Способ получения замещенных инданонов - RU2107681C1
Код документа: RU2107681C1
Описание
Изобретение относится к простому с технической точки зрения способу получения замещенных I-инданонов.
Соединения этого типа являются важными промежуточными продуктами при получении металлоценовых комплексов, так как I-инданоны можно легко превратить в соответствующие индены. Индены применяют как систему лигандов для синтеза металлоценовых компонентов (EP-A 336128). В частности, соответствующие хиральные производные циркона, содержащие мостик, имеют большое значение как высокоактивные катализаторы при полимеризации олефинов (ср. EP-A 129368: EP-A 321852). Путем изменения системы лигандов, например, путем замещения можно целенаправленно повлиять на свойства катализатора.
Далее замещенные I-инданоны имеют значение с технической точки зрения как ароматические вещества и как ценные промежуточные продукты при получении фармацевтических продуктов или других биоактивных соединений.
В литературе описано несколько способов получения замещенных I-инданонов.
I-инданоны, имеющие заместители в шестичленном кольце, могут быть получены, исходы из замещенных ароматических веществ, путем конденсирования пятичленного кольца в 2-6 стадий синтеза (J. Org. Chem., 55 (1990) 247; Bull, Soc. Chim. Fr. 6 (1969(1981).
Способы получения I-инданонов, имеющих заместители в пятичленном кольце или в обоих кольцах, также известны (J. Org. Chem. 46 (1981), 3758; J. Org. Chem. 23 (1958) 1441). Эти способы как правило имеют много стадий и приводят к низкому общему выходу целевых продуктов. Использование многих синтезов ограничено специальными производными. В других исходные материалы являются мало доступными или очень дорогими. В некоторых случаях замещение в ароматическом кольце также нельзя осуществить при помощи этих способов. Немногие известные варианты одностадийного синтеза ограничены специальными производными и дают низкий выход, так что необходимы дорогостоящие с технической точки зрения операции по очистке продуктов.
Наиболее близким по технической сущности к предлагаемому является способ получения замещенных инданонов с использованием ароматического соединения [1]. Известный способ не позволяет получать целевой продукт с высоким выходом.
Задачей изобретения является способ получения замещенных инданонов, позволяющий избежать недостатков, известных из уровня техники.
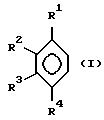
Показано, что ароматические вещества нижеследующей формулы (I) подвергают взаимодействию с имеющимися в продаже ангидридами карбоновой кислоты формулы (II) или фторидами карбоновой кислоты формулы (III) в жидком фтористом водороде и получают почти в количественном отношении инданоны формулы (IV/IVa).
Поэтому данное изобретение относится к способу получения соединения формулы (IV) или их изомеров формулы (IVa).

где
R1-R7 одинаковые или различные водород, C1-C20-алкил, C6-C10-арил, C1-C10-алкокси, или соседние остатки R1-R4 образуют вместе со связывающими их атомами до четырех колец, которые могут быть замещены низшим алкилом, заключающемуся в том, что соединение формулы (I)

где
значения R1-R4 приведены выше, подвергают взаимодействию с ангидридом карбоновой кислоты формулы (II)
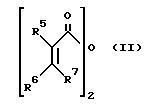
или м фторангидридом формулы (III)

