S-энантиомер замещенного 2-аминотетралина, способы его получения и фармацевтическая композиция, обладающая свойством антагониста 5-нт*001*00а-рецептора - RU2086537C1
Код документа: RU2086537C1
Чертежи

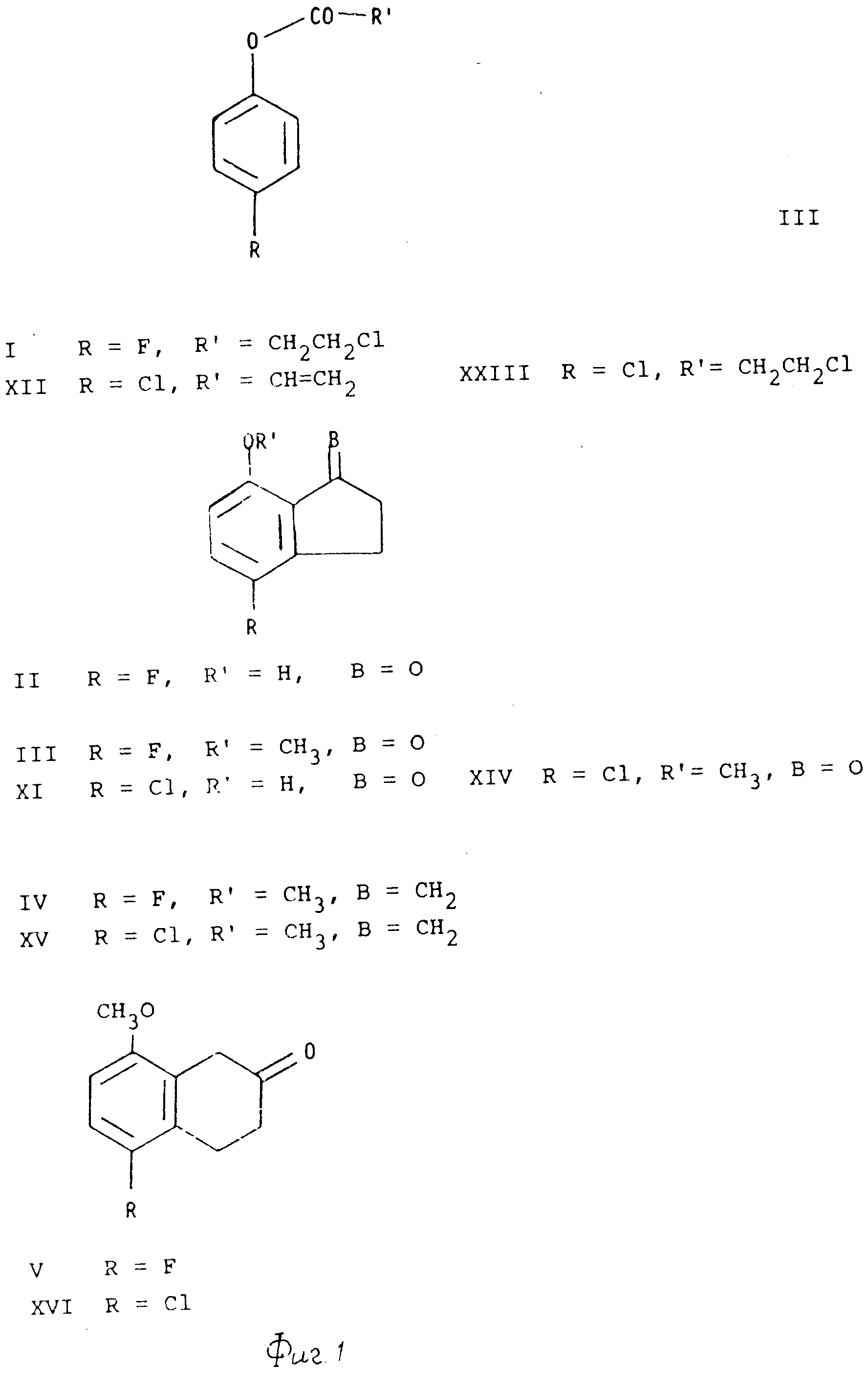
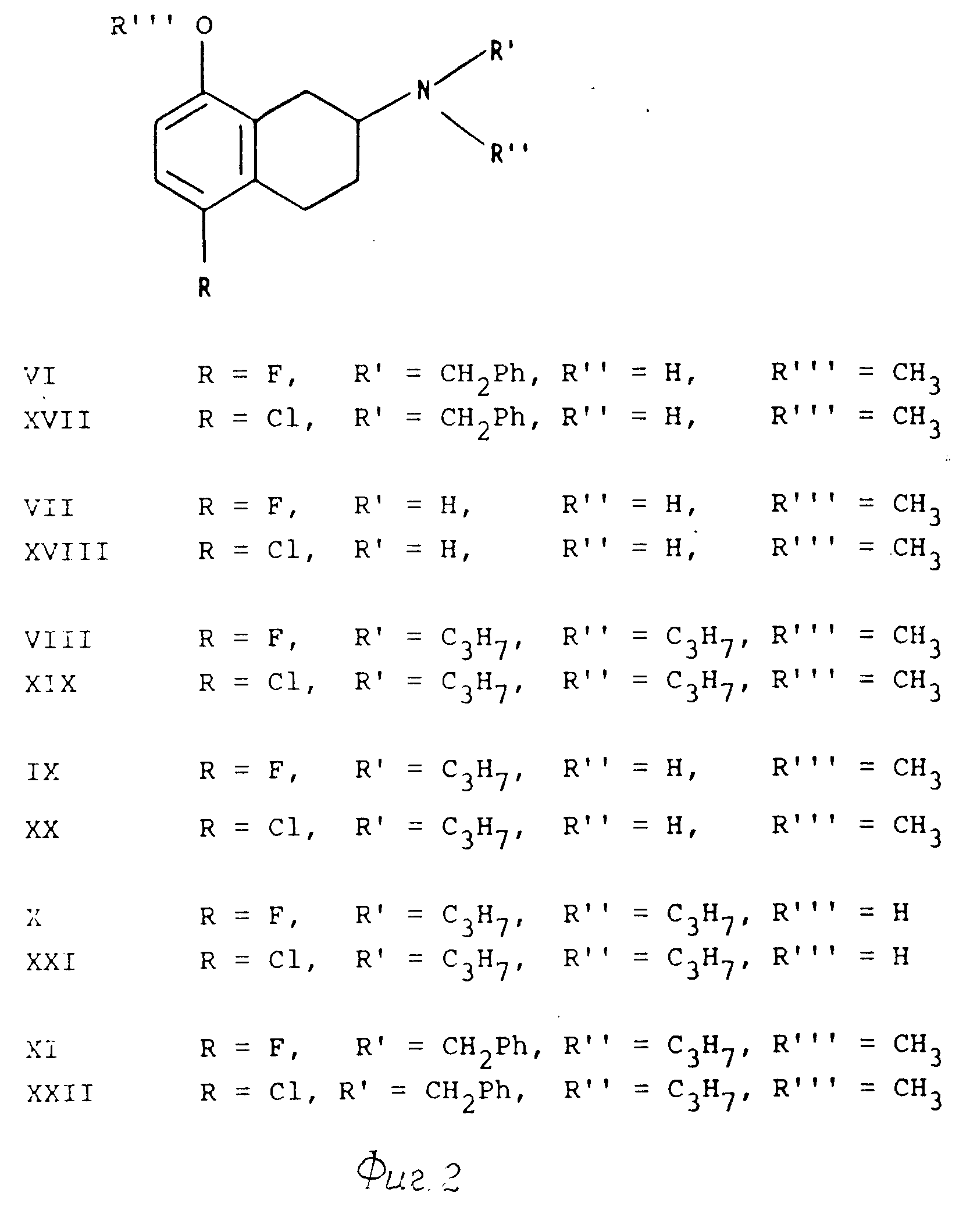
Описание
Изобретение относится к (S) энантиомерам новых замещенных-3-амино-хроманов, тиохроманов и тетралинов и их солей, способам их получения, фармацевтическим композициям, содержащим указанные терапевтически активные соединения, а также к новым промежуточным соединениям, используемым при получении терапевтически активных соединений и к использованию указанных активных соединений в терапии.
Целью изобретения является получение соединений для терапевтического использования, в частности, соединений, обладающих терапевтической активностью, направленной на центральную нервную систему (ЦНС). Другой целью настоящего изобретения является получение соединений, обладающих селективным действием на 5-гидрокси-триптаминовые рецепторы в организме млекопитающих, включая человека.
В патенте EP 0222996 раскрываются терапевтически активные 3-амино-дигидро-1-бензопираны и бензотиопираны, обладающие действием на нейроны млекопитающих.
