Лечебная жевательная резинка - RU2476076C2
Код документа: RU2476076C2
Чертежи








Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к составу жевательной резинки, включающему основу жевательной резинки, биологически активный ингредиент и одну или более вкусо-ароматических добавок или подсластителей. Изобретение также относится к способам приготовления композиций жевательной резинки.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Композиции жевательной резинки обычно включают водорастворимую основную часть, нерастворимую в воде жевательную основу резинки и вкусо-ароматические добавки. Основа жевательной резинки (резиновая основа) обычно содержит смесь эластомеров, виниловых полимеров, растворителей или пластификаторов эластомеров, эмульгаторов, наполнителей и мягчительных средств (пластификаторов). Известно, что каждый из перечисленных эластомеров, восков, растворителей эластомеров и виниловых полимеров вносит свой вклад в липкость резиновой основы.
С некоторых пор, в состав жевательных резинок начали вводить биологически активные ингредиенты. Высвобождение никотина из традиционных резинок было оценено Morjaria et al при помощи устройства, одобренного Европейской Фармакопеей, и результаты опубликованы в статье, озаглавленной "In Vitro Release of Nicotine from Chewing Gum Formulations", опубликованной в журнале Technologies, May 2004, 12-15. Профили высвобождения сравнивали с профилями Pharmagu M®, уплотняемой резинки, разработанной Компанией SPI Pharma.
В патентной заявке WO 00/35298 описана жевательная резинка, содержащая лекарственные активные вещества. Контроль высвобождения активного вещества осуществляют за счет физической модификации активного вещества при нанесении покрытия и сушке. Кроме того, в заявке особо упомянуты жевательные резинки, содержащие такие активные компоненты, как кофеин, никотин, ибупрофен, кетопрофен и напроксен.
Хорошо известным примером коммерчески доступной жевательной резинки, включающей никотин, является Nicorette™.
В патенте США 6592850 описана жевательная резинка, содержащая силденафила цитрат, который может быть использован для лечения эректильной дисфункции. В соответствии со способом изготовления, рассмотренным в указанном патенте, лекарство смешивают с основой резинки, подсластителем и вкусо-ароматической добавкой, предпочтительно в течение первых пяти минут перемешивания.
На существующем уровне техники имеется необходимость разработки улучшенных составов жевательных резинок, предназначенных для доставки в организм биологически активных ингредиентов, например, никотина.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В соответствии с первым аспектом изобретения предложена композиция жевательной резинки, включающая основу жевательной резинки, биологически активный ингредиент, полимерный материал и один или более подсластителей или вкусо-ароматических добавок, и при этом полимерный материал является амфифильным и имеет неразветвленную или разветвленную основную углерод-углеродную цепь и множество боковых цепей, присоединенных к основной цепи.
В соответствии с первым аспектом настоящего изобретения, высвобождение биологически активного ингредиента из композиции жевательной резинки происходит контролируемым образом. Природа и сила взаимодействия между активным ингредиентом и полимерным материалом определяет быстрое или замедленное высвобождение активного ингредиента. Было показано, что полимерный материал также может влиять на общее количество высвобожденного активного ингредиента, в некоторых случаях, высвобождая большее количество активного ингредиента по сравнению с известными в настоящее время жевательными резинками в течение определенного периода времени. Это означает, что в составы жевательных резинок, предлагаемых согласно настоящему изобретению, может быть введено меньшее количество активного ингредиента по сравнению с традиционными жевательными резинками.
При жевании жевательной резинки, имеющей состав, предлагаемый согласно настоящему изобретению, активный ингредиент высвобождается из жевательной резинки. Слюна покрывает ткани ротовой полости под языком (подъязычные) и боковые стенки ротовой полости; на этих участках лекарство может высвобождаться из слюны и проникать в слизистую оболочку ротовой полости. Полагают, что процесс жевания создает давление в ротовой полости, которое заставляет активный ингредиент поступать непосредственно в соматическую систему организма через слизистую оболочку ротовой полости. Это чрезвычайно ускоряет абсорбцию лекарства соматической системой по сравнению с обычными путями проникновения через желудочно-кишечный тракт.
В соответствии со вторым аспектом настоящего изобретения, предложен способ получения композиции жевательной резинки, включающий следующие стадии: (i) приготовление основы жевательной резинки путем смешивания эластомерного материала возможно с одним или более из следующих средств: пластификаторами эластомеров, мягчительными средствами, наполнителями, эмульгаторами и восками;
(ii) добавление биологически активного ингредиента к основе резинки, совместно с одним или более подсластителями или вкусо-ароматическими добавками, с образованием композиции жевательной резинки;
при этом полимерный материал, который является амфифильным и имеет неразветвленную или разветвленную основную углерод-углеродную цепь и множество боковых цепей, присоединенных к основной цепи, добавляют к основе жевательной резинки на стадии (i) и/или в композицию жевательной резинки на стадии (ii).
На стадии (i) получают основу жевательной резинки смешиванием обычных компонентов основы резинки, известных в данной области техники. Такие компоненты обычно включают эластомерный материал и, возможно, один или более следующих материалов: пластификаторов эластомеров, мягчительных средств, наполнителей, эмульгаторов и восков, которые более подробно описаны ниже.
Было показано, что предлагаемый способ позволяет изготавливать стабильные композиции жевательных резинок, имеющие равномерное распределение лекарства и прекрасные жевательные свойства.
В нашей предыдущей Патентной заявке, опубликованной под номером WO 2006/016179, указано, что полимерные материалы, описанные выше, имеют пониженную липкость и могут снижать липкость композиций жевательной резинки. Полимерные материалы содержат неразветвленную или разветвленную основную углерод-углеродную цепь полимера и множество боковых цепей, присоединенных к основной цепи. Боковые цепи получают из алкилсилилполиоксиалкилена или полиоксиалкилена. В настоящей заявке применение таких полимеров с целью контроля высвобождения биологически активных ингредиентов описано впервые.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Основа резинки
Обычно, основа жевательной резинки включает 2-90 мас.% амфифильного полимерного материала, предпочтительно 2-50%, более предпочтительно 2-25%, наиболее предпочтительно 3-20 мас.%. Полимерный материал может замещать часть или все ингредиенты основы жевательной резинки, которые вносят вклад в липкость резинки.
В альтернативном случае основа жевательной резинки не включает амфифильный полимерный материал. Напротив, амфифильный материал добавляют в композицию жевательной резинки независимо от основы жевательной резинки. Наиболее часто амфифильный полимер добавляют и к основе жевательной резинки, и в композицию жевательной резинки.
Основа жевательной резинки может включать 0-6 мас.% воска. Примеры восков, которые могут присутствовать в основе жевательной резинки, включают микрокристаллический воск, природный воск, нефтяной воск, парафиновый воск и смеси указанных материалов. Воски обычно способствуют отверждению основ жевательных резинок и увеличению срока их хранения и улучшению текстуры. Также было обнаружено, что воски смягчают смесь основы, улучшают эластичность во время жевания и влияют на сохранение вкуса. Предпочтительно основа жевательной резинки по существу не включает воска, и указанные свойства обеспечивает полимерный материал. Тем не менее в некоторых примерах осуществления изобретения, воск присутствует и наряду с амфифильным полимером способствует контролированию высвобождения активного ингредиента.
Эластомерный материал обеспечивает требуемую эластичность и текстурные свойства, а также объем. Подходящие эластомерные материалы включают синтетические и натуральные каучуки. Более конкретно, эластомерный материал выбирают из бутадиен-стирольных сополимеров и сополимеров полиизобутилена и изобутилена-изопрена. Было обнаружено, что если общее количество эластомерного материала слишком мало, то основа жевательной резинки не обладает эластичностью, жевательной текстурой и когезионной способностью, в то время как, если содержание эластомерного материала слишком велико, то основа жевательной резинки становится жесткой и резиноподобной. Типичная основа жевательной резинки содержит 10-70 мас.% эластомерного материала, более предпочтительно 10-15 мас.% Обычно полимерный материал составляет по меньшей мере 1 мас.%, предпочтительно по меньшей мере 10 мас.%, более предпочтительно по меньшей мере 50 мас.% эластомерного материала в основе жевательной резинки. В некоторых примерах осуществления, полимерный материал полностью замещает эластомерный материал в основе жевательной резинки.
Пластификаторы эластомеров (также известные как растворители эластомеров) способствуют размягчению эластомерного материала и включают метилглицериновые или пентаэритритовые эфиры канифолей или модифицированных канифолей, например, гидрированных, димеризованных или полимеризованных канифолей или смесей указанных соединений. Примеры пластификаторов эластомеров, пригодные для включения в основу жевательной резинки, предлагаемой согласно настоящему изобретению, включают пентаэритритовый эфир частично гидрированной древесной канифоли, пентаэритритовый эфир древесной канифоли, глицериновый эфир частично димеризованной канифоли, глицериновый эфир полимеризованной канифоли, глицериновый эфир канифоли талового масла, глицериновый эфир древесной канифоли и частично гидрированную древесную канифоль, и частично гидрированный метиловый эфир канифоли; терпеновые смолы, включающие политерпен, например, полимер d-лимонена и полимеры α-пинена или β-пинена и смеси указанных материалов. Пластификаторы эластомеров могут составлять до 30 мас.% основы жевательной резинки. Тем не менее предпочтительное содержание растворителя эластомера составляет 2-18 мас.%. Предпочтительно оно составляет менее 15 мас.%. В альтернативном случае растворитель эластомера может отсутствовать в композиции жевательной резинки.
Массовое отношение эластомерного плюс полимерного материала к пластификатору эластомера предпочтительно находится в диапазоне (от 1 до 50):1, предпочтительно (от 2 до 10):1.
Основа жевательной резинки предпочтительно включает нетоксичный виниловый полимер. Такие полимеры могут обладать некоторым сродством к воде и включать поливинилацетат, сополимеры этилена/винилацетата и виниллаурата/винилацетата. Предпочтительно нетоксичный виниловый полимер представляет собой поливинилацетат. Предпочтительно основа жевательной резинки содержит 15-45 мас.% нетоксичного винилового полимера. Молекулярная масса нетоксичного винилового полимера должна составлять по меньшей мере 2000. Если не указано обратное, единицей молекулярной массы, применяемой в настоящей спецификации, является г/моль.
В альтернативных примерах осуществления основа жевательной резинки не содержит виниловый полимер.
Предпочтительно основа жевательной резинки также включает наполнитель, предпочтительно порошкообразный наполнитель. Наполнители применяют для модификации текстуры основы жевательной резинки и облегчения ее обработки. Примеры типичных наполнителей включают карбонат кальция, тальк, аморфный оксид кремния и трикальцийфосфат. Предпочтительно наполнитель представляет собой оксид кремния или карбонат кальция. Размер частиц наполнителя влияет на когезионную способность, плотность и технологические характеристики основы жевательной резинки при проведении компаундирования. Было показано, что меньший размер частиц наполнителя снижает липкость основы жевательной резинки.
Количество наполнителя, присутствующего в основе жевательной резинки, обычно составляет 0-40 мас.% от массы основы жевательной резинки, более предпочтительно 5-15 мас.%
Предпочтительно основа жевательной резинки включает мягчительное средство. Мягчительные средства применяют для регулирования когезионной способности, для модификации текстуры и для получения резких переходов в расплавленное состояние при жевании продукта. Мягчительные средства обеспечивают тщательное перемешивание основы жевательной резинки. Типичные примеры мягчительных средств включают гидрированные растительные масла, ланолин, стеариновую кислоту, стеарат натрия, стеарат кальция и глицерин. Мягчительные средства обычно применяют в количествах, составляющих приблизительно от 15% до 40 мас.% от массы основы жевательной резинки, и предпочтительно в количествах, составляющих приблизительно от 20% до 35% от массы основы жевательной резинки.
Предпочтительная основа жевательной резинки включает эмульгатор. Эмульгаторы способствуют диспергированию несмешиваемых компонентов, входящих в состав жевательной резинки, с получением единой стабильной системы. Подходящие примеры эмульгаторов включают лецитин, глицерин, глицерина моноолеат, эфиры молочной кислоты жирных кислот, лактилированные эфиры жирных кислот и глицерина и пропиленгликоля, моно-, ди-, и три-стеарилацетаты, моноглицеридцитрат, стеариновую кислоту, стеарилмоноглицеридилцитрат, стеарил-2-молочную кислоту, триацетилглицерин, триэтилцитрат и полиэтиленгликоль. Эмульгатор обычно составляет приблизительно от 0% до 15%, и предпочтительно приблизительно от 4% до 6% от основы жевательной резинки.
Основную цепь полимерного материала, используемого для получения основы жевательной резинки, предлагаемой согласно настоящему изобретению, предпочтительно получают из гомополимера этиленненасыщенного углеводородного мономера или из сополимера двух или более этиленненасыщенных углеводородных мономеров. Полимеры основы, из которых получают полимерный материал, т.е. не имеющие боковых цепочек, представляют собой эластомерный материал. В целом, полимерный материал также может представлять собой эластомерный материал.
Амфифильный полимерный материал включает основную углерод-углеродную цепь полимера, обычно получаемую из гомополимера этиленненасыщенного полимеризуемого углеводородного мономера или из сополимера двух или более этиленненасыщенных полимеризуемых углеводородных мономеров. Под термином "этиленненасыщенный полимеризуемый углеводородный мономер" понимают полимеризуемый углеводород, содержащий по меньшей мере одну углерод-углеродную двойную связь, которая может подвергаться реакции аддитивной полимеризации (также известной под названием реакции роста цепи или цепной реакции) с образованием неразветвленной или разветвленной цепи углеводородного полимера, имеющего основную углерод-углеродную цепь полимера. В соответствии с одним из предпочтительных примеров осуществления, основную углерод-углеродную цепь полимера получают из гомополимера этиленненасыщенного полимеризуемого углеводородного мономера, содержащего 4 или 5 атомов углерода, например, изобутилена (2-метилпропена). В соответствии с другим примером осуществления, основная углерод-углеродная цепь полимера может быть получена из гомополимера сопряженного диенового углеводородного мономера, в частности, мономера, содержащего 4 или 5 атомов углерода, например, 1,3-бутадиена или изопрена.
Как было отмечено выше, основная углерод-углеродная цепь полимера может быть получена из сополимера двух или более этиленненасыщенных полимеризуемых углеводородных мономеров. Предпочтительно эту цепь получают из сополимера двух таких мономеров. Например, цепь может быть получена из углеводородного сополимера углеводородного мономера, содержащего одну углерод-углеродную двойную связь, и углеводородного мономера, содержащего две углерод-углеродных двойных связи. Например, основная углерод-углеродная цепь полимера может быть получена из сополимера изобутилена и изопрена. В соответствии с другим примером осуществления, основную углерод-углеродную цепь полимера получают из бутадиен-стирольного блок-сополимера. Основная цепь может представлять собой статистический, чередующийся или блок-сополимер, например, А-В или АВ-А блок-сополимер.
В альтернативном случае основная цепь амфифильного полимерного материала представляет собой сополимер по меньшей мере одного этиленненасыщенного мономера и малеинового ангидрида. Термин сополимер включает как двойные полимеры, так и тройные полимеры. Предпочтительно мономер представляет собой углеводородный мономер. Под термином "этиленненасыщенный полимеризуемый углеводородный мономер" понимают полимеризуемый углеводород, содержащий по меньшей мере одну углерод-углеродную двойную связь, способную подвергаться полимеризации с образованием углеводородного полимера с неразветвленной или разветвленной цепью, имеющего основную углерод-углеродную цепь полимера. В соответствии с одним из предпочтительных примеров осуществления, этиленненасыщенный полимеризуемый углеводородный мономер содержит 4 или 5 атомов углерода, и представляет собой, например, изобутилен (2-метилпропен). В альтернативном случае этиленненасыщенный мономер может представлять собой сопряженный диеновый углеводородный мономер, в частности мономер, содержащий 4 или 5 атомов углерода, например, 1,3-бутадиен или изопрен. В альтернативном случае этиленненасыщенный мономер может представлять собой 1-октадецен.
В соответствии с этим аспектом изобретения этиленненасыщенный мономер может быть ароматическим и/или содержать атомы, отличные от атомов водорода и углерода. Подходящие этиленненасыщенные мономеры включают стирол и винилметиловый эфир.
Молекулярная масса углеводородного полимера, из которого получают основную цепь полимерного материала, обычно находится в диапазоне от 10000 до 200000, предпочтительно от 15000 до 50000, более предпочтительно от 25000 до 45000.
Основная цепь полимерного материала обычно по природе представляет собой гидрофобный полимер. Напротив, боковые цепи могут представлять собой гидрофильные фрагменты, что дает несколько преимуществ. Гидрофобный/гидрофильный баланс гребнеобразной структуры сополимера приводит к значительным изменениям в жесткости основы жевательной резинки в сухом состоянии, что облегчает удаление ненужного переработанного материала с поверхностей. Кроме того, гидрофильные боковые цепи способствуют тому, что при жевании слюна выступает в роли растворителя эластомера, что облегчает жевание резинки. Это позволяет успешно заменять часть или все количество воска и/или растворителя эластомера полимерным материалом.
Наличие гидрофильных боковых цепей придает полимерному материалу поверхностно-активные свойства. При жевании полимерный материал, включающий гидрофильные боковые цепи, находящийся в основе жевательной резинки, обогащается поверхностно-активным веществом, образуя на резинке гидрофильное покрытие, которое не прилипает к гидрофобным поверхностям, например, асфальту и жирным камням мостовой. В присутствии воды такой полимерный материал может быть с большей легкостью удален с большинства обычных поверхностей.
Кроме того, амфифильная природа полимерного материала позволяет осуществлять успешное взаимодействие между материалом и биологически активным ингредиентом, что позволяет вводить ингредиент в состав жевательной резинки и высвобождать его во время пережевывания резинки в полости рта.
Гидрофильные боковые цепи полимерного материала предпочтительно получают из полиэтиленоксида, полиглицидола, поливинилового спирта, полистиролсульфоната или полиакриловой кислоты, наиболее предпочтительно из полиэтиленоксида. Полиэтиленоксид прочно связывается с простыми анионными поверхностно-активными веществами, например веществами, включаемыми в шампуни для волос и жидкости для мытья, с образованием электролита. В присутствии таких анионных поверхностно-активных веществ и воды, полимерный материал отторгается большинством обычных анионных поверхностей, включающих многие оксидные поверхности, хлопчатобумажные ткани и волосы. Это позволяет успешно удалять основу жевательной резинки при помощи мыльной воды.
В альтернативном случае, боковые цепи могут быть получены из полипептида, например полилизина.
В альтернативном случае, боковые цепи полимерного материала могут быть более гидрофобными, чем его основная цепь. Подходящие примеры таких материалов включают фторалканы, полисиланы, полиалкилсиланы, алкилсилилполиоксиалкилены и силоксаны, которые придают основе жевательной резинки очень низкую поверхностную энергию.
Каждая основная цепь полимерного материала может содержать множество боковых цепей, которые могут включать смеси боковых цепей, перечисленных выше, и/или иметь различные длины/молекулярные массы цепей. Тем не менее предпочтительно каждая боковая цепь имеет одну и ту же длину/молекулярную массу.
Основа жевательной резинки или состав жевательной резинки могут включать два или более полимерных материалов, рассмотренных выше.
Предпочтительно боковые цепи полимерного материала имеют следующую формулу
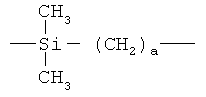
или имеют следующую формулу
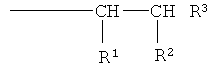
при этом R1 представляет собой Н, -C(O)OR4 или -C(O)Q, и R2 представляет собой -C(O)OR4 или -C(O)Q
при условии, что по меньшей мере один из радикалов R1 и R2 представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, имеющую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, и при этом каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода;
а равен 3 или 4, и каждый из b и с независимо равен 0 или целому числу, составляющему от 1 до 125, при условии, что сумма b+с имеет значение, находящееся в диапазоне от 10 до 250, предпочтительно от 10 до 120.
Предпочтительно боковые цепи присоединены к основной цепи полимерного материала через группу, полученную из малеинового ангидрида.
В соответствии с одним из примеров осуществления настоящего изобретения, боковые цепи, находящиеся в полимерном материале, имеют следующую формулу