где
R5-R7 приведены выше, в избытке жидкого безводного фтористого водорода. При этом алкил является линейным или разветвленным.
Кольца, образованные из соседних остатков R1-R4, могут быть замещены заместителями в значении R1-R7, включая указанные места для замещения.
Предпочтительно в формулах (IV) и (IVa) R1-R4 являются одинаковыми или различными и обозначают водород. (C1-C10)алкокси, или остатки R1 и R2, R2 и R3 или R3 и R1 образуют с соединяющими их атомами замещенной или незамещенной пятичленное или шестичленное кольцо, R5 обозначает C1-C10-алкил, R6-R7 означают водород.
Наиболее предпочтительно, когда R1-R4 являются одинаковыми или различными и обозначают водород, C1-C10алкил или остатки R1 и R2, R2 и R3 или R3 и R4 образуют с соединяющими их атомами замещенный или незамещенный шестичленный, насыщенный или ненасыщенный карбоцикл, R5 означает метил и R6 и R7 означают водород.
Образованные из соседних заместителей R1-R4 насыщенные или ненасыщенные пятичленное или шестичленное кольца (карбоцикл) могут дополнительно иметь заместители, преимущественно C11-C10алкил.
Инданоны могут образоваться в зависимости от положения групп в ароматическом кольце в форме двух структурных изомеров формулы (IV) и (IVa). В зависимости от цели применения их можно далее выделить в чистой формуле или в виде смеси. При получении металлоценовых компонентов можно использовать изомерную смесь.
Инданоны формулы (IV) и (IVa) преимущественно получают путем взаимодействия ароматических веществ формулы (I) с ангидридами формулы (II). Исходные соединения формулы (I) известны и выпускаются промышленностью. Исходные соединения формулы (II) также можно получить известными способами.
Фториды карбоновой кислоты формулы (III) можно получить из известных хлоридов карбоновой кислоты или
ангидридов карбоновой кислоты (формулы II) способом, известным из литературы, путем обмена с HF (Advanced Organic Chemistry, 1983, 399)/
При получении соединений IV/IVa во фтористый водород
можно добавить дополнительный растворитель, однако реакцию проводят преимущественно в чистом, безводном фтористом водороде.
Молярные соотношения исходных соединений, включая фтористый водород, могут колебаться в широких пределах. Преимущественно имеет молярное соотношение соединения I : II /и III/ : HF = 1 : 0,5 - 2,0 : 5 - 100; особенно 1: 0,9 - 1,2 : 20 - 50, т.е. реакцию проводят в избытке фтористого водорода.
Температура реакции составляет преимущественно 30 - 130oC, в основном 0 - 80oC.
Время реакции колеблется, как правило, между 30 мин и 50 ч, преимущественно между 1 ч и 24 ч.
Реакцию преимущественно приводят при давлении 1-15 атм.
Преимущественно готовят смесь соединений I и II /и III/ и дополнительно добавляют фтористый водород. Возможен и обратный порядок добавления.
По окончании реакции можно отогнать фтористый водород и почти в количественном отношении без существенных примесей выделить снова.
Инданоны формулы IV и IVa можно выделить, промыв раствором Na2CO3, Na HCO3 или KOH и водой от остатков кислоты, и высушить обычными средствами, таким как Na2SO4, MgSO4 или молекулярные сита. Так как реакция взаимодействия является, как правило, почти количественной, то в большинстве случаев от дальнейшей очистки можно отказаться. Однако часто рекомендуют провести фильтрацию с помощью силикагеля, окиси алюминия или фильтрующих средств, таких как, например, целиты. Если это необходимо, то можно провести дальнейшую очистку путем дистилляции, колоночной хроматографии или кристаллизации. Если это требуется, то изомеры IV и IVa можно отделить друг от друга путем колоночной хроматографии на силикагеле или окиси алюминия.
