Полимерные материалы, имеющие пониженную липкость, способы получения этих материалов и композиции жевательной резинки, содержащие такие материалы - RU2368626C2
Код документа: RU2368626C2
Чертежи


Описание
Область техники
Настоящее изобретение относится к полимерным материалам, имеющим пониженную липкость. Оно также относится к способам изготовления таких материалов и к композициям жевательной резинки, содержащим такие полимерные материалы.
Предшествующий уровень техники
Некоторые углеводородные полимеры, такие как гомополимеры диеновых мономеров, например, изопрена, демонстрируют тенденцию прилипать к другой поверхности при приведении в тесный контакт с данной поверхностью. Однако при некоторых применениях этих углеводородных полимеров свойство липкости может быть недостатком. Одним из таких применений является применение таких материалов в композициях жевательной резинки.
Композиции жевательной резинки нравятся миллионам людей во всем мире. К сожалению, многие пользователи жевательной резинки не избавляются, или не способны избавиться, от использованной жевательной резинки должным образом. Использованная жевательная резинка, от которой не избавились должным образом, вызывает проблемы благодаря ее тенденции прочно прилипать ко многим поверхностям, с которыми ей позволяют вступить в тесный контакт. Из-за этого многие места общественного пользования, особенно тротуары, были обезображены присутствием прилипших кусочков выброшенной использованной жевательной резинки. Жевательную резинку, прилипшую к поверхности, трудно удалить традиционными способами, особенно если эта резинка находилась в контакте с данной поверхностью в течение некоторого длительного периода времени. Такие поверхности можно энергично промывать с использованием воды, приложенной под высоким давлением, возможно в комбинации с соскабливанием, в попытке удалить прилипшую резинку. К сожалению, такой способ требует применения больших количеств воды, может вызвать эрозию поверхности, которую обрабатывают, требует больших затрат времени и энергии и зачастую может быть осуществлен только тогда, когда люди освободили место, подлежащее обработке, в связи с возникшим шумом и большими количествами требующейся воды. Другие типичные способы очистки поверхностей от прилипшей использованной жевательной резинки включают в себя соскабливание, возможно с применением низкотемпературного материала для того, чтобы вызвать замораживание резинки, или применение агрессивных химических веществ. Таким образом, способы очистки поверхностей для удаления с них прилипшей резинки могут быть дорогостоящими и могут создавать неудобство для населения.
Предположили, что проблем, упомянутых выше, можно избежать путем модификации свойств жевательной резинки. Например, в патенте США №5580590 описана безвредная для окружающей среды композиция жевательной резинки, которая содержит эластичный полимер на основе белка. Благодаря применению эластичного полимера на основе белка предложена композиция жевательной резинки, которую можно легче, после того как ее прожевали, удалить с физической поверхности, к которой она приклеена.
Композиции жевательной резинки типично содержат нерастворимую в воде часть гуммиосновы жевательной резинки, которая содержит один или более эластомеров, таких как полибутадиен, полиизопрен, бутадиенстирольные сополимеры, полиизобутилен и изобутилен-изопреновые сополимеры. Эти материалы демонстрируют липкость и со временем могут прочно прилипать к поверхностям. Сильная адгезия является причиной того, что выброшенная жевательная резинка не поддается отделению от поверхностей, таких как поверхности тротуаров. Липкость материала определяют как способность данного материала образовывать связь с поверхностью после кратковременного контакта при легком нажатии.
Раскрытие изобретения
Настоящее изобретение основано на открытии, что липкость углеводородных полимеров, например, природных и синтетических каучуков, может быть снижена, если данные углеводородные полимеры модифицированы путем присоединения непосредственно к их углерод-углеродным основным цепям (углерод-углеродным скелетам) определенных боковых цепей, имеющих гидрофильный характер.
Полимерные материалы, имеющие боковые цепи, содержащие полиалкиленоксидные группы, раскрыты в ЕР-А-1179564 в качестве полезных в изготовлении композиции антистатической смолы. В данном документе описан графт-сополимер, содержащий 50-95 мас.% по меньшей мере одного мономера, выбранного из конъюгированных диенов (таких как 1,3-бутадиен или изопрен) и сложных эфиров акриловой кислоты (таких как этил- или пропилакрилат), 5-50 мас.% по меньшей мере одного мономера, имеющего от 4 до 500 алкиленоксидных групп и этилен-ненасыщенную связь ("полиалкиленоксидный мономер") и 0-50 мас.% одного или более сополимеризующихся этилен-ненасыщенных мономеров. Полиалкиленоксидный мономер типично представляет собой акрилатный или метакрилатный сложный эфир полиалкиленгликоля. Таким образом, графт-сополимер синтезируют способом "прививки" (также известным в данной области как "прививка посредством макромономеров") и, соответственно, полиалкиленоксидные группы присоединяют к атомам углерода полимерного скелета полученного сополимера через группы -С(O)O-.