где R3, R4 и Q определены выше. Указанные группы получают из звеньев малеинового ангидрида или его производных, привитых на основную цепь.
Предпочтительно полимерный материал включает боковые группы карбоновой кислоты. Таким образом, предпочтительно в вышеуказанной формуле R4 представляет собой Н.
В соответствии с другим примером осуществления, боковые цепи могут иметь следующую формулу

и при этом определение Q дано выше.
В другом примере осуществления боковые цепи имеют следующую формулу

и при этом определение Q дано выше. Указанные группы происходят из материалов, на которые привиты метакриловые полимеры.
В соответствии с другим примером осуществления боковые цепи могут иметь следующую формулу

В альтернативном случае, боковые цепи могут иметь следующую формулу

Указанные группы происходят из материалов, на которые привиты акриловые полимеры.
В нижеследующей Таблице 1 указаны два полимерных материала, которые могут быть введены в новую основу жевательной резинки. Два конкретных предпочтительных полимерных материала представляют собой Р1 и Р2.
Для синтеза графт-сополимера подходит любой PIP-g-MA, имеющий подходящее распределение молекулярной массы и подходящее содержание малеинового ангидрида. В альтернативном случае, для синтеза пригодны карбоксилированные материалы PIP-g-MA, в которых используют малеиновый ангидрид после раскрытия его цикла с образованием дикислоты или монокислоты/монометилового эфира; последний пример продемонстрирован полимером Р2.
Основные цепи каждого из указанных полимеров получают из полиизопрена, на который привит малеиновый ангидрид. Уровень привитого МА в PIP-g-MA, применяемом для демонстрации предлагаемой концепции, в типичном случае составляет приблизительно 1,0 мол.%. В полимере PIP-g-MaMme тот же уровень составлял 2,7 мол.% монометилового эфира монокислоты МА. Степень сополимеризации зависит от степени функционализации полиизопрена. Например, в полимере Р1 количество прививок на цепь обычно составляет от 1 до 7, в то время как в полимере Р2 эта же величина составляет от 1 до 10.
Путем изменения длины алкиленокси-боковой цепи можно получать полимерный материал, имеющий требуемый баланс эластомерных и гидрофильных свойств. Увеличение длины алкиленокси-боковой цепи усиливает гидрофильную природу полимерного материала. Каждый из показателей b и с в вышеуказанной группе Q независимо друг от друга составляет от 0 до 125 при условии, что сумма b+с находится в диапазоне от 10 до 250. Предпочтительно сумма b+с находится в диапазоне от 10 до 120, более предпочтительно от 20 до 60, в особенности от 30 до 50, и наиболее предпочтительно от 40 до 45. Это позволяет придавать полимеру требуемую степень гидрофильности.
Не обязательно, чтобы все боковые цепи имели одни и те же значения b и с.
Так как гидрофобность боковых цепей повышается с повышением содержания углерода, предпочтительно как Y, так и Z представляют собой этиленовые группы. Аналогично, во избежание понижения гидрофильности боковых цепей, R5предпочтительно представляет собой Н или СН3.
Как указано выше, свойства полимерного материала зависят не только от характера боковых цепей, привитых на основную углерод-углеродную цепь полимера, но также и от количества привитых боковых цепей. Таким образом, в соответствии с настоящим изобретением, к основной цепи должно быть присоединено множество боковых цепей. Используемый в настоящем описании термин "множество" означает одну или более привитых боковых цепей. В соответствии с настоящим изобретением, количество боковых цепей, привитых на основную углерод-углеродную цепь полимера, обычно в среднем составляет по меньшей мере одну боковую цепь на основной углерод-углеродной цепи полимера. Реальное количество боковых цепей, привитых на основную углерод-углеродную цепь полимера, зависит от природы боковой цепи и способа прививки боковой цепи на основную цепь полимера (и применяемых для этого условий реакции). Для достижения требуемой степени гидрофильности полимерного материала, отношение количества боковых цепей к звеньям основной цепи предпочтительно находится в диапазоне от 1:350 до 1:20, но более предпочтительно от 1:100 до 1:30. Боковые цепи обычно распределены вдоль основной углерод-углеродной цепи полимера статистически, поскольку местоположение присоединения боковой цепи к основной цепи зависит от положения подходящих центров присоединения к основной цепи используемого углеводородного полимера.
Если боковые цепи присоединены к основной цепи полимера посредством привитых звеньев малеинового ангидрида, то к каждому звену малеинового ангидрида в основной цепи полимера может быть присоединено либо ноль, либо одна, либо две боковых цепи.
В одном из примеров осуществления изобретения, каждая боковая цепь включает две группы, при помощи которых она может быть присоединена к двум основным цепям с образованием поперечно-сшитой структуры. Например, до функционализации, боковая цепь полиэтиленгликоля на каждом конце обычно имеет спиртовую группу. Каждая спиртовая группа может быть привита на звено малеинового ангидрида основной цепи.
Предпочтительный полимерный материал, вводимый в основу жевательной резинки, предлагаемой согласно настоящему изобретению, который имеет боковые цепи, присоединенные непосредственно к атомам углерода основной углерод-углеродной цепи полимера, и при этом боковые цепи имеют следующую формулу:

в которой определения Y, Z, R5, b и с даны выше, может быть получен способом, который включает реакцию неразветвленного или разветвленного углеводородного полимера в растворителе и в инертной атмосфере с производным монометакрилата:

в присутствии инициатора свободнорадикальной реакции. Реакцию между углеводородным полимером и производным метакрилата проводят в соответствии со способом, описанным в WO 2006/016179.
Полимерный материал, предлагаемый согласно настоящему изобретению, в котором боковые цепи, присоединенные непосредственно к атомам углерода основной углерод-углеродной цепи полимера, имеют следующую формулу

в которой определения Y, Z, R5, а, b и с даны выше, может быть получен способом, который включает
(i) реакцию соединения, имеющего формулу
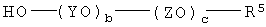
с гидридом натрия в сухом органическом растворителе в инертной атмосфере;
(ii) реакцию продукта, получаемого на стадии (i), с соединением

в котором q равен 1 или 2,
с образованием соединения II

(iii) реакцию соединения II с хлордиметилсиланом с получением соединения III

и
(iv) восстановление соединения III и реакцию продукта, α-гидродиметилсилилполиалкиленоксида, с неразветвленным или разветвленным углеводородным полимером, содержащим множество углерод-углеродных двойных связей в основной цепи углеводородного полимера в присутствии соли переходного металла.
Предпочтительно при проведении стадии (ii), продукт, получаемый на стадии (i), вводят в реакцию с 3-бромпропеном таким образом, что в формуле, указанной выше для боковой цепи, «а» равно 3.
Этот способ более подробно описан в WO 2006/016179.
Полимерный материал согласно настоящему изобретению, в котором боковые цепи, присоединенные непосредственно к атомам углерода основной углерод-углеродной цепи полимера, имеют следующую формулу

в которой один из радикалов R1 и R2 представляет собой группу -C(O)Q, а другой представляет собой группу -C(O)OR4, для которых определение Q и R4 дано выше, может быть получен способом, который включает реакцию полиизопрен-графт-малеинового ангидрида или его моноэфирного производного с соединением, имеющим формулу HO-(YO)b-(ZO)c-R5, в которой определения Y, Z, R5, b и с даны выше. Обычно, реакцию проводят в органическом растворителе, например толуоле.
В соответствии со способом, описанным выше, количество боковых цепей, присоединенных к основной цепи полимера, зависит от количества прививок малеинового ангидрида на молекуле полиизопрена, которые могут участвовать в реакции этерификации спиртом HO-(YO)b-(ZO)c-R5. Например, количество боковых цепей, имеющих вышеуказанную общую формулу, которое может быть образовано при использовании полиизопрен-графт-малеинового ангидрида, имеющего формулу

очевидно, зависит от значения у. Полиизопрен-графт-малеиновый ангидрид (PIP-g-MA) имеется в продаже. Исключительно в качестве примера можно отметить, что такой полимер PIP-g-MA, поставляемый Компанией Aldrich под номером CAS No. 139948-75-7, имеет среднюю молекулярную массу, приблизительно составляющую 25000. Мономерное отношение звеньев изопрена к звеньям малеинового ангидрида в таком графт-сополимере обычно составляет 98:1,1, что указывает на то, что при проведении реакции между таким PIP-g-MA и описанным выше спиртом можно получить приблизительно от 1 до 7 боковых цепочек на одну молекулу. Полиизопрен-графт-малеиновый ангидрид может быть приготовлен в соответствии с методиками, описанными в литературе. Например, в соответствии с методикой, описанной Visonte L.L.Y. et al, Polymers for Advanced Technologies, Vol.4, 1993, pp 490-495, полиизопрен, растворенный в о-дихлорбензоле, вводят в реакцию с малеиновым ангидридом при 180-190°С с образованием модифицированного изопрена. Увеличение продолжительности реакции с 5 до 11 часов позволяет получать различные сополимеры полиизопрен-g-малеинового ангидрида, содержащие 7, 15, 19, 26 и 29 мол.% малеинового ангидрида.
Реакцию между PIP-g-MA и полиалкиленоксиспиртом обычно проводят в органическом растворителе, например, толуоле, и обычно в присутствии активатора, например, триэтиламина, при повышенной температуре. Выход эфира в этой реакции может быть увеличен за счет удаления воды из реакционной смеси азеотропной отгонкой, поскольку толуол и вода образуют азеотропные смеси, кипящие при более низкой температуре, чем каждый из указанных компонентов. Полиалкиленоксиспирт также может быть введен в реакцию с моноэфирным производным PIP-g-MA. Например, нами были достигнуты хорошие результаты при использовании монометилового эфира, имеющего общую формулу

степень функционализации которого (т.е. n) составляла приблизительно 10, средняя молекулярная масса составляла приблизительно 25000, и температура стеклования составляла -59°С. Реакцию указанного монометилового эфира с полиалкиленоксиспиртом обычно проводят в органическом растворителе, например толуоле, при повышенной температуре. Выход эфира может быть увеличен за счет удаления воды из реакционной смеси азеотропной отгонкой. В альтернативном случае реакция может быть проведена в отсутствии растворителя, смешиванием расплава какой-либо основной цепи полиизопрена с основной цепью полиалкиленоксиспиртового графта. Несмотря на то что указанные способы требуют применения заранее образованного полиизопрена, включающего карбоксильные функциональные группы, их преимуществом является то, что они включают использование относительно быстрых и простых реакций, которые дают высокие выходы продукта.
Если основная цепь амфифильного полимерного материала представляет собой сополимер малеинового ангидрида с этиленненасыщенным мономером, то терминальными группами предшественников боковой цепи обычно являются спиртовая группа на одном конце и алкилоксигруппа на другом. Предпочтительным примером предшественника боковой цепи является МеО-РЕО-ОН. При получении полимерного материала указанным способом, такие боковые цепи реагируют со звеньями, полученными из малеинового ангидрида, при проведении алкоголиза ангидрида с образованием карбоновой кислоты и эфира карбоновой кислоты.
Реакция малеинового ангидрида со спиртом представляет собой реакцию алкоголиза, которая приводит к получению сложного эфира и карбоновой кислоты. Эта реакция также известна под названием реакции этерификации. Эта реакция протекает относительно быстро и не требует присутствия катализатора; тем не менее кислотный или основной катализатор может присутствовать.
Суммарная реакция может быть представлена нижеследующей схемой. Рх и Ру представляют собой остатки сополимера/тройного полимера, и ROH представляет собой предшественник боковой цепи.

При осуществлении способа, предшественники боковых цепей, представленные группой ROH, могут реагировать с тем же самым мономером малеинового ангидрида с образованием соединения, имеющего общую формулу

