Производное бензолацетамида, способы его получения и фармацевтическая композиция - RU2087465C1
Код документа: RU2087465C1
Чертежи

Описание
В патенте США N 4246429, соответствующем европейской заявке N 0006713, описывается ряд бензолацетамидов и тиоамидов, используемых в качестве промежуточных соединений при получении фитофармацевтических соединений. В настоящее время неожиданно было найдено, что некоторые из этих промежуточных соединений эффективно задерживают размножение вируса СПИД (HIV) и, следовательно, могут использоваться для лечения индивидуумов, инфицированных вирусом СПИД. Кроме того, были найдены достаточно близкие, но до сих пор не опубликованные соединения, которые даже в большей степени ингибируют размножение ретровируса.
В заявке Великобритании N 1423430 публикуются бензолтиоацетамидные соединения, более конкретно α-(фениламино)-3,4-диметоксибензолтиоацетамид. Названные соединения обладают антисекреторной активностью. В Аrchives Internationales de Pharmacodynamie et de Therapie, 1966, 164 (2), 321-330 публикуются алкоксибензолацетамидные производные, в частности a-[(4-этоксифенил)амино] -N-пропилбензолацетамид. Названные соединения обладают анальгетическими свойствами.
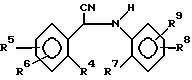
Данное изобретение относится к соединениям, используемым в медицине, имеющим формулу

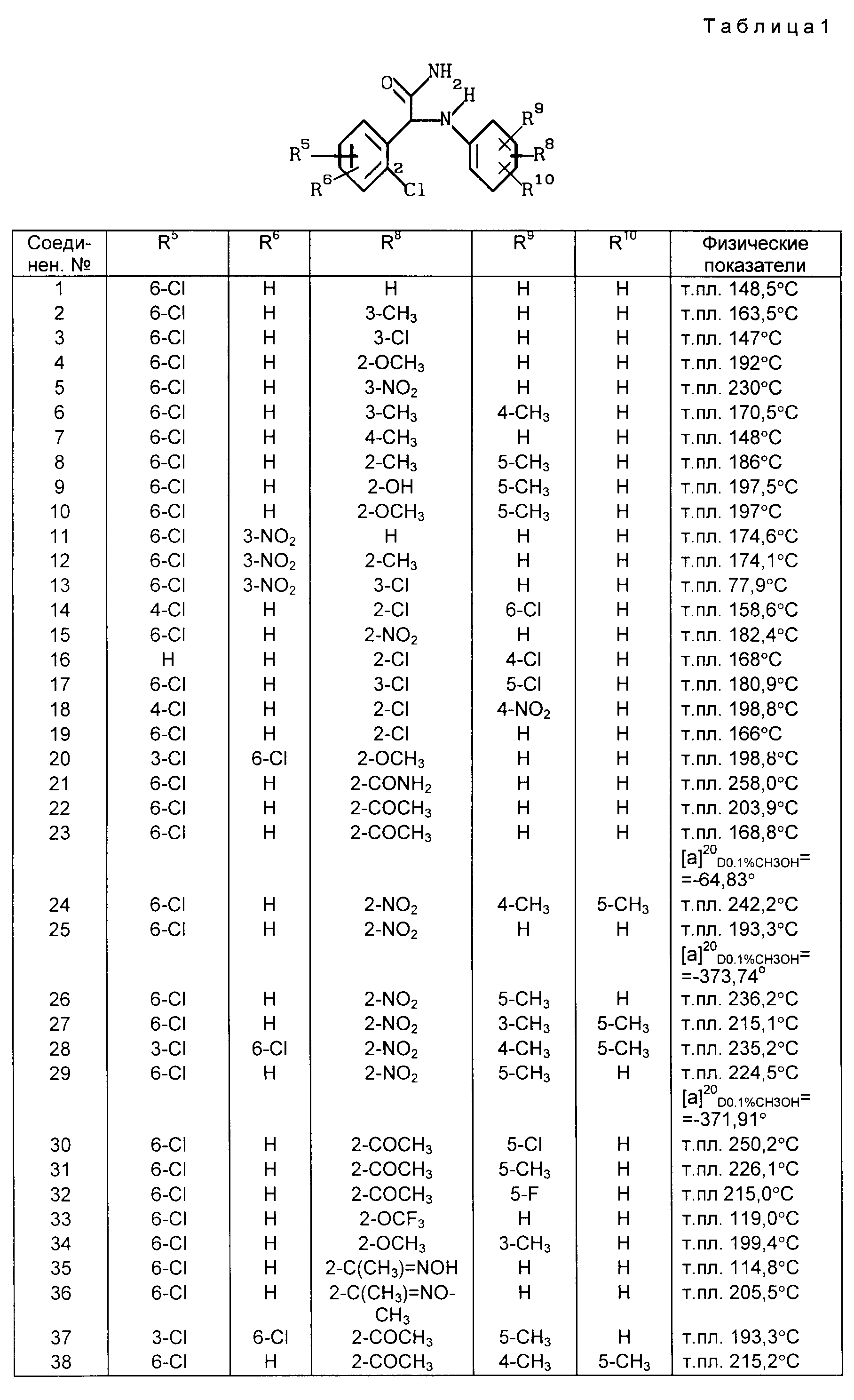
фармацевтически приемлемой кислотно-аддитивной солевой формы или ее стереохимически изомерной формы, в которой R1 и R2 каждый независимо друг от друга представляют собой водород, С1-6-алкил или С1-3-циклоалкил; или
R1 или R2 вместе с атомом азота, к которому присоединены R1 и R2, могут образовывать пирролидинильную, пиперидинильную, морфолинильную, пиперазинильную или 4-С1-4 -алкилпиперазинильную группу;
X является кислородом или серой;
R3 является водородом или С1-6-алкилом;
R4, R5 и R6 каждый независимо друг от друга являются водородом, галоидом, С1-6-алкилом, С1-6-алкилокси, нитро, трифторметилом, циано, аминометилом, карбоксилом, С1-4 -алкилоксикарбонилом, С1-4-алкилкарбонилом, аминокарбонилом или гидроксилом;
R7 представляет собой водород или галоид и R8, R9 и R10 каждый независимо друг от друга являются водородом, галоидом, С1-6-алкилом, С1-6-алкилокси, нитро, гидрокси, трифторметокси, 2,2,2-трифторэтокси, (трифторметил)карбонилом, аминокарбонилом, (циклопропил)карбонилом или радикалом С1-6-алкил-(С= Y)-, гдеY представляет собойO,N-OH,N-OCH3,N-NH2 или N-N(CH3)2 при условии, что: 1) R1 не является н-пропилом, когда R1, R2, R3, R6, R7, R9 и R10 являются водородом, R8 представляет собой 4-этокси и X представляет собой кислород; 2) X не является серой, когда R1, R2, R3, R6, R7, R9 и R10 представляют собой водород, а R4 и R5 является 3,4-диметокси.
Соединения формулы (I), в которой по крайней мере один из R1 или R2 является водородом, могут существовать также в их таутомерной форме. Названная форма, хотя она и не указана явно в приведенном выше описании, предлагается для включения в данное изобретение.
В вышеприведенных определениях термин "галоид" означает фтор, хлор, бром и йод, С1-4-алкил означает прямые или разветвленные насыщенные углеводороды и радикалы, содержащие от 1 до 4 атомов, такие как, например, метил, этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил и 1,1-диметил-этил, С1-6-алкил означает С1-4-алкил и его более высокие гомологи, содержащие 5 или 6 углеродных атомов; С3-6-циклоалкил означает циклопропил, циклобутил, циклопентил и циклогексил.
Фармацевтически приемлемые аддитивные соли, упомянутые выше, включают терапевтически активные нетоксичные аддитивные солевые формы, которые способны образовывать соединения формулы (I). Названные солевые формы обычно могут быть получены взаимодействием основной формы соединения формулы (I) с соответствующими кислотами, такими как неорганические кислоты, например гидрогалоидная кислота, например хлористоводородная, бромистоводородная и т.п. кислоты, серная кислота, азотная кислота, фосфорная кислота и подобные; или органические кислоты, такие как, например, уксусная, пропионовая, оксиуксусная, 2-оксипропионовая, 2-оксопропионовая, этандвухосновная, пропандвухосновная, бутандвухосновная, (Z)-2-бутендвухосновная, (Е)-2-бутендвухосновная, 2-оксибутандвухосновная, 2,3-диоксибутандвухосновная, 2-окси-1,2,3-пропантрехосновная, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-оксибензойная, 4-амино-2-оксибензойная кислота и подобные кислоты. Наоборот, солевая форма может быть превращена в форму свободного основания взаимодействием со щелочью. Термин "аддитивная соль" означает также гидраты и сольвентные аддитивные формы, которые могут образовывать соединения формулы (I). Примерами таких форм являются, например, гидраты, алкоголяты и т.п.
Такими частными соединениями являются упомянутые выше соединения формулы (I), в которых R4, R5 и R6 каждый независимо друг от друга являются водородом, галоидом, С1-6-алкилом, С1-6-алкилокси, нитро, гидрокси; R7 является водородом, R8, R9 и R10 каждый независимо друг от друга является водородом, галоидом, С1-6-алкилом, С1-6-алкилокси, нитро, гидрокси, трифторметокси, 2,2, 2-трифторэтокси, (трифторметил)карбонилом, аминокарбонилом, (циклопропил)карбонилом или радикалом С1-6-алкил-(С=Y)-, гдеY представляет собойO,N-OH,N-OCH3, илиN-N(CH3 )2.
Интересными соединениями для использования их в способе, соответствующем данному изобретению, являются такие соединения формулы (I), в которых каждый из R1 и R2 являются независимо водородом; и/или X является кислородом; и/или R4, R5 и R6 каждый независимо друг от друга являются водородом, галогеном, С1-6-алкилокси или нитро; и/или R7 является водородом; и/или R8, R9 и R10 каждый независимо является водородом, галоидом, С1-6-алкилом, С1-6-алкилокси, нитро, трифторметокси или С1-6-алкилкарбонилом.
Более интересными соединениями из названных являются такие соединения, в которых R1 и R2 каждый независимо друг от друга, является водородом; и/или R3 является водородом; и/или R4, R5 и R6 каждый независимо является водородом, галогеном, метокси или нитро; и/или R8, R9 и R10 каждый независимо представляет водород, галоид, метил, метокси, нитро, трифторметокси или метилкарбонил.
Особенно интересными соединениями являются такие, в которых R1 и R2 являются водородом; R4 представляет собой 2-хлор или 4-метокси, R5, R6 и R7 являются водородом; или R4 и R5 представляют собой 2,6-дихлор, 2,4-дихлор или 3,4-диметокси, R6 и R7 являются водородом; или R4, R5 и R6 представляют собой 2,6-дихлор-3-нитро или 2,3,6-трихлор; R7 является водородом; или R4, R5, R6 и R7 представляют собой 2,3,6-трихлор-4-трифторметил; и/или R8 является водородом, хлором, метилом, метокси, нитро, трифторметилом или метилкарбонилом, R9 и R10 являются водородом; или R8 и R9 представляют собой 2,4-диметил, 2,5-диметил, 2,4-дихлор, 2,6-дихлор, 3,5-дихлор, 2-гидрокси-5-хлор-2-метокси-5-хлор, 2-нитро-5-хлор, 2-нитро-5-метил, 3-метокси-5-метил, 2-метилкарбонил-5-метил, 2-метилкарбонил-5-хлор, 2-метилкарбонил-5-фтор или 2-хлор-4-нитро, R10 является водородом.