Эти
соединения определяются формулой:
где Z= O или S
R водород
R1 водород, низший алкил
R2 водород, низший алкил
R1 или R2 и вместе образуют кольцо с 4-6 атомами углерода;
R3 кислород, гидрокси, низший алкокси, арил низший алкокси, ацилокси или арилокси, если Z является S и R3 гидрокси, низший алкокси, арил низший алкокси, ацилокси или арилокси, если Z является O, и R3 в 5- или 8-положении, если Z является O;
R4 и R5 независимо являются водородом, низшим алкилом, или галогеном, и их моно- или ди-S-оксидами, если Z=S и их фармацевтически приемлемые соли.
Целью изобретения является получение новых соединений, обладающих в высокой степени сродством к 5-гидрокси-триптаминовым рецепторам в центральной нервной системе, и то же время действующих как агонисты, частичные агонисты или антагонисты к серотониновым рецепторам.
Таким образом, группа новых соединений формулы I настоящего изобретения, их соли и предшественники могут быть использованы при лечении 5-гидрокси-триптамин-опосредованных состояний, таких как депрессия, тревожные состояния, анорексия, сенильная деменция, болезнь Альцгеймера, мигрень, расстройства терморегуляции и сексуальные расстройства. В другом своем варианте, настоящее изобретение относится к использованию указанных соединений, их энантиомеров и солей в качестве болеутоляющих средств и модуляторов сердечно-сосудистой системы.
Таким образом, изобретение относится к соединениям формулы:
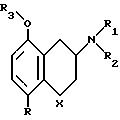
где X CH2,
R F
R1 H, C1-C6 алкил
R2 H, С1-С6 алкил
R3 H, С1-С6 алкил
C1-C6 алкил в формуле I представляет собой прямые, разветвленные и циклические алкильные группы, имеющие 1-6 атомов углерода, например, такие как, метил, этил, н-пропил, i-пропил, н-бутил, i-бутил, т-бутил, н-пентил, i-пентил, т-пентил, н-гексил, i-гексил, циклопропил, циклобутил, циклопентил, циклогексил, метилциклопропил, этилциклопропил, метилциклобутил. Предпочтительными являются группы, имеющие 1-4 атомов углерода.
В следующем своем варианте изобретение относится к фармацевтическим
композициям, содержащим в качестве
активного ингредиента соединение формулы II,
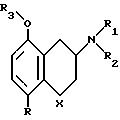
где X CH2
R F или Cl
R1 X, C1-C6 алкил
R2 X, С1-С6 алкил
R3 X, С1-С6 алкил
C1 -C6 алкил в формуле II представляет собой прямые, разветвленные и циклические алкильные группы, имеющие 1-6 атомов углерода, например, такие как метил, этил, н-пропил, i-пропил, н-бутил, i-бутил, т-бутил, н-пентил, i-пентил, т-пентил, н-гексил, i-гексил, циклопропил, циклобутил, циклопентил, циклогексил, метилциклопропил, этилциклопропил, метилциклобутил. Предпочтительными являются группы, имеющие 1-4 атомов углерода.
Особенно предпочтительными являются соединения настоящего изобретения, в которых R является фтором, а R1, R2, R3 выбирают из метила, этила и н-пропила.
Абсолютная конфигурация (+)-X-NBr была установлена с помощью рентгеновской кристаллографии (Ingeborg esoregh), неопубликованные результаты) и соответствует R.
Именно S энантиомеры соединений настоящего изобретения обладают антагонистическим действием -5HT1A-рецепторы. Это подтверждается тем фактом, что в in vivo модели (при испытаниях на крысах) (S)-X ингибирует в зависимости от дозы биохимические и поведенческие изменения, индуцированные (R)-XXIV ((S)-8-гидрокси-2-(дипропиламино)-тетралином, (S)-8-OHDPAT). Оба (S)-XXIV и (R)-XXIV, как известно, являются сильными агонистами к 5-HT1A рецептору. Напротив, рацемический X является неактивным в функциональных анализах, что должно быть является следствием того, что (R)-X проявляют фармакологические свойства, общие с другими тетралиновыми агонистами к 5-HT1A рецептору.
(S)-X (32 мкМ/кг, S, с.) не оказывал значительного воздействия на 5-НТР-уровни у крыс, которые не были предварительно обработаны резерпином, или на поведение заранее обработанных резерпином крыс. Однако он замещает (S)-XXIV из 5-HT1A рецепторов (таблица ).
