Получение мономеров из лигнина в процессе деполимеризации содержащей лигноцеллюлозу композиции - RU2739567C2
Код документа: RU2739567C2
Чертежи





Описание
Настоящее изобретение относится к способу получения мономеров из содержащей лигноцеллюлозу композиции путем деполимеризации.
Уровень техники
Сообщается, что лигнин является вторым по распространенности на Земле природным полимером после целлюлозы, составляя 15-30 масс. % лигноцеллюлозной биомассы и примерно 30% органического углерода, присутствующего в биосфере. В отличие от целлюлозы и гемицеллюлозы, двух других основных составляющих лигноцеллюлозной биомассы, лигнин не является полисахаридом. Вместе с тем утверждается, что лигнин содержит значительное количество ароматических субъединиц. Указанная структура придает лигнину плотность энергии, которая на 30% выше, чем плотность энергии целлюлозных полимеров, и делает его одним из немногих природных источников ароматических молекул. Благодаря указанным свойствам мономеры лигнина все чаще признаются основным предшественником для получения возобновляемых ароматических химических продуктов и/или топлива типа «drop-in».
Однако, несмотря на продолжительную заинтересованность в использовании лигнина, для этого существует очень немного коммерческих способов. Это связано с отсутствием практически осуществимого высокоэффективного метода деполимеризации лигнина, который может быть объединен с облагораживанием полисахаридных фракций биомассы.
В источнике S. Van den Bosch et al.: «Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps», Energy Environ. Sci. 8, 1748-1763 (2015) описано, что одной из наиболее распространенных методик деполимеризации лигнина является прямой гидрогенолиз нативного лигнина в биомассе. Сообщается, что указанный способ позволяет получить наиболее часто встречающиеся мономеры лигнина, а именно гваяциловые и сирингиловые субъединицы. Однако после гидрогенолиза указанный способ включает стадию твердофазного разделения, а именно отделение твердого гетерогенного металлического катализатора от твердых целлюлозных и гемицеллюлозных остатков (полисахаридной фракции). Сообщается, что такое разделение является практически нецелесообразным в промышленном масштабе из-за проблем с восстановлением и деактивацией катализатора и его несовместимости с ведущими методиками биопереработки. Кроме того, из-за жестких условий гидрогенолиза обычно возникают потери гемицеллюлозной и некоторой части целлюлозной фракции, или происходит их превращение в сахарные спирты. Несмотря на то, что указанные спирты могут представлять ценность, они не могут быть биологически облагорожены или превращены в фураны, то есть подвергнуты биопереработке двумя основными ее путями. Кроме того, прямой гидрогенолиз позволяет получить небольшое разнообразие мономеров.
Гидрогенолиз в отношении «экстрагированного» лигнина позволил бы избежать многих из указанных проблем, обеспечивая отдельную обработку полисахаридной и лигниновой фракций биомассы и непрерывную обработку солюбилизированного лигнина в условиях потока с применением гетерогенных катализаторов. В перспективных способах фракционирования (или предварительной обработки) в процессе биопереработки, таких как способы с применением воды, органического растворителя, такого как спирт, тетрагидрофуран (ТГФ) или γ-валеролактон (GVL), ионные жидкости (ИЖ), предварительная обработка и неферментативное осахаривание с помощью GVL, часто применяются высокие температуры и/или недорогие минеральные кислоты (например, H2SO4 и HCl), которые способствуют удалению лигнина из целлюлозы и гемицеллюлозы. Описано, что способ с применением указанных, технологий является источником для получения лигнина в большом масштабе. Однако такое применение кислоты и/или высокой температуры может привести к сильной и необратимой конденсации лигнина в процессе его экстракции, что резко сказывается на его дальнейшем облагораживании. В частности, сообщается, что простые эфирные связи лигнина расщепляются в условиях экстракции, что часто приводит к последующему образованию высокостабильных связей С-С. Предполагается, что указанный механизм включает образование высокореакционноспособного карбкатиона по α-положению боковой цепи лигнина, который вступает в реакцию с отрицательно заряженными атомами в положениях, которые стабилизированы метоксигруппами в ароматических кольцах звеньев лигнина.
Из-за вышеупомянутой конденсации гидрогенолиз экстрагированного лигнина позволяет достичь выхода примерно 5-20% из расчета по молям, что обычно в 5-10 раз ниже выхода продукта, полученного из нативного лигнина.
US 2011/268652 A1 относится к способу получения целлюлозы и по меньшей мере одного низкомолекулярного вещества многократного применения, в котором обеспечивают содержащее лигноцеллюлозу исходное вещество и подвергают его разложению с применением технологической среды.
CN 103 508 858 А относится к способу получения ароматических соединений с применением прямого каталитического крекинга промышленных лигнинов в каталитической системе с восстановительной способностью.
WO 2015/009145 А1 относится к способу фракционирования лигноцеллюлозной биомассы с целью снижения затрат на переработку, повышения делигнификации, уменьшения побочных реакций, в частности снижения разложения гемицеллюлоз, улучшения гидролиза целлюлозы и повышения нативности полученного лигнина путем осуществления фракционирования указанной биомассы с применением обрабатывающей жидкости при температуре ниже 170°C.
US 2 760 861 А относится к способу разделения и выделения целлюлозы и лигнина из лигнинсодержащих целлюлозных материалов.
Следовательно, существует необходимость в способе получения мономеров из лигнина, который может быть применен простым и эффективным образом и предпочтительно совместим с общими методиками биопереработки.
Таким образом, задачей настоящего изобретения являлось преодоление недостатков вышеупомянутых способов.
В частности, задача настоящего изобретения состояла в том, чтобы предложить способ получения мономеров из лигнина с предпочтительным выходом даже в случае применения в крупномасштабном процессе. Кроме того, указанный способ должен быть совместим с общими методиками биопереработки, что обеспечивает биологическое облагораживание или превращение в фуран гемицеллюлозной/целлюлозной фракции.
В частности, следует избегать способов, включающих жесткие условия проведения реакций.
Кроме того, следует избегать способов, включающих твердофазное разделение, в частности после гидрогенолиза.
Необходимо предложить способ, позволяющий получить высокий выход мономеров лигнина, в котором можно избежать реакций в жестких условиях, и который совместим с общими способами биопереработки.
Кроме того, необходимо предложить способ получения фрагментов гемицеллюлозных соединений и их мономеров.
Другая задача настоящего изобретения состояла в том, чтобы предложить способ получения мономеров лигнина, в котором следует избегать применения дорогостоящих и/или так называемых «замысловатых» соединений, таких как агенты на основе ферментов.
Согласно настоящему изобретению вышеуказанные задачи решают с помощью конкретного способа получения мономеров из лигнина, описанного в настоящем документе.
Краткое описание изобретения
Настоящее изобретение неожиданно решило вышеуказанные задачи посредством обеспечения нового способа получения мономеров из лигнина путем деполимеризации. В частности, был предложен способ, включающий добавление альдегида, который неожиданно позволяет получить почти теоретические выходы мономеров лигнина после гидрогенолиза экстрагированного продукта. Указанные выходы были почти на порядок выше выходов продукта, полученного из лигнина с применением такого же способа экстракции и облагораживания в отсутствие формальдегида.
Таким образом, объектом настоящего изобретения является способ получения мономеров из лигнина путем деполимеризации, включающий стадии:
a) обеспечения содержащей лигноцеллюлозу композиции,
b) нагревания композиции со стадии а) в кислых условиях совместно с альдегидом, кетоном, бороновой кислотой или соединением, выбранным из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана (с получением олигомеров из лигнина),
c) отделения продукта, полученного на стадии b),
d) превращения продукта, полученного на стадии с), в мономеры.
Лигноцеллюлоза считается наиболее широкодоступным сырьем (биомассой) на Земле. Лигноцеллюлозную биомассу можно подразделить на первичную биомассу, биомассу отходов и энергетические культуры. Первичная лигноцеллюлозная биомасса включает все существующие в природе наземные растения, такие как деревья, кустарники и траву. Лигноцеллюлозную биомассу отходов получают как малоценный побочный продукт в различных промышленных секторах, таких как аграрный (кукурузная солома, багасса сахарного тростника, солома и т.д.) и лесной (отходы лесопильного завода и бумажной фабрики).
Лигноцеллюлоза содержит гемицеллюлозу, целлюлозу и лигнин. Гемицеллюлозу и целлюлозу можно рассматривать как углеводные полимеры. Указанные углеводные полимеры содержат мономеры пяти- и шестиуглеродных сахаров и связаны с лигнином.
Лигнин можно рассматривать как ароматический полимер. Указанный ароматический полимер содержит метоксилированные фенилпропановые субъединицы, такие как гваяциловые и сирингиловые субъединицы.
Ксилан представляет собой полисахарид, который относится к гемицеллюлозам, при этом основным мономерным звеном ксилана является D-ксилоза. Целлюлоза может быть классифицирована как полисахарид, в котором основным мономерным звеном является D-глюкоза, и мономеры соединены с помощью β-1-4-связей.
В предпочтительном варианте реализации настоящего изобретения указанная содержащая лигноцеллюлозу композиция представляет собой лигноцеллюлозную биомассу, предпочтительно первичную лигноцеллюлозную биомассу, например древесину. Указанную лигноцеллюлозную биомассу предпочтительно получают из деревьев, таких как береза, бук, тополь, кедры, Дугласовы пихты, кипарисы, пихты, можжевельники, каури, лиственницы, сосны, тсуги, секвойи (redwoods), ели и тисы. Наиболее предпочтительной в качестве содержащей лигноцеллюлозу композиции является древесина твердых пород, таких как береза и/или бук.
В альтернативном предпочтительном варианте реализации настоящего изобретения указанную содержащую лигноцеллюлозу композицию получают из энергетических культур. Энергетические культуры представляют собой растительные культуры с высокими выходами лигноцеллюлозной биомассы. Кроме того, энергетические культуры быстро растут, так что лигноцеллюлозная биомасса доступна уже спустя короткий период времени, например через пару месяцев. Примеры энергетических культур включают гигантский тростник, бородач Жерара, сальное дерево, рыжик, ряску, слабительный орех, каранджу (millettia pinnata), просо прутьевидное и слоновую траву.
Стадия а) согласно настоящему изобретению представляет собой обеспечение содержащей лигноцеллюлозу композиции.
Предпочтительно указанная содержащая лигноцеллюлозу композиция является твердой при температуре 23°C. В предпочтительном варианте реализации указанную содержащую лигноцеллюлозу композицию сушат на воздухе. Например, указанную содержащую лигноцеллюлозу композицию сушат на воздухе, чтобы удалить лишнюю воду для хранения. Высушенная на воздухе композиция, содержащая лигноцеллюлозу, предпочтительно содержит менее 50 масс. %, более предпочтительно менее 30 масс. %, в частности от 0 до 20 масс. % воды.
Кроме того, в указанной содержащей лигноцеллюлозу композиции содержание лигнина предпочтительно составляет от 10 масс. % до 40 масс. %, предпочтительно от 13 масс. % до 35 масс. %, в частности от 15 масс. % до 30 масс. % из расчета на общую массу указанной содержащей лигноцеллюлозу композиции, где лигнин определяют как лигнин Класона.
Для определения лигнина Класона применяют испытание на лигнин Класона. В указанном испытании частицы древесины (0,25-0,50 г) вносили в 50 мл лабораторные стаканы с добавлением 7,5 мл 72 масс. % раствора H2SO4. Полученную смесь оставляли при комнатной температуре в течение 2 ч и перемешивали стеклянной палочкой каждые 10 минут. Затем суспензию переносили в круглодонную колбу и добавляли 290 мл воды для достижения концентрации H2SO4 3 масс. %. Стеклянную бутыль запечатывали с помощью завинчивающейся крышки и стерилизовали при 120°C в течение 1 ч в автоклаве. Полученный раствор фильтровали, и осадок промывали водой и сушили при 105°C и взвешивали для определения лигнина Класона.
Содержание лигнина Класона может быть определено с помощью следующего уравнения:
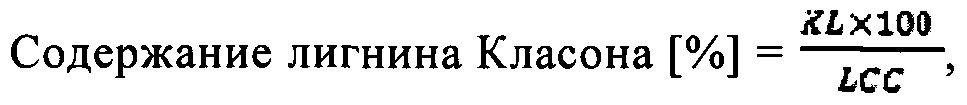
KL представляет собой лигнин Класона [г],
LCC представляет собой содержащую лигноцеллюлозу композицию [г].
Кроме того, стадия а) предпочтительно включает обеспечение указанной содержащей лигноцеллюлозу композиции в виде частиц, таких как щепа, стружка, пеллеты, бусинки, спичечная соломка (splints), гранулы, обломки (shivers), пыль и фрагменты. Например, деревья могут быть разрублены и распилены с получением указанных частиц. Кроме того, для получения указанных частиц уменьшенного и более однородного размера они предпочтительно могут быть просеяны через сито от 10 до 26 меш, предпочтительно сито от 16 до 20 меш.
В предпочтительном варианте реализации стадия а) включает суспендирование указанной содержащей лигноцеллюлозу композиции в органическом растворителе.
Органический растворитель можно рассматривать как углеродсодержащее соединение, которое предпочтительно находится в жидком состоянии при 23°C. Органический растворитель может включать один отдельный органический растворитель или смесь органических растворителей.
Кроме того, указанный органический растворитель предпочтительно имеет температуру кипения от 60°C до 250°C, предпочтительно при 1013 мбар. Указанная температура кипения не относится к какой-либо одной температуре, а может также относиться к интервалу температур, например в случае применения смеси органических растворителей. Температуру кипения предпочтительно определяют согласно Европейской фармакопеи 6.0, см. главу 2.2.12.
В предпочтительном варианте реализации указанный органический растворитель имеет растворимость в воде при 25°C более 50 масс. %, предпочтительно более 70 масс. %, в частности более 90 масс. %. Верхний предел растворимости в воде может составлять 90 масс. % или предпочтительно 100 масс. %. Растворимость в воде может быть определена путем визуального осмотра, то есть определяют долю органического растворителя по отношению к воде до того, как наблюдается образование осадка или суспензии или фазовое разделение между водой и органическим растворителем.
В предпочтительном варианте реализации настоящего изобретения второй растворитель имеет значение logKow от -3,0 до 0,8, предпочтительно от -2,5 до 0,7, более предпочтительно от -1,8 до 0,6, в частности от -1,2 до 0,5.
Значение Kow (также известное как P-значение) представляет собой коэффициент распределения (коэффициент разделения), отражающий отношение концентраций соединения в двух фазах смеси октанол/вода (гидрофобной/гидрофильной). Значение Kow определяют согласно следующей формуле