Наиболее интересными соединениями внутри данного изобретения являются такие соединения, в которых R4 и R5 представляют собой 2,6-дихлор, R6 и R7 являются водородом; или R4, R5 и R6 представляют собой 2,3,6-трихлор; R7 является водородом; R8 представляет собой 2-метокси, 2-нитро, 2-метилкарбонил, 2-трифторметокси, 3-метил, R9 и R10 являются водородом; или R8 и R9 представляют собой 2-метокси-5-метил, 2-нитро-5-хлор, 2-нитро-5-метил, 2-метокси-5-хлор-6-метилкарбонил-5-метил, 2-метилкарбонил-5-фтор или 2-метилкарбонил-5-хлор, R10 является водородом.
Предпочтительными соединениями являются:
(1-α-[(2-нитрофенил)амино]-2,
6-дихлорбензолацетамид,
(1-a-[(5-метил-2-нитрофенил)амино]-2,6-дихлорбензолацетамид,
(1-a-[(2-ацетилфенил)амино]-2,6-дихлорбензолацетамид,
(1-a-[(2-ацетил-5-метилфенил)амино] -2,6-дихлорбензолацетамид,
a-[(2-ацетил-5-хлорфенил)амино]-2,6-дихлорбензолацетамид,
a-[(5-хлор-2-нитрофенил)амино]-2,6-дихлорбензолацетамид и
a-[(2-ацетил-5-фторфенил)амино]-2,6-дихлорбензолацетамид.
Дополнительной характеристикой данного изобретения является тот факт, что некоторые соединения формулы (I) рассматриваются как новые и были специально разработаны для использования их в способе, соответствующем данному изобретению.
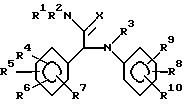
Интересная подгруппа новых соединений образует с
соединениями формулы
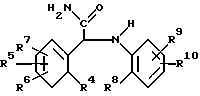
фармацевтически приемлемые кислотно-аддитивные солевые формы или их стереохимические изомерные формы, в которых R4 представляет собой галоид, С1-6-алкил, С1-6-алкокси или нитро; R5 и R6 каждый независимо друг от друга является водородом, галоидом, С1-6-алкил, С1-6-алкокси нитро, трифторметил, циано, аминометил, карбоксил, С1-4-алкоксикарбонил, С1-4-алкилкарбонил, аминокарбонил или гидрокси, R7 является водородом или галогеном; R8 представляет собой С1-6-алкилокси, нитро, трифторметил, 2,2,2-трифторэтокси, (трифторметил)карбонил, аминокарбонил, (циклопропил)карбонил или радикал С1-6-алкил-(С= Y)-, гдеY представляет собойO,N-OH,N-OCH3,N-NH2,N-N(CH3)2 при условии, что R8 не является 2-метокси, когда R4 является хлором, R5 является 6-хлор, R6, R7 и R9 являются водородом и R10 является водородом или 5-метилом.
Интересная подгруппа новых соединений образуется соединениями формулы (I) или формулы (I-а), в которых асимметричный атом углерода, несущий амидную группу, имеет абсолютно такую же конфигурацию, что и в (-)-α-[(2-нитрофенил)амино]-2,6-дихлорбензолацетамиде.
Другая подгруппа новых соединений образуется соединениями формулы (I) или формулы (I-а), в которых асимметричный атом углерода, несущий амидную группу, имеет абсолютно противоположную конфигурацию по отношению к (-)-a-[(2-нитрофенил)амино]-2,6-дихлорбензолацетамиду.
Новыми соединениями, представляющими особый интерес, являются такие соединения, в которых R4 представляет собой галоид или С1-6-алкил; R5 и R6 являются водородом, галоидом, С1-6-алкилом; R7 является водородом или хлором; R8 представляет собой С1-6-алкокси, трифторметокси, нитро или С1-6 -алкилкарбонил; R9 и R10 водород, галоген или С1-6-алкил.
Наиболее интересными новыми соединениями являются такие, в которых R4 представляет собой хлор или метил; R5 является водородом или хлором; R7 является водородом; R8 является метокси, трифторметокси, нитро или метилкарбонилом; R9 представляет собой водород, фтор или метил, а R10 водород.
Предпочтительными новыми соединениями являются такие, в которых R4 является хлором; R5 представляет собой 6-хлор или 6-метил; R6 представляет собой водород или 3-хлор; R7 является водородом; R8 представляет собой метокси, трифторметокси, нитро или метилкарбонил; R9 представляет собой водород, хлор, 5-фтор или 5-метил, а R10 -водород.
Наиболее предпочтительными соединениями являются:
(-)-a-[(2-нитрофенил)амино]-2,6-дихлорбензолацетамид,
(-)-a-[(5-метил-2-нитрофенил)амино] -2,6-дихлорбензолацетамид,
(-)-a-[(2-ацетилфенил)амино]-2,6-дихлорбензолацетамид,
(-)-a-[(2-ацетил-5-фенил)амино]-2,6-дихлорбензолацетамид,
a-[(2-ацетил-5-хлорфенил)амино]-2,6-дихлорбензолацетамид,
a-[(5-хлор-2-нитрофенил)амино]-2,6-дихлорбензолацетамид и
a-[(2-ацетил-5-фторфенил)амино]-2,6-дихлорбензолацетамид.
Соединения формулы (I) и соединения формулы (I-а), в основном, могут быть получены следующими известными в данной области приемами, такими как, например, приемы, описанные в патенте США N 4246429, и альтернативными методами, известными в данной области. Наиболее интересные способы описываются ниже более детально для соединений формулы (I). Очевидно, таким же образом предполагается включение аналогичных процессов для получения новых соединений формулы (I-а).
Соединения формулы (I), в основном, могут быть получены алкилированием соответствующих анилиновых производных формулы (II) или их соли алкилирующим агентом формулы (III) следующими известными N-алкилирующими методами.
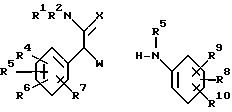
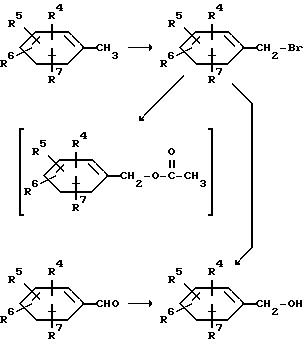
В формуле (III) и в дальнейшем W представляет собой активную уходящую группу, такую как например, галоид, например хлор, бром или йод, сульфонильную группу, например метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси, 4-метилбензолсульфонилокси, нафталинсульфонилокси и т. п. активные уходящие группы. В формуле (II) R3 представляет водород или С1-6 алкил и может представлять формил (-CHO). Названные формильные производные особенно пригодны для получения солевых форм промежуточных соединений формулы (II). В соответственно полученных продуктах формулы (I) с формильной концевой группой эта группа может быть замещена водородом посредством гидролиза или же может быть восстановлена до метильной последующими стандартными восстановительными методами.
Названная реакция N-алкилирования может проводиться стандартным методом
перемешиванием реагирующих веществ, не обязательно в реакционно инертном растворителе, таком как, например, вода, ароматическом растворителе, например бензоле, метилбензоле, диметилбензоле,
хлорбензоле, метоксибензоле и т.п. С1-6-алканоле, например метаноле, этаноле, 1-бутаноле и т.п. кетоне, например, 2-пропанское, 4-метил-2-пентароне и т.п. эфире, например этилацетате,
γ-бутиролактоне и т.п. эфире, например 1,1-оксибисэтане, тетрагидрофуране, 1,4-диоксане и т.п. диполярном апротическом растворителе, например N,N-диметилформамиде, N, N-диметилацетамиде,
диметилсульфоксиде, пиридине, 1,3-диметил-3,4,5,6- тетрагидро-2(Н)-пиримидиноне, 1,3-диметил-3-имидазолидиноне, 1,1,3,3, -тетраэтилмочевине, 1-метил-2-пирролидоне, нитробензоле, ацетонитриле и т.п.
или в смеси таких растворителей. Не обязательно может осуществляться добавление соответствующего основания, такого как, например, карбонат щелочного или щелочноземельного металла, гидрокарбонат,
гидроокись, окись, карбоксилат, алкоксид, гидрид или амид, например натрий карбонат, натрий гидрокарбонат, карбонат калия, гидроокись натрия, окись кальция, ацетат натрия, метилат натрия, гидрид
натрия, амид натрия и т.п. или органического основания, такого как, например, амин, например N,N-диэтилэтанамин, N-(1-метилэтил)-2-пропанамин, 4-этилморфолин, 1,4-диазабицикло[2,2,2]октан, пиридин и
т.п. для связывания кислоты, которая образуется в процессе реакции. Обычно, выгодно бывает осуществить превращение формильного промежуточного соединения формулы (II) сначала в его подходящую солевую
форму, такую как, например, соль щелочного или щелочноземельного металла, -взаимодействием (II) с соответствующим основанием, которое упоминалось выше, с последующим использованием названной солевой
формы в реакции с алкилирующим агентом формулы (III). Названным щелочным или щелочноземельным металлом может быть, например, натрий, калий, литий, кальций и т.п. В некоторых случаях может быть
допустимым добавление йодистых солей, предпочтительно йодидов щелочного металла, или краун-эфира, например 1,4,7,10,13,16- гексаоксациклооктадекана и т. п. Перемешивание и повышение температуры
повышает скорость реакции; в частности, реакция может проводиться при температуре дефлегмации реакционной смеси. Кроме того, благоприятным является проведение названного алкилирования в инертной
атмосфере, такой как, например, освобожденный от кислорода аргон или азот в виде газа.
Или же названное алкилирование может осуществляться в известных условиях проведения фазового переходного катализа.
Указанные условия включают перемешивание реагирующих веществ с соответствующим основанием и не обязательно в инертной атмосфере, как указывалось выше, в присутствии подходящего фазового переходного катализатора, такого, например, как триалкилфенилметиламмоний, тетраалкиламмоний, тетраалкилфосфоний, тетраарилфософний галоид, гидроокись, гидросульфат и т.п. катализаторы. Повышение температуры может способствовать повышению скорости реакции.
Эффективным вариантом для проведенных выше реакций N-алкилирования является нагревание промежуточного соединения формулы (III) в избытке анилинового производного формулы (II) в отсутствие какого-либо растворителя. Реакция проводится при перемешивании и нагревании смеси до температуры, эффективной для полного растворения реагирующих веществ.
Во всех приведенных выше случаях и в следующих далее примерах приготовления продукты реакции могут выделяться из реакционной смеси и, если необходимо, подвергаться дальнейшей очистке в соответствии с общеизвестными в данной области методиками.
Соединения формулы (I), в которых R1 и R2 являются водородом и при этом названные соединения представляются формулой (I-b), когда X равно 0, и формулой (I-с), когда X равно S, могут быть получены взаимодействием нитрила формулы (IV) с реагентом H2X (Y), а именно водой или сероводородом, в соответствующих условиях.
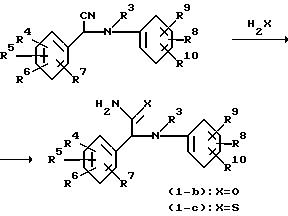
Гидролиз нитрила (IV) до соответствующего амида (I-b), где X равно 0, может быть легко осуществлен следующими известными процедурами. Предпочтительно названный гидролиз проводится при комнатной температуре в концентрированной сильной кислоте, например концентрированной серной кислоте, соляной кислоте, бромистоводородной кислоте, муравьиной кислоте, насыщенной соляной кислоте и т.п. не обязательно в присутствии небольших количеств воды.