Соответственно, в первом аспекте изобретения предложен полимерный материал с пониженной липкостью, который имеет углерод-углеродный полимерный скелет с прямой или разветвленной цепью и множество боковых цепей, присоединенных к этому скелету, где боковые цепи, которые присоединены непосредственно к атомам углерода полимерного скелета, имеют формулу
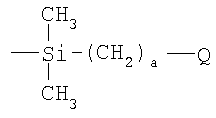
или имеют формулу
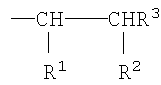
где R1 представляет собой Н, -C(O)OR4 или -C(O)Q, и R2 представляет собой -C(O)OR4 или -C(O)Q, при условии, что по меньшей мере один из R1 и R2представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, имеющую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, где каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода;
а равно 3 или 4 и каждая из переменных b и с независимо равна 0 или целому числу от 1 до 125, при условии, что сумма b+с имеет значение в диапазоне от 10 до 250, предпочтительно 10 до 120.
Полимерный материал по изобретению имеет пониженную липкость по сравнению с углеводородным полимером, который не модифицирован присутствием боковых цепей, присоединенных к нему. Кроме того, он является менее липким к некоторым поверхностям, чем немодифицированный углеводородный полимер.
Полимерный материал по изобретению имеет углерод-углеродный полимерный скелет, как правило, полученный из гомополимера этилен-ненасыщенного полимеризующегося углеводородного мономера или из сополимера двух или более этилен-ненасыщенных полимеризующихся углеводородных мономеров. Под термином "этилен-ненасыщенный полимеризующийся углеводородный мономер" авторы изобретения подразумевают полимеризующийся углеводород, содержащий по меньшей мере одну двойную углерод-углеродную связь, которая способна участвовать в реакции присоединения или реакции цепной полимеризации с образованием углеводородного полимера с прямой или разветвленной цепью, имеющего углерод-углеродный полимерный скелет. Согласно одному из предпочтительных вариантов осуществления изобретения углерод-углеродный полимерный скелет получен из гомополимера этилен-ненасыщенного полимеризующегося углеводородного мономера, содержащего 4 или 5 атомов углерода, например, изобутилен(2-метилпропен). Согласно другому варианту осуществления изобретения углерод-углеродный полимерный скелет также может быть получен из гомополимера конъюгированного диенового углеводородного мономера, особенно содержащего 4 или 5 атомов углерода, такого как бутадиен или изопрен.
Как упомянуто выше, углерод-углеродный полимерный скелет может быть получен из сополимера двух или более этилен-ненасыщенных полимеризующихся углеводородных мономеров. Предпочтительно он получен из сополимера двух таких мономеров. Например, он может быть получен из углеводородного сополимера углеводородного мономера, имеющего одну двойную углерод-углеродную связь, и углеводородного мономера, имеющего две двойные углерод-углеродные связи. Например, углерод-углеродный полимерный скелет может быть получен из сополимера изобутилена и изопрена. Согласно другому варианту осуществления изобретения углерод-углеродный полимерный скелет получен из бутадиенстирольного блок-сополимера.
Особенно предпочтительным в настоящем изобретении является то, что полимерный материал имеет по существу линейный углерод-углеродный полимерный скелет, полученный из углеводородного полимера с прямой или разветвленной цепью, который представляет собой эластомер при температурах окружающей среды. Эластомерные полимеры имеет эластичные свойства при температурах выше их температуры стеклования (Tg), и некоторые синтетические эластомеры проявляют эти и другие свойства, ассоциированные с природным каучуком, который представляет собой полимер, полученный из цис-изопреновых (2-метил-1,3-бутадиеновых) мономерных звеньев. В настоящем изобретении полимерный материал предпочтительно имеет по существу линейный углерод-углеродный полимерный скелет, полученный из эластомерных полимеров, выбранных из полибутадиеновых, полиизопреновых, бутадиенстирольных блок-сополимеров, полиизобутиленовых и изобутилен-изопреновых сополимеров, более предпочтительно - полибутадиеновых, полиизопреновых, полиизобутиленовых и изобутилен-изопреновых сополимеров, и наиболее предпочтительно - полиизопрена, который может представлять собой природный каучук или синтетически производимый полиизопрен. Под термином "по существу линейный", который использован в данном описании, авторы изобретения подразумевают, что углерод-углеродный скелет не содержит в значительной степени разветвлений с длинной цепью.
Углеводородный полимер, из которого получен углерод-углеродный полимерный скелет полимерного материала по изобретению, типично будет иметь молекулярную массу в диапазоне от 15000 до 50000, предпочтительно от 25000 до 40000, для гарантии того, что полимерный материал не является чрезмерно жестким.
Как указано выше, полимерный материал по изобретению содержит множество боковых цепей, присоединенных непосредственно к атомам углерода углерод-углеродного полимерного скелета. Боковые цепи имеют формулу
или имеют формулу
где R1 представляет собой Н, -C(O)OR4 или -C(O)Q и R2 представляет собой -C(O)OR4 или -C(O)Q, при условии, что по меньшей мере один из R1 и R2представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, имеющую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, где каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода;
а равно 3 или 4 и каждая из переменных b и с независимо равна 0 или целому числу от 1 до 125, при условии, что сумма b+с имеет значение в диапазоне от 10 до 250, предпочтительно от 10 до 120.
