Бициклические карбоновые кислоты, ингибирующие биологическую активность лейкотриена b4, и фармацевтическая композиция - RU2137765C1
Код документа: RU2137765C1
Чертежи





Описание
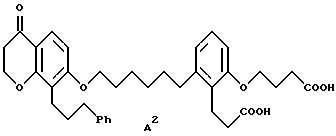
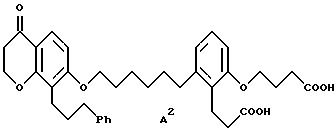
Объектом изобретения являются бициклические карбоновые кислоты формул A, B и C:
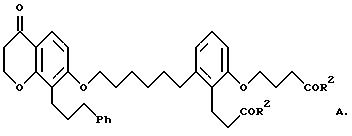
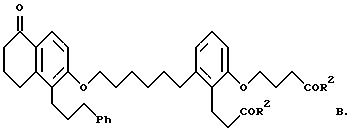
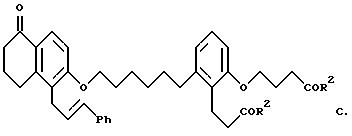
где R2 в каждом случае независимо друг от друга означает гидроксильную группу, низшую алкоксильную группу, и Ph означает фенил, и для соединения формулы C - к его геометрическому изомеру, и в случае, когда R2 означает гидроксильную группу, к фармацевтически применимым солям этих соединений с основанием.
Дополнительно R2 может означать NR3R4, где R3 и R4 независимо друг от друга означают водород или низший алкил.
Cоединения формул A, B и C являются сильнодействующими антагонистами лейкотриена B4 и поэтому пригодны для лечения воспалительных болезней, например, псориаза, ринита, хронических обструктивных легочных заболеваний, воспаления пищеварительного тракта, астмы, острого респираторного дистресс-синдрома, муковисциоза, аллергии, артрита, например, ревматоидного артрита, дерматита, например, контактного дерматита, гастропатии, вызванной нейросекреторным приобретенным иммунодефицитом, подагры, ишемии/нарушения кровеснабжения и травматических повреждений, таких, как повреждение спинного мозга.
Объектом данного изобретения являются соединения формул A, B и C и их фармацевтически применимые соли как таковые и используемые в качестве терапевтически активных веществ, получение таких соединений, лекарства, содержащие эти соединения, и изготовление таких лекарств, а также применение соединений формул A, B и C и их фармацевтически применимых солей для лечения или профилактики болезней или для улучшения здоровья, особенно для лечения или предотвращения воспалительных заболеваний, таких, как псориаз, ринит, хронические обструктивные легочные заболевания, воспаление пищеварительного тракта, астма, острый респираторный дистресс-синдром, муковисциоз, аллергия, артрит, например, ревматоидный артрит, дерматит, например контактный, гастропатия, вызванная нейросекреторным приобретенным иммунодефицитом (НСПИД), подагра, ишемия/нарушение кровeснабжения и травматические повреждения, например повреждение спинного мозга.
Другим объектом изобретения является фармацевтическая композиция, ингибирующая биологическую активность лейкотриена B4, а также способы применения соединений формул A, B и C.
Еще
одним объектом изобретения является промежуточное соединение 2-(3-фенилпропенилиден)-1,3-циклогександион, имеющее формулу
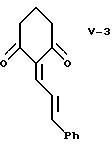
где Ph означает фенил,
и промежуточное соединение 3-(2-цианэтокси)-2-(3-фенилпропил)-2-циклогексен-1-он формулы

где Ph обозначает фенил.
Настоящее изобретение относится также к способу ингибирования биологической активности лейкотриена B4, который заключается во введении в организм пациента, который требует такое ингибирование, эффективного количества соединения формул A, B и C.
В данном описании используются следующие определения общих терминов, независимо от того, используются они в отдельности или в сочетании.
Используемый термин "низший алкил" означает линейный или разветвленный углеводородный насыщенный радикал, содержащий 1-7 атомов углерода, предпочтительно 1-4 атома углерода, например, метил, этил, пропил, изопропил, бутил, трет. -бутил, неопентил, пентил, гептил и т.п. Термин "низший алкоксильный" радикал означает алкилэфирную группу, в которой алкил обозначен выше, например, метокси, этокси, пропокси, пентокси и т.п.
Используемый термин "отщепляющаяся" группа означает галоид, предпочтительно бром и йод; низший алкилсульфонилоксигруппу, например, метилсульфонилоксигруппу, трифторметилсульфонилоксигруппу или т.п.; арилсульфонилоксигруппу, например, п-толуолсульфонилоксигруппу или т.п.
Кислоточувствительная гидроксизащитная группа означает предпочтительно тетрагидропиранил, 1-этоксиэтил, 1-метил-1-метоксиэтил и т.п.
Щелочной металл означает предпочтительно литий, натрий, калий и цезий.
Используемый термин "гидроксизащитная группа", удаляемая путем гидрирования, означает предпочтительно бензил, п-метоксибензил, трифенилметил и т.п.
Предпочтительные группы соединений представлены формулами A, B и C.
Наиболее
предпочтительными соединениями согласно изобретению являются:
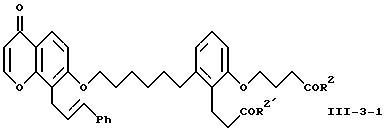
2-(3-карбоксипропокси)-6-[6-[[3,4-дигидро-4-оксо-8-(3-фенилпропил) - 2H-1-бензопиран-7-ил] окси] гексил] бензолпропановая кислота
(соединение формулы A);
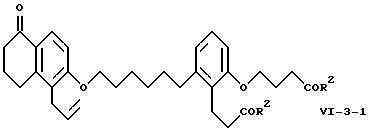
2-(3-карбоксипропокси)-6-[6-[[5,6,7,8-тетрагидро-5-оксо-1-(3- фенилпропил)-2-нафталинил] -окси]гексил]бензолпропановая кислота (соединение формулы B); и
(Е)-2-(3-карбоксипропокси)-6-[6-[[5,6,7,8-тетрагидро-5-оксо-1-(3- фенил-2-пропенил)-2-нафталинил]окси]гексил]бензолпропановая кислота (соединение формулы C).
Соединения настоящего
изобретения формул A, B и C могут быть получены способом, который включает
а) для получения соединений формул A или B, где R2=R2' и означает низший алкокси или водород,
взаимодействие соединения формулы
или
с соединением формулы
где R2' обозначает низший алкокси или водород и L обозначает отщепляющуюся группу,
или
б) для получения соединений формулы A, где R2 имеет указанные выше значения, каталитическое гидрирование соединения формулы
где R2 имеет указанные выше значения,
или
в) для получения соединений формулы B, где R2 имеет указанные выше значения, каталитическое гидрирование соединения формулы C, где R2 имеет указанные выше значения, или
г) для получения соединений формулы C, где R2 имеет указанные выше значения, взаимодействие соединения формулы
где R имеет указанные выше значения,
с соединением
PhL II-4
где Ph означает фенил и L обозначает отщепляющуюся группу, или
д) для получения соединений формул A, B и C, где R2 обозначает гидроксильную группу, омыление соединения A, B или C, где R2 означает низший алкокси, и
е) для получения соединений формул A, B и C, где R2 означает -NR3R4 и R3 и R4 имеют указанные выше значения, превращение соединения формул A, B или C, где R2 означает гидроксильную группу, в соответствующее соединение, где R2 означает -NR3R4,
ж) если желательно, превращение соединения формул A, B и C в его фармацевтически применимую соль.
Условия реакции для вышеуказанных способов а)-е) и для получения промежуточных соединений указаны подробнее ниже на реакционных схемах I-VI.
Реакционная схема 1
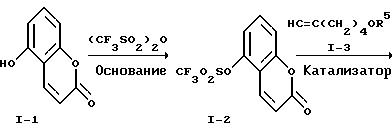
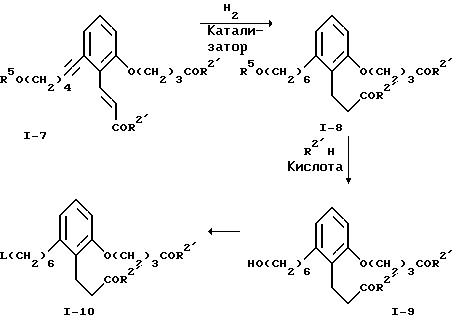
где R2' обозначает низший алкокси, R5 обозначает кислоточувствительную гидроксизащитную группу, L обозначает отщепляющуюся группу и М обозначает щелочной металл.
В реакционной схеме I, 5-гидроксикумарин, известное соединение формулы I-1, превращается в соответствующий эфир трифторметансульфокислоты I-2 обработкой ангидридом трифторметансульфокислоты в присутствии амина в качестве основания. Можно использовать любой амин в качестве основания. Предпочтительны пиридин или триэтиламин. Превращение предпочтительно проводят в среде растворителя - дихлорметана при 0-25oC. Соединение формулы I-2 можно выделить обычными методами, например, хроматографией или перекристаллизацией.
Соединение формулы I-2 подвергают взаимодействию с ацетиленовым соединением формулы I-3, которое является известным, в присутствии палладиевого катализатора и амина в качестве основания с получением соединения формулы I-4. Предпочтительно, чтобы это превращение проводилось с применением в качестве катализатора дихлорбис-(трифенилфосфин)палладия (II) и в качестве основания - триэтиламина в среде диметилформамида при 80-100oC. Соединение формулы I-4 выделяют с использованием обычных хроматографических методов.