Нитрил (IV) может быть легко превращен в тиоамид (I-с), в котором X является S реакцией с сероводородом в соответствующем растворителе, например пиридине, моно-, ди- или триметилированном пиридине и подобных растворителях, и в присутствии соответствующего основания, такого как амин, например N, N-диэтилэтанамин, 1-метилморфолин, N-(1-метилэтил)-1-метилэтанамин и т.п. Эта последняя реакция может быть легко проведена при комнатной температуре, а в некоторых примерах даже при более низких температурах, таких, например, как между 0oC и комнатной температурой. Тиоамидные соединения формулы (I-с) могут быть легко превращены в соответствующие амиды формулы (I-b) взаимодействием с окислительным агентом, таким, например, как перекись водорода в воде, не обязательно при перемешивании реакционно инертного органического сорастворителя.
Соединения формулы (I), в которых X является 0 и по крайней мере один или несколько из R8, R9 и R10 представляют собой группы, оттягивающие электроны, такие, например, как галоид, нитро или С1-6 -алкокси, и при этом названные соединения представлены формулой (I-d), -могут быть получены N-арилированием промежуточного соединения формулы (VI) соответствующим бензольным производным формулы (VII).
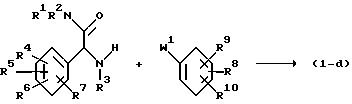
В формуле (VII) W1 представляет собой активную уходящую группу, такую, например, как галоид, C1-6-алкилокси, арилокси, (C1-6-алкил или арил) сульфонилокси, (C1-6-алкил или арил) сульфонил, C1-6-алкилтио или нитро, предпочтительно фтор, хлор, нитро, 4-метилбензолсульфонилокси, метокси или метилтио. Указанная реакция арилирования может быть легко проведена осуществлением описанных здесь ранее способов для реакции алкилирования промежуточного соединения (II) промежуточным соединением (III). Более конкретно, реагирующие вещества могут подвергаться перемешиванию, преимущественно при несколько повышенной температуре и, в частности, при температуре дефлегмирования в присутствии основания, как это было определено в упомянутой выше реакции алкилирования, в соответствующем растворителе, таком, например, как диполярный апротический растворитель, например N,N-диметилформамид, N, N-диметилацетамид, диметилсульфоксид, 1-метил-2-пирролидон, ацетонитрил, пиридин, гексаметилфосфор триамид; спирт, например 1-бутанол; эфир, например тетрагидрофуран, 1,1'-оксибисэтан, 1,4-диоксан и т.п. и смесях растворителей. К упомянутой реакции арилирования в равной степени могут быть применимы условия фазового переходного катализа.
Соединения формулы (I), в которых X представляет собой 0, и названное соединение, представленное формулой (I-е), могут быть получены также амидированием соответствующих карбоксильных кислот или их подходящих активных функциональных производных формулы (VIII). В формуле (VIII) L может представлять гидрокси, С1-6-алкокси, фенокси (не обязательно далее замещенные), 1Н-имидазолил, (С1-6-алкил или фенил)оксикарбонилокси, галоид и т.п. активные удаляющиеся группы.

Названное получение амидов формулы (I-е) может быть легко осуществлено посредством реакций амидирования и трансамилирования. Например, указанные амиды могут быть получены взаимодействием соответствующей карбоксильной кислоты (L представляет собой ОН) с амином (IX) в присутствии реагента, способного активизировать реакции амидирования. Типичными примерами таких реагентов являются, например, дициклогексилкарбодиимид, 2-хлор-1-метилпиридиний йодид, пятиокись фосфора, 1,1'-карбонил-бис-[1Н-имидазол] 1, 1'- сульфонил-бис-[1Н-имидазол] и т.п. реагенты.
Или же названные карбоксильные кислоты могут быть превращены в их соответствующее активное функциональное производное, такое, например, как галоидный ацил, симметричный или смешанный гидрид, эфир, амид, ацилированный азид и т.п. производные, перед реакцией с амином HNR1R2. Названные активные функциональные производные могут быть получены следующими известными способами, например взаимодействием карбоксильной кислоты с галогенирующим реагентом, таким как, например, тионил хлорид, трихлорид фосфора и т.п. или взаимодействием названной карбоксильной кислоты с галоидным ацилом, такими как ацетил хлорид, этилхлорформиат и т.п. Конкретно, интересным способом для получения амидов, в которых R3 является водородом, является взаимодействие соответствующего производного карбоксильной кислоты с карбонатобразующим реагентом, таким как, например, дихлорид карбоновой кислоты, трихлорметил хлорформиат, 1,1'-карбонил-бис-[1Н-имидазол] ди(С1-6-алкил)карбонат и т.п. получая, таким образом, циклический ангидрид формулы (VIII-a), с последующим взаимодействием циклического ангидрида с амином R1R2NH.
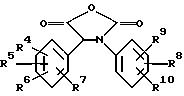
Названные активные функциональные производные карбоксильных кислот могут быть произведены на месте или, если необходимо, могут быть выделены и далее очищены перед их взаимодействием с амином HNR1R2.
Амидирование названных активных функциональных производных удобно проводить перемешиванием реагирующих веществ, не обязательно в соответствующем реакционно инертном растворителе, таком как, например, галогенированный углеводород, например дихлорметан, трихлорметан и т.п. ароматический углеводород, например, бензол, метилбензол и т.п. эфир, например 1,1'-оксибисэтан, тетрагидрофуран и т.п. или биполярный апротический растворитель, например N, N-диметилформамид, N, N-диметилацетамид, пиридин и т.п. В некоторых случаях возможно применение избытка одного из реагентов в качестве растворителя. Вода, кислота, спирт или амин, которые могут выделяться в процессе реакции, должны быть удалены из реакционной смеси известными методами, например такими, как азеотропная дистилляция, комплексованием, солеобразованием или подобными методами. В некоторых примерах, в частности, может быть приемлемо добавление подходящего основания, такого как, например, амин, N,N-диэтилэтанамин, 4-этилморфолин, пиридин или N, N-диметил-4-пиридинамин. Для того, чтобы повысить скорость реакции, названная реакция амидирования может быть благоприятно проведена при несколько повышенной температуре, в частности при температуре дефлегмирования реакционной смеси.
Соединения формулы (I) могут быть превращены также друг в друга с использованием известных реакций превращения функциональных групп. Например, соединения, в которых один из радикалов R8, R9 и R10 представляет собой радикал С1-6-алкил-С(=Y)-, где Y является N-ОН, N-OCH3 илиN-N(CH3)2, могут быть получены в соответствии с известными способами из соответствующих соединений, в которых Y представляетO взаимодействием с гидроксиламином, O-метилгидроксиламином, гидразином или ди(метил)гидразином, или с их соответствующей аддитивной солевой формой.
Соединения данного изобретения имеют по меньшей мере один асимметричный атом углерода в своей структуре, а именно углеродный атом, несущий амидную или тиоамидную группу. Названный хиральный центр и любой другой хиральный центр, который может быть представлен, может выявлен стереохимическими идентификаторами R и S.
Чистые стереохимически изомерные формы соединений формулы (I) могут быть получены применением известных в данной области методов. Диастереоизомеры могут быть выделены физическими методами, такими как селективная кристаллизация и использование хроматографических способов, например, противоточным распределением, жидкостной хроматографией и т.п. и энантиомеры могут быть отделены друг от друга с использованием известных разделяющих методов, таких как, например, селективная кристаллизация их диастереоизомерных солей хиральными кислотами. Стереохимически чистые изомерные формы могут быть извлечены также из соответствующих чистых стереохимически изомерных форм соответствующих исходных материалов при условии, что реакции осуществляются стереоспецифически. Предпочтительно в том случае, когда требуется получить конкретный стереоизомер, названное соединение должно быть синтезировано с использованием стереоспецифического способа получения. Эти способы преимущественно применяют энантиомерно чистые исходные материалы. Стереохимические изомерные формы соединений формулы (I) предполагаются как входящие в объем данного изобретения. Соединения формулы (I), полученные в соответствии с вышеописанными процессами, обычно представляют собой рацемические смеси энантиомеров, которые могут быть отделены один от другого последующими известными разделительными методами. Рацемические соединения формулы (I), которые являются в определенной степени основными, могут быть превращены в соответствующие диастереоизомерные солевые формы взаимодействием с соответствующей хиральной кислотой. Названные диастереоизомерные солевые формы впоследствии разделяются, например, селективной или фракционной кристаллизацией, и из них выделяются энантиомеры с использованием щелочного или кислотного гидролиза.
Предпочтительным методом выделения энантиомерных форм соединений формулы (I) является жидкостная хроматография, использующая хиральную стационарную фазу, такую как подходящее производное целлюлозы, например три(диметилкарбамоил)целлюлозу (Chiracel OD®), и аналогичные стационарные фазы.
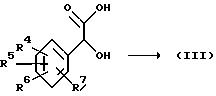
В качестве альтернативы упомянутому выше разделению соединений формулы (I) следует указать также разделение рацемических промежуточных
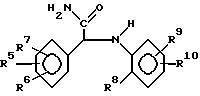
соединений. Особенно удобными промежуточными соединениями для этой цели являются аминокислоты формулы

которые могут быть легко получены из соответствующих бензол(тио)ацетамидных соединений формулы (I) кислотным или, предпочтительно, щелочным гидролизом, например действием водным раствором основания, такого как гидроокись калия, в смеси с подходящим органическим растворителем, таким, например, как алканол, например метанол, этанол и т.п. Полученные таким образом аминокислоты формулы (VIII-b) могут быть легко разделены образованием соответствующих диастереоизомерных солевых форм реакцией с подходящим хиральным основанием, таким как фенилэтанамин, нафтилэтанамин, хинонин и другие алкалоидные основания. Очевидно, что названные аминокислоты могут быть разделены также методом жидкостной хроматографии с использованием соответствующей хиральной стационарной фазы.
Энантиомерные формы аминокислот формулы (VIII-b) превращаются в энантиомерные формы бензол(тио)ацетамидных соединений формулы (I) в соответствии с описанными выше способами превращения промежуточных соединений формулы (VII) в соединения формулы (I).
Другие интересные новые промежуточные соединения или производные рацемических соединений формулы (I) для разделения методом
жидкостной хроматографии представляют собой, например, иминоэфиры формулы (VIII-c) и производные формулы (VIII-d)
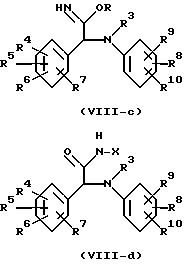
Некоторые из промежуточных соединений формулы (VIII-c) и (VIII-d) являются особенно интересными благодаря их значительно более высокой растворимости, что позволяет увеличить нагрузку рацемического вещества на хиральную стационарную фазу. Промежуточные соединения формулы (VIII-c) могут быть получены из нитридов формулы (IV) алкоголизом спиртов ROH, где R представляет собой С1-6-алкил или фенил, в присутствии безводной соляной кислоты. После разделения жидкостной хроматографией выделенные энантиомеры формулы (VIII-c) превращаются в соответствующие энантиомерные амиды формулы (I-b) гидролизом иминоэфиров в водной кислотной среде в карбоксильную кислоту и дальнейшим превращением, как это было описано выше.