где

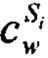
Значение Kow (P-значение) обычно применяют в виде десятичного логарифма как logKow(logP).

Примеры органического растворителя представляют собой спирты, содержащие от 1 до 6 атомов углерода, такие как метанол, этанол, пропанол, изопропанол, бутанол и полиэтиленгликоль, циклические простые эфиры, такие как тетрагидрофуран и диоксан, нитрилы, такие как ацетонитрил, карбоновые кислоты, такие как муравьиная и уксусная кислота, карбоксамиды, такие как диметилформамид и диметилацетамид, лактоны, такие как γ-валеролактон, лактамы, такие как N-метил-2-пирролидон и диметилсульфоксид. Предпочтительными являются спирты, такие как метанол и этанол, циклические простые эфиры, такие как тетрагидрофуран и диоксан, и лактоны, такие как γ-валеролактон. Особенно предпочтительными являются диоксан и тетрагидрофуран, в частности диоксан.
В предпочтительном варианте реализации указанный органический растворитель может содержать воду. Предпочтительно указанный органический растворитель содержит менее 50 объемных процентов воды, предпочтительно менее 30 объемных процентов воды, в частности от 0 до 10 объемных процентов воды.
Предпочтительно суспензия может содержать от 3 до 15 мл, предпочтительно от 5 до 12 мл, в частности от 6 до 10 мл органического растворителя на один грамм указанной содержащей лигноцеллюлозу композиции.
Стадия b) включает нагревание композиции со стадии а) в кислых условиях совместно с альдегидом, кетоном, бороновой кислотой или соединением, выбранным из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана.
Нагревание композиции со стадии а) рассматривается как подведение тепла к композиции, обеспеченной на стадии а), для повышения температуры композиции, обеспеченной на стадии а), от начальной температуры до более высокой конечной температуры. В предпочтительном варианте реализации настоящего изобретения композицию на стадии а) обеспечивают при температуре от 15°C до 25°C, которая считается начальной температурой. В предпочтительном варианте реализации на стадии b) может быть применена температура от 35 до 140°C, предпочтительно от 40 до 130°C, в частности от 50 до 120°C, которая считается конечной температурой. Особенно предпочтительно, чтобы была применена температура от 70 до 100°C, в частности примерно 80°C.
Для обеспечения кислых условий к композиции со стадии а) добавляют одно или более кислотных соединений. Кислотное соединение можно рассматривать как химическое соединение, которое при добавлении в воду снижает значение рН до менее 7. Примеры кислотных соединений представляют собой органические карбоновые кислоты, такие как уксусная кислота, и минеральные кислоты, причем минеральные кислоты являются предпочтительными. Минеральные кислоты рассматриваются как кислоты, которые не содержат атома углерода. Примеры минеральных кислот представляют собой хлористоводородную кислоту, бромистоводородную кислоту, иодистоводородную кислоту, серную кислоту, фосфорную кислоту, борную кислоту и кремниевую кислоту. Предпочтительными являются хлористоводородная кислота, серная кислота и фосфорная кислота, более предпочтительными хлористоводородная кислота и фосфорная кислота, в частности хлористоводородная кислота.
В предпочтительном варианте реализации для обеспечения кислых условий может быть применено от 1 до 10 ммоль, предпочтительно от 2 до 9 ммоль, в частности от 3 до 7 ммоль указанного кислотного соединения на грамм указанной содержащей лигноцеллюлозу композиции.
Кроме того, на стадии b) альдегид, кетон, бороновую кислоту или соединение, выбранное из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, нагревают совместно с указанной содержащей лигноцеллюлозу композицией в кислых условиях.
Предпочтительно на стадии b) совместно с указанной содержащей лигноцеллюлозу композицией в кислых условиях нагревают альдегид или кетон.
Более предпочтительно применяют альдегид.
Альдегид представляет собой органическое соединение, которое в общем случае может быть представлено следующей структурой:
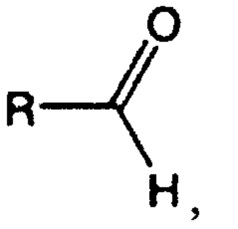
где R представляет собой водород или органический остаток, например содержащий от 1 до 10 атомов углерода.
Термин «органический остаток» в общем случае относится к остатку, известному в органической химии. Предпочтительно скелет органического остатка содержит преимущественно атомы углерода, атомы азота и/или кислорода.
Атом остатка R, который ковалентно связан с атомом углерода, имеющим двойную связь с атомом кислорода, представляет собой атом углерода.
В предпочтительном варианте реализации настоящего изобретения R может представлять собой ароматический остаток или алифатический остаток.
Ароматический остаток содержит по меньшей мере одну кольцевую систему, преимущественно содержащую атомы углерода, азота, серы или кислорода, где указанная кольцевая система содержит согласно правилу Хюккеля 4n+2 (n=0, 1, 2…) делокализованных электронов в сопряженных двойных связях, свободные электронные пары или вакантные p-орбитали.
В предпочтительном варианте реализации ароматический остаток относится к остатку с ароматической каркасной структурой, при этом кольцевые атомы указанной ароматической каркасной структуры представляют собой атомы углерода. В альтернативном предпочтительном варианте реализации указанный ароматический остаток может быть замещен одним или несколькими заместителями.
Заместители предпочтительно могут быть независимо выбраны из одного или более следующих заместителей: алкильные группы, содержащие от 1 до 4 атомов углерода, галоген, нитро, нитрил, карбоксильная группа, сложные эфиры карбоновых кислот и амид карбоновой кислоты, метокси и этокси.
Примеры ароматических остатков представляют собой фенил, o-толил и n-толил. Предпочтительным является фенил, так что соответствующий альдегид представляет собой бензальдегид.
В более предпочтительном варианте реализации R представляет собой алифатический остаток. Алифатический остаток представляет собой неароматическое углеводородное соединение, которое помимо атомов углерода и водорода может содержать, например, еще и атомы кислорода, серы и азота. Указанный алифатический остаток может быть замещенным или незамещенным. То же, что описано выше в отношении ароматического остатка, может быть применено к указанным заместителям.
В предпочтительном варианте реализации R может представлять собой незамещенный, разветвленный или циклический алифатический остаток, содержащий от 3 до 6 атомов углерода. Примерами являются циклопропил, изопропил и трет-бутил.
Более предпочтительно R может представлять собой замещенный или незамещенный линейный алифатический остаток, содержащий от 1 до 6 атомов углерода, в частности незамещенный линейный алифатический остаток, содержащий от 1 до 6 атомов углерода.
Примерами являются метил, этил, пропил и бутил, в частности метил, так что указанный альдегид представляет собой ацетальдегид.
В альтернативном особенно предпочтительном варианте реализации R представляет собой водород, так что указанный альдегид представляет собой формальдегид.
Кетон представляет собой органическое соединение, которое в общем случае может быть представлено следующей структурой:

где R1 и R2 независимо представляют собой органический остаток.
Для термина «органический остаток» справедливо описанное выше.
В более предпочтительном варианте реализации R1 и R2 независимо представляют собой алифатические остатки. Опять же, для алифатических остатков справедливо описанное выше.
Более предпочтительно R1 и R2 независимо могут представлять собой замещенный или незамещенный линейный или разветвленный алифатический остаток, содержащий от 1 до 6 атомов углерода, в частности незамещенный линейный алифатический остаток, содержащий от 1 до 3 атомов углерода.
В особенно предпочтительном варианте реализации R1 и R2 представляют собой метил, так что указанный кетон представляет собой ацетон.
В альтернативном особенно предпочтительном варианте реализации R1 представляет собой метил, и R2 представляет собой этил, так что указанный кетон представляет собой бутан-2-он.
Бороновая кислота представляет собой соединение, которое в общем случае может быть представлено следующей структурой:
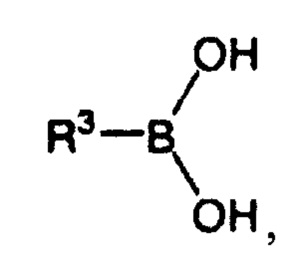
где R3 представляет собой водород или органический остаток, например содержащий от 1 до 10 атомов углерода.
Для термина «органический остаток» справедливо описанное выше.
В более предпочтительном варианте реализации R3 представляет собой ароматический остаток. Опять же, для ароматических остатков справедливо описанное выше.
Более предпочтительно R3 может представлять собой замещенный или незамещенный ароматический остаток. Для заместителей справедливо описанное выше.
Предпочтительно, чтобы R3 представлял собой n-толил или фенил, так что указанная бороновая кислота представляет собой n-толилбороновую кислоту или фенилбороновую кислоту. В частности, R3 представляет собой фенил, так что указанная бороновая кислота представляет собой фенилбороновую кислоту.
2-Метоксипропен может быть представлен следующей структурой:

Диметилкарбонат может быть представлен следующей структурой:
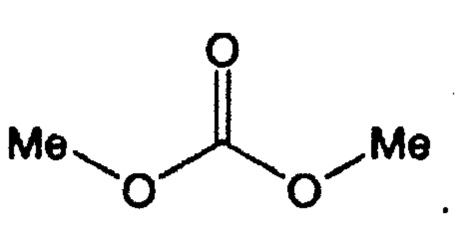
2,2-Диметоксипропан может быть представлен следующей структурой:

В предпочтительном варианте реализации настоящего изобретения указанная содержащая лигноцеллюлозу композиция и указанный альдегид, кетон, бороновая кислота или соединение, выбранное из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, присутствуют в массовом отношении от 25:1 до 1:1, предпочтительно от 20:1 до 1,25:1, более предпочтительно от 15:1 до 1,5:1, в частности от 10:1 до 2:1, в котором масса альдегида, кетона, бороновой кислоты или соединения, выбранного из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, берется из расчета на массу формальдегида.
Под термином «из расчета на массу формальдегида» подразумевается следующее. Молекулярная масса формальдегида составляет 30 г/моль. Альдегид, отличный от формальдегида, или кетон имеет молекулярную массу x г/моль. Например, молекулярная масса ацетальдегида составляет 44 г/моль, а молекулярная масса ацетона составляет 58 г/моль. Таким образом, чтобы содержать такое же количество реакционноспособных альдегидных или кетоновых групп, альдегид, отличный от формальдегида, или кетон должен находиться в массе, которая в x/30 раз больше, чем масса формальдегида.
Например, указанная содержащая лигноцеллюлозу композиция и формальдегид присутствуют в массовом отношении 25:1, если обеспечивают 25 г содержащей лигноцеллюлозу композиции и 1 г формальдегида.
В случае если указанный альдегид представляет собой ацетальдегид, указанная содержащая лигноцеллюлозу композиция и ацетальдегид, имеющий молекулярную массу 44 г/моль, присутствуют в массовом отношении 25:1, если обеспечивают 25 г содержащей лигноцеллюлозу композиции и 1,47 г альдегида. 1,47 соответствует отношению молекулярной массы ацетальдегида к молекулярной массе формальдегида.
Таким образом, в случае если указанная содержащая лигноцеллюлозу композиция и ацетальдегид присутствуют в массовом отношении 12,5:1 (что соответствует 25:2), обеспечивают 25 г содержащей лигноцеллюлозу композиции и 2,93 г ацетальдегида.
В случае когда указанный кетон представляет собой ацетон, указанная содержащая лигноцеллюлозу композиция и ацетон, имеющий молекулярную массу 58 г/моль, присутствуют в массовом отношении 25:1, если обеспечивают 25 г содержащей лигноцеллюлозу композиции и 1,93 г кетона. 1,93 соответствует отношению молекулярной массы ацетона к молекулярной массе формальдегида.
В альтернативном предпочтительном варианте реализации может быть применено от 1 до 50 ммоль, предпочтительно от 3 до 35 ммоль, более предпочтительно от 5 до 25 ммоль, в частности от 10 до 20 ммоль указанного альдегида, кетона, бороновой кислоты или соединения, выбранного из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, на грамм указанной содержащей лигноцеллюлозу композиции.
Кроме того, на стадии (b) реакционная смесь предпочтительно может быть подвергнута механическому движению, такому как перемешивание.
В предпочтительном варианте реализации стадия b) может быть проведена в течение периода от 0,5 до 12 часов, предпочтительно от 0,75 до 10 часов, более предпочтительно от 1 до 8 часов, в частности от 1,5 до 5,5 часов.
При осуществлении стадии b) расщепляются связи между лигниновой фракцией и целлюлозной или гемицеллюлозной фракциями в указанной содержащей лигноцеллюлозу композиции. Кроме того, связи внутри лигнина также расщепляются, таким образом получают «фрагменты лигнина», которые в качестве альтернативы можно рассматривать как так называемые «олигомеры из лигнина».
Кроме того, на стадии b) могут расщепляться связи внутри гемицеллюлозной/целлюлозной фракции, так что помимо прочего получают так называемые «фрагменты ксилана или глюкана». В случае когда на стадии b) применяли формальдегид, указанные фрагменты ксилана могут содержать диформилксилозу, которая представлена следующей формулой:

Данные анализов в отношении диформилксилозы представлены на фиг. 6.
На стадии с) способа согласно настоящему изобретению полученные фрагменты лигнина отделяют от остаточной смеси со стадии b).
Стадия с) предпочтительно может включать следующие подстадии:
c1) разделение фрагментов лигнинсодержащей фазы и остатка,
с2) нейтрализация указанных фрагментов лигнинсодержащей фазы и удаление растворителя,
c3) обработка продукта, полученного на стадии с2), растворителем и удаление нерастворимых частей.
Стадия c1) предпочтительно может включать фильтрование смеси. Указанная смесь предпочтительно может быть подвергнута фильтрованию путем создания вакуума со стороны фильтрата. Кроме того, отфильтрованный осадок предпочтительно может быть подвергнут промыванию. Подходящая жидкость для промывания может представлять собой, например, ацетон или диоксан. Как правило, указанный отфильтрованный осадок может содержать целлюлозу или гемицеллюлозу, а также нерастворимое вещество, содержащееся в указанной содержащей лигноцеллюлозу композиции. Фильтрат можно рассматривать как фрагменты лигнинсодержащей фазы.
В качестве альтернативы предпочтительная стадия c1) может включать центрифугирование смеси со стадии b) и декантацию фрагментов лигнинсодержащей фазы от остатка.
На стадии с2) фрагменты лигнинсодержащей фазы со стадии c1), предпочтительно жидкие фрагменты лигнинсодержащего фильтрата, предпочтительно могут быть нейтрализованы путем добавления щелочного неорганического соединения. Щелочные неорганические соединения могут представлять собой, например, гидроксиды, карбонаты, гидрокарбонаты, фосфаты, гидрофосфаты и сульфаты щелочных и щелочноземельных металлов. В предпочтительном варианте реализации в качестве щелочного неорганического компонента применяют гидроксид кальция и/или карбонат кальция. Более предпочтительными являются карбонаты щелочных металлов, такие как карбонат натрия или карбонат калия, в частности карбонат натрия. После нейтрализации указанный растворитель предпочтительно может быть удален из полученной смеси, например при повышенной температуре, такой как температура от 40 до 60°C, и/или при пониженном давлении, таком как давление от 20 до 100 мбар.
На стадии с3) продукт, полученный на стадии с2), может быть обработан растворителем, таким как тетрагидрофуран или диоксан, предпочтительно тетрагидрофураном. Обработка продукта, полученного на стадии с2), предпочтительно может быть проведена при механическом движении, таком как перемешивание. Кроме того, обработка растворителем предпочтительно может быть проведена при температуре от 20°C до 25°C. После указанной обработки растворителем полученная смесь может быть подвергнута фильтрованию для удаления нерастворимых соединений, так что обеспечивается раствор, содержащий фрагменты лигнина. Указанные фрагменты лигнина могут быть получены путем удаления растворителя, предпочтительно тетрагидрофурана, из фильтрата. В качестве альтернативы фильтрат, содержащий фрагменты лигнина, может быть подвергнут дополнительной переработке без дополнительной обработки.
На стадии с), предпочтительно на подстадиях c1)-c3), часть, содержащая фрагменты лигнина, предпочтительно дополнительно содержит фрагменты ксилана. Например, фильтрат, содержащий фрагменты лигнина, или фрагменты лигнина, полученные при удалении растворителя из фильтрата, предпочтительно могут дополнительно содержать фрагменты ксилана.
На стадии d) фрагменты лигнина, полученные на стадии с), могут быть превращены в мономеры, то есть в мономеры из лигнина.
Превращение, которое можно рассматривать как разновидность деполимеризации, может быть выполнено, например, путем окисления и последующего подвергания окисленного продукта воздействию кислых условий, таких как водная муравьиная или уксусная кислота.
Более предпочтительно превращение фрагментов лигнина со стадии с) в мономеры из лигнина может быть выполнено путем подвергания указанных фрагментов лигнина со стадии с) гидрогенолизу.
Как правило, гидрогенолиз представляет собой реакцию, причем указанная реакция включает расщепление одинарных связей углерод-углерод или углерод-гетероатом, при этом указанная реакция также может быть интерпретирована как «лизис» посредством водорода. Указанный гетероатом обычно представляет собой кислород, азот или серу. Обычно гидрогенолиз проводят в присутствии катализатора и водорода.
В предпочтительном варианте реализации превращение фрагментов лигнина, полученных на стадии с), предпочтительно в виде раствора, такого как фильтрат, полученный на стадии с3), проводят в присутствии катализатора.
Подходящие катализаторы для гидрогенолиза представляют собой, например, катализаторы на основе благородных металлов, предпочтительно на угле. Примерами являются Ru/C, Pd/C, Pt/C и Rh/C, предпочтительно Ru/C, более предпочтительно 5 масс. % Ru/C. Предпочтительно, чтобы фрагменты лигнина и указанный катализатор присутствовали в массовом отношении от 100:1 до 3:1, более предпочтительно от 50:1 до 5:1.
В предпочтительном варианте реализации превращение указанных фрагментов со стадии с) проводят в автоклаве, таком как реактор Парра, предпочтительно путем применения механического движения, такого как перемешивание.
Кроме того, на стадии d) превращение путем гидрогенолиза может быть проведено в присутствии водорода, предпочтительно в присутствии сжатого водорода. Для указанной цели, например, в реакторе Парра может быть создано давление водорода от 2 до 60 бар, предпочтительно от 7 до 50 бар, более предпочтительно от 10 до 40 бар. В процессе указанного превращения может быть произведено дальнейшее повышение давления водорода не более чем в семь раз, предпочтительно в два или три раза.
В предпочтительном варианте реализации настоящего изобретения стадия d) может быть проведена при температуре от 100°C до 300°C, предпочтительно от 150°C до 290°C, более предпочтительно от 200°C до 280°C.Особенно предпочтительно, чтобы могла быть применена температура примерно 250°C.
Кроме того, предпочтительно, чтобы стадия d) могла быть проведена в течение периода от 2 до 24 часов, предпочтительно от 3 до 21 часа, более предпочтительно от 4 до 18 часов, в частности от 5 до 16 часов.
В предпочтительном варианте реализации катализатор удаляют из смеси, которая была подвергнута реакции гидрогенолиза, предпочтительно путем фильтрования. Кроме того, растворитель предпочтительно может быть удален из полученной смеси, например при повышенной температуре, такой как температура от 40 до 60°C, и/или при пониженном давлении, таком как давление от 20 до 100 мбар.
В предпочтительном варианте реализации органический растворитель, применяемый в способе согласно настоящему изобретению, может быть использован повторно. Это относится, например, к растворителю, удаляемому на стадии с2), или к растворителю, в котором растворяют фрагмент лигнина, чтобы подвергнуть гидрогенолизу (стадия d).
Кроме того, предпочтительно, чтобы мономеры из лигнина, полученные в результате превращения фрагментов лигнина со стадии с), могли быть отделены друг от друга, например, путем дистилляции. Указанные мономеры могут быть выделены.
В предпочтительном варианте реализации на стадии (d) могут быть получены фрагменты ксилана (помимо мономеров из лигнина). Предпочтительно указанные фрагменты ксилана содержат диформилксилозу. Указанные фрагменты ксилана могут быть отделены и при необходимости выделены.
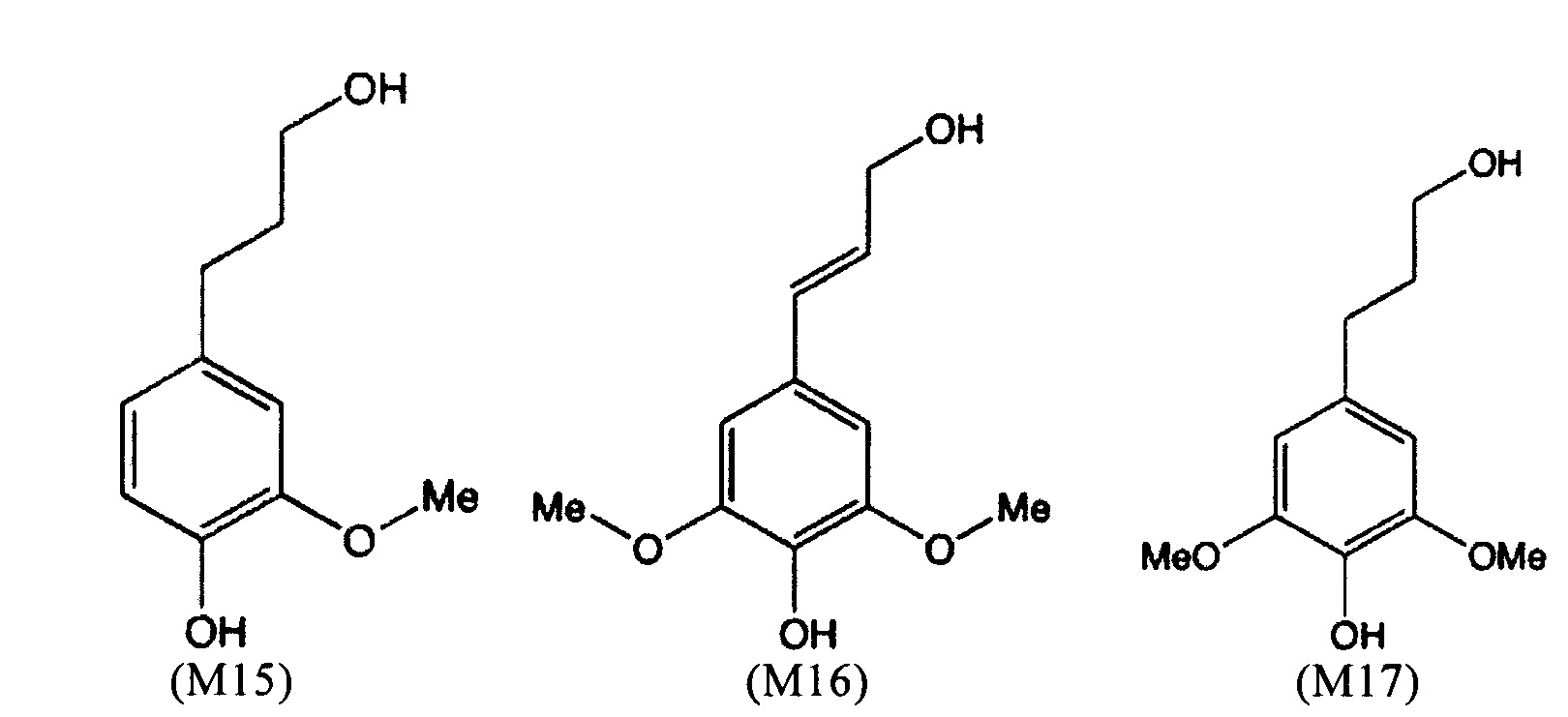
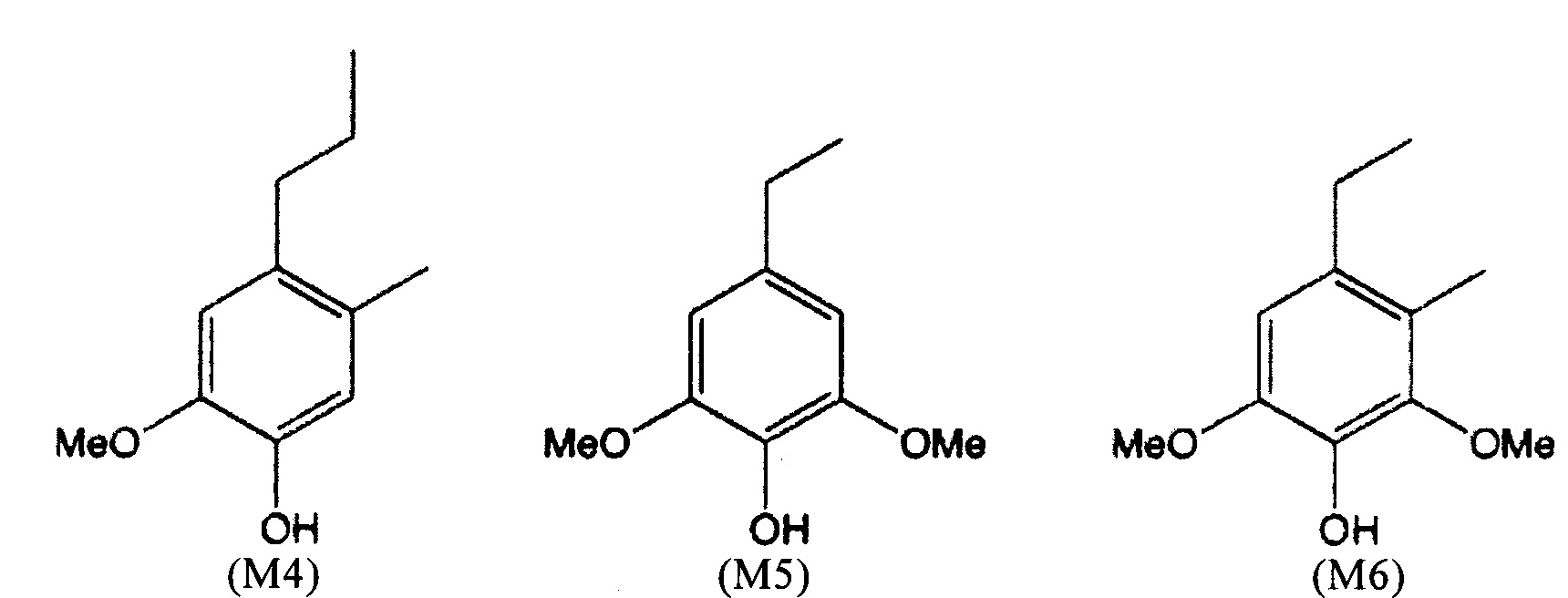
Примеры мономеров лигнина, которые могут быть получены по способу согласно настоящему изобретению, могут быть представлены следующими формулами (М1-М24):







