Рацемические и оптические активные производные тетралина и 7-гидрокситетралина - RU2073673C1
Код документа: RU2073673C1
Чертежи

Описание
Настоящее изобретение касается замещенных тетралинов, 7-гидрокситетралинов и близких им соединений формулы (1), представленной ниже, которые благодаря ингибированию 5-липоксигеназного фермента и/или блокированию лейкотриеновых рецепторов полезны для предупреждения или лечения астмы, аритмии, псориаза, язвы, инфаркта миокарда и связанных с ними болезненных состояний у млекопитающих. Настоящее изобретение касается также фармацевтических композиций и промежуточных соединений, используемых для синтеза указанных соединений формулы (1).
Крефт и др. в патенте США N 4 661 596 описывают соединения, представляющие собой дизамещенные нафталины, дигидронафталины или тетралины, имеющие формулу:

в которой пунктирные лини обозначают возможные двойные связи, Ra представляет собой 2-пиридил, 2-хинолил, 2-пиразинил, 2-хиноксалинил, 2-тиазолил, 2-бензотиазолил, 2-оксазолил, 2-бензоксазолил, 1-алкил-2-имидазолил или 1-алкил-2-бензимидазолил, и Rb представляет собой гидрооксигруппу, низший алкокси или перфторалкил. Как и соединения настоящего изобретения, эти соединения ингибируют липоксигеназный фермент и антагонизируют действия лейкотриена D4 и в связи с этим полезны для предупреждения и лечения астмы. Используемая в данном описании химическая номенклатура соответствует номенклатуре, принятой Международным Союзом теоретической и прикладной химии "Номенклатура органической химии", 1979 г. Изд. "Пергамон Пресс", Нью-Йорк, 1979 г.
Настоящее изобретение касается рацемических или оптических активных соединений, имеющих структурную
формулу:

в которой
n равно 1; Х представляет собой СН2; X' представляет собой СН2; Y и Y', взятые вместе, образуют карбонильную группу, или Y и Y', взятые в отдельности, Y представляет собой водород и Y' представляет собой гидрооксигруппу; Z представляет собой СН2; Z' представляет собой СН; R представляет собой 2-пиридил или 2-хинолил; и R' представляет собой (С1 C3)алкил.
С учетом легкости получения и ценной биологической активности предпочтительными являются соединения формулы (1), в которой n, X, X', Y,Y' и R', Z и Z1 имеют значения, определенные выше, и R1 представляет собой C2 или C3 алкил. Самыми предпочтительными являются рацемические
или оптически активные соединения, имеющие относительную стереохимическую формулу:
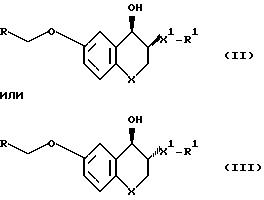
особенно те рацемические или оптически активные соединения формулы (II) или (III), в которых Х и Х1 каждый представляет собой CH2, R представляет собой 2-пиридил или 2-хинолил и R1 представляет собой н-пропил, а также пара оптически активных энантиомерных соединений формулы (III), в которой Х и Х1 каждый представляет собой CH2, R представляет собой 2-хинолил и R1 представляет собой н-пропил.
Настоящее изобретение описывает также фармацевтические композиции для ввода в организм млекопитающих животных, которые содержат соединение формулы (I) и фармацевтически приемлемый носитель; и способ ингибирования 5-липоксигеназного фермента и/или блокирования лейкотриеновых D4 рецепторов в организме млекопитающих животных с целью предупреждения или лечения астмы (особенно у мужчин), артрита, псориаза, язвы желудочно-кишечного тракта или инфаркта миокарда.
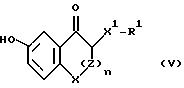
И, наконец, настоящее изобретение касается ценных
промежуточных соединений структурной формулы:

Настоящее изобретение практически легко осуществимо.
Соединения формулы (1), в которых Y + Y' карбонил, или Y представляет собой Н и Y' представляет собой ОН, и Х1 представляет собой CH2, независимо от того, являются ли они геометрическими (цис-транс) или оптическими изомерами, получаются путем химических превращений, показанных на схемах 1 и 2, в которых n, X, Z, Z1, R и R1 определены выше. Ниже подробно обсуждаются различные химические превращения, представленные на данных схемах, а также химические превращения, необходимые для получения соединения формулы (1) с другими значениями и Y, Y' и X1, и способы разделения цис-транс и оптических изомеров.
Конденсация согласно схеме 1 обычно осуществляется с фенольной группой в защищенной форме, как это показано, причем металл является предпочтительной защищающей группой. Согласно предпочтительным условиям используется молярный избыток требуемого альдегида и молярный избыток вторичного амина, такого как пирролидин или пиперидин в качестве основания. Такое основание облегчает конденсацию за счет образования енаминового промежуточного соединения.


Данная реакция обычно осуществляется в инертном к реализационной смеси растворителе, причем особенно подходящим растворителем для данной цели являются низшие спирты, такие как метанол. Температурные условия данного химического превращения не являются критическими, обычно они составляют от 0 до 70oC, и особенно подходящей является комнатная температура.
Под термином "инертный к реакционной среде растворитель" имеется в виду растворитель, который не взаимодействует с исходными веществами, химическими реагентами, промежуточными продуктами или конечными продуктами таким образом, чтобы отрицательно влиять на выход желаемого продукта.
С-Алкилирование, согласно схеме 1, осуществляется путем превращения сначала кетона (А) в его литиевую соль, обычно in situ, путем воздействия в основном одного молярного эквивалента сильного пространственно затрудненного основания, такого как диизопропиламид лития, осуществляемого обычно при низкой температуре (например, примерно от -40 до -80oC, обычно при температуре бани сухой лед-ацетон). Данную соль, в свою очередь, подвергают взаимодействию с алкилирующим реагентом, предпочтительно высоко реакционноспособным иодидом, обычно в молярном избытке, в присутствии молярного избытка гексаметилфосфорамида, теперь уже при более высокой температуре (например, примерно от 0 до 40oC). Последние указанные реагенты удобным образом вводятся в холодный раствор литиевой соли, и температуре дают возможность повышаться до комнатной по мере протекания реакции. Реакции получения соли и алкилирования обычно осуществляются в одном и том же инертном к реакционной смеси растворителе (например, тетрагидрофуране). Для специалистов в данной области должно быть ясно, что любые свободные гидрокси- или карбоксигруппы в алкилирующем реагенте должны быть в защищенной форме.
Преобразования с помощью каталитического гидрирования (дебензилирование, ввод Н2 в двойную связь) согласно схемам 1 и 2 осуществляются в обычных условиях, обычно в инертном к реакционной смеси растворителе, и предпочтительно с использованием в качестве катализатора благородного металла, и в условиях умеренной температуры (например, примерно от 0 до 70oC и под давлением водорода (например, под давлением примерно от 1 до 10 атмосфер). Хотя в отдельных случаях могут быть желательны более высокие давления, указанные умеренные давления позволяют использовать значительно меньшие затраты труда и менее дорогостоящее оборудование.
Подходящими для данной цели металлическими катализаторами являются платина, палладий, рений, родий и рутений, либо на носителе, либо без носителей, а также известные их каталитические соединения, такие как оксиды, хлориды и т.д. Примерами подходящих носителей катализаторов являются углерод, двуокись кремния, сульфат бария. Катализаторы могут быть приготовлены непосредственно в условиях рабочего процесса путем предварительного восстановления подходящей соли каталитического соединения.
Примерами предпочтительных катализаторов являются 5% палладдий на угле, 5% платина на угле, 5% родий на угле, хлорид платины, хлорид палладия, оксид платины и оксид рутения.
Наиболее предпочтительным катализатором согласно изобретению является палладий на угле. Подходящими растворителями для процесса гидрирования являются низшие алканолы, этилацетат и тетрагидрофуран.
Простые метиловые эфиры [соединения формулы (С)] согласно схеме 1 деблокируются с образованием соответствующего фенольного производного также общепринятыми способами; так, например, они деблокируются с использованием концентрированных НВr или ВВr3.
Фенольное алкилирование, как показано на схеме 2, представляет собой обычную реакцию нуклеофильного замещения.
Данная реакция обычно осуществляется в присутствии основания достаточной крепости и взятого в количестве, по меньшей мере достаточном для нейтрализации кислотного побочного продукта (НХ2).
В тех субстратах, которые содержат алифатическую спиртовую группу (например, соединение IV, в котором Y2 представляет собой Н и Y3 представляет собой ОН), обычно используются основания достаточной крепости для превращения данной группы в анион, в количестве не большем, чем достаточное для превращения более кислотного фенола в соль. Когда любой из данных реагентов содержит группу с кислотностью, аналогичной или превышающей кислотность соединения нуклеофильного замещения, то также потенциально участвующие в процессе группы желательнее всего вводить в защищенной форме (например, гетероароматическая фенольная группа в виде метокси- или бензилоксигруппы, карбоксильная группа в виде сложного метилового или бензилового эфира, удаляемые путем гидролиза или гидрогенолиза согласно подробно описанным здесь примерам).
Данные реакции нуклеофильного замещения осуществляются в инертном к реакционной среде растворителе, предпочтительно таком, который имеет меньшую кислотность, чем вытесняющий фенол, спирт или меркаптан. Наиболее предпочтительными являются полярные апротонные растворители, такие как диметилформамид или ацетон, обычно с молярным избытком более легко доступного из двух реагентов. Температура не является критически важным фактором, обычно удовлетворительной является температура в пределах примерно от 10 до 70oC, и наиболее желательна комнатная температура. Согласно одному из предпочтительных вариантов, фенол, спирт или меркаптан необратимо превращается в анион с помощью основания, такого как гидрид натрия. Согласно другим предпочтительным вариантам, в качестве основания используется К2CO3 в присутствии NaI; или используется Cs2CO3 в присутствии CsI.
Реакция "восстановления" согласно схеме 2 требует восстановления кетона во вторичный спирт, для чего доступны ряд реагентов избирательного действия. В случае, когда отсутствуют какие-либо другие группы, восстанавливаемые с помощью LiAlH4 (такие как карбокси- или метоксикарбонил), данный реагент вполне подходит для данной цели. С другой стороны, когда присутствуют такие группы, в качестве восстанавливающего реагента предпочтительным является NaBH4. В любом случае гидридные восстановления обычно осуществляются в инертном к реакционной смеси растворителе (таким как тетрагидрофуран в случае LiAlH4, метанол или сочетание метанол/тетрагидрофуран в случае NaBH4). В любом случае температура не является критической, обычно вполне удовлетворительна температура в пределах от 0 до 50oС, и наиболее предпочтительна комнатная температура. Данный этап восстановления обеспечивает возможность получения смеси цис- и трансизомеров (как иллюстрировано формулами (II) и (III), и при данном гидридном восстановлении обычно наблюдается данный результат. Если особенно желателен один из этих изомеров, то обычно определяются такие условия восстановления, которые благоприятствуют образованию желаемого изомера. Так, например, восстановление NaBH4 в присутствии хлорида цезия обычно благоприятствует образованию цис-изомера. Каталитическое гидрирование обычно также является желательным способом восстановления, и, как правило, оно осуществляется в условиях, которые несколько более жесткие, чем описанные выше (например, более продолжительное время, более высокое содержание катализатора, более высокие температура и/или давление).
Гидрирование осуществляется предпочтительно на таких субстратах, как
которые не содержат никакой другой легко гидрируемой группы. Катализатор Pd/C благоприятствует образованию цис-изомера. Однако в результате изменения условий и замены катализатора эту тенденцию можно модифицировать и даже изменить на обратную.
В случае, когда, согласно данному изобретению, образуются и цис и транс-изомер одновременно, обычно они могут разделяться стандартными химическими приемами (например путем избирательной или фракционной кристаллизации, хроматографии и т.д.).
Те кетоновые соединения формулы (I) или (IV), в которых Y и Y1 или Y2 и Y3 образуют карбонильную группу, содержат асимметричный атом углерода в альфа-положении, которое смежно с карбонильной группой, и, следовательно, являются рацемическими соединениями, способными к разделению на оптически активные энантиомеры путем, например, превращения рацемата в диастереоизомерные соли с оптически активной кислотой, которые обычно способны разделяться путем фракционной кристаллизации.
Как возможный вариант, если данный субстрат содержит карбоксильную группу, то образуются разделяемые диастереоизомерные соли с оптически активным органическим амином. Оптическая активность может индуцироваться также использованием оптически активного реагента на данной стадии, в результате чего образуется асимметричный атом углерода, например в результате использования оптически активного катализатора типа катализатора Уилкинсона, или благородного металла, нанесенного на оптически активный носитель, при осуществлении гидрирования. Оптически активные кетоны также могут быть получены путем обычного повторного окисления оптически активного спирта, описываемого в следующем абзаце, например, путем окисления Джоунса, пример которого приводится ниже.