В промежуточных соединениях формулы (VIII-d) X представляет собой радикал формулы -CH2ОН или -CH2N(CH3). Названные промежуточные соединения могут быть легко получены из амидов формулы (I-b) реакцией с формальдегидом или [(CH3)2= CH2]+Cl- последующими известными способами. Термическая обработка выделенных энантиомеров дает соответствующие энантиомерные амиды формулы (I-b).
Некоторые промежуточные соединения и исходные вещества, использованные в приведенных выше методах получения представляют собой известные соединения, которые могут быть синтезированы в соответствии с известными методиками получения названных или аналогичных соединений. Некоторые промежуточные соединения являются менее известными или новыми, и, следовательно, некоторые методы получения будут описаны далее более детально.
Промежуточные соединения (III), в которых X является О, могут быть получены из производных α-гидроксибензолуксусной кислоты формулы (Х) взаимодействием с галогенирующим агентом, таким как, например, пятихлористый фосфор, фосфорилхлорид, трихлорид фосфора, трибромид фосфора, тионил хлорид и т.п. или с другим активирующим агентом, таким, например, как галоидный сульфонил. Названная реакция может проводиться при повышенной температуре, в частности при температуре дефлегмирования реакционной смеси, в избытке галогенированного реагента в качестве растворителя, который не обязательно может быть разбавлен подходящим реакционно-инертным растворителем, таким как ароматический углеводород, галогенированный углеводород, эфир и т.п. растворители. Полученный таким образом галоидный бензолацетил легко превращается в нужный бензацетамид выливанием реакционной смеси в водный или спиртовой раствор, содержащий амин формулы HNR1R2.
Промежуточные соединения формулы (IV-a) могут быть получены реакцией соответствующего бензальдегида (XII) с анилином (XI) в присутствии цианидной соли и подходящего растворителя.
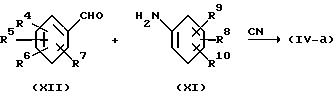
В качестве примера цианидных солей можно назвать (IV-a) цианиды щелочных и щелочноземельных металлов, например цианид натрия и калия. Подходящими растворителями являются, например, вода, спирты, например метанол, этанол и т.п. карбоксильные кислоты, например уксусная кислота и т.п. или смесь таких растворителей. Названная реакция обычно проводится перемешиванием при комнатной температуре и, если нужно, небольшим нагреванием реагентов, например в интервале между 40 и 60oC, в частности при 50oC. В некоторых случаях благоприятным является проведение названной реакции в присутствии соли металла, такой, например, как безводный хлорид цинка и т.п. в неводном растворителе, в частности в ледяной уксусной кислоте (Chem. Ber, 1965, 98, 3902).
Или же промежуточные соединения формулы (IV-a) могут быть
получены превращением N-арилбензамида (XIII) в хлорид имидоида (XIII-a) галогенированным реагентом, взаимодействием названного галоидного имидоила с цианидной солью и восстановлением полученного таким
образом α-иминонитрила (XIII-b)

Названный галоидный имидоил может быть получен взаимодействием (XIII) с галогенирующим агентом, таким как, например, пентахлорид фосфора, фосфорил хлорид, трихлорид фосфора, трибромид фосфора, тионил хлорид и т.п. в подходящем растворителе, таком, например, как галогенированный углеводород, например дихлорметан, трихлорметан, эфире, например тетрагидрофуране, 1,1'-оксибисэтане, 1,4-диоксане и т.п. или в смеси таких растворителей, или в избытке галогенирующего реагента, не обязательно в смеси с одним или несколькими названными растворителями.
Реакция замещения галоида цианом проводится в реакционноинертном растворителе с цианидной солью, такой, как в способе получения (IV-a) из альдегида (XII). Преимущественно названное замещение проводится в условиях фазового переходного катализа в двухфазной системе растворителей (Synthesis, 1978, р. 894). Затем полученное соединение (XIII-b) восстанавливается до (IV-a) в присутствии соответствующего восстановительного агента, такого, например, как борогидрид натрия, борогидрид лития, цианоборогидрид натрия, литий алюминий гидрид и т.п. восстановительные агенты.
Ряд промежуточных соединений формулы (IV) предлагаются как новые соединения. Интересная
подгруппа новых промежуточных соединений формулы (IV) образуется промежуточными соединениями формулы:
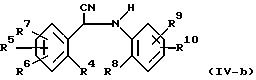
и ее стереохимической изомерной формой, в которой
R4 представляет собой галоид, С1-6-алкил, С1-6-алкокси, гидрокси или нитро;
R5 и R6 каждый независимо друг от друга представляет собой водород, галоид, С1-6-алкил, С1-6-алкокси, нитро, трифторметил, циано, аминометил, карбоксил, С1-4 -алкоксикарбонил, С1-4-алкилкарбонил, аминокарбонил или гидрокси, является водородом или галоидом;
R8 представляет собой С1-6-алкилокси, нитро, трифторметилокси, 2,2,2-трифторэтокси, (трифторметил)карбонил, аминокарбонил, (циклопропил)карбонил или радикал С1-6-алкил-(С=Y)-, гдеY- представляет собойO,N-OH,N-OCH3,N-NH2 или N-N(CH3)2, R9 и R10 каждый независимо является водородом, галоидом, С1-6-алкилом, С1-6-алкокси, нитро, гидрокси, трифторметилокси, 2,2,2-трифторэтокси, (трифторметил)карбонилом, аминокарбонилом, (циклопропил)карбонилом или радикалом С1-6-алкил (С=Y)-, гдеY представляет собойO,N-OH,N-OCH3, N-NH2 илиN-N(CH3)2, при условии, что R8 не является 2-метокси, когда R4 является хлором, R5 является 6-хлор, R6, R7 и R9 являются водородом, а R10 является водородом или 5-метилом.
Промежуточные соединения формулы (IV) могут быть просто получены из соответствующих производных карбоксильных кислот, где α имеет значение, определенное в формуле (VII), взаимодействием с амином HNR1R2 (IX).

Или же промежуточные соединения (VI), где R1 и R2 являются водородом, могут быть получены гидролизом промежуточного соединения (XV) при использовании последующих способов, описанных для получения соединения (I-b).
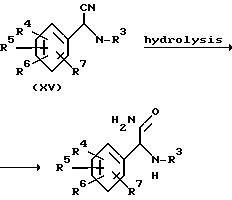
Альтернативный метод превращения промежуточных соединений формулы (XV) в промежуточное соединение формулы (VI-а) включает перемешивание промежуточного соединения формулы (XV) в спирте, таком как метанол, в присутствии кетона, такого как ацетон или циклогенсанон (R-(C=O)-R), и каталитического количества основания, такого как метилат натрия и т.п. Полученное таким образом циклическое промежуточное соединение (XVI), которое может перегруппироваться в промежуточное соединение формулы (XVII), подвергается затем гидролизу в промежуточное соединение формулы (VI-а) нагреванием в воде.

Очевидно, что промежуточные соединения формулы (XV) могут легко получаться из бензальдегидов формулы (XII) взаимодействием с соответствующим амином R3-NH2 и цианидом, как при получении промежуточных соединений (IV-а).
Некоторые анилины формулы (XI) являются новыми и были специально приготовлены для использования в данном изобретении. Например, промежуточные соединения формулы (XI-а), в которых R8 представляет собой С1-6-алкил-(С= О)-, могут быть получены из нитрила (XVIII-a) или карбоксильной кислоты формулы (XVIII-b).
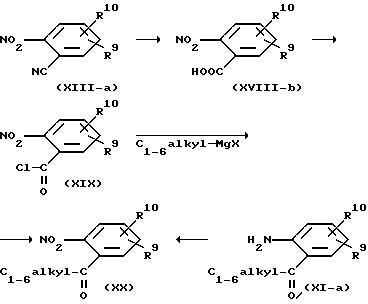
через хлористый ацетил (XIX), который взаимодействует с органометаллическим реагентом, таким как С1-6-алкил магний галоид или магниевая соль диалкил-2-С1-6-алкил-1,3-пропандиоата. Последнее соединение дает производное малонового эфира, которое превращается в (XX) кислотным гидролизом и сопутствующим декарбоксилированием. Восстановление нитрогруппы дает промежуточное соединение (XI-а).
Промежуточные соединения (XI-b), в которых R8 представляет собой (циклопропил)карбонил, могут быть получены из 2-фторбензолнитрила (XXI) взаимодействием с реактивом Гриньяра, полученным из циклопропан бромида с последующим нуклеофильным ароматическим замещением фтора и гидрогенолизом бензильной группы.

Альдегиды формулы (XII) обычно получаются из соответствующих толуольных производных (XXII) бромированием, гидролизом и окислением.
Соединение формулы (I) показывает антиретровирусные свойства по отношению к вирусу человеческого иммунодефицита (HIV СПИД), известному также как LAV, HILV-III или ARV, который является этилогическим агентом синдрома приобретенного иммунодефицита (AIDS) у людей.
Вирус HIV предпочтительно инфицирует человеческие Т-4 клетки и разрушает их или изменяет их нормальную функцию, особенно координацию иммунной системы. В результате инфицированный пациент имеет постоянно снижающееся число клеток Т-4, которые, кроме того, ведут себя ненормально. Таким образом, иммунологическая защитная система не в состоянии бороться с инфекциями и новообразованиями и инфицированный HIVом человек обычно умирает от патогенных инфекций, таких как пневмония, или от злокачественных опухолей. Другие состояния, связанные с HIV инфекцией, включают тромбоцитопению, Kaposi's саркому и инфекцию центральной нервной системы, характеризующуюся прогрессирующей демиелинизацией, приводящей к слабоумию и таким симптомам, как прогрессирующая дисартрия, атаксия и дезориентировка. HIV инфекция может также сопровождаться периферийной невропатией, прогрессирующей общей лимфаденопатией (PGL) и родственным AID комплексом (ARC).
Благодаря их антиретровирусным свойствам, особенно их свойствам против HIV вируса, соединения формулы (I), их фармацевтические приемлемые соли и их стереохимические изомерные солевые формы являются полезными для лечения людей, инфицированных HIV, и для профилактики людей. Обычно соединения данного изобретения могут быть полезными для лечения теплокровных животных, инфицированных вирусами, наличие которых осуществляется или зависит от фермента обратной транскриптазы.
Состояния, которые могут быть предотвращены или вылечены соединениями данного изобретения, особенно состояния, связанные с HIV и другими патогенными ретровирусами, включают AIDS, родственный AID комплекс (ARC), прогрессирующая общая лимфаденопатия (PGL), а также хронические заболевания центральной нервной системы, вызванные ретровирусами, такими, например, как вызванное HIV вирусом слабоумие и множественный склероз.
Кроме того, установлено, что промежуточные соединения формулы (IV) также показывают антиретровирусные свойства, в частности против HIV.
