Способ гидроформилирования - RU2674698C1
Код документа: RU2674698C1
Описание
Настоящее изобретение относится к способу гидроформилирования.
Известно, что альдегиды могут быть получены взаимодействием олефин-ненасыщенного соединения с монооксидом углерода и водородом в присутствии катализатора на основе комплекса родия с органофосфитным лигандом, и что предпочтительные способы включают непрерывное гидроформилирование и рецикл раствора катализатора, как описано, например, в патентах США 4148830; 4717775 и 4769498. Такие альдегиды находят широкое применение и подходят, например, в качестве промежуточных соединений для гидрирования до алифатических спиртов, для альдольной конденсации с получением пластификаторов и для окисления с получением алифатических кислот.
Несмотря на преимущества таких способов гидроформилирования с применением жидкостного рецикла катализатора на основе комплекса родия с фосфорорганическим лигандом, основной проблемой является стабилизация катализатора и фосфорорганического лиганда. Потеря катализатора или каталитической активности вследствие нежелательных реакций весьма дорогих родиевых катализаторов отрицательно влияет на получение требуемого альдегида. Разложение фосфорорганического лиганда, используемого в процессе гидроформилирования, может приводить к образованию вредных соединений, например, отравляющих фосфорорганических соединений, ингибиторов или кислотных побочных продуктов, которые могут снижать каталитическую активность родиевого катализатора. Затраты на производство альдегидного продукта увеличиваются при снижении производительности катализатора.
Гидролитическая нестабильность гидролизуемых органофосфитных лигандов является основной причиной разложения лиганда и дезактивации катализатора в процессах гидроформилирования, катализируемых комплексом родия с фосфорорганическим лигандом. Все органофосфиты в некоторой степени подвержены гидролизу, при этом скорость гидролиза, в целом, зависит от стереохимической природы органофосфита. Как правило, чем массивнее стерическое окружение атома фосфора, тем медленнее протекает гидролиз. Например, третичные триорганофосфиты, такие как трифенилфосфит, более подвержены гидролизу, чем диорганофосфиты, например, описанные в US 4737588, и органополифосфиты, например, описанные в US 4748261 и US 4769498. Все указанные реакции гидролиза неизбежно приводят к образованию фосфорсодержащих кислотных соединений, которые катализируют реакции гидролиза. Например, в результате гидролиза третичного органофосфита образуется сложный диэфир фосфоновой кислоты, который может гидролизоваться до сложного моноэфира фосфоновой кислоты, который, в свою очередь, может гидролизоваться до H3PO3 (фосфорной кислоты). Кроме того, гидролиз сопутствующих продуктов побочных реакций, например, между сложным диэфиром фосфоновой кислоты и альдегидом или между некоторыми органофосфитными лигандами и альдегидом, может приводить к образованию нежелательных сильных альдегидокислот, например, н-C3H7CH(OH)P(O)(OH)2.
Даже весьма благоприятные стерически затрудненные органобисфосфиты, которые слабо подвержены гидролизу, могут взаимодействовать с альдегидным продуктом с образованием отравляющих органофосфитов, например, органомонофосфитов, которые являются каталитическими ингибиторами и которые гораздо сильнее подвержены гидролизу и образованию указанных альдегидокислотных побочных продуктов, например, гидроксиалкилфосфоновых кислот, как показано, например, в US 5288918 и US 5364950. Кроме того, гидролиз органофосфитных лигандов может рассматриваться как автокаталитический процесс с учетом образования таких фосфорсодержащих кислотных соединений, например, H3PO3, альдегидокислот, таких как гидроксиалкилфосфоновые кислоты, H3PO4 и т.п., и без должной проверки каталитическая система, используемая в непрерывном процессе гидроформилирования с жидкостным рециклом, со временем становится все более и более кислотной. Возможное накопление недопустимого количества фосфорсодержащих кислотных соединений может вызывать полное разложение присутствующего органофосфита, приводя к совершенной неэффективности (дезактивации) катализатора гидроформилирования и возможной потере ценного металла родия, например, вследствие осаждения и/или отложения на стенках реактора. Например, в US 5741944 описан содержащий буфер экстрактор, который может быть использован для удаления кислотных соединений по мере их образования, однако указанную экстракцию проводят за пределами системы реактора, и в некоторых случаях может быть перегружена. Снижение содержания кислоты не проводят при высокой температуре и в течение многих часов пребывания в реакторе, поэтому некоторое разложение может происходить до нейтрализации кислоты. Кроме того, оксикислотные буферы на основе натрия склонны к отложению твердых соединений на основе Na (главным образом, из нейтрализованных кислотных соединений), которые вызывают серьезные технологические проблемы, включая остановку производственной установки.
Предложено множество способов для сохранения стабильности катализатора и/или органофосфитного лиганда. Например, в US 5288918 предложено добавление в реакционную зону добавки, усиливающей каталитическую активность, такой как вода и/или слабокислотное соединение; в US 5364950 предложено добавление в реакционную зону эпоксида для стабилизации органофосфитного лиганда; и в US 5741944 описано добавление в экстрактор буфера на основе соли оксикислоты, необязательно с аминными добавками, для удаления кислотных соединений из раствора катализатора. Дополнительное усовершенствование содержащего буфер экстрактора описано в WO 2012/064586, где для удаления солей металлов, образованных из буфера на основе соли оксикислоты, добавлена стадия промывания водой перед возвратом раствора катализатора в реакционную зону.
В US 5744649 описана экстракция и удаление кислотных соединений с применением не содержащей буфера воды, т.е. «исключительно водного экстрактора». Однако для поддержания требуемого эффективного рН раствора катализатора необходим очень большой поток деионизированной воды, что приводит к увеличению потерь продукта, лиганда и катализатора вследствие уноса или растворения в водной фазе. Для восстановления дезактивированного катализатора или для предотвращения образования комплексов кислотных примесей с активным катализатором могут быть необязательно использованы аминные добавки. Амины также могут действовать для «доставки» нейтрализованной кислоты в исключительно водный экстрактор. Описано, что амин предпочтительно переходит в органическую фазу и, следовательно, по существу не поступает в водную фазу. В документе '649 описано, в частности, что «происходит экстракция кислотных соединений в воду, как описано в настоящем документе, в отличие от простого поглощения и/или нейтрализации при сохранении в реакционной среде». Для обеспечения эффективности для вышеуказанных целей необходимы относительно высокие концентрации (до 10 масс. %) аминных добавок. Однако столь высокие концентрации аминов могут вызывать проблемы раздела фаз в экстракторе, например, образование эмульсий и увеличение потерь каталитического металла.
В US 4567306 описано применение аминов для нейтрализации кислотных соединений для сохранения активности катализатора. Не описано, что происходит с указанными аминами (за исключением их испарения вместе с продуктом), и не описан способ удаления образующихся солей. В конечном итоге, указанные соли будут накапливаться вплоть до выпадения в осадок.
В US 8110709 заявлено применение аминов для улавливания кислотных примесей, затем применение ионообменной колонки для удаления образованных солей аминов. Аналогично, в US 7495134 описано добавление добавок на основе вторичных аминов для осаждения кислотных солей, которые удаляют фильтрованием.
В некоторой степени допустим гидролиз нежелательных фосфорсодержащих соединений. В US 5288918 описано, что важно гидролизовать некоторые продукты разложения лиганда, которые действуют как каталитические яды или ингибиторы. Гидролиз может быть проведен без существенного гидролиза необходимого гидролизуемого лиганда посредством тщательного регулирования эффективного рН системы, поскольку указанные продукты разложения лиганда в определенных диапазонах рН разлагаются быстрее, чем необходимые лиганды. В US 5741944 описано, что предпочтительный диапазон рН в зоне удаления кислоты составляет от 4,5 до 7,5 и наиболее предпочтительно от 5,6 до 7,0. Если эффективный рН ниже указанного значения, то происходит гидролиз всех фосфорсодержащих сложных эфиров; однако если он выше указанного значения, то скорость гидролиза каталитического яда слишком мала, и катализатор становится отравленным.
Известные в данной области техники экстракторы, содержащие буфер, основаны на металлических солях оксикислот, таких как NaxHyPO4. Буфер обычно получают заранее и подают в концентрации >0,1 ммоль/л в противоточный экстрактор, где происходит нейтрализация кислот и их удаление в условиях тщательно регулируемого рН. Из уровня техники предполагается, что регулирование рН в водной буферной фазе соответствует эффективному регулированию кислотности в реакционной зоне. К сожалению, несмотря на описание, изложенное в WO 2012/064586, наблюдается медленное накопление засоряющих материалов на основе солей натрия. Добавление к воде аминов в указанных концентрациях, без буфера на основе соли оксикислоты, приводит к неприемлемо высоким значениям рН и к образованию тяжелых соединений в реакционной жидкости. Для обеспечения достаточной буферной емкости необходимы высокие концентрации аминов, таких как пиридин, триалкиламины и т.п., что приводит к недопустимо высоким значениям рН водной фазы (>9).
Несмотря на ценность известных в данной области техники материалов, продолжается поиск альтернативных способов и еще более совершенных и более эффективных способов стабилизации используемого родиевого катализатора и органофосфитного лиганда. Необходимо обеспечить способ снижения или исключения сильнокислотных соединений в реакционной зоне гидроформилирования для минимизации разложения лиганда при снижении степени отравления фосфита, без засорения, наблюдаемого при использовании буферов на основе солей металлов.
Раскрытие сущности изобретения
В настоящем изобретении предложен способ, включающий: (1) проведение в реакционной зоне реакции гидроформилирования с применением реакционной жидкости, содержащей (a) фосфорсодержащее кислотное соединение, (b) катализатор на основе комплекса металла с фосфорорганическим лигандом, который содержит металл группы 8, 9 или 10, связанный в комплекс с фосфорорганическим лигандом, и, необязательно, (c) свободный фосфорорганический лиганд; (2) приведение в контакт по меньшей мере части реакционной жидкости с водорастворимым амином для нейтрализации по меньшей мере некоторого количества фосфорсодержащего кислотного соединения и получения нейтрализованного фосфорсодержащего кислотного соединения; (3) по меньшей мере частичное выделение в экстракционной зоне по меньшей мере одного нейтрализованного фосфорсодержащего кислотного соединения из реакционной жидкости; и (4) удаление нейтрализованного фосфорсодержащего кислотного соединения из экстракционной зоны с водным потоком, выходящим из экстракционной зоны; при условии, что количество амина является таким, что концентрация амина в реакционной зоне составляет не более 0,075 ммоль на литр жидкости реакции гидроформилирования.