где
(M1) представляет собой 4-этил-2-метоксифенол, соответствующий гваяцилэтану,
(М2) представляет собой 4-этил-2-метокси-5-метилфенол, соответствующий метилированному гваяцилэтану,
(М3) представляет собой 2-метокси-4-пропилфенол, соответствующий гваяцилпропану,
(М4) представляет собой 2-метокси-5-метил-4-пропилфенол, соответствующий метилированному гваяцилпропану,
(М5) представляет собой 4-этил-2,6-диметоксифенол, соответствующий сирингилэтану,
(М6) представляет собой 4-этил-2,6-диметокси-3-метилфенол, соответствующий метилированному сирингилэтану,
(М7) представляет собой 2,6-диметокси-4-пропилфенол, соответствующий сирингилпропану,
(М8) представляет собой 2,6-диметокси-3-метил-4-пропилфенол, соответствующий метилированному сирингилпропану,
(М9) представляет собой 4-(3-гидроксипропил)-2,6-диметоксифенол, соответствующий сирингилпропанолу,
(М10) представляет собой 4-(3-гидроксипропил)-2,6-диметокси-3-метилфенол, соответствующий метилированному сирингилпропанолу,
(M11) представляет собой 2-метоксифенол,
(M12) представляет собой 2-метокси-4-метилфенол,
(М13) представляет собой 2,6-диметоксифенол,
(M14) представляет собой 2,6-диметокси-4-метилфенол,
(M15) представляет собой 4-(3-гидроксипропил)-2-метоксифенол,
(M16) представляет собой (E)-4-(3-гидроксипроп-1-енил)-2,6-диметоксифенол,
(M17) представляет собой 4-(3-гидроксипропил)-2,6-диметоксифенол, соответствующий сирингилпропанолу (стереоизомеру М9),
(M18) представляет собой 4-этил-2-метокси-3-метилфенол, соответствующий изомеру метилированного гваяцилэтана,
(M19) представляет собой 4-этил-2-метокси-3,5-диметилфенол, соответствующий дважды метилированному гваяцилэтану,
(М20) представляет собой 4-пропил-2-метокси-3-метилфенол, соответствующий изомеру метилированного гваяцилпропана,
(М21) представляет собой 4-пропил-2-метокси-3,5-диметилфенол, соответствующий дважды метилированному гваяцилпропану,
(М22) представляет собой 4-этил-2,6-диметокси-3,5-диметилфенол, соответствующий дважды метилированному сирингилэтану,
(М23) представляет собой 4-пропил-2,6-диметокси-3,5-диметилфенол, соответствующий дважды метилированному сирингилпропану,
(М24) представляет собой 4-(3-гидроксипропил)-2,6-диметокси-3,5-диметилфенол, соответствующий дважды метилированному сирингилпропанолу.
На стадии превращения фрагментов лигнина в мономеры, предпочтительно путем гидрогенолиза, фрагменты ксилана могут оставаться без изменений. В предпочтительном варианте реализации фрагменты из ксилана могут быть отделены от мономеров лигнина, например, путем дистилляции или путем кристаллизации.
В одном из вариантов реализации стадии согласно настоящему изобретению фрагменты ксилана могут быть дополнительно превращены в ксилозу. Указанное превращение предпочтительно может включать реакцию указанных фрагментов ксилана в воде в кислых условиях. Для указанных кислых условий справедливо упомянутое выше. Предпочтительные кислоты представляют собой хлористоводородную кислоту и серную кислоту, в частности серную кислоту.
В одном из вариантов реализации указанное превращение фрагментов ксилана проводят в 1-7 масс. %, предпочтительно 2-5 масс. %, более предпочтительно примерно 3 масс. % растворе серной кислоты в воде.
В одном из вариантов реализации указанное превращение фрагментов ксилана проводят в автоклаве, таком как реактор Парра, предпочтительно путем применения механического движения, такого как перемешивание.
В одном из вариантов реализации настоящего изобретения указанное превращение фрагментов ксилана может быть проведено при температуре от 60°C до 200°C, предпочтительно от 80°C до 180°C, более предпочтительно от 100°C до 150°C.Особенно предпочтительно, чтобы можно было применять температуру примерно 120°C.
Кроме того, предпочтительно, чтобы указанное превращение фрагментов ксилана могло быть проведено в течение периода от 15 до 240 минут, предпочтительно от 30 до 180 минут, более предпочтительно от 45 до 120 минут, в частности примерно 60 минут.
Таким образом, в другом аспекте настоящее изобретение относится к получению фрагментов ксилана. Предпочтительный фрагмент ксилана представляет собой диформилксилозу.
Таким образом, другим объектом настоящего изобретения является способ получения фрагментов ксилана путем деполимеризации, включающий стадии
a) обеспечения содержащей лигноцеллюлозу композиции,
b) нагревания композиции со стадии а) в кислых условиях совместно с альдегидом, кетоном, бороновой кислотой или соединением, выбранным из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, предпочтительно альдегидом, в частности формальдегидом,
с') отделения фрагментов ксилана от смеси, полученной на стадии b).
Для условий реакций и компонентов на стадиях а) и b) справедливо описанное выше в отношении стадий а) и b).
Предпочтительно, чтобы на стадии b) альдегид или кетон нагревали совместно с композицией со стадии а) в кислых условиях.
На стадии с') фрагменты ксилана отделяют от остаточной смеси со стадии b).
Стадия с') предпочтительно может включать следующие подстадии:
c1) разделение фрагментов ксилансодержащей фазы и остатка,
с2) нейтрализация указанных фрагментов ксилансодержащей фазы и удаление растворителя,
с3) обработка продукта, полученного на стадии с2), растворителем и удаление нерастворимых частей,
с'4) подвергание смеси со стадии с3) гидрогенолизу и последующее отделение фрагментов ксилана.
Для условий реакций и компонентов на подстадиях c1)-c3) справедливо описанное выше в отношении подстадий c1)-c3).
На стадии с4) смесь, полученная на стадии с3), предпочтительно может быть подвергнута гидрогенолизу.
Для гидрогенолиза смеси, полученной на стадии с3), могут быть применены условия и компонент, описанные выше. Фрагменты ксилана могут быть отделены от смеси, полученной путем гидрогенолиза, например, путем дистилляции или путем кристаллизации.
В предпочтительном варианте реализации в случае, когда альдегид, применяемый на стадии b), представляет собой формальдегид, фрагменты ксилана могут быть подвергнуты дистилляции при повышенной температуре и/или при пониженном давлении, предпочтительно при температуре примерно 120°C и давлении примерно 0,1 мбар. Полученный фрагмент ксилана представляет собой диформилксилозу. Диформилксилоза может быть идентифицирована с помощью различных спектров, полученных в результате применения соответствующих способов определения, как представлено на фигуре 6.
Фрагменты ксилана необязательно могут быть дополнительно превращены в ксилозу. Для указанного превращения фрагментов ксилана в ксилозу могут быть применены условия и компоненты, описанные выше.
Другим объектом настоящего изобретения является применение формальдегида для деполимеризации содержащей лигноцеллюлозу композиции. В предпочтительном варианте реализации формальдегид применяют для получения мономеров из лигнина путем деполимеризации лигноцеллюлозной биомассы.
В другом предпочтительном варианте реализации формальдегид, ацетальдегид или пропиональдегид применяют для получения фрагментов ксилана, в частности диформилксилозы или других аддуктов кетона и ксилозы или альдегида и ксилозы, кетона и ксилана или альдегида и ксилана, путем деполимеризации лигноцеллюлозной биомассы.
В другом предпочтительном варианте реализации формальдегид, ацетальдегид или пропиональдегид применяют для получения фрагментов глюкана, в частности глюкозы, глюкоолигомеров, аддуктов кетона и глюкозы или альдегида и глюкозы, кетона и глюкана или альдегида и глюкана, путем деполимеризации лигноцеллюлозной биомассы.
В предпочтительном варианте реализации мономеры из лигнина (фрагменты, полученные из лигнина) применяют для получения смол, включая заменители фенолформальдегидных смол и термореактивные смолы.
В предпочтительном варианте реализации мономеры из лигнина (фрагменты, полученные из лигнина) применяют для получения транспортного топлива.
В предпочтительном варианте реализации мономеры из лигнина (фрагменты, полученные из лигнина) применяют для получения мономеров для получения полимеров, включая стиролы, метакрилаты и сложные полиэфиры.
Что касается соединений и условий для деполимеризации указанной содержащей лигноцеллюлозу композиции, они соответствуют описанным в способе согласно настоящему изобретению.
Настоящее изобретение далее будет проиллюстрировано со ссылкой на следующие примеры.
Примеры:
I. Материалы
Все коммерческие химические продукты являлись аналитическими реагентами, и их применяли без дополнительной очистки. 5% рутений (Ru) на углеродной матрице (Ru/C), гваякол (2-метоксифенол, 98%), 4-этилгваякол (>97%), 4-пропилгваякол (>99%), сирингол (2,6-диметоксифенол, 99%) и пропилбензол (98%) были приобретены у компании Sigma Aldrich. Метанол (>99%), тетрагидрофуран (ТГФ, >99%) и дихлорметан (>99%) были приобретены у компании ABCR. 4-пропанолгваякол (3-(4-гидрокси-3-метоксифенил)-1-пропанол, >98%), 1,4-диоксан (98%) были приобретены у компании TCI chemicals. Образцы древесины бука (Fagus sylvatica) были собраны в Золликофене (Швейцария) в октябре 2014 года и были высушены на воздухе для дальнейшего хранения. До экспериментов размер буковой щепы уменьшали для того, чтобы она проходила через сито, величина меш для которого составляет 18.
II. Аналитические способы
(1) Анализ состава биомассы
Анализ состава биомассы и субстрата после экстракции лигнина проводили согласно способу, принятому Технической ассоциацией в целлюлозно-бумажной промышленности (TAPPI). Частицы древесины (0,25-0,50 г) вносили в 50 мл лабораторные стаканы с добавлением 7,5 мл раствора H2SO4 с концентрацией 72 масс. %. Полученную смесь оставляли при комнатной температуре в течение 2 часов и перемешивали стеклянной палочкой каждые 10 минут. Затем указанную суспензию переносили в круглодонную колбу и добавляли 290 мл воды для достижения концентрации H2SO4 3 масс. %. Стеклянную бутыль запечатывали при помощи завинчивающейся крышки и стерилизовали при 120°C в течение 1 часа в автоклаве. Полученный раствор фильтровали и фильтрат применяли для анализа сахаров на ВЭЖХ-хроматографе, осадок промывали водой и сушили при 105°C и взвешивали для определения лигнина Класона.
Для анализа предварительно обработанного раствора указанную суспензию после экстракции лигнина фильтровали и промывали 30 мл воды и анализировали с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Для гидролиза возможных олигомеров или ацеталированных сахаров до мономерных сахаров 20 мкл концентрированной серной кислоты добавляли к 1 мл фильтрата и нагревали до 120°C в течение 1 часа в автоклаве. Полученную смесь анализировали с помощью ВЭЖХ.
Анализ сахаров проводили с применением ВЭЖХ-системы Agilent Infinity 1260, оборудованной рефрактометрическим детектором и колонкой Bio-Rad Aminex НРХ-87Р, при 80°C с применением воды в качестве подвижной фазы и при скорости потока 0,6 мл/мин.
Анализ на определение фурфурола и гидроксиметилфурфурола (HMF) проводили с применением ВЭЖХ-системы Agilent Infinity 1260, оборудованной детектором ультрафиолетового излучения и колонкой Bio-Rad Aminex НРХ-87Н, при 80°C с применением 5 мМ H2SO4 в воде в качестве подвижной фазы и при скорости потока 0,6 мл/мин.
Анализ состава частиц древесины бука был следующим:

(2) Анализ мономеров лигнина
Для анализа мономеров лигнина после гидрогенолиза 1 мл полученного раствора непосредственно отбирали для проведения анализов без какой-либо дополнительной обработки кроме добавления 100 мкл подготовленного внутреннего стандарта (8 мг декана в 5 мл диоксана). Раствор (~1,1 мл) анализировали при помощи газового хроматографа (ГХ) (серия Agilent 7890 В), оборудованного колонкой НР5 и пламенно-ионизационным детектором (ПИД). Температура впрыска составляла 573 K (примерно 300°C). Программа температуры в колонке составляла: 313 K (примерно 40°C, 3 мин), от 30 K/мин (примерно -243°C/мин) до 373 K (примерно 100°C), от 40 K/мин (примерно -233°C) до 573 K (примерно 300°C) и 573 K (примерно 300°C, 5 мин). Температура обнаружения составляла 573 K (примерно 300°C). Коэффициенты чувствительности продуктов были получены с применением оценок из расчета на эффективное число атомов углерода по причине отсутствия коммерческих стандартов. Выход мономера рассчитывали следующим образом:



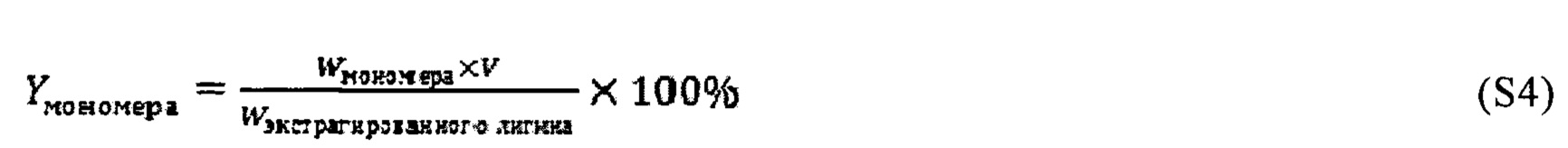
В указанных уравнениях
Wдекана в образце (мг): масса декана в качестве внутреннего стандарта в каждом анализируемом образце;
MWдекана (мг ммоль-1): молекулярная масса декана (142 мг ммоль-1);
nдекана (ммоль): молярное количество декана в каждом анализируемом образце;
nмономера (ммоль): молярное количество мономера в анализируемом образце;
Амономера в образце: площадь пика мономера в ГХ-ПИД-хроматограмме;
Адекана в образце: площадь пика декана в ГХ-ПИД-хроматограмме;
ECNдекана: эффективное число атомов углерода (10) в молекуле декана;
ECNмономера: эффективное число атомов углерода в молекуле мономера лигнина;
Wмономера (мг): молекулярная масса дегидратированных мономерных звеньев (гваяцилглицерин (196 мг ммоль-1) или дегидратированный сирингилглицерин (226 мг ммоль-1)) в зависимости от анализируемого мономера;
Yмономера: выход мономера из расчета на массу экстрагированного лигнина;
Wэкстрагированного лигнина (мг): Масса экстрагированного лигнина;
V (мл): общий объем образца, 1 мл которого применяли для анализа при помощи ГХ.
Идентификацию пиков мономеров на ГХ-ПИД-хроматограммах проводили первоначально при помощи газовой хроматографии с масс-спектрометрией (ГХ-МС) с применением газового хроматографа Agilent 7890 В, оборудованного капиллярной колонкой HP5-MS и масс-спектрометрическим детектором Agilent 5977А. Пики на ГХ-МС-хроматограмме появляются в тех же порядках, что и на ГХ-ПИД-хроматограмме по причине применения аналогичной капиллярной колонки. Применяли следующие рабочие режимы: температура впрыска при 523 К (примерно 250°C), программа температуры для колонки 323 К (примерно 50°C, 1 мин), от 15 К/мин (примерно -258°C/мин) до 573 К (примерно 300°C) и 573 К (примерно 300°C, 7 мин) и температура обнаружения 563 К (примерно 290°C).
(3) ЯМР-анализ лигнина
Высушенный образец растворяли в 600-1000 мкл D-хлороформа и переносили в пробирки для ЯМР. ЯМР-спектры получали на Bruker Avance III 400МГц спектрометре. Пик растворителя хлороформа применяли в качестве внутреннего эталона (δC, 77,2 ppm; δH, 7,24 ppm).
(4) Анализ гель-проникающей хроматографии (ГПХ)
Высушенный образец растворяли в 600-1000 мкл ТГФ и ГПХ-анализ проводили с применением ВЭЖХ-системы Agilent Infinity 1260, оборудованной рефрактометрическим детектором и колонкой Agilent PLgel MIXED С, при 40°C с применением ТГФ в качестве подвижной фазы и при скорости потока 1 мл/мин.
III. Идентификация и количественное определение мономера
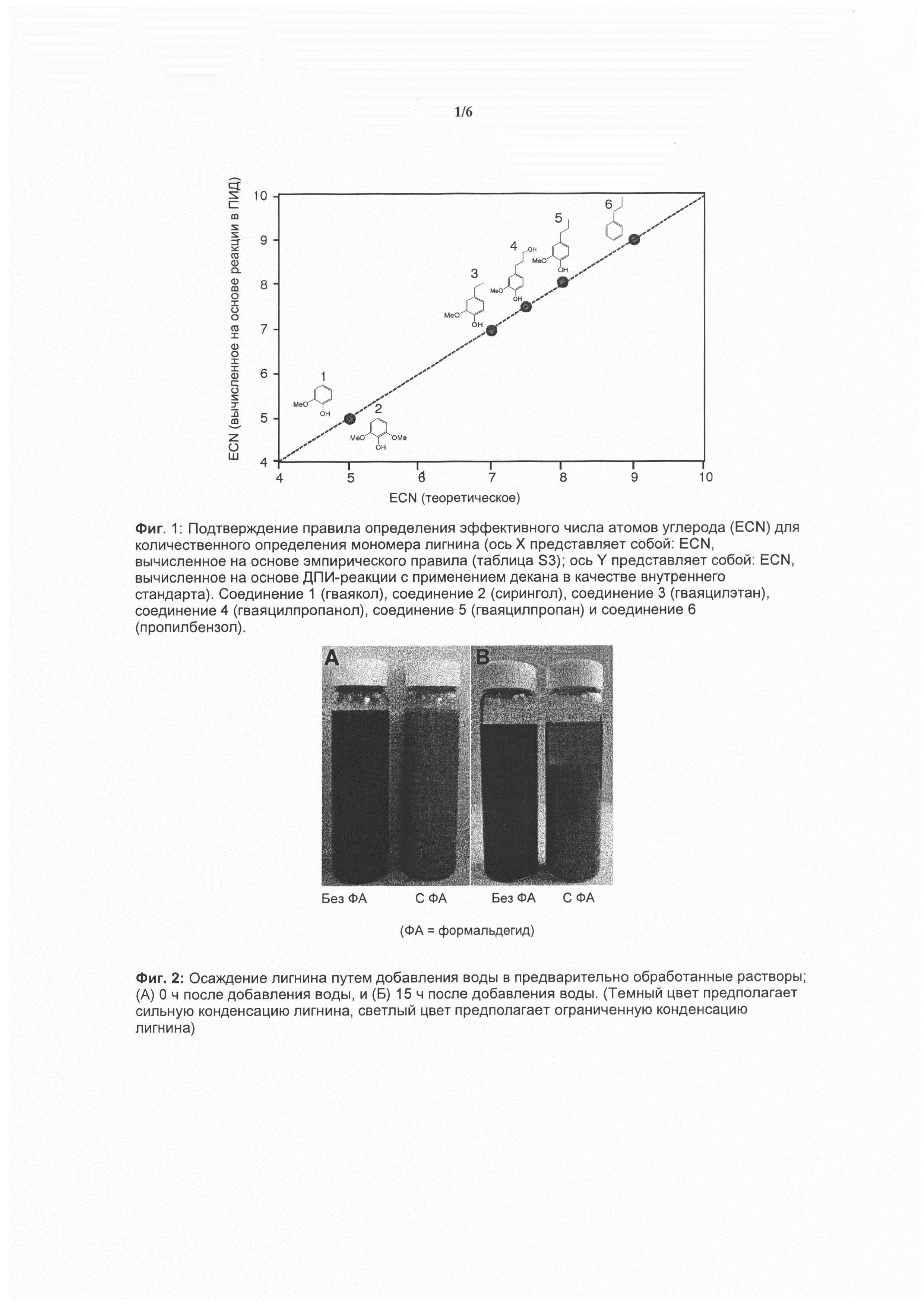
Валидация способа определения эффективного числа атомов углерода:
Правило определения эффективного числа атомов углерода (ECN) широко применяется для количественного определения углеродсодержащих продуктов, исходя из их реакций в ГХ-ПИД-хроматографе в случае, когда исходное стандартное соединение недоступно или доступно только в ограниченных количествах. ECN представляет собой сумму вкладов отдельных атомов углерода, модифицированную по вкладами их функциональных групп. Вследствие структурных различий в разных молекулах точность указанного правила может варьироваться. Для подтверждения точности указанного правила при его применении к количественной оценке мономера лигнина, измеряли эффективные числа атомов углерода в шести различных модельных соединениях лигнина, исходя из их известного количества и их реакций в ГХ-хроматографе по сравнению с реакциями декана. ECN, вычисленные на основе реакций в ПИД с применением декана в качестве внутреннего стандарта, сравнивали с теоретическим значением ECN, рассчитанным на основе указанного эмпирического правила. Для этой цели приведена ссылка на фиг. 1. Согласно эмпирическому правилу атомы углерода, связанные только с атомами водорода и углерода, добавляют 1 единицу к ECN, атомы углерода в метоксигруппе (простом эфире), как предполагается, не привносят вклад в реакцию в ПИД, атомы углерода, связанные с первичной гидроксильной группой, добавляют 0,5-0,6 единиц к ECN, и углерод, связанный с фенольным гидроксилом, обычно добавляет к ECN 0,5 единицы. Единственная несогласованность, которая была обнаружена авторами настоящего изобретения, заключается в том, что углерод, связанный с фенольным гидроксилом в мономерах лигнина, не привносит вклад в ECN. Вклад углерода мономеров лигнина в ECN (соответствующий приращениям для расчета ECN) приведен в таблице ниже:

Исходя из указанного скорректированного ECN-правила, ECN, вычисленные экспериментально, сопоставляли с теми числами, расчет которых основан на ECN-правиле с ошибками меньшими, чем 1% для всех соединений. Была продемонстрирована высокая точность при применении декана в качестве внутреннего стандарта и с применением коэффициента ECNмономера/ECNдекана (уравнение S2) для количественного определения мономеров лигнина. Основываясь на указанных результатах, авторы настоящего изобретения следовали эмпирическому правилу применительно к модифицированным фенольным углеродам, связанным с гидроксильной группой, с вкладом 0 для вычисления ECN мономеров лигнина, полученных из биомассы. Все эффективные числа атомов углерода мономеров лигнина (ECNмономеров), применяемые для количественного определения, перечислены в следующей таблице:

IV. Методика проведения эксперимента
1. Предварительное испытание: Экстракция лигнина (стадии а) и b)) с визуальным сравнением
Пример 1 согласно настоящему изобретению:
В стеклянный флакон вместимостью 50 мл вносили 1 г частиц высушенной на воздухе древесины бука, 9 мл диоксана, 420 мкл раствора HCl (36,5-37% в воде) (180-185 мг HCl и 315 мкл воды) и 1 мл раствора формальдегида (ФА) (36,5% в воде) (400 мг ФА и 690 мкл воды). Реакцию проводили на масляной бане, установленной на 80°C, в течение пяти часов и реакционную смесь перемешивали с помощью якоря магнитной мешалки при скорости перемешивания 300 об/мин. После завершения реакции суспензию фильтровали и промывали ацетоном до тех пор, пока фильтрат не становился бесцветным. Затем фильтрат нейтрализовывали путем добавления раствора бикарбоната натрия (~420 мг в 5 мл воды) и дополнительно разбавляли водой для осаждения «фрагментов лигнина».
Сравнительный пример 1 (без добавления формальдегида)
В стеклянный флакон вместимостью 50 мл вносили 1 г частиц высушенной на воздухе древесины бука, 9 мл диоксана, 420 мкл раствора HCl (36,5-37% в воде) (180-185 мг HCl и 315 мкл воды) и 690 мкл воды. Реакцию проводили на масляной бане, установленной на 80°C, в течение пяти часов и реакционную смесь перемешивали с помощью якоря магнитной мешалки при скорости перемешивания 300 об/мин. После завершения реакции суспензию фильтровали и промывали ацетоном до тех пор, пока фильтрат не становился бесцветным. Затем фильтрат нейтрализовывали путем добавления раствора бикарбоната натрия (~420 мг в 5 мл воды) и дополнительно разбавляли водой для осаждения экстрагированного лигнина. Снимки делали сразу после добавления воды и через 15 часов после добавления воды.
Как можно видеть из фигуры 2, цвет «фрагментов лигнина», полученных по примеру 1 согласно настоящему изобретению, является светлым, что свидетельствует об ограниченной конденсации, тогда как более темный цвет «фрагментов лигнина», полученных по сравнительному примеру 1, указывает на нежелательную сильную конденсацию.
2. Получение мономеров (стадии a)-d))
Примеры 2.1-2.6 согласно настоящему изобретению (стадии а)-с))
В стеклянный флакон вместимостью 50 мл вносили 1 г частиц высушенной на воздухе древесины бука, 9 мл диоксана, 420 мкл раствора HCl (36,5-37% в воде) (180-185 мг HCl и 315 мкл воды) и 1 мл раствора формальдегида (ФА) (36,5% в воде) (400 мг ФА и 690 мкл воды). Реакцию проводили на масляной бане, установленной на соответствующую заданную температуру, и реакционную смесь перемешивали в течение соответствующих заданных часов с помощью якоря магнитной мешалки при скорости перемешивания 300 об/мин. После завершения реакции суспензию фильтровали и промывали ацетоном до тех пор, пока фильтрат не становился бесцветным. Затем фильтрат нейтрализовывали путем добавления раствора бикарбоната натрия (~420 мг в 5 мл воды). Растворитель удаляли в ротационном испарителе, установленном на 60°C. Высушенный остаток растворяли в 25 мл тетрагидрофурана (ТГФ) для экстракции лигнина, при этом соль и углеводы оставались в виде осадка.
Примеры 2.1-2.6 согласно настоящему изобретению (стадия d); гидрогенолиз):
В реактор Парра высокого давления вместимостью 50 мл вносили 20 мл полученного раствора лигнина в ТГФ совместно с 100 мг катализатора (5 масс. % Ru/C). Содержимое реактора перемешивали с применением якоря магнитной мешалки, и реактор нагревали с помощью высокотемпературной нагревательной ленты (Omega), подключенной к источнику переменного питания, управляемому посредством пропорционально-интегрально-дифференцирующего (ПИД) регулятора температуры (Omega) с термопарой K-типа, которая измеряла температуру реакции через термокарман. После закрытия реактор продували 3 раза, а затем создавали давление водорода 40 бар. Реактор нагревали до соответствующей заданной температуры, а затем выдерживали при указанной температуре в течение соответствующего заданного времени пребывания. После завершения реакции реактор охлаждали с помощью внешнего потока сжатого воздуха при комнатной температуре. Образец полученной жидкости отбирали для анализа при помощи ГХ.
Пример 2.7
Условия для экстракции лигнина, описанные в примерах 2.1-2.6, соответствующим образом применяли, как описано выше. После завершения реакции суспензию фильтровали и промывали посредством 10 мл диоксана. Затем фильтрат нейтрализовывали с применением раствора бикарбоната натрия (~420 мг в 5 мл воды). К нейтрализованному раствору добавляли диоксан для достижения 25 мл и центрифугировали для удаления любых осажденных солей.
В реактор Парра высокого давления вместимостью 50 мл вносили 20 мл полученного раствора лигнина в диоксане совместно с 100 мг катализатора (5 масс. % Ru/C), и остальную процедуру проводили, как описано выше, при этом реактор нагревали до соответствующей заданной температуры, а затем выдерживали при указанной температуре в течение соответствующего заданного времени пребывания.
Пример 2.8
Применяли условия, описанные в примерах 2.1-2.6 (стадии a)-d)), при этом после удаления растворителя высушенный остаток растворяли в 25 мл метанола, после чего подвергали гидрогенолизу.
Пример 2.9
Применяли условия, описанные в примерах 2.1-2.6 (стадии a)-d)), при этом при экстракции лигнина вносили только 1/10 формальдегида.
Пример 2.10
Применяли условия, описанные в примерах 2.7, при этом для стадий а)-с), а также для стадии d) применяли γ-валеролактон (GVL).
На фиг. 3а представлена ГХ-хроматограмма и на фиг. 3b представлен масс-спектр мономеров из лигнина (ароматических соединений), полученных по способу согласно настоящему изобретению. На фиг. 4 представлены данные в отношении ЯМР-характеристики мономеров из лигнина. Что касается всех указанных данных, можно видеть, что способ согласно настоящему изобретению позволяет получить десять различных ароматических соединений, которые были определены.
Сравнительный пример 2.1 (стадия а)-с) без добавления формальдегида)
В стеклянный флакон вместимостью 50 мл вносили 1 г частиц высушенной на воздухе древесины бука, 9 мл диоксана, 420 мкл раствора HCl (36,5-37% в воде) (180-185 мг HCl и 315 мкл воды) и 690 мкл воды. Реакцию проводили на масляной бане, установленной на 80°C, в течение пяти часов и реакционную смесь перемешивали с помощью якоря магнитной мешалки при скорости перемешивания 300 об/мин. После завершения реакции суспензию фильтровали и промывали ацетоном до тех пор, пока фильтрат не становился бесцветным. Затем фильтрат нейтрализовывали путем добавления раствора бикарбоната натрия (~420 мг в 5 мл воды). Растворитель удаляли в ротационном испарителе, установленном на 60°C. Высушенный остаток растворяли в 25 мл ТГФ для экстракции лигнина, при этом соль и углеводы оставались в виде осадка.
Стадия d) (гидрогенолиз):
В реактор Парра высокого давления вместимостью 50 мл вносили 20 мл полученного раствора лигнина в ТГФ совместно с 100 мг катализатора (5 масс. % Ru/C), и остальную процедуру проводили, как в примерах 2.1-2.9, при этом реактор нагревали до 200°C, а затем выдерживали при указанной температуре в течение 6 часов.
Сравнительный пример 2.2 (только стадия d))
1 г древесной муки смешивали с 20 мл тетрагидрофурана и 200 мг катализатора, представлявшего собой 5 масс. % Ru/C. Полученную суспензию вносили в реактор Парра высокого давления вместимостью 50 мл, и остальную процедуру проводили, как в примерах 2.1-2.9, при этом реактор нагревали до 250°C, а затем выдерживали при указанной температуре в течение 15 часов.
Сравнительный пример 2.3
Процедуру проводили, как в сравнительном примере 2.2, при этом вместо тетрагидрофурана применяли метанол.
Количество экстрагированного лигнина определяли путем вычитания количества лигнина Класона в экстрагированном остатке из количества лигнина Класона в необработанной древесине.
В таблице 1 представлены конкретные условия для экстракции и гидрогенолиза и выходы мономеров из лигнина (ароматических соединений), полученных с применением соответствующих конкретных условий. Из указанной таблицы можно видеть, что в каждом из примеров 2.1-2.10 согласно настоящему изобретению демонстрируется значительно более высокий выход мономеров из расчета на экстрагированный лигнин, чем в сравнительном примере 2.1, где стадию экстракции проводили в отсутствие формальдегида.
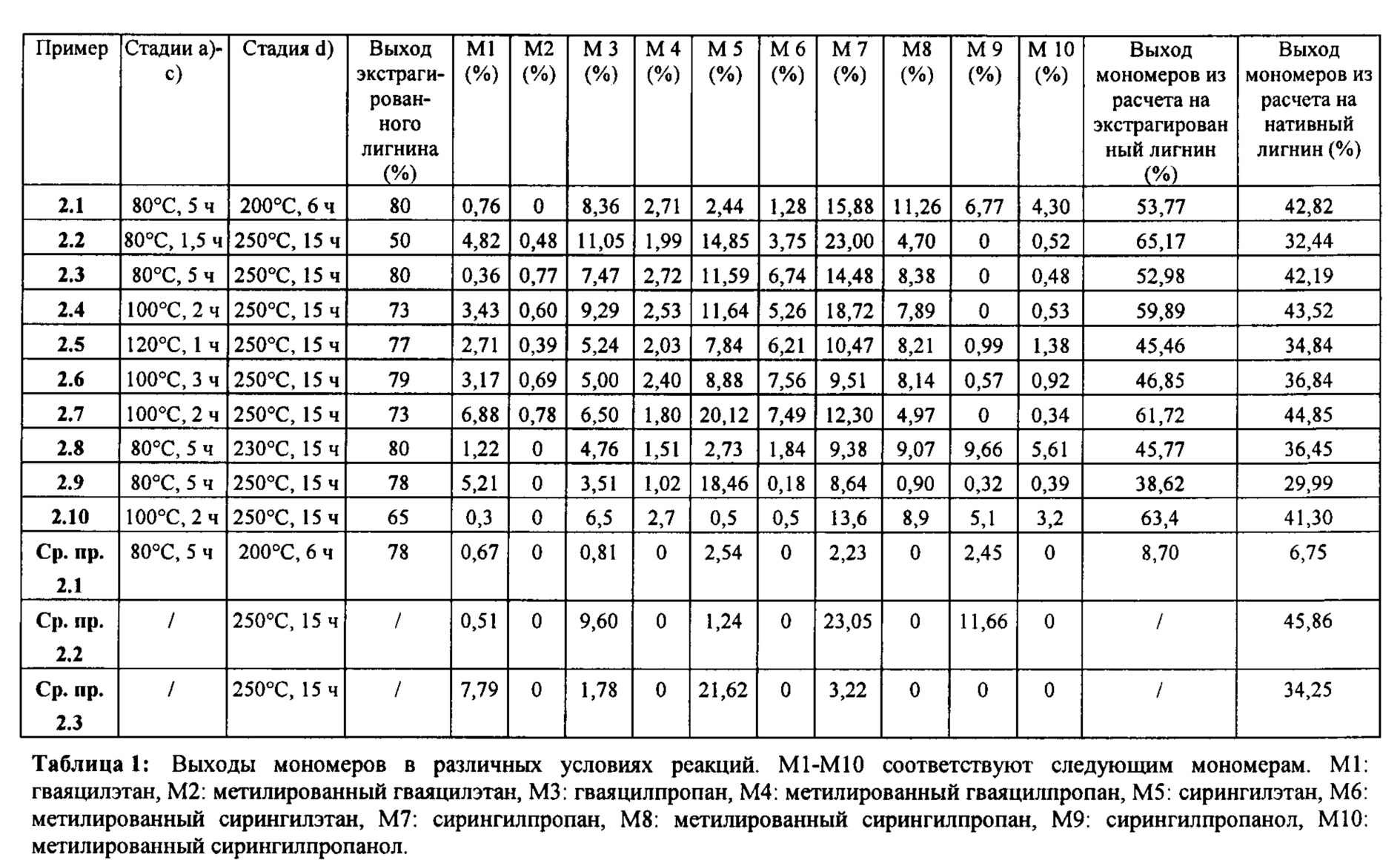
Кроме того, из сравнительных примеров 2.2 и 2.3 можно видеть, что при осуществлении прямого гидрогенолиза образца обеспечивается высокий выход мономеров из лигнина. Однако выход не является единственным решающим фактором. Прямой гидрогенолиз имеет недостатки, описанные выше.
Кроме того, согласно фиг. 5 при прямом гидрогенолизе, примененном в обоих сравнительных примерах 2.2 и 2.3, получают лишь небольшое количество различных ароматических соединений. Способ согласно настоящему изобретению, напротив, позволяет получить большее разнообразие ароматических соединений.
Пример 3
Примеры Пр. 3.1-Пр. 3.10; стадии а)-с) согласно настоящему изобретению
Пр. 3.1 В стеклянный флакон вместимостью 50 мл вносили 1 г частиц высушенной на воздухе древесины березы, 9 мл диоксана, 420 мкл раствора HCl (36,5-37% в воде) (180-185 мг HCl и 315 мкл воды) и 1 мл раствора формальдегида (ФА) (36,5% в воде) (400 мг ФА и 690 мкл воды). Реакцию проводили на масляной бане, установленной на 90°C, в течение 3,5 часов, и реакционную смесь перемешивали с помощью якоря магнитной мешалки при скорости перемешивания 300 об/мин. После завершения реакции суспензию фильтровали и промывали ацетоном до тех пор, пока фильтрат не становился бесцветным. Затем фильтрат нейтрализовывали путем добавления раствора бикарбоната натрия (~420 мг в 5 мл воды). Растворитель удаляли в ротационном испарителе, установленном на 60°C. Высушенный остаток растворяли в 25 мл ТГФ для экстракции лигнина, при этом соль и углеводы оставались в виде осадка.
Пр. 3.2 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом ацетальдегида совместно с 690 мкл воды.
Пр. 3.3 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом пропиональдегида совместно с 690 мкл воды.
Пр. 3.4 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом бензальдегида совместно с 690 мкл воды.
Пр. 3.5 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом ацетона совместно с 690 мкл воды.
Пр. 3.6 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом 2-бутанона совместно с 690 мкл воды.
Пр. 3.7 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом фенилбороновой кислоты совместно с 690 мкл воды.
Пр. 3.8 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом 2-метоксипропена совместно с 690 мкл воды.
Пр. 3.9 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом диметилкарбоната совместно с 690 мкл воды.
Пр. 3.10 проводили, как Пр. 3.1, при этом раствор формальдегида заменяли молярным эквивалентом 2,2-диметоксипропана совместно с 690 мкл воды.
Контрольные примеры К. пр. 3.1 и К. пр. 3.2
В стеклянный флакон вместимостью 50 мл вносили 1 г частиц высушенной на воздухе древесины березы, 9 мл диоксана, 420 мкл раствора HCl (36,5-37% в воде) (180-185 мг HCl и 315 мкл воды) и 690 мкл воды. Реакцию проводили на масляной бане, установленной на 90°C, в течение 3,5 часов и реакционную смесь перемешивали с помощью якоря магнитной мешалки при скорости перемешивания 300 об/мин. После завершения реакции суспензию фильтровали и промывали ацетоном до тех пор, пока фильтрат не становился бесцветным. Затем фильтрат нейтрализовывали путем добавления раствора бикарбоната натрия (~420 мг в 5 мл воды). Растворитель удаляли в ротационном испарителе, установленном на 60°C. Высушенный остаток растворяли в 25 мл ТГФ для экстракции лигнина, при этом соль и углеводы оставались в виде осадка.
Стадия d) гидрогенолиза
20 мл каждого из полученных растворов лигнина в ТГФ из примеров Е3.1-Е3.10, а также СЕ3.1 и СЕ3.2, описанных выше, каждый совместно с 100 мг катализатора (5 масс. % Ru/C) вносили в реактор Парра высокого давления вместимостью 50 мл, соответственно. Содержимое реактора перемешивали с применением якоря магнитной мешалки, и реактор нагревали с помощью высокотемпературной нагревательной ленты (Omega), подключенной к источнику переменного питания, управляемому посредством ПИД-регулятора температуры (Omega) с термопарой K-типа, которая измеряла температуру реакции через термокарман. После закрытия реактор продували 3 раза, а затем создавали давление водорода 40 бар. Реактор нагревали до 250°, а затем выдерживали при указанной температуре в течение 15 часов. После завершения реакции реактор охлаждали с помощью внешнего потока сжатого воздуха при комнатной температуре. Образец полученной жидкости отбирали для ГХ-анализа.
В таблице 2 представлены конкретные примеры в отношении и выходы мономеров из лигнина (ароматических соединений), полученных путем добавления конкретных альдегидов, кетонов, бороновых кислот и соединений, выбранных из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана.