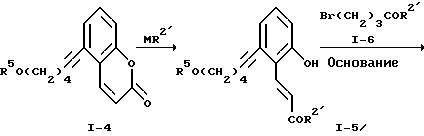
Соединение формулы I-4 превращают в соответствующий гидроксилсодержащий эфир коричной кислоты формулы I-5 путем алкоголиза лактонного цикла с использованием низшего алкоксида щелочного металла в среде низшего алканола. Это превращение проводят, применяя низший алкоксид лития, натрия или калия. Предпочтительно это превращение осуществлять в среде метанола или этанола в присутствии метоксида натрия или этоксида натрия при 60-80oC. Соединение формулы I-5 выделяют обычными хроматографическими методами или перекристаллизацией.
Алкилирование соединения формулы I-5 осуществляют бромсодержащим эфиром формулы I-6, являющимся известным соединением. Процесс проводят в присутствии основания, например, карбоната щелочного металла, например, карбоната натрия или калия, при температуре от примерно 25 до примерно 110oC в среде полярного апротонного растворителя, такого, как ацетонитрил, N,N-диметилформамид, 2-бутанон, диметилсульфоксид и т.п. Получаемое соединение формулы I-7 выделяют методом хроматографии.
Каталитическое гидрирование соединения формулы I-7 приводит к получению соответствующего насыщенного соединения формулы I-8. Это гидрирование проводят при обычных условиях. Более конкретно, предпочтительно применять катализатор на основе переходного металла на носителе, например, 5% или 10% палладия, нанесенного на уголь. Также предпочтительно проводить гидрирование при комнатной температуре и при давлении водорода в 1 атм. Предпочтительными растворителями при гидрировании служат низшие алканолы, например, метанол или этанол, или сложные эфиры, например, этилацетат или т.п. Можно также использовать смеси этих растворителей.
Удаление защитной группы R5 в соединении формулы I-8 для получения соответствующего спирта формулы I-9 проводят с использованием кислого катализатора. Предпочтительно этот процесс удаления защитной группы проводить в среде низшего алканола, например, метанола или этанола. Подходящими кислыми катализаторами являются органические сульфокислоты или их соли с аминами при 20-80oC. Особенно предпочтительно осуществлять это превращение с использованием п-толуолсульфокислоты в метаноле. Соединение формулы I-9 выделяют обычными хроматографическими методами.
Соединение формулы I-9 превращают в соответствующее производное I-10, используя стандартные методы, известные в области превращения гидроксильных групп в отщепляющиеся. Эти методы включают обработку галоидирующими реагентами, например, N-бромсукцинимид/трифенилфосфином или N-хлорсукцинимид/трифенилфосфином, в среде дихлорметана. Иначе соединение формулы I-9 можно превратить в соответствующий эфир сульфокислоты формулы I-10 обычными методами, например, обработкой алкил- или арилсульфонилхлоридом и органическим амином. Предпочтительно обрабатывать соединение формулы I-9 метансульфонилхлоридом и триэтиламином в среде дихлорметана, простого эфира или этилацетата при 0-25oC. Эти эфиры метансульфокислоты в свою очередь можно превратить в соответствующие йодиды формулы I-10 обработкой йодидом щелочного металла в среде полярного апротонного растворителя. Предпочтительно осуществлять это превращение, используя йодид натрия, в ацетонитриле при 20-80oC. Соединения формулы I-10 выделяют обычными методами экстракции.
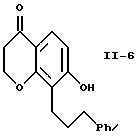
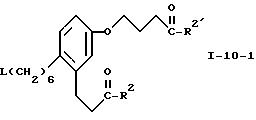
Реакционная схема II
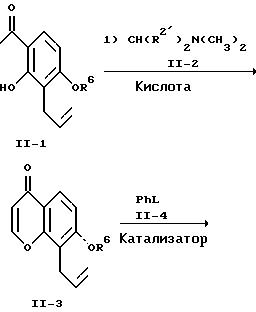
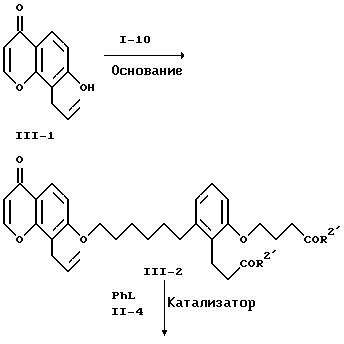
где R6 означает гидроксизащитную группу, удаляемую гидрированием, и R2', L и М имеют указанные выше значения.
На схеме II орто-гидроксиацетофенон формулы II-1, являющийся известным соединением, обрабатывают известным формамидацеталем формулы II-2 при 120-160oC в среде ароматического углеводородного растворителя, предпочтительно ксилола, с образованием промежуточного соединения, которое не очищают, а немедленно подвергают циклизации путем обработки кислотой с образованием хромона II-3. Предпочтительными кислотами для проведения этой циклизации служат органические сульфокислоты, например, п-толуолсульфокислота. Предпочтительными растворителями для осуществления этой циклизации являются низшие алканолы, например, метанол и этанол, при 60-80oC. Хромон формулы II-3 выделяют обычными хроматографическими методами или перекристаллизацией.
Хромон формулы II-3 конденсируют с производным бензола формулы II-4, являющимся известным соединением, в присутствии основания, палладиевого катализатора и четвертичной аммониевой соли с образованием соединения формулы II-5. Примерами производных бензола являются известные соединения типа иодбензола, фенилтрифторметансульфоната и т.п. Предпочтительно эту конденсацию осуществлять с использованием в качестве основания ацетата щелочного металла и тетраалкиламмонийгалогенида в качестве четвертичной аммониевой соли. Особенно предпочтительно, чтобы основанием служил ацетат натрия и четвертичной аммониевой солью - тетраэтиламмонийхлорид. Для проведения конденсации предпочтительным катализатором служит ацетат палладия (II). Эту конденсацию предпочтительно осуществлять при 25-100oC в полярном апротонном растворителе, например, N, N-диметилформамиде. Соединение формулы II-5 выделяют обычными хроматографическими методами или перекристаллизацией.
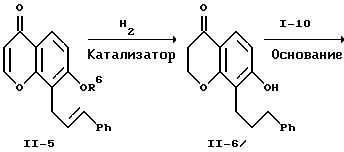
Каталитическое гидрирование хромона формулы II-5 с сопутствующим восстановительным отщеплением защитной группы R6 приводит к получению гидроксихроманона формулы II-6. Гидрирование проводят при обычных условиях. Катализатор на основе переходного металла на носителе является предпочтительным, например, 5% или 10% палладия на угле или древесном угле. Предпочтительно, чтобы это гидрирование проводилось при комнатной температуре и давлении водорода, равном 1 атм. Предпочтительными растворителями для проведения гидрирования являются низшие алканолы, например, метанол или этанол, или сложные эфиры, например, этилацетат. Можно также применять смеси этих растворителей. Это гидрирование можно проводить в две стадии, на первой удаляют защитную группу в присутствии палладия на угле и затем восстанавливают двойные связи, применяя обычный катализатор - никель Ренея. Соединение формулы II-6 можно выделить обычными методами хроматографии или перекристаллизацией.
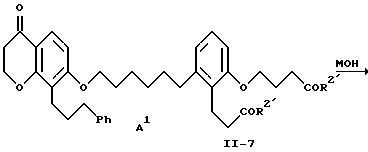
Гидроксихроманон формулы II-6 подвергают взаимодействию с соединением формулы I-10 (схема I) в присутствии основания, например, карбоната щелочного металла, такого как карбонат натрия или калия, при температуре от примерно 25 до примерно 110oC в полярном апротонном растворителе, например, ацетонитриле, N, N-диметилформамиде, 2-бутаноне, диметилсульфоксиде и т.п. В качестве основания можно также использовать гидрид щелочного металла, например, гидрид натрия, в этом случае предпочтительны такие инертные растворители, как тетрагидрофуран, простой эфир, толуол или N,N-диметилформамид. Кроме того, можно использовать способ по патенту США 4931574. В этом случае соединения формул II-6 и I-10 подвергают взаимодействию в присутствии карбоната щелочного металла, предпочтительно карбоната калия, и катализатора переноса фаз, предпочтительно трис[2-(2-метоксиэтокси)этил]амина (TDA-1), в ароматическом углеводородном растворителе, предпочтительно толуоле, при 80-110oC. Полученный диэфир формулы II-7 можно выделить обычными методами, например, хроматографией, и его можно превратить в соответствующую дикислоту формулы A2 путем омыления, используя гидроксид щелочного металла, например, гидроокись лития, натрия или калия, в смеси воды и смешивающегося с водой растворителя, например, метанола, этанола или тетрагидрофурана, при температуре от примерно 25 до примерно 60oC. Предпочтительно осуществлять это омыление при комнатной температуре в среде водного тетрагидрофурана, используя гидроксид лития. Соединения формулы A2 можно выделить обычными методами, например, перекристаллизацией или хроматографией.
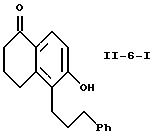
Реакционная схема III
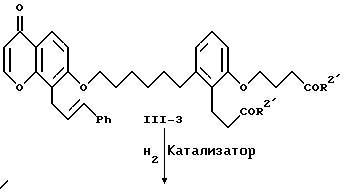
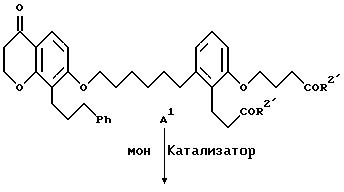
где L, М и R2' имеют указанные выше значения.