Неожиданно было обнаружено, что способ согласно настоящему изобретению обеспечивает возможность регулирования разложения лиганда без увеличения образования тяжелых соединений и при снижении засорения в сравнении со способами с применением буферов на основе солей металлов.
Подробное описание изобретения
Описанный способ включает приведение в контакт водорастворимого амина с жидкостью реакции гидроформилирования. Реакционная жидкость содержит (1) фосфорсодержащее кислотное соединение, (2) катализатор на основе комплекса металла с фосфорорганическим лигандом, который содержит металл, связанный в комплекс с фосфорорганическим лигандом, и, необязательно, (3) свободный фосфорорганический лиганд. Реакционная жидкость может быть получена в зоне реакции гидроформилирования. Экстракционную зону преимущественно используют совместно с реакционной зоной как часть системы выделения продукта. Применение амина может обеспечивать образование в экстракционной зоне водного исходящего потока с приемлемым диапазоном рН, и обеспечивает приемлемую буферную емкость экстракционной зоны. Показатель рН водного потока, выходящего из экстракционной зоны, регулируют посредством экстракции амина и нейтрализованного фосфорсодержащего кислотного соединения из органической фазы экстракционной зоны и образования буферного водного раствора in situ.
Все ссылки на периодическую таблицу элементов и различные группы в данном контексте относятся к версии, опубликованной в CRC Handbook of Chemistry and Physics, 72-е изд. (1991-1992) CRC Press, на страницах I-10.
Если не указано иное или явно не следует из контекста, то все доли и проценты основаны на массе, и все методы испытаний являются такими, как на момент подачи настоящей заявки. Для целей патентной практики США содержание любых упомянутых патентов, патентных заявок или публикаций включено в настоящий документ посредством ссылки в полном объеме (или их эквивалентные версии США включены таким же образом посредством ссылки), особенно в отношении описания определений (в той степени, в которой они не противоречат определениям, специально представленным в настоящем описании) и общих знаний в данной области техники.
В данном контексте термины в единственном числе, «по меньшей мере один» и «один или более» использованы взаимозаменяемо. Термины «содержит», «включает» и их варианты, использованные в описании и формуле изобретения, не имеют ограничивающего значения. Так, например, водная композиция, которая содержит частицы «одного» гидрофобного полимера, может означать, что композиция содержит частицы «одного или более» гидрофобных полимеров.
В данном контексте указание числовых диапазонов посредством конечных точек включает все значения, входящие в указанный диапазон (например, от 1 до 5 включает 1, 1,5, 2, 2,75, 3, 3,80, 4, 5 и т.д.). Для целей настоящего изобретения следует понимать, согласно общему пониманию специалистов в данной области техники, что числовой диапазон включает и охватывает все возможные поддиапазоны, входящие в указанный диапазон. Например, диапазон от 1 до 100 включает от 1,01 до 100, от 1 до 99,99, от 1,01 до 99,99, от 40 до 60, от 1 до 55 и т.д. Кроме того, указание в настоящем документе числовых диапазонов и/или числовых значений, включая такие указания в формуле изобретения, можно понимать как включающие термин «примерно». В таких случаях термин «примерно» относится к числовым диапазонам и/или числовым значениям, которые являются по существу такими же, как значения, указанные в настоящем документе.
В данном контексте термин «ppm масс.» означает количество частей на миллион по массе.
Для целей настоящего изобретения термин «углеводород» включает все возможные соединения, имеющие по меньшей мере один атом водорода и один атом углерода. Указанные возможные соединения также могут содержать один или более гетероатомов. В широком аспекте возможные углеводороды включают ациклические (с гетероатомами или без них) и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические органические соединения, которые могут быть замещенными или незамещенными.
В данном контексте термин «замещенный» включает все возможные заместители органических соединений, если не указано иное. В широком аспекте, допустимые заместители включают ациклические и циклические, разветвленные и неразветвленные, карбоциклические и гетероциклические, ароматические и неароматические заместители органических соединений. Иллюстративные заместители включают, например, алкил, алкилокси, арил, арилокси, гидроксиалкил, аминоалкил, в которых количество атомов углерода может составлять от 1 до 20 или более, предпочтительно от 1 до 12, а также гидрокси, галоген и амино. Возможные заместители могут представлять собой один или более заместителей, и могут быть одинаковыми или различными для соответствующих органических соединений. Настоящее изобретение никоим образом не ограничено возможными заместителями органических соединений.
В данном контексте термин «гидроформилирование» включает, но не ограничивается ими, все процессы гидроформилирования, которые включают превращение одного или более замещенных или незамещенных олефиновых соединений или реакционной смеси, содержащей одно или более замещенных или незамещенных олефиновых соединений, в один или более замещенных или незамещенных альдегидов или в реакционную смесь, содержащую один или более замещенных или незамещенных альдегидов. Альдегиды могут быть асимметричными или неасимметричными.
Термины «реакционная жидкость», «реакционная среда» и «раствор катализатора» использованы в данном контексте взаимозаменяемо, и могут включать, но не ограничиваются ей, смесь, содержащую: (a) катализатор на основе комплекса металла с фосфорорганическим лигандом, (b) свободный фосфорорганический лиганд, (c) альдегидный продукт, образованный в реакции, (d) непрореагировавшие реагенты, (e) растворитель для указанного катализатора на основе комплекса металла с фосфорорганическим лигандом и указанного свободного фосфорорганического лиганда и, необязательно, (f) одно или более фосфорсодержащих кислотных соединений, образованных в реакции (которые могут быть гомогенными или гетерогенными, и указанные соединения включают соединения, налипшие на поверхности технологического оборудования). Реакционная жидкость может включать, но не ограничивается ими, (a) жидкость в реакционной зоне, (b) поток жидкости, направляемый в зону разделения, (c) жидкость в зоне разделения, (d) поток рецикла, (e) жидкость, выводимую из реакционной зоны или зоны разделения, (f) выведенную жидкость, обработанную водным раствором, (g) обработанную жидкость, возвращаемую в реакционную зону или зону разделения, (h) жидкость во внешнем охладителе и (i) продукты разложения лиганда и их соли.
Для целей настоящего изобретения термин «тяжелые соединения» означает соединения, которые имеют температуру кипения выше, чем температура кипения требуемого альдегидного продукта(-ов).
В данном контексте термин «экстрактор» означает любую подходящую емкость или контейнер, например, любую емкость, подходящую для применения в качестве жидкость-жидкостного экстрактора, который обеспечивает подходящее средство для полного контакта между реакционной жидкостью и водным раствором.
Для целей настоящего изобретения термин «зона экстракции» означает систему оборудования, которая содержит по меньшей мере один экстрактор. Зона экстракции может содержать несколько экстракторов, расположенных параллельно, последовательно или в обоих вариантах.
Термин «водный поток, выходящий из зоны экстракции» относится к исходящему потоку из зоны экстракции, который в качестве своего источника содержит водную фазу, которая обеспечивает последующий контакт раствора катализатора с водным раствором в зоне экстракции.
Для целей настоящего изобретения термин «реакционная зона» означает систему оборудования, которая содержит по меньшей мере один реактор и которая обеспечивает подачу по меньшей мере части жидкого исходящего потока в зону разделения продукта и катализатора, которая может содержать зону экстракции. Термин «первый реактор» относится к первому реактору в реакционной зоне.
«Гидролизуемые фосфорсодержащие лиганды» представляют собой лиганды, содержащие трехвалентный фосфор, которые содержат по меньшей мере одну связь P-Z, где Z представляет собой кислород, азот, хлор, фтор или бром. Примеры включают, но не ограничиваются ими, фосфиты, фосфинофосфиты, бисфосфиты, фосфониты, бисфосфониты, фосфиниты, фосфорамидиты, фосфинофосфорамидиты, бисфосфорамидиты, фторфосфиты и т.п. Лиганды могут содержать хелатные структуры и/или могут содержать несколько фрагментов P-Z, такие как полифосфиты, полифосфорамидиты и т.д., а также смешанные фрагменты P-Z, такие как фосфит-фосфорамидиты, фторфосфит-фосфиты и т.п.
Термин «комплекс» в данном контексте означает координационное соединение, образованное посредством объединения одной или более молекул или атомов с избытком электронов, способных к независимому существованию, с одной или более молекулами или атомами с недостатком электронов, которые также способны к независимому существованию. Например, фосфорорганические лиганды, которые могут быть использованы согласно настоящему изобретению, могут иметь один или более донорных атомов фосфора, каждый из которых имеет одну доступную или неподеленную пару электронов, способную образовывать координационную связь независимо или возможно совместно (например, посредством хелатообразования) с металлом. Монооксид углерода, который в узком смысле слова также классифицируют как лиганд, также может присутствовать и координироваться с металлом. Конечная композиция комплексного катализатора также может содержать дополнительный лиганд, например, водород или анион, соответствующий координационным центрам или ядерному заряду металла. Иллюстративные дополнительные лиганды включают, например, галоген (Cl, Br, I), алкил, арил, замещенный арил, ацил, CF3, C2 F5, CN, (R)2PO и RP(O)(OH)O (где все R являются одинаковыми или различными и представляют собой замещенный или незамещенный углеводородный радикал, например, алкил или арил), ацетат, ацетилацетонат, SO4, PF4, PF6, NO2, NO3, CH3, CH2=CHCH2, CH3CH=CHCH2, C6H5CN, CH3CN, NH3, пиридин, (C2H5)3N, моноолефины, диолефины и триолефины, тетрагидрофуран и т.п. Комплексные соединения предпочтительно не содержат никаких дополнительных органических лигандов или анионов, которые могут отравлять катализатор или оказывать неблагоприятное действие на характеристики катализатора. В реакциях гидроформилирования, катализируемых комплексом металла с органофосфитным лигандом, предпочтительно, если активные катализаторы не содержат галогена и серы, напрямую связанных с металлом, хотя указанное условие может не быть абсолютно необходимым.
Количество доступных координационных центров у переходных металлов известно в данной области техники и зависит от конкретного выбранного переходного металла. Каталитические соединения могут содержать комплексную каталитическую смесь мономерных, димерных форм или форм более высокой нуклеарности, которые предпочтительно характеризуются по меньшей мере одной фосфорорганической молекулой, связанной в комплекс, на одну молекулу металла, например, родия. Например, предусмотрено, что каталитические соединения предпочтительного катализатора, используемого в реакции гидроформилирования, могут быть связаны в комплекс с монооксидом углерода и водородом, помимо одного или более фосфорорганических лигандов(-а).