Кроме того, воздействие (R)-XXIV (мкМ/кг, S. с.) на поведение резерпинизированных крыс полностью блокируется предварительной обработкой (S)-2 (10 мкМ/кг, s с. вводили за 10 мин до испытания). Предварительная обработка с использованием (S)-X за 2 ч до испытания ослабляет (R)-XXIV-индуцированное поведение, но блокады не наблюдается, когда (S)-X вводят за 4 ч до (S)-XXIV. Этот антагонизм является также эффективным после предварительной обработки галопериндолом, антагонистом к D-рецептору (2 мг/кг, i.p).
По методу Liu, Y.
Mellin, C. Bjork, L. Svensson, B. Csoregh, J. Helander, A. Kenne, L. Anden, N.-E. Hack sell, U. I. Med. chem. 1989. 32,
2311-3218
Формулы отдельных соединений настоящего изобретения,
представлены в приведенной ниже схеме формул III(см. фиг.1,2).
Фармацевтические препараты.
Согласно настоящему изобретению, соединения формулы 1 могут быть введены перорально, ректально или путем инъекции, в виде фармацевтических композиций, содержащих активный ингредиент либо в виде свободного основания, либо в виде фармацевтически приемлемой нетоксичной соли, например, гидрохлорида, гидробромида, лактата, ацетата, фосфата, сульфата, сульфамата, цитрата, тартрата, оксалата и т. п. в фармацевтически приемлемой лекарственной форме. Указанной формой может быть твердый, полутвердый или жидкий препарат. Обычно активное вещество составляет 0,1 99% по массе препарата, в частности, в пределах от 0,5 до 20 мас. для инъекций, и 0,2 50 мас. для перорального введения.
В целях получения препарата, содержащих соединение формулы 1, в одноразовых лекарственных формах для перорального введения, выбранное соединение может быть смешано с твердым наполнителем, таким как лактоза, сахароза, сорбит, маннит, крахмалы (например, картофельный крахмал, кукурузный крахмал или амилопектин); производные целлюлозы; связующим, таким как желатин или поливинилпирролидон: и замасливателем, таким как старат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и т.п. а затем спрессовано в таблетки. Если полученные таким образом формы необходимо покрыть оболочкой, то для этих целей могут быть использованы концентрированные растворы сахара, содержащие, например, аравийскую камедь, желатин, тальк, диоксид титана, и т.п. Альтернативно таблетки могут быть покрыты полимером, выбранным специалистом в растворенном в быстро улетучивающемся органическом растворителе или смеси органических растворителей. В целях быстрого распознавания таблеток, содержащих различные активные ингредиенты или различные количества активных ингредиентов, к указанным покрытиям могут быть добавлены красители.
Для получения мягких желатиновых капсул, активные вещества могут быть смешаны, например, с растительным маслом полиэтиленгликолем. Жесткие желатиновые капсулы могут содержать активные компоненты в виде гранул, изготовленных с использованием наполнителей, указанных выше для таблеток, например, таких, как лактоза, сахароза, сорбит, маннит, крахмалы (картофельный крахмал, кукурузный крахмал или аминопектин), производные целлюлозы или желатин. Жесткие желатиновые капсулы могут быть также наполнены жидкостями или наполовину жидкости составами, содержащими лекарственное средство.
Разовые лекарственные формы для ректального введения могут быть изготовлены в виде растворов или суспензий или в виде суппозиториев, содержащих активные ингредиенты в смеси с инвертными жирными основаниями, или они могут быть изготовлены в виде желатиновых капсул для ректального введения в смеси с растительным или парафиновым маслом.
Жидкие препараты для перорального введения могут быть изготовлены в виде сиропов или суспензий, например, в виде растворов, содержащих от около 0,2% до около 20 мас. активного ингредиента, и сбалансированных добавлением сахара и/или смесей этанола, воды, глицерина или пропиленгликоля, необязательно, такие жидкие препараты могут содержать красители, ароматизаторы, сахарин и карбоксиметил-целлюлозу в качестве загустителя, или другие наполнители, хорошо известные специалистам.
Растворы для парентерального введения, например, для инъекций, могут быть получены в виде водных растворов водорастворимой фармацевтически приемлемой соли активного соединения, предпочтительно в концентрации от около 0,5 до 10% по массе. Указанные растворы могут также содержать стабилизирующие агенты, и эти приготовленные растворы могут быть введены в разовые ампулы с различной дозировкой.
Приемлемые суточные дозы соединений настоящего изобретения, предназначенные для лечения людей, составляют около 0,01-100 мг/кг веса тела для парентерального введения.
Способы синтеза.