В альтернативном случае, на один мономер малеинового ангидрида в реакцию вступает только один предшественник боковой цепи. При этом оказывается, что в звене, полученном из малеинового ангидрида, остается свободная карбоксильная группа, которая может образовывать соответствующее производное при проведении последующих этапов синтеза. Эта группа также может быть подвергнута депротонированию с образованием ионной основной цепи полимерного материала.
В соответствии со способом согласно настоящему изобретению, предшественниками боковой цепи могут быть гидроксильные группы, находящиеся на каждом из терминальных участков молекулы, и каждая терминальная группа реагирует со звеньями, полученными из малеинового ангидрида, находящимися в различных основных цепях, с получением поперечно-сшитого полимерного материала.
После проведения реакции предшественников боковой цепи с исходным сополимером или тройным полимером, все непрореагировавшие звенья, полученные из малеинового ангидрида, находящиеся в основной цепи, могут быть подвергнуты реакции раскрытия цикла. Эта реакция может включать гидролиз или протекать в присутствии основания. Полученный продукт может быть ионизируемым (ионогенным). Эта дополнительная стадия реакции особенно полезна, если в основной цепи полимера имеется большое количество групп малеинового ангидрида, например, в чередующемся сополимере.
Композиция жевательной резинки
Композиция жевательной резинки включает основу жевательной резинки, один или более подсластителей или вкусо-ароматических добавок и биологически активный ингредиент. Обычно композиция жевательной резинки включает как подсластитель, так и вкусо-ароматическую добавку. Композиция жевательной резинки также может включать другие вещества, включающие нутрицевтические активные ингредиенты, травяные экстракты, стимулирующие вещества, душистые вещества, сенсибилизирующие вещества, вещества, обеспечивающие охлаждающее, разогревающее или раздражающее действие, микрокапсулированные вещества, абразивы, отбеливающие вещества и окрашивающие вещества.
Количество основы жевательной резинки в готовой композиции жевательной резинки обычно находится в диапазоне 5-95 мас.% в пересчете на массу готовой композиции, при этом предпочтительные количества находятся в диапазоне 10-50 мас.%, более предпочтительно 15-25 мас.%
Биологически активный ингредиент
Биологически активный ингредиент представляет собой любое вещество, которое модифицирует химический или физический процесс, происходящий в организме человека или животного. Предпочтительно биологически активный ингредиент представляет собой фармацевтически активный ингредиент, который, например, выбирают из группы, включающей лекарства против агрегации тромбоцитов, лекарства, воздействующие на эректильную дисфункцию, противоотечные средства, анестезирующее средства, оральные контрацептивы, противораковые химиотерапевтические средства, психотерапевтические средства, сердечно-сосудистые средства, нестероидные противовоспалительные препараты (НПВП), доноры NO для лечения стенокардии, неопиоидные болеутоляющее средства, антибактериальные лекарства, антациды, диуретики, противорвотные средства, антигистаминные средства, противовоспалительные средства, средства против кашля, противодиабетические средства (например, инсулин), опиоиды, гормоны и сочетания указанных средств. Предпочтительно активный ингредиент представляет собой стимулирующее вещество, например, кофеин или никотин. В альтернативном случае активный ингредиент представляет собой болеутоляющее средство. Другой пример активного ингредиента представляет собой инсулин.
В одном из примеров осуществления изобретения, биологически активный ингредиент представляет собой нестероидный противовоспалительный препарат (НПВП), например, диклофенак, кетопрофен, ибупрофен или аспирин. В альтернативном случае активный ингредиент представляет собой парацетамол (который в общем случае не классифицируется как НПВП).
В другом примере осуществления изобретения, биологически активный ингредиент представляет собой витамин, минерал или другую пищевую добавку.
Биологически активный ингредиент может представлять собой противорвотное средство, например доласетрон. В альтернативном случае, биологически активный ингредиент представляет собой лекарство, воздействующее на эректильную дисфункцию, например, силденафила цитрат.
В общем случае, композиция жевательной резинки включает 0,01-20 мас.% активного ингредиента, более предпочтительно 0,1-5 мас.%. Композиция жевательной резинки может быть выполнена в виде дозированной формы, подходящей для перорального введения. Масса дозированной формы предпочтительно находится в диапазоне 0,5-4,5 г, например, составляет приблизительно 1 г. В общем случае, композиция жевательной резинки включает 1-400 мг биологически активного ингредиента, более часто 1-10 мг, в зависимости от типа активного ингредиента. Если активный ингредиент представляет собой, например, никотин, то композиция жевательной резинки обычно включает 1-5 мг никотина. Если активный ингредиент представляет собой нестероидный противовоспалительный препарат, например, ибупрофен, то композиция обычно включает 10-100 мг активного ингредиента.
В общем случае, композицию жевательной резинки следует пережевывать в течение времени, составляющего до одного часа, хотя чаще достаточно 30 минут. Предпочтительно в течение 30 минут пережевывания, по меньшей мере 40%, более предпочтительно по меньшей мере 45%, наиболее предпочтительно по меньшей мере 50% активного ингредиента, присутствующего в составе жевательной резинки, высвобождается в ротовую полость. В зависимости от типа активного ингредиента и его предполагаемого назначения, высвобождение может происходить в течение относительно более длительного или относительно более непродолжительного периода. Например, для некоторых активных ингредиентов предпочтительно медленное или замедленное высвобождение, которое способствует уменьшению побочных эффектов, вызываемых активным ингредиентом. Это предпочтительно в случае силденафила цитрата, описанного в патенте США 6592850. В таких случаях предпочтительно не более 50% активного ингредиента высвобождается в течение 15 минут пережевывания, и высвобождение активного ингредиента все еще продолжается в течение промежутка времени, составляющего от 15 до 30 минут с момента начала пережевывания.
В альтернативном случае, может быть предпочтительна более высокая скорость высвобождения. Например, для курильщиков, проходящих никотин-заместительную терапию, более предпочтительной является большая скорость высвобождения никотина, удовлетворяющая их потребность в никотине. В таких случаях, предпочтительно, если в течение первых 10 минут пережевывания высвобождается 25-100% активного ингредиента. Более часто в течение первых 10 минут пережевывания высвобождается 35-65% активного ингредиента. Преимуществом композиции жевательной резинки с быстрым высвобождением, которая позволяет производить полное высвобождение никотина в течение разумного промежутка времени, является то, что потребитель должен покупать и пережевывать меньшее количество резинки (т.е. меньше подушечек резинки или подушечек с меньшей массой). В альтернативном случае, в композицию жевательной резинки должно быть добавлено меньшее количество активного ингредиента, что, в том числе, также выгодно и изготовителю.
Подсластитель может быть выбран из группы, включающей широкий спектр материалов, включающих водорастворимые искусственные подсластители, водорастворимые вещества и подсластители на основе дипептидов, а также их смеси. Предпочтительно подсластитель представляет собой сорбит. Вкусо-ароматические добавки могут быть выбраны из группы, включающей синтетические вкусо-ароматические добавки, представляющие собой жидкости и/или масла, полученные из растений, листьев, цветков, плодов (и т.д.), а также сочетания указанных средств.
Подходящие подсластители и вкусо-ароматические добавки более подробно описаны в патенте США 4518615.
Кроме основы жевательной резинки, подсластителя и вкусо-ароматической добавки, композиция жевательной резинки согласно настоящему изобретению может включать дополнительный амфифильный полимерный материал (т.е. дополнительно к полимерному материалу, который может присутствовать в основе жевательной резинки). Предпочтительно этот дополнительный полимерный материал, если таковой имеется, составляет 1-20%, более предпочтительно 3-15 мас.% от массы композиции жевательной резинки. Он может быть растворим или нерастворим в воде.
Способ приготовления композиции жевательной резинки
Способ обычно включает приготовление композиции жевательной резинки смешиванием основы жевательной резинки с биологически активным ингредиентом, подсластителем и вкусо-ароматическими добавками. Стандартные способы приготовления композиций жевательных резинок описаны в публикации Formulation and Production of Chewing and Bubble Gum. ISBN: 0-904725-10-3, в которой описано изготовление жевательных резинок, имеющих покрытия и жидкие центральные части.
Обычно композиции жевательной резинки получают смешиванием в расплавленном состоянии основы жевательной резинки, подсластителя и вкусо-ароматических добавок с последующим охлаждением смеси. Такой способ может быть использован в соответствии с настоящим изобретением.
Изобретали обнаружили, что осуществление приготовления при контролируемой температуре способствует введению биологически активного ингредиента в композицию жевательной резинки.
Для приготовления как основы жевательной резинки, так и композиции жевательной резинки в лаборатории может быть использован микросмеситель НААКЕ MiniLab Micro Compounder (Thermo Fisher Corporation).
В случае приготовления основы жевательной резинки, ингредиенты обычно смешивают друг с другом при их последовательном добавлении при температуре, находящейся в диапазоне 80-120°С, обычно приблизительно составляющей 100°С. После образования основы жевательной резинки, материал экструдируют из смесителя MiniLab.
Следует отметить, что смеситель MiniLab Compounder не следует использовать для смешивания больших объемов жевательной резинки. В таких случаях следует использовать промышленное устройство, например, Z-лопастной миксер.
Для равномерного смешивания компонентов композиции жевательной резинки может потребоваться нагревание до температуры, составляющей приблизительно 100°С (например, находящейся в диапазоне 80-120°С). Это может представлять собой проблему, если биологически активный ингредиент чувствителен к температуре, т.е. нестабилен при таких высоких температурах. Если активный ингредиент чувствителен к температуре, то стадию (ii) способа предпочтительно проводят в виде двух отдельных операций. Первая операция должна представлять собой смешивание, во время которого основу жевательной резинки смешивают с одним или более подсластителями и/или вкусо-ароматическими добавками и нагревают. Полученную смесь затем охлаждают до температуры, при которой активный ингредиент стабилен, и активный ингредиент добавляют в охлажденную смесь, возможно, совместно с одним или более другими подсластителями и вкусо-ароматическими добавками, получая композицию жевательной резинки. Амфифильный полимерный материал, определение которого дано выше в соответствии с первым аспектом изобретения, добавляют либо на стадии формирования основы жевательной резинки, либо на стадии (ii), когда композиция жевательной резинки уже получена. Полимерный материал может быть добавлен при проведении обеих указанных стадий.
Предпочтительно смесь нагревают до температуры, находящейся в диапазоне 80-120°С, обычно приблизительно 100°С. В общем случае, смесь охлаждают до температуры, находящейся в диапазоне 40-80°С, предпочтительно 50-70°С.
После завершения смешивания, композиция жевательной резинки может быть подвергнута экструзии.
Биологически активный ингредиент может быть добавлен в твердом, расплавленном или жидком состоянии. В общем случае никотин добавляют, например, в виде масла, несмотря на то, что предпочтительным является применение никотина в твердом состоянии (например, никотина на ионообменной смоле, например, на смоле Полакрилекс™). Перед добавлением активного ингредиента на стадии (ii), активный ингредиент может быть предварительно смешан с полимерным материалом и/или подсластителем. Предпочтительно подсластитель представляет собой сорбит.
На любой из стадий способа смесь может быть подвергнута перемешиванию для улучшения ее гомогенности.
Стадия (ii) может включать применение прессования для придания формы композиции жевательной резинки.
Дозированная форма композиции жевательной резинки может быть получена экструзией жевательной резинки и приданием экструдату желаемой формы. Масса дозированной формы обычно находится в диапазоне 0,5-2,5 г, обычно приблизительно 1 г. Дозированная форма может быть изготовлена в форме цилиндра или сферы или в виде подушечки.
Обычно, композиция жевательной резинки включает 5-95 мас.%, предпочтительно 10-50 мас.%, более предпочтительно 15-45% основы жевательной резинки. В композицию жевательной резинки также может быть добавлен дополнительный полимерный материал в количестве, составляющем 1-15%, более предпочтительно 3-15% композиции жевательной резинки.
Стадии получения композиции жевательной резинки могут быть проведены последовательно в одном и том же устройстве, или они могут быть проведены на различных участках; в этом случае способ может включать попеременное осуществление охлаждения и нагревания.
В соответствии с указанным способом, основа жевательной резинки может обладать любым из предпочтительных признаков, рассмотренных выше.
Один из примеров осуществления настоящего изобретения относится к амфифильному полимерному материалу, имеющему неразветвленную или разветвленную основную углерод-углеродную цепь и множество боковых цепей, присоединенных к основной цепи, применяемому для введения биологически активного ингредиента в организм человека или животного.
Описания амфифильного материала и биологически активного ингредиента даны выше при рассмотрении аспектов настоящего изобретения.
Введение может быть осуществлено пероральным, внутривенным, перректальным и парентеральным способами, ингаляцией, топическим нанесением, введением в глаза, назальным введением или через слизистую оболочку ротовой полости.
Амфифильный полимерный материал может быть введен в композицию совместно с биологически активным материалом, что приводит к получению композиции, подходящей для предполагаемого способа введения. Композиции могут быть приготовлены способами, известными специалистам в данной области техники; композиции могут быть приготовлены таким образом, что они обеспечивают контролируемое высвобождение, например, быстрое высвобождение или замедленное высвобождение соединений, применяемых согласно настоящему изобретению. Предпочтительно композиция представляет собой фармацевтическую композицию.
Фармацевтически приемлемые носители, пригодные для введения в такие композиции, хорошо известны в данной области техники. Композиции согласно настоящему изобретению могут содержать 0,1-99 мас.% биологически активного соединения. В общем случае, композиции согласно настоящему изобретению приготавливают в виде дозированной формы. Предпочтительно дозированная форма содержит активное соединение в количестве 1-500 мг. Наполнители, применяемые при приготовлении указанных композиций, представляют собой наполнители, известные в данной области техники.