Понятно, что термин «рН» надлежащим образом определен только для водных систем. В данном контексте термин «эффективный рН» относится к рН водного экстракта органической фазы для отображения степени кислотности/щелочности органической фазы.
Буферы представляют собой смеси кислот и оснований. Для целей настоящего изобретения буфер представляет собой водный раствор, состоящий из смеси слабой кислоты и ее сопряженного основания или из смеси слабого основания и его сопряженной кислоты.
Водорастворимый амин используют в количестве, достаточном для поддержания рН в водном потоке, выходящем из зоны экстракции, на требуемом уровне. Преимущественно, амин используют в положительном количестве, составляющем не более 0,075 ммоль/л реакционной жидкости, предпочтительно не более 0,05 ммоль/л и еще более предпочтительно не более 0,030 ммоль/л, по результатам измерения в реакционной жидкости в реакционной зоне. Преимущественно, количество используемого амина составляет по меньшей мере 0,005 ммоль/л реакционной жидкости, предпочтительно по меньшей мере 0,015 ммоль/л и еще более предпочтительно по меньшей мере 0,025 ммоль/л. Преимущественно, амин используют в количестве, составляющем от 0,005 до 0,075 ммоль/л реакционной смеси, предпочтительно от 0,015 до 0,05 ммоль/л и еще более предпочтительно от 0,025 до 0,030 ммоль/л. Могут быть использованы смеси аминов. Концентрация амина в реакционной жидкости в реакционной зоне может быть измерена обычными способами, общеизвестными специалистам в данной области техники, включая, например, газовую хроматографию и жидкостную хроматографию.
Преимущественно, водорастворимый амин имеет два следующих свойства: 1) является слабощелочным во избежание образования тяжелых соединений в реакционной зоне; и 2) является водорастворимым во избежание накопления в реакционной жидкости. Щелочность или основность водорастворимого амина обычно записывают как pKa сопряженной кислоты, который преимущественно составляет от 5 до 11 при температуре зоны экстракции. Значение pKa предпочтительно составляет от 6,0 до 9,5 и наиболее предпочтительно от 6,5 до 9,0. Потенциальный амин может быть проверен на образование тяжелых соединений посредством нагревания альдегидного продукта с указанным амином при повышенной температуре. Подходящие амины демонстрируют образование менее 1 г тяжелых соединений на литр экспериментального раствора в день при температуре гидроформилирования. Количество образованных тяжелых соединений может быть легко определено с помощью газовой или жидкостной хроматографии, как известно специалистам в данной области техники. Амин преимущественно выбран из одного или более из следующих классов.
Один из классов аминов имеет структуру:
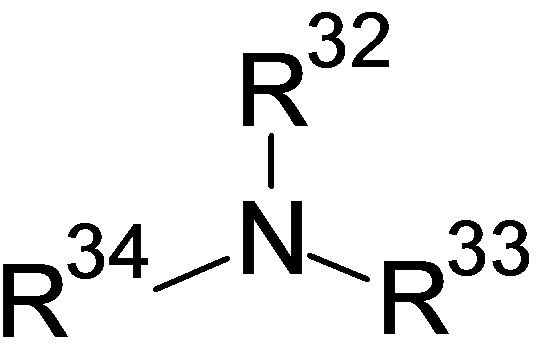
где R32, R33 и R34, каждый независимо, представляют собой H или алкильные, или арильные заместители, так что не более чем один из R32, R33 и R34 представляет собой водород, предпочтительно ни один из них не представляет собой водород, и при этом по меньшей мере один является электроноакцепторным заместителем, и предпочтительно по меньшей мере 2 являются электроноакцепторными заместителями. Электроноакцепторные алкильные или арильные заместители включают алкил-замещенный или незамещенный арил, галогенированные, алкоксилированные, алкилалкоксилированные или карбоксилированные арильные группы, бета-алкокси или бета-алкоксиалкилы (такие как бета-гидроксиэтил, бета-гидрокси-альфа-метилэтил, бета-гидрокси-бета-метилэтил и их этоксилированные и/или пропоксилированные аддукты). Примеры предпочтительных аминов данного класса включают триэтаноламин, метилдиэтаноламин, этилдиэтаноламин, диметилэтаноламин и три(2-гидроксипропил)амин, а также их этоксилаты.
Другой класс аминов имеет структуру, представленную формулой (X):

где каждый R36 независимо представляет собой C1-C4 алкил, гидроксил (и их этоксилированные и пропоксилированные аддукты), алкилалкокси или галоген, x равен 1-3, и z равен 0-6. Предпочтительно, присутствуют несколько фрагментов R36, которые могут быть различными и, наиболее предпочтительно, несколько из них расположены с каждой стороны азотного фрагмента. Предпочтительные примеры представляют собой 4-гидрокси-2,2,6,6-тетраметилпиперидин и его производные.
Другой класс подходящих аминов представляют собой пиридины и родственные циклические структуры, представленные формулой (XI), где R36 является таким, как описано выше, и q равен 0-5.

Предпочтительно, по меньшей мере один R36 не представляет собой H, и более предпочтительно, не являющийся H заместитель находится в орто-положении. Еще более предпочтительно, не являющиеся H заместители находятся в обоих орто-положениях.
Другой класс подходящих слабоосновных аминов включает имидазолы, пиразолы, индазолы, 1,2,3-триазолы, 1,2,4-триазолы, 2,1,3-триазолы, 4,1,2-триазолы, 1,2-диазины, 1,3-диазины, 1,4-диазины, 1,3,5-триазины и бензимидазолы, которые имеют заместители, такие как описаны для R32, которые увеличивают водорастворимость для удовлетворения требования разделения раствора воды и катализатора, описанного ниже. Примеры указанных аминов описаны в US 7351339 и в одновременно рассматриваемой предварительной заявке на патент США с серийным номером 61/790642, поданной 15 марта 2013 года.
Для целей настоящего изобретения термин «растворимость в воде» амина определяют как отношение растворимости между водной фазой в зоне экстракции и органической фазой раствора катализатора в зоне экстракции, и преимущественно составляет по меньшей мере 2:1, предпочтительно по меньшей мере 100:1 и наиболее предпочтительно по меньшей мере 200:1. Указанное отношение может быть определено добавлением амина к 1:1 смеси воды и раствора катализатора, перемешиванием, выдерживанием времени для разделения фаз с последующим анализом двух слоев для определения концентрации в двух фазах обычными способами, такими как газовая хроматография, как известно специалистам в данной области техники. Указанные амины являются полярными аминами, в которых полярные фрагменты (обычно электроноакцепторные фрагменты) усиливают растворимость в воде и снижают основность аминной функциональной группы.
Процесс гидроформилирования и условия его осуществления хорошо известны. Проведение реакции гидроформилирования включает приведение в контакт в реакционной зоне CO, H2 и по меньшей мере одного олефина в присутствии катализатора гидроформилирования в условиях гидроформилирования, достаточных для образования по меньшей мере одного альдегидного продукта. В качестве компонентов катализатор содержит переходный металл и гидролизуемый фосфорорганический лиганд. Необязательные компоненты для добавления в реакционную зону включают эпоксид и/или воду.
Водород и монооксид углерода могут быть получены из любого подходящего источника, включая установки крекинга нефти и нефтеперерабатывающие заводы. Предпочтительным источником водорода и CO являются смеси синтез-газа.
Синтез-газом (от выражения синтетический газ) называют газообразную смесь, которая содержит различные количества CO и H2. Способы его получения хорошо известны. Водород и CO, как правило, представляют собой основные компоненты синтез-газа, но синтез-газ может содержать CO2 и инертные газы, такие как N2 CH4 и Ar. Отношение H2 к CO варьируется в широких пределах, но обычно составляет от 1:100 до 100:1 и предпочтительно от 1:10 до 10:1. Синтез-газ имеется в продаже и зачастую используется в качестве источника топлива или в качестве промежуточного соединения для получения других химических соединений. Наиболее предпочтительное отношение H2:CO для химического производства составляет от 3:1 до 1:3, обычно должно составлять от 1:2 до 2:1 для большинства применений гидроформилирования.
Замещенные или незамещенные олефин-ненасыщенные реагенты, которые могут быть использованы в процессе гидроформилирования, включают оптически активные (прохиральные и хиральные) и оптически неактивные (ахиральные) олефин-ненасыщенные соединения, содержащие от 2 до 40, предпочтительно от 3 до 20 атомов углерода. Указанные соединения подробно описаны в US 2010/006980. Такие олефин-ненасыщенные соединения могут иметь концевую или внутреннюю ненасыщенность и могут быть неразветвленными, разветвленными или циклическими структурами, а также олефиновыми смесями, такими как смеси, получаемые при олигомеризации пропена, бутена, изобутена и т.д. (например, так называемый димерный, тримерный или тетрамерный пропилен и т.п., как описано, например, в US 4518809 и 4,528,403).
В процессе гидроформилирования преимущественно используют растворитель. Может быть использован любой подходящий растворитель, который не оказывает неблагоприятного влияния на процесс гидроформилирования. В качестве иллюстрации, подходящие растворители для процессов гидроформилирования, катализируемых родием, включают растворители, описанные, например, в патентах США 3527809; 4148830; 5312996; и 5929289. Неограничивающие примеры подходящих растворителей включают насыщенные углеводороды (алканы), ароматические углеводороды, воду, простые эфиры, простые полиэфиры, альдегиды, кетоны, нитрилы, спирты, сложные эфиры и продукты конденсации альдегидов. Конкретные примеры растворителей включают: тетраглим, пентаны, циклогексан, гептаны, бензол, ксилол, толуол, диэтиловый эфир, тетрагидрофуран, бутиральдегид и бензонитрил. Органический растворитель также может содержать растворенную воду вплоть до предела насыщения. Иллюстративные предпочтительные растворители включают кетоны (например, ацетон и метилэтилкетон), сложные эфиры (например, этилацетат, ди-2-этилгексилфталат, 2,2,4-триметил-1,3-пентандиолмоноизобутират), углеводороды (например, толуол), нитроуглеводороды (например, нитробензол), простые эфиры (например, тетрагидрофуран (ТГФ)) и сульфолан. В процессах гидроформилирования, катализируемых родием, может быть предпочтительно использовать в качестве основного растворителя альдегидные соединения, соответствующие получаемым альдегидным продуктам и/или побочным жидким продуктам конденсации альдегидов с более высокой температурой кипения, например, которые могут быть образованы in situ в процессе гидроформилирования, как описано, например, в US 4148380 и US 4247486. В конечном итоге, основной растворитель обычно может дополнительно содержать альдегидные продукты и тяжелые соединения, что обусловлено природой непрерывного процесса. Количество растворителя не является особенно критичным и должно лишь быть достаточным для обеспечения реакционной среды с требуемой концентрацией переходного металла. Как правило, количество растворителя варьируется от примерно 5 процентов до примерно 95 процентов по массе от общей массы реакционной жидкости. Могут быть использованы смеси растворителей.