Соединения настоящего изобретения могут быть получены из п-фторфенола или п-хлорфенола с использованием соответствующих 3-хлорпропионатов или акрилатов (например, соединений 1, XII, и XXIII) и с помощью перегруппировки Фриса с последующим замыканием кольца по типу Фриделя-Крафтса, в результате чего получают 7-гидрокси-1-инданоны (например, соединения II и XXIII). Эти инданоны подвергают 0-метилированию с получением простых метиловых эфиров (например, соединений III и XIV), и с помощью реакции Виттига превращают в соответствующие 2-метилен-инданы (например, соединения IV и XV), которые подвергают окислительной перегруппировке при катализе нитратом таллия (III) с получением 5-фторо- или 5-хлоро-8-метокси-2-тетралонов (например, соединений Y и XVI). Эти тетралоны превращают в 2-бензиламино-тетралины (например, соединения VI и XVII) путем восстановления с использованием их оснований Шиффа, полученных при конденсации с бензиламином. После этого, получение 5-фторо- или 5-хлоро-8-метокси-2-диалкиламино тетралинов (например, соединений VIII и XIX) может быть осуществлено несколькими способами.
Первый способ заключается в получении моноалкиламинотетралинов (например, соединений IX и XX) путем гидрогенизационного удаления бензильной группы в соответствующих соединениях бензилалкиламинотетралинов (например, соединений XI и XXII), а затем путем исчерпывающего алкилирования получают соответствующие диалкиламинотетралины.
Второй способ заключается в том, что сначала путем восстановительного удаления бензильного заместителя получают 2-аминотетралины (например, соединения VII и XVII), которые затем подвергают диалкилированию с получением указанных диалкиламинотетралинов.
Последняя стадия синтеза включает в себя удаление O-метильной группы путем кипячения с концентрированной бромистоводородной кислотой с получением соответствующих 5-фторо- или 5-хлоро-8-гидрокси-2-диалкиламино тетранов (например, соединений X и XXI).
Соединения настоящего изобретения получают либо в виде свободных оснований, либо в виде их солей. Каждое основание может быть превращено в соответствующую кислую аддитивную, предпочтительно, с использованием терапевтически приемлемой кислоты или ионообменника. Соли, полученные в соответствии с настоящим изобретением, могут быть превращены в соответствующие свободные основания, например, путем использования более сильного основания или ионита. Поскольку между свободным основанием и его солями существует настолько тесная химическая взаимосвязь, то, если даже идет речь об одной из указанных форм соединений, всегда подразумевается и другая его форма. Соединения настоящего изобретения иногда могут быть получены после кристаллизации в виде их гидратов с различным содержанием воды.
Так как большинство из промежуточных амино-соединений, также как и конечные продукты являются восприимчивыми к окислению, то обычно реакции проводят в присутствии азота. Предпочтительно, если соединения настоящего изобретения хранятся в виде их аддитивных солей, и в первую очередь в виде гидрохлоридов или гидробромидов.
Соединения настоящего изобретения, содержащие асимметрический атом углерода, смежный с амино-группой (которая может быть неалкилированной, моноалкилированной или диалкилированной), могут быть разделены на их оптические антиподы стандартными способами. В качестве примера, можно указать на разрешение рацемического соединения VI с помощью L-винной кислоты.
Пример 1. 4'-фторофенил-3-хлоропропионат (I).
3-Хлорпропионилхлорид (391,5 г) и 3 капли триэтиламина добавляли, перемешивая при 60oC, к 4-фторфенолу (308,3 г). После нагревания при 100oC в течение 1,5 ч, и дистилляции получали 544,7 г соединения (I) с т.кип. 126-130oC, 10 мм рт.ст.
4-Фторо-7-гидрокси-1-инданол (II).
Соединение (I) (219 г) медленно добавляли к безводному AlCl3 (720 г) в присутствии азота, и перемешивая, выдерживали при комнатной температуре. Перемешивание прекращали, когда смесь становилась слишком вязкой. После нагревания до 120oC в течение 1 ч, начинали перемешивать снова, и кроме того, повышали температуру до 180oC (2 ч). После охлаждения и добавления избыточного количества воды продукт перегоняли с водяным паром. После экстракции хлороформом и выпаривания получали 127 г неочищенного соединения (II).
4-фторо-7-метокси-1-инданона (III).
В атмосфере азота к 300 г измельченного в порошок карбоната кальция в 1,5 л ацетона добавляли 121 г соединения (II), перемешивания при этом, а затем добавляли 83 мл диметилсульфата. После нагревания до температуры перегонки в течение 2 ч, растворитель отгоняли, добавляли воду, а смесь нагревали с обратным холодильником еще в течение 1 ч. После экстракции метиленхлоридом и выпаривания получали 125 г неочищенного соединения (III), которое затем очищали путем кристаллизации из EtOAc, т.пл. 118-120oC.