Композиции, предназначенные для перорального введения, включают известные фармацевтические формы, пригодные для такого введения, например, таблетки, драже, пастилки, водные или масляные суспензии, дисперсные порошки или гранулы, эмульсии, твердые или мягкие капсулы или сиропы и эликсиры. Композиция также может представлять собой жевательную резинку, рассмотренную при описании первого аспекта настоящего изобретения. Композиции могут содержать одно или более веществ, например, подсластителей, вкусо-ароматических добавок, окрашивающих веществ и консервирующих веществ, введение которых позволяет приготавливать фармацевтически изысканные препараты, обладающие приятным вкусом.
Композиции, предназначенные для перорального введения, также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешивают с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, в которых активный ингредиент смешивают с водной или масляной средой, например арахисовым маслом, жидким парафиновым или оливковым маслом.
В альтернативном случае, композиция может быть приготовлена в форме водной или масляной суспензии.
Фармацевтические композиции могут быть приготовлены в виде стерильных водных или масляных суспензий, применяемых для инъекций. Указанные суспензии могут быть приготовлены в соответствии со способами, известными в данной области техники, с использованием подходящих диспергирующих или смачивающих веществ, а также суспензирующих веществ.
Композиции для топического введения также могут быть использованы в соответствии с настоящим изобретением. Активное соединение может быть диспергировано в фармацевтически приемлемом креме, мази или геле.
Далее изобретение будет более подробно описано при помощи следующих Примеров со ссылками на сопроводительные графические материалы, в которых:
На Фиг.1 показано совокупное высвобождение никотина из коммерчески доступной жевательной резинки и жевательной резинки, содержащей Р1, определенное при помощи ВЭЖХ;
На Фиг.2 показано совокупное высвобождение кофеина под действием искусственной слюны, определенное при помощи ВЭЖХ, из образца жевательной резинки, содержащего Р1, в сравнении с контрольным образцом жевательной резинки, не содержащим Р1;
На Фиг.3 показана временная зависимость высвобождения кофеина из двух образцов, показанных на Фиг.2;
На Фиг.4 показано сравнение совокупного высвобождения ибупрофена, определенного при помощи ВЭЖХ, под действием искусственной слюны из жевательной резинки, содержащей Р1, и контрольной жевательной резинки;
На Фиг.5 показано сравнение совокупного высвобождения никотина, определенного при помощи ВЭЖХ, из жевательной резинки, содержащей Р1, и контрольной жевательной резинки, каждая из которых изготовлена с использованием никотинового полакрилекса;
На Фиг.6 показано сравнение совокупного высвобождения кофеина, определенного при помощи ВЭЖХ, под действием искусственной слюны из жевательной резинки, содержащей Р1, и контрольной жевательной резинки;
На Фиг.7 показано высвобождение коричного альдегида из жевательных резинок; и
На Фиг.8 показано высвобождение ибупрофена из образцов.
Сравнительный Пример А: Реакция полиизопрен-графт-малеинового ангидрида с метиловым эфиром полиэтиленгликоля (получение Р1)
PIP-g-MA (3,50 кг, полиизопрен-графт-малеиновый ангидрид, предоставленный Kuraray, марка LIR-403), CAS № 139948-75-7, средняя Mw которого приблизительно составляла 25000, а типичная степень сополимеризации МА приблизительно составляла 1,0 мол.%, и метиловый эфир полиэтиленгликоля (ПЭГМЭ) (2,67 кг, приобретенный у Компании Aldrich), средняя молекулярная масса которого составляла 2000, взвешивали и помещали в воздухонепроницаемый реактор емкостью двадцать литров, снабженный рубашкой и верхней мешалкой. Для растворения исходных материалов в реактор добавляли толуол (8,15 кг), и через сосуд пропускали ток газообразного азота.
Затем сосуд нагревали на масляной бане, установленной на 140°С и соединенной с рубашкой реактора, до температуры кипения толуола (115-116°С). Для удаления воды из метилового эфира полиэтиленгликоля и толуола при помощи азеотропной перегонки, между сосудом и выпускным отверстием для азота устанавливали ловушку Дина-Старка и конденсатор. Таким образом, при протекании реакции, вода собиралась в ловушке Дина-Старка.
Реакционную смесь кипятили всего приблизительно в течение 37,5 часов. Реакция может быть катализирована добавлением кислоты или основания. Продукт очищали порциями по 2 л добавлением еще теплого (50°С) материала в 3 литра деионизированной воды. В случае каждой партии продукта воду удаляли фильтрованием, и процедуру промывки графт-сополимера деионизированной водой и удаления воды фильтрованием повторяли еще пять раз. Продукт сушили в вакууме при 50°С в течение 1 недели.
1H ЯМР спектр регистрировали на ЯМР-спектрометре Delta/GX 40 при 400 МГц в CDCl3 (дейтерированный хлороформ). Получали Р1.
Сравнительный Пример В: Реакция монометилового эфира полиизопрен-графт-монокислоты с метиловым эфиром полиэтиленгликоля (получение Р2)
Метиловый эфир полиэтиленгликоля вводили в реакцию с PIP-g-MaMme (монометиловым эфиром полиизопрен-графт-монокислоты, поставляемым Kuraray Co. Ltd., марка LIR-410). Степень функционализации (т.е. количество групп карбоновой кислоты в одной молекуле) указанного PIP-g-MaMme была равна 10, а молекулярная масса составляла приблизительно 25000.
Метиловый эфир полиэтиленгликоля (ПЭГМЭ) (2,60 кг, приобретенный у Компании Aldrich), средняя молекулярная масса которого составляла 2000, взвешивали и помещали в воздухонепроницаемый реактор емкостью двадцать литров, снабженный рубашкой и верхней мешалкой. ПЭГМЭ расплавляли нагреванием до 60°С, и в реактор добавляли PIP-g-MaMme (3,20 кг), а затем толуол (7,35 кг), и во время смешивания материалов через сосуд пропускали ток газообразного азота.
Затем сосуд нагревали на масляной бане, установленной на 140°С и соединенной с рубашкой реактора, до температуры кипения толуола (115-116°С). Для удаления воды из метилового эфира полиэтиленгликоля и толуола при помощи азеотропной перегонки, между сосудом и выпускным отверстием для азота устанавливали ловушку Дина-Старка и конденсатор. Таким образом, при протекании реакции, вода собиралась в ловушке Дина-Старка.
Реакционную смесь кипятили всего приблизительно в течение 98,5 часов. Реакция также может быть катализирована добавлением кислоты или основания. Продукт очищали порциями по 2 л, обычно добавлением еще теплого (50°С) материала в 3 литра деионизированной воды. В случае каждой партии продукта воду удаляли фильтрованием, и процедуру промывки графт-сополимера деионизированной водой и удаления воды фильтрованием повторяли еще пять раз. Продукт сушили в вакууме при 50°С в течение 1 недели.
1H ЯМР спектр регистрировали на ЯМР-спектрометре Delta/GX 40 при 400 МГц в CDCl3 (дейтерированный хлороформ). Получали Р2.
Физические свойства полимеров приведены в Таблице 2.
Сравнительный Пример С: ПЭГ-сополимеризация полимеров с малеиновым ангидридом по основной цепи
Сополимеры малеинового ангидрида
Поли(изобутилен-чередующийся-малеиновый ангидрид):
Две молекулярные массы (Мn: 6000, 60000 г/моль, в соответствии с маркировкой поставщика), оба полимера были поставлены Компанией Sigma-Aldrich.
Поли(малеиновый ангидрид-чер-1-октадецен):
Молекулярная масса 30-50000 г/моль (в соответствии с маркировкой поставщика), поставлен Компанией Sigma-Aldrich.
Тройные полимеры этилена-малеинового ангидрида
Эти полимеры представляют собой статистические сополимеры этилена, малеинового ангидрида и другого мономера.
Поли(этилен-со-бутилакрилат-со-малеиновый ангидрид)
Это сополимер этилена (91 мас.%), N-бутилакрилата (6%) и малеинового ангидрида (3%). Указанный материал был поставлен Компанией Sigma-Aldrich (молекулярная масса и информация о свойствах не предоставлена).
Поли(этилен-со-винилацетат-со-малеиновый ангидрид)
Это сополимер этилена, винилацетата и малеинового ангидрида. Указанный полимер был поставлен Компанией Arkema; он продается под торговой маркой Orevac (использовали марку 9304).
Предшественники боковых цепей
Во всех случаях прививка (графт) представляла собой метоксиполиэтиленгликоль (МПЭГ), также известный, как метиловый эфир полиэтиленгликоля (ПЭГМЭ). Материал был поставлен двумя поставщиками, Компанией Sigma-Aldrich и Компанией Clariant (торговая марка Polyglykol M 2000S). В обоих случаях, поставщики указывали, что молекулярная масса полимеров составляла 2000, и они, как полагают, имеют очень близкие химические структуры и свойства. Полимеры А, С-Е и G (Таблица 3) были синтезированы с использованием материала, поставленного Компанией Aldrich, остальные - с использованием материала, поставленного Компанией Clariant.
Графт-сополимеры
Под термином "графт-сополимер" имеется в виду "полимерный материал", и эти два термина используют взаимозаменяемо.
Несколько графт-сополимеров были синтезированы прививочной сополимеризацией МПЭГ на основные цепи полимеров, как описано выше.
Как видно из Таблицы 3, часто не весь МА вступает в реакцию. Например, в случае образцов Полимеров А-Е реагирует только часть малеинового ангидрида, находящегося в основной цепи чередующегося сополимера. При этом в основных цепях остается некоторое количество циклов малеинового ангидрида, которые впоследствии могут быть раскрыты (см. раздел, посвященный эмульгированию). Следует отметить, что в некоторых случаях, не все звенья малеинового ангидрида, предназначенные для вступления в реакцию с МПЭГ, могут действительно вступать в эту реакцию.
Синтез графт-сополимеров
Полимер А:
Поли(изобутилен-чередующийся-малеиновый ангидрид) (Мn: 6000 г/моль, 40 г) и метиловый эфир полиэтиленгликоля (Mn: 2000 г/моль, 50 г) растворяли в реакционной колбе в смеси ДМФА (100 мл) и толуола (100 мл). Колбу нагревали до температуры кипения в атмосфере азота в течение 24 ч, при постоянном удалении возможно присутствующей в реакции воды азеотропной перегонкой с извлечением ее в устройство Дина-Старка. Полученный раствор полимера охлаждали и осаждали диэтиловым эфиром, полимер извлекали фильтрованием и сушили для удаления следов растворителя. Успешную прививочную полимеризацию МПЭГ по основной цепи подтверждали при помощи инфракрасной спектроскопии, наблюдая на спектрометре Bruker изменения в области поглощения фрагментов малеинового ангидрида при 1700-1850 см-1.
Полимер В:
Полимер В был синтезирован аналогично Полимеру А с использованием в качестве графт-материала метилового эфира полиэтиленгликоля (Мn: 2000 г/моль, 110 г). Общее время проведения реакции составляло 36 ч. Полимер характеризовали так же, как и полимер А.
Полимер С:
Полимер С был синтезирован аналогично Полимеру А с использованием в качестве основной цепи поли(изобутилен-чередующегося-малеинового ангидрида) (Мn: 60000 г/моль, 40 г). Полимер характеризовали так же, как и полимер А.
Полимер D:
Полимер D был синтезирован аналогично Полимеру А с использованием в качестве основной цепи поли(малеиновый ангидрид-чередующегося-1-октадецена) (Мn: 30-50000 г/моль, 50 г) и метилового эфира полиэтиленгликоля (Мn: 2000 г/моль, 30 г) в качестве графт-материала. В качестве реакционного растворителя использовали толуол (200 мл); в этом случае, полимер осаждали из раствора водой. Амфифильные свойства полученного графт-сополимера привели к получению низкого выхода (25% от теоретического). Полимер характеризовали так же, как и полимер А.
Полимер Е:
Полимер Е был синтезирован аналогично Полимеру D, с тем лишь исключением, что раствор этого полимера не осаждали водой; напротив, реакционный растворитель отгоняли в вакууме. Таким образом, этот материал выделяли с более высоким выходом, чем полимер D, и он может быть использован в тех случаях, когда избыток ПЭГ в готовом продукте не является критическим параметром. Полимер характеризовали так же, как и полимер А.
Полимер F:
Полимер F был синтезирован аналогично Полимеру D с использованием поли(малеиновый ангидрид-чередующегося-1-октадецена) (Мn: 30-50000 г/моль, 20 г) и метилового эфира полиэтиленгликоля (Мn: 2000 г/моль, 136 г) в качестве графт-материала. В качестве реакционного растворителя использовали толуол (500 мл); полимер осаждали из раствора гексаном. Реакцию проводили всего в течение 36 ч. Полимер характеризовали так же, как и полимер А. Избыток ПЭГ может быть удален из полимера диализом или аналогичным способом.
Полимер G:
Полимер G был синтезирован аналогично Полимеру А с использованием в качестве основной цепи поли(этилен-сополимера-бутилакрилата-сополимера-малеинового ангидрида) (40 г) и метилового эфира полиэтиленгликоля (Мn: 2000 г/моль, 30 г) в качестве графт-материала. В качестве реакционного растворителя использовали смесь ксилола (100 мл) и толуола (100 мл); в этом случае полимер осаждали из раствора этанолом. Полимер характеризовали так же, как и полимер А.
Полимер Н:
Полимер Н был синтезирован аналогично Полимеру А с использованием в качестве основной цепи поли(этилен-сополимера-винилацетата-сополимера-малеинового ангидрида) (40 г) и метилового эфира полиэтиленгликоля (Мn: 2000 г/моль, 13 г) в качестве графт-материала. В качестве реакционного растворителя использовали смесь ксилола (125 мл) и толуола (125 мл); в этом случае, полимер осаждали из раствора этанолом. Полимер характеризовали так же, как и полимер А.
Полимер I:
Полимер I был синтезирован аналогично Полимеру Н с использованием метилового эфира полиэтиленгликоля (Мn:2000 г/моль, 39 г) в качестве графт-материала. Для удаления ПЭГ из полимера, полимер после фильтрования тщательно промывали дополнительным количеством этанола. Полимер характеризовали так же, как и полимер А.
Сравнительный Пример D: Испытания по высвобождению лекарства из лечебных жевательных резинок
Экспериментальная методика
Каждый предварительно сформованный кусочек жевательной резинки взвешивали перед пережевыванием, и измеренную массу записывали для оценки общего содержания лекарства.
Использовали устройство для пережевывания 'ERWEKA DRT-1', поставляемое Компанией АВ FIA, работа которого заключается в попеременном сжатии и перекручивании жевательной резинки между двумя плетеными сетками. Для установки в камере пережевывания температуры, аналогичной температуре пережевывания in vivo, камеру снабжали водяной рубашкой, температуру воды в которой устанавливали равной 37°С, и скорость пережевывания устанавливали равной 40 «жеваний» в минуту. Щель между «челюстями» устанавливали равной 1,6 мм.
40 мл искусственной слюны (полученной из водных растворов различных солей, рН которой приблизительно равен 6 - см. Таблицу 4 ниже) помещали в камеру пережевывания, на дно которой помещали пластиковую сетку. В центр сетки помещали кусок жевательной резинки известной массы и поверх помещали вторую пластиковую сетку.
Процедура для анализа профилей высвобождения активных ингредиентов из жевательной резинки
Если не указано обратное, параметры, показанные в Таблице 5, даны для пережевывания.
Перед проведением каждого опыта, камеру, содержащую искусственную слюну и жевательную резинку, оставляли на 5 минут для уравновешивания системы при 37°С. Затем проводили пережевывание жевательной резинки. Затем при проведении опытов по высвобождению, из камеры для испытаний периодически (спустя 5, 10, 15, 20, 25, 30, 40, 50 и 60 минут) извлекали образец объемом 0,5 мл.
Затем все образцы анализировали при помощи ВЭЖХ с использованием обычной системы Perkin Elmer HPLC Series 200, снабженной автоматической пипеткой, насосом и детектором на диодной матрице. Обработку данных и управление устройствами осуществляли при помощи программного обеспечения Totalchrom v 6.2. Колонки и подвижные фазы выбирали в зависимости от ингредиентов следующим образом:
Параметры ВЭЖХ анализа ибупрофена: Колонка: Hypersil C18 BDS, 150×4,6 мм; Подвижная фаза: Ацетонитрил/0,05% водная ортофосфорная кислота в отношении 60/40, 1 мл/мин; УФ-детектор, длина волны - 220 нм.