Иллюстративные комплексы металла с фосфорорганическим лигандом, которые могут быть использованы в таких реакциях гидроформилирования, включают катализаторы на основе комплекса металла с фосфорорганическим лигандом. Указанные катализаторы, а также способы их получения хорошо известны в данной области техники и включают те, которые описаны в патентах, упомянутых в настоящем документе. В целом, такие катализаторы могут быть получены заранее или могут быть получены in situ и могут содержать металл в комплексной комбинации с фосфорорганическим лигандом, монооксидом углерода и необязательно водородом. Комплексные соединения лиганда могут присутствовать в мононуклеарных, динуклеарных формах и/или формах более высокой нуклеарности. Однако точная структура катализатора неизвестна.
Катализатор на основе комплекса металла с фосфорорганическим лигандом может быть оптически активным или оптически неактивным. Металлы могут включать металлы групп 8, 9 и 10, выбранные из родия (Rh), кобальта (Co), иридия (Ir), рутения (Ru), железа (Fe), никеля (Ni), палладия (Pd), платины (Pt), осмия (Os) и их смесей, при этом предпочтительные металлы представляют собой родий, кобальт, иридий и рутений, более предпочтительно родий, кобальт и рутений, особенно родий. Могут быть использованы смеси указанных металлов. Возможные фосфорорганические лиганды, которые образуют комплексы металла с фосфорорганическим лигандом и свободный фосфорорганический лиганд, включают моно-, ди-, три- и высшие полифосфорорганические лиганды. В катализаторе на основе комплекса металла с фосфорорганическим лигандом и/или в свободном лиганде могут быть использованы смеси лигандов, и указанные смеси могут быть одинаковыми или различными.
Фосфорорганические соединения, которые могут служить в качестве лиганда для катализатора на основе комплекса металла с фосфорорганическим лигандом и/или свободного лиганда, могут быть ахирального (оптически неактивного) или хирального (оптически активного) типа, и они хорошо известны в данной области техники. Предпочтительны ахиральные фосфорорганические лиганды.
Среди фосфорорганических лигандов, которые могут служить в качестве лиганда для катализатора на основе комплекса металла с фосфорорганическим лигандом, представляют собой моноорганофосфитные, диорганофосфитные, триорганофосфитные и органополифосфитные соединения. Указанные фосфорорганические лиганды и способы их получения хорошо известны в данной области техники.
Иллюстративные моноорганофосфиты могут включать соединения, имеющие формулу:

где R10 представляет собой замещенный или незамещенный трехвалентный углеводородный радикал, содержащий от 4 до 40 атомов углерода или более, такой как трехвалентные ациклические и трехвалентные циклические радикалы, например, трехвалентные алкиленовые радикалы, такие как радикалы, полученные из 1,2,2-триметилолпропана, и т.п., или трехвалентные циклоалкиленовые радикалы, такие как радикалы, полученные из 1,3,5-тригидроксициклогексана, и т.п. Указанные моноорганофосфиты более подробно описаны, например, в US 4567306.
Иллюстративные диорганофосфиты могут включать соединения, имеющие формулу:

где R20 представляет собой замещенный или незамещенный двухвалентный углеводородный радикал, содержащий от 4 до 40 атомов углерода или более, и W представляет собой замещенный или незамещенный одновалентный углеводородный радикал, содержащий от 1 до 18 атомов углерода или более.
Иллюстративные замещенные и незамещенные одновалентные углеводородные радикалы, представленные обозначением W в указанной выше формуле (II), включают алкильные и арильные радикалы, тогда как иллюстративные замещенные и незамещенные двухвалентные углеводородные радикалы, представленные обозначением R20, включают двухвалентные ациклические радикалы и двухвалентные ароматические радикалы. Иллюстративные двухвалентные ациклические радикалы включают, например, алкилен, алкилен-окси-алкилен, алкилен-S-алкилен, циклоалкиленовые радикалы и алкилен-NR24-алкилен, где R24 представляет собой водород или замещенный или незамещенный одновалентный углеводородный радикал, например, алкильный радикал, имеющий от 1 до 4 атомов углерода. Более предпочтительные двухвалентные ациклические радикалы представляют собой двухвалентные алкиленовые радикалы, такие как радикалы, более подробно описанные, например, в патентах США 3415906 и 4567302, и т.п. Иллюстративные двухвалентные ароматические радикалы включают, например, арилен, бисарилен, арилен-алкилен, арилен-алкилен-арилен, арилен-окси-арилен, арилен-NR24-арилен, где R24 является таким, как описано выше, арилен-S-арилен, арилен-S-алкилен и т.п. Более предпочтительно, R20 представляет собой двухвалентный ароматический радикал, такой как радикал, более подробно описанный, например, в патентах США 4599206, 4717775, 4835299, и т.п.
Пример более предпочтительного класса диорганофосфитов представляют собой соединения формулы:

где W является таким, как описано выше, каждый Ar является одинаковым или различным и представляет собой замещенный или незамещенный арильный радикал, каждый y является одинаковым или различным и имеет значение 0 или 1, Q представляет собой двухвалентную мостиковую группу, выбранную из -C(R33)2-, -O-, -S-, -NR24-, Si(R35)2 и -CO-, где каждый R33 является одинаковым или различным и представляет собой водород, алкильный радикал, имеющий от 1 до 12 атомов углерода, фенил, толил и анизил, R24 является таким, как описано выше, каждый R35 является одинаковым или различным и представляет собой водород или метильный радикал, и m имеет значение 0 или 1. Такие диорганофосфиты более подробно описаны, например, в патентах США 4599206; 4717775; и 4835299.
Иллюстративные триорганофосфиты могут включать соединения, имеющие формулу:

где каждый R46 является одинаковым или различным и представляет собой замещенный или незамещенный одновалентный углеводородный радикал, например, алкильные, циклоалкильные, арильные, алкарильные и аралкильные радикалы, которые могут содержать от 1 до24 атомов углерода. Иллюстративные триорганофосфиты включают, например, триалкилфосфиты, диалкиларилфосфиты, алкилдиарилфосфиты, триарилфосфиты и т.п., такие как, например, триметилфосфит, триэтилфосфит, бутилдиэтилфосфит, диметилфенилфосфит, трифенилфосфит, тринафтилфосфит, бис(3,6,8-три-трет-бутил-2-нафтил)метилфосфит, бис(3,6,8-три-трет-бутил-2-нафтил)циклогексилфосфит, трис(3,6-ди-трет-бутил-2-нафтил)фосфит, бис(3,6,8-три-трет-бутил-2-нафтил)фенилфосфит и бис(3,6,8-три-трет-бутил-2-нафтил)(4-сульфофенил)фосфит и т.п. Наиболее предпочтительный триорганофосфит представляет собой трифенилфосфит. Указанные триорганофосфиты более подробно описаны, например, в патентах США 3527809 и 5277532.
Иллюстративные органополифосфиты содержат два или более третичных (трехвалентных) атомов фосфора и могут включать соединения, имеющие формулу:

где X представляет собой замещенный или незамещенный n-валентный органический мостиковый радикал, содержащий от 2 до 40 атомов углерода, каждый R57 является одинаковым или различным и представляет собой двухвалентный органический радикал, содержащий от 4 до 40 атомов углерода, каждый R58 является одинаковым или различным и представляет собой замещенный или незамещенный одновалентный углеводородный радикал, содержащий от 1 до 24 атомов углерода, a и b могут быть одинаковыми или различными, и каждый имеет значение от 0 до 6, при условии, что сумма a+b равна от 2 до 6, и n равен a+b. Следует понимать, что если a имеет значение 2 или более, то каждый радикал R57 может быть одинаковым или различным. Каждый радикал R58 также может быть одинаковым или различным в любом данном соединении.
Иллюстративные n-валентные (предпочтительно двухвалентные) органические мостиковые радикалы, представленные выше обозначением X, и иллюстративные двухвалентные органические радикалы, представленные обозначением R57, включают ациклические радикалы и ароматические радикалы, такие как алкиленовые, алкилен-Qm-алкиленовые, циклоалкиленовые, ариленовые, бисариленовые, арилен-алкиленовые и арилен-(CH2)y-Qm-(CH2)y-ариленовые радикалы и т.п., где каждый Q, y и m являются такими, как описано выше в отношении формулы (III). Более предпочтительные ациклические радикалы, представленные выше обозначениями X и R57, представляют собой двухвалентные алкиленовые радикалы, а более предпочтительные ароматические радикалы, представленные выше обозначениями X и R57, представляют собой двухвалентные ариленовые и бисариленовые радикалы, такие как радикалы, более подробно описанные, например, в патентах США 4769498; 4774361; 4885401; 5179055; 5113022; 5202297; 5235113; 5264616 и 5364950, и 5527950. Иллюстративные предпочтительные одновалентные углеводородные радикалы, представленные выше каждым радикалом R58, включают алкильные и ароматические радикалы.
Иллюстративные предпочтительные органополифосфиты могут включать бифосфиты, такие как соединения формул (VI)-(VIII), представленных ниже:
где каждый R57, R58 и X в формулах (VI)-(VIII) является таким же, как описано выше для формулы (V). Предпочтительно, каждый R57 и X представляет собой двухвалентный углеводородный радикал, выбранный из алкилена, арилена, арилен-алкилен-арилена и бисарилена, где каждый радикал R58 представляет собой одновалентный углеводородный радикал, выбранный из алкильных и арильных радикалов. Органофосфитные лиганды таких формул (V)-(VIII) описаны, например, в патентах США 4668651; 4748261; 4769498; 4774361; 4885401; 5113022; 5179055; 5202297; 5235113; 5254741; 5264616; 5312996; 5364950; и 5391801.