Указанные выходы вычисляли из расчета на количество лигнина Класона в 1 г березы (18% лигнина Класона).
Как можно видеть из таблицы 2, все примеры, в которых применяли альдегиды (Пр. 3.1-Пр. 3.4), кетоны (Пр. 3.5 и Пр. 3.6), бороновую кислоту (Пр. 3.7) и соединения, выбранные из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана (Пр. 3.8-Пр. 3.10), позволяют получить статистически значительно более высокий общий выход мономеров из лигнина по сравнению с контрольными примерами (К. пр. 3.1 и К. пр. 3.2).
Реферат
Настоящее изобретение относится к способу получения мономеров, способу получения фрагментов ксилана, применению формальдегида и применению мономеров. Способ получения мономеров из лигнина путем деполимеризации включает стадии: a) обеспечения содержащей лигноцеллюлозу композиции, b) нагревания композиции со стадии а) в кислых условиях совместно с альдегидом, кетоном, бороновой кислотой или соединением, выбранным из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, c) отделения фрагментов лигнина от смеси, полученной на стадии b), d) превращения фрагментов лигнина, полученных на стадии с), в мономеры. Способ получения фрагментов ксилана путем деполимеризации включает стадии: a) обеспечения содержащей лигноцеллюлозу композиции, b) нагревания композиции со стадии а) в кислых условиях совместно с альдегидом, кетоном, бороновой кислотой или соединением, выбранным из 2-метоксипропена, диметилкарбоната и 2,2-диметоксипропана, и с') отделения фрагментов ксилана от смеси, полученной на стадии b). Мономеры, полученные из лигнина, применяются для получения смол, транспортного топлива или мономеров для получения полимеров. Технический результат – разработка способа получения мономеров из лигнина, который может быть применен простым и эффективным образом даже в случае применения в крупномасштабном процессе и предпочтительно совместим с общими методиками биопереработки. 4 н. и 13 з.п. ф-лы, 6 ил., 2 табл., 23 пр.
Формула






Комментарии