Параметры ВЭЖХ анализа кофеина: Колонка - Polaris C18-A 58, 250×4,6 мм. Подвижная фаза - Ацетонитрил/0,05% водная ортофосфорная кислота в отношении 60/40. Скорость истечения - 1 мл/мин, УФ-детектор, длина волны - 270 нм.
Параметры ВЭЖХ анализа никотина: Колонка - Hypersil Gold C18 58,250×4,6 мм. Подвижная фаза - Ацетонитрил/0,05М водный дигидрофосфат аммония, рН 8.5 (рН регулировали добавлением гидроксида аммония) в отношении 30/70. Скорость истечения - 1 мл/мин. УФ-детектор, длина волны - 260 нм.
Параметры анализа коричного альдегида: Колонка - Varian Polaris 5u C18-A 250×4,6 м. Подвижная фаза - Ацетонитрил/0,05% ортофосфорная кислота (60/40). Скорость истечения - 1 мл/мин. Обнаружение - УФ 250 нм. Объем впрыска - 5 микролитров.
Для подтверждения воспроизводимости результатов, для каждого образца применяли по два впрыска в ВЭЖХ колонку.
Приготовление основы жевательной резинки и жевательной резинки
Химические вещества
Карбонат кальция (СаСО3), этерифицированная канифоль, гидрированное растительное масло (ГРМ), полиизобутилен (ПИБ), поливинилацетат (PVAc), глицеромоностеарат (ГМС), микровоск, сорбит жидкий, сорбит твердый и масло мяты перечной представляли собой пищевые материалы, которые были поставлены Компанией Gum Base Company. Ибупрофен (марки 40) был поставлен Компанией Albemarle; кетопрофен, напроксен и (-)-никотиновое масло были поставлены Компанией Sigma-Aldrich. Никотиновый полакрилекс был поставлен Компанией Siegfried.
Смешивание жевательной резинки и основы жевательной резинки
Основу жевательной резинки и жевательную резинку смешивали в микросмесителе HAAKE MiniLab (т.е. в лабораторном смесителе) производства Thermo Fisher Corporation. Шнеки устанавливали таким образом, что они вращались в одном направлении со скоростью 80 оборотов/мин.
При получении основы жевательной резинки, ингредиенты обычно смешивали друг с другом, добавляя их при проведении четырех различных операций при 100°С. При проведении каждой операции, ингредиенты совместно загружали в HAAKE MiniLab и перемешивали в течение установленного периода времени перед проведением следующей операции. После выполнения финальной операции, материал экструдировали из смесителя MiniLab.
Жевательную резинку также готовили в MiniLab аналогичным многостадийным способом. При проведении первой стадии часть основы жевательной резинки обычно направляют обратно в смеситель вместе с сорбитом и перемешивают при 100°С. Смеситель оставляют охлаждаться до 60°С и затем добавляют смесь активного ингредиента/P1 с соответствующими компонентами. Если активный ингредиент и другие добавки стабильны при 100°С, то перемешивание ингредиентов может быть предпочтительно проведено при этой температуре. После завершения смешивания, жевательную резинку экструдируют из смесителя MiniLab.
Пример 1: Приготовление жевательной резинки, содержащей никотин
Цель
Смешивание композиции жевательной резинки, содержащей 2 мг никотина, и никотина, предварительно смешанного с полимерным материалом, в хлороформе.
Экспериментальная часть
3 г Р1 (приготовленного, как описано в Сравнительном Примере А) растворяли в 5 мл хлороформа и затем добавляли 0,1 мл никотинового масла. Смесь тщательно перемешивали, затем выливали в чашку Петри и оставляли сушиться в вытяжном шкафу в течение ночи. Смесь оставляли еще на 3 часа в вакуумной печи при комнатной температуре для удаления следов хлороформа. Смесь никотин/Р1 затем смешивали с основой жевательной резинки R3 в процессе добавления подсластителя с получением композиции, приведенной в нижеследующей Таблице 6. В указанной Таблице каждая операция относится к конкретному этапу, при проведении которого смешивают соединения, указанные в графе "химические вещества".
Жевательную резинку экструдировали в виде гомогенной белой ленты, которую затем формовали в виде цилиндрических подушечек жевательной резинки раскатыванием между двумя стеклянными поверхностями для придания ей цилиндрической формы.
Результаты
Для проведения анализа высвобождения лекарства использовали ВЭЖХ. Жевательную резинку, включающую 2 мг никотина, "пережевывали" в 40 мл слюны (см. Сравнительный Пример С, "Экспериментальная часть", в которой описан применяемый способ). Максимальное теоретическое высвобождение при условии, что весь никотин высвобождается в 40 мл искусственной слюны, составляло 50 мкг/мл. С жевательной резинкой, включающей Р1, сравнивали жевательную резинку Nicorette™. Образцы для ВЭЖХ анализа отбирали каждые 5 минут. На Фиг.1 показано сравнение высвобождения никотина из жевательных резинок Nicorette™ и Р1. Наблюдали большую скорость высвобождения никотина из жевательной резинки, содержащей Р1. В результате, по наблюдениям, общее высвобождение никотина из жевательной резинки, содержащей Р1, было выше как спустя 5 минут, так и в остальное время проведения эксперимента.
Пример 2: Приготовление контрольной жевательной резинки, содержащей никотин
Цель
Смешивание жевательной резинки, содержащей 2 мг никотина, но не содержащей полимерного материала, с лекарством, предварительно смешанным с микровоском, заменяющим полимерный материал, с целью сравнения влияния полимерного материала на высвобождение никотина относительно высвобождения из обычной жевательной резинки.
Экспериментальная часть
3 г микровоска растворяли в 5 мл хлороформа. Для растворения воска раствор нужно было оставить на ночь на встряхивателе для перемешивания. Спустя указанное время большую часть микровоска диспергировали в хлороформе, после чего добавляли 0,1 мл никотина. Смесь тщательно перемешивали, затем выливали в чашку Петри и оставляли сушиться в вытяжном шкафу в течение ночи. Смесь оставляли еще на 3 часа в вакуумной печи при комнатной температуре для удаления следов хлороформа. Смесь никотин/микровоск затем смешивали с основой жевательной резинки S3 при проведении этапа добавления подсластителя при помощи низкотемпературной процедуры приготовления композиции, композиция которой указана ниже.
Жевательную резинку экструдировали в виде гомогенной белой ленты, которую затем формовали в виде цилиндрических подушечек жевательной резинки массой 1 г жевательной резинки раскатыванием между двумя стеклянными поверхностями для придания ей цилиндрической формы.
Пример 3: Кофеин
Образцы, композиция которых указана в Таблице 10, готовили с использованием способов, аналогичных описанным в Примерах 1 и 2, но при этом вводили кофеин, а не никотин.
Основа жевательной резинки представляла собой стандартную основу жевательной резинки R3, указанную в Таблице 5.
На Фиг.2 и Фиг.3 показано, что предлагаемые полимерные материалы усиливают высвобождение кофеина из образцов жевательной резинки; при этом высвобождение из образца, содержащего Р1, происходит быстрее и в большем количестве, чем из контрольного образца, не содержащего Р1.
Пример 4: Приготовление основы жевательной резинки, содержащей различные НПВП препараты
Цель
Смешивание основ жевательной резинки, содержащих ибупрофен, кетопрофен и напроксен.
Экспериментальная часть
Сначала при помощи смесителя для получения жевательной резинки готовили смеси из лекарств, ГРМ и амфифильного полимерного материала (Р1). Были сделаны некоторые изменения в способах приготовления, применяемых в приведенных ранее Примерах.
Перед добавлением в смеситель, амфифильный полимерный материал расщепляли на мелкие кусочки; полимер и ГРМ добавляли попеременно в небольших количествах для обеспечения равномерного распределения этих двух материалов.
Смесь ГРМ/полимер оставляли перешиваться по меньшей мере на 10 минут и затем охлаждали. Перед добавлением лекарства, смесь охлаждали до 63°С, поскольку известно, что при этой температуре большинство активных ингредиентов, включающих ибупрофен, вполне стабильны, по меньшей мере в течение времени, необходимого для изготовления жевательных резинок, рассматриваемых в настоящем описании.
Лекарство добавляли постепенно, в течение 5-10 минут, получая равномерное распределение вещества.
Для получения каждой из жевательных резинок, смесь ГРМ/полимер/лекарство смешивали в течение по меньшей мере 30 минут; на этом этапе для некоторых образцов (например, 20 мг ибупрофена в смеси Р1) смесь была еще вполне прозрачной (оценивали через смотровое отверстие, не открывая смеситель), что указывает на то, что смесь не содержит большого количества лекарства, так что при необходимости перемешивание продолжали в течение времени, составляющего до 1 часа, до появления белого цвета.
Затем жевательную резинку экструдировали и для определения количества лекарства в экструдатах применяли анализ1H ЯМР. Его проводили сравнением отношений пиков лекарств и пиков полиизопрена (в Р1) в полученных спектрах. Сигналы ароматических групп ибупрофена, кетопрофена и напроксена обычно наблюдают при 7,1 м.д., 7,7 м.д. и 7,5 м.д., соответственно. В полиизопрене пик атома водорода, присоединенного к двойной связи, наблюдают приблизительно при 5,1 м.д. Указанный сигнал полиизопрена получают от одного атома водорода, находящегося в одном из повторяющихся звеньев полимера, получая приблизительно 367 сигналов на одну цепь полимера. Пример вычисления для смеси, содержащей ибупрофен, выглядит следующим образом:
Отношение сигналов ибупрофен: Р1=2: 8,754, что соответствует 1 молю ибупрофена на каждые (8,754/367)=0,024 моля Р1. Для ибупрофена Fw=206 и для Р1 Мn=30000. Таким образом, массовое отношение составляет 1: 3,47, то есть 23,7 мг ибупрофена на 1,5 г части основы жевательной резинки. Результаты, полученные для всех трех лекарств, представлены в нижеследующей Таблице.
Все смеси лекарство/масло/полимер затем смешивали с основой жевательной резинки, приготовленной из композиции ′R3′, как описано ниже.
Пример 5: Приготовление жевательной резинки, содержащей амфифильный полимерный материал и ибупрофен, смешанные в общей массе
Цель
Добавление амфифильного полимерного материала и лекарства, заранее смешанных в смесителе (без растворителя) при проведении финального этапа приготовления жевательной резинки. Это позволяет производить смешивание композиции жевательной резинки при 100°С, что является нормой при изготовлении жевательных резинок. Это может способствовать улучшению когезионных свойств готовой жевательной резинки.
Смешивание Р1 с ибупрофеном:
Композиция основы жевательной резинки "R3", используемая в этой композиции, приведена в Таблице 6.
Этот продукт экструдировали в виде белого пластичного твердого вещества. Перемешивание большей части жевательной резинки при 100°С, по-видимому, улучшает такие характеристики жевательной резинки, как эластичность.
Пример 6: Приготовление жевательной резинки при добавлении ибупрофена при проведении финального этапа приготовления
Цель
Приготовление жевательной резинки, при котором финальную операцию выполняют при 60°С (во избежание разрушения чувствительных активных ингредиентов) затруднительно, поскольку смешивание при более низких температурах может приводить к получению жевательной резинки с плохими когезионными свойствами и жевательными характеристиками. Таким образом, цель этого Примера состоит в исследовании возможности добавления лекарства при проведении финального этапа приготовления, что позволяет проводить остальные операции смешивания, как обычно, при 100°С. В этом случае, в каждом 1 г кусочке жевательной резинки диспергировали 20 мг ибупрофена.
Композиция основы жевательной резинки "R3", используемая в этой композиции, приведена в Таблице 6.
Экструдированная белая жевательная резинка затем может быть успешно подвергнута формованию, но она не настолько податлива, как материал, полученный в Примере 7.
Пример 7: Приготовление жевательной резинки при смешивании полимерного материала и ибупрофена в общем объеме с добавлением сорбита - 20 мг
Цель
Смешивание полимерного материала и лекарства в виде другой композиции при 60°С, с целью получения жевательной резинки, обладающей лучшими когезионными свойствами, чем смеси, полученные при низкотемпературном смешивании.
Экспериментальная часть
Ибупрофен заранее смешивали с полимерным материалом и добавляли при проведении операции добавления сорбита и вкусо-ароматической добавки.
Смешивание полимерного материала с ибупрофеном:
Эту смесь готовили как на основе Р1, так и на основе Р2. Перемешивание производили в течение 10 мин, после чего продукт тестировали и, при необходимости, перемешивали еще 10 мин.
Количество ибупрофена, присутствующего в каждой смеси, вычисляли из отношений пиков1Н ЯМР при помощи способа, рассмотренного в Примере 4:
В обеих смесях применяли одну и ту же основу жевательной резинки R3, включающую Р1 (Таблица 6).
Условия смешивания для получения полностью приготовленной жевательной резинки показаны в Таблице 18:
Оба продукта экструдировали в виде пластичных белых твердых веществ. Для сравнения высвобождения ибупрофена из рассматриваемой жевательной резинки и контрольной жевательной резинки, не содержащей Р1, но изготовленной в соответствии с аналогичным способом и имеющей аналогичную композицию, см. Пример 9.
Пример 8: Приготовление контрольных образцов: жевательная резинка, содержащая ибупрофен, но не содержащая амфифильный полимерный материал
Цель
Приготовление жевательных резинок, применяемых в качестве контрольных образцов в экспериментах по высвобождению, включающее добавление ибупрофена при проведении финальной операции приготовления, и без добавления каких-либо полимерных материалов.
Композиция, содержащая 20 мг ибупрофена в 1 г жевательной резинки, с использованием основы жевательной резинки S3 (Таблица 8).
Пример 9: Приготовление контрольного образца с микровоском и ибупрофеном. которые были предварительно смешаны в объеме и были добавлены в конце
Цель
Приготовление контрольного образца жевательной резинки, содержащей вместо Р1 микровоск, для сравнения с образцом Примера 7.
Экспериментальная часть
Смесь получали как в Примере 7, но вместо Р1 использовали микровоск. Поскольку количество применяемого жидкого сорбита было уменьшено, количества для приготовления этой смеси были несколько увеличены таким образом, что общая масса смеси составляла 8 г.
Применяли композицию основы жевательной резинки "S3", представленную в Таблице 8.
Затем эту основу использовали для получения готовой жевательной резинки:
Продукт экструдировали в виде пластичного белого твердого материала, который формовали в соответствии с обычным способом.
Высвобождение ибупрофена из жевательной резинки, содержащей Р1, приготовление которой описано в Примере 7, затем сравнивали с высвобождением из контрольной жевательной резинки в соответствии с процедурой, описанной в Сравнительном Примере С. Результаты показаны на Фиг.4. Как видно из представленных данных, высвобождение ибупрофена в течение первых 30 мин (время пережевывания, близкое к реальному) из жевательной резинки, содержащей Р1, происходит значительно быстрее. Общие количества ибупрофена, высвобожденного из жевательных резинок, приблизительно уравниваются не ранее чем спустя 60 мин.
Пример 10: Приготовление жевательной резинки с использованием никотинового полакрилекса
Цель
Готовили жевательную резинку, в которой никотин присутствует в виде никотинового полакрилекса. Этот материал состоит из свободного основания никотина, адсорбированного на катионообменой смоле Amberlite IRP64. В партии полакрилекса, используемой в этом примере, 21,5 мас.% материала составлял никотин. Указанный полакрилекс часто используют вместо никотинового масла, поскольку с ним удобнее работать, и эта форма повышает стабильность никотина. Было рассчитано, что готовая жевательная резинка, полученная в этом примере, содержит 2 мг никотина на 1 г жевательной резинки.