Конкретные иллюстративные примеры таких органофосфитных лигандов включают следующие: 2-трет-бутил-4-метоксифенил(3,3'-ди-трет-бутил-5,5'-диметокси-1,1'-бифенил-2,2'-диил)фосфит, метил(3,3'-ди-трет-бутил-5,5'-диметокси-1,1'-бифенил-2,2'-диил)фосфит, 6,6'-[[3,3'-бис(1,1-диметилэтил)-5,5'-диметокси-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]диоксафосфепин, 6,6'-[[3,3',5,5'-тетракис(1,1-диметилэтил)-[1,1'-бифенил]-2,2'-диил]бис(окси)]бис-дибензо[d,f][1,3,2]-диоксафосфепин, (2R,4R)-ди[2,2'-(3,3',5,5'-тетракис-трет-бутил-1,1-бифенил)]-2,4-пентилдифосфит, (2R,4R)-ди[2,2'-(3,3'-ди-трет-бутил-5,5'-диметокси-1,1'-бифенил)]-2,4-пентилдифосфит, 2-[[2-[[4,8-бис(1,1-диметилэтил)-2,10-диметоксидибензо-[d,f]-[1,3,2]диоксофосфепин-6-ил]окси]-3-(1,1-диметилэтил)-5-метоксифенил]метил]-4-метокси, метиленди-2,1-фенилен, тетракис[2,4-бис(1,1-диметилэтил)фениловый]эфир фосфорной кислоты и [1,1'-бифенил]-2,2'-диил-тетракис[2-(1,1-диметилэтил)-4-метоксифениловый]эфир фосфорной кислоты.
Катализаторы на основе комплекса металла с фосфорорганическим лигандом могут быть в гомогенной или гетерогенной форме. Например, полученные заранее катализаторы на основе комплекса родия с гидридо-карбонил-фосфорорганическим лигандом могут быть получены и введены в реакционную смесь гидроформилирования. Более предпочтительно, катализаторы на основе комплекса родия с фосфорорганическим лигандом могут быть получены из предшественника родиевого катализатора, который может быть введен в реакционную среду для in situ образования активного катализатора. Например, предшественники родиевого катализатора, такие как дикарбонил-ацетилацетонат родия, Rh2O3, Rh4(CO)12, Rh6(CO)16, Rh(NO3)3 и т.п., могут быть введены в реакционную смесь вместе с фосфорорганическим лигандом для in situ образования активного катализатора. В предпочтительном варианте реализации дикарбонил-ацетилацетонат родия используют в качестве родиевого предшественника и приводят во взаимодействие с фосфорорганическим лигандом в присутствии растворителя с получением каталитического предшественника на основе комплекса родия с фосфорорганическим лигандом, который вводят в реактор вместе с избытком (свободного) фосфорорганического лиганда для in situ образования активного катализатора. В любом случае, достаточно, чтобы монооксид углерода, водород и фосфорорганический лиганд представляли собой лиганды, которые способны образовывать комплекс с металлом, и чтобы активный катализатор на основе комплекса металла с фосфорорганическим лигандом присутствовал в реакционной смеси в условиях, используемых для реакции гидроформилирования. Карбонил и фосфорорганические лиганды могут быть связаны в комплекс с родием либо до, либо in situ во время процесса гидроформилирования.
В качестве иллюстрации, предпочтительная композиция предшественника катализатора состоит по существу из солюбилизированного предшественника комплекса родия с карбонильным и органофосфитным лигандом, растворителя и необязательно свободного органофосфитного лиганда. Предпочтительная композиция предшественника катализатора может быть получена посредством получения раствора дикарбонил-ацетилацетоната родия, органического растворителя и органофосфитного лиганда. Фосфорорганический лиганд легко вытесняет один из карбонильных лигандов в предшественнике комплекса ацетилацетоната родия, о чем свидетельствует выделение газообразного монооксида углерода.
Соответственно, катализатор на основе комплекса металла с фосфорорганическим лигандом преимущественно содержит металл, связанный в комплекс с монооксидом углерода и фосфорорганическим лигандом, и указанный лиганд связан (в комплекс) с металлом хелатным и/или нехелатным образом.
Могут быть использованы смеси катализаторов. Количество катализатора на основе комплекса металла с фосфорорганическим лигандом, присутствующего в реакционной жидкости, должно быть лишь таким минимальным количеством, которое необходимо для обеспечения данной концентрации металла, предусмотренной для применения, и которое будет обеспечивать основу для по меньшей мере каталитического количества металла, необходимого для катализа конкретного процесса гидроформилирования, как описано, например, в вышеупомянутых патентах. В целом, концентрация каталитического металла, например, родия, в диапазоне от 10 ppm масс. до 1000 ppm масс. в пересчете на свободный металл в реакционной среде, достаточна для большинства процессов, при этом обычно предпочтительно использовать от 10 до 500 ppm масс. металла и более предпочтительно от 25 до 350 ppm масс. металла.
Помимо катализатора на основе комплекса металла с фосфорорганическим лигандом, в реакционной среде также может присутствовать свободный фосфорорганический лиганд (т.е. лиганд, не связанный в комплекс с металлом). Свободный фосфорорганический лиганд может соответствовать любому из вышеуказанных фосфорорганических лигандов, рассмотренных выше. Предпочтительно, чтобы свободный фосфорорганический лиганд был таким же, что и фосфорорганический лиганд в используемом катализаторе на основе комплекса металла с фосфорорганическим лигандом. Однако указанные лиганды не обязательно должны быть одинаковыми в любом рассматриваемом процессе. В процессе гидроформилирования согласно настоящему изобретению может быть использовано от 0,1 моль или менее до 100 моль или более свободного фосфорорганического лиганда на моль металла в реакционной среде. Предпочтительно, процесс гидроформилирования проводят в присутствии от 1 до 50 моль фосфорорганического лиганда на моль металла, присутствующего в реакционной среде. Более предпочтительно, для органополифосфитов используют от 1,1 до 4 моль органополифосфитного лиганда на моль металла. Указанные количества фосфорорганического лиганда представляют собой сумму количества фосфорорганического лиганда, связанного (в комплекс) с присутствующим металлом, и количества свободного присутствующего фосфорорганического лиганда. При необходимости в реакционную среду процесса гидроформилирования в любое время и любым подходящим способом может быть введено дополнительное количество фосфорорганического лиганда, например, для поддержания заданного содержания свободного лиганда в реакционной среде.
В одном из вариантов реализации родиевым катализатором может быть пропитана любая твердая подложка, такая как неорганические оксиды (т.е. оксид алюминия, диоксид кремния, диоксид титана или диоксид циркония), углерод, мембраны, тонкие пленки или ионообменные смолы, он может быть нанесен или внедрен в поры цеолита, стекла или глины, нерастворимой полимерной подложки, или также может быть растворен в жидкой пленке, покрывающей поры указанного цеолита или стекла.
Иллюстративные способы гидроформилирования, катализируемые комплексом металла с фосфорорганическим лигандом, в которых может протекать гидролитическое разложение, включают способы, описанные, например, в патентах США 4148830; 4593127; 4769498; 4717775; 4774361; 4885401; 5264616; 5288918; 5360938; 5364950; 5491266 и 7196230. Соединения, содержащие фрагмент P-Z, которые вероятно подвергается гидролитическому разложению, включают органофосфониты, фосфорамидиты и фторфосфониты, такие как описаны в WO 2008/071508, WO 2005/042458 и в патентах США 5710344, 6265620, 6440891, 7009068, 7145042, 7586010, 7674937 и 7872156. Соответственно, преимущественно используемые технологии гидроформилирования могут соответствовать любым известным технологиям, таким как, например, газовый рецикл, жидкостный рецикл и их комбинации. Предпочтительные способы гидроформилирования представляют собой те, которые включают жидкостный рецикл катализатора.
В одном из вариантов реализации настоящего изобретения в процесс по существу не добавляют никакого буфера на основе соли металла. В одном из вариантов реализации настоящего изобретения в процесс по существу не добавляют никакого буфера на основе натриевой соли оксикислоты.
В способе согласно настоящему изобретению используют стадию водной экстракции вместе с добавлением небольших количеств водорастворимого, но относительно слабоосновного амина. Одна из функций амина заключается в нейтрализации кислотных примесей. Нейтрализованные кислоты представляют собой соли, например, соли аммония. Необходимо удалять указанные соли для предотвращения их накопления, что может приводить к засорению и протеканию побочных реакций солей. Предпочтительный способ удаления избытка аминной добавки и нейтрализованных кислотных частиц заключается в применении экстрактора, в котором реакционную жидкость и водную фазу приводят в контакт. В одном из вариантов реализации настоящего изобретения для удаления по меньшей мере части указанных солей также может быть использовано фильтрование и ионообменные смолы, такие как описаны в US 7495134; US 6153800; и US 8110709.
Преимущественно, амин может выполнять по меньшей мере одну из следующих двух функций: 1) он может нейтрализовать кислоты, например, в реакционной зоне, для уменьшения разложения лиганда и катализатора; и 2) он может регулировать рН на стадии экстракции. Преимущественно, стадия экстракции может выполнять по меньшей мере одну из следующих трех функций: 1) удаление нейтрализованных кислотных соединений (в виде солей или кислот) из системы, 2) обеспечение воды для разложения отравляющего фосфита и 3) удаление избытка амина для предотвращения накопления амина во избежание избыточного образования тяжелых соединений. Комбинация трех характеристик обеспечивает самоуравновешивающуюся систему, в которой исключены экстремальные значения эффективного рН и образования тяжелых соединений при сохранении возможности регулируемого гидролиза отравляющего фосфита.
Амин может быть добавлен в процесс по существу в любой точке, при условии достижения требуемой концентрации амина. Например, амин преимущественно добавляют в процесс по меньшей мере в одной из реакционных зон и/или зон экстракции. В одном из вариантов реализации водорастворимый амин добавляют в процесс более чем в одном месте. В одном из вариантов реализации настоящего изобретения амин добавляют в воду, подаваемую в зону экстракции. В одном из вариантов реализации настоящего изобретения амин добавляют в первый реактор. Водорастворимый амин в двух точках добавления может быть одинаковым или различным.
В одном из вариантов реализации настоящего изобретения амин добавляют, главным образом или полностью, в реакционную зону, а скорость добавления водорастворимого амина в реакционную жидкость в реакционной зоне изменяют для регулирования рН водного потока, выходящего из зоны экстракции, для регулирования кислотности в реакционной зоне. В другом варианте реализации настоящего изобретения амин добавляют, главным образом, в зону экстракции, а скорость добавления водорастворимого амина в зону экстракции изменяют для регулирования рН водного потока, выходящего из зоны экстракции. В одном из вариантов реализации настоящего изобретения амин вводят в зону экстракции в составе входящего водного потока. Происходит in situ образование аминно-аммониевого буфера по мере поступления кислоты в зону экстракции вместе с органической фазой, например, реакционной жидкостью из реакционной зоны.