5-фторо-8-метокси-2-тетралон (V).
NaH (6,8 г) суспендированного в диметилсульфоксиде (40 мл) в присуствии азота нагревали до 80oC в течение 1 ч. После чего частями добавляли еще 40 мл диметилсульфоксида, а затем 80 г бромида метилтрифенилфосфония. После размещения в течение 15 мин, добавляли 20 г соединения (III), частично суспендированного в 40 мл диметилсульфоксида, и полученную смесь нагревали до 70oC в течение ночи. Смесь выливали на лед+гексан, и после экстракции гексаном получали 17,6 г неочищенного 4-фторо-7-метокси-1-метилениндана (IV), который растворяли в 70 мл MeOH. Этот раствор добавляли к размещенному раствору тригидрата тринитрата талия (43,9 г) в 400 мл MeOH и HC(OCH3)3. После размешивания в течение 1 мин, сразу добавляли 200 мл хлороформа. После промывания водным бикарбонатом натрия, сушки и концентрирования, получали неочищенный продукт, который очищали с помощью хроматографии (силикагель, диэтиловый эфиргексан, 1;1). Полученную смесь соединения V и и его диметилкеталь гидролизовали с 1 М NCl-диэтиловым эфиром и получали 11,8 г соединения V.
IH ЯМР (хлороформ-d1) δ 7,02-6,64 (м, 2H); 3,79 (c, 3H); 3,51 (c, 2H); 3,10 (т, 2H); 2,56 (т, 2H).
(±
-2-бензиламино-5-фторо-8-метокситетралин (±) VI
К 500 мл бензола и 11,8 г соединения V в присутствии азота добавляли бензиламин (13,4 мл).
После выдерживания смеси при температуре перегонки в течение ночи, воду удаляли путем азеотропной дистилляции. Неочищенную реакционную смесь выпаривали, а затем растворяли в 500 мл метанола. pH доводили до 3-4 путем добавления HCl/MeOH.
Затем добавляли NaCNBH3 (2,36 г) и смесь перемешивали в атмосфере азота в течение 2 ч, pH поддерживали при 3-4 путем добавления HCl/MeOH или триэтиламина. После концентрирования добавляли 5 М водной HCl, а осадок отфильтровывали и промывали диэтиловым эфиром. Конверсию в свободное основание (±)-VI осуществляли путем обработки с использованием IMNaOH/диэтилового эфира. После очистки с помощью хроматографии на окиси алюминия (элюируя смесью диэтилового эфира и петролейного эфира, 1:2) получали 12,4 г свободного основания (±)-VI.
Разделение (±)-VI/
(+)-L-винную кислоту (9,08 г) добавляли к (±)-VI (17,25 г) в 1050 мл горячего 95% этанола. Раствор выдерживали в течение ночи
при комнатной
температуре, и полученные кристаллы (7,25 г) перекристаллизовывали из EtOH. После обработки с использованием 5 М NaOH получали свободный амин, который экстрагировали диэтиловым эфиром.
Полученное
соединение превращали в гидрохлорид, после перекристаллизации которого из MEOH/диэтилового эфира получали 3,88 г (+)-VI.HCI.
IH ЯМР (метанол-du); d 7,60-7,40 (м,
5H); 7,
05-6,65 (м, 2H); 4,35 (c, 2H); 3,82 (с, 3H); 3,7-1,6 (м, 7H). Оптическое вращение ([альфаb 22, MeOH, как во всех последующих определениях): +61,4 (с, 1.0). Энантиомерный избыток, определенный с
помощью капиллярной ГХ (R)-2-метокси-2-фенилацетамидов (ее), составляет 99,7%
Свободный амин (11,77 г) выделяли из маточных растворов от получения (+)-VI и добавляли (-)-D-винную кислоту (6,
19 г). Эту процедуру использовали для получения (-)-VI HCl (4,93 г), в основном, аналогичным образом.
(+)-2-Амино-5-фторо-8-метокситетралина гидрохлорид [(+)-VII•HCl]
(+)-VI H 1,63 г растворяли в 100 мл MeOH. После каталитической гидрогенизации в присутствии Pd/C получали 1,14 г (+)-VII•HCl, т.пл. 261 - 263oC. Оптическое вращение: +67,8 (с, 1,
0).