Ввиду своих фармакологических свойств рассматриваемые соединения могут быть приготовлены в ряде различных фармацевтических форм с целью их приема. Эти фармацевтические формы или композиции рассматриваются как новые и, следовательно, составляют другой аспект данного изобретения. Еще один аспект изобретения составляет также получение названных композиций. Для приготовления фармацевтических композиций данного изобретения эффективное количество активного ингредиента при тщательном перемешивании смешивается с фармацевтически приемлемым носителем. Желательно, чтобы эти фармацевтические композиции соответствовали унитарным дозированным формам для приема оральным способом, ректальным, чрезкожно или для парентеральных инъекций. Например, для приготовления композиций в оральной дозированной форме может быть использована любая из обычно применяемых фармацевтических сред, такая как, например, вода, гликоли, масла, спирты и т.п. в случае жидких препаратов для орального приема такие как суспензии, сиропы, элексиры и растворы, или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие, раздробляющие агенты и т. п. в случае порошков, пилюль, капсул и таблеток. Вследствие легкости использования для приема таблетки и капсулы являются наиболее благоприятной дозированной формой для орального приема. В композициях для парентерального приема носитель обычно представляет собой стерилизованную воду, по крайней мере в большей части, хотя могут быть использованы и другие ингредиенты, например способствующие растворению. Например, могут быть приготовлены растворы для инъекций, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Могут быть приготовлены суспензии для инъекций, в которые введены соответствующие жидкие носители, суспендирующие агенты и т.п. В композициях, пригодных для чрезкожного применения, носитель, не обязательно включает агент, усиливающий проникающую способность, и/или подходящий увлажняющий агент, не обязательно в сочетании с соответствующими добавками.
Особенно благоприятными при создании упомянутых фармацевтических композиций в единичных дозированных формах являются легкость их приема и единообразие дозировок. Под единичной дозированной формой подразумеваются физические дискретные единицы, пригодные в качестве унитарных доз. При этом рассчитывается, что каждая единица содержит предварительно определенное количество активного ингредиента, достаточное для создания желаемого терапевтического эффекта в сочетании с требуемым фармацевтическим носителем. Примерами таких единичных дозированных форм являются таблетки (включая таблетки с отметками и покрытые таблетки), капсулы, пилюли, порошки в обертках, облатки, растворы для инъекции или суспензии и т.п. и их многочисленные сегрегации.
Данное изобретение касается также способа лечения теплокровных животных, страдающих от названных вирусных заболеваний, приемом антивирусного количества соединений формулы (I), его фармацевтически приемлемой кислотно-аддитивной соли или ее стереоизомерной формы. Специалисту в области лечения HIV инфекции легко определить эффективное ежедневное количество из представленных здесь результатов испытаний. Обычно предполагается, что эффективное ежедневное количество может составлять от 0,01 до 50 мг/кг веса тела, более предпочтительно от 0,1 до 10 мг/кг веса тела. Это может соответствовать приему требуемой дозы в виде двух, трех, четырех или большего числа субдоз через соответствующие интервалы в течение всего дня. Названные субдозы могут быть приготовлены в виде единичных дозированных форм, содержащих, например, от 1 до 1000 мг, в частности от 5 до 200 мг активного ингредиента на единичную дозированную форму.
Очевидно, что названное эффективное дневное количество может быть уменьшено или повышено в зависимости от реакции подвергаемого лечению субъекта и/или в зависимости от оценки врачом прописываемых соединений данного изобретения.
Экспериментальная часть
А. Получение промежуточных соединений
Пример 1. К перемешиваемой смеси из 43,8 частей 2,6-дихлорбензальдегида и 325
частей уксусной кислоты по каплям добавлялось 35,3 части 2-метоксибензамина. Через 15 минут по каплям добавлялся раствор из 20,3 частей цианида калия в 35 частях воды при поддержании температуры ниже
30oC. Перемешивание продолжалось в течение 20 минут при комнатной температуре. Осадок отфильтровывался и перекристаллизовывался из 2-пропанола (2х). Продукт отфильтровывался и сушился,
давая 35,3 частей 2-(2,6-дихлорфенил)-2-[(2-метоксифенил)амино]ацетонитрила; т.пл. -117,5oC (промежуточное соединение 1).
Пример 2. К перемешиваемой и охлаждаемой (на ледяной бане) смеси 44 частей 2,6-дихлорбензальдегида и 500 частей уксусной кислоты добавлялись 27,6 частей 2-нитробензамина, 50 частей хлорида цинка (II) и 16,3 частей цианида калия. Перемешивание при 50oC продолжалось в течение 17 часов. После охлаждения реакционная смесь выливалась в 1000 частей воды. Осадок отфильтровывался и растирался в порошок с 2,2'-оксибиопропаном. Продукт отфильтровывался, промывался 2,2'-оксибиопропаном и сушился, давая 45,9 частей 2-(2,6-дихлорфенил)-2-[(2-нитрофенил)амино] ацетонитрила; т. пл. -194,8oC (промежуточное соединение 2).
Пример 3. Смесь 8,75 частей 2,6-дихлорбензальдегида, 5,4 частей 1-(2-аминофенил)-1-этанона и 105 частей уксусной кислоты перемешивалась в течение 1/2 часа при комнатной температуре. Добавлялось 3,26 части цианида калия, и перемешивание продолжалось в течение 20 часов. Реакционная смесь разбавлялась водой. Осадок отфильтровывался, промывался водой и перекристаллизовывался из ацетонитрила. Продукт отфильтровывался и высушивался, образуя 961 частей (71,3% ) (±)-α-[(2-ацетилфенил)амино]-2,6- дихлорбензолацетонитрила; т.пл. - 178,7oC (промежуточное соединение 3).
Пример 4.
а) Смесь 2 частей 4-этил-2-нитробензолкарбонитрила, 31,6 частей этанола, 27,8 частей перекиси водорода (30%) и 1,7 мл 6 н. гидроокиси натрия перемешивалась в течение 1 часа при комнатной температуре. Реакционная смесь разбавлялась водой, и продукт экстрагировался дихлорметаном (3х). Соединенные экстракты промывались водой, сушились, фильтровались и выпаривались. Остаток кристаллизовался из 2,2'-оксибиопропана. Продукт отфильтровывался и высушивался, образуя 1,3 частей (60,9%) 4-этил-2-нитробензамида; т.пл. -175,0oC (промежуточное соединение 4).
b) К перемешиваемой и нагреваемой (80oC) смеси из 29,2 частей промежуточного соединения 4 и 234 частей серной кислоты (75%) по порциям добавлялись 53 части нитрита натрия. Общая смесь перемешивалась в течение 10 минут при комнатной температуре и затем разбавлялась водой. Осадок отфильтровывался, промывался водой и высушивался, образуя 26 частей (88,8%) 4-этил-2-нитробензойной кислоты; т.пл. -111,2oC (промежуточное соединение 5).
с) Смесь из 25,5 частей промежуточного соединения 5 и 259,2 частей тионилхлорида перемешивалась в течение 5 часов при температуре дефлегмации. Реакционная смесь выпаривалась, а остаток испарялся с метилбензолом (2х), образуя 27,8 частей (100%) 4-этил-2-нитробензолкарбонилхлорида (промежуточное соединение 6).
d) К перемешиваемой смеси 3,4 частей магниевых стружек, 2,0 частей этанола и 2,2 частей трихлорметана добавлялось по каплям 35,5 частей 1,1'-оксибисэтана. При температуре дефлегмации по каплям добавлялась смесь 22,4 частей диэтил-1,3-пропандиоата, 11,9 частей этанола и 10,7 частей 1,1'-оксибисэтана, после 3 часов перемешивания раствор 27,8 частей промежуточного соединения 6 и 10,7 частей 1,1'-оксибисэтана. Перемешивание при температуре дефлегмации продолжалось в течение 1 часа. После охлаждения по каплям добавлялось 91,4 частей серной кислоты (8%). Органический слой отделялся, а водный слой экстрагировался метилбензолом (2х). Объединенные органические слои промывались водой, сушились, фильтровались и выпаривались. Остаток кипятился с обратным холодильником в течение 2 часов в смеси из 84 частей уксусной кислоты, 18,4 частей серной кислоты и 45 частей воды. После охлаждения общая смесь разбавлялась водой и экстрагировалась 2,2'-оксибиспропаном (2х). Соединенные экстракты промывались NaOH (10%) и водой, сушились, фильтровались и выпаривались, образуя 22 части (87,6% ) 1-(4-этил-2-нитрофенил)этанона (промежуточное соединение 7).
e) Смесь 22 частей промежуточного соединения 7,2 частей раствора тиофена в метаноле 4% и 316 частей метанола подвергалась гидрогенизации при нормальном давлении и 50oC в присутствии 2 частей угольно-палладиевого катализатора 10% После того, как было растворено рассчитанное количество водорода, катализатор отфильтровывался, а фильтрат упаривался. Остаток очищался на хроматографической колонке (силикагель; CH2Cl2 80:20). Элюент нужной фракции выпаривался, образуя 16 частей (89,1% ) 1-(4-этил-2-этилфенил)этанона (промежуточное соединение 8).
f) Смесь 5,25 частей (2,6-дихлорфенил)метанона, 4,08 частей промежуточного соединения 8 и 105 частей уксусной кислоты перемешивалась при комнатной температуре в течение 2 часов. Добавлялось 1,96 частей цианида калия, и перемешивание продолжалось в течение 20 часов. Реакционная смесь выливалась в воду и продукт экстрагировался дихлорметаном. Экстракт промывался NaOH (10%) и водой, сушился, фильтровался и выпаривался. Остаток очищался на хроматографической колонке (силикагель; CH2Cl2 80:20). Элюент нужной фракции выпаривался, а остаток кристаллизовался из 2,2'-оксибиопропана, образуя 4,28 частей (49,3% ) (±)-a-[(2-ацетил-5-этилфенил)амино]-2,6-дихлорбензолацетонитрила; т.пл. 111,3oC (промежуточное соединение 9).
Пример 5.
а) К раствору 117 частей (4-трифторметилфенил)метанона в 136 частях метанола порционно добавлялось 13,1 частей тетрагидробората натрия в атмосфере азота при охлаждении льдом. После перемешивания в течение 18 часов при комнатной температуре реакционная смесь выливалась в воду, которая подкислялась 20%-ной соляной кислотой. Продукт экстрагировался 2,2'-оксибиопропаном (2х), и объединенные экстракты промывались водой, сушились, фильтровались и выпаривались, образуя 130 частей (100% ) 4-(трифторметил)бензолметанола (промежуточное соединение 10).
b) Смесь 125 частей промежуточного соединения 10 и 821 части 1-метил-2-пирролидона подвергалась гидрогенизации при нормальном давлении и комнатной температуре в присутствии 2 частей угольно-палладиевого катализатора на древесном угле 10% После того, как было растворено рассчитанное количество водорода, катализатор отфильтровывался и фильтрат упаривался. Остаток перегонялся (1•105 Па, 90-100oC), давая 75 частей (66,0%) 1-трифторметил-4-метилбензола (промежуточное соединение 11).