Амин преимущественно удаляют из процесса с водной фазой, которую выводят из зоны экстракции. Таким образом, в процесс должно быть добавлено дополнительное количество амина для поддержания требуемой концентрации амина. Количество добавляемого амина может быть определено посредством определения рН в зоне водной экстракции, например, измерением рН водного потока, выходящего из зоны экстракции, например, остаточного потока из экстрактора. Преимущественно, количество добавляемого амина достаточно для поддержания рН в указанном водном потоке, выходящем из зоны экстракции, на уровне от 4,5 до 9,0, предпочтительно от 5,6 до 8,0, более предпочтительно от 6,0 до 7,5 и наиболее предпочтительно от 6,3 до 7,2. Иногда в течение коротких периодов времени могут быть использованы относительно высокие значения рН от 7,0 до 9,0 для уменьшения значительного разложения лиганда, например, при нарушении технологических параметров в случае наличия значительного гидролиза лиганда, но в течение более продолжительных периодов это приведет к медленному накоплению отравляющего фосфита. В альтернативном варианте в течение коротких промежутков времени могут быть использованы относительно низкие значения рН (от 4,5 до 6,0) для максимальной активности и превращения олефина (вследствие минимальной концентрации отравляющего фосфита) за счет более высокого расхода лиганда. Указанная ситуация может иметь место при более низком качестве сырья или материалов, содержащих высокие концентрации вторичных или внутренних олефинов, для которых необходимы более активные катализаторы для сохранения производительности. Маловероятно, что указанный сценарий является экономичным для продолжительных периодов времени, что обусловлено расходами в результате разложения лиганда, но возможность быстрого возврата в предпочтительный диапазон рН посредством простого увеличения скорости добавления амина демонстрирует гибкость настоящего изобретения. Поскольку амин удаляют в экстракторе, то повышение и понижение рН экстрактора легко регулировать посредством изменения скорости добавления амина в процесс для обеспечения указанной процедуры смягчения, не нарушая процесс гидроформилирования. Необходимо избегать значений рН выше 9 вследствие низкой активности катализатора (из-за высокого содержания отравляющих фосфитов) и избыточного образования тяжелых соединений.
Измерение рН может быть проведено с помощью любых средств, известных специалистам в данной области техники, включая, например, обычное титрование или имеющиеся в продаже рН-метры с надлежащей калибровкой. Для целей настоящего изобретения сделано предположение, что кислотность органической фазы или «эффективный рН» органической фазы коррелирует с наблюдаемым рН остатков экстрактора.
В способе согласно настоящему изобретению по меньшей мере часть водорастворимого амина удаляют с водным слоем или фазой зоны экстракции и, следовательно, амин не накапливается в органической фазе. Поскольку водорастворимый амин склонен присутствовать в водной фазе, происходит его непрерывное удаление, и амин не накапливается в органическом слое или фазе. Одна из стадий способа согласно настоящему изобретению включает по меньшей мере частичное выделение в зоне экстракции по меньшей мере одного нейтрализованного фосфорсодержащего кислотного соединения из реакционной жидкости с получением водного потока, выходящего из зоны экстракции, и обработанной жидкости реакции гидроформилирования. Выделение включает приведение в контакт реакционной жидкости с водным раствором в зоне экстракции, в которой происходит экстракция. Приведение в контакт в зоне экстракции обеспечивает не только удаление свободных фосфорсодержащих кислотных соединений из реакционной жидкости, содержащей катализатор на основе комплекса металла с фосфорорганическим лигандом, но и удаление нейтрализованного фосфорсодержащего кислотного соединения. Обработанная реакционная жидкость может быть возвращена в реакционную зону. Основная часть полярной аминной добавки переходит в водную фазу в виде свободного амина или аммониевой соли в растворе.
Водный раствор, подаваемый в зону экстракции, преимущественно содержит большую часть воды, предпочтительно деионизированной или дистиллированной воды. Питательная вода может содержать следовые примеси, добавки или консерванты, например, добавки против коррозии, которые не влияют на катализатор гидроформилирования. Некоторые из указанных добавок могут обладать собственным буферным эффектом, но в одном из вариантов реализации настоящего изобретения они обеспечивают менее 10% общей нейтрализации кислоты, происходящей в экстракторе. Как указано выше, в одном из вариантов реализации настоящего изобретения весь или часть амина может быть добавлена в водный раствор, подаваемый в зону экстракции.
Способ, которым аминосодержащую реакционную жидкость из реакционной зоны и питательную воду приводят в контакт в зоне экстракции, а также количество водного раствора, температура, давление и время контакта не являются узко критичными и должны лишь быть достаточными для получения требуемого результата. Снижение одного из указанных условий может быть компенсировано увеличением одного или более других условий, при этом следствие также верно. В целом, для большинства случаев подходит температура жидкости от 10°С до 120°С, предпочтительно от 20°С до 80°С и более предпочтительно от 25°С до 60°С, хотя при необходимости может быть использована более низкая или высокая температура. Преимущественно, приведение в контакт в зоне экстракции проводят при давлении в диапазоне от внешнего давления до существенно более высокого давления, чем давление в реакторе, а время контакта может варьироваться от нескольких секунд или минут до нескольких часов или более. В целом, предпочтительно пропускать реакционную жидкость через водный раствор в колонне экстрактора противоточным способом. В колонне могут быть использованы ситчатые тарелки, вибрирующие тарелки, структурированные или неструктурированные насадки и т.п.
Водный поток, выходящий из зоны экстракции, преимущественно удаляют из процесса, и он может быть утилизирован или использован в соответствии со способами, известными специалистам в данной области техники.
Успешность удаления фосфорсодержащих кислотных соединений из реакционной жидкости может быть определена измерением скорости разложения (расходования) фосфорорганического лиганда, присутствующего в реакционной среде гидроформилирования. Скорость расходования может варьироваться в широком диапазоне, например, от менее 0,06 до 5 грамм на литр в сутки, и ее регулируют в соответствии с требуемым компромиссом между стоимостью лиганда и частотой обработки для удержания гидролиза на уровне ниже автокаталитического уровня. Предпочтительно, водную экстракцию проводят так, чтобы скорость расхода необходимого фосфорорганического лиганда, присутствующего в реакционной среде гидроформилирования, сохранялась на приемлемом уровне, например, менее 0,5 грамм лиганда на литр в сутки и более предпочтительно менее 0,1 грамм лиганда на литр в сутки, и наиболее предпочтительно менее 0,06 грамм лиганда на литр в сутки. По мере протекания нейтрализации и экстракции фосфорсодержащих кислотных соединений в водный раствор в зоне экстракции, рН водной фазы, выходящей из зоны экстракции, постепенно снижается, и скорость подачи водорастворимого амина в реакционную зону может быть увеличена для компенсации указанного эффекта.
Удаление по меньшей мере некоторого количества фосфорсодержащих кислотных соединений, например, H3PO3, H3PO4, альдегидокислот, таких как гидроксиалкилфосфоновые кислоты, такие как гидроксибутилфосфоновая кислота и гидроксипентилфосфоновая кислота, и т.п., из системы гидроформилирования обеспечивает возможность регулирования кислотности реакционной среды гидроформилирования, тем самым стабилизируя используемый фосфорорганический лиганд посредством предотвращения или снижения его гидролитического разложения. Не ограничиваясь теорией, полагают, что добавление водорастворимого амина в процесс и обеспечение возможности его прохождения через весь процесс способствует нейтрализации амином кислот по мере их образования. Поскольку водорастворимый амин присутствует на ранних стадиях процесса, то необходимо гораздо меньшее количество амина, по сравнению с известным уровнем техники, при сохранении неожиданно очень эффективного регулирования рН и, следовательно, активности и характеристик разложения лиганда, без заметного увеличения образования тяжелых соединений. При добавлении амина в зону экстракции также происходит нейтрализация кислоты вследствие миграции некоторого количества амина в органическую фазу и/или миграции кислоты в щелочную водную фазу, и общее разделение значительно способствует удалению кислотных соединений (в виде свободной кислоты или нейтрализованной соли) в водную фазу.
В одном из вариантов реализации настоящего изобретения могут быть использованы эпоксидные добавки для уменьшения содержания сильнокислотных примесей, как описано в заявке на патент WO, номер заявки PCT/US13/058714, поданной 9 сентября 2013 года. Эпоксидные добавки могут быть добавлены непрерывно или «по мере необходимости». Полученный эпоксидный аддукт также удаляют с помощью не содержащего буфера экстрактора, и указанное удаление усиливается в присутствии небольшого количества водорастворимых аминов согласно настоящему изобретению. Предпочтительные эпоксиды представляют собой водорастворимые или малорастворимые в воде эпоксиды (их растворимость увеличивается при взаимодействии с кислотными соединениями), поэтому указанные аддукты эффективно выводят из системы, например, вместе с водным потоком, выходящим из зоны экстракции.
Процесс гидроформилирования может быть проведен периодическим, непрерывным или полунепрерывным способом и может включать применение любого способа жидкостного и/или газового рецикла катализатора. Конкретный способ гидроформилирования для получения альдегидов из олефин-ненасыщенного соединения, а также реакционные условия и ингредиенты способа гидроформилирования не являются критическими особенностями настоящего изобретения.
В предпочтительном варианте реализации реакционная жидкость гидроформилирования содержит любую жидкость, полученную из любого соответствующего процесса гидроформилирования, которая содержит по меньшей мере некоторое количество четырех различных главных ингредиентов или компонентов, т.е. альдегидный продукт, катализатор на основе комплекса металла с фосфорорганическим лигандом, свободный фосфорорганический лиганд и растворитель для указанного катализатора и указанного свободного лиганда. Композиции реакционной смеси гидроформилирования могут и обычно содержат дополнительные ингредиенты, такие как ингредиенты, специально используемые в процессе гидроформилирования, или образованные in situ во время указанного процесса. Примеры таких дополнительных ингредиентов включают непрореагировавший олефиновый исходный материал, газообразный монооксид углерода и водород, инертные примеси, которые поступают в систему вместе с сырьем, такие как метан, диоксид углерода и т.п., и образованные in situ побочные продукты, такие как насыщенные углеводороды и/или непрореагировавшие изомеризованные олефины, соответствующие олефиновым исходным материалам, продукты разложения лиганда и высококипящие жидкие побочные продукты конденсации альдегида, а также другие инертные материалы типа сорастворителя или углеводородных добавок, в случае их использования.