Согласно вышесказанному, согласно одному из вариантов осуществления изобретения полимерный материал имеет множество боковых цепей, присоединенных непосредственно к атомам углерода углерод-углеродного полимерного скелета, имеющих формулу
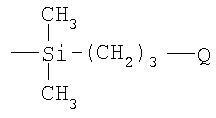
где Q является таким, как определено выше.
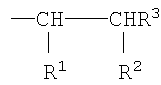
Согласно другому варианту осуществления изобретения полимерный материал имеет множество боковых цепей, присоединенных непосредственно к атомам углерода углерод-углеродного полимерного скелета, которые имеют формулу
где R1, R2 и R3 являются такими, как определено выше. Согласно предпочтительному варианту осуществления изобретения боковые цепи имеют вышеуказанную формулу, где R1 представляет собой Н, R3 представляет собой -СН3 и R2 представляет собой группу -C(O)Q, в которой Q является таким, как определено выше. Согласно другому предпочтительному варианту осуществления изобретения боковые цепи имеют вышеуказанную формулу, в которой R3 представляет собой Н и в которой одна из групп R1 и R2 представляет собой группу -C(O)Q, а другая из этих групп представляет собой -C(O)OR4, в которой R4 и Q являются такими, как определено выше. Группа Q, которая представлена в упомянутых выше боковых цепях, имеет формулу
-O-(YO)b-(ZO)c-R5,
в которой Y, Z, R5, b и с являются такими, как определено выше.
Полимерный материал по изобретению содержит, как описано выше, гидрофобный углерод-углеродный полимерный скелет, на который привито множество боковых цепей, которые, благодаря содержанию в них алкиленоксигрупп, имеют гидрофильную природу. Комбинация гидрофобного скелета с гидрофильными боковыми цепями, привитыми на скелет, дает амфифильное вещество, имеющее свойства, которые зависят от количества и характера боковых цепей, привитых на гидрофобный полимерный скелет, т.е. в тех случаях, когда количество алкиленоксигрупп в полимерном материале снижается, гидрофобный характер полимерного скелета начинает доминировать над объемными свойствами полимерного материала, тогда как в тех случаях, когда количество алкиленоксигрупп в полимерном материале увеличивается, полимерный материал становится в большей степени гидрофильным. Кроме того, когда длина цепи алкиленокси в привитых боковых цепях увеличивается, объемные свойства полимерного материала имеют тенденцию становиться более похожими на свойства соответствующего полиалкиленового полимера. Поэтому согласно настоящему изобретению можно производить полимерный материал, имеющий желаемый баланс эластомерных и гидрофильных свойств. По этой причине, каждый из коэффициентов b и с в группе Q, определенной выше, независимо равен от 0 до 125, при условии, что сумма b+с лежит в пределах диапазона от 10 до 250. Предпочтительно b+с находится в диапазоне от 10 до 120.
Конечно, в настоящем изобретении нет необходимости, чтобы все боковые цепи имели одинаковые значения b и с. Очевидно, что в полимерном материале по изобретению разные боковые цепи, привитые на углерод-углеродный полимерный скелет, могут иметь разные значения b и с, при условии, что b лежит в пределах диапазона от 0 до 125 и с лежит в пределах диапазона от 0 до 125, и при условии, что сумма b+с лежит в пределах диапазона от 10 до 250. Предпочтительно, чтобы гарантировать, что боковые цепи в полимерном материале обеспечат предпочтительную степень гидрофильности в полимерном материале и, следовательно, большее снижение липкости к поверхностям, значение суммы b+с в боковых цепях должно находиться в пределах диапазона от 10 до 120, более предпочтительно от 20 до 60, особенно от 30 до 50 и еще более предпочтительно от 40 до 45. Хотя адгезивность или липкость зависит от поверхностных свойств самой поверхности, с которой полимерный материал может находиться в тесном контакте, наиболее предпочтительно в настоящем изобретении, чтобы значение b+с находилось в диапазоне от 40 до 45 для того, чтобы полимерный материал проявлял пониженную адгезивность и пониженную липкость к ряду твердых поверхностей и, как следствие, легче удалялся в присутствии воды с некоторых твердых поверхностей. Как обсуждалось выше, присутствие полиоксиалкиленовой функциональной группы в боковых цепях будет придавать гидрофильность боковым цепям. Каждая из алкиленовых групп Y и Z в группе Q, определенной выше, независимо содержит от 2 до 4 атомов углерода, и примеры таких алкиленовых групп включают в себя этилен, пропилен, триметилен и тетраметилен. Однако, поскольку гидрофобность в боковых цепях увеличивается с содержанием углерода, предпочтительно, чтобы как Y, так и Z представляли собой этиленовые группы, так что присутствие множества боковых цепей, содержащих полиоксиэтиленовые группы, будет вызывать большее увеличение гидрофильности полимерного материала по сравнению с углеводородным полимером, не содержащим такие боковые цепи. Группа R5 в Q, как определено выше, представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода. Предпочтительно, для того, чтобы не приуменьшать гидрофильной природы боковых цепей, R5 не будет представлять собой группу, которая сама вносит значительную степень гидрофобности в боковую цепь, и поэтому R5 предпочтительно представляет собой Н или -СН3, более предпочтительно Н.
