Лиганды х-рецептора ретиноевой кислоты - RU2146241C1
Код документа: RU2146241C1
Чертежи
Описание
Изобретение
относится к новым лигандам Х- рецептора ретиноевой кислоты (RXR). В частности, предметом настоящего изобретения являются соединения формулы I:
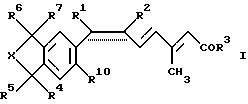
где необязательно присутствует связь, обозначенная пунктиром; и если связь, обозначенная пунктиром, присутствует, то R1 обозначает низший алкил и R2 обозначает галоген или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5-8-членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один атом серы, кислорода или азота, причем, если кольцо является ароматическим, то связь, обозначенная пунктиром, представляет собой часть мезомерной системы; или, если связь, обозначенная пунктиром, отсутствует, то R1 и R2 вместе представляют собой метилен, образуя цис-замещенное циклопропиловое кольцо; R3 обозначает гидрокси или низший алкокси; R4, R5, R6 и R7 независимо друг от друга обозначают водород или низший алкил; X обозначает группу (>CR8R9 )n, где n равно 1, 2 или 3; и R8, R9 независимо друг от друга обозначают водород или низший алкил; R10 обозначает водород, алкил или алкокси;
и фармацевтически приемлемые соли карбоновых кислот формулы I.
В контексте данного описания понятие "низший" обозначает группы, содержащие 1-4 атома углерода. Низшие алкильные группы могут иметь прямую или разветвленную цепь. Предпочтительными являются метил и этил. Понятие "галоген" включает фтор, хлор, бром и йод, причем бром является предпочтительным. Примерами 5-8-членных карбоциклических колец, образованных R1 и R2 вместе с атомами углерода, к которым они присоединены, являются бензол, циклопентен, циклогексен, циклогептен, среди которых предпочтительным является бензол. Примерами 5-8-членных гетероциклических колец, образованных R1 и R2 вместе с атомами углерода, к которым они присоединены, являются тиофен, фуран, дигидрофуран и пиридин, среди которых предпочтительным является тиофен. Группы R4-R7 предпочтительно обозначают низший алкил, наиболее предпочтительно метил. X предпочтительно обозначает этилен. R10 предпочтительно обозначаeт водород. Если обозначенная пунктиром связь присутствует и является частью мезомерной системы ароматического кольца, то это означает, что дополнительная связь рассматривается как делокализованная над кольцом, как это принято в классической модели ароматичности.
Соединения формулы I, где R3 обозначает гидрокси, образуют соли с фармацевтически приемлемыми основаниями, такие как соли щелочных металлов, например соли натрия или калия, или соли аммония или соли замещенного аммония, такие как соли триметиламмония, которые подпадают под объем настоящего изобретения.
Одним из предметов изобретения являются соединения формулы Iа:
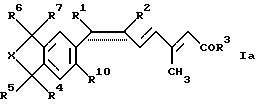
где R1 обозначает низший алкил и R2 обозначает галоген или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5-8- членное карбоциклическое кольцо или 5-8-членное гетероциклическое кольцо, содержащее один атом серы, кислорода или азота, причем если кольцо является ароматическим, то обозначенная пунктиром связь между атомами углерода, присоединенными к R1 и R2, представляет собой часть мезомерной системы; R3-R7, R10 и X имеют значения, указанные для формулы I;
и фармацевтически приемлемые соли карбоновых кислот формулы Ia.
Другим предметом изобретения предпочтительно являются
соединения формулы Ib:
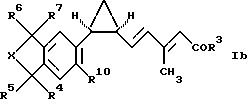
где R3-R7, R10 и X имеют значения, указанные для формулы I;
и фармацевтически приемлемые соли карбоновых кислот формулы Ib.
Особенно предпочтительными соединениями формулы Ia являются соединения формулы
Ia(1):
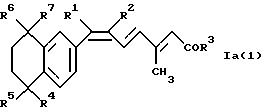
где R1-R7 имеют значения, указанные для формулы Ia. Наиболее предпочтительны соединения формулы Ia(1), где все R4-R7 обозначают метил, a R1-R3 имеют значения, указанные для формулы Ia. Наиболее предпочтительны соединения формулы Ia(1), где все R4-R7 обозначают метил и R3 обозначает гидроксил, а R1, R2 имеют значения, указанные для формулы Ia.
Особенно предпочтительными соединениями формулы Ib являются соединения формулы Ib(1):
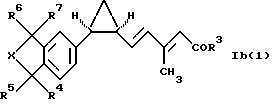
где R1-R7 имеют значения, указанные для формулы Ib. Наиболее предпочтительны соединения формулы Ib(1), где все R4-R7 обозначают метил и R3 имеет то же значение, что и в формуле Ib.
Под объем изобретения также подпадают фармацевтически приемлемые соли карбоновых кислот соединений формулы Ia(1) и Ib(1).
Соединения по настоящему изобретению проявляют высокую степень избирательности по отношению к семейству RXR-рецепторов. Они пригодны в качестве антипролиферативных агентов и могут применяться при дерматологических и онкологических показаниях. В частности, соединения по настоящему изобретению ингибируют пролиферацию себоцитов и, таким образом, могут применяться для лечения угрей.
В международной заявке WO 95/04036 описаны избирательные в отношении RXR производные тетрагидронафталина, которые могут содержать боковую цепь алифатической триеновой кислоты. В GB-А-2122200 описаны производные тетрагидронафтилдиметилоктатриеновой кислоты, которые пригодны для лечения неоплазм и дерматозов.
Кроме того, соединения по настоящему изобретению в дозах, в которых они неактивны сами по себе, увеличивают активность соединений, связывающихся с рецепторами ретиноевой кислоты (RAR). Примером таких RAR- избирательных ретиноидов является полностью транс-ретиноевая кислота. Поэтому введение соединений по настоящему изобретению в сочетании с RAR-избирательным ретиноидом позволяет использовать намного более низкие дозы RAR-избирательного ретиноида в случае таких показаний, при которых применяют RAR-избирательные ретиноиды. Одним из таких показаний является лечение лейкемии человека.
Соединения по настоящему изобретению также могут применяться в комбинации с лигандами других ядерных рецепторов того же самого суперсемейства, которые образуют гетеродимеры с RXR, например, соединениями витамина D или тироидными гормонами, усиливая при этом их действие.
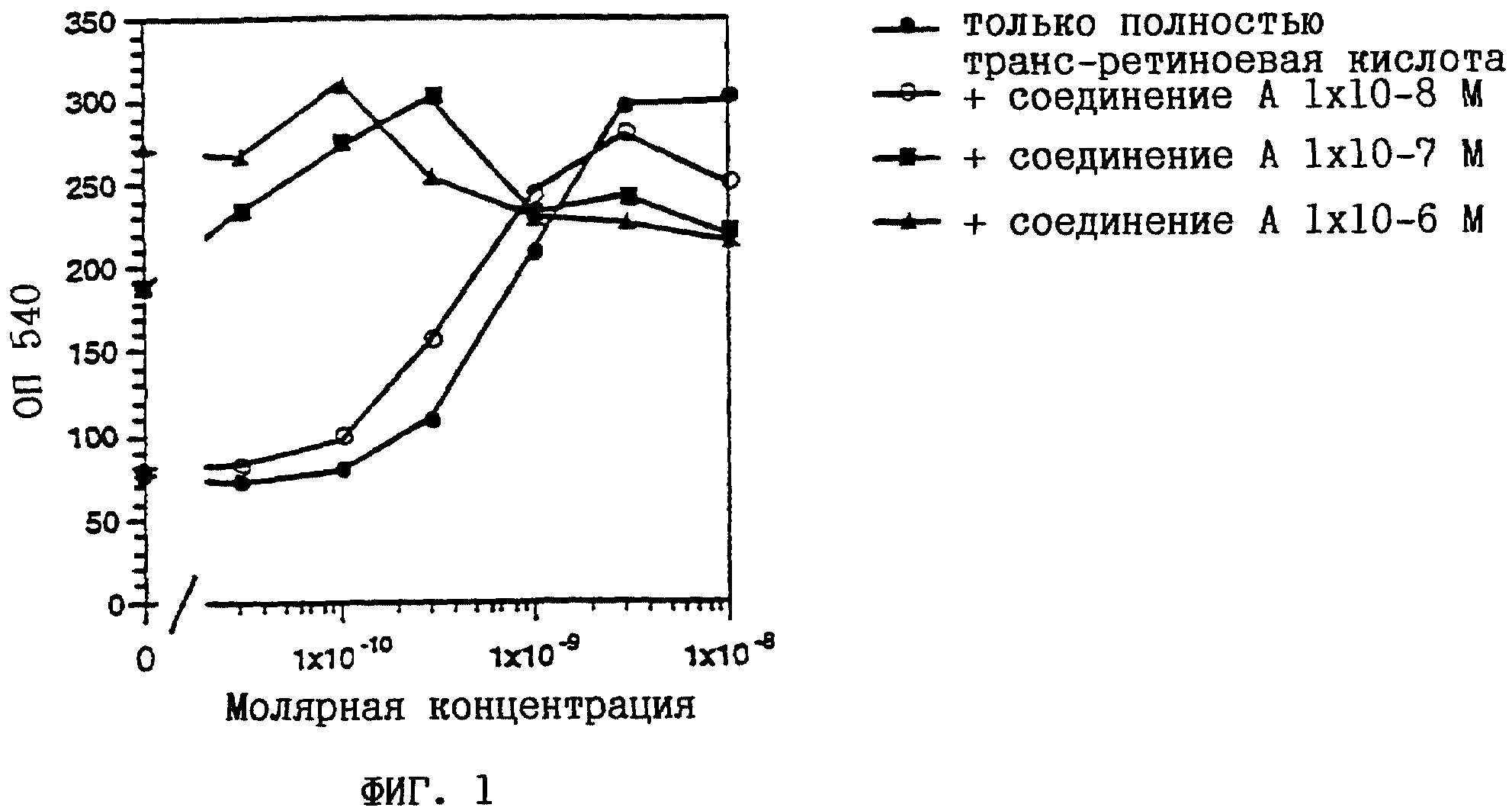
Клетки HL-60 представляют собой линию клеток лейкемии спинного мозга человека, которая исключительно чувствительна к индуцируемой ретиноидами дифференцировке (см. у Breitman и др., Proc. Nat. Acad. Sci. USA 1989, 86, 7129-7133), и поэтому могут использоваться в качестве модели при тестировании активности, индуцирующей дифференцировку клеток. Индукция дифференцировки клеток HL-60 представляет собой стандартную модель для лечения лейкемии человека. Дифференцировку HL-60 клеток определяли путем измерения их окислительного потенциала при восстановлении нитросинего тетразолия (NBT). Клетки HL-60 выращивали в среде RPMI 1640, дополненной 10%-ной ФТС (фетальная телячья сыворотка), 2 мМ L-глутамина, 1 мМ пирувата натрия, 1 % заменимых аминокислот, 50 ед/мл пенициллина и 50 мкг/мл стрептомицина. Было обнаружено, что клетки лишены микоплазмы. 30000 клеток/100 мкл RPMI/ФТС высевали в плоскодонные лунки для микротитрования. Одновременно добавляли 10 мкл ретиноидного раствора, разбавленного дополненной средой, для получения конечных концентраций в диапазоне от 10-11 до 10-6М (маточные растворы с концентрацией 10-2 М в этаноле выдерживали при -20oC в защищенном от света месте). Через 3 дня среду удаляли с помощью многоканальной пипетки и замещали 100 мкл раствора NBT (1 мг/мл в ЗФР (забуференном фосфатом физиологическом растворе) с 200 нМ форболмиристатацетата). После дополнительной инкубации в течение 1 часа при 37oC раствор NBT удаляли и добавляли 100 мкл 10%-ного ДСН (додецилсульфата натрия) в 0,01Н HCl. Количество восстановленного NBT определяли фотометрически при 540 нм с использованием автоматического планшет-ридера. Вычисляли средние значения по 3 лункам. Среднеквадратичная ошибка составляла 5-10%.
