может ли она быть замещена на «боеголовку» так, чтобы «боеголовка» образовала ковалентную связь с идентифицированным Цис-остатком в сайте связывания протеазы ВГС. «Боеголовка» (акриламид) была надстроена в трехмерном пространстве на матрицу (Формула VIIIB-1), а остальная часть обратимого ингибитора была оставлена без изменений. Затем проверяли структуру полученного соединения с целью определить, способна ли «боеголовка» достичь одного из идентифицированных Цис-остатков в сайте связывания.

Для того чтобы проверить гибкость «боеголовки» и позиций боковой цепи, была создана стандартная молекулярно-динамическая симуляция «боеголовки» и позиций боковых цепей с целью выяснить, находится ли «боеголовка» в пределах 6 ангстремов от любого из идентифицированных Цис-остатков в сайте связывания. Такой подход подтвердил, что позиция R1 на матрице находилась возле Cys159. Затем эта позиция была подвергнута финальной фильтрации, критерием которой было формирование продукта акриламидной реакции между предполагаемым ингибитором и Cys159, в том числе формирование связи длиной менее 2 ангстремов, с использованием стандартной молекулярно-динамической симуляции. Этим ограничительным условиям удовлетворяло Соединение 12.
Как будет показано ниже, синтезированное Соединение 12 является сильным ингибитором протеазы ВГС.
Синтез Соединения 12
(2S,4R)-1-(2-акриламидоацетил)-4-(7-метокси-2-фенилхинолин-4-ил окси)-N-((1R,2S)-1-(фенилсульфонилкарбамоил)-2-винилциклопропил)пирролидин-2-карбоксамид:

Титульное соединение было подготовлено в соответствии с этапами и промежуточными соединениями, описанными ниже.
Промежуточное соединение 8. В-1

Этил[[(2S,4R)-1-[(1,1-диметилетокси)карбонил]-4-[(7-метокси-2-фенил-4-хинолинил)окси]-2-пирролидинил]карбонил]амино]-2-этенил-(1R,2S)-циклопропанкарбоксидат: К раствору (1R, 2S)-1-амино-2-винилциклопропан карбоновой кислоты этиловый эфир толуолсульфокислоты (2,29 г, 7,0 ммоль) и N-Вос (2S, 4R)-(2-фенил-7-метокси хинолин-4-оксо)пролина (3,4 г, 7,3 ммоль) в 100 мл дихлорметана добавили HATU (3,44 г, 9,05 ммоль), а затем DIEA (3,81 мл, 21,9 ммоль) при перемешивании. Смесь перемешивали при комнатной температуре в течение двух часов. После полного израсходования исходных материалов реакционную смесь промывали солевым раствором в два раза и сушили над MgSO4. После удаления растворителя сырой продукт подвергался хроматографии на силикагеле (гексан:EtOAc = 2:1). Было получено 3,45 г титульного соединения: Rf 0,3 (EtOAc; гексан = 2:1); МС m/z: 602,36 (M+H+).
Промежуточное соединение 8. В-2

1-([[(2S,4R)-1-[(1,1-диметилетокси)к:арбонил]-4-[(7-метокси-2-фенил-4-хинолинил)окси]-2-пирролидинил]карбонил]амино]-2-этенил-(1R,2S)-циклопропанкарбоновая кислота: К раствору продукта Промежуточного соединения 8. В-1 (1,70 г, 2,83 ммоль) в 140 мл ТГФ/Н2O /МеОН (9:5:1.5) добавили лития гидроксид моногидрат (0,95 г, 22,6 ммоль). После перемешивания при комнатной температуре в течение 24 ч реакционную смесь нейтрализовали 1,0 N НСl. Органические растворители выпаривали в вакууме, а оставшуюся водную фазу подкисляли до рН~3 с помощью 1,0 N НСl и экстрагировали этилацетатом. Органический слой промывали солевым раствором и сушили над безводным сульфатом магния. После удаления растворителя было получено 1,6 г титульного соединения: Rf 0,2 (EtOAc:MeOH = 10:1); MC m/z: 574,36 (M+H+).
Промежуточное соединение 8. В-3