Экспериментальная часть
1 г Р1 тщательно смешивали при помощи шпателя с 0,154 г никотинового полакрилекса в алюминиевом флаконе. Смесь никотин/Р1 смешивали с основой жевательной резинки R3 (Таблица 6) при проведении операции добавления подсластителя, применяя более низкотемпературный способ с внесением изменений в композицию, как указано ниже.
Жевательную резинку экструдировали в виде гомогенной белой ленты, которую затем раскатывали между 2 стеклянными поверхностями, получая цилиндрические подушечки жевательной резинки массой 1 г.
Для сравнения высвобождения никотина из рассматриваемой жевательной резинки и контрольной жевательной резинки, не содержащей Р1, но изготовленной в соответствии с аналогичным способом и имеющей аналогичную композицию, см. Пример 11.
Пример 11: Приготовление контрольной жевательной резинки, содержащей никотиновый полакрилекс
Цель
Приготовление контрольного образца жевательной резинки, содержащего никотиновый полакрилекс, с использованием микровоска вместо Р1, для сравнения с материалом Примера 10. Полагали, что готовая жевательная резинка, получаемая в этом Примере, содержит 2 мг никотина в 1 г жевательной резинки.
Экспериментальная часть
1 г микровоска тщательно смешивали при помощи шпателя с 0,154 г никотинового полакрилекса в алюминиевом флаконе. Для первоначального размягчения воска требовалось небольшое нагревание (приблизительно до 30°С). Затем смесь никотин/микровоск смешивали с основой жевательной резинки S3 (композиция указана в Таблице 7) при проведении операции добавления подсластителя, применяя более низкотемпературный способ, с внесением изменений в композицию, как указано ниже.
Жевательную резинку экструдировали в виде белой ленты, которую затем раскатывали между 2 стеклянными поверхностями, получая цилиндрические подушечки жевательной резинки массой 1 г.
На Фиг.5 показано совокупное высвобождение никотина из представленной контрольной жевательной резинки и сравнительной жевательной резинки, содержащей Р1, под действием искусственной слюны, определенное при помощи способа, описанного в Сравнительном Примере С. В течение большей части эксперимента, скорость высвобождения никотина из жевательной резинки, содержащей Р1, была значительно выше, чем скорость высвобождения из контрольной резинки. В результате, общее высвобожденное количество никотина из жевательной резинки, содержащей амфифильный полимер Р1, значительно выше количества никотина, высвобожденного из контрольной резинки как в начальный момент времени, так и в любой момент проведения эксперимента. Общее количество никотина, высвобожденного из жевательной резинки, содержащей Р1, приблизительно в два раза превышает количество никотина, высвобожденное из контрольной резинки, в течение 60 мин.
Пример 12: Приготовление жевательной резинки, содержащей кофеин Цель
Приготовление жевательной резинки, содержащей 47 мг кофеина в 1 г готовой жевательной резинки. Для контроля скорости высвобождения активного ингредиента, композиция включает Р1. Указанная жевательная резинка принципиально отличается от материала, описанного в примере 3, тем, что кофеин смешивали с Р1 и добавляли при проведении финальной операции приготовления жевательной резинки, в отличие от описанного выше примера, в котором Р1 вводили в основу жевательной резинки.
Экспериментальная часть
1 г Р1 тщательно смешивали при помощи шпателя с 0,78 г кофеина в алюминиевом флаконе. Затем смесь кофеин/Р1 смешивали с основой жевательной резинки R3 (Таблица 5) при проведении операции добавления подсластителя, финальной операции приготовления композиции, композиция которой указана в Таблице 24.
Жевательную резинку экструдировали в виде гомогенной пластичной белой ленты, которую затем раскатывали между 2 стеклянными поверхностями, получая цилиндрические подушечки жевательной резинки массой 1 г.
Для сравнения высвобождения кофеина из рассматриваемой жевательной резинки и контрольной жевательной резинки, не содержащей Р1, но изготовленной в соответствии с аналогичным способом и имеющей аналогичную композицию, см. Пример 13.
Пример 13: Приготовление контрольной жевательной резинки, содержащей кофеин
Цель
Приготовление образца жевательной резинки, содержащего кофеин, но не содержащего Р1, для сравнения с материалом Примера 12; вместо Р1 в основу жевательной резинки вводили микровоск. Полагали, что готовая жевательная резинка, полученная в этом Примере, содержит 47 мг кофеин в 1 г жевательной резинки.
Экспериментальная часть
Кофеин смешивали с основой жевательной резинки S3 (композиция указана в Таблице 7) в процессе добавления подсластителя, последней операции приготовления композиции, состав которой указан в Таблице 25.
Жевательную резинку экструдировали в виде пластичной белой ленты, которую затем раскатывали между 2 стеклянными поверхностями, получая цилиндрические подушечки жевательной резинки массой 1 г.
На Фиг.6 показано совокупное высвобождение кофеина из контрольной жевательной резинки и сравнительной жевательной резинки, содержащей Р1, под действием искусственной слюны, определенное способом, описанным в Сравнительном Примере С. Скорость высвобождения кофеина из жевательной резинки, содержащей Р1, была равна или превышала скорость высвобождения кофеина из контрольного образца в течение всего эксперимента. В частности, наблюдали более высокую скорость высвобождения кофеина из жевательной резинки, содержащей Р1, в течение первых 20 мин эксперимента. В результате было обнаружено, что общее количество кофеина, высвобожденного из жевательной резинки, содержащей амфифильный полимер Р1, на 16% превышало количество кофеина, высвобожденного из контрольной жевательной резинки при отборе первого образца (5 мин), и по меньшей мере на 20% превышало количество кофеина, высвобожденного из контрольной жевательной резинки в остальное время проведения эксперимента.
Пример 14: Применение амфифильных графт-сополимеров для опосредования высвобождения химического вещества из жевательной резинки
14.1 Цели
Иллюстрация применения графт-сополимеров для изготовления жевательной резинки, которые опосредуют высвобождение химического вещества (в данном случае, коммерчески доступной вкусо-ароматической добавки, коричного альдегида). Тщательный контроль высвобождения вкусо-ароматической добавки в лечебной жевательной резинке представляет собой важный фактор, обеспечивающий маскировку вкуса некоторых активных ингредиентов.
14.2 Методика
Химические вещества
Карбонат кальция (СаСО3, этерифицированная канифоль, гидрированное растительное масло (ГРМ, гидрированное соевое масло), полиизобутилен (ПИБ, имеющий молекулярную массу, составляющую 51000 г/моль), поливинилацетат (PVAc, имеющий молекулярную массу, составляющую 26000 г/моль), глицеромоностеарат (ГМС), микрокристаллический воск (микровоск, имеющий т.пл. 82-90°С), жидкий сорбит и твердый сорбит представляли собой пищевые материалы, поставляемые Компанией Gum Base Company. Коричный альдегид (98+%) был поставлен Компанией Fisher-Scientific UK.
Изготовление жевательной резинки
Композиция основы жевательной резинки представлена в нижеследующей Таблице:
Материалы основ жевательных резинок смешивали в микросмесителе Нааке Minilab производства Thermo Electron Corporation, который представляет собой малосерийный лабораторный смеситель/экструдер. Ингредиенты смешивали друг с другом в процессе четырех стадий; экструзию жевательной резинки проводили только после завершения последней стадии. Основу жевательной резинки смешивали при 100°С.
Жевательную резинку готовили в соответствии с данными следующей Таблицы:
Жевательную резинку смешивали на том же самом оборудовании, что и основу, и экструдировали после выполнения завершающего этапа. Жевательную резинку смешивали при 60°С. При выполнении операции 1, жидкий сорбит и порошок сорбита предварительно смешивали перед их добавлением в жевательную резинку.
Экспериментальная методика
См. Сравнительный Пример D.
Образцы сравнивали со стандартными образцами (приготовленными в искусственной слюне), имеющими концентрации в диапазоне 0,02-1,00 мг/мл. Время удержания коричного альдегида, определенное на используемом оборудовании, было равно 4,9 мин; таким образом, для определения количества высвобожденного коричного альдегида использовали пик, имеющий указанное время удержания. Образцы подвергали пережевыванию два или три раза, и в каждом случае определяли согласующиеся кривые высвобождения. Определение каждого образца на установке ВЭЖХ проводили дважды, что показало высокую воспроизводимость результатов.
14.3 Результаты
Были приготовлены жевательные резинки, включающие полимеры A-D и F-I, которые подвергали пережевыванию в искусственной слюне; высвобожденное количество коричного альдегида анализировали при помощи ВЭЖХ. Также были приготовлены контрольные образцы (S3), в которых графт-сополимеры были заменены микровоском, и их анализировали тем же способом (Фиг.7).
Было обнаружено, что высвобождение коричного альдегида из контрольных образцов (S3) довольно стабильно и достигает приблизительно 60% высвобождения спустя 60 минут. Несмотря на то что профили высвобождения двух жевательных резинок, содержащих графт-сополимеры (Н и I), аналогичны профилям материала, содержащего микровоск, профили высвобождения коричного альдегида из большинства этих материалов имеют либо более ранний и высокий максимум, либо более поздний и низкий максимум. Например, полимер Н высвобождает в жевательную резинку лишь 40% коричного альдегида в течение 60 мин; в то время как контрольный образец высвобождает 50% коричного альдегида. Напротив, оказалось, что высвобождение коричного альдегида из жевательной резинки, включающей D, достигает плато, соответствующего высвобождению приблизительно 70% коричного альдегида, в течение времени, составляющего менее 30 мин. Скорость высвобождения из жевательной резинки, содержащей С, меньше, но максимум высвобождения сравним с указанным или был чуть выше.
14.4 Заключение
Изменяя строение основной цепи и степень сополимеризации (и, таким образом, гидрофильность) амфифильного полимера, можно изменять профиль высвобождения химических веществ из жевательной резинки, в рассматриваемом случае - коричного альдегида. По-видимому, скорость высвобождения определяется рядом факторов, включающих химическое строение основной цепи и степень сополимеризации, что приводит к изменениям во взаимодействии со слюной и другими компонентами жевательной резинки. Таким образом, в настоящем описании рассмотрены системы графт-сополимеров, потенциально имеющие широкий диапазон скоростей высвобождения, пригодные для введения в композиции жевательных резинок.
Пример 15: Применение амфифильных графт-сополимеров для опосредования высвобождения активного ингредиента
15.1 Цели
Демонстрация применения амфифильных графт-сополимеров для доставки и высвобождения активных ингредиентов посредством рассмотрения профилей высвобождения ибупрофена из твердых смесей полимеров и ибупрофена, т.е. смесей, включающих инкапсулированный ибупрофен. Термин «инкапсулированный» означает, что на активный ингредиент физически нанесено покрытие или оболочка из графт-сополимера. Такой инкапсулированный материал затем диспергируют в жевательной резинке при помощи способов, рассмотренных в описании настоящего изобретения, что делает его более приемлемым для использования потребителем.
15.2 Методика
Материалы
Ибупрофен (марки 40) был поставлен Компанией Albemarle.
Получение твердых смесей полимера и ибупрофена
Порошкообразный графт-сополимер и ибупрофен взвешивали и помещали в мензурку таким образом, что содержание ибупрофена составляло 1 мас.%. Компоненты подвергали предварительному смешиванию шпателем, получая приблизительно гомогенную смесь, и затем смешивали и экструдировали в микросмесителе Haake Minilab при 60°С. В случае Полимера В использовали 3,96 г полимера и 0,04 г ибупрофена; в случае Полимера С использовали 2,97 г полимера и 0,03 г ибупрофена.
Методика испытания
Образцы инкапсулированного ибупрофена (приблизительно 1 г материала, имеющего известную массу) помещали между двумя пластиковыми стеками и подвергали механическому пережевыванию в искусственной слюне. Методика пережевывания инкапсулированного ибупрофена была идентична методике, применяемой для оценки жевательной резинки, содержащей коричный альдегид, описанной выше; образцы отбирали, спустя 5 мин, 10 мин, 15 мин, 20 мин, 25 мин, 30 мин, 40 мин, 50 мин и 60 мин. Затем образцы готовили для анализа ВЭЖХ фильтрованием через 10-миллиметровый акродисковый шприцевой фильтр PTFE. Образцы анализировали на установке для ВЭЖХ, описанной в Сравнительном Примере D.
Образцы инкапсулированного ибупрофена подвергали пережевыванию два или три раза, и в каждом случае определяли согласующиеся кривые высвобождения. Определение каждого образца на установке ВЭЖХ проводили дважды, что показало высокую воспроизводимость результатов.
15.3 Результаты
Для инкапсулирования ибупрофена применяли два различных полимера, которые также подвергали пережевыванию, и профили высвобождения ибупрофена отслеживали при помощи ВЭЖХ (Фиг.8).
Высвобождение ибупрофена в раствор во время пережевывания происходило из обеих смесей полимер/ибупрофен; при этом в слюну высвобождались сходные общие количества ибупрофена - приблизительно 60% от общего максимального количества - содержание, при котором наблюдаемое высвобождение достигало плато в обоих рассматриваемых примерах. Интересно отметить, что высвобождение ибупрофена происходило гораздо быстрее из полимера В, чем из полимера С, поскольку, несмотря на то, что оба полимера имели химически сходные основные цепи, количество МПЭГ привитого на основную цепь в полимере В, было значительно выше. Таким образом, возможное объяснение состоит в том, что повышение гидрофильности полимеров способствует дезинтеграции инкапсулированных образцов, что приводит к быстрейшему высвобождению во время пережевывания/размалывания (эти полимеры представляют собой твердые вещества).
15.4 Заключение
Ибупрофен подвергали инкапсулированию в двух образцах графт-сополимеров, после чего производили его высвобождение при пережевывании образцов в искусственной слюне. Высвобождение ибупрофена из графт-сополимера В происходило быстрее, чем из графт-сополимера С; первый полимер также содержал большее количество ПЭГ и был более гидрофильным. По-видимому, регулирование гидрофильности амфифильных графт-сополимеров позволяет изменять скорость высвобождения ибупрофена.
Реферат
Изобретение относится к пищевой промышленности. Композиция жевательной резинки содержит основу жевательной резинки, биологически активный ингредиент, полимерный материал и один или более подсластителей или вкусоароматических добавок. Полимерный материал является амфифильным и имеет неразветвленную или разветвленную углерод-углеродную основную цепь. Также к основной цепи присоединена одна или более боковых цепей определенной структурной формулы. Способ получения композиции жевательной резинки предусматривает приготовление основы жевательной резинки путем смешивания эластомерного материала с пластификаторами и/или мягчительными средствами, и/или наполнителями, и/или эмульгаторами, и/или восками. После чего к основе жевательной добавляют вышеуказанный полимерный материал. Затем в основу вносят биологически активный ингредиент совместно с одним или более подсластителями или вкусоароматическими добавками. Изобретение позволяет получить жевательную резинку с повышенной скоростью высвобождения биологически активного ингредиента из композиции жевательной резинки. 2 н. и 22 з.п. ф-лы, 8 ил., 27 табл., 14 пр.
Формула