Как указано выше, свойства полимерного материала зависят не только от характера боковых цепей, привитых на углерод-углеродный полимерный скелет, но также от количества привитых боковых цепей. Согласно изобретению существенным является то, что множество боковых цепей присоединено к скелету. Подразумевают, что используемый в данном описании термин "множество" имеет свое нормальное значение, т.е. много, и поэтому исключает соединения, которые содержат одну или всего несколько привитых боковых цепей, которые, как ожидают, не будут иметь свойств, отличающихся от свойств углеводородного полимера, из которого получен углерод-углеродный полимерный скелет. Количество боковых цепей, привитых на углерод-углеродный полимерный скелет, согласно настоящему изобретению, типично будет выражаться в виде среднего арифметического по меньшей мере одной боковой цепи на углерод-углеродный полимерный скелет. Фактическое количество боковых цепей, привитых на углерод-углеродный полимерный скелет, зависит от природы боковой цепи и способа, с помощью которого боковую цепь прививают на полимерный скелет (и условий реакции, используемых в данном изобретении). Для того чтобы достичь желаемой степени гидрофильности в полимерном материале, предпочтительно, чтобы среднее количество привитых боковых цепей на основную цепь полимера находилось в диапазоне от 5 до 10, т.е. соотношение основной цепи к боковым цепям составляло от 1:5 до 1:10. Очевидно, что боковые цепи не обязательно должны располагаться с регулярными интервалами вдоль углерод-углеродного полимерного скелета, поскольку местоположение присоединения боковой цепи к скелету будет зависеть от положений подходящих мест присоединения в скелете углеводородного полимера, используемого для изготовления материала. Например, если углеводородный полимер представляет собой полимер, который содержит двойные углерод-углеродные связи в полимерном скелете, например полиизопрен, то эти или некоторые из этих двойных углерод-углеродных связей могут быть использованы в реакции, с помощью которой боковые цепи могут быть присоединены к скелету.
Полимерный материал по настоящему изобретению, имеющий боковые цепи, присоединенные непосредственно к атомам углерода в углерод-углеродном полимерном скелете, где боковые цепи имеют формулу
-СН2СН(СН3)-С(O)-O-(YO)b-(ZO)с-R5,
в которой Y, Z, R5, b и с являются такими, как определено выше, может быть получен способом, который включает взаимодействие углеводородного полимера с прямой или разветвленной цепью, в растворителе и в инертной атмосфере, с монометакрилатным соединением
CH2=C(CH3)C(O)O-(YO)b-(ZO)c-R5
в присутствии инициатора свободных радикалов. Реакцию между углеводородным полимером и метакрилатным соединением осуществляют в подходящем растворителе, и в этом смысле подходящий растворитель представляет собой растворитель, который является растворителем для реагентов и для используемого инициатора свободных радикалов. Типично, растворитель будет представлять собой неполярный органический растворитель, например толуол.
Может быть использован любой инициатор свободных радикалов, при условии, что он растворим в используемом растворителе, и при условии, что он способен отнимать метиленовые атомы водорода от скелета углеводородного полимера для инициации реакции прививки. Хорошие результаты в настоящем изобретении были получены с использованием бензоилпероксида в качестве инициатора свободных радикалов.
Для снижения вероятности образования поперечных связей между полимерами в реакционной смеси предпочтительно осуществлять способ по изобретению в разбавленном растворе. Типично, каждый углеводородный полимер и каждое монометакрилатное соединение будут использовать в концентрации, которая составляет менее 11 мас.%.
Реакцию осуществляют в инертной атмосфере. Типично, этого можно достичь путем продувки реакционного сосуда, содержащего раствор реагентов и инициатор, азотом в течение нескольких минут. Реакционную смесь типично нагревают до температуры выше температуры окружающей среды для ускорения реакции, и реакцию можно осуществлять в течение периода времени вплоть до нескольких дней с перемешиванием до тех пор, пока не достигнут завершения. Типично, реакцию осуществляют при температуре в диапазоне от 60 до 130°С, предпочтительно от 60 до 65°. Реакцию можно типично осуществлять в течение 20-150 часов. Предпочтительно ее осуществляют в течение 20-50 часов. По прошествии этого времени реакцию можно гасить путем быстрого охлаждения реакционного сосуда, например до 0°С, или путем быстрого разбавления реакционной смеси растворителем.
Полученный графт-сополимер может быть извлечен путем удаления, выпаривания, части растворителя и затем добавления метанола для того, чтобы вызвать осаждение желаемого графт-сополимера. Этот осадок, когда собран, может быть типично промыт несколько раз в метаноле при 60°С и затем высушен под вакуумом для удаления оставшегося растворителя.