На схеме III известный гидроксихроманон формулы III-1 алкилируют соединением формулы II-6, как показано на реакционной схеме II для превращения соединения формулы II-6 в соединение формулы II-7. Соединение формулы III-2 выделяют обычной хроматографией и фенилируют с образованием соединения формулы III-3, как показано на схеме II для превращения соединения формулы II-3 в соединение формулы II-5. Соединение формулы III-3 выделяют обычной хроматографией. Каталитическое гидрирование соединения формулы III-3 приводит к образованию соответствующего соединения формулы II-7, выделяемого хроматографией. Это гидрирование проводят при условиях, показанных на схеме II для превращения соединения формулы II-5 в соединение формулы II-6. Омыление соединения формулы II-7 приводит к получению соответствующей дикислоты формулы А2 как показано на схеме II.
Реакционная схема IV
где R2', R6 и L имеют указанные выше значения.
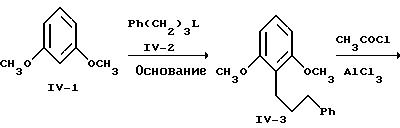
На реакционной схеме IV 1,3-диметоксибензол, известное соединение формулы IV-1, превращают в соединение формулы IV-3 сначала обработкой сильным основанием и затем алкилирующим агентом формулы IV-2, который представляет известные соединения, такие как 3-бром-1-фенилпропан, 3-йод-1-фенилпропан, 3-[(метилсульфонил)окси] -1-фенилпропан и т.п. Предпочтительно, чтобы основание, применяемое при алкилировании, было из ряда литийорганических соединений, например, метиллитий, фениллитий, н-бутиллитий и т.п., и чтобы алкилирование проводилось в среде инертного простого эфира. Особенно предпочтительно проводить алкилирование, используя н-бутиллитий в среде тетрагидрофурана при температуре от -20oC до комнатной. Соединение формулы IV-3 выделяют обычным методом хроматографии.
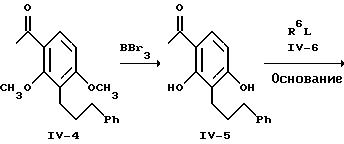
Ацетилирование соединения формулы IV-3 проводят при обычных условиях реакции Фриделя-Крафтса. Конкретно обработку ацетилхлоридом и алюминийхлоридом проводят в дихлорметане с получением соответствующего ацетофенона формулы IV-4, выделяемого хроматографией. Обработка соединения формулы IV-4 при стандартных условиях деметилирования, например, с использованием трехбромистого бора в растворе дихлорметана при температуре от -50oC до комнатной, приводит к образованию соответствующего дигидроксиацетофенона формулы IV-5, выделяемого обычной хроматографией или перекристаллизацией.
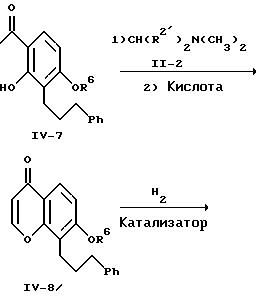
Дигидроксиацетофенон формулы IV-5 подвергают взаимодействию с соединением формулы IV-6, являющимся известным, в присутствии основания с образованием соединения формулы IV-7. Среди различных соединений формулы IV-6, которые можно применять, предпочтительны бензилхлорид или бензилбромид. Предпочтительно проводить это алкилирование, применяя в качестве основания карбонат калия, в среде ацетона или ацетонитрила при 20-80oC. Обработка соединения формулы IV-7 известным формамидацеталем формулы II-2, с последующей циклизацией в присутствии кислоты, как показано на реакционной схеме II для превращения соединения формулы II-1 в соединение формулы II-3, приводит к образованию хромона IV-8. Этот хромон IV-8 обычно выделяют обычным методом хроматографии или перекристаллизацией. Каталитическое гидрирование хромона IV-8 с сопутствующим восстановительным удалением арилметилэфирных фрагментов R6 приводит к получению хроманона формулы II-6. Это гидрирование-восстановление осуществляют как показано на реакционной схеме II для превращения соединения формулы II-5 в хроманон II-6.
Реакционная схема V
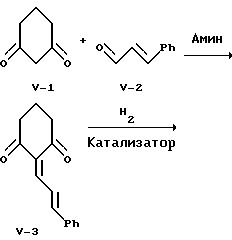
На реакционной схеме V 1,3-циклогександион, известное соединение формулы V-1, конденсируют с известным соединением - коричным альдегидом формулы V-2 в присутствии катализатора - вторичного амина с образованием диендиона формулы V-3, выделяемого кристаллизацией. Предпочтительно эту альдольную конденсацию осуществлять, используя в качестве катализатора циклический вторичный амин, например, пиперидин, в среде этанола при температуре от 0 до 30oC.
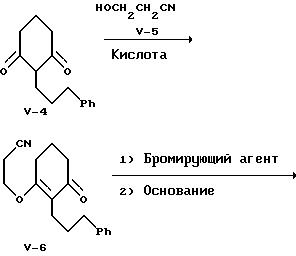
Каталитическое гидрирование диендиона формулы V-3 проводят, используя в качестве катализатора палладий на угле, в среде этилацетата с образованием соответствующего циклогександиона формулы V-4. Предпочитают проводить это гидрирование при комнатной температуре и при давлении водорода в 1 атм.
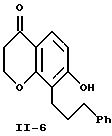
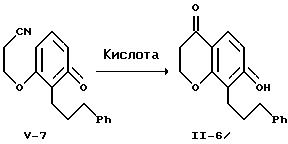
Соединение формулы V-4 обрабатывают известным соединением - 3-гидроксипропионитрилом формулы V-5 в присутствии кислого катализатора с получением енольной формулы простого эфира формулы V-6, выделяемой обычной хроматографией. Предпочтительно проводить эту реакцию, используя в качестве катализатора органическую сульфокислоту, например, п-толуолсульфокислоту, в среде инертного углеводородного растворителя, например, бензола или толуола, при 80-120oC.
Ароматизация енолэфира формулы V-6 c образованием соответствующего фенола формулы V-7 достигается бромированием с последующим дегидробромированием основанием. Бромирование можно осуществить любым обычным бромирующим реагентом, например, бромом, N-бромсукцинимидом и т.п., в среде инертного растворителя. Предпочтительно это бромирование проводить в растворе дихлорметана при температуре от -10 до 5oC с использованием 1,3-дибром-5,5-диметилгидантоина. Дегидробромирование можно провести в инертном растворителе, применяя стерически затрудненный третичный амин. Предпочтительно проводят дегидробромирование, используя в качестве основания 1,4-диазабицикло[2.2.2] октан, в растворе толуола при 25-110oC. Фенол формулы V-7 выделяют хроматографией.
Циклизацию фенола формулы V-7 проводят обработкой сильной кислотой с образованием гидроксихроманона формулы II-6, выделяемого хроматографией или кристаллизацией. Эту циклизацию можно осуществить с применением известных сильных кислот, например, серной, соляной, фосфорной и т.п. Предпочтительно осуществлять эту циклизацию, используя 85%-ную фосфорную кислоту, в растворе уксусной кислоты при 100-150oC.
Реакционная схема VI
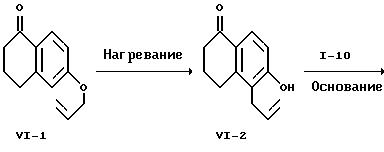
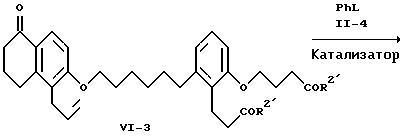
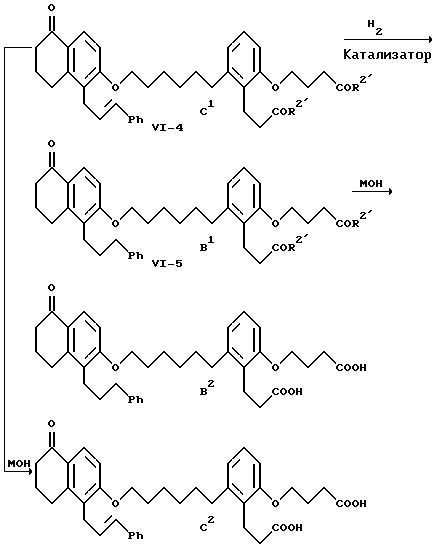
где R2', L и М имеют указанные выше значения.
На схеме VI аллиловый простой эфир формулы VI-1, известное соединение, подвергают термолизу для осуществления перегруппировки Клайзена. Предпочтительно проводить этот термолиз при 180-230oC без растворителя или в растворителе с достаточно высокой точкой кипения, например, в N,N-диэтиланилине. Желаемый изомерный нафталинон формулы VI-2 можно выделить перекристаллизацией. Нафталинон формулы VI-2 алкилируют соединением формулы I-10 с образованием соединения формулы VI-3, как показано на реакционной схеме II для превращения соединения формулы II-6 в соединение формулы II-7. Соединение формулы VI-3 выделяют обычной хроматографией и фенилируют с образованием продукта формулы VI-4, как показано на схеме II в случае превращения соединения формулы II-3 в соединение формулы II-5. Соединение формулы VI-4 выделяют обычной хроматографией. Каталитическое гидрирование соединения формулы VI-4 приводит к получению соединения формулы VI-5, которое выделяют хроматографией. Гидрирование проводят при условиях, показанных на схеме III, в случае превращения соединения формулы III-3 в соединение формулы II-7. Омыление соединения формулы VI-5 приводит к образованию дикислоты формулы B2, выделяемой перекристаллизацией, и это омыление осуществляют, как показано на схеме II для превращения соединения формулы II-7 в соединение формулы A2. Альтернативно омыление соединения формулы IV-4 в тех же условиях приводит к получению соответствующей дикислоты формулы C2, выделяемой хроматографией или перекристаллизацией.