Гидроксисоединения формулы (I) и (IV), где V (или Y2 представляют собой водород и Y1 (или Y3) представляет собой ОН, содержат два таких асимметричных атома углерода, соответствующих двум рацематам и четырем оптически активным соединениям. Один из этих рацематов является указанным выше цис-изомером и другой является транс-изомером. Каждый из этих рацематов способен к разделению на пару энантиомеров через диастереоизомерные соли, как подробно разъяснено выше. Однако желательно превращение данного рацемического спирта в соответствующие диастереоизомерные сложные эфиры или уретаны, образуемые с оптически активными кислотой или изоцианатом. Такие производные соединения с ковалентной связью обычно подвергаются различным способам разделения (например, хроматографии), отличным от способов разделения диастереизомерных солей.
Эти диастереоизомерные сложные эфиры образуются из спиртов и оптически активных кислот стандартными способами, обычно способами, включающими активацию кислоты, например хлорангидрида, смешанного ангидрида с алкилхлорформатом, или с дегидративным агентом сочетания, таким как дициклогексилкарбодиимид.
Предпочтительной оптической активной кислотой в данном случае является S-O-ацетилминдальная кислота. После разделения образующихся диастереоизомерных сложных эфиров путем, например, хроматографических методов, они гидролизуются общепринятыми способами, например водной кислотой или водным основанием, и в результате получаются энантиомерные оптически активные спирты.
Пролекарственные сложные эфиры данного
изобретения получаются способами, аналогичными
способам, используемым для синтеза сложных эфиров, как описано выше. Сложные эфиры с альфа-аминокислотами, включающими природные L-аминокислоты, обычно
получаются из подходящей аминокислоты, в которой
альфа-аминогруппа, группы заместителя NH2 или NH (например, лизин, орнитин, аргинин, гистидин, триптофтан), гидроксигруппы (серин,
гомосерин, треонин, тирозин), меркаптогруппы (цистеин) и
карбоксигруппы (глутаминовая кислота, аспарагиновая кислота) находятся в защищенной форме (например N-бензилоксикарбонил, О- и S-бензил),
которые обычно удаляются путем каталитического гидрирования в
следующей стадии. Аналогичным образом, в случае сложных эфиров с первичным или вторичным аминозаместителями данные кислоты будут
образовывать сопряженные соединения с защищенными аминогруппами. Такая
защита, безусловно, не является обязательной в случае с кислотами, содержащими третичные аминозаместители. И, наконец, карбокси
замещенные сложные эфиры наилучшим образом получаются из циклического
ангидрида

Что касается биологической активности соединений, отвечающих данному изобретению, то известно, что арахидоновая кислота метаболизируется в организме млекопитающих животных двумя различными путями, один из которых приводит к образованию простагландинов и тромбоксанов и другой приводит к различным окислительным продуктам, называемым лейкотриенами, которые обозначаются буквенно-цифровым сочетанием, например Б4, С4 и D4. Первый этап данного окислительного пути это окисление арахидоновой кислоты под воздействием 5-липоксигеназного фермента, фермента, который обычно ингибируется соединениями /1/, отвечающими данному изобретению, блокируя таким образом синтез всех лейкотриенов. Он как таковой обеспечивает механизм, позволяющий использовать данные соединения для лечения или предупреждения астмы (когда LTC4 и LTD4 являются медиаторами при воспалениях), артрита (когда LTB4 является медиатором при воспалении), псориаза (когда LTB4 является медиатором), язв (когда LTC4 и LTD4 являются медиаторами) и инфаркта миокарда (когда LTB4 является медиатором). Повышение этой ингибирующей фермент активности проявляется в обычной способности данных соединений антагонизировать лейкотриен D4 (то есть блокировать рецепторы LTD4). Обычно данные соединения антагонизируют также лейкотриен В4. Информацию по лейкотриенам можно получить в публикации: Bailey и др. Ann. Reports Med.Chem. стр.203 217 1982 г.).
Активность соединений формулы (1) в условиях "ин витро" определяется при следующем испытании. Клетки RBL-1, сохраняемые в форме монослоя, выращиваются в течение 1 или 2 дней в тканевой культуре в минимальной основной среде (Eagle) с солью Earl плюс 15% эмбриональной бычьей сыворотки, дополненной антибиотическим (противогрибковым раствором (GIBCO). Клетки один раз промываются RPMI 1640 (GIBCO) и повторно суспендируются в RPMI 1640 плюс 1 микромолярный глютатион до клеточной плотности 1 х 10 клеток/мл. 0,5 мл этой клеточной суспензии инкубируется при 30> 198>C вместе с 0,001 мл диметоксисульфоксидного раствора лекарства в течение 10 минут. Реакция начинается при одновременном вводе 0,005 мл (14С) арахидоновой кислоты в этаноле и 0,002 мл А23187 в диметилсульфоксиде до получения конечных концентрация соответственно 5,0 и 7,6 микроМол. После инкубирования в течение 5 минут при температуре 30oС реакция прекращается при вводе 0,27 мл ацетонитрила/уксусной кислоты (100/0,3) и данная среда осветляется путем центрифугирования. Осуществляется анализ распределения продукта путем инжекции 0,2 мл осветвленного поверхностного слоя от центрифугирования в колонку жидкостной хроматографии высокого разрешения. Разделение радиоактивных продуктов осуществляется в радиальной колонке РАХ CN (внутренним диаметром 5 мм, Waters) с использованием системы растворителей ацетонитрил (Н2O) уксусная кислота (0,1%) с линейным ацетонитрильным градиентом от 35 до 70% в течение 15 минут со скоростью 1 мл/мин. Количественное определение осуществляется с использованием регистрирующего устройства радиоактивности Бертольда, снабженного встроенным интегратором и ячейкой потока объемом 0,2 мл. Омнифлер 2,4 мл/мин (NEN) с колонкой для потока. Суммарные единичные дозы для каждого продукта рассчитываются как процент от общих суммарных доз и затем сопоставляются со средними контрольными значениями. Данные результаты выражаются как "процент подавления" и выражаются в виде кривых зависимости от логарифма концентрации лекарства. Значения 1С50 определяются графически (см.таблицу).
Для соединения известного из патента США 4 661 596 значение 1С50 ингибирования фермента липоксигеназы составляет 50 микроМол, что примерно в 5 10 раз меньше активности, проявляемой соединениями настоящего изобретения.
Проводили испытание
рецептора лейкотриена 4 (LTD4) с определением способности соединения конкурировать с радиомеченным LTD4 для специфических
точек рецептора LTD4 на легочных оболочках морской свинки. В этом испытании
морские свинки в возрасте 3 4 недели акклиматизировались в стандартных условиях в течение 3 дней, после чего они
умерщвлялись. Конечный возраст свинок составлял 24 31 день. Морских свинок оглушали
ударом по задней части шеи и обескровливали путем выреза сонной артерии. Раскрывали полость грудной клетки и легкое
удаляли, промывали в 50 мМол. буфера Трис (рН 7,0) и помещали в чистый буферный
раствор. В данной и во всех последующих операциях все ткани и буферные растворы выдерживались на льду в процессе
препарирования, и все операции центрифугирования осуществлялись при температуре 4oС. Ткани бронхов и соединительные ткани извлекались из легких. Данная ткань взвешивалась и помещалась в 50
мл поликарбонатные пробирки с буферным раствором в соотношении 1 г ткани/3 мл
буферного раствора. Ткань гомогенизировалась посредством устройства Тиссьюмайзера Текмана при полной скорости в течение
30 секунд и центрифугировалась во вращающемся устройстве Sovall SS-34 со
скоростью вращения 3250 об/мин в течение 15 минут. Поверхностный слой центрифугировался со скоростью 1900 об/мин в течение 10
минут. Полученный спрессованный осадок снова суспендировался в буферном
растворе с использованием гомогенизатора Тиссьюмайзера со средней скоростью (позиция 75) в течение 10 секунд. Эта новая
суспензия снова центрифугировалась со скоростью 19000 об/мин в течение 10 минут.
Образующийся осадок снова суспендировался с использованием устройства Тиссьюмайзера с небольшой скоростью (позиция 50)
в течение 10 секунд в 1 мл буферного раствора на грамм исходной ткани. Эта
конечная суспензия перемешивалась при температуре 4oC с одновременным отбором аликвот в полипропиленовые пробирки
и выдерживалась при -70oC. В полистирольную пробирку вводились
следующие компоненты:
(1) 25 мкл одного из следующих:
А. Диметилсульфоксид (для определения общего
связывания).
В. 1 микроМол.LTD4 (для определения неспецифического связывания).
С. 30 наноМол 100 микроМол соединения в диметилсульфоксиде.
(2) 0,025 мл 3Н-LTD4 (с удельной активностью 30 60 Ci/мМол) в 50 мМол буфера Трис (рН 7,0) + 10 микроМол L-цистеина (12000 15000 срm/0,025 мл).
(3) 0,2 мл разбавленного мембранного препарата (1 Мг/мл) (препарат разбавлялся в 50 микроМол. буферного раствора Трис + MgCl2, так чтобы в 200 микрол. белка достигалась концентрация MgCl2 10 микроМол).
Эти
реакционные пробирки инкубировались при 25oC в течение 30 минут. В каждую пробирку
вводили 4 мл холодного буферного раствора Трис + 10 микроМол MgCl2. Содержимое их быстро
фильтровалось через фильтр Ватмана GF/C в разделительном устройстве Yeda. Фильтр трехкратно
промывался четырьмя миллилитрами буферного раствора Трис-MgCl2. Этот фильтр переносился в
сцинтилляционную ампулу. В ампулу вводилась ультрафлюоресцентная сцинтилляционная жидкость. Ампула
закрывалась, подвергалась завихряющему движению и подвергалась детектированию в течение трех часов.
Процент специфического связывания рассчитывался с помощью следующей формулы:
SB (X NSB)/(TB
NSB),
где Х cpm отсчетов в минуту образца;
NSB cpm отсчетов в минуту
неспецифического связывания;
ТВ срm отсчетов в минуту общего связывания.
Процент
специфического связывания представлен графически как функция концентрации соединения. 1С50 является концентрацией, при которой имеет место 50% SB. Кi рассчитывается по следующей формуле:
Ki (IC50)/[1+(L/Kd)]
где L концентрация вводимого лиганда
(микроМол) срm ввода/срm 1 микроМол 3Н-LTD4;
Кd 1 микроМол (константа диссоциации).
Человеческие нейтрофильные лейкоциты используются для измерения конкуренции испытываемых молекул с (3Н)-LТB4 для связывания в рецепторе LTB4. В данном испытании нейтрофилы отделяются от гепаринизированной человеческой периферической крови (обычно 100 мл) с использованием градиента Hypagye-Ficoll (плотностью 1,095 г/мл). Для повторного суспендирования клеток используется уравновешенный солевой раствор Хэнка (HBSS), содержащий 0,1 г/100 мл альбумина бычьей сыворотки (HBSS BSA). Этот одноэтапный процесс Hypague-Ficoll дает высокочистую популяцию нейтрофил (более чем 95%). Клеточная жизнеспособность определяется по эксклюзии красителя трипанового синего (должно быть более 95%), и функциональное единство нейтрофил определяется по восстановлению нитросинего тетразолиевого (должно быть более чем на 85% положительным). Подвергаемые испытанию соединения растворяются в диметилсульфоксиде до концентрации 100 микроМол. Эти растворы разбавляются в 500 раз с использованием HBSS-BSA. 100 микроМолярная концентрация лекарства достигается путем ввода разбавления образца в аликвоте (0,5 мл) в реакционную пробирку. Приготавливается ряд разбавленных растворов 1 3 и 1 5 (принятым образом), и алюквата (0,5 мл) этих разбавленных растворов вводится в инкубационную пробирку. В боросиликатные пробирки (12 х 75 мм) вводится (3Н) LTBA/NEN: удельная радиоактивность более чем 180 Сi/мМол; 0,005 мл в абсолютном этаноле). Затем вводится 0,5 мл лекарственного раствора (см.выше). Реакция связывания инфицируется путем добавления 0,5 мл охлажденных льдом нейтрофил с клеточной плотностью (5 х 106 клеток/мл) и продолжается при температуре 4oC в течение 30 минут. Инкубация прекращается путем быстрой фильтрации через фильтр Ватман GF/C стекло с отделением свободного от связанного радиоактивного меченного лиганда. Эти фильтры трехкратно промываются охлажденным льдом HBSS, высушиваются, помещаются в 4 мл ультрафлюор и подвергаются индикации. Общее связывание определяется как СРМ, присутствующий на фильтре (клеточно ассоциированный), когда радиоактивно меченный лиганд инкубируется с нейтрофилами при отсутствии какого-либо конкурирующего агента. Неспецифическое связывание получается путем инкубации клеток с радиоактивно меченным лигандом плюс 1 микроМол. нерадиоактивно меченного LTB4. Специфическое связывание является суммарным связыванием СРМ, скорректированным на неспецифическое связывание СРМ. Каждая пробирка корректируется на неспецифическое связывание. Точки полумаксимального смещения радиоактивного меченого лиганда определяются путем графического анализа полулогарифмической кривой процента специфического связывания (при отсутствии какого-либо конкурента) в зависимости от концентрации.