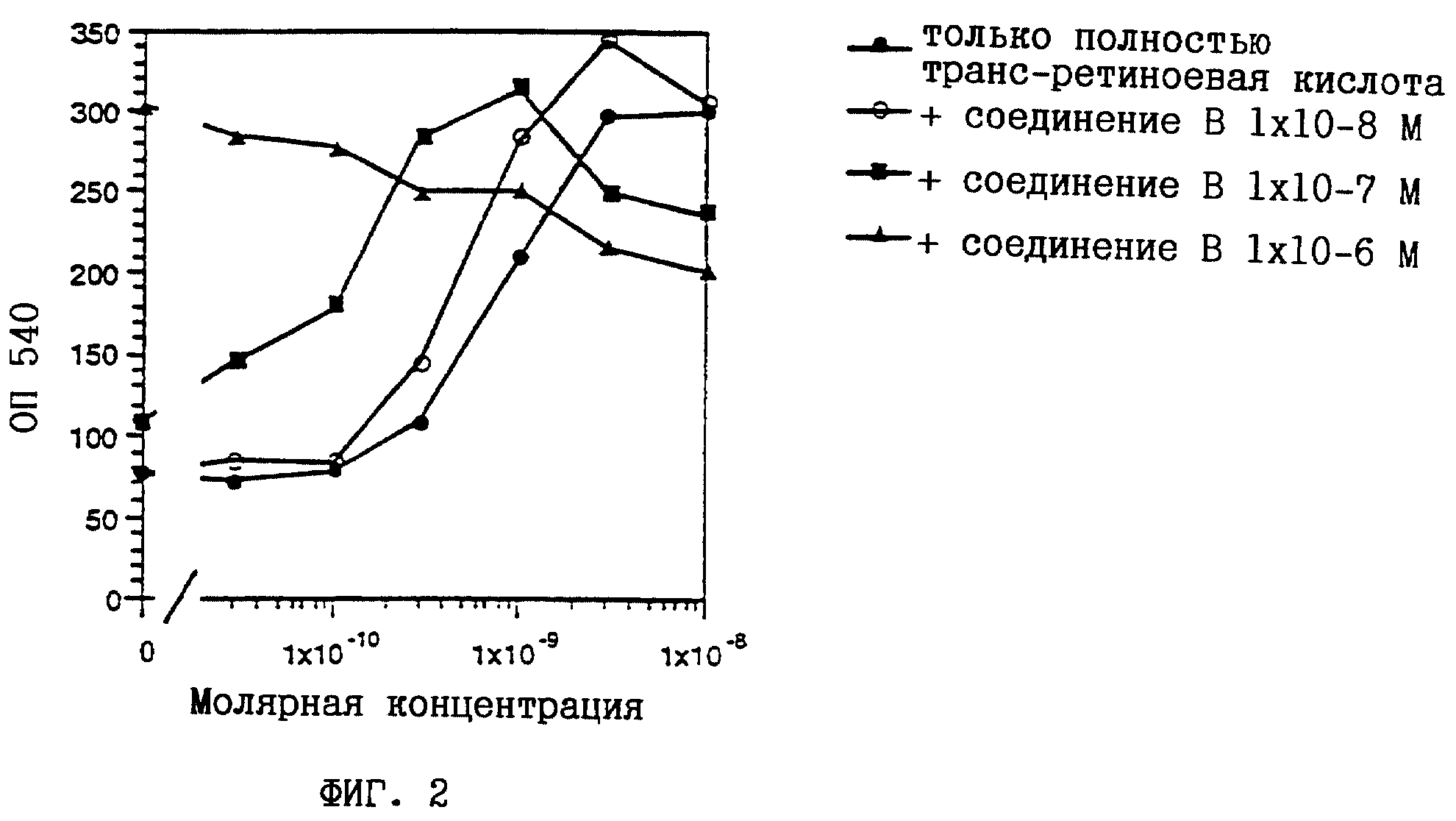
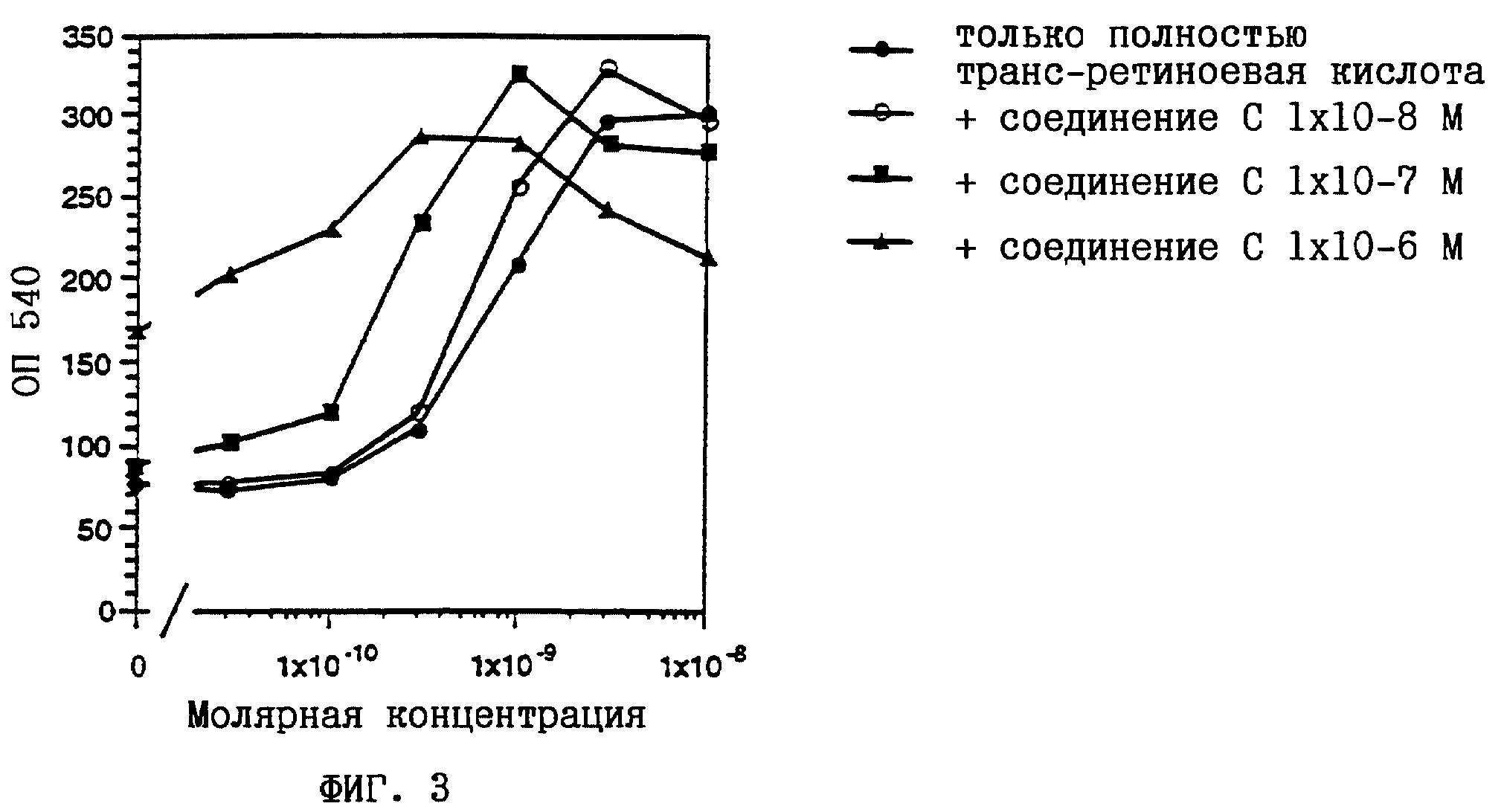
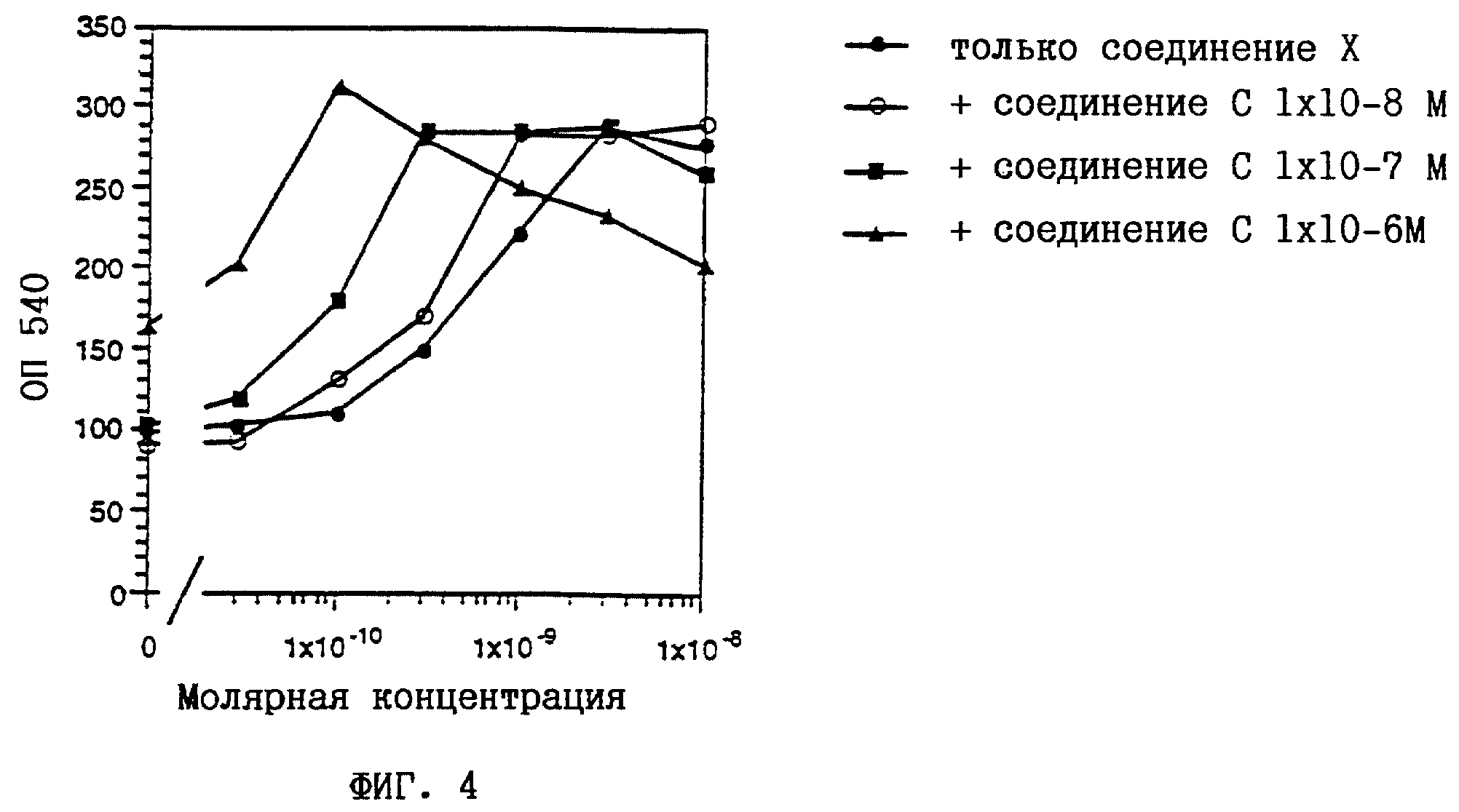
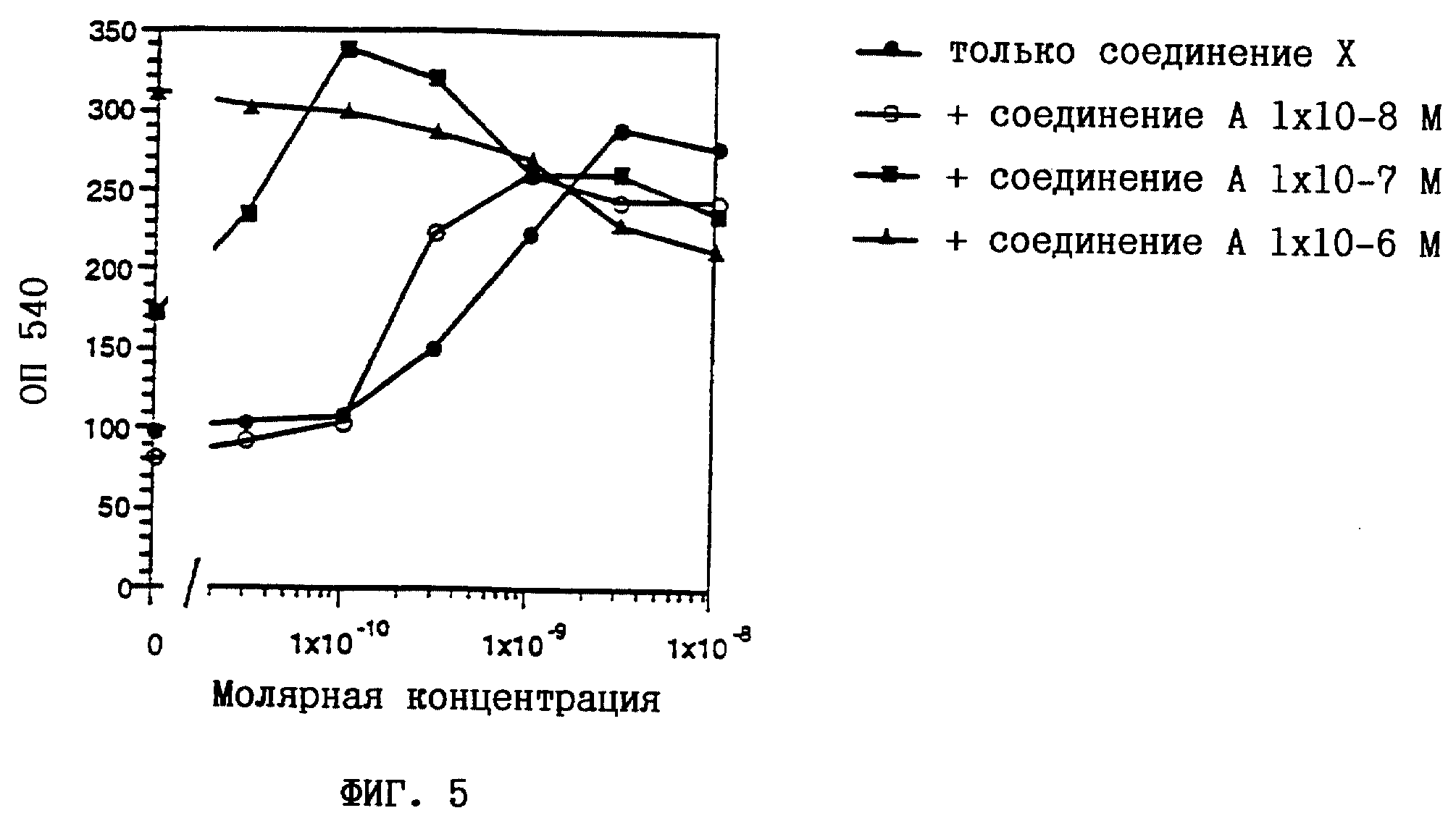
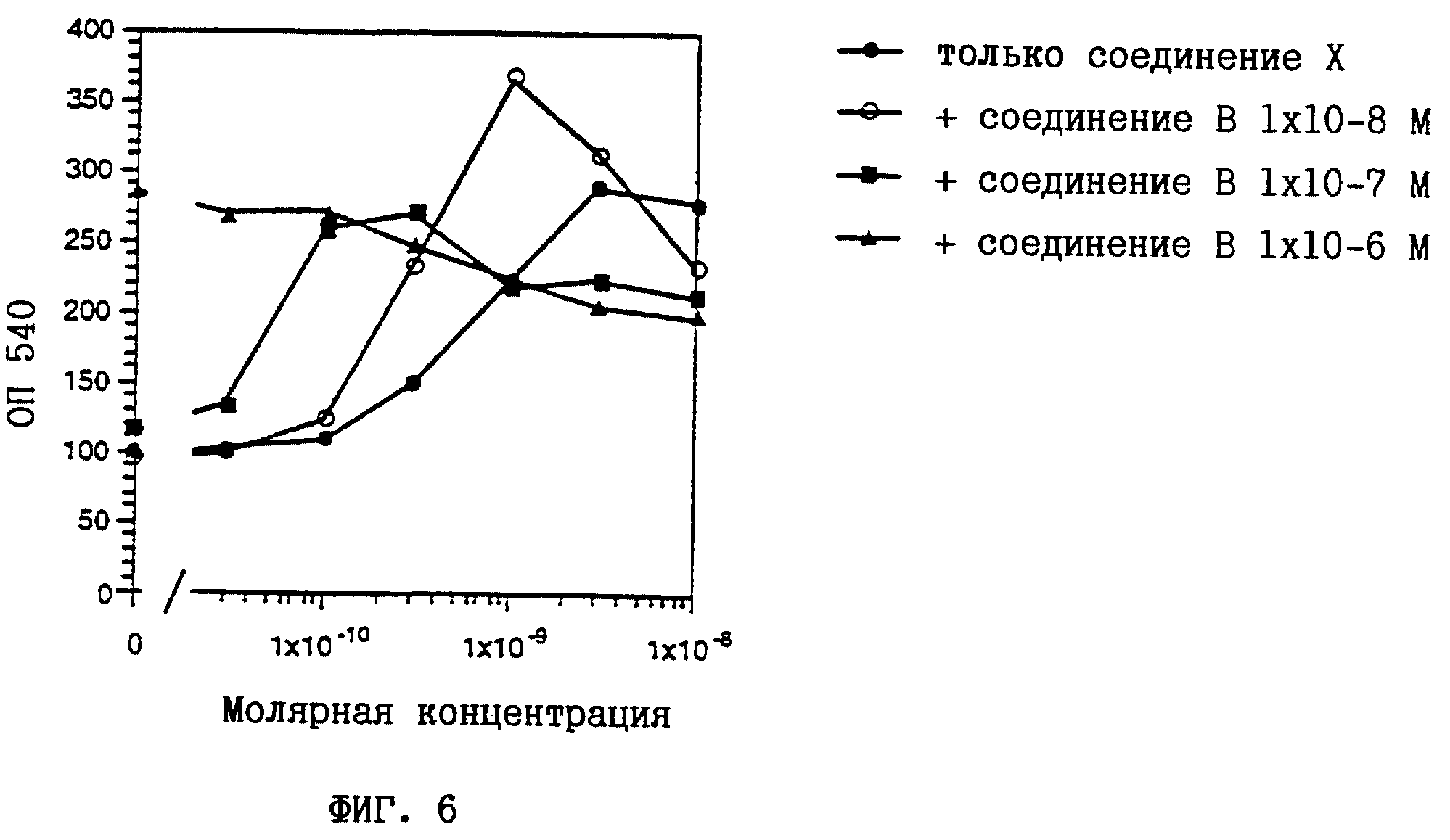
На фиг. 1-6 показано воздействие полностью транс-ретиноевой кислоты индивидуально и в сочетании с соединениями по настоящему изобретению на индукцию дифференцировки клеток HL-60.