Соединения формул A, B и C, где R2 означает -NR3R4 и R3 и R4 означают водород или низший алкил, могут быть получены из дикислот обычными методами, известными специалистам в данной области.
Изобретение также относится к солям соединений формул A, B и C, когда они содержат функциональные кислотные группы, что приводит к образованию солей с основанием. Соли соединений формул A, B и C, которые содержат карбоксильные группы, получают взаимодействием с нетоксичным, фармакологически применимым основанием. В общем использование любого основания, которое образует соль с карбоновой кислотой и фармакологические свойства которого не оказывают вредного физиологического воздействия, находится в рамках данного изобретения.
Подходящие основания включают, например, гидроокиси и карбонаты щелочных и щелочноземельных металлов и т.п., например, гидроокись кальция, гидроокись натрия, карбонат натрия, карбонат калия или т.п., аммиак, первичные, вторичные и третичные амины, такие как моноалкиламины, диалкиламины, триалкиламины, например, метиламин, диэтиламин, триэтиламин или т.п., азотсодержащие гетероциклические амины, например, пиперидин или т.п. Полученная таким образом соль является функциональным эквивалентом соответствующего соединения формул A, B и C, где R2 означает гидроксил, и специалисту очевидно, что разнообразные соли, охватываемые данным изобретением, должны только отвечать требованию, заключающемуся в том, что основание, используемое при получении соответствующих солей, должно быть нетоксичным и физиологически приемлемым.
Ниже показана полезная активность соединений формулы I в качестве антагонистов лейкотриена B4 (LTB4).
Методика.
Реакция связывания LTB4 рецептором.
Исследование реакции связывания можно проводить в лунках на микротитровальном планшете. Выделенные нейтрофилы человека в растворе соли Гея инкубируют на льду в течение 45 мин, используя 0,5 нМ3H-LTB4 в присутствии или в отсутствие испытуемых соединений. Испытания заканчивают добавлением 12 мл ледяного 50 мМ трисбуфера (pH 7,4) с последующей быстрой фильтрацией под вакуумом через GF/C фильтры. Радиоактивность определяют сцинтилляционным счетчиком. Неспецифическое связывание определяют как связывание, не проявляющееся при 100-кратном избытке немеченого LTB4. Специфическое связывание определяется как разница между общим и неспецифическим связыванием. Нелинейный анализ данных осуществляют с использованием LIGAND (Munson and Rodbard, 1980). Кi (константы ингибирования) определяют по уравнению Cheng-Prusoff (Cheng and Prusoff, 1973).
При использовании представителей соединений формул A, B или C по изобретению получают результаты ингибирования связывания3H-LTB4, приведенные в табл. 1.
Бронхостеноз у морских свинок, in vivo.
Проводят анестезирование самцов морских свинок (Hartley) весом 300-500 г внутрибрюшинно уретаном (2 г/кг) и вставляют в яремную вену полиэтиленовый катетер для введения лекарства. Давление в трахее записывают при помощи канюли, помещенной в трахее и соединенной с датчиком давления Gould P231D. После хирургической подготовки животных некоторое время ожидают стабилизации легочных функций. Затем животных парализуют сукцинилхолином (1,2 мг/кг, внутривенно) и осуществляют механическую искусственную вентиляцию легких (респиратор Harvard) с частотой 40 вдохов/мин и приливно-отливным объемом 2,5 см3. Испытуемое соединение вводят перорально за 2 ч до введения лейкотриена B4. Внутривенно вводят пропанолол (0,1 мг/кг) за 5 мин до введения лейкотриена B4. Затем внутривенно вводят животным промежуточную вызывающую сужение бронхов дозу лейкотриена B4 (200 мкг/кг).
Усредняют для 6 контрольных животных и 6 животных, которым ввели лекарство, разницу в давлении (см H2О) искусственного дыхания, измеренного
начале опыта и в пиковый момент. Процент ингибрирования рассчитывают по формуле:
((Контрольные - Получившие лекарство)/Контрольные)•100.
При использовании представителей соединений формул A, B и C по изобретению получены результаты, приведенные в табл. 2.
В практике использования изобретения доза соединения формул A, B, C или его соли, и частота приема зависит от эффективности и длительности действия конкретного соединения формул A, B, C или его соли и способа его введения, а также от степени и природы болезни и возраста млекопитающего, подвергающегося лечению и т.д. Оральные дозы соединения формул A, B или C или его соли колеблются от 2 мг до 2 г в день, предпочтительно от примерно 2 мг до 1 г в день в виде одноразовой дозы или нескольких доз.
Нижеследующие примеры иллюстрируют данное изобретение.
Соединение формул A, B или C или соль, или фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формул A, B или C или его соль вводится методами, хорошо известными в данной области техники. Таким образом, соединения формул A, B или C или их соли могут вводиться либо сами по себе, либо с другими фармацевтическими агентами, например, антигистаминами, ингибиторами выделения медиатора, метилксантинами, бета-агонистами или антиастматическими гормонами, например, преднизоном и преднизолоном, перорально, парентерально, ректально или путем ингаляций, например, в виде аэрозоля, мелкоизмельченного порошка или распыляемого раствора. Для орального введения их можно применять в виде таблеток, капсул, например, в смеси с тальком, крахмалом, молочным сахаром или другими инертными ингредиентами, т. е. фармацевтически применимыми носителями, или в виде водных растворов, суспензий, эликсиров или водно-спиртовых растворов, например, в смеси с сахаром или другими подсластителями, ароматизирующими веществами, красителями, загустителями и другими обычными фармацевтическими наполнителями. Для парентерального применения их можно применять в виде растворов или суспензии, например, в виде водного раствора или суспензии, или раствора в арахисовом масле, с использованием наполнителей и носителей, обычно применяемых при этом способе введения. При использовании в виде аэрозолей их можно растворить в подходящем фармацевтически приемлемом растворителе, например, этиловом спирте или смеси растворителей, и смешать с фармацевтически приемлемым пропеллентом. Такие аэрозольные композиции упаковывают в контейнер под давлением, снабженный аэрозольным клапаном для выделения композиции, находящейся под давлением. Предпочтительно, чтобы аэрозольный клапан был дозирующим, т. е. таким, который при работе выпускает заданную эффективную дозу аэрозольного состава. Для местного применения соединения назначаются в виде мази, крема, лосьона, порошка, геля или т.п. Подходящими носителями для местного применения являются глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воски, жидкие парафины, жидкие жирные спирты, стеролы, полиэтиленгликоли, производные целлюлозы и т.п.
Следует иметь в виду, что используемая в настоящем описании формула C обозначает также геометрические изомеры. Геометрические изомеры можно разделить на соответствующие E- и Z-изомеры, используя известные методы, как описано ниже в примерах.
В следующих примерах "обычное выделение" включает в себя три экстракции конкретным растворителем. Органические экстракты соединяют, промывают водой и насыщенным соляным раствором; сушат над безводным сульфатом магния, фильтруют и концентрируют под давлением водяного аспиратора. Остаток высушивают до постоянного веса под высоким вакуумом при 45oC. Все реакции, кроме гидрирования, проводят в инертной атмосфере азота или аргона.
Пример 1.
Получение 7-(фенилметокси)-8-(2-пропенил)-4H-1-бензопиран-4-она.
Смесь 5,0 г (17,73 ммоля) 1-[2-гидрокси-4-(фенилметокси)-3-(2- пропенил)фенил] этанона, 2,3 г (19,48 ммоля) диметилформамиддиметилацеталя и 5,0 мл ксилола перемешивают и нагревают на масляной бане при 120-130oC при отгонке метанола в течение 2,5 ч с помощью колонки Vigreux (3 дюйма = 7,62 см). Затем температуру бани повышают до 150-160oC и реакционную смесь перемешивают при этой температуре еще 30 мин. Смесь охлаждают и концентрируют при 60oC/высокий вакуум. К вязкому красно-коричневому маслянистому остатку добавляют 3,7 г (19,48 ммоля) моногидрата п-толуолсульфокислоты и 50 мл этанола. Полученный раствор перемешивают и нагревают с обратным холодильником 24 ч, затем охлаждают и разбавляют водой. Обычное выделение простым эфиром приводит к получению технического продукта, который перекристаллизовывают из смеси гексан-этилацетат. Получают 3,5 г (67,6%) 7-(фенилметокси)-8-(2-пропенил)-4H-1-бензопиран-4-она в виде желтого твердого вещества с температурой плавления 90-92oC.
Элементный анализ. Вычислено для C19H16O3, %: C 78,06; H 5,52. Найдено, %: C 77,97; H 5,56.
Пример 2.
Получение (Е)-7-(фенилметокси)-8-(З-фенил-2-пропенил)-4H-1- бензопиран-4-она.