Для оценки соединений формулы (1) в условиях "ин виво" они испытываются путем так называемого анализа на летальность PAF.
Материалы для испытания.
Мыши: самцы разновидности CD1, все примерно одинакового веса (примерно по 26 г), по 12 штук на группу.
Носитель для дозирования лекарства, вводимого орально: EES (5% этанол, 5% эмульфор, 90% солевой раствор). Выдерживается при комнатной температуре.
Лекарства: для обычного отбора дозой 50 мг/кг 20 мг лекарства растворяются в 4 мл EES с использованием ультразвукового воздействия в ультразвуковой ванне или измельчения в измельчающем устройстве Ten Broech для растворения лекарства, если это необходимо. Если растворимость все еще остается проблемой, то лекарство используется в форме суспензии.
Носитель для внутривенной инъекции; солевой раствор вместе с 2,5 мг/мл бычьего сывороточного альбумина (BSA, Cигма A 4378) и 0,05 мг/мл пропранолола (Сигма H 0884). Ежедневно приготавливается свежая порция и выдерживается при комнатной температуре. Фактор активирования промбоцитов (РАF): приготавливается 10 микроМол. основной раствор путем растворения 1 мн PAF (Calbiochem 429460) в 0,18 мл этанола. Этот раствор выдерживается при -20oC и разбавляется в носителе в день использования. Концентрация используемого PAF регулируется таким образом, чтобы при вводе дозой 0,1 мл/10 г тела умерщвлялось примерно 80% необработанных контрольных организмов. Это обычно составляет 0,028 г/кг (разбавление основного раствора от 1 до 2034). Раствор приготавливается в стеклянных емкостях и используется посредством стеклянных шприцов для уменьшения до минимума адгезии PAF. Он выдерживается при комнатной температуре. Положительный контроль: Используется фенидон в количестве 25 мг/кг (его приблизительная доза ED50).
Метод.
За 45 минут до инъекции PAF мыши обрабатываются путем орального ввода лекарства в количестве 0,1 мл/10 г тела. Через 35 40 минут их помещают под воздействие обогревающей лампы для расширения хвостовой вены для инъекции PAF. PAF вводится путем внутривенной инъекции в количестве 0,1 мл/10 г веса тела, и смерть обычно наступает в течение 30 минут, редко по прошествии 60 минут. Результаты выражаются как процент смертности в сравнении с контрольными результатами. Ввиду того, что данный анализ чувствителен к эндогенным катехоламинам (например, защита мышей бета- агонистами), то для разрешения этой возможной проблемы используется пропранолол. Он помогает также, если мыши акклиматизируются к комнатным условиям до испытания и если комнатные шумы и температура сохраняются умеренными и постоянными. Расстояние от нагревательной лампы должно устанавливаться таким, чтобы расширение сосудов происходило без создания видимого стресса на мышей. Голодание мышей должно быть исключено.
Вариации.
1. Время для орального дозирования может быть изменено.
2. Внутривенный ввод дозированного лекарства возможен за счет совместной инъекции лекарства и PAF в том же объеме и в том же носителе, что указаны выше. Для совместной инъекции PAF приготавливается в двойной концентрации от желаемой концентрации в солевом растворе вместе с BSA и пропанололом, как указано выше, и лекарство приготавливается в двойной концентрации от желаемой концентрации в том же носителе. Оба препарата смешиваются в равных объемах непосредственно перед инъекцией.
Для использования с целью профилактики или лечения астмы, артрита, псориаза и язвы желудочного тракта у млекопитающих животных, в том числе человека, соединение формулы (1) дается в количестве, ингибирующем 5-липоксигеназу и/или блокирующем лейкотриеновый рецептор, составляющем примерно 0,5 50 мг/кг/день в виде единичной или разделенных дневных доз. Более предпочтительный дозированный предел составляет от 2 до 20 мг/кг/день, хотя в отдельных случаях по решению лечащего врача могут потребоваться дозы за указанными пределами. Предпочтительным способом введения лекарства является оральный, но в отдельных случаях предпочтителен парэнтеральный ввод (например внутримышечный, внутривенный, внутрикожный, например в тех случаях, когда впитывание лекарства при оральном вводе снижается под влиянием заболевания, или когда пациент не в состоянии проглотить лекарство.
Соединения, отвечающие данному изобретению, обычно вводятся в форме фармацевтических композиций, включающих по меньшей мере одно из соединений формулы (1), вместе с фармацевтически пригодным носителем или разбавителем. Такие композиции обычно приготавливаются общеизвестным образом с использованием твердых или жидких носителей или разбавителей, как принято для подходящего способа ввода: для орального ввода форме таблеток, твердых или мягких желатиновых капсул, суспензий, гранул, порошков и т.д. и для парэнтерального ввода в форме инъекционных растворов или суспензий и т.д.
Нижеследующие примеры приводятся для иллюстрации данного изобретения.
Пример 1.
6-(2-Хинолил)метокси-4-хроманон.
Смесь 6-окси-4-хроманона (10,0 г, 0,0609 моля), 2-хлорметилхинолина (11,9 г, 0,0670 моля), иодида натрия (109 г, 0,0670 моля), карбоната калия (25,3 г, 0,183 моля) и ацетона (200 мл) нагревается с обратным холодильником в течение ночи в атмосфере N2. По прошествии 17 часов смесь становится светлее и анализ методом тонкослойной хроматографии (10% этилацетата) CH2Cl2 показывает полное превращение исходного продукта в несколько менее полярный продукт. Смесь охлаждается, фильтруется и фильтрат концентрируется в вакууме. Остаточный продукт концентрирования растворяется в этилацетате (400 мл), промывается Н2О и солевым раствором, высушивается над MgSO4 и концентриpуется в вакууме до темнокоричневого масла. После очистки в колонке с силикагелем при элюировании смесью 10% этилацетата (CH2Cl2) получается желаемый продукт в виде беловатого твердого вещества, 15,3 г (82%), точка плавления 112 114oC; анализ методом тонкослойной хроматографии (этилацетат:CH2Cl2 в соотношении 1:9), Rf 0,30.
Пример 2.
3-Оксиметилен-6-(2-хинолил)метокси-4-хроманон.
В раствор желаемого продукта предыдущего примера (7,00 г, 0,0229 моля) и избытка этилформата (35 мл) в толуоле (80 мл) при комнатной температуре вводят отдельными порциями в течение 5 минут 2,2 г (0,0458 моля) 50% гидрида натрия в минеральном масле. Желтовато-зеленая смесь перемешивается при комнатной температуре в течение 5 минут, затем добавляется 2 капли этанола для инициирования реакции. В течение 5 минут смесь превращается в красно-оранжевую с выделением газа и является слабо экзотермической. Эта смесь перемешивается при комнатной температуре в течение 1 часа, после чего, как показывает анализ методом тонкослойной хроматографии (5% CH3OH/CH2Cl2), исходный материал полностью превращается в более полярный продукт. Эта реакционная смесь вливается в 400 мл ледяной воды, величина рН доводится до 5 посредством 2 нормальной HCl, и она экстрагируется этилацетатом (500 мл). Органический слой промывается водой и солевым раствором, высушивается над MgSO4 и концентрируется в вакууме до образования пастообразного желтого твердого вещества. После повторного перемешивания с гексаном с целью удаления минерального масла получается желаемый продукт с выходом 85% тонкослойная хроматография (CН3OH CH2Cl2 1:19), Rf 0,40.
Пример 3.
3-Диазо-6-(2-хинолил)метокси-4-хроманон.
В растворе конечного продукта предыдущего примера (7,60 г, 0,023 моля) и сухого триэтиламина (6,4 мл, 0,046 моля) в сухом СН2Cl2 (100 мл) при температуре -30oС (баня с смесью сухой лед ацетон) вводят по каплям в течение 20 минут раствор азида толиза (4,5 г, 0,023 моля) в CH2Cl2 (25 мл). Реакционная смесь медленно нагревается до комнатной температуры в течение ночи при перемешивании. По прошествии 18 часов, как показывает тонкослойная хроматография (20% этилацетата (CH2CLl2) происходит полное исчезновение исходного материала и образование менее полярного продукта. Эта смесь обрабатывается 1 нормальной NaOH (100 мл) и перемешивается в течение 10 минут. После обработки солевым раствором слои разделяются и органический слой разбавляется 200 мл этилацетата. Затем метиленхлорид удаляется в вакууме. Этилацетатный остаточный продукт промывается водой и солевым раствором, высушивается над MgSO4 и концентрируется в вакууме, и в результате получается конечный желаемый продукт в виде темно-желтого твердого вещества, 6 г (90%); анализ методом тонкослойной хроматографии, (этилацетат: CH2Cl2 1:4), Rf 0,27.
Пример 4.
3-Циклогексилокси-6-(2-хинолил)метокси-4-хроманон.
В суспензию конечного продукта предыдущего примера (1,50 г, 4,53 моля) и циклогексанола (1,7 мл, 16,4 ммоля) в сухом толуоле (25 мл) при температуре 70oС вводится 5 мг димера ацетата родия (2-вал). В ходе реакции быстро выделяется N2 и смесь становится гомогенной. Анализ методом тонкослойной хроматографии (20% этилацетата (CH2Cl2) показал образование менее полярного продукта и наличие лишь следов исходного материала. Реакционная смесь концентрируется в вакууме. Остаточный продукт растворяется в этилацетате (100 мл), промывается водой и солевым раствором, высушивается над MgSO4 и концентрируется в вакууме до образования масла янтарного цвета. После хроматографического разделения в колонке с силикагелем при элюировании смесью 10% этилацетата (CH2 Cl2 получается желаемый продукт в виде желтого осадка, 0,59 г (32%); анализ тонкослойной хроматографией (этилацетат:CH2Cl2 в соотношении 1:4) Rf 0,68. ИК спектр (KBr) 2940, 1700, 1490 см-1, масса-спектр (m/e) 403, 1780 (М+). Осуществляя процесс таким же образом, но с использованием октилмеркаптана вместо циклогексанола, получается 3-октилтио-6-(2-хинолил)метокси-4-хроманон.
Пример 5.
Цис и транс-3-циклогексилокси-6-(2-хинолил)метокси-4-хроманол.
В раствор конечного продукта предыдущего примера (580 мг, 1,44 моля) в метаноле (30 мл) при температуре 0 5oC вводят 56 мг (1,45 ммоля) боргидрида натрия. Реакционная смесь нагревается до комнатной температуры при перемешивании. По прошествии 1 часа, как показала тонкослойная хроматография (20% этилацетата) CH2Cl2), происходит полная конверсия исходного материала с образованием двух более полярных продуктов. Смесь концентрируется в вакууме. Остаточный продукт концентрирования растворяется в этилацетате, промывается водой и солевым раствором, высушивается над MgSO4 и концентрируется в вакууме, и в результате получается желто-белое твердое вещество. После разделения в хроматографической колонке на силикагеле с элюированием смесью 20% этилацетата (CH2 Cl2 получается конечный менее полярный цис-продукт в виде желтой пены (450 г) и более полярный конечный транс-продукт в виде светло-желтого масла (30 мг). Общий выход 82% Этот цис-изомер перекристаллизовывается из толуола-гексана с образованием 417 мг желто-белых игольчатых кристаллов, температура плавления 127 130oC, и транс-изомер перемешивается с гексаном с образованием 11 мг белого твердого вещества, с температурой плавления 63 65oC.
Цис-изомер. Спектр ИК (КВr) 1500, 2940 см-1.
Масс-спектр (m/e) 405, 1922 (М+).
Элементарный анализ.
Рассчитано по формуле C25H27NO4.
C, 74,05; H, 6,71; N, 3,45%
Найдено: C, 74,07;
H, 6,69; N, 3,38%
Транс-изомер. ИК спектр (КВr) 1495, 2940 см-1.
Масс-спектр (m/e) 405, 1980 (M+).
При осуществлении процесса таким же образом 3-октилтиоаналог предыдущего примера превращается в смесь цис- и транс-3-октилтио-6-(2-хинолил)метокси-4-хроманола.
Пример 6.
3-(1-Метилэтокси)-6-(2-хинолил)метокси-4-хроманон.
Осуществляя процессы таким же образом, как описано в примере 4, конечный продукт примера 3 (1,12 г) и изопропиловый спирт превращаются в соединение, которое после хроматографического разделения дает желаемый конечный продукт, 1,48 г (81% ), с температурой плавления 85oC; анализ методом тонкослойной хроматографии (этилацетат:CH2Cl2 в соотношении 1:9), Rf 0,35.
Пример 7.
Цис- и транс-3-(1-метилэтокси)-6-(2-хинолил)метокси-4-хроманол.
Осуществляя процессы таким же образом, как в примере 5, конечный продукт предыдущего примера (1,38 г) превращается в желаемые конечные соединения после их хроматографического разделения.
Цис-изомер. 1,19 г (86% ), температура плавления 116 118oC, менее полярный. ИК спектр (KBr) 1490 см-1. Масс-спектр (m/e) 365, 1360 (M+).