с) Смесь 29,4 частей промежуточного соединения 11 и 4,1 части трихлорида сурьмы перемешивалась в течение 6 часов при 60oC в условиях пропускания хлора. Реакционная смесь разбавлялась 50 частями воды и перемешивалась еще 15 минут при 60oC. Продукт экстрагировался 2,2'-оксибиопропаном, и экстракт промывался водой, сушился, фильтровался и выпаривался. Остаток растворялся в гексане, и общий продукт фильтровался и выпаривался. Остаток перегонялся (1,3•105 Па, 100-105oC), давая 29 частей (61,1%) 1,3, 4-трихлор-5-трифторметил-4-метилбензола (промежуточное соединение 12).
d) Смесь 29 частей промежуточного соединения 12, 30 частей N-бромсукциимида, 2,5 частей перекиси бензоила и 795 частей тетрахлорметана перемешивалась в течение 6 часов при температуре дефлегмации. Реакционная смесь промывалась водой (2х), сушилась, фильтровалась и выпаривалась. Остаток кипятился в гексане, и общая смесь фильтровалась и выпаривалась, образуя 40 частей (100%) 2-бромметил-1,3,4-трихло-5-трифторметилбензола (промежуточное соединение 13).
e) Смесь 40 частей промежуточного соединения 13, 600 частей воды и 27,6 частей карбоната калия перемешивалась в течение 21 часа при температуре дефлегмации. После охлаждения осадок отфильтровывался и перекристаллизовывался из гексана. Продукт отфильтровывался и сушился, давая 15 частей (48,8%) 2,3,6-трихлорметилбензолметанола (промежуточное соединение 14).
f) К перемешиваемой смеси, содержащей 80 частей воды, 79,12 частей серной кислоты (конц. ) и 1 часть бензилтриэтиламмоний хлорида добавлялось 6,6 частей бихромата калия и по каплям раствор, содержащий 15 частей промежуточного соединения 14 в 160 частях дихлорметана. Перемешивание при комнатной температуре продолжалось в течение 4 часов. Реакционная смесь разбавлялась 400 частями воды, и продукт экстрагировался дихлорметаном (2х). Соединенные экстракты промывались водой, сушились, фильтровались и выпаривались, давая 16 частей (100%) [2,3,6-трихлор-4-(трифторметил)фенил]метанона (промежуточное соединение 15).
g) Смесь 4,1 части 1-(2-аминофенил)этанона, 11 частей промежуточного соединения 15 и 105 частей уксусной кислоты перемешивалась при комнатной температуре в течение 2 часов. Было добавлено 2,6 частей цианида калия, и перемешивание при комнатной температуре было продолжено в течение 18 часов. Реакционная смесь экстрагировался дихлорметаном (2х). Соединенные экстракты промывались Na2CO3 (5% ), сушились, фильтровались и выпаривались, давая 16 частей (100%) 2,3,6-трихлор-4-(трифторметил)-a-[[2-(метилкарбонил)фенил]амино]бензолацетонитрила (промежуточное соединение 16).
Пример 6.
а) К перемешиваемой смеси 10 частей магния и 72 частей тетрагидрофурана добавлялось порциями 50 частей бромциклопропана в условиях поддержания температуры ниже 10oC. При температуре дефлегмации добавлялся раствор из 50 частей 2-фторбензолкарбонитрила в 72 частях тетрагидрофурана. Перемешивание при температуре дефлегмации продолжалось до завершения реакции. Реакционная смесь охлаждалась льдом (0-5oC) и разлагалась смесью льда, воды и уксусной кислоты. Продукт экстрагировался трихлорметаном (3х), и объединенные экстракты промывались водой (2х), сушились, фильтровались и выпаривались. Остаток перегонялся (1,3 Па, 78-103oC) (2х), образуя 19,8 частей (29,4%) циклопропил-2-(фторфенил)метанона; т.кип. 100-103oC (при 1,3 Па) (промежуточное соединение 17).
b) К раствору из 50 частей промежуточного соединения 17, 34,5 частей бензолметанамина и 218 частей метилбензола добавлялось 45 частей карбоната натрия. Общая смесь перемешивалась в течение 5 дней при температуре дефлегмации. Реакционная смесь фильтровалась, а органический слой отделялся, промывался водой и NaCl (насыщ.), высушивался, фильтровался и упаривался. Остаток растирался в порошок в метаноле, давая 33 части (41,6%) циклопропил-[2-[(фенилметил)амино]фенил]метанона (промежуточное соединение 18).
с) Смесь из 23 частей промежуточного соединения 18 и 223 частей тетрагидрофурана подвергалась гидрогенизации при нормальном давлении и комнатной температуре в присутствии 3 частей палладиевого катализатора на древесном угле 10% После того, как было растворено рассчитанное количество водорода, катализатор отфильтровывался, а фильтрат упаривался. Остаток очищался на хроматографической колонке (силикагель; гексан/(C2H5)2 10:90). Элюент нужной фракции выпаривался, образуя 14,1 частей (95,6%) (2-аминофенил)циклопропилметанона (промежуточное соединение 19).
d) К перемешиваемому раствору из 5 частей промежуточного соединения 19 в 105 частях уксусной кислоты добавлялись 6,5 частей (2,6-дихлорфенил)метанола и спустя 1/2 часа 2,4 части цианида калия. Общая смесь перемешивалась при комнатной температуре в течение 41 часа и затем выливалась в воду. Осадок отфильтровывался и перекристаллизовывался из смеси метанола и ацетонитрила, образуя 8,5 частей (79, 4%) (±)-2,6-дихлор-a-[[2-(циклопропилкарбонил)фенил] амино]-бензолацетонитрила; т.пл. 144,1oC (промежуточное соединение 20).
В. Получение конечных соединений
Пример 7. К 200 частям смеси концентрированной серной кислоты и воды (10:1 по объему) по порциям добавлялось 28,5 частей промежуточного соединения 1. После перемешивания в течение 18 часов при
комнатной температуре реакционная смесь выливалась в 200 частей воды со льдом. Осадок фильтровался и растворялся в трихлорметане. Этот раствор промывался водой, сушился, фильтровался и выпаривался.
Остаток кристаллизовался из 2-пропанола. Продукт отфильтровывался и сушился, давая 17,2 частей 2-(2,6-дихлорфенил)-2-[(2-метоксилфенил)амино]ацетамида; т.пл. -192oC (соединение 4).
Пример 8. К перемешиваемым и охлаждаемым (на ледяной бане) 450 частям концентрированной серной кислоты порционно добавлялось 44 части промежуточного соединения 2 в условиях поддержания температуры ниже 15oC. Образующийся раствор перемешивался при комнатной температуре в течение 3 часов и затем выливался в 1000 частей воды со льдом. Осадок фильтровался, промывался водой, растирался в порошок с 2,2'-оксибиспропаном и перекристаллизовывался из уксусной кислоты. Продукт отфильтровывался и сушился, образуя 24 части 2-(2,6-дихлорфенил)-2-[(2-нитрофенил)амино] ацетамида. Разбавление маточного раствора водой дает вторую фракцию продукта осаждения. Продукт отфильтровывается, промывается петролейным эфиром и сушится, образуя дополнительно 6,6 части 2-(2, 6-дихлорфенил)-2-[(2нитрофенил)амино]ацетамида. Общий выход 30,6 частей; т.пл. 182,4oC (соединение 15).
Пример 9.
а) Смесь 3 частей промежуточного соединения 3 и 50,2 частей смеси концентрированной серной кислоты и воды (10:1 по объему) перемешивалась при комнатной температуре в течение 3 часов. Реакционная смесь выливалась в смесь вода-лед. Осадок отфильтровывался, промывался водой и перекристаллизовывался из ацетонитрила. Продукт отфильтровывался и высушивался, образуя 1,9 частей (59,9% ) (±)-a-[(2-ацетилфенил)амино]-2, 6- дихлорбензолацетамида; т. пл. 203,9oC (соединение 22).
b) 0,9 части соединения 22 разделялось на хроматографической колонке (Chiracel OD®; гексан/C2H5OH 80:20). Элюент нужной фракции испаряли, а остаток суспендировали в 2,2-оксибиспропане. Продукт отфильтровывался и высушивался, образуя 0,164 части (18,2%) (-)-α -[(2-ацетилфенил)амино]-2,6-дихлорбензолацетамида; т. пл. 168,8oC; [α]= -64, 83° (конц. 0,1% в CH3OH) (соединение 23).
Пример 10. К перемешиваемому раствору из 2 частей соединения 22 в 3,5 частях 2-пропанола добавлялось 0,59 части пиридина и 0,5 части моногидрохлорида гидроксиламина. После перемешивания в течение 2 часов при температуре дефлегмации реакционная смесь выливалась в воду. Продукт экстрагировался дихлорметаном, и экстракт промывался водой (2х), высушивался, фильтровался и выпаривался. Затем остаток растирался в порошок в петролейном эфире и перекристаллизовывался из ацетонитрила. Продукт отфильтровывался и сушился, давая 1,2 части (±)-(Е)-2,6-дихлор-α-[[2-[1-(гидроксиламино)этил]фенил]амино] бензо лацетамида; т.пл. 114,8oC (соединение 35).
Пример 11. К перемешиваемому раствору из 2 частей соединения 22 в 3,5 частях 2-пропанола добавлялось 0,59 частей пиридина и 0,6 частей моногидрохлорида метоксиамина. После перемешивания в течение 4,5 часов при температуре дефлегмации реакционная смесь охлаждалась и выливалась в воду. Осадок отфильтровывался, промывался водой и перекристаллизовывался из ацетонитрила образуя 1,2 части (54,6%) (±)-(Е)-2, 6-дихлор-a-[[2-[1-(метоксиимино)этил]фенил]амино]бензолацетамида; т.пл. 205,5oC (соединение 36).
Пример 12. К перемешиваемому раствору из 1,8 части соединения 22 в 3,12 частях 2-пропанола добавлялось 0,52 части пиридина и 0,32 части моногидрата гидразина. После перемешивания в течение 12 часов при температуре дефлегмации реакционная смесь экстрагировалась дихлорметаном. Экстракт промывался водой (2х), высушивался, фильтровался и выпаривался. Остаток очищался на хроматографической колонке (силикагель; CH2Cl2/CH3OH 95:5). Элюент нужной фракции выпаривался, а остаток растирался в порошок в 1,1'-оксибисэтане и перекристаллизовывался из ацетонитрила. Продукт отфильтровывался и сушился, давая 0,6 части (32,2%) (± )-(Е)-2,6-дихлор-a-[[2-(1-(гидразиноэтил)фенил] амино]бензолацетамида; т.пл. 175,7oC (соединение 43).
Пример 13.
a) К перемешиваемому и нагреваемому (60oC) раствору из 120 частей соединения (15) в 2100 частях уксусной кислоты добавлялись 252 части соляной кислоты. Перемешивание продолжалось в течение 22 часов при 100oC. Реакционная смесь концентрировалась, осадок отфильтровывался и очищался на хроматографической колонке (ВЭЖХ; силикагель; CH2Cl2/CH3OH 99:1). Элюент нужной фракции выпаривался, давая 48,8 частей (40,6%) (±)-2,6-дихлор-a-[(2-нитрофенил)амино]бензолуксусной кислоты (промежуточное соединение 21).
b) К раствору из 5,1 частей промежуточного соединения 21 в 66,8 частей тетрагидрофурана добавлялось 1,7 части N,N-диэтилэтанамида и по каплям -1,8 части этилхлорформиата. После перемешивания в течение 20 минут при комнатной температуре реакционная смесь выливалась в 90 частей NH4OH (конц.). Растворитель выпаривался, а остаток растворялся в 298 частях трихлорметана. Общая смесь отфильтровывалась, промывалась водой (2х), сушилась, фильтровалась и выпаривалась. Остаток кристаллизовался из ацетонитрила, образуя 1,8 части (35,2%) 2,6-дихлор-a-[(2-нитрофенил)амино]бензолацетамида (соединение 15).