Реакционные условия процесса гидроформилирования могут включать любые подходящие условия гидроформилирования, используемые для получения оптически активных и/или оптически неактивных альдегидов. Используемые условия реакции гидроформилирования зависят от типа требуемого альдегидного продукта. Например, общее давление газообразного водорода, монооксида углерода и исходного олефина в процессе гидроформилирования может составлять от 1 до 69000 кПа. Однако, в целом, предпочтительно проводить процесс при общем давлении газообразного водорода, монооксида углерода и исходного олефина менее 14000 кПа и более предпочтительно менее 3400 кПа. Минимальное общее давление ограничено, главным образом, количеством реагентов, необходимых для достижения требуемой скорости реакции. Более конкретно, парциальное давление монооксида углерода в процессе гидроформилирования предпочтительно составляет от 1 до 6900 кПа и более предпочтительно от 21 до 5500 кПа, тогда как парциальное давление водорода предпочтительно составляет от 34 до 3400 кПа и более предпочтительно от 69 до 2100 кПа. В целом, молярное соотношение газообразных H2:CO может составлять от 1:10 до 100:1 или выше, и более предпочтительное молярное соотношение составляет от 1:10 до 10:1. В целом, процесс гидроформилирования может быть проведен при любой пригодной к эксплуатации температуре реакции. Преимущественно, процесс гидроформилирования проводят при температуре реакции от -25°С до 200°С, предпочтительно от 50°С до 120°С.
Процесс гидроформилирования может быть проведен с применением одного или более подходящих реакторов, таких как, например, реактор с неподвижным слоем, реактор с жидким слоем, реактор с пробковым потоком, смесительный реактор непрерывного действия (CSTR) или суспензионный реактор. Оптимальный размер и форма реактора зависят от типа используемого реактора. Используемая реакционная зона может представлять собой одну емкость или может содержать две или более отдельных емкостей, расположенных последовательно или параллельно. Реакционные стадии могут быть осуществлены посредством постепенного добавления одного из исходных материалов к другому. Кроме того, реакционные стадии могут быть объединены посредством совместного добавления исходных материалов. Если полное превращение не является необходимым или достижимым, то исходные материалы могут быть выделены из продукта, например, перегонкой, и исходные материалы затем возвращают в реакционную зону.
Зона экстракции, используемая согласно настоящему изобретению, может представлять собой одну емкость или может содержать две или более отдельных емкостей. В одном из вариантов реализации настоящего изобретения реакционная емкость может быть использована в качестве экстрактора, например, если процесс проводят в периодическом режиме.
Процедура рецикла, в целом, включает непрерывное или периодическое выведение части жидкой реакционной среды, содержащей катализатор и альдегидный продукт, из реактора гидроформилирования, т.е. реакционной зоны, и выделение из нее альдегидного продукта с помощью композиционной мембраны, такой как описана в US 5430194 и US 5681473, или более стандартным и предпочтительным способом перегонки, т.е. испарительного разделения, в одну или более стадий при нормальном, пониженном или повышенном давлении, в зависимости от обстоятельств, в отдельной зоне перегонки; не испарившийся остаток, содержащий металлический катализатор, возвращают в реакционную зону, как описано, например, в US 5288918. Конденсация испарившихся материалов и последующее их разделение и выделение, например, с помощью дополнительной перегонки, могут быть проведены любым стандартным способом, и неочищенный альдегидный продукт может быть, при необходимости, направлен на дальнейшую очистку и выделение изомеров, гидрирование, окисление и/или конденсацию, а выделенные реагенты, например, олефиновый исходный материал и синтез-газ, могут быть возвращены в зону гидроформилирования (реактор) любым требуемым способом. Рафинат, содержащий металлический катализатор, выделенный в результате указанного мембранного разделения, или не испарившийся остаток, содержащий металлический катализатор, выделенный в результате указанного испарительного разделения, может быть возвращен в зону гидроформилирования (реактор) любым требуемым, стандартным способом.
Материалы конструкции не являются особенно критичными для настоящего изобретения и могут быть легко подобраны специалистами в данной области техники. Процесс гидроформилирования может быть проведен, например, в эмалированном реакторе, в реакторе из нержавеющей стали или в реакционном оборудовании аналогичного типа. Реакционная зона может быть оснащена одним или более внутренними и/или внешними теплообменниками(-ом) для регулирования нежелательных температурных колебаний или для предотвращения возможных «разбросов» температуры реакции.
В целом, предпочтительно проводить процесс гидроформилирования в непрерывном режиме. Непрерывные процессы гидроформилирования, с применением или без применения рецикла олефина и/или катализатора, хорошо известны в данной области техники.
Используемая зона разделения может представлять собой одну емкость или может содержать две или более отдельных емкостей. В одном из вариантов реализации смесь альдегидных продуктов может быть отделена от других компонентов неочищенной реакционной смеси, в которой образована альдегидная смесь, любым походящим способом, таким как, например, экстракция растворителем, кристаллизация, перегонка, испарение, разделение на пленочном испарителе, испарение с падающими пленками, разделение фаз, фильтрование и т.п., или любыми их комбинациями. Может быть необходимо удалять альдегидные продукты из неочищенной реакционной смеси по мере их образования с помощью улавливающих агентов, как описано в WO 88/08835. Реакционная зона(-ы) и зона(-ы) разделения, используемые согласно настоящему изобретению, могут быть расположены в одной емкости или в разных емкостях. Например, технология реактивного разделения, такая как реактивная перегонка, реактивное мембранное разделение и т.п., может быть проведена в реакционной зоне(-ах). Один из способов отделения альдегидной смеси от других компонентов неочищенной реакционной смеси представляет собой мембранное разделение, которое описано, например, в патентах США 5430194 и 5681473.
Более конкретно, перегонка и выделение требуемого альдегидного продукта из реакционной жидкости, содержащей катализатор на основе комплекса металла с фосфорорганическим лигандом, может быть проведена при любой подходящей температуре. В целом, предпочтительно проводить указанную перегонку при относительно низких температурах, таких как ниже 150°С и более предпочтительно при температуре от 50°С до 140°С. В целом, предпочтительно также проводить указанную перегонку альдегидов при пониженном давлении, например, при общем давлении газов, которое существенно ниже общего давления газов, используемого при гидроформилировании, с участием низкокипящих альдегидов (например, C4 - C6) или под вакуумом, с участием высококипящих альдегидов (например, C7 или более). Например, обычная практика включает воздействие на жидкую среду продукта реакции, выведенную из реактора гидроформилирования, пониженного давления для испарения существенной части непрореагировавших газов, растворенных и жидкой среде, которая теперь имеет гораздо более низкую концентрацию синтез-газа, чем реакционная среда, подаваемая в зону перегонки, например, испаритель/разделитель, где происходит перегонка требуемого альдегидного продукта. В целом, для большинства целей достаточным является давление перегонки от вакуумного давления до общего давления газов 340 кПа.
Иллюстративные оптически неактивные альдегидные продукты включают, например, пропиональдегид, н-бутиральдегид, изобутиральдегид, н-валеральдегид, 2-метил-1-бутиральдегид, гексаналь, гидроксигексаналь, 2-метил-1-гептаналь, нонаналь, 2-метил-1-октаналь, деканаль, адипальдегид, 2-метилглутаральдегид, 2-метиладипальдегид, 3-гидроксипропиональдегид, 6-гидроксигексаналь, алкенали, например, 2-, 3- и 4-пентеналь, алкил-5-формилвалерат, 2-метил-1-нонаналь, 2-метил-1-деканаль, 3-пропил-1-ундеканаль, пентадеканаль, 3-пропил-1-гексадеканаль, эйкозаналь, 2-метил-1-трикозаналь, пентакозаналь, 2-метил-1-тетракозаналь, нонакозаналь, 2-метил-1-октакозаналь, гентриаконтаналь, 2-метил-1-триаконтаналь и т.п.
Иллюстративные оптически активные альдегидные продукты включают (энантиомерные) альдегидные соединения, полученные способом асимметричного гидроформилирования согласно настоящему изобретению, такие как, например, S-2-(п-изобутилфенил)пропиональдегид, S-2-(6-метокси-2-нафтил)пропиональдегид, S-2-(3-бензоилфенил)пропиональдегид, S-2-(3-фтор-4-фенил)фенилпропиональдегид и S-2-(2-метилацетальдегид)-5-бензоилтиофен.
Конкретные варианты реализации изобретения
Все доли и проценты в следующих примерах выражены относительно массы, если не указано иное. Давление представляет собой абсолютное давление, если не указано иное.
Общий способ
Используют систему реактора жидкостного рецикла, которая состоит из двух реакторов смешения объемом 1 литр, выполненных из нержавеющей стали и соединенных последовательно. Каждый реактор оснащен вертикально установленной мешалкой и кольцевым трубчатым рассекателем, расположенным вблизи дна реактора. Каждый рассекатель содержит множество отверстий достаточного размера для обеспечения требуемого потока газа в жидкое тело в реакторе. Рассекатели используют для подачи олефина и/или синтез-газа в реактор, и она также могут быть использованы для возврата непрореагировавших газов в каждый реактор. Каждый реактор имеет оболочку из кремнийорганического масла для регулирования температуры реактора. Реакторы 1-2 дополнительно соединены трубопроводами для перекачивания непрореагировавших газов и трубопроводами для перекачивания части жидкого раствора, содержащего альдегидный продукт и катализатор, из реактора 1 в реактор 2. Таким образом, непрореагировавший олефин из реактора 1 подвергают дополнительному гидроформилированию в реакторе 2. Каждый реактор содержит также пневматический регулятор уровня жидкости для поддержания требуемого уровня жидкости. Реактор 2 имеет спускное отверстие для удаления непрореагировавших газов.
Часть жидкого реакционного раствора непрерывно перекачивают из реактора 2 в испаритель, который состоит из нагретой емкости при пониженном давлении. Выходящий из испарителя поток направляют в разделитель газа-жидкости, расположенный в нижней части испарителя, где испаренный альдегид отделяют от нелетучих компонентов жидкого реакционного раствора. Испаренный альдегидный продукт конденсируют и собирают в приемнике продукта. Пневматический регулятор уровня жидкости обеспечивает регулирование уровня нелетучих компонентов в разделителе, включая уровень раствора катализатора, подлежащего рециклу. Жидкий поток, выходящий из разделителя, направляют в экстрактор, где поток питательной воды приводят в контакт с жидким потоком, выходящим из разделителя, для удаления кислотных соединений. Экстрактор содержит контактную область насадочной колонны и зону разделения фаз, т.е. отстойник. В отстойнике происходит образование водного слоя и отдельного органического слоя. Органический слой, который содержит катализатор, подлежащий рециклу, перекачивают из отстойника по рециркуляционному трубопроводу в реактор 1.