Элементарный анализ.
Рассчитано согласно формуле: С22H23NO4
C, 72,31; H,
6,34; N, 3,83%
Найдено: C, 71,95; H,6,01; N,3,76%
Транс-изомер. 0,09 г, температура плавления 102 103oC, более полярный. ИК спектр (КВr) 1500 см-1. Масс-спектр
(m/e) 365, 1360 (М+).
Пример 8.
2-Бутил-3,4-дигидро-7-метокси-1(2Н)-нафталинон.
В раствор (при температуре -78oC) литийдиизопропиламида (из 4,37 мл (31,2 ммоля) диизопропиламина в 28 мл тетрагидрофурана и 11,9 мл (29,8 ммоля) 2,5 Мол н-бутиллития медленно вводят (в течение 15 минут) раствор 5,00 г (28,4 ммоля) 3, 4-дигидро-7-метокси-1(2Н)-нафталинона в 10 мл тетрагидрофурана. Полученная реакционная смесь перемешивается в течение 10 минут при температуре -78oC. Эту охлаждающую баню затем заменяют баней лед-вода (с температурой 0oC), после чего быстро вводят 3,98 мл (35 ммолей) н-бутилиодида. Затем вводят гексаметилфосфорамид (10,4 мл, 60 ммолей) и полученный раствор перемешивается при 25oC в течение 2 часов. Эта реакционная смесь вводится в смесь 200 мл насыщенного хлористого аммония с 300 мл простого эфира. Органический слой отделяется, промывается насыщенным хлористым аммонием (200 мл), насыщенным хлористым натрием (200 мл), высушивается над сульфатом магния и выпаривается с образованием масла, которое очищается в хроматографической колонке на силикагеле (250 г) с элюированием смесью 5% простого эфира-гексана, и в результате получается 1,6 г (24%) желаемого конечного продукта в виде масла. Спектр1H-ЯМР (COCl3) дельта (ч/млн): 0, 92 (шир.т. СН3), 1,1 2,7(м. 9Н), 2,87 (м.CH2), 3,80 (OCH3), 7,0(м.2ArH) и 7,41(д.J 2 Гц, ArH).
Пример 9.
2-Бутил-3, 4-дигидро-7-окси-1(2Н)-нафталинон.
Смесь 19,1 г (82,4 ммоля) конечного продукта из предыдущего примера в 77 мл ледяной уксусной кислоты и 77 мл концентрированной бромистоводородной кислоты нагревается с обратным холодильником в течение 3 часов с отбором небольшого объема (примерно 30 мл) дистиллата. Реакционная смесь охлаждается, вводится в 1 литр смеси лед-вода и экстрагируется три раза простым эфиром порциями по 200 мл. Соединенные эфирные экстракты промываются 1 литром воды и 500 мл насыщенного раствора бикарбоната натрия, высушиваются над сульфатом магния и выпариваются до получения масла, которое отверждается при отстаивании, и в результате получается 17,2 г (96%) конечного желаемого продукта, который перекристаллизовывается из смеси простой эфир-гексан; температура плавления продукта 55 58oC; ИК спектр (СНСl3) 3352, 3580, 1671 см-1.
Спектр1H-ЯМР (CDCl3) дельта (ч/млн): 0,90(м.CH3), 1,1-2,7 (м.9Н); 2,90 (м.CH2, 7,1 (м. 2ArH) и 7,75 (шир.с. 1ArH).
Элементарный анализ.
Рассчитано согласно формуле: С14H18O2
: C, 77,03; H, 8,31%
Найдено: C, 77,25; H, 8,
25%
Пример 10.
2-Бутил-3,4-дигидро-7-(2-хинолил)-метокси-1(2Н)-нафталинон.
Cмесь 4,35 г (20,0 ммолей) конечного продукта предыдущего примера, 4,27 г (20 ммолей) хлоргидрата 2-хлорметилхинолина, 16,3 г (50 ммолей) карбоната цезия и 200 мг (0,769 ммоля) иодида цезия в 43 мл ацетона нагревается с обратным холодильником в течение 21 часа. Реакционная смесь охлаждается, разбавляется 43 мл простого эфира и фильтруется. Фильтрат выпаривается с образованием масла, которое очищается в хроматографической колонке на силикагеле (120 г) при элюировании дихлорметаном, и в результате получается конечный желаемый продукт в виде масла (5,55 г). Это очищенное масло кристаллизуется путем перемешивания с гексаном, и в результате получается 3,22 г (45%) кристаллического продукта, температура плавления 49 51oC.
Масс-спектр (m/e) 359 (M+), 303, 142 и 115. Спектр ИК (СНСl3) 1670, 1600, 1568 см-1.
Спектр1H-ЯМР (СDCl3) дельта (ч/млн): 0,90 (м. CH3), 1,1 2,7(м. 9Н), 2,85 (м.CH2), 5,34 (с.OCH2) и 7,1 8,2 (м. 9АrH).
Элементарный анализ.
Рассчитано согласно формуле: С24H25NO2:
C, 80,18;
H, 7,01; N, 3,90%
Найдено: C, 80,44;
H,7,08; N, 3,76%
Пример 11. Цис- и транс-2-бутил-1,2,3,4-тетра-гидро-7-(2-хинолил)метокси-1-нафтол.
В растворе (ОoC) 2,00 г (5,57 ммоля) конечного продукта предыдущего примера в 40 мл метанола вводят 1,26 г боргидрида натрия. Реакционная смесь перемешивается в течение 2 часов при ОoC и затем концентрируется во вращающемся выпарном аппарате. Остаточный продукт концентрирования растворяется в смеси простого эфира и насыщенного NaCl. Органический слой высушивается над сульфатом магния и выпаривается до образования масла, которое очищается в колонке жидкостной хроматографии умеренного давления на силикагеле с элюированием смесью простой эфир: толуол в соотношении 1:3, и элюируются в последовательном порядке 1,0 г (50%) цис-изомера и 770 мг (38%) транс-изомера, оба в виде масла. Оба изомера кристаллизуются из смеси простой эфир/гексан.
Цис-изомер. Температура плавления 78,5 80oС. Масс-спектр (m\e) 361 (M+), 342, 286, 143, 142 и 115. Спектр ИК (СНСд3) 3590, 3400, 1609, 1600, 1572 см-1.
Спектр1Н-ЯМР (СDCl3, 300 МГц) дельта (ч/млн): 0,89(т. J 7 Гц, CH3), 1,2 1,7(м. 9Н), 2,55 2,82 (м. CH2), 4,53 (д. J 4,0 Гц, CH), 4,73 (ОН), 5,33 (с. CH2O), 6, 85 (д.д. J 8, 2 Гц; ArH), 6,98 (м. ArH), 7, 49(д.д. J 8, 8 Гц, АrН), 7,62(д. J 8 Гц,ArH), 7,68 (д.д. J 8, 8 Гцб ArH), 7,77 (д. J 8 Гц, АrH).
Элементарный анализ.
Рассчитано согласно формуле: С24
H27NO2:
C, 79,74; H, 7,53; N, 3,87%
Найдено: C, 79,44; H, 7,42; N, 3,81%
Транс-изомер. Температура плавления
70 72oС. Масс-спектр (m/e) 361
(М+, 286, 143, 142 и 115. ИК спектр (СНСl3) 3580, 3435, 1605, 1600, 1575 см-1.
Спектр1Н-ЯМР (CDCl3, 300МГц) дельта (ч/млн): 0, 87 (т. J 8 Гц, СН3), 1,1 1,8 (м. 8Н), 1,97 (м. 1Н), 2,66 (м. СН2), 4,32 (т. J 6,98 Гц, СН), 5,33 (с. ОСН2), 6,83 (д.д. J 8, 2 Гц, ArH), 6,96 (д. J 8 Гц, ArH), 7,15 (д. J 2 Гц, ArH), 7,49 (д.д. J 8, 8 Гц, ArH), 7,63 (д. J 8 Гц, ArH), 7,66 (д.д. 8; 8 Гц, ArH), 7,77 (д. J 8 Гц, ArH), 8,03 (д. J 8 Гц, ArH), 8,13 (д. J 8 Гц, ArH).
Элементарный анализ.
Рассчитано согласно формуле: С24H27NO2:
C, 79,74; H, 7,53; N, 3,87%
Найдено: C, 79,38; H,
7,42; N, 3,79%
Пример
12.
Диастереоизомерные транс-2-Бутил-1,2,3,4-тетрагидро-7-(2-хинолил)-метокси-1 -нафтил-R-O-ацетилманделаты.
В раствор (OoC) 764 мг (2,12 ммоля) транс-изомера конечного продукта предыдущего примера, 493 мг (2,54 ммоля) (R/-(-)-O-ацетилминдальной кислоты и 305 мг (2,5 ммоля) 4-(N,N-диметиламино) пиридина в 4 мл дихлорметана добавляют 480 мг (2, 32 ммоля) дициклогексилкарбодиимида. По прошествии 5 минут реакционная смесь нагревается и перемешивается при 25oC в течение 3 часов. Образующийся осадок удаляется путем фильтрации и фильтрат выпаривается с образованием масла, которое очищается методом жидкостной хроматографии умеренного давления на силикагеле с элюированием смесью 20 50% простого эфира гексана, и в результате получаются продукты А и В. Каждый из них кристаллизуется из смеси простой эфир-гексан, и в результате получается 436 г (39%) диастереоизомера А и 466 мг (41%) диастереоизомера В.
Диастереоизомер А. Температура плавления 94 94oC; спектр1H-ЯМР (CDCl3), 300 МГц, дельта (ч/млн): 0,86 (т. J 7 Гц, СН3), 1,1 2,1 (м. 9Н), 2,18 (с. СН3CO), 2,66 (м.СН2), 4,98 (картина АВ, ОСН2), 5,75 (д. J 6 Гц, СН), 5,88 (с. СН), 6,34 (д. J 2 Гц, ArH), 6,77 (д.д. J 8, 2 Гц, ArH), 6,93 (д. J 8 Гц, ArH), 7,1 7, 6 (м. 7 ArH), 7,71 (д.д. J 8, 8 Гц, ArH), 7,81 (д. J 8 Гц, ArH), 8,07 (д. J 8 Гц, ArH), и 8,16 (д. J 8 Гц, ArH).
Диастереоизомер В. Температура плавления 70 81oС. Спектр1H-ЯМР (CDCl3, 300 МГц) дельта (ч/млн): 0,72 (т. J 7 Гц, СН3), 0,8 1,9 (м. 9Н), 2,18 (с. СH3CO), 2,63 (м. СН2), 5,31 (картина АВ; ОСН2 ), 5, 77 (д. J 6 Гц, СН), 5,87 (с. СН), 6,85 (д.д. J 8,2 Гц, ArH); 6,93 (д. J 2 Гц, ArH)- 6,95 (д. J 8 Гц, ArH), 7,3 (м. 2 ArH)- 7,45 (м. 2 ArH), 7,67 (м. 2 ArH), 7,79 (д. J 8 Гц, ArH), 8,05 (д. J 8 Гц, ArH) и 8,15 (д. J 8 Гц, ArH).
Пример 13.
(-)-Трас-2-Бутил-1,2,3,4-тетрагидро-7-(2-хинолил)метокси-1-нафтол.
Смесь 405 мг (0,75 ммоля) диастереоизомера А из предыдущего примера и 832 мг (6,03 ммоля) безводного карбоната калия в 6,25 мл метанола, 6,25 мл тетрагидрофурана и 1,5 мл воды перемешивается в течение 15 часов при температуре 25oC. Эта реакционная смесь затем вводится в 100 мл насыщенного хлорида натрия и экстрагируется три раза простым эфиром порциями по 30 мл. Соединенные эфирные экстракты высушиваются над сульфатом магния и выпариваются до образования масла. Это масло кристаллизуется из смеси простой эфир-гексан, и в результате получается 160 мг (59%) желаемого конечного продукта, температура плавления 59 61oC.
(α)= -26,3° (CH3OH, c 0, 001). Спектр1H-ЯМР (СDCl3, 300 МГц), дельта (ч/млн): 0,89 (т. J 7 Гц, CH3, 1,1 2,1 (м. 9Н), 2,68 (м. CH2), 4,33 (д.д. J 6, 6 Гц, CH), 5,36 (с. OCH2), 6,83 (д.д. J 8; 2 Гц, ArH), 6,97 (д. J 8 Гц, ArH), 7,17 (д. J 2 Гц, ArH), 7,50 (д.д. J 8; 8 Гц, ArH), 7,65 (д. J 8 Гц, ArH), 7,69 (д.д. J 8 Гц, ArH), 7,79 (д. J 8 Гц, ArH), 8,04 (д. J 8 Гц, ArH) и 8,15 (д. J 8 Гц, ArH).
Пример 14.
(+)-Транс-2-Бутил-1,2,3,4-тетрагидро-7-(2-хинолил)метокси-1-нафтол.