Полимерный материал согласно настоящему изобретению, где боковые цепи, присоединенные непосредственно к атомам углерода в углерод-углеродном полимерном скелете, имеют формулу

в которой Y, Z, R5, a, b и с являются такими, как определено выше, может быть получен способом, который включает:
(1) взаимодействие соединения формулы
HO-(YO)b-(ZO)c-R5
с гидридом натрия в сухом органическом растворителе в инертной атмосфере;
(2) взаимодействие продукта, полученного на стадии (1), с соединением
СН2=СН-(СН2)q-Br,
где q означает 1 или 2,
с получением соединения II
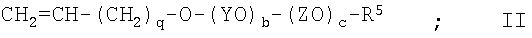
(3) взаимодействие соединения II с хлордиметилсиланом с получением соединения III
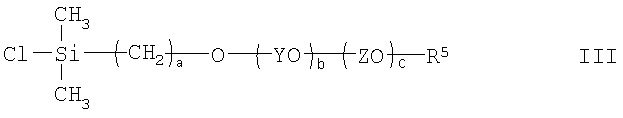
и
(4) восстановление соединения III и взаимодействие продукта α-гидродиметилсилилполиалкиленоксида с углеводородным полимером с прямой или разветвленной цепью, содержащим множество двойных углерод-углеродных связей в углеводородном полимерном скелете, в присутствии соли переходного металла.
Предпочтительно на стадии (2) выше продукт, полученный на стадии (1), подвергают взаимодействию с 3-бромпропеном, так что в формуле, приведенной выше для боковой цепи, «а» равно 3.
Очевидно, что реакция гидросилилирования, как описано выше (стадия 4), включает присоединение силановых соединений по двойным углерод-углеродным связям в углеводородном полимере. Реакции гидросилилирования обычно катализируются переходными металлами или их солями, особенно теми, которые образуют богатые электронами комплексы, например Pt(O), Pd(O), Rh(I), Ni(O) и Co(I). Авторы изобретения достигли хороших результатов, используя в этой реакции платинохлористоводородную кислоту (H2PtCl6) в качестве катализатора.
Согласно предпочтительному варианту осуществления изобретения на стадии (1) выше добавляют гидрид натрия к раствору в органическом растворителе, таком как толуол или тетрагидрофуран, а в отсутствие воды поли(этиленоксид)монометиловый эфир в инертной атмосфере, такой как аргон, при комнатной температуре. После нагревания раствора, типично с перемешиванием и типично при примерно 60°С в течение примерно 12 часов, добавляют аллилбромид и реакцию продолжают в течение до 2 дней. Реакцию между α-аллил-ω-метилполиэтиленоксидом и хлордиметилсиланом можно затем осуществлять в растворе толуола в присутствии платинохлористоводородной кислоты в течение нескольких часов при примерно 60°С, и продукт затем восстанавливают, используя LiAlH4. Затем осуществляют реакцию гидросилилирования, как описано выше, предпочтительно используя полиизопрен в качестве углеводородного полимера, содержащего множество двойных углерод-углеродных связей.
Поскольку способ, описанный выше, основан на присоединении по углерод-углеродной двойной связи, то он возможен благодаря выбору ненасыщенного углеводородного полимера, содержащего желаемое количество ненасыщенных связей, с получением полимерного материала, имеющего заданное количество боковых цепей, присоединенных к полимерному скелету.
Полимерный материал согласно настоящему изобретению, где боковые цепи, присоединенные непосредственно к атомам углерода в углерод-углеродном полимерном скелете, имеют формулу
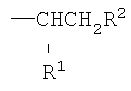
в которой один из R1 и R2 представляет собой -C(O)Q, а другой представляет собой -C(O)OR4, где Q и R4 являются такими, как определено выше, может быть получен способом, который включает взаимодействие полиизопрен-графт-малеинового ангидрида или его моноэфирного производного с соединением НО-(YO)b-(ZO)c-R5, в котором Y, Z, R5, b и с являются такими, как определено выше. Типично, реакцию осуществляют в органическом растворителе, таком как толуол.
В способе, описанном выше, количество боковых цепей, присоединенных к полимерному скелету, будет зависеть от количества малеиновых ангидридных прививок на полиизопреновой молекуле, которая может участвовать в реакции этерификации со спиртом HO-(YO)b-(ZO)c-R5. Например, при использовании полиизопрен-g-малеинового ангидрида формулы
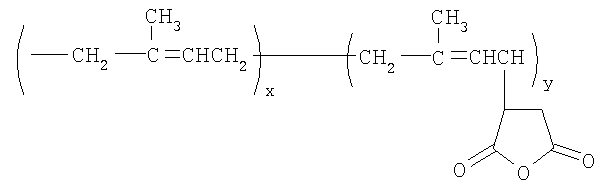
количество боковых цепей, имеющих общую формулу, приведенную выше, которые могут быть образованы, будет безусловно зависеть от значения «y». Полиизопрен-графт-малеиновый ангидрид (PIP-g-MA) имеется в продаже. Один такой PIP-g-MA, имеющий САS №139948-75-7, доступен от компании Aldrich, имеет среднюю молекулярную массу примерно 25000, вязкость по Брукфилду 10-50 сП (в виде 20 мас.% раствора в толуоле) при 30°С и плотность 0,92 г/мл (при 25°С). Соотношение изопреновых мономерных звеньев к мономерным звеньям малеинового ангидрида в этом графт-сополимере составляет 98:2, что указывает на то, что реакция между этим PIP-g-MA и спиртом, описанная выше, может давать примерно 7 боковых цепей на молекулу. Полиизопрен-графт-малеиновый ангидрид может быть получен согласно методикам, описанным в литературе. Например, согласно Visonte L.L.Y. с соавт. (Polymers for Advanced Technologies, Vol 4, 1993, pp 490-495), полиизопрен, растворенный в орто-дихлорбензоле, подвергали взаимодействию с малеиновым ангидридом при 180-190°C с получением модифицированного изопрена. Различные сополимеры полиизопрен-g-малеинового ангидрида с 7, 15, 19, 26 и 29 мол.% малеинового ангидрида получали путем увеличения времени реакции от 5 до 11 часов.