Пример
14
a) Через перемешиваемый раствор, содержащий 5 частей промежуточного соединения 20, 2,2 частей N,N-диэтилэтанамида и 98 частей пиридина, пропускали газообразный сероводород до завершения
реакции, а затем газообразный азот в течение 1 часа. Реакционная смесь выливалась в 200 частей воды, и продукт экстрагировался дихлорметаном. Экстракт сушился, фильтровался и выпаривался. Остаток
растворялся в трихлорметане, и смесь промывалась разбавленной соляной кислотой и выпаривалась. Остаток отфильтровывался и сушился в вакууме при 70oC, давая 4,3 частей (81%) (±)-2,
6-дихлор-a-[(2-циклопропилкарбонил)фенил] амино]бензолэтантиоамид; т.пл. 222,0oC (соединение 58).
b) К перемешиваемому и охлаждаемому (0oC) раствору из 1 части соединения 58 в 18,8 частей N,N-диметилформамида добавляли 4 части раствора 2 н. гидроокиси натрия и 1,3 части перекиси водорода 30% После перемешивания при комнатной температуре в течение ночи осадок отфильтровывался, промывался водой и кристаллизовался из ацетонитрила. Продукт отфильтровывался и сушился в вакууме при 70oC, давая 0,56 части (60%) (±)-2, 6-дихлор-a-[(2-циклопропилкарбонил)фенил]амино]бензолацетамида; т.пл. 196,4oC (соединение 48).
Пример 15.
a) К раствору из 3,7 частей гидрокарбоната натрия в 225 частях воды добавлялись 154 части формальдегида (35% водн.) и 102,0 частей соединения 15. После перемешивания в течение 5 часов при температуре дефлегмации и последующего охлаждения реакционная смесь разбавлялась 1000 частями воды. Общая смесь перемешивалась в течение 1/2 часа при комнатной температуре. Осадок отфильтровывался, промывался водой (3х) и кристаллизовался из метанола. Общая смесь фильтровалась в горячем виде, а фильтрат перемешивался в течение 16 часов при комнатной температуре. Выкристаллизованный продукт отфильтровывался, промывался метанолом (2х) и сушился в вакууме при 40oC, давая 76,2 частей (68,6% ) (±)-2,6-дихлор-N-(гидроксиметил)-a-[(2-нитрофенил)амино]бензолацетамида; т.пл. -175,0oC.
b) Полученное таким образом производное разделялось методом жидкостной хроматографии с использованием хиральной стационарной фазы (Chiralcel ΟΙ®). Хроматографическая система была деаэрирована этанолом. Элюент, состоящий из этанола, подогревался до температуры около 35oC и прокачивался через систему. Рацемическое соединение растворялось в минимальном количестве этанола, нагретого до 30-40oC, и впрыскивалось в хроматографическую систему. Нужные фракции собирались (УФ-определение 240 нм), и растворитель испарялся, давая отдельные энантиомерные производные.
c) Суспензия из 0,53 части полученного остатка и 4 частей 4-метил-2-пентанона сначала перемешивалась в течение 4 часов при температуре дефлегмации, а затем в течение 18 часов при температуре 25oC. Высадившийся продукт отфильтровывался, а фильтрат перемешивался далее при той же температуре. После фильтрации высадившийся продукт соединялся с предыдущей порцией и промывался 0,8 части 4-метил-2-пентанона. Продукт отфильтровывался и сушился в вакууме при 40oC, давая первую фракцию из 0,17 части (34,9%) (-)-2,6-дихлор-a-[(2-нитрофенил)амино] бензолацетамида. Фильтрат упаривался при 40oC и охлаждался при перемешивании на ледяной бане, давая вторую часть в 0,15 (30,8%) (-)-2,6-дихлор-a-[(2-нитрофенил)амино] бензолацетамида. Общий выход: 0,32 части (65,7%) (-)-2,6-дихлор-a-[(2-нитрофенил)амино]бензолацетамида (искл.оп. 100% ) (соединение 25).
Пример 16
a) К перемешиваемому раствору из 3,37 частей N,N,N,
N-тетраметилдиаминометана в 44 частях метилбензола по каплям добавлялся раствор из 2,59 частей ацетилхлорида в 26 частях метилбензола в течение 30 минут при 20-25oC в атмосфере азота. После
окончания реакционная смесь перемешивалась еще в течение 20 минут. К предыдущей смеси добавлялось 10,21 частей соединения 15 и 78 частей метилбензола, и смесь перемешивалась в течение 5 часов при
80oC. Реакционной смеси давали охладиться до 20oC, осажденный продукт отфильтровывали и сушили в вакууме при 40oC, получая 11,66 частей (97,8%)
N-[(диметиламино)метил]-2,6-дихлор-a-[(2-нитрофенил)амино]бензолацетамида.
b) Полученное таким образом производное разделялось методом жидкостной хроматографии с использованием хиральной стационарной фазы (Chiralcel ΟΙ®). Хроматографическая система была деаэрирована смесью гексана и этанола (80:20 по объему). Элюент, состоящий из смеси гексана и этанола (80:20 по объему), подогревался до температуры около 35oC и прокачивался через систему. Рацемическое соединение растворялось в минимальном количестве этанола, нагретого до 30-40oC, и впрыскивалось в хроматографическую систему. Нужные фракции собирались (УФ-определение 240 нм), и растворитель испарялся, давая отдельные энантиомерные производные.
c) 0,2 части нужной фракции и 8 частей 4-метил-2-пентанона кипятились с обратным холодильником в течение 5 часов на масляной бане при 120oC. Охлаждали до комнатной температуры, высадившийся продукт отфильтровывали, промывали 2,4 частей 4-метил-2-пентанона и сушили в вакууме при 40oC. Фильтрат упаривали при 50oC и очищали на хроматографической колонке (ВЭЖХ; силикагель; CH2Cl2/CH3OH 99:1 _→ 90:10), давая 0,1 части (58,8%) (-)-2,6-дихлор-α-[(2-нитрофенил)амино]бензолацетамида (искл.с.ш. 82%) (соединение 25).
Пример 17.
a) К перемешанной смеси из 585 частей хлорметана и 72 частей метанола, насыщенной газообразным хлористым водородом, добавлялось 96,66 частей промежуточного соединения 2. Реакционная смесь перемешивалась в течение 6,5 часов при 0-5oC во время пропускания газообразного хлористого водорода. После охлаждения до -10o C добавлялось 240 частей метанола и 260 частей дихлорметан. Реакционная смесь нейтрализовывалась и подщелачивалась 365 частями N, N-диэтилэтанамина до pH 9. После выпаривания в вакууме при 50oC остаток перемешивался в 668 частях тетрагидрофурана. Осадок отфильтровывался, промывался тетрагидрофураном, и фильтрат выпаривался до сухого состояния в вакууме при 50oC. Остаток кипятился в 280 частях 2-пропанола в атмосфере азота. Продукт оставляли кристаллизоваться (2 часа, 0-5oC), отфильтровывали и промывали 56 частями 2-пропанола, получая 70,62 частей (66,5% ) (±)-О-метил-2,6-дихлор-a-[(2-нитрофенил)амино]бензолэтанимидата.
b) Полученное таким образом производное разделялось методом жидкостной хроматографии с использованием хиральной стационарной фазы (Chiralcel OI®). Хроматографическая система обезвоживалась этанолом. Элюент, состоящий из этанола, подогревался до температуры около 35oC и прокачивался через систему. Рацемическое соединение растворялось в минимальном количестве этанола, нагретого до 30-40oC, и впрыскивалось в хроматографическую систему. Нужные фракции собирались (УФ-определение 240 нм), и растворитель испарялся, давая отдельные энантиомерные производные.
c) 0,1 части полученного выше остатка растворялось в 5,6 частей дихлорметана при 20oC. Общая смесь нагревалась до 45oC, и добавлялось несколько капель метанола, насыщенного 9,8 н. газообразным хлористым водородом. После перемешивания в течение 2 часов при температуре дефлегмации (45-50oC) реакционная смесь выпаривалась до сухого состояния, образуя 0,085 части (85%) (-)-2,6-дихлор-α-[(2-нитрофенил)амино] бензолацетамида (искл. с. л. 84% ) (соединение 25).
Пример 18.
a) К перемешанному и охлажденному (15oC) раствору из 350 частей 2,6-дихлорбензальдегида и 1600 частей уксусной кислоты добавлялся по каплям раствор 162,8 частей цианида калия в воде в течение 30 минут при 15-20oC. После перемешивания в течение 16 часов при 20oC добавлялось 1000 частей дихлорметана и 1000 частей воды. Отделенный водный слой трижды промывался дихлорметаном. Объединенный органический слой промывался водой, сушился, фильтровался и выпаривался при 40oC, образуя 396 частей 2,6-дихлор-a-гидроксибензолацетонитрила.
b) 396 частей приведенного выше продукта соединялось с 80 частями метанола и перемешивалось при 20oC. Добавлялось 800 частей метанола, насыщенного аммиаком, и полученная смесь кипятилась с обратным холодильником в течение 4 часов (40oC _→ 60oC). После выпаривания в вакууме остаток растворяли в 520 частях дихлорметана и 200 частях воды. Отделенный органический слой дважды промывался 200 частями воды, сушился и охлаждался до 10oC. Общая смесь подкислялась 280 частями 2-пропанола, насыщенного соляной кислотой. Выкристаллизовавшийся продукт отфильтровывался, растворялся в 390 частях дихлорметана и 200 частях воды и насыщался аммиаком до pH > 9. Отделенный органический слой промывался 200 частями воды, сушился, фильтровался и выпаривался, образуя 245 частей α-амино-2,6-дихлорбензолацетонитрила.
c) Полученное таким образом производное разделялось методом жидкостной хроматографии с использованием хиральной стационарной фазы (Chiralcel OI®). Хроматографическая система обезвоживалась смесью гексана и этанола (80:20 по объему). Элюент, состоящий из смеси гексана и этанола (80:20 по объему), прокачивался через систему. Рацемическое соединение растворялось в минимальном количестве этанола и впрыскивалось в хроматографическую систему. Нужные фракции собирались (УФ-определение 240 нм), и растворитель испарялся, давая отдельные энантиомерные производные.
d) Выделенное энантиомерное производное перемешивалось в 52 частях дихлорметана при 10-15oC. Добавлялся 2-пропанол, насыщенный соляной кислотой, и после перемешивания в течение 1 часа при 20oC осадившийся продукт отфильтровывался, промывался 26 частями дихлорметана и растворялся в 260 частях дихлорнатана и 100 частях воды. Раствор насыщался аммиаком до pH > 9. Выделенный органический слой промывался, высушивался и растворялся в 280 частях дихлорметана. После охлаждения до 0oC по каплям добавлялось 26,8 частей серной кислоты. Общий раствор перемешивался в течение 18 часов при 20oC, и добавлялось 100 частей воды. Реакционная смесь обрабатывалась гидроокисью аммония при <20oC. Отделенный водный слой дважды экстрагировался дихлорметаном. Объединенный экстракт промывался водой, сушился, фильтровался и выпаривался при <45oC, образуя 8,1 частей (82,6%) α-амино-2, 6-дихлорбензолацетамида.
e) Смесь 0,092 части a-амино-2,6-дихлорбензолацетамида, 0,032 части 2-фторнитробензола и 1 части 1,3-диметил-2-имидазолидинона перемешивалась в течение 6 часов при 140oC. Продукт экстрагировался, и экстракт высушивался, фильтровался и выпаривался при 100oC и 133-266 Па. Остаток очищался на хроматографической колонке (ВЭЖХ; силикагель; CH2Cl2/CH3OH 99:1 --> 90:10), давая 0,12 части (-)-2,6-дихлор-a-[(2-нитрофенил)амино] бензолацетамида (искл.ошиб. 90%).