Осуществляя процессы таким же образом, как описано в предыдущем примере, диастереоизомер В. продукт примера 13 (0,46 г) превращается в желаемый конечный кристаллизованный продукт, 0,13 г, (54%), температура плавления 58 - 59o С.
(α)= +23,6° (CH3ОН, с=0, 001). Спектр1H-ЯМР идентичен спектру (-)-изомера предыдущего примера.
Пример 15.
2-Бутил-3,4-дигидро-7-(2-пиридил)-метокси-1(2Н)-нафталинон.
Осуществляя процесс таким же образом как в примере 10, конечный продукт примера 9 (5,70 г, 34,3 ммоля) и хлоргидрат 2-пиколилхлорида (5,63 г, 34,3 ммоля) превращаются в данное желаемое соединение, 4,37 г (41%), температура плавления 56 60oC. Масс-спектр (m/e) 309 (М+), 253, 93 и 92. Спектр ИК (СНСl3) 1677, 1608, 1594, 1573 см-1. Спектр1H-ЯМР (CDCl3) дельта (ч/млн): 0,98 (м. CH3, 1,1 2,7 (м. 9Н), 2,96 (м. CH2, 5,25 (с. CH2O), 7,05 7,9 (м. 6 ArH) и 8,3 (шир. д. J 6 Гц, ArH).
Элементарный
анализ. Рассчитано согласно формуле: C20H23NO2:
C, 77,64; H, 7,49; N, 4,35%
Найдено: C, 77,93; N, 7,42; N, 4,50%
Пример 16.
Цис- и транс-2-Бутил-1,2,3,4-тетрагидро-7-(2-пиридил)метокси-1-нафтол
Осуществляя процесс таким же образом, как и в примере 11, конечный продукт предыдущуго
примера (2,29 г, 7,41 ммоля)
превращается в данные желаемые соединения.
Цис-изомер. 0,96 г (42%), температура плавления 101 103oC; менее полярный. Масс-спектр (m/e) 311 (М+), 236, 199, 94, 93 и 92.
ИК-спектр (СНСl3) 3592, 1610, 1594, 1574 см-1. Спектр1H-ЯМР (CDCl3, 300 МГц) дельта (ч/млн): 0,87 (м. CH3, 1,1 1,9 (м. 9Н), 2,5 2,8 (м. CH2), 4,51 (шир. с. CH), 5,13 (с. CH2O), 6,80 (д. J 8 Гц, ArH), 6,91 (шир.с. ArH), 6,97 (шир. д. J 8 Гц, ArH), 7,14 (д.д. J 8, 8 Гц, ArH), 7,44 (д. J 8, Гц, ArH), 7,63 (д.д. J 8; 8 Гц, ArH) и 8,51 (д. J 5 Гц, ArH).
Элементарный анализ.
Рассчитано согласно формуле: C20J25
NO2:
C, 77,14;
H, 8,09; N 4,50.
Найдено: C, 77,31; H 7,94; N, 4,46%
Транс-изомер. 1,12 г (49%), температура плавления 62 64oC; более полярный.
Масс-спектр (m/e) 311 (M+
), 292, 236, 199, 94, 93 и 92. ИК Спектр (СНСl3) 3584, 3414, 1609, 1594, 1574 см-1. Спектр1H-ЯМР (CDCL3, 300 МГц)
дельта (ч/млн): 0,89 (м. CH3),
1,1 2,1 (м. 9Н), 2,67 (м. CH2), 4,32 (шир.с. CH), 5,15 (с. OCH2), 6,79 (д.д. J 8, 2 Гц, ArH), 6,96 (д. J 8 Гц, ArH), 7,11 (д. J 2 Гц,
ArH), 7,17 (д.д. J 8, 8 Гц, ArH), 7,48 (д. J
8 Гц, ArH), 7,66 (д.д. J 8, 8 Гц, ArH) и 8,53 (д. J 5 Гц, ArH).
Пример 17.
3-Изопропил-6-бензилокси-4-хорманон.
Осуществляя процедуру примера 8, 6-бензилокси-4-хорманон и изопропилиодид превращается в данный желаемый продукт.
Пример 18.
3-Изопропил-6-окси-4-хроманон.
Конечный продукт предыдущего примера (10 г) подвергается гидрогенизации при 44 psig (3,08 атм) в присутствии 1 г 5% Pd/C при комнатной температуре в 200 мл CH3ОН и 100 мл тетрагидрофурана до тех пор, пока не потребляется одномолярный эквивалент Н2. Катализатор восстанавливается путем фильтрации над диатомовой землей и конечный продукт извлекается путем выпаривания фильтрата досуха.
Пример 19.
3-Изопропил-6-(2-хинолил)-метокси-4-хроманон.
Осуществляя процедуру таким же образом, как в примере 1, продукт предыдущего примера превращают в данное желаемое соединение.
Пример 20.
Цис- и транс-3-изопропил-6-(2-хинолил)метокси-5-хроманол.
Осуществляя процедуру таким же образом, как в примере 5, продукт из предыдущего примера превращают в данный желаемый продукт.
Пример 21.
Осуществляя процедуру согласно примеру 1 или примеру 10, используя подходящее замещенное хлорметилом гетероциклическое соединение вместо 2-хлорметилхинолина, конечный продукт примера 9 превращается в нижеследующие дополнительные продукты: 2-Бутил-3, 4-дигидро-7-(3-пиридил)метокси-1(2Н)-нафталинон; 2-бутил-3,4-дигидро-7-(4-пиридил)метокси-1-(2Н)-нафталинон; 2-бутил-3,4-дигидро-7-(2-пиразинил)метокси-1(2Н)-нафталинон; 2-бутил-3, 4-дигидро-7-(6-фтор-2-хинолил)метокси-1(2Н)-нафталинон; 2-бутил-3,4-дигидро-7-(2-бензотиазолил)метокси-1(2Н)-нафталинон; 2-бутил-3,4-дигидро-7-(2-пирамидинил)метокси-1(2Н)-нафталинон; и 2-бутил-3, 4-дигидро-7-(2-хиназолинил)метокси-1(2Н)-нафталинон.
Осуществляя процедуру по способу примера 11, эти соединения превращают в соответствующий цис- и транс-2-бутил-1,2,3, 4-тетрагидро-7-(замещенный)метокси-1-нафтол.
Пример 22.
3-Пентилиден-6-(2-хинолил)метокси-4-хроманон.
В смесь 0,112 моля конечного продукта из примера 1 и 0,169 моля пентаналя в 100 мл метанола при температуре 25oC добавляют 14, 1 мл (0,169 моля) пирролидина. Полученный раствор перемешивается в течение 60 часов при температуре 25oC, охлаждается до 0oC и фильтруется и в результате получается конечное соединение.
Используя в данной процедуре соответствующий альдегид вместо пентаналя, получают следующие продукты: 3-(2-Циклопентилэтилиден)-6-(2-хинолил)метокси-4-хроманон; 3-(2-этоксипропилиден)-6-(2-хинолил)метокси-4-хроманон; и 3-(4-метоксикарбонилбутилиден)-6-(2-хинолил)-метокси-4-хроманон.
Пример 23.
3-Пентил-6-(2-хинолил)метокси-4-хроманон.
Смесь 25,2 г конечного продукта предыдущего примера и 2 г 5% Pd/C(50%) H2O в 1 л этилацетата подвергается гидрогенизации при 35 psi (2,45 атм) до тех пор, пока не потребляется 1 молярный эквивалент Н2 (примерно 18 часов). Реакционная смесь фильтруется через диатомовую землю с этилацетатной промывкой, соединенный фильтрат и промывка выпариваются досуха и в результате получается конечный желаемый продукт.
Осуществляя процедуру таким же образом, другие продукты предыдущего примера превращаются в следующие продукты: 3-(2-Циклопентилэтил)-6-(2-хинолил)метокси-4-хроманон; 3-(3-этоксипропил)-6-(2-хинолил)метокси-4-хроманон; и 3-(4-метоксикарбонил)бутил-6-(2-хинолил)метокси-4-хроманон.
Осуществляя процедуру таким же образом, как в примере 11, продукты данного примера подвергаются дальнейшему превращению в соответствующие цис- и транс-2-(замещенные)-6-(2-хинолил)метокси-4-хроманолы.
Пример 24.
3-(4-Карбоксибутил)-6-(2-хинолил)-метокси-4-хроманон.
В раствор 630 мг замещенного 4-(метоксикарбонил)-бутилом продукта предыдущего примера в 100 мл метанола и 25 мл тетрагидрофурана добавляют 10 мл 5-нормальной NaOH. Реакционная смесь нагревается в паровой бане в течение 10 минут. Летучие вещества выпариваются в вакууме, и остаточный продукт выпаривания растворяется в воде и подкисляется разбавленной HCl до достижения величины рН=5. Выпавший осадок конечный желаемый продукт извлекается путем фильтрации и высушивается в воздухе.
Пример 25.
Цис-3-(4-Карбоксибутил)-6-(2-хинолил)метокси-4-хроманол.
Осуществляя процесс гидрогенизации согласно примеру 18, но с двойным количеством катализатора и при температуре 40o C и с потреблением 1 молярного эквивалента Н2, конечный продукт предыдущего примера превращается в конечный продукт данного примера.
Пример 26.
7-Бензилокси-3,4-дигидро-4-(циклопентил)метокси-1-бензоксепин5-(2Н)-он.
В раствор 1,5 г (циклопентил)метанола в 50 мл тетрагидрофурана вводят 720 мг 50%-ного гидрида натрия. После перемешивания до тех пор, пока практически полностью не улетучивается H2 (примерно 30 минут), вводится раствор 4,5 г сырого 7-бензилокси-4-бром-3,4-дигидро-1-бензоксепин-5-(2Н)-она. Реакционная смесь перемешивается при комнатной температуре в течение 5 часов. Тетрагидрофуран выпаривается в вакууме, и остаточный продукт выпаривания растворяется в этилацетате и промывается водой. Этилацетатный слой высушивается над сульфатом натрия и выпаривается в вакууме, и в результате получается желаемое конечное соединение.
Пример 27.
3, 4-Дигидро-7-окси-4-(циклопентил)метокси-1-бензоксепин-5-(2Н)-он.
Смесь 2 г продукта предыдущего примера, 200 мг 10% Pd/C и 50 мл метанола подвергается гидрогенизации во встряхивающем аппарате Парра при 50 psig (3,5 атм) в течение 2,5 часов. Катализатор удаляется путем фильтрации, фильтрат выпаривается в вакууме и в результате получается конечный продукт, который используется без очистки в последующем этапе.
Пример 28.
Цис- и транс-2,3,4, 5-Тетгаридро-4-(циклопентил)метокси-1-бензоксепин5,7-диол.
В раствор 3,5 г продукта предыдущего примера в 100 мл тетрагидрофурана вводят 1 г гидрида лития-алюминия. Реакционная смесь перемешивается при комнатной температуре в течение 15 минут, затем резко охлаждается водой, подкисляется до величины рН=4 разбавленной соляной кислотой и экстрагируется этилацетатом. Этилацетатный слой высушивается над сульфатом натрия и выпаривается в вакууме, и в результате получается смесь конечных продуктов, которая разделяется в хроматографической колонне на силикагеле с элюированием смесью дихлорметан/простой эфир.
Пример 29.
(±)-Транс-2, 3,4,5-Тетрагидро-4-(циклопентил/-метокси-7- (2-хинолил)метокси-1-бензоксепин-5-ол.
В раствор 840 мг транс-изомера конечного продукта предыдущего примера в 25 мл диметилформамида добавляют 154 мг 50%-ного NaH. После перемешивания в течение 20 минут вводят 570 мг 2-хлорметилхинолина. Реакционная смесь перемешивается при комнатной температуре в течение 1 часа, вливается в воду и экстрагируется этилацетатом. Этилацетатный слой высушивается над сульфатом натрия и выпаривается в вакууме и в результате получается желаемое конечное соединение. Аналогичным образом получается соответствующий цис-продукт.
Пример 30.
3-(Октилсульфинил)-6-(2-хинолил)метокси-4-хроманон.
В раствор 3-(октилтио) продукта примера 4 (0,57 ммоля) и CH2Cl2 (25 мл) при температуре 0oC вводят 125 мг (0,57 ммоля) мета-хлорнадбензойной кислоты. Реакционная смесь перемешивается при 0oC в течение 2 часов, затем разбавляется СH2Cl2 (50 70 мл), промывается насыщенным NaHCO3, Н2О, солевым раствором, высушивается над Na2SO4 и концентрируется в вакууме и в результате получается конечный желаемый продукт.
Пример 31.
3-(октансульфонил)-6-(2-хинолил)метокси-4-хроманон.
В неполный раствор 3-(октилтио) продукта примера 4 (1,20 ммоля) в 50 мл горячего метанола вводят раствор KHSO3 (2,20 г, 3,58 ммоля) в Н2 (20 мл). Реакционная смесь перемешивается при комнатной температуре в течение 30 минут, затем разбавляется Н2 (200 мл) и этилацетатом (250 мл). Органический слой промывается 2 х Н2О и солевым раствором, высушивается над Na2 SO4 и отгоняется досуха, и в результате получается конечный желаемый продукт.