Реакцию между PIP-g-MA и полиалкиленоксиспиртом типично осуществляют в органическом растворителе, таком как толуол, и типично в присутствии активатора, например триэтилемина, при повышенной температуре. Выход сложного эфира в этой реакции может быть увеличен путем удаления воды из реакционной смеси посредством азеотропной перегонки, поскольку толуол и вода образуют азеотропные смеси, которые кипят при более низкой температуре, чем любой из компонентов. Полиалкиленоксиспирт может быть также подвергнут взаимодействию с моноэфирным производным PIP-g-MA. Например, авторы изобретения достигли хороших результатов, используя карбоксилированный полиизопрен, доступный от компании Kuraray Co. Ltd. в виде LIR-410. Этот карбоксилированный полиизопрен имеет общую формулу

и имеет функциональность 10, молекулярную массу примерно 25000, вязкость расплава 180 Па·с при 38°С, удельную массу 0,92 г/см3 и температуру стеклования -59°С. Реакцию LIR-410 со спиртом типично осуществляют в органическом растворителе, таком как толуол, при повышенной температуре. Как описано выше в случае реакции с использованием PIP-g-MA, выход сложного эфира может быть увеличен путем удаления воды из реакционной смеси азеотропной перегонкой. Эти способы, хотя они требуют применения предварительно образованного полиизопрена, имеющего функциональность по карбоксильным группам, имеют преимущество в том, что они вовлекают относительно простую и быструю реакцию и дают высокие выходы.
Принимая во внимание низкую липкость полимерного материала по изобретению, можно получить композицию жевательной резинки, которая имеет пониженную адгезию к поверхностям и которая поэтому легче удаляется с поверхностей, к которым она прилипает, путем включения полимерного материала по изобретению в композицию жевательной резинки. Поэтому согласно дополнительному аспекту в настоящем изобретении предложена композиция жевательной резинки, демонстрирующая пониженную адгезию к поверхностям, содержащая нерастворимую в воде гуммиоснову в количестве, достаточном для образования композиции жевательной резинки, и подсластитель, где по меньшей мере часть нерастворимой в воде гуммиосновы содержит полимерный материал, как описано в данном изобретении выше.
Композиции жевательной резинки обычно содержат растворимую в воде часть и нерастворимую в воде часть гуммиосновы жевательной резинки. Растворимая в воде часть композиции типично содержит компоненты, такие как один или более подсластителей, вкусоароматических агентов, окрашивающих агентов, подкислителей и наполнителей, и может дополнительно содержать другие вещества, такие как стабилизаторы и/или антиоксиданты. Нерастворимая в воде часть гуммиосновы композиции типично содержит, помимо одного или более эластомеров, ответственных за свойство разжевываемости жевательной резинки, эластомерные пластификаторы, масла и нерастворимые в воде наполнители.
Эластомеры, которые подходят для обеспечения свойства разжевываемости композиций жевательной резинки, хорошо известны в данной области. Они включают в себя, но не ограничиваются этим, природный каучук, природные жевательные резинки и синтетические эластомерные полимеры, такие как полибутадиен, полиизопрен, бутадиенстирольные сополимеры, полиизобутилен и изобутилен-изопреновые сополимеры. В композиции жевательной резинки по настоящему изобретению по меньшей мере часть эластомера в композиции замещена полимерным материалом, описанным выше, предпочтительно полимерным материалом, имеющим эластомерные свойства. Типично, полимерный материал по настоящему изобретению составляет по меньшей мере 1 мас.%, предпочтительно по меньшей мере 10 мас.%, более предпочтительно по меньшей мере 50 мас.% эластомерного компонента в композиции жевательной резинки. Он также входит в рамки изобретения для применения полимерного материала по изобретению в качестве полной замены эластомерного компонента в жевательной резинке.
Гуммиоснова композиции жевательной резинки может, как известно в данной области, содержать один или более пластификаторов для смягчения эластомерного компонента в композиции для обеспечения необходимого уровня разжевываемости и желаемого разжевывания данной жевательной резинки. Пластификаторы, которые традиционно используют для модификации свойств эластомера в композициях жевательной резинки, включают в себя сложные эфиры природных смол. Примеры сложных эфиров природных смол, которые могут быть использованы, включают в себя глицериновые сложные эфиры древесной смолы или гидрированной древесной смолы и пентаэритролэфиры древесной смолы или гидрированной древесной смолы. Такие материалы могут быть использованы, как общепринято в данной области, в количестве вплоть до примерно 70 мас.% гуммиосновы. Также в композицию жевательной резинки можно включать один или более других материалов, которые традиционно используют для смягчения или модификации физических свойств композиции, такие как глицерин, лецитин и глицерилмоностеарат. Такие материалы, если используются, будут включены в композицию в количестве, которое типично может составлять вплоть до примерно 15 мас.% композиции жевательной резинки.