Способ согласно изобретению отличается в основном тем, что различные замещенные I-инданоны путем простого и быстрого синтеза (одностадийный процесс) получают селективно при почти количественно выходе. Поэтому дорогостоящая очистка производных не является необходимой. Еще одно преимущество заключается в том, что фтористый водород, используемый в качестве катализатора, можно в значительной степени неоднократно использовать, так как во время реакции вода не образуется. Это имеет еще одно технически важное преимущество, а именно отсутствие коррозии, вызываемой фтористоводородной кислотой.
Поэтому этот способ представляет собой очень выгодный с экономической и экологической точки зрения способ получения замещенных I-инданонов. Заместители в пятичленном и шестичленном кольцах могут при этом широко варьироваться. Поэтому и новые производные I-инданонов также являются доступными.
Инданоны формулы IV/IVa применяют преимущественно для получения металлоценовых комплексов, которые используют как высокоактивные компоненты катализатора при полимеризации олефинов. Для этого инданоны, преимущественно в виде изомерной смеси, вначале восстанавливают известными способами с помощью восстановителей, таких, как NaBH4 или LiAIH4 в соответствующие инданоны, и затем их дегидратируют с помощью кислот, таких, как серная, щавельная n-толуолсульфоновая кислоты, или путем обработки обезвоживающими веществами, такими, как сульфат магния, сульфат натрия, окись алюминия, силикагель или молекулярные сита до получения инденов.
Замещенные инданоны можно получить в виде изомеров с двойными связями. Их можно подвергнуть очистке от побочных продуктов путем дистилляции, колоночной хроматографии или кристаллизации. Изомеры можно использовать непосредственно как смесь для синтеза соответствующих металлоценовых комплексов.
Пример 1. 6,
7-бензо-2-метилиндан-1-он (I) и 4,5-бензо-2-метилиндан-1-он (Ia)
12,6 г (98 ммоль) нафталина и 15,8 г (103 моль) ангидрида метакриловой кислоты смешивают в автоклаве из стали повышенного
качества вместимостью 250 мл со 100 г (5 моль) безводной HF (фтористоводородной кислоты) и перемешивают 18 ч при 50oC. Затем фтористый водород отгоняют, остаток вносят в уксусный эфир и
нейтрализуют разбавленным раствором КОН. Отделенную водную фазу дважды экстрагируют уксусным эфиром. Объединенные органические фазы сушат с помощью MgSO4и отделяют при пониженном давлении
от растворителя. Получают 19,0 г (99% теор.) светло-коричневого масла. Селективность по I и Ia составляет 58 и 39%.
1H-ЯМР - спектры (100 Мгц, CDCl3):1:9,15 (m, 1H), 7,40 - 8,10 (m, 5H), 3,47 (dd, 1H), 2,62 - 2,95 (m, 3H), 1,37 (d, 3H), 1a:7,4 - 8,0 (m, 6H), 3,70 (dd, 1H), 2,75 - 3,10 (m, 2H), 1,40 (d, 3H).
Масс-спектр: 196 М+, правильное разложение.
Пример 2. 6,7-бензо-2-метилиндан-1-он (1) и 4,5-бензо-2-метилиндан-1-он (1a).
В автоклав вместимостью 2 л помещают 120 г (0,94 моль) нафталина и 153 мл (1,03 моль) ангидрида метакриловой кислоты и медленно смешивают при комнатной температуре с 1000 г HF. Эту смесь медленно нагревают до 60oC и выдерживают при этой температуре 18 ч. Фтористый водород затем конденсируют при 30 - 35oC и выделяют снова. Осадок вносят в этилацетат, дважды промывают водой, дважды насыщенным раствором NaHCO3 и один раз раствором NaCl. После фильтрации через силикагель и удаления растворителя при пониженном давлении получают 180 г (98% теор.) чистой изомерной смеси. Селективность по 1 и 1a составляет 60 и 40%.
Пример 3. 5,7-диизопропил-2-метилиндан-1-он (2) и 4,6-диизопропил-2-метилиндан-1-он (2a). 15,6 (96 ммоль) 1,3-диизопропилбензола и 15,8 г (103 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 с HF и выделяют. Получают 22 г (99% теор.) светло-коричневого масла. Селективность по 2 и 2a составила 66% и 30%.