На фиг. 1-6
соединение А представляет собой (2E,4E)-3-метил-5-[3-(5,5,8,8- тетраметил-5,6,7,8-тетрагидронафталин-2-ил)тиофен-2-ил] пента-2,
4- диеновую кислоту (пример 2);
соединение В представляет собой (2E, 4E)-3-метил-5-[(1RS, 2RS)-2-(5,5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)циклопропил] пента-2,4-диеновую кислоту
(пример 6);
соединение С представляет собой (2E,4E)-3-метил-5-[2- (5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)фенил] пента- 2,4-диеновую кислоту (пример 1);
соединение X
представляет собой пара-(5,5,8,8-тетраметил-5,6,7,8-тетрагидро-2- нафталинкарбоксамидо)бензойную кислоту (см., например, патент США 4703110).
Из полученных результатов очевидно, что действие соединений по настоящему изобретению и полностью-транс-ретиноевой кислоты является более сильным, чем просто аддитивное.
Следовательно, соединения по настоящему изобретению могут найти применение при лечении состояний, опосредуемых ретиноидными рецепторами. Например, соединения по настоящему изобретению могут применяться для лечения и предупреждения дерматологических заболеваний, таких как угри или псориаз. Соединения по настоящему изобретению обладают пониженной токсичностью или тератогенностью по сравнению с классическими ретиноидными соединениями, избирательными по отношению к RAR, такими как полностью транс- ретиноевая кислота. Соединения по настоящему изобретению могут применяться совместно с RAR-избирательными ретиноидами, снижая тем самым дозу таких ретиноидов и риск возникновения побочных воздействий, связанных с таким лечением.
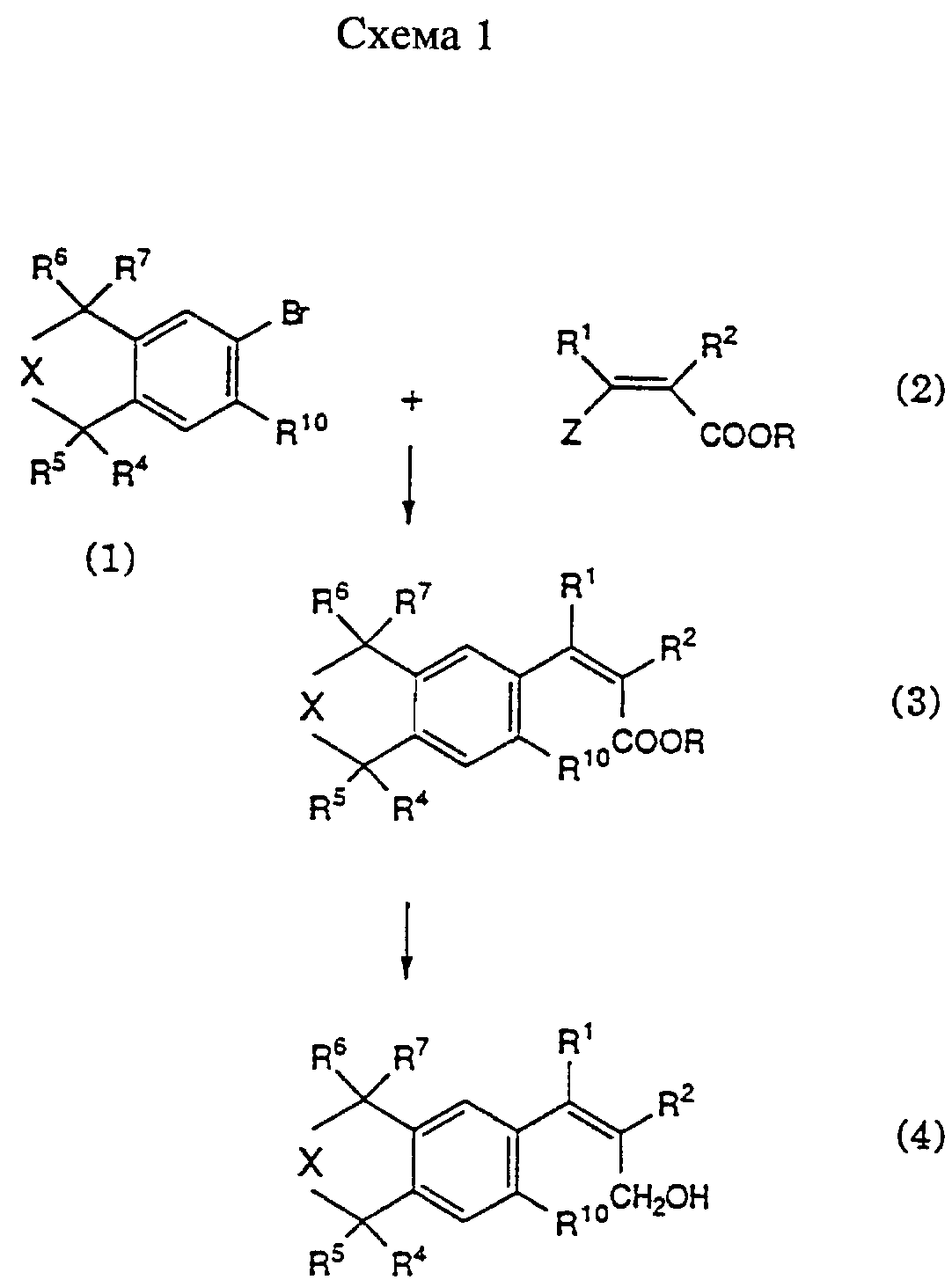
В соответствии с настоящим изобретением соединения формулы 1 могут быть получены путем
взаимодействия соединения формулы II

с соединением формулы III
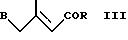
где А обозначает формил и В обозначает триарилфосфоний или ди-(низший алкокси)фосфинил; или А обозначает триарилфосфоний или ди-(низший алкокси)фосфинил и В обозначает формил; R обозначает низший алкокси; и R1, R2, R4, R5, R6, R7 и R10 имеют значения, указанные выше;
с образованием соединения формулы I, где R3 обозначает низший алкокси, и при необходимости путем гидролиза низшей алкоксигруппы R3 в полученном таким образом соединении формулы I.
Взаимодействие соединения формулы II с соединением формулы III может быть осуществлено в соответствии с хорошо известными методами по реакции Виттига или Хорнера.
В том случае, если одно из соединений формул II и III содержит триарил (предпочтительно группу трифенилфосфония) (реакция Виттига), то взаимодействие может быть осуществлено в присутствии связывающего кислоту агента, например сильного основания, такого как, например, бутиллитий, гидрид натрия или натриевая соль диметилсульфоксида, но прежде всего в присутствии этиленоксида, который может быть замещен низшим алкилом, такого как эпоксибутан, необязательно в растворителе, например в простом эфире, таком как диэтиловый эфир или тетрагидрофуран, или в ароматическом углеводороде, таком как бензол, в диапазоне температур от комнатной и до температуры кипения реакционной смеси. Анион для группы фосфония может представлять собой неорганический анион, такой как хлорид или бромид или бисульфат, или органический анион, такой как тозилат.