(-)-2-Амино-5-фторо-8-метокситетралина гидрохлорид [(-)-VII•HCl]
Повторяли процедуру, описанную для получения (+)-VII•HCl, но исходя из (-)-VI•HCl.
Выход 99% т.пл. 262 264oC; оптическое вращение: -67,2 (с 1,0).
(+)-5-фторо-8-метокси-2-дипропиламино тетралина гидрохлорид [(+)-VIII•HCl]
1-Иодопропан (0,89
мл) добавляли к смеси (+)-VII•HCl (1,01 г) и размельченного в порошок карбоната кальция в 30 мл MeCN в присутствии азота. Смесь размешивали при комнатной температуре в течение 10 дней, во
время
которых добавляли две части от 0,3 мл 1-иодопропана. После добавления диэтилового эфира, фильтрации и выпаривания неочищенный продукт очищали с помощью хроматографии на окиси алюминия (элюируя
смесью
диэтилового эфира и петролейного эфира, 1:4), и получали в результате 0,90 г (+) -VIII•HCl, и после еще одного элюирования получали 0,23 г (+)-5-фторо-8-метокси-2-пропиаминотетралина
гидрохлорида, (+)-XI•HCl.
Выход 71% После повторной перекристаллизации получали чистый (+)-VIII, т.пл. 134 135oC. Оптическое вращение: 78,9 (с 1,0).
(-)-5-фторо-8-метокси-2-(дипропиламино) тетралина гидрохлорид [(-)-VII, HCl]
Повторяли процедуру, описанную для получения (+)-VIII•HCl, но исходя из (-)-VII•HCl. Выход 80%
т.пл.
134 135oC. Оптическое вращение: -78,4 (с 1,0).
(+)-5-фторо-8-гидрокси-2-дипропиламино тетралина гидробробромид [(+)-X•HBr]
Все оборудование тщательно
промывали концентрированной серной кислотой перед началом реакции. (+)-VIII•HCl (98 мг) добавляли к (47%) водному раствору HBr и нагревали с обратным холодильником в течение 2 ч в присутствии
азота. Кислоту отгоняли в вакууме при 100oC. Затем, дважды добавляли диэтиловый эфир, с последующим выпариванием. После перекристаллизации из MeOH/диэтилового эфира получали 75 мг (69%)
(+)-X•HBr, т.пл. 186 - 187oC. Оптическое вращение: +69,4 (с.0,9).
(-)5-фторо-8-гидрокси-2-(дипропиламино) тетралина гидробромид[(-)-X•HBr]
Повторяли
процедуру, описанную для (+)-X•HBr. Выход 85% т.пл. 186 - 187oC. Оптическое вращение: -69,3 (с. 1,0).
(+)-2-(N-бензил-N-пропиламино)-5-фторо-8-метокситетралина
гидрохлорид, (+)-XI•HCl
0,13 мл 1-иодопропан добавляли к смеси (+)-VI (0,41 г) и порошкообразного карбоната кальция (1,22 г) в 10 мл CH3CN Смесь размешивали при комнатной
температуре в присутствии азота в течение 31 дня. В течение этого периода добавляли частями еще 1,6 мл 1-иодопропана. После обработки, проведенной аналогично описанию для (+)-VIII получали 211 мг
(+)-XI-HCl, т.пл. 152 153oC путем кристаллизации из смеси метанола и диэтилового эфира. Оптическое вращение: +78,7 (с.1,0).
(-)-2-(N-бензил-N-пропиамино)-5-фторо-8-метокситетралина гидрохлорид, (-)-XI•HCl
Указанное соединение синтезировали способом, аналогичным описанному для (+)-XI•HCl, но
используя
(-)-VI в качестве исходного материала. После кристаллизации из EtOH-диэтилового эфира получали полугидрат, т.пл. 124 - 130oC. Оптическое вращение: -79,6 (с.1,0).
(-)-5-фторо-8-метокси-пропиламинотетралина гидрохлорид, (-)-IX•HCl/
(-)-XI•HCl (1,93 г) растворяли в 100 мл MeOH и гидрогенизировали в присутствии Pd/C. Выход (-)-IX•HCl
оставлял 0,75 г, т. пл. 208 210oC (EtOH/диэтиловый эфир). Оптическое вращение: -71,5 (с, 1,0).
(+)-HI•HCl получали аналогичным образом, но исходя из (+)-X•HC, т.пл. 207 210oC (EtOH/диэтиловый эфир).
(-)-VIII HCl (-)-IX•HCl
Реакцию осуществляли способом, в основном, аналогичным описанному выше алкилированию (-)-VI
с
использованием иодопропана.