Пример 32.
7,8-Дигидро-7-метил-3-(2-хинолил)метокси-5(6Н)-хинолон.
Осуществляя процедуру согласно примеру 1, 7, 8-дигидро-3-окси-7-метил-5(6Н)-хинолон и 2-хлорметилхинолон превращаются в конечный желаемый продукт с выходом 67% температура плавления 141 - 144oC. Масс-спектр (m/e). рассчитано: 318, 1365; найдено: 318, 1325.
Пример 33.
6-Бутил-7,8-дигидро-7-метил-3-(2-хинолил)метокси-5(6Н)-хинолон.
Осуществляя процедуру по способу 8, конечный продукт предыдущего примера превращается в желаемый конечный продукт данного примера.
Пример 34.
Цис- и транс-6-бутил-7, 8-дигидро-7-метил-3-(2-хинолил)метокси-5(6Н)-хинолон.
Осуществляя процедуру согласно примеру 2, конечный продукт предыдущего примера превращается в смесь конечных продуктов данного примера. Этот продукт представляет собой смесь 6,7-цис и 6,7-транс-изомеров, хотя не исключена возможность того, что этот продукт включает один из этих изомеров.
Пример 35.
Цис-6-Бутил-5,6,7,8-тетрагидро-с- и транс-7-метил-3-(2-хинолил)метокси-r-5-хинолол и транс-6-Бутил-5,6,7,8-тетрагидро-с и транс-7-метил-3-(2-хинолил)метокси-r-5-хинолол.
Осуществляя процедуру согласно примеру 5, продукт предыдущего примера превращается в конечные желаемые продукты данного примера, названия которым даны согласно существующей номенклатуре Международного Союза теоретической и прикладной химии, (IUPAC Nomenclature of Organic Chemistry, 1979. Ed. стр. 477 478). Каждый из этих продуктов представляет собой смесь двух соединений, одно из которых имеет 7-метильную группу цис(с) относительно (r) к 5-оксигруппе и другое имеет 7-метильную группу транс (t) относительно (r) к 5-оксигруппе. Однако не исключена возможность того, что каждый из этих продуктов включает один или другой из этих с-7 или t-7 изомеров.
Пример 36.
6(8Н)-Оксиметилен-7-метил-3-(2-хинолил)метокси-5(7Н)-хинолон.
Осуществляя процедуру согласно примеру 2, конечный продукт примера 61 превращается в конечный желаемый продукт данного примера с выходом 99% анализ методом тонкослойной хроматографии (CH2 Cl2 этанол 19 1) Rf 0,6.
Пример 37.
8(8Н)-Диазо-7-метил-3-(2-хинолил)-метокси-5(7Н)-хинолон.
Осуществляя процедуру согласно примеру 3, конечный продукт предыдущего примера превращается в конечный желаемый продукт данного примера с выходом 99% анализ методом тонкослойной хроматографии (CH2Cl2 этанол в соотношении 19 1) Rf 0,25.
Пример 38.
Цис- и транс-7,8-Дигидро-7-метил-6-пентокси-3-(2-хинолил)метокси-5(6Н)-хинолон.
(См. пример 34 для пояснения состава re-изомера).
Осуществляя процедуру согласно примеру 4, конечный продукт предыдущего примера и пентанол превращаются в смесь конечных продуктов данного примера.
Пример 39.
5,6,7,8-Тетрагидро-цис- и транс-7-метил-с-6-фенокси-3-(2-хинолил)метокси-r-5-хинолил и 5,6,7,8-тетрагидро-цис- и транс-7-метил-t-6-фенокси-3-(2-хинолил)метокси-r-5-хинолил.
(См. пример 35 для пояснения номенклатуры и состава изомера).
Осуществляя процедуру согласно примеру 5, конечные продукты предыдущего примера превращаются в конечные продукты данного примера.
Пример 40.
Цис-3-Циклогексилокси-6-(2-хинолил)-метокси-4-хроманил-N, N- диметилглицинат.
Цис-изомер конечного продукта примера 5 (0,22 ммоля) растворяется в CH2Cl2 (3 мл). Затем в раствор добавляется в последовательном порядке 4-(Диметиламино)-пиридин (0,043 г, 0,35 ммоля), N, N-диметилглицинхлоргидрат (0,038 г, 0,26 ммоля) и дициклогексилкарбодиимид (0,050 г, 0,26 ммоля) и смесь перемешивается в течение 18 часов. Реакционная смесь резко охлаждается равным объемом воды и побочный продукт циклогексилмочевина удаляется путем фильтрации. Органический слой в фильтрате отделяется, промывается насыщенным NaHCO3, водой и солевым раствором, высушивается над Na2SO4 и отгоняется до получения конечного продукта данного примера. Таким же способом получаются соответствующие сложные эфиры глицината из транс-изомера конечного продукта примера 11, а также из конечных продуктов примеров 7, 11, 13, 14 и 16.
Пример 41.
Дихлоргидрат цис-3-циклогексилокси-6-(2-хинолил)метокси-4-хроманил-N,N- диметилглицината.
К конечному продукту предыдущего примера (0,10 г, 0,19 ммоля), растворенному в 5 мл абсолютного этанола, добавляют 0,475 мл (0,475 ммоля) 1-нормальной HCl и смесь перемешивается в течение нескольких минут, затем отгоняется досуха и в результате получается желаемое конечное соединение данного примера.
Пример 42.
Дихлоргидрат транс-2-бутил-1,2,3, 4-тетрагидро-7-(2-хинолил)метокси-1-нафтил-4- пиперидинобутирата.
К раствору 935 мг (4,52 ммоля) хлоргидрата 4-пиперидиномасляной кислоты, 733 мг (6,02 ммоля) 4-(N, N-диметиламино)пиридина и 1,36 г (3,77 ммоля) транс-изомера конечного продукта примера 11 в 7,5 мл CH2Cl2 при температуре 0oC добавляют 852 мг (4,14 ммоля) дициклогексилкарбодиимида. Полученная реакционная смесь перемешивается в течение 15 часов при температуре 25oС, затем фильтруется и фильтрат выпаривается досуха. Полученный остаточный продукт выпаривается растворяется в 100 мл этанола. Вводится 1 нормальная соляная кислота (7,54 мл), и реакционная смесь концентриpуется досуха во вращающемся выпарном аппарате, и в результате получается конечный продукт данного примера.
Пример 43.
3,4-Дигидро-7-(2-хинолил)метокси-1(2Н)-нафталинон.
Осуществляя процедуру примера 10, из 5,00 г (30,9 ммолей) 7-окси-3,4-дигидро-1(2Н)нафталинона и 9,91 г (46,3 ммоля) хлоргоидрата 2-хлорметилхинолина получают 3,5 г (37%) конечного желаемого соединения.
Масс-спектр (m/e) 303 (M+; 286, 274, 142 и 115.
Спектр1H-ЯМР (СDCl3, 300 МГц), дельта (ч/мин): 2,08 (м. 2Н), 2,60 (т. J 7 Гц, CH2),2,87 (т. J 6 Гц, CH2), 5,39 (с. OCH2), 7,16 (д.J 2 Гц, ArH), 7,52 (д.д. J 8; Гц, ArH), 7,6 7,75 (м. 4ArH), 7,79 (д. J 8 Гц, ArH), 8,07 (д. J 8 Гц, ArH) и 8,16 (д. J 8 Гц, ArH).
Пример 44.
Осуществляя процедуру согласно примеру 22, используя соответствующий альдегид в качестве исходного реагента, конечный продукт предыдущего примера превращают в следующие соединения: 2(4H)-Гексилиден-7-(2-хинолил)метокси-1(3Н)-нафталинон; 2(4Н)-циклопентилметилен-7-(2-хинолил)метокси-1(3Н)-нафталинон; и 2(4Н)-(2, 2-дифторбутилиден)-7-(2-хинолил)-метокси-1(3Н)-нафталинон.
Пример 45.
Осуществляя процедуру согласно примеру 23, продукты предыдущих примеров превращают в следующие
соединения:
3,4-Дигидро-2-гексил-7-(2-хинолил)метокси-1(2Н)-нафталинон;
3,4-дигидро-2-циклопентилметил-7-(2-хинолил)-метокси-1(2Н)-нафталинон; и
3,4-дигидро-2-(2,
2-дифторбутил)-7-(2-хинолил)-метокси-1(2Н)-нафталинон.
Пример 46.
Осуществляя процедуру согласно примеру 5, продукты предыдущего примера превращают в следующие
соединения:
Цис- и транс-1,2,3,4-тетрагидро-2-гексил-7-(2-хинолил)метокси-1-нафтол;
цис- и транс-1,2,3,4-тетрагидро-2-циклопентилметил-7-(2-хинолил)- метокси-1-нафтол; и
цис- и транс-1,2,3,4-тетрагидро-2-(2,2-дифторбутил)-7-(2-хинолил)метокси-1-нафтол.
Пример 47.
2-Бутилиден-6-метокси-1-индинон.
В смесь 9,66 г (59,6 ммолей) 6-метокси-1-инданона и 4,29 г (59,6 ммолей) масляного альдегида в 10 мл этанола при температуре 0oC добавляют 9,66 мл 4% КОН в этанольном растворе. Реакционная смесь перемешивается в течение 1 часа и затем вводится в 300 мл воды, и величина рН быстро доводится до 2 посредством 1-нормальной соляной кислоты. Полученная смесь экстрагируется 3 х 300 мл простого эфира, и экстракты соединяются, высушиваются над MgSO4, выпариваются, остаточный продукт выпаривания перемешивается с простым эфиром, и в результате получается конечное соединение, отвечающее данному примеру.
Пример 48.
2-Бутил-6-метокси-1-инданон.
Осуществляя процедуру согласно примеру 18, конечный продукт из предыдущего примера превращается в конечное желаемое соединение данного примера.
Пример 49.
2-Бутил-6-окси-1-инданон.
Осуществляя процедуру согласно способу получения препарата 3, как описано ниже, 8,00 г (31,7 ммоля) конечного продукта предыдущего примера превращается в конечное соединение данного примера.
Пример 50.
2-Бутил-6-(2-хинолил)метокси-1-инданон.
Осуществляя процедуру согласно примеру 10, 14,7 ммоля конечного продукта предыдущего примера и 4,80 г (22,4 ммоля) хлоргидрата 2-хлорметилхинолина превращаются в конечное соединение данного примера. Используя вместо хлоргидрата 2-хлорметилхинолина молярный эквивалент хлоргидрата 2-, 3- или 4-пиколилхлорида, согласно данной процедуре получают соответственно 2-бутил-6-(2-, 3- и 4-пиридил)-метокси-1-инданоны.
Пример 51.
Цис- и транс-2-бутил-6-(2-хинолил)метокси-1-инданол.
Осуществляя процедуру согласно примеру 5, конечный продукт предыдущего примера превращают в конечные продукты данного примера.
Пример 52.
3-Пентилиден-6-метокси-1-(пара-толуолсульфонил)-2,3-дигидро4(1Н)-хинолинон.
Осуществляя процедуру примера 22, 25,0 г (75,5 ммолей) 6-метокси-1-толуолсульфонил-2, 3-дигидро-4(1Н)хинолинона (J.Am. Chem.Soc. Vol. 71, р.1901, 1949 г и 9,7 г (113 ммолей) пентанала превращают в конечный продукт данного примера.
Пример 53.
6-Метокси-1-(пара-толуолсульфонил)-3-пентил-2,3-дигидро-4(1Н)- хинолинон.
Осуществляя процедуру согласно примеру 23, продукт предыдущего примера превращают в конечный продукт данного примера.
Пример 54.
6-Окси-3-пентил-2,3-дигидро-4(1Н)хинолин.
Смесь 10 г конечного продукта предыдущего примера в 35 мл уксусной кислоты и 35 мл концентрированного НВr нагревается с обратным холодильником в течение 8 часов, после чего она вводится в смесь лед-вода, и в результате осаждается конечный продукт данного примера.
Пример 55.
6-Окси-3-пентил-1-(пара-толуолсульфонил)-2,3-дигидро-4(1Н)- хинолон.
В раствор 8,66 ммолей конечного продукта предыдущего примера в 13 мл пиридина медленно вводят 1,65 г (8,66 ммолей) паратолуолсульфонилхлорида. Реакционная смесь перемешивается в течение 1 часа, затем вводится в 200 мл 1-нормальной НСl и экстрагируется этилацетатом. Органический слой промывается свежим 1- нормальным HCl и затем насыщенным NaCl, высушивается над MgSO4 и отгоняется, и в результате получается конечный желаемый продукт.
Пример 56.