В том случае, если одно из соединений формул II и III содержит диалкоксифосфинильную группу (реакция Хорнера), то взаимодействие может быть осуществлено в присутствии основания и предпочтительно в присутствии инертного органического растворителя, например в присутствии гидрида натрия в бензоле, толуоле, диметилформамиде, тетрагидрофуране, диоксане либо 1,2- диметоксиалкане, или же алкоголята натрия в алканоле, например метилата натрия в метаноле, в диапазоне температур от 0oC до температуры кипения реакционной смеси. В предпочтительном варианте осуществления изобретения соединения формулы I получают путем взаимодействия соединения формулы II, где А обозначает формил, с соединением формулы III, где В обозначает ди-(низший алкокси)фосфинил.
Полученный таким образом эфир карбоновой кислоты формулы I может быть гидролизован известным способом, например путем обработки щелочами, в частности путем обработки водно-спиртовым раствором гидроксида натрия или калия в диапазоне температур от комнатной до температуры кипения реакционной смеси.
Полученная таким образом карбоновая кислота формулы I может быть выделена известным способом в свободном виде или в виде соли, например соли щелочного металла, в частности в виде натриевой или калиевой соли.
Соединения формулы II являются новыми соединениями и также представляют собой предмет настоящего изобретения. Соединения формулы II могут быть получены в соответствии с приведенными ниже схемами 1, 2 и 3, где R обозначает низший алкил, Z обозначает бром или йод и R1, R2, R4-R7 и R10 имеют значения, указанные выше.
Соединения формулы II, в которых присутствует обозначенная пунктиром связь, R1 обозначает низший алкил и R2 обозначает водород; или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5-8-членное ароматическое или неароматическое кольцо, которое может содержать один атом серы, кислорода или азота, могут быть получены согласно схеме 1. На схеме 1 соединение формулы (1) сначала превращают в его литиевую или магниевую соль, затем путем замещения металла превращают в цинксодержащее производное и далее путем реакции, катализируемой переходным металлом (предпочтительно палладием), подвергают сочетанию с соединением формулы (2), получая соединение формулы (3). Эфирная группа карбоновой кислоты в соединении формулы (3) может быть восстановлена, например, с помощью гидрида металла, такого как гидрид диизобутилалюминия, с образованием соединения формулы (4), гидроксиметильная группа которого может быть трансформирована, например, путем обработки окислителем, таким как диоксид марганца, с получением соединения формулы II, в котором А обозначает формил; R1 обозначает низший алкил и R2 обозначает водород; или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5-8-членное ароматическое или неароматическое кольцо, которое может содержать один атом серы, кислорода или азота.
Соединения формулы II, в которых R1 обозначает низший алкил и R2 обозначает галоген, могут быть получены согласно схеме 2. В соответствии со схемой 2 соединение формулы (5) подвергают реакции Виттига-Хорнера с три(низший алкил)фосфоноацетатом, получая соединение формулы (6). Затем сложноэфирную группу соединения формулы (6) превращают в формильную группу путем двухстадийного процесса восстановления и окисления, как описано выше, с получением соединений формул (7) и (8) соответственно. Соединение формулы (8) может быть галогенировано посредством галогенирования - дегидрогалогенирования, например, путем обработки свободным галогеном, таким как Br2, с последующим дегидрогалогенированием сильным основанием, таким как 1,8-диазабицикло[5.4.0] ундек-7-ен (ДБУ), с получением соединения формулы II, в котором А обозначает формил, R1 обозначает низший алкил и R2 обозначает галоген.
Соединения формулы II, в которых связь, обозначенная пунктиром, отсутствует, и R1 и R2 вместе обозначают метилен, могут быть получены согласно схеме 3. В соответствии с указанной схемой проводят катализируемое палладием взаимодействие соединения формулы (1) с пропаргиловым спиртом, получая ацетиленовое соединение (9). Гидроксильная группа в пропаргиловом спирте необязательно может быть защищена, например, триметилсилильной группой. Восстановление тройной связи в соединении (9) приводит к получению соединения (10), которое превращают в соединение (11) по реакции Симмонса-Смита. Соединение формулы (11) может быть окислено с использованием известных в данной области техники методов, например, с помощью хлорхроматпиридиния, или путем окислительной реакции Сверна- или Десс- Мартина с получением соединения формулы II, в котором А обозначает формил, связь, обозначенная пунктиром, отсутствует и R1 и R2 вместе обозначают метилен.
Соединения формулы II, в которых А обозначает триарилфосфониевую или ди-(низший алкокси)фосфинильную группу, могут быть получены из соединений формулы II, где А обозначает формил, восстановлением формильной группы до гидроксиметильной группы известным способом, например, с помощью металлогидридного комплекса, такого как NaBH4, последующим замещением гидроксильной группы на атом брома или хлора, например, обработкой бромирующим или хлорирующим агентом, таким как оксихлорид фосфора или трибромид фосфора, и взаимодействием полученного таким образом бромида или хлорида с триарилфосфином или с три-(низший алкил)фосфитом. Все эти реакции могут быть осуществлены известным способом.
Соединения формулы I и их соли могут применяться в виде фармацевтических композиций.
Композиции для системного применения могут быть получены, например, путем добавления соединения формулы I или его соли в качестве активного ингредиента к нетоксичным инертным твердым или жидким носителям, которые являются традиционными для таких композиций.
Композиции могут быть введены энтерально, парентерально или местно. Композиции в виде таблеток, капсул, драже, сиропов, суспензий, растворов и суппозиториев пригодны, например, для энтерального введения.
Композиции в виде растворов для инфузии или инъекции пригодны для парентерального введения.
При энтеральном и парентеральном введении соединения формулы I могут назначаться взрослым пациентам в количествах приблизительно 1-100 мг, предпочтительно 5-30 мг/день.
Для местного применения действующие вещества обычно используют в виде мазей, настоек, кремов, растворов, лосьонов, аэрозолей, суспензий и т.п. Предпочтительны мази и кремы, а также растворы. Эти композиции, предназначенные для местного применения, могут быть получены смешением активных ингредиентов с нетоксичными инертными твердыми или жидкими носителями, которые пригодны для местного лечения и которые используются в таких композициях.
Для местного применения обычно пригодны приблизительно 0,1-5%-ные, предпочтительно 0,3-2%- ные растворы, а также приблизительно 0,1-5%-ные, предпочтительно 0,3-2%-ные мази или кремы.
При необходимости с композициями может быть смешан антиокислитель, например токоферол, N-метил-γ-токоферамин, а также бутилированный гидроксианизол или бутилированный гидрокситолуол.
Ниже изобретение проиллюстрировано на примерах.
Пример 1
(2E. 4E)-3-метил-5-[2-(5.5.8.8-тетраметил- 5.6.7.8-тетрагидронафталин-2-ил)фенил] пента-2.4-диеновая кислота
А. 5,4 г
6-бром- 1,1,4,4-тетраметилтетралина растворяли в 40 мл тетрагидрофурана (ТГФ) и при -78oC по каплям добавляли 29 мл 1,5М раствора третичного бутиллития в пентане. Через 30 минут добавляли
раствор, содержащий 2,7 г безводного хлорида цинка в 80 мл ТГФ. После 30-минутного перемешивания при -78oC этот раствор медленно добавляли ко второй реакционной смеси, которую выдерживали
при 0oC и готовили следующим образом: 0,7 г хлорида бис (трифенилфосфин) палладия (II) суспендировали в 60 мл ТГФ и добавляли 1,8 мл 20%-ного раствора диизобутилалюминийгидрида в толуоле.
Через 15 минут черную реакционную смесь охлаждали до 0oC и по каплям добавляли раствор, содержащий 5 г этил-2-йодбензоата в 70 мл ТГФ. После 15- минутного перемешивания при 0oC
добавляли первый раствор. Реакционную смесь перемешивали в течение ночи при комнатной температуре, сливали на ледяную воду, подкисляли 2H соляной кислотой и несколько раз экстрагировали этилацетатом.
Объединенные органические фазы промывали водой, сушили над сульфатом натрия и упаривали. Маслянистый остаток хроматографировали (силикагель, гексан/этилацетат = 19:1), получая после перекристаллизации
из гексана 2,6 г этилового эфира 2-(5,5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)бензойной кислоты в виде белых кристаллов, tпл79-81oC.
Б. Полученный продукт (2,6 г) растворяли в 70 мл ТГФ и по каплям при 0oC добавляли 32 мл 20%-ного раствора диизобутилалюминийгидрида в толуоле. После 2-часового перемешивания при 0oC по каплям добавляли 50 мл смеси метанола и воды (1: 1) с последующим добавлением 25 мл 6H соляной кислоты. Реакционную смесь экстрагировали этилацетатом, промывали водой, сушили и упаривали, получая 2,4 г бесцветного масла, которое кристаллизовали на холоде. После перекристаллизации из гексана получали 2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2- ил)бензиловый спирт, tпл 111-113oC.
В. Полученный спирт (2,1 г) растворяли в 50 мл метиленхлорида и добавляли 8,5 г диоксида марганца. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем фильтровали и упаривали, получая 2,6 г бесцветного масла, которое кристаллизовали из гексана и получали 2,0 г 2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2- ил)бензальдегида, tпл 85-87oC.
Г. 0,7 г дисперсии гидрида натрия (50%-ная в минеральном масле) трижды промывали пентаном, сушили и суспендировали в 30 мл ТГФ. При 0oC медленно добавляли раствор, содержащий 3,0 г этилового эфира 4-(диэтоксифосфинил)-3-метилкротоновой кислоты в 30 мл ТГФ. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, вновь охлаждали до 0oC и по каплям добавляли раствор, содержащий 2,2 г указанного выше альдегида в 15 мл ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 3 часов, затем сливали на ледяную воду и экстрагировали простым эфиром. Органические фазы промывали водой, сушили и упаривали, получая 5,9 г желтого масла, которое очищали путем фильтрации через силикагелевую колонку (растворитель гексан/этилацетат = 19:1), получая 2,9 г бесцветного масла.