Лиганд A
Сравнительный эксперимент A (не является вариантом реализации изобретения)
Реакцию гидроформилирования проводили в течение 60 дней по общему способу, описанному выше, за исключением того, что поток питательной воды заменяли потоком, состоящим из 0,04 М водного раствора натрий-фосфатного буфера с рН 6,8. В систему реактора загружали 2 литра раствора катализатора, содержащего: (a) дикарбонил-ацетилацетонат родия (280 ppm родия), (b) лиганд A (0,68 масс. %; 3 мол. экв. на моль родия) и (c) смесь растворителей, содержащую 15% по массе UCAR FILMER IBT (2,2,4-триметил-1,3-пентандиола моноизобутират) производства компании The Dow Chemical Company, и 85% по массе смешанных C5 альдегидов (н-валеральдегид и 2-метилбутиральдегид в массовом соотношении примерно 30:1). Затем реакторы нагревали до 75°С под потоком монооксида углерода и водорода. Давление в реакторах 1 и 2 поддерживали при 160 и 110 фунт/кв.дюйм изб. давления, соответственно. Поток смешанных бутеновых олефинов (состоящий из примерно 18% 1-бутена, 37% транс-2-бутена, 30% цис-2-бутена, 5% изобутена и 10% н-бутана) подавали в реактор 1 со скоростью 1,74 грамм моль на литр объема реактора в час. Систему испарителя эксплуатировали при 11 фунт/кв.дюйм абс. давления и при 102-104°С.
За 20 дней наблюдали значительное накопление натрия в органическом слое отстойника (до измеренного значения 20 ppm). Произошло единичное забивание трубопровода.
Пример 1
Использовали способ сравнительного эксперимента A, который проводили в течение 42 дней, за исключением того, что не использовали натрий-фосфатный буферный раствор, а рН остаточного потока водного экстрактора регулировали при среднем значении 6,0 посредством добавления триэтаноламина (TEA) (в виде 12 масс. % водного раствора) непосредственно в реактор 1 с помощью шприцевого насоса. Первоначально TEA добавляли в реактор 1 со скоростью 0,002 ммоль/л раствора катализатора, и указанная скорость равна ожидаемой молярной скорости образования кислоты в результате разложения лиганда. Периодически измеряли фактическое образование кислоты в результате разложения лиганда с помощью ионной хроматографии и жидкостной хроматографии при высоком давлении, и соответствующим образом регулировали скорость добавления TEA.
В течение 42 дней суммарное общее количество TEA, добавленного в реактор 1, составило 20 миллимоль TEA/литр раствора катализатора, что в молярном выражении эквивалентно измеренному общему количеству кислоты, образованной в результате разложения лиганда.
Наблюдали превосходный материальный баланс TEA на основании экспериментальных данных водного потока, выходящего из отстойника. Для общего периода 42 дней измеренный расход лиганда был сопоставим с его расходом в контрольной системе, эксплуатированной в таких же условиях с применением обычного водного экстрактора с натрий-фосфатным буфером.
В течение 42 дней проводили многочисленные отборы аналитических проб и технологические замеры, которые сравнивали со способом сравнительного эксперимента A.
В отличие от сравнительного эксперимента A, не наблюдали неблагоприятного влияния на скорость гидроформилирования, селективность в отношении нормальных и изо-валеральдегидов или скорость образования продуктов разложения лиганда. Кроме того, в отличие от сравнительного примера A, не наблюдали новых промежуточных фосфорсодержащих соединений с применением31P ядерного магнитного резонанса (ЯМР) в процессе гидроформилирования и, неожиданно, не наблюдали засорения (о чем свидетельствует отсутствие закупоривания трубопровода). В процессе гидроформилирования TEA, подаваемый при указанных выше условиях, поступал в водный поток, выходящий из экстрактора, в виде свободного TEA или солей TEA, удаляя кислотные соединения, образованные в результате разложения лиганда. Кроме того, не наблюдали увеличения содержания натрия в органическом слое отстойника.
Пример 2
По окончании 60 дней сравнительного эксперимента A поток раствора натрий-фосфатного буфера заменяли питательным потоком, состоящим только из воды, и начинали схему добавления TEA, использованную в примере 1. Со временем содержание натрия в органической фазе снижалось с периодическими всплесками, обусловленными растворением твердых солей. Через 40 дней эксплуатации систему по существу не содержала натрия. Данный пример демонстрирует, что применение только водного экстрактора и TEA обеспечивает удаление солей, осажденных в сравнительной системе, при сохранении хороших характеристик катализатора, без необходимости в остановке процесса для очистки.
Сравнительный эксперимент B (не является вариантом реализации изобретения)
Использовали способ примера 1, за исключением того, что в реактор 1 не подавали TEA и не использовали экстрактор. Через 190 дней экстрагировали раствор катализатора, содержащий более 4000 ppm гидроксибутилфосфоновой кислоты (HBPA, кислотный продукт разложения лиганда, содержание которого может быть легко измерено с помощью ионной хроматографии), с помощью двух порций воды, снизив количество HBPA в органической фазе до 1000 ppm. Экстракцию проводили в реакторе после остановки подачи сырья и охлаждения до температуры окружающей среды. Не обнаружили образования эмульсий или потерь родия в водных экстракционных слоях. Однако указанное количество остаточной HBPA является недопустимо высоким.
Сравнительный эксперимент C (не является вариантом реализации изобретения)
Обрабатывали органическую фазу, полученную в сравнительном эксперименте B, 1% водным раствором триэтаноламина (TEA) (0,2 масс. % раствора катализатора, 0,12 ммоль/л, рН > 9,5), затем провели окончательное промывание водой для полного удаления HBPA из органической фазы. Однако наблюдали значительное образование эмульсии и потерю 1,5 ppm родия в промывочной воде.
Сравнительный эксперимент D (не является вариантом реализации изобретения)
Полученную органическую фазу, содержащую промытый TEA катализатор, из сравнительного эксперимента C испытывали на активность и обнаружили, что вследствие очень высокого рН промывочного водного TEA (>9,5) полученный раствор катализатора быстро накапливает отравляющий фосфит, что обусловливает лишь 25% активности относительно свежего катализатора. Эффективный рН промытого раствора катализатора слишком высок для обеспечения возможности гидролиза отравляющего фосфита.
Сравнительные эксперименты B-D демонстрируют, что применение известного уровня техники дает неудовлетворительные результаты. Сравнительный эксперимент B демонстрирует, что простая водная экстракция неэффективна для удаления кислот до приемлемых значений. Сравнительный эксперимент C демонстрирует, что хотя добавление избыточного амина эффективно для удаления кислоты, имеют место другие нежелательные последствия, такие как потеря родия и образование эмульсии, которые недопустимы в промышленном производстве. Кроме того, сравнительный эксперимент D демонстрирует, что обработанный материал сравнительного эксперимента C демонстрирует неприемлемо низкую активность для гидроформилирования.
Пример 3
Повторяли способ примера 1, за исключением того, что проводили группы испытаний по 30 дней с различными средними значениями рН (измеренными в водном потоке, выходящем из экстрактора) и получали значения активности катализатора (на основании значений кинетической модели лиганда, построенной для свежего катализатора при таких же условиях) и норм расхода лиганда. Средние значения рН регулировали посредством изменения скорости подачи TEA в реактор 1. Относительные нормы расхода лиганда определяли относительно центральной точки (рН 6,8), которой присвоили значение 1. Взаимосвязь активности катализатора и расходом лиганда представлена в таблице 1. Поскольку активность и норма расхода напрямую влияют на стоимость получения альдегида и связаны со средним рН в экстракторе, то может быть определена требуемая скорость реакции и скорость расхода лиганда для данного процесса, и затем может быть выбрано среднее значение рН для достижения требуемых результатов.
Таблица 1
Пример 4
Использовали способ сравнительного эксперимента A, за исключением того, что не использовали раствор натрий-фосфатного буфера, а TEA (в виде 700 ppm масс. водного раствора; рН 9,3) подавали в экстрактор с постоянной скоростью 12,6 г раствора в час. В течение 46 дней с помощью ионной хроматографии периодически измеряли содержание кислоты, образованной в результате разложения лиганда, и рассчитывали расход лиганда на основании анализов жидкостной хроматографии при высоком давлении. Скорость разложения лиганда переменна; поэтому концентрация кислоты также изменялась, и молярное соотношение TEA:кислота составляло от 1,7:1 до 2,8:1, а рН водного потока, выходящего из экстрактора, составлял от 6,4 до 7,2. Общее количество TEA, загруженного в экстрактор, составило 66 ммоль, что в молярном выражении эквивалентно измеренному общему количеству кислоты, образованной в результате разложения лиганда.
В отличие от способа сравнительного примера A, не наблюдали неблагоприятного влияния на скорость расхода лиганда, скорость гидроформилирования, образование тяжелых соединений или селективность в отношении продукта. Важно, что не наблюдали никаких признаков наличия нерастворимых материалов в органическом слое отстойника, таких как засорение или закупоривание трубопровода/фильтра.
Реферат
Настоящее изобретение относится к способу гидроформилирования. Способ включает следующие стадии: проведение в реакционной зоне реакции гидроформилирования с применением реакционной жидкости, содержащей (a) фосфорсодержащее кислотное соединение, (b) катализатор на основе комплекса металла с фосфорорганическим лигандом, который содержит металл группы 8, 9 или 10, связанный в комплекс с фосфорорганическим лигандом, и, необязательно, (c) свободный фосфорорганический лиганд; приведение в контакт по меньшей мере части реакционной жидкости с водорастворимым амином с нейтрализацией по меньшей мере некоторого количества фосфорсодержащего кислотного соединения и получением нейтрализованного фосфорсодержащего кислотного соединения; по меньшей мере частичное выделение в экстракционной зоне по меньшей мере одного нейтрализованного фосфорсодержащего кислотного соединения из реакционной жидкости; и удаление нейтрализованного фосфорсодержащего кислотного соединения из экстракционной зоны с водным потоком, выходящим из экстракционной зоны. При этом количество амина является таким, что концентрация амина в реакционной зоне составляет не более 0,075 ммоль на литр жидкости реакции гидроформилирования. Предлагаемый способ позволяет снизить или исключить сильнокислотные соединения в реакционной зоне гидроформилирования для минимизации разложения лиганда при снижении степени отравления фосфита. 10 з.п. ф-лы, 1 табл., 4 пр.
Формула
Документы, цитированные в отчёте о поиске
Способ рекуперации катализатора гидроформилирования,содержащего родий и фосфины с сульфокислыми группами
Комментарии