В композицию жевательной резинки принято включать один или более подсластителей. Количество используемого подсластителя будет зависеть, конечно, от уровня сладости, желаемого в конечном продукте, и сладости используемого подсластителя. Например, могут быть использованы искусственные подсластители, такие как аспартам. Сладость может быть обеспечена, конечно, подсластителями, которые придают, помимо сладости, объем конечной композиции. Примеры объемных подсластителей, которые традиционно используют в изготовлении жевательной резинки, включают в себя сахариды, такие как сахароза, декстроза, ксилоза, и гидролизаты крахмала, такие как кукурузный сироп, а также несахариды, такие как полиолы сорбит, ксилит, маннит и гидрированные гидролизаты крахмала. Объемные подсластители могут быть использованы в количестве вплоть до 80 мас.% композиции жевательной резинки и, более типично, в количестве от примерно 20 до примерно 70 мас.%.
Композиция жевательной резинки по настоящему изобретению будет типично содержать один или более других ингредиентов, которые являются традиционными в данной области, таких как наполнители, вкусоароматические агенты, воски, окрашивающие агенты, камеди, стабилизаторы, эмульгаторы и антиоксиданты. Такие материалы могут быть использованы в настоящем изобретении в соответствии с методами, хорошо известными в области изготовления жевательной резинки.
Композиция жевательной резинки по изобретению может быть изготовлена согласно известным методикам. Например, способ изготовления будет обычно включать нагревание ингредиентов гуммиосновы вместе в смесителе для расплавления эластомерной части гуммиосновы и для образования гомогенной смеси компонентов гуммиосновы. Расплавленную гуммиоснову затем смешивают с другими компонентами, и после того, как их тщательно перемешали, полученную массу можно удалить из смесителя и сформовать в желаемую форму, например прокатыванием в слои и нарезанием на желаемый размер или путем заливки в таблетки. Резинка может быть затем опудрена сахарной пудрой или покрыта карамелью согласно методикам, известным специалисту.
Пример 1
К 100 мл толуола в круглодонной колбе добавляли 0,434 г (1,14×10-5 моль) полиизопрена, имеющего молекулярную массу 38000 и микроструктуру 98,8% цис-1,4 и 1,006 г (5,03×10-4 моль) метоксиполи(этиленгликоль)-монометакрилата, имеющего молекулярную массу 2000. Количества используемых реагентов соответствовали соотношению полиизопрена к полиэтиленгликолю 1:2.
Смесь перемешивали, используя магнитную мешалку, в течение 2 мин при 25-30°С до тех пор, пока полиизопрен не растворялся. Добавляли бензоилпероксид (0,025 г; 1,03×10-4 моль) и реакционный сосуд продували Na в течение 5 мин для обеспечения инертной атмосферы. Сосуд нагревали до 60°С и поддерживали при этой температуре при перемешивании в течение 48,5 часов.
По завершении реакции реакционную смесь быстро охлаждали до 0°С и 85-90% толуола вапаривали под вакуумом. К оставшейся смеси добавляли метанол и сразу образовывали коллоидный раствор, при помощи которого суспендировали продукт. Полученный полимерный материал содержал полимерный скелет, полученный из полиизопрена, имеющий множество привитых на него боковых цепей, имеющих формулу
-СН2СН(СН3)С(O)O(СН2СН2О)nСН3).