Д. Полученное масло (2,0 г) растворяли в 50 мл этанола и добавляли раствор, содержащий 4, 1 г гидроксида калия в 20 мл воды. После добавления 20 мл ТГФ раствор нагревали до 40oC в течение 4 часов. Охлажденную реакционную смесь сливали на ледяную воду, подкисляли 2H соляной кислотой и экстрагировали этилацетатом. Органические фазы промывали водой, сушили и упаривали, получая кристаллический продукт. После перекристаллизации из смеси этилацетата и гексана получали 1,6 г (2E,4E)-3-метил-5-[2-(5,5,8,8-тетраметил -5,6,7,8 -тетрагидронафталин -2-ил) фенил] пента - 2,4 -диеновой кислоты в виде белых кристаллов, tпл 180-182oC.
Пример
2
(2E. 4E)-3-метил-5-[3- (5.5.8.8-тетраметил-5.6.7.8-тетрагидронафталин-2-ил)тиофен-2-ил] пента-2.4-диеновая кислота
Это соединение (tпл 188-189oC)
синтезировали аналогично примеру 1, используя в качестве исходных продуктов 6-бром-1,1,4,4-тетраметилтетралин и метил -3-йод -2- тиофенкарбоксилат.
Пример 3
(2E.
4E)-3-метил-5-[2-(5.5.8.8- тетраметил-5.6.7.8-тетрагидронафталин-2-ил)тиофен-3-ил]пента-2.4- диеновая кислота
Это соединение (tпл 195-197oC) синтезировали аналогично
примеру 1, используя в качестве исходных продуктов 6- бром-1,1,4,4-тетраметилтетралин и метил-2-йод-3-тиофенкарбоксилат.
Пример 4
(2E. 4E)-3-метил-5-[3-(5.5,8,
8-тетраметил-5.6.7.8- тетрагидронафталин-2-ил)тиофен-4-ил]пента-2.4-диеновая кислота
Это соединение (tпл 192-193oС) синтезировали аналогично примеру 1, используя в
качестве исходных продуктов 6-бром-1,1,4,4- тетраметилтетралин и метил-3 -йод-4 -тиофенкарбоксилат.
Пример 5
а) 7,30 мл триэтилового эфира фосфонуксусной кислоты растворяли в
100 мл абсолютного ТГФ. При 0oC добавляли 3,90 г KOtBu и смесь перемешивали при этой температуре в течение 15 минут. По каплям в течение 30 минут при комнатной температуре добавляли 4,20 г
1- (5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)этанона, растворенного в 30 мл ТГФ, и перемешивание продолжали при 40oC в течение 16 часов. Затем реакционную смесь сливали на ледяную
крошку/NH4Cl, экстрагировали EtOEt, промывали соляным раствором и водой и сушили над Na2SO4. После выпаривания растворителя и последующей быстрой хроматографии
(силикагель, гексан/AcOEt = 97:3) получали 3,70 г этилового эфира (Е)-3-(5,5,8,8- тетраметил-5,6,7,8-тетрагидронафталин-2-ил)бут-2-еновой кислоты в виде желтоватого масла (ГХ-чистота 98%).
б) 3,70 г этилового эфира (Е)-3-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)бут-2- еновой кислоты растворяли в 38 мл абсолютного ТГФ. После охлаждения до -75oC добавляли с помощью шприца в течение 5 минут 26,7 мл 1,2М диизобутилалюминийгидрида (ДИБАГ) (в толуоле). Через 10 минут реакционную смесь нагревали до 0oC и выдерживали при этой температуре в течение 1/2 часа. Поскольку ТСХ показала, что осталось некоторое количество исходного продукта, то при 0oC добавляли дополнительные 3,0 мл ДИБАГ. Через 15 минут реакцию в смеси прекращали с помощью ледяной крошки/HCl и экстрагировали EtOEt. Органический экстракт промывали раствором NaHCO3 и соляным раствором, сушили над Na2SO4 и растворители выпаривали в вакууме. Полученный (Е)-3-(5,5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)бут-2-ен-1-ол (3,42 г) использовали на следующей стадии без дополнительной очистки.
в) 3,42г (Е)-3- (5,5,8, 8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)бут-2-ен-1-ола растворяли в 23 мл CH2Cl2 и обрабатывали 16,1 г MnO2. Реакционную смесь интенсивно перемешивали в течение ночи при температуре окружающей среды и затем фильтровали через целит. После выпаривания растворителя и последующей быстрой хроматографии (силикагель, гексан/AcOEt = 92: 8) получали 2,86 г (Е)-3- (5,5, 8,8-тетраметил-5,6,7,8-тетрагидронафта-лин-2-ил)бут-2-еналя в виде светло-желтого масла.
г) 2,86 г (Е)-3-(5,5,8,8-тетраметил- 5,6,7,8-тетрагидронафталин-2-ил)бут-2-еналя растворяли в 16 мл CH2Cl2 и охлаждали до -75oC. По каплям добавляли 1,1 экв. Br2 (0,63 мл), растворенного в 3 мл CH2Cl2, и смесь выдерживали при этой температуре в течение 10 минут. ТСХ показала образование дибромида. Затем одной порцией добавляли 4,98 мл ДБУ и повышали температуру до 0oC. Через 1/2 часа реакционную смесь сливали на ледяную крошку/HCl, экстрагировали EtOEt, дважды промывали соляным раствором, сушили над Na2SO4 и растворители выпаривали в вакууме. После быстрой хроматографии (силикагель, гексан/AcOEt = 96:4) получали 0,95 г нестойкого (Е)-2-бром-3-(5,5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)бут-2-еналя в виде желтых кристаллов, tпл 115-117oC. Расположение двойной связи было подтверждено с помощью ядерного эффекта Оверхаузера.
д) 290 мг NaH (50%-ный в минеральном масле) суспендировали в 8 мл абсолютного ТГФ и при 0oC обрабатывали 1,69 г этилового эфира 4-(диэтоксифосфинил)-3- метилкротоновой кислоты. После прекращения выделения H2 (через 1/2 часа при 0oC) добавляли 1,04 г (Е)-2-бром-3-(5,5,8,8- тетраметил-5,6,7,8-тетрагидронафталин-2-ил)бут-2-еналя, растворенного в 8 мл абсолютного ТГФ, и перемешивание продолжали в течение 3/4 часа при 0oC и в течение 1 часа при комнатной температуре. Затем реакцию в смеси прекращали с помощью ледяной крошки и экстрагировали EtOEt. После двукратной промывки соляным раствором, сушки над Na2SO4 и выпаривания растворителя получали неочищенный продукт, который очищали с помощью быстрой хроматографии (силикагель, гексан/AcOEt = 98:2), получая 527 мг этилового эфира (2E,4E,6E)-6-бром-3-метил-7- (5, 5,8,8-тетраметил- 5,6,7,8-тетрагидронафталин-2-ил)окта-2,4,6-триеновой кислоты в виде желтых кристаллов, tпл 97-98,5oC.
е) 472 мг этилового эфира (2E,4E, 6E-6-бром-3-метил-7-(5,5,8,8-тетраметил -5,6,7,8- тетрагидронафталин -2-ил) окта -2,4,6-триеновой кислоты растворяли в 10 мл ЕtOH/ТГФ (1:1) и обрабатывали 2,65 мл 2H водного NaOH. Реакционную смесь перемешивали в темноте в течение 22 часов при температуре окружающей среды. Затем ее сливали на ледяную крошку, экстрагировали AcOEt, промывали соляным раствором и водой и сушили над Na2 SO4. После выпаривания растворителя и двукратной кристаллизации (гексан/AcOEt = 8:2) окончательно получали 168 мг (2E,4E,6E)-6-бром-3-метил-7-(5,5,8,8-тетраметил-5,6,7, 8- тетрагидронафталин-2-ил)окта-2,4,6-триеновой кислоты в виде желтых кристаллов, tпл 189-190oC.
Пример 6
а) 114 г неочищенного 2-бром- 5,5,8,
8-тетраметил-5,6,7,8-тетрагидронафталина (ГХ-чистота: 76,5%) растворяли в 250 мл пиперидина и последовательно обрабатывали 4,80 г ((Ph)3P)4Pd, 0,95 г CuI и 1,35 г (Ph)3
P. Затем внутреннюю температуру повышали до 90-95oC и через капельную воронку в течение 4 часов добавляли 150 мл пропаргилокситриметилсилана. Спустя еще 1 час реакционную смесь сливали на
ледяную крошку/конц. HCl и интенсивно перемешивали, пока ТСХ не показала, что весь силиловый эфир уже отщеплен. Добавляли EtOEt, слои разделяли и органическую фазу промывали водой и соляным раствором.
Путем сушки над Na2SO4, выпаривания растворителя и очистки с помощью быстрой хроматографии (силикагель, гексан/AcOEt = 85:15) получали 60,6 г 3- (5,5,8,8-тетраметил-5,6,7,
8-тетрагидронафталин-2-ил)проп-2-ин- 1-ола в виде коричневато-желтых кристаллов, tпл 84-85oC (ГХ- чистота > 96%).