N-(1,1-диметилетокси)карбонил)-(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R,2S)-циклопропанкарбоксамид: К раствору продукта Промежуточного соединения 8.В-2 (1,24 г, 2,16 ммоль) в 20 мл ДМФ добавили HATU (0,98 г, 2,58 ммоль) и DIEA (1,43 мл, 8,24 ммоль), смеси перемешивали в течение одного часа, прежде чем добавить раствор из бензолсульфонамида (1,30 г, 8,24 ммоль), DMAP (1,0 г, 8,24 ммоль) и DBU (1,29 г, 8,4 ммоль) в 15 мл ДМФ. Перемешивание продолжали дополнительно в течение четырех часов. Реакционную смесь разбавляли этилацетатом и промывали водным буфером NaOAc (pH ~ 5, 2×10 мл), раствором NaHCO3 и солевым раствором. После сушки над MgSO4 и удаления растворителя чистый продукт осаждали, добавляя одну порцию DCM. Фильтрат концентрировали и остаток подвергали хроматографии на силикагеле с использованием гексана/EtOAc (1:1-1:2). Всего было получено 0,76 г титульного соединения: Rf 0,3 (EtOAc гексан = 3:1), MC m/z: 713,45 (М+Н+), 735,36 (M+Na+).
Промежуточное соединение 8. В-4

(4R)-4-[(7-метокси-2-фенил-4-хинолинил)окси]-L-пролил-1-амино-2-этенил-N-(фенилсульфонил)-(1R,2S)-циклопропанкарбоксамид: К раствору продукта Промежуточного соединения 8. В-3 в 30 мл DCM добавили по каплям 15 мл ТФК. Смесь перемешивали при комнатной температуре в течение двух часов. После удаления растворителей добавляли порцию 20 мл DCM с последующим выпариванием досуха. Этот процесс добавления DCM с последующим выпариванием повторяли четыре раза. Толуол (20 мл) добавляли, а затем удаляли выпариванием досуха. Два повторения этого цикла позволили получить остаток, который застыл в 0,9 г белого порошка, как ТФК-соль титульного соединения. Маленький образец ТФК-соли нейтрализовали NaHCO3, чтобы получить титульное соединение: Rf0,4 (DCM:MeOH = 10:1); МС m/z: 613,65 (M+H+).
Промежуточное соединение 8. В-5

(2S,4R)-1-(2-t-бутоксикарбониламиноацетил)-4-(7-метокси-2-фенилхинолин-4-ил окси)-N-((1R,2S)-1-(фенилсульфонилкарбамоил)-2-винилциклопропил)пирролидин-2-карбоксамид: К раствору продукта Промежуточного соединения 8. В-4 (0,10 г, 0,15 ммоль) и N-Boc-глицина (0,035 г, 0,20 ммоль) в 3,0 мл ацетонитрила добавили HATU (85,1 мг, 0,22 ммоль) и DIEA (0,09 мл, 0,5 ммоль) при комнатной температуре при перемешивании. Реакционную смесь перемешивали в течение 2 ч. ЖХ-МС и ТСХ анализ показали завершение реакции связывания. Далее добавляли 20 мл этилацетата и смесь промывали буфером (рН~4, AcONa/АсОН), NaHCO3 и солевым раствором и сушили над Na2SO4. После удаления растворителя сырой продукт подвергали хроматографии на силикагеле (элюенты: EtOAc/гексан). В общей сложности 0,11 г соединения титульного был получен растворителя, the crude product was subject to chromatography on silica gel (eluents: EtOAc/гексан). Всего было получено 0,11 г титульного соединения: Rf 0,2 (EtOAc гексан = 2:1); МС m/z: 770,3 (М+Н+).
Промежуточное соединение 8. В-6

[0001] (2S,4R)-1-(2-аминоацетил)-4-(7-метокси-2-фенилхинолин-4-ил окси)-N-((1R,2S)-1-(фенилсульфонилкарбамоил)-2-винилциклопропил)пирролидин-2-карбоксамид: Продукт Промежуточного соединения 8. В-5 (0,11 г, 0,13 ммоль) растворяли в 2 мл 4 N НСl в диоксане и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. После удаления растворителей порцию 3 мл DCM добавляли с последующим выпариванием досуха. Этот процесс добавления DCM с последующим выпариванием досуха повторяли три раза, чтобы получить Промежуточное соединение 6 в виде его НСl соли (0,10 г). МС m/z: 670.2 (М+Н+).
Соединение 12 (CNX-221)