Смесь 8,76 г (30 мМ) 7-(фенилметокси)-8-(2-пропенил)-4H-1- бензопиран-4-она (предыдущий пример), 6,7 г (32,84 ммоля) иодбензола, 5,11 г (30,84 ммоля) тетраэтиламмонийхлорида, 8,94 г (91,22 ммоля) безводного ацетата натрия и 64 мл сухого N.N-диметилформамида перемешивают при комнатной температуре и очищают в атмосфере аргона. Ацетат палладия (II) (0,38 г; 1,7 ммоля) добавляют при перемешивании при комнатной температуре в течение 24 ч. Смесь темного цвета разбавляют водой и выделяют простым эфиром по обычной методике (эфирные экстракты дополнительно промывают 12%-ным водным раствором бисульфита натрия). Перекристаллизация технического продукта из ацетонитрила приводит к получению 6,13 г (55,5%) конечного продукта в виде коричневого твердого вещества. Образцы для анализа получают отдельно в виде желтых кристаллов, температура плавления 131-133oC.
Элементный анализ. Вычислено для C25H20O3, %: C 81,50; H 5,47. Найдено, %: C 81,34; H 5,10.
Пример 3.
Получение 2,3-дигидро-7-гидрокси-8-(3-фенилпропил)-4H-1- бензопиран-4-она.
Смесь 6,1 г (16,6 ммоля) (Е)-7-(фенилметокси)-8-(3-фенил-2-пропенил)-4H-1-бензопиран-4-она из предыдущего примера, 1 г 10%-ного палладия на угле, 100 мл метанола и 300 мл этилацетата перемешивают при комнатной температуре в атмосфере водорода, пока не израсходуется приблизительно одна треть от теоретического объема газообразного водорода. Катализатор отфильтровывают под вакуумом и концентрируют фильтрат в вакууме. Остаток растворяют в 150 мл метанола и добавляют 0,5 г никеля Ренея. Продолжают гидрирование при тщательном контроле с помощью тонкослойной хроматографии. Когда восстановление практически завершится, катализатор отфильтровывают под вакуумом, и фильтрат концентрируют в вакууме. Твердый остаток (4,75 г) соединяют с 6,4 г, полученными в другом опыте (23,9 ммоля), и хроматографируют на силикагеле. Элюирование смесью гексан-этилацетат приводит к получению 10,73 г (93,9%) конечного продукта в виде бесцветного твердого вещества, температура плавления 110-112oC.
Пример 4.
Получение 1,3-диметокси-2-(3-фенилпропил)-бензола.
Раствор 8,70 г (63 ммоля) 1,3-диметоксибензола в 164 мл безводного тетрагидрофурана перемешивают при -20oC, одновременно по каплям в течение 20 мин добавляют 1,6М н-бутиллития в гексане (42,1 мл; 67,2 ммоля). Раствор перемешивают при -20oC в течение 3 ч и затем оставляют нагреваться до -5oC, после чего в течение 15 мин добавляют 15,66 г (63,6 ммоля) 1-йод-3-фенилпропана. Реакционную смесь перемешивают при -5oC в течение 1 ч и затем при комнатной температуре в течение 3 дней. После повторного охлаждения до -5oC реакционную смесь разлагают путем добавления 1,5 H водного раствора серной кислоты. Добавляют воду, и смесь обрабатывают простым эфиром по обычной методике. Остаток обрабатывают 100 мл гексана и смесь отфильтровывают. Удаление растворителя в вакууме приводит к получению 15,28 г (94,7%) конечного соединения в виде желтого масла.
Пример 5.
Получение 1-[2,4-диметокси-3-(3-фенилпропил)фенил]этанона.
Раствор 15,28 г (59,6 ммоля) 1,3-диметокси-2-(3-фенилпропил)-бензола из предыдущего примера и 4,68 г (59,6 ммоля) ацетилхлорида в 306 мл дихлорметана перемешивают при -5-0oC и добавляют 7,95 г (59,6 ммоля) хлористого алюминия. Полученную смесь перемешивают при -5- 0oC в течение 2 ч и затем оставляют нагреваться до комнатной температуры перед выливанием на лед. Обработка простым эфиром по обычной методике приводит к получению продукта, который хроматографируют на силикагеле. Элюирование смесью 7:3 гексан-простой эфир приводит к получению 10,0 г (56,3%) конечного продукта в виде бледно-желтого масла.
Пример 6.
Получение 1-[2,4-дигидрокси-3-(3-фенилпропил)фенил]этанона.
Раствор 10,0 г (33,5 ммоля) 1-[2,4-диметокси-3-(3-фенилпропил) фенил] этанона из предыдущего примера в 250 мл дихлорметана перемешивают при -50oC, в то время как в течение 15 мин добавляют 67 мл (67 ммоля) 1М трехбромистого бора в дихлорметане. Реакционную смесь перемешивают при -50oC в течение 1 ч и при комнатной температуре в течение 3 дней, прежде чем выливают на лед. Обработка смесью 9:1 дихлорметана и метанола по обычной методике приводит к получению продукта, который хроматографируют на силикагеле. Элюирование смесью гексан-простой эфир приводит к получению 6,69 г (74%) конечного соединения в виде твердого вещества. Перекристаллизация образца из смеси простой эфир-гексан приводит к получению бесцветного твердого вещества с температурой плавления 120-122oC.
Элементный анализ. Вычислено для C17H18О3, %: C 75,53; H 6,71. Найдено, %: C 75,31; H 6,73.
Пример 7.
Получение 1-[2-гидрокси-4-(фенилметокси)-3-(3-фенилпропил)фенил]этанона.
Смесь 6,69 г (24,7 ммоля) 1-[2, 4-дигидрокси-3-(3-фенилпропил) фенил]этанона из предыдущего примера, 5,35 г (31,3 ммоля) бензилбромида, 14,9 г (0,108 моля) безводного карбоната калия, 115 мл сухого N,N-диметилформамида и 230 мл ацетона нагревают с обратным холодильником при перемешивании в течение 8 ч. После охлаждения суспензию фильтруют под вакуумом и твердый остаток хорошо промывают ацетоном. Фильтрат и промывные воды соединяют и концентрируют при пониженном давлении с образованием желтого масла, которое хроматографируют на силикагеле. Получают 5,57 г (62,6%) желаемого моноэфира в виде бледно-желтого твердого вещества. Перекристаллизация образца из смеси гексан-этилацетат позволяет получить конечное соединение в виде бесцветных иголок с температурой плавления 115-116oC.
Элементный анализ. Вычислено для C24H24О3, %: C 79,97; H 6,71. Найдено, %: C 79,97; H 6,80.
Пример 8.
Получение 7-(фенилметокси)-8-(3-фенилпропил)-4H-1-бензопиран-4-она.
Используя процедуру примера 1, 1-[2-гидрокси-4-(фенилметокси)-3- (3-фенилпропил)фенил] этанон из предыдущего примера превращают в конечное соединение, получают бесцветное твердое вещество с температурой плавления 106-107,5oC (перекристаллизация из смеси гексан-этилацетат) с выходом 56,7%.
Элементный анализ. Вычислено для C25H22О3, %: C 81,05; H 5,99. Найдено, %: C 81,20; H 5,99.
Пример 9.
Получение 2, 3-дигидро-7-гидрокси-8-(3-фенилпропил)-4H-1-бензопиран-4-она.
Проводят каталитическое гидрирование 7-(фенилметокси)-8-(3-фенилпропил)- 4H- 1-бензопиран-4-она из предыдущего примера над 10% палладия на угле при комнатной температуре и давлении 1 атм в среде смеси 1:1 метанолэтилацетат. Технический продукт очищают хроматографией на силикагеле, используя в качестве элюента cмеси простой эфир-дихлорметан. Получают конечное соединение в виде бесцветного твердого вещества с температурой плавления 110-112oC (перекристаллизация из смеси гексан-простой эфир), выход составляет 44,9%.
Элементный анализ. Вычислено для C18H18О3, %: C 76,57; H 6,43. Найдено, %: C 76,42; H 6,43.
Пример 10.
Получение 2-оксо-2H-1-бензопиран-5-илового эфира трифторметансульфоновой кислоты.
Смесь 1,62 г (10 ммолей) 5-гидроксикумарина и 10 мл сухого пиридина в 25 мл дихлорметана перемешивают при охлаждении на ледяной бане, в то время как по каплям добавляют 4,5 г (16 ммолей) ангидрида трифторметансульфоновой кислоты. Смесь перемешивают на холодe в течение 30 мин и затем оставляют нагреваться до комнатной температуры и перемешивают еще 30 мин, прежде чем выливают в 3Н соляную кислоту. Обработка простым эфиром по обычной методике приводит к получению желтого твердого вещества. После быстрой хроматографии на силикагеле с использованием в качестве элюента смеси 2:1 гексан-этилацетат получают 2,6 г (88,4%) 2-оксо-2H-1-бензопиран-5-илового эфира трифторметансульфоновой кислоты в виде белого твердого вещества с температурой плавления 104-105oC.
Элементный анализ. Вычислено для C10H5F3O5S, %: C 40,83; H 1, 71. Найдено, %: C 40,65; H 1,59.
Пример 11.
Получение рац-5-[6-[(тетрагидро-2H-пиран-2-ил)окси]-1-гексинил]- 2H-1-бензопиран-2-она.