б) 30,3 г 3-(5,5,8,8-тетраметил-5,6,7, 8- тетрагидронафталин-2-ил)проп-2-ин-1-ола растворяли в 500 мл абсолютного EtOH и гидрировали при температуре окружающей среды и при давлении H2 1 атм. Тремя порциями добавляли катализатор (Lindlar тип А, 30 г). Превращение исходных продуктов определяли с помощью ГХ. После приблизительно 11 часов катализатор отфильтровывали и спиртовой раствор (EtOH) упаривали досуха, получая 30,9 г (Z)-3-(5,5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)проп-2-ен-1-ола в виде масла оранжевого цвета (ГХ-чистота: 91,6%), которое применяли на следующей стадии без дополнительной очистки.
в) В течение 20 минут кипятили с обратным холодильником 12,5 г цинкового порошка (активированного с помощью промывки HCl, H2О, EtOH, ацетона и EtOEt) и 1,89 г свежеочищенного CuCl в 120 мл абсолютного EtOEt. После охлаждения добавляли 18,0 г (Z)-3-(5,5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)проп-2-ен-1-ола, растворенного в 40 мл EtOEt, a затем 15,2 мл CH2 I2. Смесь выдерживали при температуре дефлегмации в течение 16 часов. После этого ее сливали на ледяную крошку, экстрагировали EtOEt, промывали водой и сушили над Na2SO4. Путем выпаривания растворителя и очистки с помощью быстрой хроматографии (силикагель, гексан/AcOEt = 90:10) получали 11,28 г (1RS,2SR)-2-(5,5,8,8-тетраметил-5,6,7, 8- тетрагидронафталин-2-ил)циклопропилметанола в виде желтоватого масла (ГХ-чистота: 97,6%).
г) 5,25 мл свежеперегнанного оксалилхлорида растворяли в 130 мл CH2Cl2 и охлаждали до -62oC. Медленно добавляли (температура повышалась до -52oC) 9,5 мл абсолютного ДМСО. Через 10 минут по каплям при -60oC добавляли 14,35 г (1RS.2SR) -2- (5,5,8,8-тетраметил- 5,6,7,8-тетрагидронафталин-2-ил)циклопропилметанола, растворенного в 30 мл CH2Cl2. Через 15 минут добавляли 38,7 мл NEt3 и удаляли охлаждающую баню. Через 1 час реакцию в смеси прекращали с помощью ледяной крошки, экстрагировали EtOEt, промывали соляным раствором и водой, сушили над Na2SO4 и упаривали под вакуумом. После быстрой хроматографии (силикагель, гексан/AcOEt = 94: 6) получали 12,52 г (1RS, 2SR)-2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2- ил)циклопропилкарбальдегида в виде светло-желтого масла.
д) 2,34 г NaH (50% -ный в минеральном масле) суспендировали в 120 мл абсолютного ДМФ и при 0oC обрабатывали 16,8 г этилового эфира 4-(диэтоксифосфинил)-3-метилкротоновой кислоты. После прекращения выделения H2 (приблизительно через 1/2 часа при 0oC) медленно добавляли при 0oC 12,5 г (1RS.2SR)-2-(5, 5,8,8-тетраметил-5,6,7,8- тетрагидронафталин-2-ил)циклопропилкарбальдегида, растворенного в 25 мл абсолютного ДМФ. Перемешивание продолжали в течение 30 минут. Затем реакционную смесь сливали на EtOH/H2O (8:2) и экстрагировали гексаном. Гексановый слой сушили над Na2SO4 и упаривали под вакуумом. После быстрой хроматографии (силикагель, гексан/AcOEt = 98,5: 1, 5) получали 8,85 г этилового эфира (2E,4E)-3-метил-5-[(1RS,2RS)-2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин -2-ил)циклопропил] пента-2,4-диеновой кислоты в виде бесцветного масла (с чистотой приблизительно 94% по данным1H-ЯМР) и кроме этого 8,46 г E/Z-смеси ((2E,4E)/(2Z,4E)) = приблизительно 2:1.
е) 8,84 г этилового эфира (2E,4E)-3-метил-5- [(1RS,2RS)-2-(5,5,8, 8-тетраметил-5,6,7,8-тетрагидронафталин-2- ил)циклопропил] пента-2,4-диеновой кислоты (с приблизительно 94%- ной степенью чистоты по данным1H-ЯМР) растворяли в 100 мл ТГФ/EtOH (1:1) и обрабатывали 40 мл 3H водного NaOH. Реакционную смесь выдерживали в темноте при температуре окружающей среды в течение 3 дней. Затем ее сливали на ледяную крошку, экстрагировали EtOEt, промывали водой и сушили над Na2SO4. После выпаривания растворителя и двукратной кристаллизации (гексан/AcOEt = 8:2 и 7:3) получали 4,25 г (2E, 4E)-3-метил-5-[(1RS, 2RS)-2-(5,5,8,8-тетраметил-5, 6,7,8- тетрагидронафталин-2-ил)циклопропил]пента-2,4-диеновой кислоты в виде белых кристаллов, tпл 150-151oC.
Пример 7
Аналогично стадиям д) и е) из
примера 6 получали
(2E. 4E)-3-метил-5-[(1S. 2S)-2- (5.5.8.8-тетраметил-5.6.7.8-тетрагидронафталин-2-ил)циклопропил] пента-2.4-диеновую кислоту в виде белых кристаллов (tпл
108-110oC, aDRT=+161o (CHCl3, с=0,8%)) из (1R,2S) -2- (5,5,8,8-тетраметил- 5,6,7,8-тетрагидронафталин-2-ил)циклопропилкарбальдегида и этилового эфира
4-(диэтоксифосфинил)-3-метилкротоновой кислоты по реакции Виттига-Хорнера с последующим разделением изомеров с двойной связью и катализируемым основанием гидролизом. Необходимый альдегид синтезировали,
используя в качестве ключевой стадии энантиоселективное циклопропанирование, описанное в Chemistry Letters 1992, 61, и последующее окисление по Сверну, как описано ниже.
1,67 г (Z)-3-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин- 2-ил)проп-2-ен-1-ола растворяли в 36 мл CH2Cl2. При 0oC добавляли 7,62 мл раствора Et2Zn (1M [в гексане]), а затем через 15 минут добавляли 1,33 мл L-(+)-диэтилтартрата, растворенного в 18 мл CH2Cl2. Перемешивание продолжали в течение 45 минут. Затем реакционный сосуд охлаждали до -22oC и добавляли вторую порцию, содержащую 13,8 мл раствора Et2Zn (1M [в гексане]), после чего добавляли 2,23 мл CH2I2. Реакционному сосуду давали нагреться до +18oC в течение 16 часов. После прекращения реакции с помощью ледяной крошки/раствора NH4Cl смесь экстрагировали EtOEt, промывали разбавленной HCl и водой, сушили над Na2SO4 и упаривали досуха. После быстрой хроматографии (силикагель, гексан/AcOEt = 87: 13) получали 1,057 г (1R,2S)-2-(5,5,8,8- тетраметил-5,6,7, 8-тетрагидронафталин-2-ил)циклопропилметанола с 99%-ной чистотой по данным ГХ. Абсолютную конфигурацию определяли по аналогии с методикой, приведенной в Chemistry Letters 1992, 61, однако она не была строго доказана; оптическую чистоту определяли на следующей стадии.
381 мл свежеперегнанного оксалилхлорида растворяли в 13 мл CH2Cl2 и охлаждали до -65oC. Медленно добавляли 687 мл абсолютного ДМСО, растворенного в 4 мл CH2Cl2. Через 10 минут по каплям при -65oC добавляли 1,04 г (1R, 2S)-2-(5,5,8, 8-тетраметил- 5,6,7,8-тетрагидронафталин-2-ил)циклопропилметанола, растворенного в 11 мл CH2Cl2. Через 10 минут добавляли 2,79 мл NEt3 и удаляли охлаждающую баню. Через 1 час реакцию в смеси прекращали с помощью ледяной крошки, экстрагировали EtOEt, промывали соляным раствором и водой, сушили над Na2SO4 и упаривали в вакууме. После быстрой хроматографии (силикагель, гексан/AcOEt = 9:1) получали 876 мг (1R,2S)-2-(5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2- ил)циклопропилкарбальдегида в виде светло-желтого масла с 99%-ной чистотой по данным ГХ.
Оптическую чистоту определяли следующим образом (ср. J. Org. Chem. 46, 5159, 1981).
22 мг (1R, 2S)-2- (5,5,8,8-тетраметил-5,6,7, 8-тетрагидронафталин-2- ил)циклопропилкарбальдегида растворяли в 0,5 мл толуола и последовательно обрабатывали 30 мг MgSO4•2H2O, 1 мг пара-TsOH и 16 мл D-(-)-2, 3-бутандиола. Смесь выдерживали в течение 110 минут при 50oC. Затем ее сливали на ледяную крошку, экстрагировали EtOEt, промывали водой, сушили над Na2SO4 и упаривали в вакууме. ГХ- анализ показал соотношение диастереомеров 91,9/3,90, соответствующее значению е.е. 92%.1H-ЯМР-спектр полностью соответствовал результатам этого анализа.
Пример 8
Аналогично примеру 7, но используя на стадии циклопропанирования D-(-)- диэтилтартрат, получали
(2E. 4E)-3-метил-5-[(1R.
2R)-2-(5.5.8.8- тетраметил-5.6.7.8-тетрагидпонафталин-2-ил)циклопропил] пента-2.4- диеновую кислоту в виде белых кристаллов, tпл 93-96oC, aDRT= -156 (CHCl3,
c=0,9%).