(2S,4R)-1-(2-акриламидоацетил)-4-(7-метокси-2-фенилхинолин-4-ил окси)-N-((1R,2S)-1-(фенилсульфонилкарбамоил)-2-винилциклопропил)пирролидин-2-карбоксамид(I-27); Титульное соединение было получено путем связывания Промежуточного соединения 8. В-6 и акриловой кислоты при помощи HATU с последующей реакцией связывания, описанной для Промежуточного соединения 8. В-5. Всего было получено 0,10 г титульного соединения 87%: Rf0,5 (5% МеОН в DCM); MC m/z: 724,3 (М+Н+).
Данные биохимического и клеточного анализов
Соединение 12 тестировали во время анализа репликона, описанного в Примере 4. Соединение 12 имеет ЕС50 204 нМ, согласно анализу, тогда как обратимо действующее Соединение 11 имело ЕС50 более чем 3000 нМ, согласно анализу.
FRET-анализ протеазы ВГС для дикого типа и мутированных NS3/4A 1b ферментов (IC50)
Протокол представляет собой модифицированный FRET-анализ (v_02), описанный в работе In Vitro Resistance Studies of ВГС Serine Protease Inhibitors, 2004, JBC, vol. 279, No.17, pp 17508-17514. Специфическая активность соединения оценивалась против A156S, А156Т, D168A и D168V мутантов протеазы ВГС NS3/4A 1b фермента таким образом: 10Х сток NS3/4A протеазного фермента производства Bioenza (Маунтин-Вью, Калифорния) и 1.13Х 5-FAM/QXL™520 FRET пептидного субстрата производства Anaspec (Сан-Хосе, Калифорния) было подготовлено в 50 мМ HEPES, рН 7,8, 100 мМ NaCl, 5 мМ ДТТ и 20% глицерина. 5 мкл каждого фермента предварительно инкубировали в Coming (#3573) 384-луночных, черных, не обработанных планшетах для микротитрования (Corning, Нью-Йорк) в течение 30 мин при 25°С с 0,5 мкл объема 50% ДМСО и серийно разбавленным соединением, приготовленным в 50% ДМСО. Протеазные реакции были начаты с добавлением 45 мкл FRET субстрата и контролировались в течение 120 мин. при λех487/λem514 при помощи Quad4 монохроматоров на планшет-ридере Synergy4 производства BioTek (Винуски, Вермонт). В конце каждого оценочного анализа прогрессирующие кривые для каждой лунки рассматривались относительно линейной кинетики реакции и статистики пригодности (R2, абсолютная сумма квадратов). Начальная скорость (от 0 минут до 30+ минут) из каждой реакции определялась по наклону зависимости относительных единиц флуоресценции vs время (в минутах), а затем строили кривую против концентрации ингибитора с целью оценить IC50 от log[Ингибитор] vs Ответ, модель Variable Slope в приложении GraphPad Prism производства GraphPad Software (Сан-Диего, Калифорния). Результаты представлены в Табл. 12.
Данные свидетельствуют о том, что Соединение 12, которое содержит «боеголовку», ковалентно связанную с протеазой ВГС, является сильным ингибитором дикой или мутантной протеазы ВГС, тогда как обратимо действующее Соединение 11 таковым не является.
Идеи всех патентов, опубликованных приложений и ссылок, процитированных здесь, включены в данный документ посредством ссылок в полном объеме. Данное изобретение было описано и проиллюстрировано на конкретных Примерах его воплощения, однако любой специалист в данной области понимает, что возможны некоторые изменения формы и деталей, без отклонения от объема изобретения, охватываемого прилагаемой формулой изобретения.
Реферат
Изобретение относится к области биотехнологии и может быть использовано для разработки необратимого ингибитора, ковалентно связывающего целевой полипептид. Указанный способ включает: а) представление структурной модели обратимого ингибитора или модели сайта связывания в целевом полипептиде, b) идентификацию Цис-остатка в сайте связывания целевого полипептида, с) предоставление структурной модели предполагаемого необратимого ингибитора, который потенциально ковалентно связывается с целевым полипептидом, где ингибитор содержит «боеголовку», или предоставление структурной модели группы-«боеголовки», d) определение, находится ли реакционноспособная группа «боеголовки» предполагаемого ингибитора в пределах расстояния образования