Смесь 1,47 г (5 ммолей) 2-оксо-2H-1-бензопиран-5-илового эфира трифторметансульфоновой кислоты из предыдущего примера, 1,0 г (5,5 ммоля) рац-6-[(тетрагидро-2H-пиран-2-ил)окси] -1-гексина, 75 мг йодистой меди, 0,3 г (0,428 ммоля) дихлор-бис(трифенилфосфин)палладия (II), 7,5 мл триэтиламина и 35 мл сухого N,N-диметилформамида перемешивают и нагревают при 100oC в течение 24 ч. Реакционную смесь охлаждают, выливают в воду и обрабатывают простым эфиром по обычной методике. Темно-коричневый маслянистый остаток быстро хроматографируют на силикагеле. Элюирование смесью 2:1 гексан-этилацетат приводит к получению 1,09 г (67%) рац-5-[6-[(тетрагидро-2H-пиран-2-ил)окси]- 1-гексинил]-2H-1-бензопиран-2-она в виде оранжевого масла.
Пример 12.
Получение метилового эфира рац-(Е)-3-[2-гидрокси-6-[6-[(тетрагидро- 2H-пиран-2-ил)окси]-1-гексинил]фенил]-2-пропеновой кислоты.
Раствор 1,09 г (3,3 ммоля) рац-5-[6-[(тетрагидро-2H-пиран-2-ил)окси]- 1-гексинил] -2H-1-бензопиран-2-она из предыдущего примера и 1,8 мл (7,9 ммоля) 25%-ного метанольного раствора метоксида натрия в 5 мл метанола перемешивают и нагревают с обратным холодильником в течение 24 ч и затем концентрируют при пониженном давлении. Остаток обрабатывают 1H соляной кислотой и обрабатывают этилацетатом по обычной методике (органические экстракты дополнительно промывают насыщенным водным раствором бикарбоната натрия). Остаток очищают быстрой хроматографией на силикагеле, используя в качестве элюента смесь 2:1 гексан-простой эфир. Получают 0,7 г (59%) метилового эфира рац-(E)-3-[2-гидрокси-6-[(тетрагидро-2H-пиран-2-ил)окси] -1-гексинил]- фенил]-2-пропеновой кислоты в виде желтого масла. Обработка образца, полученного таким образом, гексаном приводит к получению бесцветного твердого вещества с температурой плавления 66-67,5oC.
Элементный анализ. Вычислено для C21H26O5, %: C 70,37; H 7,31. Найдено, %: C 70,24; H 7,33.
Пример 13.
Получение этилового эфира рац-(Е)-4-[2-(3-метокси-3-оксо-1-пропенил)- 3-[6-[(тетрагидро-2H-пиран-2-ил)окси]-1-гексинил]-фенокси]бутановой кислоты.
Смесь 7,16 г (20 ммолей) метилового эфира рац-(Е)-3-[2-гидрокси- 6-[6-(тетрагидро-2H-пиран-2-ил)окси] -1-гексинил] фенил] -2-пропеновой кислоты из предыдущего примера, 4,37 г (22,4 ммоля) этил-4-бромбутирата, 8,32 г (60,29 ммоля) безводного гранулированного карбоната калия и 50 мл сухого диметилсульфоксида перемешивают при комнатной температуре в течение 23 ч. Полученную смесь разбавляют простым эфиром и обрабатывают по обычной методике. Получают 9,73 г этилового эфира рац-(Е)-4-[2-(3-метокси-3-оксо-1-пропенил)-3-[6-[(тетрагидро-2H- пиран-2-ил)окси]-1-гeкcинил]-фенокси]бутановой кислоты в виде желтого масла, содержащего примерно 10% этил-4-бромбутирата (данные ЯМР). Это вещество используют без дальнейшей очистки.
Пример 14.
Получение метилового эфира рац-2-(4-этокси-4-оксобутокси)- 6-[6-[(тетрагидро-2H-пиран-2-ил)окси]гексил]бензолпропановой кислоты.
9,73 г (примерно 20 ммолей) образца технического этилового эфира рац-4-[2-(3-метокси-3-оксо-1-пропенил)-3-[6-[(тетрагидро-2H-пиран-2- ил)окси]-1-гексинил]-фенокси]бутановой кислоты из предыдущего примера гидрируют в 275 мл этилацетата над 0,75 г 10%-ного палладия на угле при комнатной температуре и давлении 1 атм в течение 24 ч. Метиловый эфир рац-2-(4-этокси-4-оксобутокси)-6-[6-[(тетрагидро-2H-пиран-2- ил)окси] гексил] бензолпропановой кислоты в виде масла выделяют фильтрацией катализата и концентрированием фильтрата с количественным выходом (9,74 г).
Пример 15.
Получение метилового эфира 2-(6-гидроксигексил)-6-(4-метокси-4- оксобутокси)бензолпропановой кислоты.
Раствор 9,74 г (примерно 20 ммолей) метилового эфира рац-2-(4-этокси-4-оксобутокси)-6-[6-[(тетрагидро-2H-пиран-2- ил)окси]гексил]бензолпропановой кислоты из предыдущего примера и 0,53 г моногидрата п-толуолсульфокислоты в 270 мл метанола перемешивают и нагревают с обратным холодильником в течение 21,5 ч. Большую часть растворителя удаляют под вакуумом и остаток растворяют в простом эфире. Эфирный раствор промывают насыщенным раствором бикарбоната натрия и обрабатывают обычным способом с получением масла. Это вещество хроматографируют на 200 г силикагеля, используя в качестве элюента смесь гексан-этилацетат. Получают 7,03 г (92,5%) метилового эфира 2-(6-гидроксигексил)-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты в виде бесцветного масла.
Пример 16.
Получение метилового эфира 2-(4-метокси-4-оксобутокси)-6-[6- [(метилсульфонил)окси]-гексил]бензолпропановой кислоты.
Раствор 7,03 г (18,5 ммоля) метилового эфира 2-(6-гидроксигексил)-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты из предыдущего примера и 22,5 мл триэтиламина в 67,5 мл этилацетата перемешивают при охлаждении на ледяной бане, в то время как по каплям в течение 10 мин добавляют 6,75 мл (87,25 ммоля) метансульфонилхлорида. Полученная плотная суспензия перемешивается при 0-5oC в течение 10 мин и затем выдерживается в течение 21 ч при 0-5oC. Смесь обрабатывают 100 мл воды и 100 мл простого эфира при охлаждении. Обработка простым эфиром по обычной методике (органические экстракты дополнительно промывают 1H водным раствором соляной кислоты и насыщенным водным раствором бикарбоната натрия) приводит к получению 8,45 г (99,7%) метилового эфира 2-(4-метокси-4- оксобутокси)-6-[6-[(метилсульфонил)окси]-гексил] бензолпропановой кислоты в виде бледно-желтого масла, который используют без дальнейшей очистки.
Пример 17.
Получение метилового эфира 2-(6-иодгексил)-6-(4-метокси-4- оксобутокси)бензолпропановой кислоты.
Смесь 10,81 г (примерно 23,37 ммоля) технического метилового эфира 2-(4-метокси-4-оксобутокси)-6-[6-[(метилсульфонил)окси] - гексил]бензолпропановой кислоты из предыдущего примера, 7,01 г (46,7 ммоля) безводного йодистого натрия и 44 мл сухого ацетонитрила перемешивают при комнатной температуре в течение 17 ч и затем нагревают с обратным холодильником в течение 3,5 ч. После охлаждения смесь разбавляют 200 мл простого эфира и фильтруют под вакуумом. Твердое вещество промывают тщательно простым эфиром. Соединяют фильтрат и промывные воды и промывают 12%-ным водным раствором бисульфита натрия, затем обрабатывают по обычной методике. Получают 11,14 г (97,3%) метилового эфира 2-(6-иодгексил)-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты в виде желтого масла.
Пример 18.
Получение метилового эфира 2-[6-[(3, 4-дигидро-4-оксо-8-(3-фенилпропил)- 2H-1-бензопиран-7-ил)окси]гексил]-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты.
Смесь 12,25 г (25 ммоля) метилового эфира 2-(6-иодгексил)-6-(4- метокси-4-оксобутокси)бензолпропановой кислоты из предыдущего примера, 7,1 г (25,2 ммоля) 2,3-дигидро-7-гидрокси-8-(3-фенилпропил)- 4H-1-бензопиран-4-она (пример 9), 8,3 г (60 ммолей) безводного карбоната калия и 60 мл сухого ацетонитрила перемешивают и нагревают с обратным холодильником в течение 22 ч. После охлаждения смесь разбавляют простым эфиром и фильтруют под вакуумом. Твердое вещество хорошо промывают простым эфиром. Соединяют фильтрат и промывные воды и концентрируют под вакуумом с получением 16,5 г желтого масла. Это вещество хроматографируют на силикагеле, используя в качестве элюента смесь гексан-этилацетат. Получают 13,75 г (85,4%) чистого диметилового эфира, указанного в названии, в виде масла.
Пример 19.
Получение 2-(3-карбоксипропокси)-6-[6-[[3,4-дигидро-4-оксо-8-(3- фенилпропил)-2H-1-бензопиран-7-ил]окси]гексил]бензолпропановой кислоты.
Диэфир из предыдущего примера (13,75 г; 21,35 ммолей) омыляют при перемешивании в 500 мл тетрагидрофурана, содержащего 60 мл 3Н водного гидроксида лития, при комнатной температуре в течение 24 ч. Тетрагидрофуран удаляют под вакуумом, остаток растворяют в воде и подкисляют 3Н водным раствором соляной кислоты. Смесь обрабатывают этилацетатом по обычной методике с получением белого твердого вещества. Это вещество очищают быстрой хроматографией на силикагеле, используя в качестве элюента смесь 96:2,5:1 хлороформ-метанол-уксусная кислота. Перекристаллизация соединенных фракций чистой дикислоты из ацетонитрила приводит к получению 10,4 г (79%) дикислоты, указанной в названии, в виде бесцветного твердого вещества с температурой плавления 104-106oC.