1H-ЯМR - спектр (360 МГц,
CDCl3: изомерная смесь 7,49 (d) 7,36 (d), 7,13 (S), 7,10 (S), 4,15 (sept.), 3,25 - 3,40 (m) 3,10 (sept.), 2,90 - 3,00 (m), 2,60 - 2,73 (m), 1,22 - 1,30 (m)
Масс - спектр: 230
М+, правильное разложение.
Пример 4. 5,7-диизопропил-2-метилиндан-1-он (2) и 4,6-диизопропил-2-метилиндан-1-он (2a).
Подвергают взаимодействию 15,6 г (96 ммоль) 1,3-диизопропилбензола с 15,8 г (103 ммоль) ангидрида метакриловой кислоты (аналогично примеру 3). Сырую смесь подвергают хроматографии на 700 г силикагеля 60 с помощью смеси гексана и этилацетата (20:1), состав которой во время хроматографии изменен в соотношении 10:1, элюируют вначале 14,0 г (63 % теор.) инданона 2 и затем 6,2 г ( 28 % теор.) инданона 2a. Соединения получают в виде масла от бесцветного до желтоватого цвета.
1H-ЯМR - спектр 2 (360 Мгц, CDCl3): 7,13 (S, 1H), 7,10 (S, 1H), 4,15 (sept. , 1H), 3,30 (m, 1H), 2,95 (sept., 1H); 2,65 (m, 2H), 1,23 - 1,32 (m, 15H).
1H-ЯМR - спектр 2a (360 Мгц, CDCl3): 7,49 (d, 1H), 7,36 (d, 1H), 3,35 (m, 1H), 3,09 (sept., 1H), 2,95 (sept., 1H), 2,70 (m, 2H), 1,24 - 1,33 (m, 15H).
Пример 5. 2,5-диметилиндан-1-он (3) и 2,6-диметилиндан-1-он (3a) 9,21 г (100 ммоль) толуола и 15,4 г (100 ммоль) ангидрида метакриловой кислоты смешивают в автоклаве вместимостью 250 мл со 100 г (5 моль) HF и перемешивают в течение 4 ч при 50oC. Способ осуществляют аналогично примеры 1. Получают 15,2 г (95%) смеси продукта в виде светло-коричневого масла. Селективность по 3 и 3а составляет 85 % и 6 %.
1H-ЯМR - спектр (100 Мгц, DMCO): 7,14 - 7,59 (m), 3,15 - 3,50 (m), 2,45 - 2,80 (m), 2,4 (S), 1,12 - 1, 27 (d).
Масс-спектр: 160 М+, правильное разложение.
Пример 6. 5-изобутил-2-метилинден-1-он (4).
13,4 г (100 ммоль) изобутилбензола и 15,4 г (100 ммоль) ангидрида метакриловой кислоты смешивают в автоклаве вместимостью 250 мл со 100 г (5 моль) HF и перемешивают в течение 5 ч. при 50oC. Способ проводят аналогично примеру 1. Получают 19,4 г (96 %) продукта 4 в виде коричневого масла. После фильтрации через силикагель с уксусным эфиром получают 18,5 г (92%) чистого инданона 4 в виде желтоватого масла.
1H-ЯMR - спектр (100 Мгц, CDCl3): 7,7 (m), 7,2 (m), 3,35 (q), 2,70 (m), 2,58 (d), 1,95 (q), 1,25 (d), 0,93 (d).
Масс-спектр: 202 M+, правильное разложение.
Пример 7. 2,5,7-триметил-1-инданон (5) и 2,4,6-триметил-1-инданон (5а).
10,6 г (100 ммоль) m-ксилола (99%) и 15,4 г (100 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 с HF в течение 8 ч. при комнатной температуре. Получают 18 г (примерно 100%) продукта 5 + 5а в виде коричневого масла. Дистилляция сырого продукта при 80-84oC (0,1 мбар позволяет выделить 16,0 г (92 %) изомерной смеси 5 и 5а в виде от бесцветного до слегка желтоватого масла.
Молярное соотношение 5 и 5а
составляет 1:1:
1H-ЯMR - спектр (300 Мгц, CDCl3): 7,38 (S, 1H), 7,22 (S, 1H), 7,07 (S, 1H), 6,89 (S, 1H), 3,18-3,32 (m, 2H), 2,46-2,74 (m, 7H), 2,35-2,38 (2S, 6H), 2,29
(S, 3H), 1,30 (d, 3H), 1,26 (d, 3H).
Масс-спектр: 174 M+, правильное разложение.
Пример 8. 2-метилиндан-1-он (6).
7,8 г (100 ммоль) бензола и 15,4 г (100 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 с HF в течение 4 ч. при комнатной температуре. Получают 13,7 г (94%) продукта 6 в виде коричневого масла. Сырой продукт подвергают хроматографии с 200 г силикагеля 60. С помощью растворителя, состоящего их смеси хлористого метилена и гексана 1:1, получают 12,2 г (84%) инданона 6 в виде бесцветного масла.
1H-ЯMR - спектр (100 Мгц, CDCl3: 7,5 (m), 3,33 (q), 2,73 (m), 1,30 (d).