в которой R1 представляет собой Н, -C(O)OR4 или -C(O)Q, R2 представляет собой -C(O)OR4 или -C(O)Q,
при условии, что по меньшей мере один из радикалов R1 и R2 представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, содержащую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, в которой каждый из Y и Z независимо представляет собой алкиленовую группу, содержащую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, содержащую от 1 до 4 атомов углерода;
каждый из b и с независимо равен 0 или целому числу от 1 до 125, при условии, что сумма b+с составляет от 10 до 250, предпочтительно от 10 до 120.

в которой один из радикалов R1 и R2 представляет собой -C(O)Q, и другой радикал представляет собой -C(O)OR4, где Q и R4 определены в п.1.
при этом полимерный материал, который является амфифильным и имеет неразветвленную или разветвленную основную углерод-углеродную цепь и одну или более боковых цепей, присоединенных к основной цепи, где боковые цепи имеют формулу
в которой R1 представляет собой Н, -C(O)OR4 или -C(O)Q, R2 представляет собой -C(O)OR4 или -C(O)Q,
при условии, что по меньшей мере один из радикалов R1 и R2 представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, содержащую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, в которой каждый из Y и Z независимо представляет собой алкиленовую группу, содержащую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, содержащую от 1 до 4 атомов углерода;
каждый из b и с независимо равен 0 или целому числу от 1 до 125, при условии, что сумма b+с составляет от 10 до 250, предпочтительно от 10 до 120,
добавляют к основе жевательной резинки на стадии (i) и/или в композицию жевательной резинки на стадии (ii).
Документы, цитированные в отчёте о поиске
Жевательная резинка с покрытием, содержащая никотин, способ ее получения и ее применение
Комментарии