Элементный анализ. Вычислено для C37H44O8, %: C 72,06; H 7,19. Найдено, %: C 71,74; H 7,27.
Пример 20.
Получение 3,4-дигидро-6-гидрокси-5-(2-пропенил)-1(2H)нафталинона.
Раствор 12,51 г (61,93 ммоля) 3,4-дигидро-6-(2-пропенилокси)- (2H)нафталинона в 125 мл N, N-диэтиланилина перемешивают и нагревают на масляной бане с температурой 225-230oC в течение 20,5 ч. Полученный раствор темно-янтарного цвета охлаждают и выливают в 300 мл холодной 3Н HCl. Смесь обрабатывают простым эфиром по обычной методике с образованием 12,29 г желтого твердого вещества, которое является смесью 5- и 7-аллил-изомеров оксинафталинонов. Это вещество перекристаллизовывают из этилацетата с получением чистого 5-аллильного изомера с выходом 60% в несколько этапов. Образец для анализа представляет собой желтое твердое вещество с температурой плавления 145-148oC.
Элементный анализ. Вычислено для C13H14О2 , %: C 77,20; H 6,98. Найдено, % : C 76,97; H 7,00.
Пример 21.
Получение метилового эфира 2-(4-метокси-4-оксобутокси)-6-[6-[[5,6,7, 8- тетрагидро-5-оксо-1-(2-пропенил)-2-нафталинил] окси] гексил]бензолпропановой кислоты.
Смесь 3,5 г (17,3 ммоля) 3,4-дигидро-6-гидрокси-5-(2-пропенил)- 1(2H)нафталинона (из примера 20), 8,5 г (17,35 ммоля) метилового эфира 2-(6-иодгексил)-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты (пример 17), 5,8 г (42,0 ммоля) безводного карбоната калия и 40 мл безводного ацетонитрила перемешивают и нагревают с обратным холодильником в течение 24 ч. Полученную смесь охлаждают, разбавляют простым эфиром и фильтруют под вакуумом. Твердое вещество тщательно промывают простым эфиром, затем соединяют фильтрат и промывные воды и концентрируют в вакууме. Остаток подвергают хроматографической очистке на силикагеле, используя в качестве элюента смесь гексан-этилацетат. Конечный диэфир получают с количественным выходом (9,73 г) в виде бледно-желтого масла.
Пример 22.
Получение метилового эфира (Е)-2-(4-метокси-4-оксобутокси)-6- [6-[[5,6,7, 8-тетрагидрo-5-оксо-1-(3-фенил-2-пропенил)-2-нафталинил] окси] гексил]бензолпропановой кислоты.
Смесь 9,73 г (17,23 ммоля) метилового эфира 2-(4-метокси-4- оксобутокси)-6-[6-[[5,6,7, 8-тетрагидро-5-оксо-1-(2-пропенил)-2- нафталинил]окси]гексил] бензолпропановой кислоты из примера 21, 3,84 г (18,86 ммоля) йодбензола, 2,93 г (17,72 ммоля) тетраэтиламмонийхлорида, 5,13 г (52,25 ммоля) безводного ацетата натрия и 37 мл сухого N,N-диметилформамида перемешивают при комнатной температуре и очищают в атмосфере аргона. Добавляют ацетат палладия (II) (0,216 г; 0,96 ммоля) и продолжают перемешивание при комнатной температуре в течение 22 ч. Темно-коричневую смесь обрабатывают простым эфиром и водой и фильтруют через целит. Фильтрат обрабатывают простым эфиром по обычной методике (эфирный экстракт дополнительно промывают 12%-ным водным раствором бисульфита натрия) с получением 11,54 г технического продукта. Это вещество подвергают хроматографической очистке на 350 г силикагеля, используя в качестве элюента смесь гексан-этилацетат. Получают 8,63 г (78%) олефина, указанного в названии, в виде бледно-желтого масла.
Пример 23.
Получение метилового эфира 2-(4-метокси-4-оксобутокси)-6-[6-[[5,6,7,8- тетрагидро-5-оксо-1-(3-фенилпропил)-2-нафталинил] окси] гексил] бензолпропановой кислоты.
Метиловый эфир (Е)-2-(4-метокси-4-оксобутокси)-6-[6-[[5,6,7,8-тетрагидро- 5-оксо-1-(3-фенил-2-пропенил)-2-нафталинил] окси]гексил]бензолпропановой кислоты (0,54 г; 0,84 ммоля) из предыдущего примера гидрируют в 25 мл этилацетата над 0,1 г 10%-ного палладия на угле при комнатной температуре и давлении 1 атм в течение 2 ч. Катализатор отфильтровывают и фильтрат концентрируют с получением 0,52 г (96%) диэфира, указанного в названии, в виде бледно-желтого масла.
Пример 24.
Получение 2-(З-карбоксипропокси)-6-[6-[[5,6,7, 8-тетрагидро-5-оксо- 1-(3-фенилпропил)-2-нафталинил]окси]гексил]бензолпропановой кислоты.
Метиловый эфир 2-(4-метокси-4-оксобутокси)-6-[6-[[5,6,7, 8-тетрагидро- 5-оксо-1-(3-фенилпропил)-2-нафталинил] окси] гексил]бензолпропановой кислоты из предыдущего примера (0,52 г; 0,8 ммоля) омыляют при перемешивании в 22 мл тетрагидрофурана, содержащего 2, 6 мл 3Н водного раствора гидроксида лития при комнатной температуре в течение 24 ч. Смесь концентрируют в вакууме. Остаток растворяют в воде и подкисляют 3Н водным раствором соляной кислоты. Обработка простым эфиром по обычной методике приводит к получению вязкого маслянистого остатка, который подвергают быстрой хроматографической очистке на силикагеле, используя в качестве элюента смесь 90:10:5 толуол-этилацетат-уксусная кислота. Перекристаллизация соединенных чистых фракций из смеси гексан-этилацетат приводит к получению дикислоты, указанной в названии, с выходом 73% (0,36 г) в виде бесцветного твердого вещества с температурой плавления 89-91oC.
Элементный анализ. Вычислено для C38H46O7, %: C 74,24; H 7,54. Найдено, %: C 74,15; H 7,75.
Пример 25.
Получение метилового эфира 2-(4-метокси-4-оксобутокси)-6-[6-[(4-оксо- 8-(2-пропенил)-4H-1-бензопиран-7-ил)окси]гексил]бензолпропановой кислоты.
К суспензии 202 мг (5,05 ммоля) 60%-ной дисперсии гидрида натрия в минеральном масле (предварительно промытой пентаном) в 3 мл безводного N,N-диметилформамида добавляют раствор 0,77 г (3,81 ммоля) 7-гидрокси-8-(2-пропенил)-4H-1-бензопиран-4-она в 12 мл безводного N,N-диметилформамида в течение 1 мин. Смесь перемешивают при комнатной температуре в течение 15 мин, после чего в течение 1 мин добавляют раствор 2,1 г (4,29 ммоля) метилового эфира 2-(6-иодгексил)-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты (пример 17) в 15 мл безводного N, N-диметилформамида. Полученную смесь перемешивают при комнатной температуре в течение 3 ч, затем обрабатывают ледяной водой и обрабатывают этилацетатом по обычной методике. Маслянистый продукт хроматографируют на силикагеле, используя в качестве элюента смесь гексан-этилацетат. Получают 1,7 г (79,1%) соединения, указанного в названии, в виде желтого масла.
Пример 26.
Получение метилового эфира 2-(4-метокси-4-оксобутокси)-6-[6-[(4-оксо-8- (3-фенил-2-пропенил)-4H-1-бензопиран-7-ил)окси] гексил]бензолпропановой кислоты.
Используя методику примера 22, метиловый эфир 2-(4-метокси-4-оксобутокси)- 6-[6-[(4-оксо-8-(2-пропенил)-4H-1-бензопиран-7-ил)окси]гексил]бензолпропановой кислоты из предыдущего примера превращают в соединение, указанное в названии, получают бледножелтое масло с выходом 73%.
Пример 27.
Получение метилового эфира 2-[6-[(3, 4-дигидро-4-оксо-8-(3-фенилпропил)- 2H-1-бензопиран-7-ил)окси]гексил]-6-(4-метокси-4-оксобутокси)бензолпропановой кислоты.
Используя методику примера 23, метиловый эфир 2-(4-метокси-4- оксобутокси)-6-[6-[(4-оксо-8-(3-фенил-2-пропенил)-4H-1-бензопиран-7- ил)окси]гексил] бензолпропановой кислоты подвергают каталитическому гидрированию с получением соединения, указанного в названии, в виде масла.
Пример 28.
Получение 2-(3-фенилпропенилиден)-1,3-циклогександиона.
Раствор 11,2 г (0,1 моля) 1, 3-циклогександиона в 110 мл этанола перемешивают при комнатной температуре при добавлении 1 мл пиперидина и последующем добавлении по каплям в течение 2 мин 13 мл (0,103 моля) транскоричного альдегида. Полученную смесь перемешивают при комнатной температуре в течение 2 ч и затем на ледяной бане в течение 1 ч. Полученную желтую суспензию фильтруют под вакуумом, и твердое вещество сушат под высоким вакуумом, получая 13,9 г (61,5%) диона, указанного в названии. Перекристаллизация образца этого вещества из этилацетата приводит к получению желтого твердого вещества с температурой плавления 115,5-118oC.