Пример 2. (±)-5-фоторо-8-гидрокси-2-(дипропиламино тетралина гидробромид, (+)-X.
Получали аналогично получению гидробромидов (+)-X и (-)-X из соединения (±-VI, т.пл. 201 203oC после кристаллизации из EtOH/диэтилового эфира. Промежуточное соединение (±-VII превращали без предварительной очистки в (± )-VIII, которое кристаллизировали с 1/4 молекулы воды из EtOH/диэтилового эфира, т.пл. 146 147oC.
Пример 3. (±)-5-хлоро-8-гидрокси-2-(дипропиламино) тетралина гидрохлорид, (±)-XXI•HCl получали аналогично получению (±-X, но исходя из 4'-хлорфенил-3-хлоропропионата (XXIII), полученного с помощью реакции 4-хлорофенола и 3-хлоро-пропионилхлоридом. Соединение XXIII является бесцветной жидкостью с т. кип. 155 159oC при 10 мм рт.ст. выход 94% (±)-XXI•HCl имеет т.пл. 229 231oC (после перекристаллизации из смеси MeOH и диэтилового эфира).
4-Хлоро-7-гидрокси-1-инданон (XIII) получали из XXIII способом, в основном, аналогичным описанному для фторосоединения II.
Кристаллы собирали непосредственно путем перегонки с водяным паром, т.пл. 119 121oC; выход 74%
4-Хлоро-7-метокси-1-инданон (XIV) получали способом, аналогичным описанному для
получения III. Т.пл. 139 141oC. Выход 82%
5-Хлоро-8-метокси-2-тетралон (XVI) получали с использованием метилениндана XV (88% выход) в соответствии со способом, описанным для
синтеза соединения V. Выход из XV составлял 98%
IH ЯМР (метанол-d4): d 7,25 (д, 1H; 6,70 (д, 1H); 3,81 (м, 3H); 3,51 (с, 2H); 3,20 (т, 2H); 2,57 (т, 2H).
(± )-5-Хлоро-8-метокси-2-(пропиламино) тетралина гидрохлорид, (+)-XVI.
5,1 (±)-XVI растворяли в 250 мл сухого бензола, и добавляли 2,8 г 1-пропиламина. Смесь нагревали с обратным холодильником в течение 2 ч в аппарате Дина-Старка. После концентрации реакционной смеси полученный амин растворяли в 200 мл EtOH и гидрогенизировали в присутствии PtO2. После обработки и превращения в свободный амин с использованием 5 М смеси NaOH и диэтилового эфира проводили хроматографию на окиси алюминия, элюируя диэтиловым эфиром, и получали 4,45 г соответствующего гидрохлорида, после того как в диэтиловый эфир добавили HCl. Т. пл. 244 245oC. После кристаллизации из метанола/диэтилового эфира получали полугидрат.
(± )-5-хлоро-8-метокси-2-(дипропиламино) тетралина гидрохлорид, (+)-XIX.
1-Иодопропан (0,84 мл) добавляли к смеси гидрохлорида соединения XVI (2,00 г) и порошкового карбоната кальция 5, 48 г в 20 мл диметилцианида в присутствии азота. Смесь размешивали при комнатной температуре в течение 8 дней. В течение этого времени добавляли еще 0,66 мл (частями) иодопропана. После обработки, аналогичной описанной для фторо-аналога, получали 61% (±) XIX в виде гидрохлорида. Т.пл. 160 161oC. Это соединение использовали для получения (±)-XXI (выход 59%) путем дегидробромирования, осуществляемого описанным выше способом.
Реферат
Использование: в качестве препаратов, обладающих терапевтической активностью, направленной на центральную нервную систему. Сущность изобретения: продукт: (-)-5-фтор-8-гидрокси-2-(дипропиламино) тетралина гидробромид, выход 85%, т.пл. 186-187oC. Оптическое вращение: - 69,3/с, 1 (+) -5-фтор-8 гидрокси-2-(дипропиламино) тетралина гидробромид, выход 69%, т.пл. 186-187oC. Оптическое вращение: + 69,4 (с, 0,9). 2 с. и 6 з.п. ф-лы, 1 табл., 2 ил.
Формула
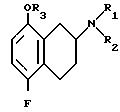
где R1 водород или С1 С6-алкил;
R2 водород или С1 С6-алкил;
R3 водород или С1 С6-алкил, который является прямой, разветвленной или циклической алкильной группой, имеющей 1 6 атомов углерода,
или его фармацевтически приемлемая соль.