Пример 9
Этиловый эфир (2E.4E)-3-метил-5-[2-(5.5.8.8- тетраметил-5,6.7.8-тетрагидронафталин-2-ил)циклопент-1-енил] пента-2.4-диеновой кислоты
1,59 г 2-бром-5,
5,8,8-тетраметил- 5,6,7,8-тетрагидронафталина растворяли в 12 мл ТГФ и обрабатывали при -75oC 4,37 мл 1,5М н-бутиллития (в гексане). Через 15 минут добавляли 850 мг тщательно высушенного
ZnCl2, растворенного в 9 мл ТГФ, и смесь перемешивали при -75oC в течение 1/2 ч.
Параллельно этому в другой колбе, содержащей 12 мл ТГФ, суспендировали 210 мг ((Ph)3P)2PdCl2 и восстанавливали, добавляя с помощью шприца 497 мл ДИБАГ (1,2М [в толуоле]). После перемешивания в течение 1 ч при 0oC к образовавшемуся черному раствору Pd0 добавляли 1,70 г этилового эфира (2E,4E)-5-(2- бромциклопент-1-енил) -3-метилпента-2,4-диеновой кислоты, растворенного в 6 мл ТГФ, а затем добавляли полученный согласно описанной выше методике раствор арилцинка, который переносили с помощью иглы с раздвоенным концом. Полученную смесь выдерживали при температуре окружающей среды в течение 1 ч, затем сливали на ледяную крошку и экстрагировали с помощью EtOEt. После промывки насыщенным раствором NaCl, сушки над Na2SO4 и упаривания досуха получали неочищенный продукт, который очищали с помощью быстрой хроматографии (силикагель, гексан/AcOEt = 96:4) и после окончательной кристаллизации из гексана получали 1,372 г указанного в заголовке соединения в виде желтоватых кристаллов, tпл 83-86oC.
Необходимый для этой реакции этиловый эфир (2E,4E)- 5-(2-бромциклопент-1-енил)-3-метилпента-2,4-диеновой кислоты синтезировали следующим образом.
2, 03 г NaH (50%-ный в минеральном масле) суспендировали в 120 мл ДМФ. При 0oC добавляли 12,9 г этилового эфира 4-(диэтоксифосфинил)-3-метилбут-2-еновой кислоты. Смесь перемешивали в течение 15 минут при 0oC и в течение 30 минут при комнатной температуре. После повторного охлаждения до 0oC по каплям добавляли 5,72 г 2-бромциклопент-1-енкарбальдегида, растворенного в 11 мл ДМФ, и проводили реакцию в течение 10 минут при 0oC и в течение 2 ч при комнатной температуре. Затем смесь сливали на ледяную крошку, экстрагировали с помощью EtOEt, промывали насыщенным раствором NaCl, сушили над Na2SO4 и упаривали досуха. После очистки остатка с помощью быстрой хроматографии (силикагель, гексан/AcOEt = 97: 3) и кристаллизации из гексана/следовые количества AcOEt окончательно получали 3,408 г чистого этилового эфира (2E,4E)-5-(2-бромциклопент-1-енил)-3- метилпента-2,4-диеновой кислоты в виде желтоватых кристаллов, tпл 85-86oC.
Пример 10
(2E. 4E)-3-метил-5-[2-(5.5.8.8-тетраметил- 5.6.7.8-тетрагидронафталин-2-ил)циклопент-1-енил]пента-2.4- диеновая кислота
1,32 г
этилового эфира (2E, 4E)-3-метил-5-[2- (5,5,8,8-тетраметил-5,6,7,8-тетрагидронафталин-2-ил)циклопент-1- енил]пента-2,4-диеновой кислоты растворяли в 13 мл ТГФ/EtOH (1:1) и обрабатывали 5,6 мл 3H NaOH.
Смесь выдерживали в течение 48 ч при температуре окружающей среды и затем сливали на ледяную крошку/HCl. После экстракции с помощью AcOEt, промывки водой, сушки над Na2SO4,
выпаривания растворителя и перекристаллизации из AcOEt получали 803 мг указанного в заголовке соединения в виде желтых кристаллов, tпл 195-196oC (разложение).
Пример 11
Аналогично примеру 10 получали
(2E. 4E)-3-метил-5-[2-(5,5.8.8- тетраметил-5.6.7.8-тетрагидронафталин-2-ил) пиклогепт-1-енил] пента- 2.4-диеновую кислоту
в виде
желтых кристаллов, tпл 159-160oC; и
(2E. 4E)-3-метил-5-[2-(5.5.8.8-тетраметил-5,6.7.8- тетрагидронафталин-2-ил)циклогекс-1-енил]пента-2.4-диеновую кислоту
в
виде желтоватых кристаллов, tпл 202-203oC.
Пример А
Желатиновые капсулы с твердым покрытием могут быть приготовлены следующим образом.
Ингредиенты - мг/капсулу
1. Высушенный распылением порошок, содержащий 75% соединения формулы 1 - 20
2. Диоктилсульфосукцинат натрия - 0,2
3. Натрийкарбоксиметилцеллюлоза
- 4,8
4. Микрокристаллическая целлюлоза - 86,0
5. Тальк - 8,0
6. Стеарат магния - 1,0
Всего - 120
Высушенный распылением порошок, в состав которого входят
активный ингредиент, желатин и микрокристаллическая целлюлоза и который имеет средний размер частиц активного ингредиента <1 мкм (измеренный с помощью автокореллирующей спектроскопии),
увлажняют водным раствором натрийкарбоксиметилцеллюлозы и диоктилсульфосукцината натрия и перемешивают. Полученную массу гранулируют, сушат, просеивают и полученный гранулят смешивают с
микрокристаллической целлюлозой, тальком и стеаратом магния. Порошком заполняют капсулы размера 0.
Пример Б
Таблетки могут быть приготовлены следующим образом.
Ингредиенты - мг/таблетку
1. Соединение формулы 1 в виде тонкоизмельченного порошка - 20
2. Порошкообразная лактоза - 100
3. Белый кукурузный крахмал - 60
4.
Повидон КЗО - 8
5. Белый кукурузный крахмал - 112
6. Тальк - 16
7. Стеарат магния - 4
Всего - 320
Тонкоизмельченный активный ингредиент смешивают с
лактозой и с порцией кукурузного крахмала. Смесь увлажняют водным раствором повидона К30 и перемешивают. Полученную массу гранулируют, сушат и просеивают. Гранулят смешивают с оставшимся ингредиентами
- кукурузным крахмалом, тальком и стеаратом магния и прессуют в таблетки соответствующего размера.
Пример В
Желатиновые капсулы с мягким покрытием могут быть приготовлены
следующим образом.
Ингредиенты - мг/капсулу
1. Соединение формулы 1 - 5
2. Триглицерид - 450
Всего - 455
При перемешивании в атмосфере инертного
газа, защищая от света, растворяют 10 г соединения формулы 1 в 90 г триглицерида со средней длиной цепи. Раствор обрабатывают по методике, общепринятой для обработки массы, предназначенной для
заполнения капсул с мягким желатиновым покрытием, содержащих 5 мг активного ингредиента.
Пример Г
Лосьон может быть приготовлен следующим образом.
Ингредиенты
1. Тонкоизмельченное соединение формулы 1 - 1,0 г
2. Карбопол 934 - 0,6 г
3. Гидроксид натрия - q.s. до pH 6
4. Этанол, 94%-ный - 50,0 г
5.
Деминерализованная вода - до 100,0 г
Активный ингредиент вводят в смесь 94%-ный этанол/вода, защищая от света. Смешивают с карбополом 934 до завершения гелеобразования и с помощью гидроксида
натрия регулируют значение pH.
Реферат
Изобретение относится к новым лигандам Х-рецептора ретиноевой кислоты формулы I, где R1 - R7, R10, Х и связь, обозначенная пунктиром, имеют значения, указанные в описании. Фармацевтическая композиция, обладающая антипролиферативным действием, содержащая соединение формулы I и фармацевтический носитель. Соединение формулы I избирательно связывается с ретиноидными рецепторами RXR, которое пригодно в качестве антипролиферативного агента при дерматологических и онкологических показаниях. 2 с. и 16 з.п.ф-лы, 6 ил.
Формула
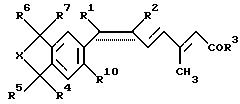
где необязательно присутствует связь, обозначенная пунктиром, и если связь, обозначенная пунктиром, присутствует, то R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5 - 8-членное карбоциклическое кольцо или 5 - 8-членное гетероциклическое кольцо, содержащее один атом серы, кислорода или азота, причем если кольцо является ароматическим, то связь, обозначенная пунктиром, представляет собой часть мезомерной системы, или, если связь, обозначенная пунктиром, отсутствует, то R1 и R2 вместе представляют собой метилен, образуя цис-замещенное циклопропиловое кольцо;
R3 обозначает гидрокси или низший алкокси;
R4, R5, R6 и R7 независимо друг от друга обозначают водород или низший алкил;
Х обозначает группу (>CR8R9)n, где n равно 1, 2 или 3, и R8, R9 независимо друг от друга обозначают водород или низший алкил;
R10 обозначает водород, алкил или алкокси.
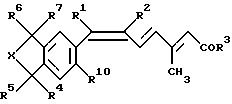
где R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5 - 8-членное карбоциклическое кольцо или 5 - 8-членное гетероциклическое кольцо, содержащее один атом серы, кислорода или азота, причем если кольцо является ароматическим, то двойная связь между атомами углерода, присоединенными к R1 и R2, представляет собой часть мезомерной системы;
группы R3 - R7, R10 и Х имеют значения, указанные для формулы I.
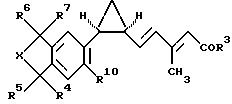
где R3 - R7, R10 и Х имеют значения, указанные для формулы I.
10.08.94 - по пп.1-3, 8-15 и 18;
05.07.95 - по пп.4-7, 16 и 17.
Комментарии