Элементный анализ. Вычислено для C15H14О2, %: C 79,62; H 6,24. Найдено, %: C 79,46; H 6,34.
Пример 29.
Получение 2-(3-фенилпропил)-1,3-циклогександиона.
Смесь 6,8 г (30 ммолей) 2-(3-фенилпропенилиден)-1,3-циклогександиона из предыдущего примера, 0,7 г 10%-ного палладия на угле и 150 мл этилацетата перемешивают при комнатной температуре в атмосфере водорода, пока не прекратится поглощение газа. Катализатор отфильтровывают под вакуумом и концентрируют фильтрат под вакуумом с получением 6,7 г (97%) диона, указанного в названии, в виде белого твердого вещества, которое используют без дальнейшей очистки.
Пример 30.
Получение 3-(2-цианэтокси)-2-(3-фенилпропил)-2-циклогексен-1-она.
Смесь 6,7 г (29 ммолей) 2-(3-фенилпропил)-1,3-циклогександиона из предыдущего примера, 9,9 мл (0,145 моля) 3-гидроксипропионитрила, 0,3 г моногидрата п-толуолсульфокислоты и 110 мл толуола перемешивают и нагревают с обратным холодильником, используя для удаления воды ловушку Дина-Старка, в течение 3 ч. Смесь охлаждают, разбавляют этилацетатом и промывают насыщенным водным раствором бикарбоната натрия. Обработку завершают по обычной методике, получая 9,3 г желтого масла. Это вещество хроматографируют на силикагеле. Элюирование смесью гексан-этилацетат приводит к получению 5,9 г (71,6%) соединения, указанного в названии, в виде желтого масла.
Пример 31.
Получение 3-(2-цианэтокси)-2-(3-фенилпропил)фенола.
К перемешиваемому раствору 6,7 г (23,7 ммоля) 3-(2-цианэтокси)-2-(3-фенилпропил)-2-циклогексен-1-она (предыдущий пример) в 75 мл дихлорметана, охлажденного до 0-5oC, добавляют 6,8 г (23,8 ммоля) 1,3-дибром-5,5-диметилгидантоина. После перемешивания в течение 30 мин на холодe добавляют 50 мл насыщенного водного раствора бисульфита натрия. Обработка дихлорметаном по обычной методике приводит к получению 9,3 г желтого масла. Это вещество растворяют в 125 мл толуола и добавляют 7,1 г (63,4 ммоля) 1,4-диазабицикло[2.2.2] октана. Смесь перемешивают и нагревают с обратным холодильником в течение 30 мин, затем охлаждают и обрабатывают 2н. соляной кислотой. Обработка этилацетатом по обычной методике приводит к получению оранжевого масла, которое хроматографируют на силикагеле. Элюирование смесью гексан-этилацетат приводит к получению 3,5 г (52,6%) фенола, указанного в названии, в виде желтого масла.
Пример 32.
Получение 2,3-дигидро-7-гидрокси-8-(3-фенилпропил)-4H-1-бензопиран-4-она.
Смесь 3,5 г (12,5 ммоля) 3-(2-цианэтокси)-2-(3-фенилпропил)фенола (предыдущий пример), 16 мл 85%-ной фосфорной кислоты и 7,7 мл уксусной кислоты перемешивают и нагревают при 125oC в течение 23 ч. После охлаждения смесь разбавляют водой и обрабатывают этилацетатом по обычной методике с получением 4 г красного масла. Это вещество хроматографируют на силикагеле. Элюирование смесью гексан-этилацетат позволяет получить 2,3 г (65,2%) соединения, указанного в названии, в виде желтого твердого вещества.
Пример 33.
Получение (Е)-2-(3-карбоксипропокси)-6-[6-[[5,6,7, 8-тетрагидро-5-оксо-1- (3-фенил-2-пропенил)-2-нафталинил]окси]гексил]бензолпропановой кислоты.
Метиловый эфир (Е)-2-(4-метокси-4-оксобутокси)-6-[6-[[5,6,7, 8-тетрагидро- 5-оксо-1-(3-фенил-2-пропенил)-2-нафталинил] окси]гексил]бензолпропановой кислоты (из примера 22) омыляют, используя методику примера 19, с получением дикислоты, указанной в названии, с выходом 82% в виде бесцветного твердого вещества с температурой плавления 102-104oC, перекристаллизованного из смеси гексан-этилацетат.
Элементный анализ. Вычислено для C38H44O7, %: C 74,49; H 7,24. Найдено, %: C 74,28; H 7,14.
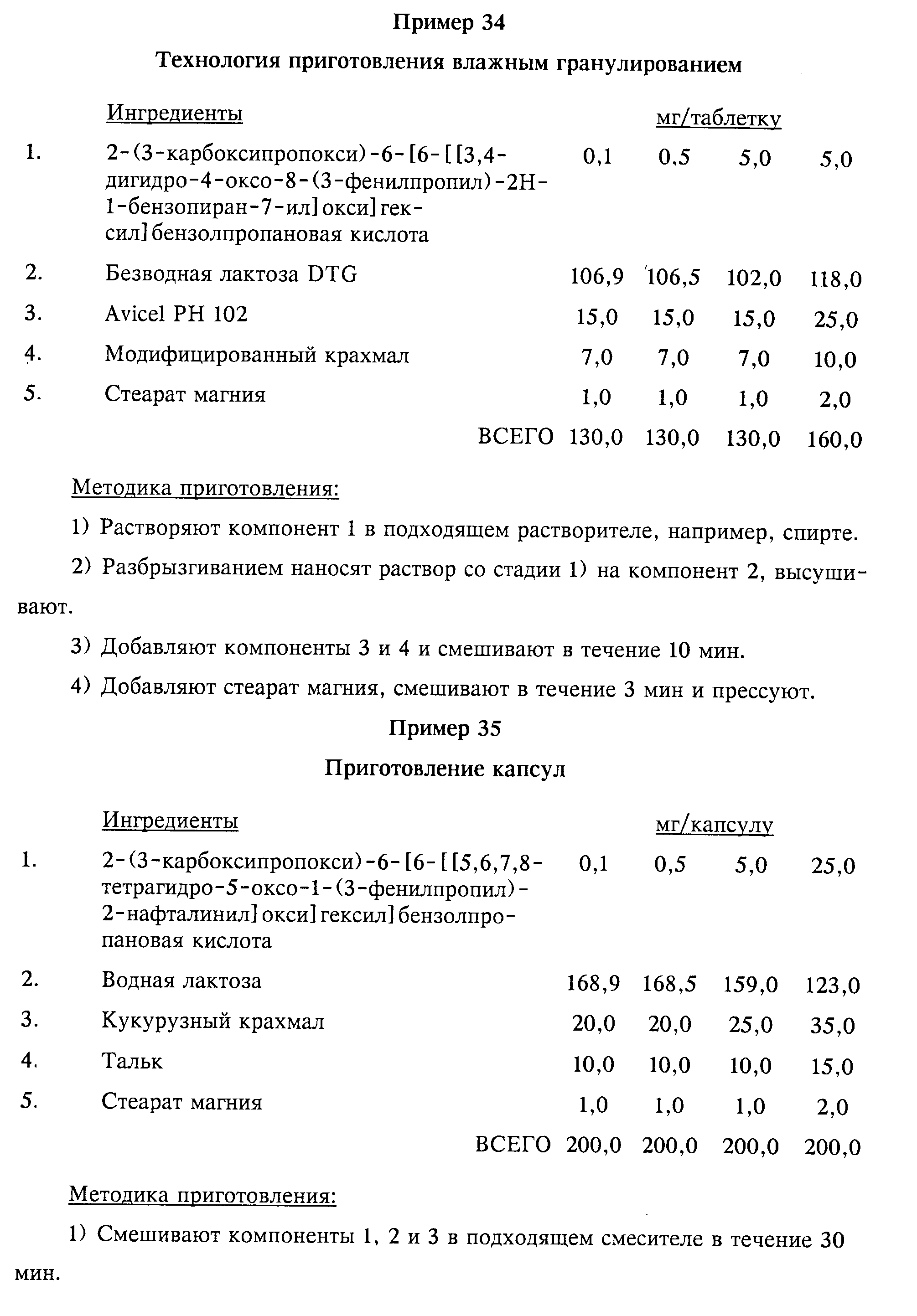
Примеры 34-39 приведены в конце описания.
Реферат
Бициклические карбоновые кислоты формул А, В и С, где R2 - гидроксил или низший алкоксил, являются сильнодействующими антагонистами лейкотриена В4 и поэтому пригодны для лечения воспалительных болезней. 4 c. и 5 з.п. ф-лы, 2 табл.
Формула
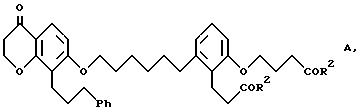
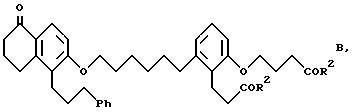
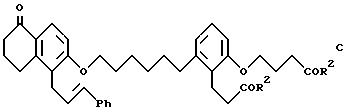
где R2 в каждом случае независимо друг от друга означают гидроксильную группу или низшую алкоксильную группу, Ph означает фенил и для соединения формулы С - его геометрический изомер, и в случае, когда R2 означает гидроксильную группу, фармацевтически применимая соль этого соединения с основанием.
Комментарии