Осуществляя процедуру согласно примерам 1 или 10, конечный продукт предыдущего примера взаимодействует с подходящим гетероариметилхлоридом и в результате получаются
соответствующие
6-(2-хинолил, 2-пиридил, 3-пиридил, 4-пиридил, 2-хиноксалинил, 2-пиримидинил, 6-фтор-2-хинолил, 5-фтор-2-бензотиазолил и 1-фталазинил)метокси производные. Эти соединения, в свою
очередь, гидролируются
(1 г) путем нагревания с обратным холодильником в течение 8 9 часов в смеси 7,5 мл уксусной кислоты и 4 мл концентрированной HCl, с последующим разбавлением равным объемом воды,
доведением величины рН
до 8,0 посредством 6- нормального NaOH и экстракции CH2Cl2. Органический слой высушивается над MgSO4, и отгоняется, и в результате получаются
желаемые кетоновые
продукты:
2,3-Дигидро-3-пентил-6-(2-хинолил)метокси-4-(1Н)-хинолон;
2,3-дигидро-3-пентил-6-(2-пиридил)метокси-4-(1Н)-хинолон;
2,
3-дигидро-3-пентил-6-(3-пиридил)метокси-4(1Н)-хинолон;
2,3-дигидро-3-пентил-6-(4-пиридил)метокси-4(1Н)-хинолон;
2,3-дигидро-3-пентил-6-(2-хиноксалинил)метокси-4(1Н)-хинолон;
2,3-дигидро-3-пентил-6-(2-пиримидинил)метокси-4(1Н)-хинолон;
2,3-дигидро-3-пентил-6-(6-фтор-2-хинолил)-метокси-4(1Н)-хинолон;
2,
3-дигидро-3-пентил-6-(5-фтор-2-бензотиазолил)-метокси-4(1Н)-хинолон;
и 2,3-дигидро-3-пентил-6-(1-фталазинил)метокси-4(1Н)-хинолон.
Пример 57.
Осуществляя процедуру согласно примеру 5, продукты предыдущего примера превращают в соответствующие цис- и транс-1,2,3,4-тетрагидро-3-пентил-6-(замещенные)метокси-4-хинолоны.
Пример 58.
3-Бутил-6-(2-хинолил)метокситиохроман-4-он
Осуществляя последовательные этапы согласно примерам 8 10, 7-метокситиохроман-4-он превращается в конечный продукт данного примера.
Пример 59.
Цис- и транс-3-бутил-6-(2-хинолил)-метокситиохроман-4-он-1-оксид.
В раствор 30 ммолей конечного продукта предыдущего примера в 100 мл CH2Cl2 при температуре от -5 до 9oC медленно вводят 31 ммолей мета-хлорнадбензойной кислоты. Смесь перемешивается до тех пор, пока крахмальная-КI испытательная бумага не становится негативной (несколько часов), затем быстро охлаждается в 100 мл воды. Органический слой отделяется, промывается насыщенным NaHCO3, высушивается над MgSO4 и отгоняется, и в результате получаются конечные продукты данного примера.
Используя 70 ммолей мета-хлорнадбензойной кислоты и осуществляя реакцию в течение более длительного периода времени (16 часов) при комнатной температуре, получают соответствующий сульфон (1,1-диоксид).
Пример 60.
Осуществляя процедуру согласно примеру 5, конечные продукты предыдущих трех примеров превращаются в соответствующие цис- и транс-тиохроман-4-олы.
Пример 61.
7,8-Дигидро-7-метил-3-(2-хинолил)-метокси-5(6Н)-хинолон.
Осуществляя процедуру согласно примеру 1, 7,8-дигидро-3-окси-7-метил-5(6Н)-хинолон и 2-хлорметилхинолин превращаются в конечный продукт данного примера с выходом 67% температура плавления 141 - 144oC. Масс-спектр (m/e), рассчитано: 318, 1365; найдено: 318, 1325.
Получение 1.
4-(2-Цианоэтокси)анизол.
4-Метоксифенол (248 г), КОН (5,6 г) и акрилонитрил (397 мл) растворяют в 1 литре трет-бутанола и нагревают с перемешиванием при температуре 75oC в течение 5 часов. Затем смесь охлаждается до комнатной температуры и отгоняется в вакууме до образования твердого остаточного продукта, который повторно суспензируется в простом эфире, и нерастворимые вещества извлекаются путем фильтрации. Последние растворяются в 2 л этилацетата, промываются последовательно 1 л каждого из следующих агентов: водой, насыщенным NaHCO3 и насыщенным NaCl, высушиваются над MgSO4 и повторно отгоняются до образования очищенного конечного продукта, 199,4 г, температура плавления 62 64oC.
Получение 2.
6-Метокси-4-хроманон.
Конечный продукт предыдущего примера (199 г) смешивается с 240 мл Н2О и 480 мл концентрированной НСl и нагревается с обратным холодильником в течение ночи. Реакционная смесь охлаждается до комнатной температуры и твердые вещества извлекаются путем фильтрации. Последние растворяются в 2 л этилацетата, промываются 200 мл мл воды, высушиваются над MgSO4 и отгоняются в вакууме, и в результате получается промежуточный продукт 3-(4-метоксифенокси)пропионовая кислота, 195 г, температура плавления 105 107oС. Последняя вводится в 600 мл горячей перемешанной полифосфорной кислоты, поддерживаемой при температуре 75oC, и смесь перемешивается в течение 2 часов. Температура повышается до максимума 89oC в течение первого получаса, затем снижается до температуры бани, равной 75oC. Реакционная смесь быстро охлаждается в 3,2 л льда и воды и экстрагируется 1,2 л этилацетата. Органический экстракт последовательно промывается (по 600 мл каждым) водой, насыщенным NaHCO3 и насыщенным NaCl, высушивается над MgSO4 и отгоняется до образования 180 г твердых продуктов, которые растворяются в 400 мл CH2 Сl2, обрабатываются активированным углем и повторно отгоняются до образования аналогичного количества твердых веществ. Последние перекристаллизовываются из простого изопропилового эфира с образованием очищенного конечного продукта, 120 г, температура плавления 46 48oC, который идентичен промышленно выпускаемому продукту.
Получение 3.
6-Окси-4-хроманон.
Раствор 36 г продукта предыдущего примера получения препарата в 290 мл уксусной кислоты и 290 мл 48%-ной бромистоводородной кислоты нагревается с обратным холодильником в течение 3 часов. Реакционная смесь охлаждается и отгоняется в вакууме до образования сырого продукта, который разбавляется водой (6 л), охлаждается до температуры 0 5oC, и конечный продукт извлекается путем фильтрации, 25,7 г (80%), температура плавления 133 - 136oC. При желании этот продукт дополнительно очищается путем пропускания через хроматографическую колонку с силикагелем, с использованием смеси этилацетат/гексан в качестве элюента.
Получение 4.
Смесь 25 г продукта предыдущего примера получения препарата, 26,5 г бромистого бензила и 28 г карбоната калия в 150 мл ацетона нагревается с обратным холодильником в течение ночи. Реакционная смесь охлаждается и фильтруется с удалением карбоната калия. Фильтрат выпаривается, и остаточный продукт выпаривания растворяется в этилацетате и промывается водой. Этилацетатный слой высушивается над сульфатом натрия и выпаривается в вакууме, и в результате получается сырой продукт, который очищается путем перекристаллизации из хлористого метилена/гексана, и в результате получается 29 г конечного желаемого продукта, температура плавления 107 108oC.
Спектр1H-ЯМР (ацетон-d6) дельта (ч/млн): 2,7 (т.2Н), 4,4 (т. 2Н), 5,08 (с. 2Н), 7,2 7,5 (м. 3Н).
Получение 5.
3-Оксиметилен-6-бензилокси-4-хроманон.
В раствор 172,5 г продукта предыдущего примера приготовления препарата в 1,7 л толуола, содержащего 168 мл этилформата и 3,5 мл этанола, вводят отдельными порциями 66 г 50%-ного гидрата натрия. Реакционная смесь перемешивается при комнатной температуре в течение 1 часа, затем вливается в 1,5 л смеси льда с водой и подкисляется до величины рН=4 разбавленной соляной кислотой. Водный слой экстрагируется несколькими порциями этилацетата. Органические слои смешиваются, высушиваются над сульфатом натрия и выпариваются в вакууме, и в результате получается сырой продукт, который перемешивается с гексаном для удаления гидридного масла. Полученный продукт кристаллизуется при выдержке, температура плавления продукта 82 85o C.
Получение 6.
3-Диазо-6-бензилокси-4-хроманон.
В раствор 35,3 г конечного продукта предыдущего примера получения препарата в 250 мл дихлорметана, содержащего 25,2 г триэтиламина, при температуре -10oC добавляется по каплям раствор 24,4 г азида тозила в 100 мл дихлорметана. После прекращения ввода реакционная смесь нагревается до комнатной температуры и перемешивается в течение ночи. Реакционная смесь промывается водой, высушивается над сульфатом натрия и выпаривается в вакууме, и в результате получается сырой продукт, который очищается в хроматографической колонке на силикагеле с элюированием дихлорметаном, и в результате получается 21 г продукта, температура плавления 100 103oC.
Спектр1H-ЯМР (СDCl3) дельта (ч/млн): 5,02 (д. J 4, 2H), 6,7 7,5 (м. 10Н).
Получение 7.
4-(4-Метоксифенокси)масляная кислота.
4-Метоксифенил вводится в раствор NaOC2 H5, приготовленный путем растворения 2,3 г Na в 50 мл этанола. По прошествии 5 минут вводится гамма-бутиролактон и смесь нагревается с обратным холодильником в течение ночи. Этанол отгоняется, и остаточный продукт отгонки нагревается при 155oC в течение ночи, затем охлаждается, разбавляется водой и подкисляется до величины рН=3 разбавленной соляной кислотой. Этот продукт извлекается путем фильтрации, получается 19,5 г продукта с температурой плавления 103 - 104oC.
Получение 8.
3-4-Дигидро-7-метокси-1-бензоксепин-5(2Н)-он
34 г продукта предыдущего примера получения препарата растворяется в 300 мл полифосфорной кислоты в нагревается при 100oC в
течение 1
часа. Реакционная смесь охлаждается, вливается в воду и экстрагируется простым эфиром, и в результате получается сырой продукт. Этот сырой продукт очищается путем отгонки, температура
кипения продукта
100oC при 0,5 мм.
Получение 9.
3,4-Дигидро-7-окси-1-бензоксепин-5-(2Н)-он.
Смесь 19,23 г конечного продукта предыдущего примера получения препарата, 95 мл 48% бромистоводородной кислоты и 95 мл уксусной кислоты нагреваются с обратным холодильником в течение 4 часов. Реакционная смесь охлаждается и выпаривается в вакууме, и в результате получается сырой продукт, который очищается в хроматографической колонке на силикагеле, с элюированием дихлорметаном, и в результате получается 8,3 г конечного продукта, с температурой плавления 116 120oC.
Спектр1H-ЯМР (CDCl3) дельта (ч/млн): 2,0 2,45 (м. 2Н), 2,95 (т. J 7, 2H), 4,20 (т. J 7, 2Н), 6,8 7,1 (м. 3Н), 7,4 (с. 1Н).
Получение 10.
7-Бензилокси-3,4-дигидро-1-бензоксепин-5(2Н)-он.
Смесь 6,5 г продукта предыдущего примера получения препарата, 4,3 мл бромистого бензила, 6,3 г карбоната калия и 40 мл ацетона нагреваются с перемешиванием с обратным холодильником в течение ночи. Реакционная смесь охлаждается и фильтруется и в результате удаляются неорганические вещества. Фильтрат выпаривается в вакууме, и остаточный продукт выпаривания растворяется в этилацетате и промывается водой. Этилацетатный слой высушивается над сульфатом натрия и выпаривается в вакууме, и в результате получается сырой продукт, который очищается путем перекристаллизации из простого изопропилового эфира, и в результате получается 8,4 г конечного продукта с температурой плавления 62 63oC.
Получение 11.
7-Бензилокси-4-бром-3,4-дигидро-1-бензоксепин-5/2H/-он.
В раствор 6,3 г конечного продукта предыдущего примера получения препарата в 25 мл уксусной кислоты добавляют раствор 3,76 г брома в 25 мл уксусной кислоты. Реакционная смесь перемешивается в течение 3 минут, и летучие вещества отгоняются в вакууме до образования остаточного продукта, который растворяется в этилацетате и промывается водой. Этилацетатный слой высушивается и выпаривается и в результате получается 8,2 г продукта, который используется без очистки в последующем этапе.
Получение 12.
3-Бромо-6-метокси-4-хинолон
В раствор 6-метокси-4-хроманона (35 г) в простом
этиловом эфире (1,6 л) при температуре
5- 10oC вводят по каплям в течение 30 минут 10,6 мл брома. Смесь перемешивается при 5 10oC в течение 30 минут и затем нагревается до
комнатной температуры. По прошествии 2 часов,
как показан анализ методом тонкослойной хроматографии (CH2Cl2), происходит образование менее полярных продуктов и остаются лишь
следы исходного материала. Реакционная смесь
промывается водой (1 л), насыщенным NaHCO3 (500 мл) и солевым раствором (500 мл), высушивается над MgSO4 и выпаривается в вакууме до
образования желтого твердого вещества. Сырой
продукт очищается в хроматографической колонке на силикагеле (методом испарительной хроматографии) с использованием 2,4 кг тонкого силикагеля, при
элюировании градиентной системой, состоящей из
гексана-дихлорметана в соотношении 3:1, затем системой, состоящей из гексана-дихлорметана в соотношении 2:1 и, наконец, системой 30%
гексана-дихлорметан. В результате получается конечный желаемый
продукт в виде желтого твердого вещества с выходом 80%
Получение 13.