Масс-спектр: 146 M+, правильное разложение.
Пример 9. 2,4,5,6-тетраметилиндан-1-он (7).
12 г (100 ммоль) 1,2,3-триметилбензола и 15,4 г (100 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 с HF в течение 6 ч. при комнатной температуре и выделяют. Получают 18,0 г (96%) инданона 7 в виде коричневого масла. Дистилляция сырого продукта при 0,05 Торр. (98-104oC дала 17,4 г (93 %) чистого соединения 7 в виде бесцветного масла.
1H-ЯMR - спектр (100 Мгц, CDCl3): 7,2 (S, 1H), 3,20 (m, 14), 2,4-2,8 (m, 11H), 1,35 (d).
Масс-спектр: 188 M+, правильное разложение.
Пример 10. 5-фенил-2-метилиндан-1-он (8).
15,4 г (100 ммоль) бифенила и 16 г (104 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 со 100 г (5 моль) HF в течение 60 ч. при 70oC. Аналогично примеру 1 получают 23 г (8) с чистотой 90% (93% теор).
Пример 11. 8-метил-4,5,7,8-тетрагидроциклопента (e) аценафтатилен-9-он (9)
30,84 г (200 ммоль) аценфтена и 35 г (228 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично
примеру 1 с 50 г (2,5 моль) HF в течение 20 ч. при 20oC. Аналогично примеру А выделяют 44 г (9) с чистотой 92% (90% теор.).
Пример 12.
2-метил-3, 9-дигидро-2H-циклопента (b)-флуорен-1-он (10) и 2-метил-2,10-дигидро-1H-циклопента (a)флуорен-3-он (10a).
33,24 г (200 ммоль) флуорена и 35 г (228 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 с 50 г (2,5 моль) HF в течение 25 ч. при 70oC. Аналогично примеру 1 выделяют 46 г (10) и (10а). Чистота (10) и (10а) составляет 94% (выход: 91, 5% теор.), при этом молярное соотношение (10) к (10а) составляло 2:1.
Пример 13.
16-метил-6,7,15,16-тетрагидроциклопента (a) фенантрен-17-он (II) и 9-метил-5,6,9, 10-тетрагидро-циклопента (b) фенантран-8-он (IIa).
18 г (100 ммоль) 9,10-дигидрофенантрена и 16 г (104 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 с 90 г (4,5 моль) HF в течение 3 ч. при 70oC. Аналогично примеру 1 выделяют 24,7 г (II) и (IIa).
Чистота II и IIa составляет 92% (выход: 91% теор.), при этом молярное соотношение II к IIa составляет 6:4.
Пример 14. 5-метокси-2-метилиндан-1-он (12)
10,8 г (100 ммоль) анизола и 16 г (104 ммоль) ангидрида метакриловой кислоты
подвергают взаимодействию аналогично примеру 1 со 100 г (5 моль) HF в течение 3 ч. при 70oC. Аналогично примеру 1 получают 17 г (12) с чистотой (68%) (выход: 65% теор.).
Пример 15. 5,6-диметокси-2-метилиндан-2-он (13)
13,32 г 9100 ммоль) вератрола и 16 г (104 ммоль) ангидрида метакриловой кислоты подвергают взаимодействию аналогично примеру 1 со 100 г (5
моль) HF в течение 18 ч. при 30o. Аналогично примеру 1 получают 20,3 г (13) с чистотой 96% (выход: 93,5 % теор.).
Реферат
Изобретение относится к способу получения замещенных инданонов формулы (IV) и их изомеров формулы (IVa)

где R1-R7 преимущественно обозначают водород или алкид или соседние остатки R1-R4 образуют кольцо, получают одностадийным способом путем взаимодействия соединения (I)
с ангидридом карбоновой кислоты формулы (II)

или с фторангидридом формулы (III)

Формула


где R1 - R7 - одинаковые или различные, водород, С1 - С20-алкил, С6 - С10-арил, С1 - С10-алкокси, или соседние остатки R1 - R4 образуют вместе со связывающими их атомами до четырех колец, которые могут быть замещены низшим алкилом,
с использованием ароматического соединения, отличающийся тем, что в \\1 качестве ароматического соединения используют соединение формулы I

где значения R1 - R4 приведены выше, которое подвергают взаимодействию с ангидридом карбоновой кислоты формулы II
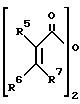
или с фторангидридом формулы III

где значения R5 - R7 приведены выше,
в избытке жидкого безводного фтористого водорода.
Комментарии