связи с Цис-остатком в сайте связывания целевого полипептида, или идентификацию замещаемой позиции обратимого ингибитора, к которой может быть присоединена группа-«боеголовка», способная образовывать ковалентную связь с Цис-остатком в сайте связывания целевого полипептида, e) формирование ковалентной связи между атомом серы Цис-остатка в сайте связывания целевого полипептида и реакционноспособной группой «боеголовки» или присоединение группы-«боеголовки» к замещаемой позиции обратимого ингибитора. Изобретение позволяет снизить финансовые и временные трудозатраты при разработке структуры необратимого ингибитора. 3 н. и 38 з.п. ф-лы, 11 ил., 12 табл., 8 пр.
Формула
A) представление структурной модели обратимого ингибитора, связанного с сайтом связывания в целевом полипептиде, где обратимый ингибитор образует нековалентные контакты с сайтом связывания;
B) идентификацию Цис-остатка в сайте связывания целевого полипептида, причем любой атом Цис-остатка, отличный от водорода, расположен в пределах приблизительно 20Å от любого атома, отличного от водорода, обратимого ингибитора, когда обратимый ингибитор связан с сайтом связывания;
C) предоставление по меньшей мере одной структурной модели предполагаемого ингибитора, который потенциально ковалентно связывается с целевым полипептидом, где каждый предполагаемый ингибитор содержит «боеголовку», связанную с замещаемой позицией обратимого ингибитора, причем «боеголовка» содержит реакционноспособную химическую функциональную группу и, при необходимости, линкер, и где химическая функциональная группа «боеголовки» способна вступать в реакцию с Цис-остатком, образуя тем самым ковалентную связь между химической функциональной группой и Цис-остатком;
D) определение, находится ли реакционноспособная химическая функциональная группа «боеголовки» предполагаемого ингибитора в пределах расстояния образования связи с Цис-остатком в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания;
E) для предполагаемого ингибитора, который содержит «боеголовку», находящуюся в пределах расстояния образования связи с Цис-остатком в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания, формирование ковалентной связи между атомом серы Цис-остатка в сайте связывания и реакционноспособной химической функциональной группой «боеголовки», когда предполагаемый ингибитор связан с сайтом связывания, где длина ковалентной связи менее 2,1 Å указывает на то, что предполагаемый ингибитор является ингибитором, который ковалентно связывается с целевым полипептидом; и
причем способ осуществляют in silico.
F) выяснение, закрыт ли сайт связывания, когда сформирована ковалентная связь между атомом серы Цис-остатка в сайте связывания и реакционноспособной химической функциональной группой «боеголовки».
X - связь или бивалентная С1-С6 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, в которой при необходимости одна, две или три метиленовых единицы углеводородной цепи независимо заменены на: -NR-, -О-, -С(О)-, - ОС(О)-, -С(O)O-, -S-, -SO-, -SO2-, -C(=S)-, -C(=NR)-, -N=N- или -C(=N2)-;
L - ковалентная связь или бивалентная C1-8 насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, в которой одна, две или три метиленовых единицы L при необходимости и независимо заменены на циклопропилен, -NR-, - N(R)C(O)-, -C(O)N(R)-, -N(R)SO2-, -SO2N(R)-, -O-, -C(O)-, -OC(O)-, -С(O)O-, -S-, -SO-, -SO2-, -C(=S)-, -C(=NR)-, -N=N- или -C(=N2)-;
Y - водород, C1-6алифатическая группа, при необходимости замещенная оксо, галогеном или CN, или 3-10-членное моноциклическое или бициклическое, насыщенное, частично ненасыщенное или арильное кольцо, имеющее 0-3 гетероатома, независимо выбранных из азота, кислорода или серы, и где указанное кольцо замещено 1-4 группами, независимо выбранными из -Q-Z, оксо, NO2, галогена, CN или C1-6алифатической группы, где:
Q - ковалентная связь или бивалентная C1-6насыщенная или ненасыщенная, прямая или разветвленная углеводородная цепь, в которой одна или две метиленовые единицы Q при необходимости и независимо заменены на -NR-, -S-, -О-, -С(О)-, -SO- или -SO2-; и
Z - водород или C1-6алифатическая группа, при необходимости замещенная оксо, галогеном или CN;
каждая R-группа - независимо водород или при необходимости замещенная группа, выбранная из C1-6алифатической группы, фенила, 4-7-членного гетероциклического кольца, имеющего 1-2 гетероатома, независимо выбранных из азота, кислорода или серы, или 5-6-членного моноциклического гетероарильного кольца, имеющего 1-4 гетероатома, независимо выбранных из азота, кислорода или серы.
А) предоставление структурной модели обратимого ингибитора, связанного с сайтом связывания в целевом полипептиде, где обратимый ингибитор образует нековалентные контакты с сайтом связывания;
B) идентификацию Цис-остатка в сайте связывания целевого полипептида, причем любой атом Цис-остатка, отличный от водорода, расположен в пределах приблизительно 20Å от любого атома, отличного от водорода, обратимого ингибитора, когда обратимый ингибитор связан с сайтом связывания;
C) предоставление по меньшей мере одной структурной модели группы-«боеголовки», причем «боеголовка» содержит реакционноспособную химическую функциональную группу, способную вступать в реакцию с Цис-остатком и образовывать ковалентную связь между атомом серы Цис-остатка в сайте связывания и реакционноспособную химическую функциональную группу группы-«боеголовки»;
D) идентификацию замещаемой позиции обратимого ингибитора, к которой может быть присоединена группа-«боеголовка», при необходимости посредством линкера, так, чтобы связь, образованная между атомом серы Цис-остатка в сайте связывания и реакционноспособной химической функциональной группой группы-«боеголовки», имела длину связи менее 2,1Å;
E) присоединение группы-«боеголовки», при необходимости посредством линкера, к замещаемой позиции обратимого ингибитора; и
причем способ осуществляют in silico.
A) предоставление структурной модели сайта связывания в целевом полипептиде, где обратимый ингибитор может образовывать нековалентные контакты с сайтом связывания;
B) идентификацию Цис-остатка в сайте связывания целевого полипептида, причем любой атом Цис-остатка, отличный от водорода, расположен в пределах приблизительно 20Å от любого атома, отличного от водорода, обратимого ингибитора, когда обратимый ингибитор связан с сайтом связывания;
C) предоставление по меньшей мере одной структурной модели предполагаемого ингибитора, который потенциально ковалентно связывается с целевым полипептидом, где каждый предполагаемый ингибитор содержит «боеголовку», связанную с замещаемой позицией обратимого ингибитора, причем «боеголовка» содержит реакционноспособную химическую функциональную группу и, при необходимости, линкер, и где химическая функциональная группа «боеголовки» способна вступать в реакцию с Цис-остатком, образуя тем самым ковалентную связь между химической функциональной группой и Цис-остатком;
D) определение, находится ли реакционноспособная химическая функциональная группа «боеголовки» предполагаемого ингибитора в пределах расстояния образования связи с Цис-остатком в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания;
E) для предполагаемого ингибитора, который содержит «боеголовку», находящуюся в пределах расстояния образования связи с Цис-остатком в сайте связывания целевого полипептида, когда предполагаемый ингибитор связан с сайтом связывания, формирование ковалентной связи между атомом серы Цис-остатка в сайте связывания и реакционноспособной химической функциональной группой «боеголовки», когда предполагаемый ингибитор связан с сайтом связывания, где длина ковалентной связи менее 2,1 Å указывает на то, что предполагаемый ингибитор является ингибитором, который ковалентно связывается с целевым полипептидом; и
причем способ осуществляют in silico.
Комментарии