1-Амино-5-метилциклогекс-1-ен-3-он.
5-Метил-1,3-циклогександион (40 г, 0,32 моля) растворяется в 500 мл бензола при 70oC. Раствор нагревается с обратным холодильником в течение 2 часов, в течение которых через реакционную смесь пропускается в виде пузырьков NH3 , и образующаяся вода собирается в ловушке Дина и Старка. Затем смесь охлаждается до 0oC и конечный продукт извлекается путем фильтрации, выход 39,8 г; температура плавления 165 169oC.
Спектр1H-ЯМР (диметилсульфоксид-d6) дельта (ч/млн): 0,98 (с. 3Н), 1,6 1,88 (2Н), 2,14 2, 38 (2Н), 3,14 3,6 (1Н), 4,93 (с. 1Н), 6,2 7,2 (м. 2Н).
Получение 14.
7,8-Дигидро-7-метил-3-нитро-5(6Н)-хинолон.
Нитромалонангидрид натрия (Org.Synth.Coll. Том 4, стр. 844; 42,4 г,269 моля) растворяется в 200 мл диметилформамида, и полученный раствор, высушенный над молекулярными ситами типа 4А, извлекается путем фильтрации со 100 мл того же растворителя, что и промывка. К соединенным фильтрату и промывке добавляется пиридин (91 мл, 89 г, 1,13 моля), и смесь охлаждается до -5oC. Вводится по каплям хлористый тозил (53 г, 0,277 моля) в 200 мл диметилформамида, при этом поддерживается температура от -5 до -8oC, и реакционная смесь нагревается до комнатной температуры. Конечный продукт предыдущего примера получения препарата (33,6 г, 0,270 моля), растворенный путем нагревания в 200 мл диметилформамида и введенный в стационарный поток, направляемый в реакционную смесь, который затем перемешивается в течение 18 часов при комнатной температуре, затем вводится в 2 литра смеси лед-вода и экстрагируется (2 х 1 л) этилацетатом. Органические слои смешиваются, высушиваются над MgSO4 и отгоняются, и в результате получается конечный желаемый продукт, 33 г (61% с температурой плавления 64 67oC).
Получение 15.
3-Амино-7, 8-дигоидро-7-метил-5(6Н)-хинолон.
Конечный продукт предыдущего примера получения препарата (27 г) вводится в склянку Парра на 250 мл с 830 мл абсолютного этанола и 9,0 г 10% Pd/C. Затем эта смесь перемешивается в аппарате Парра под давлением водорода (Н2) 50 psi (3,5 атм) в течение 2 часов при комнатной температуре. Катализатор извлекается путем фильтрации над диатомовой землей, и фильтрат концентрируется досуха. Оставшийся коричневый твердый продукт затем подвергается испарительной хроматографии путем первоначального растворения в CH3OH, с вводом 50 мл сухого силикагеля с размером зерен 32 63 мкм и с концентрированием досуха. Полученный в результате продукт затем вводится в колонку испарительной хроматографии 30 см х 15 см с силикагелем, через которую пропускается 1% триэтиламин в CH2Cl2 изопропаноле (19:1). Колонка элюируется той же системой растворителя. Содержащие продукт средние фракции собираются и отгоняются с образованием конечного желаемого продукта. Масо-спектр (m/e), рассчитанный: 176, 0950; найдено: 176, 0944; анализ методом тонкослойной хроматографии (19:1, CH2 Cl2 C2H5OH), Rf 0,32.
Получение 16.
7, 8-Дигидро-7-метил-5(6Н)-хинолон-6-диазонийгексафторфосфат.
Конечный продукт предыдущего примера получения препарата (15,26 г) при комнатной температуре помещается в 500 мл 3-горлую колбу, снабженную механической мешалкой, капельной воронкой и отводной трубкой, помещенной с задней части вытяжного колпака. Затем вводится 6,93 мл ледяной уксусной кислоты. Затем вводится сразу 159 мл 3,48 нормальной НСl, после чего реакционная смесь превращается в прозрачный густо- красный раствор. Затем последний охлаждается до 0oC, и в течение этого времени в растворе осаждается некоторое количество твердого вещества. К этой суспензии, имеющей все еще температуру 0oC, добавляется 5,98 г NaNO2 в 35 мл Н2О по каплям в течение 5 10 минут, и полученная смесь перемешивается при 0oC в течение 30 минут. К этой смеси, находящейся все еще при температуре 0oC, добавляется 15,24 мл HPF6 (60 вес. в Н2) в течение 5 минут. Тотчас образуется светлокоричневый осадок. После прекращения этого добавления интенсивное перемешевание продолжается в течение 10 15 минут. Полученный твердый продукт фильтруется, промывается 2 х 25 мл холодной Н2O, 2 х 25 мл простого эфира и затем высушивается в вакууме в течение ночи над Р2О5, и в результате получается 25,62 г (89%) конечного желаемого продукта, температура плавления 175 176,5oC.
Получение 17.
7,8-Дигидро-3-окси-7-метил-5(6Н)-хинолон.
Конечный продукт предыдущего примера (25,62 г) вводится в виде порция по 0,5 г в 500 мл кипящей 5%-ной H2 SO4 в течение такого времени (в данном случае 2,5 часа), которое исключает чрезмерное вспенивание за счет улетучивания N2. Реакционная смесь нагревается с обратным холодильником еще в течение 40 минут, затем охлаждается до 0oC и величина рН руководится до 6 посредством 6-нормального NaOH (в данном случае требуется 160 мл). Реакционная смесь экстрагируется этилацетатом 3 х 250 мл. При первой экстракции эмульсия разбивается путем фильтрации через диатомовую землю. Органические экстракты соединяются, высушиваются над MgSO4, отгоняются до образования твердых веществ, и остаточный продукт отгонки растворяется в CH3OH, перемешивается в силикагелем, отгоняется и подвергается испарительной хроматографии, как и в предыдущем примере, с использованием в качестве элюента CH2Cl2-изопропанола (19 1), и в результате получается конечный желаемый продукт, 9,2 г (67%), с температурой плавления 210,5 212oC.
Получение 18.
3-Бензилокси-7, 8-дигидро-7-метил-5(6Н)-хинолон.
Осуществляя процедуру примера получения препарата 4, продукт предыдущего примера получения препарата превращают в конечный желаемый продукт данного примера с выходом 78% с температурой плавления 80,5 8105oC. Масс-спектр (m/e), рассчитано: 267, 1259; найдено: 267, 1261.
Получение 19.
2-Хлорметилхиноксалин.
2-Метилхиноксалин (8,94 г) смешивают с 50 мл ССl4 и 6,5 г Na2СО в 125- миллилитровом химическом стакане. Эту смесь нагревают до 68oC и затем вводят Сl2 через обратную воронку, так чтобы Cl2 барботировал очень медленно. Эта процедура продолжается в течение 1 часа, и затем реакционная смесь охладжается до 20oС в ледяной бане и распределяется между раствором насыщенного NaHCO3 и простым эфиром. Эфир отделяется, высушивается над MnSO4 и концентрируется досуха. Остаточный продукт концентрирования тотчас пропускается через испарительную колонку (20 см), наполненную силикагелем размером частиц 32 63 мкм (диаметр колонки 8 см) с использованием в качестве элюента смеси простой эфир гексан в соотношении 1 1. После пропускания 1 л собираются 250 мл фракций. Фракции 3 50 смешиваются и концентрируются, и в результате получается 2,58 г (23% ) конечного продукта в виде желтого твердого вещества; анализ методом тонкослойной хроматографии (этилацетат CH2Cl2 в соотношении 3 7), Rf 0,65. Спектр1H-ЯМР (CDCl3) дельта (ч/млн): 4,86 (с. 2Н), 7,74 7,78 (м. 2Н), 8,02 8,16 (м. 2Н), 9,01 (м. 1Н).
Получение 20.
2-Бром-3,4-дигидро-7-метокси-1(2Н)-нафталинон.
К раствору 25 г (0,142 моля) 7-метокси-3,4-дигидро-1(2Н)-нафталинона в 1 л простого эфира при температуре 10oC добавляют по каплям (при поддержании температуры реакционной смеси примерно 10oC (37,9 г (0,237 моля) брома. Реакционный раствор концентрируется во вращающемся выпарном аппарате, и остаточный продукт концентрирования кристаллизуется из простого эфира, и в результате получается 31,6 г (87%) конечного соединения данного примера, температура плавления 79 80oC.
Масс-спектр (m/e) 256 и 254 (M+, 174, 173, 148, 131, 120, 115 и 103. ИК Спектр (СНСl3) 1680, 1610 см-1.
спектр1H-ЯМР (СDCl3) дельта (ч/млн): 2,2 2,7 (м. 2Н), 2,9 3,5 (м. 2Н), 3,95 (с. OCH3), 4,78 (т. J= 4 Гц, СНВr), 7,0 - 7,4 (м. 2АrH) и 7,58 (шир.с. ArH).
Элементарный анализ.
Рассчитано согласно формуле: C11H11BrO2 • 1/2 H2O:
C, 50,89; H, 4,46%
Найдено: C, 50,71; Н, 4,36.
Получение 21.
6-Бензилокси-3-метилен-4-хроманон.
Раствор 9,2 г 6-бензилокси-4-хроманона, хлоргидрата диметиламина и 1,3 г параформальдегида в 100 мл уксусной кислоты нагревается в паровой бане в течение пяти часов. Летучие продукты отгоняются в вакууме, и остаточный продукт отгонки очищается на силикагеле при элюировании СН2Cl2, и в результате получается 3,7 г продукта, Rf (CH2Cl2) 0,5. Спектр1H-ЯМР (CDCl3) дельта (ч/млн): 4,85 (с. 2Н), 5,05 (с. 2Н), 5,55 (с. 1Н), 6,30 (с. 1Н), 6,80 7, 60 (м. 8Н).
Получение 22.
3-Бромо-2-(бромометил)-6-метилперидин и 3-бромо-6-(бромометил)-2-метилпиридин.
В круглодонную колбу на 25 мл, снабженную мешалкой и холодильником, в инертной атмосфере вводят 1,4 г (7, 35 ммолей) 3-бром-2,6-лютидина, 1,21 г (6,77 ммолей) N-бромсукцинимида, 4,5 мл тетрахлорида углерода и 10 мг (0,04 ммоля) перекиси бензоила. Полученная смесь нагревается в течение ночи. Анализ методом тонкослойной хроматографии, осуществляемый в этот момент, показывает, что исходный материал все еще присутствует, и добавляют 0,7 г (3,9 ммоля) N-бромсукцинимида, и реакционная смесь нагревается с обратным холодильником еще в течение 4 часов. Полученный осадок фильтруется и промывается 2 х 50 мл ССl4 (горячим). Фильтрат концентрируется до образования масла, и затем сырой продукт очищается методом испарительной хроматографии на силикагеле (200 г) при использовании в качестве элюента смеси гексан CH2Cl2 в соотношении 3 1, и в результате получается два указанных конечных соединения, отвечающих данному примеру, с выходом 218 мг (11%) 2-(бромометилового) производного и с выходом 285 мг (14%) 6-бромометилового) производного; анализ методом тонкослойной хроматографии (гексан - CH2Cl2 в соотношении 3:1), Rf 0,07 и Rf 0,13 соответственно.
2-(Бромометиловое)производное.
Спектр1H-ЯМР (Диметилсульфоксид-d6 (дельта (ч/млн): 7,99 (д. J 7,8 Гц, (1Н), 7,19 (д. J 7,8 Гц, 1Н), 4,71 (с. 2Н), 2,46 (с. 3Н).
6-(Бромометиловое) производное.
Спектр1H-ЯМР (Диметилсульфоксид-d6) дельта (ч/млн): 8,00 (д. J 7-8 Гц, 1Н), 7,32 (д. J 7,8 Гц, 1Н), 4,63 (с. 2 Гц), 2,56 (с. 3Н).
Реферат
Использование: в медицине. Сущность: продукт-производные тетралина ф-лы: R-CH2-O-тетралин-(YY')-CH-R', где R - 2- пиридил, 2-хинолил, R'-C1-C3-алкил, Y и Y' - вместе образуют карбонильную группу или взятые отдельно Y-водород, Y'-гидроксил, производные 7-гидрокси-тетралина. 1 табл.
Формула

где R 2-пиридил, 2-хинолил; R1 (C1-C3)- алкил; Y и Y1 взятые вместе, образуют карбонильную группу или, взятые отдельно, Y - водород, Y1 -гидроксил.

где R1, Y, Y′ имеют указанные значения.
Комментарии