Все другие соединения, приведенные в табл. 1 и 2, были получены в соответствии со способами, описанными в примерах 7-18.
С. Фармакологический пример
Пример 19. Для оценки in vitro
действия антивирусных агентов HIV использовался быстрый, чувствительный автоматизированный аналитический способ. Преобразованная HIV-1-Т4-клеточная линия МТ-4, которая, как было показано ранее
(Koyanagu и др. //Int. J. Cancer, 1985, 36, 445-451), является высокочувствительной и восприимчивой к HIV инфекции, служит в качестве мишеневой клеточной линии. Конечной точкой служило ингибирование
эффекта заболевания клетки, вызванного HIV вирусом. Жизнеспособность клеток, инфицированных как HIV вирусом, так и ложно инфицированных, анализировалась спектрофотометрически через снижение на месте
3-(4,5-диметилтиазол2-ил)-2,5-дифенилтетразол бромида (МТТ). 50% -ная цитотоксическая доза (CD50 мкг/мл определялась как концентрация соединения, которая снижает абсорбцию ложно
инфицированного контрольного образца на 50% Процент защиты, достигаемый соединением по отношению к HIV инфицированным клеткам, рассчитывался по следующей формуле:
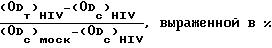
при этом (ODт)HIV представляет собой оптическую плотность, измеренную при данной концентрации испытуемого соединения в HIV инфицированных клетках; (ODс)моск является оптической плотностью, измеренной для контрольных клеток, не подвергшихся воздействию ложной инфекции; все значения оптических плотностей определялись при 540 нм. Доза, при которой достигается 50%-ная защита в соответствии с приведенной выше формулой, обозначалась как 50%-ная эффективная доза (ED50 в мкг/мл). Отношение CD50 к ED50 обозначалось как селективный показатель (SI). Было показано, что все соединения приведенной выше таблицы имеют селективный показатель, больший, чем 1, то есть эффективно подавляют вирус HIV-1 при дозах ниже цитологической дозы. Конкретные значения указанных показателей перечислены в табл.3.
Реферат
Использование: в качестве лекарственного препарата. Соединение имеет формулу
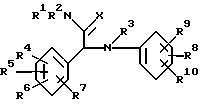
фармацевтически приемлемой кислотно-аддитивной солевой формы или ее стереохимически изомерной формы, в которой R1 и R2 каждый раз независимо являются водородом, С1-6-алкилом или С3-6-циклоалкилом; или R1 или R2 вместе с атомом азота, к которому присоединены названные R1 и R2, могут образовывать пирролидинил, пиперидинил, морфолинил, пиперазинил или 4-С1-4-алкилпиперазинильную группу; X является кислородом или серой; R3 является водородом или С1-6 алкилом; R4, R5 и R6 каждый независимо друг от друга являются водородом, галогеном, С1-6-алкилом, С1-6 -алкилокси, нитро, трифторметилом, циано, аминометилом, циано, аминометилом, карбоксилом, С1-4-алкилоксикарбонилом, С1-4-алкилкарбонилом, аминокарбонилом или гидроксигруппой; R7 представляет собой водород или галоид; R8, R9 и R10 каждый независимо друг от друга являются водородом, галоидом, С1-6-алкилом, С1-6 -алкилокси, нитро, гидрокси, трифторметокси, 2,2,2-трифторэтокси, (трифторметил)карбонилом, аминокарбонилом, (циклопропил)карбонилом или радикалом С1-6-алкил-(С=Y)-, где =Y представляет собой =O, =N, -OH, =N-OCH3, = N-NH2 или =N-N(CH3)2 при условии, что: 1) R1 не является н-пропилом, когда R1, R2, R3, R6, R7, R9 и R10 являются водородом, R8 представляет собой 4-этокси и X представляет собой кислород; 2) X не является серой, когда R1, R2, R3, R6, R7, R9 и R10 представляют собой водород, а R4 и R5 представляют 3,4-диметоксигруппу. 6 с. и 8 з.п.ф-лы, 3 табл.
Формула

или его фармацевтически приемлемая кислотно-аддитивная соль, или его стереоизомер,
где R1 и R2 каждый независимо представляет собой водород, C1-C6-алкил или С3-С6- циклоалкил;
Х О или S;
R4, R5 и R6 каждый независимо представляет собой водород, С1-С6-алкил или С1-С6- алкокси, нитро, трифторметил, галоген, циано, аминометил, карбоксил, С1-С4-алкилоксикарбонил, С1-С4-алкилкарбонил, аминокарбонил или гидрокси;
R7 представляет собой водород или галоген;
R8, R9 и R10 каждый независимо представляет собой водород, С1-С6-алкил, С1-С6-алкокси, галоген, нитро, гидрокси, трифторметокси, 2,2,2-трифторэтокси, (трифторметил)карбонил, аминокарбонил, (циклопропил)карбонил или радикал формулы С1-С6-алкил (СY)-,
где Y представляет собой О, N-OH, N-OCH3, N NH2, N - N(CH3)2, при условии, что 1) когда R2, R4, R5, R6, R7, R9 и R10 представляют собой водород, R8 представляет собой 4-этокси и X кислород, то R1 отличен от н-пропила; 2) когда R1, R2, R6, R7, R8, R9 и R10 представляют собой водород и R4 и R5 представляют собой 3,4-тиметокси, то X отличен от серы.
его фармацевтическая приемлемая кислотно-аддитивная солевая форма или его стереохимически изомерная форма, где R4 представляет собой галоид, С1-С6-алкил, С1 -С6-алкокси или нитро; R5 и R6 каждый независимо представляют собой водород, галоген, С1-С6-алкил, C1-С6-алкокси, нитро, трифторметил, циано, аминометил, карбоксил, С1-С6-алкилоксикарбонил, С1-С6-алкилкарбонил, аминокарбонил или гидрокси; R7 является гологеном или водородом;
R8 представляет собой С1-С6-алкилокси, нитро, трифторметокси, 2,2,2-трифторэтокси, (трифторметил)карбонил, аминокарбонил, (циклопропил)карбонил, или радикал С1-С6-алкил -(С Y)-, гдеY представляет собойО,N-ОН,N-ОСН3N-NH2 или N N(CH3)2 и R9 и R10 каждый независимо друг от друга представляют собой водород, галоид, С1-С6-алкил, С1-С6-алкилокси, нитро, гидрокси, трифторметокси, (циклопропил)карбонил или радикал С1-С6-алкил (С=Y)-, гдеY равняется O,N-OH,N-OCH3,N-NH2
илиN-N(CH3)2, при условии, что R8 не равно 2-метокси, когда R4 представляет собой хлор, R5 является 6-хлор; R6, R7 и R9 являются водородом, а R10 является водородом или 5-метилом.
R8 представляет собой С1-С6-алкокси, трифторметокси, нитро или С1-С6-алкилкарбонил; R9 и R10 представляет собой водород, галоид или С1-С6 -алкил.
его фармацевтически приемлемая кислотно-аддитивная солевая форма или его стереохимически изомерная форма, где R4 представляет собой галоид, С1-С6-алкил, С1-С6-алкокси, нитро или гидрокси; R5 и R6 каждый независимо один от другого представляют собой водород, галоген, С1-С6-алкил, С1-С6-алкокси, нитро или гидрокси; R7 является С1-С6-алкилокси, нитро, трифторметилокси, 2,2,2-трифторэтокси, (трифторметил) карбонилом, аминокарбонилом, (циклопропил) карбонилом или радикалом С1-С6-алкил-(С=Y-, гдеY представляет собойО,N -ОН, N -ОСН3, илиN N(СН3)2 и R8 и R9 каждый независимо один от другого является водородом, галоидом, С1-С6-алкилом, С1 -С6- алкилокси, нитро, гидрокси, трифторметокси, 2,2,2-трифторэтокси, (трифторметил)карбонилом, аминокарбонилом, (циклопропил)карбонилом или радикалом С1-С6-алкил-(С= Y)-, где Y представляет собойO,N-OH,N-ОСН3 илиN-N (СН3)2, при условии, что R7 не равно 2-метокси, когда R4 является хлором; R5 является 6-хлором; R6 и R8 представляет собой водород и R9 является водородом или 5-метилом.
где R4, R5, R6 каждый независимо представляет водород С1-С6-алкил или С1-С6-алкилокси, нитро, трифторметил, галоген, циано, аминометил, карбоксил, С1-С4 -алкилоксикарбонил, С1-С4-алкилкарбонил, аминокарбонил или гидрокси; R7
водород или галоген; R8, R9 и R10 каждый независимо означает водород, С1-С6-алкил, С1-С6-алкилокси, нитро, гидрокси, трифторметокси, 2,2,2-трифторэтокси, (трифторметил) карбонил, аминокарбонил, (циплопропил)карбоноил или радикал формулы С1-С6-алкил(С=Y), где Y означает О, N-ОН, N-ОСН3, N-NH2, N-N(CH3)2, отличающийся тем, что нитрил формулы

где R4-R10 как указано выше,
обрабатывают концентрированной серной кислотой, возможно, в присутствии небольшого количества воды с последующим выделением полученного продукта или переводом его в энантиомерно чистую форму разделением рацемической смеси методом жидкостной хроматографии на хиральной стационарной фазе или переводом его в фармацевтически приемлемую соль.

где R4, R5, R6 каждый независимо представляет водород, С1-С6- алкил или С1-С6-алкилокси, нитро, трифторметил, галоген, циано, аминометил, карбоксил, С1 -С4-алкилоксикарбонил, С1-С4-алкилкарбонил, аминокарбонил или гидрокси, R7
водород или галоген; R8, R9 и R10 каждый независимо означает водород, С1-С6-алкил, С1-С6-алкокси, нитро, гидрокси, трифторметокси, 2,2,2-трифторэтокси, (трифторметил)- карбонил, аминокарбонил, (циклопропил)карбонил или радикал формулы С1-С6-алкил (С=Y)-, где Y означает О, N-OH, N-OCH3, N-NH2, N=N(CH3)2, отличающийся тем, что нитрил формулы IV подвергают взаимодействию с сероводородом в соответстсвующем растворителе в присутствии основания с последующим выделением полученного продукта или обработкой его окислительным агентом с получением рацемата соединения Iв, который затем подвергают разделению методом жидкостной хроматографии на хиральной стационарной фазе с получением энантиомерных чистых форм, или переводят в фармацевтически приемлемую соль.
10.07.90 соединения формулы I
R1, R2, R3 и R7 как указано в п.1 формулы; R4 R6 и R8- R10 каждый независимо означает водород, галоген, С1-С6-алкил, С1-С6-алкокси, нитро, гидрокси;
22.03.91 cоединения формулы I,
где R8, R9 и R10 каждый независимо означает трифторметокси, 2,2, 2-трифторэтокси, (трифторметил)карбонил, аминокарбонил, (циклопропил)карбонил или радикал (С1-С6)-алкил (С= Y), где Y означает О, N-OH, N-OCH3, N-NH2, N-N(CH3)2, при условии, что 1) R1 не является н-пропилом, когда R3 R7, R9 и R10 означают водород; R8 означает 4-этокси; Х означает кислород и 2) Х не является серой, когда R1 R3, R6 R10 означает водород, R4 и R5 означает 3,4-диметокси.
где R4, R7 R10 как указано в п.6, формулы R5 и R6 каждый независимо означает водород, галоген, С1-С6-алкил, С1-С6-алкокси, нитро, гидрокси.
Комментарии