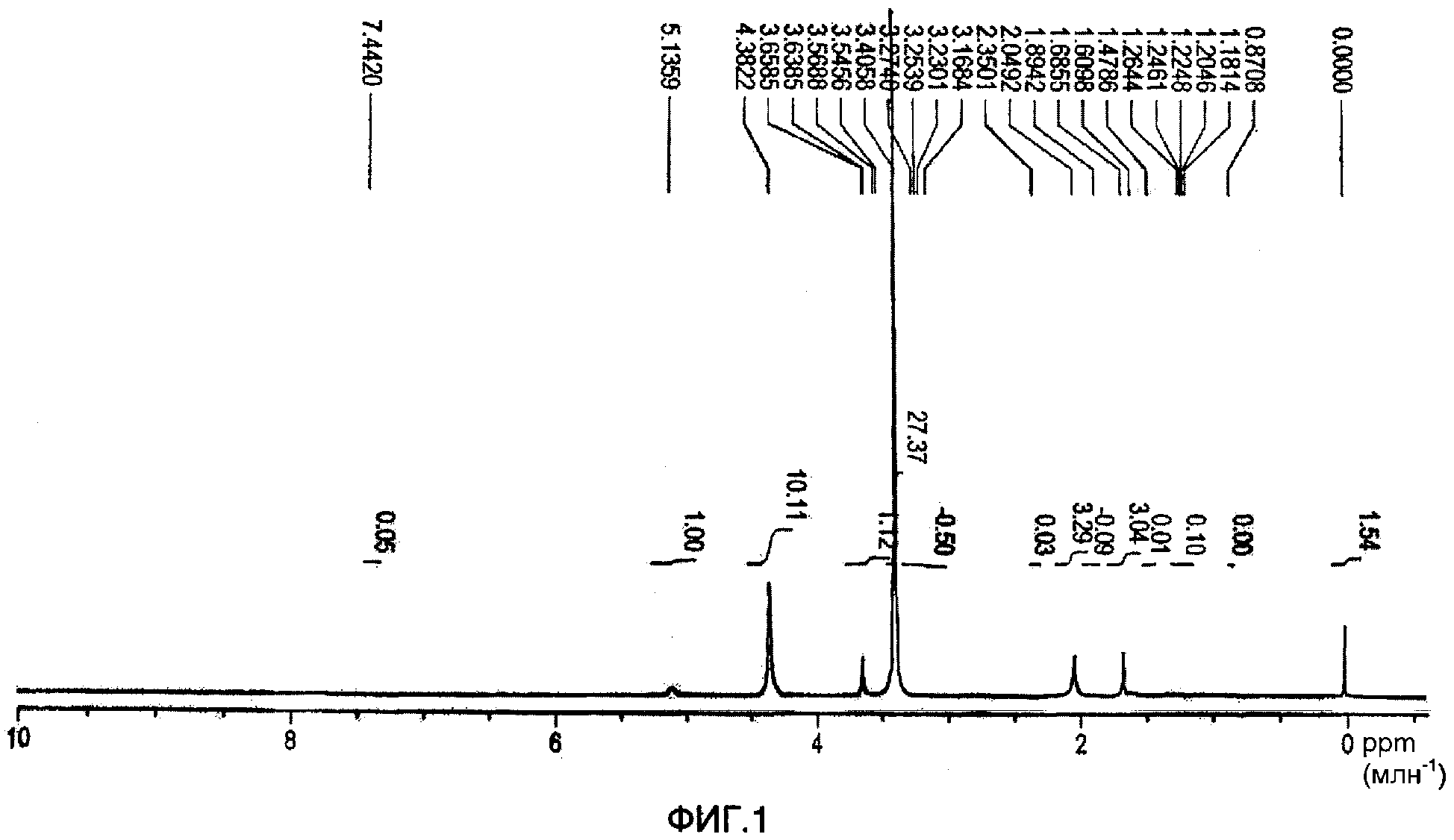
Суспензию графт-сополимеров центрифугировали для извлечения твердого продукта и этот продукт затем промывали в метаноле при 60°С (три раза) и сушили под вакуумом.1H-ЯМР спектр продукта показан на фиг.1. ЯМР спектр получали с использованием 400 МГц ЯМР спектрофотометра Bruker DSX, работающего при 400,14 МГц со встроенным твердотельным зондом (HP WB 73A MAS 4BL СР VTN). Одиночный импульс использовали для измерения спектра, где количество сканирований равнялось 8, 90° длительность импульса составляла 3 мс, db равнялось 2, задержка рециркуляции составляла 2 секунды, и было собрано 6144 числовых данных. Спектры регистрировали для скоростей вращения 0,5 и 9,6 кГц. ЯМР спектр, показанный на фиг.1, включает в себя интенсивный сигнал при 3,41 млн-1. Предполагают, что это обусловлено присутствием метанольного загрязнения в тестируемом образце и поэтому этим следует пренебречь. Из этого1H-ЯМР спектра и1Н-ЯМР спектров, полученных для полиизопрена и для полиэтиленгликоля, согласно которым могут быть получены параметры интегрирования, было рассчитано среднее количество боковых цепей (имеющих молекулярную массу примерно 2000) на скелет, полученный из полиизопрена (имеющий молекулярную массу примерно 38000), которое составлет 8,4.
Методы тестирования
А. Набухание в метаноле
Полиэтиленгликоль является гидрофильным и, следовательно, не обладает хорошей растворимостью в органических растворителях. Поэтому решили исследовать влияние полярного растворителя (метанола) на полиизопрен и на полимерный продукт, полученный в примере 1 выше.
0,5 г полиизопрена помещали в склянку, в которую затем добавляли 5 мл метанола (тест 1).
0,5 г графт-сополимера, полученного в примере 1 выше, помещали во вторую склянку, в которую затем добавляли 5 мл метанола (тест 2).
После добавления метанола в тесте 1 полиизопрен не демонстрировал набухания с метанолом, оставаясь бесцветным.
После добавления метанола в тесте 2 графт-сополимер значительно разбухал в такой степени, что полимерные цепи образовывали полупрозрачную коллоидную суспензию.
Пример 2
Взаимодействие полиизопрен-графт-малеинового ангидрида и поли(этиленгликопь)метилового эфира с катализатором триэтиламином
В этом примере полиэтиленгликоль подвергали взаимодействию с полиизопрен-графт-малеиновым ангидридом (PIP-g-MA), доступным от компании Aldrich. Этот PIP-g-MA, который имеет общую формулу
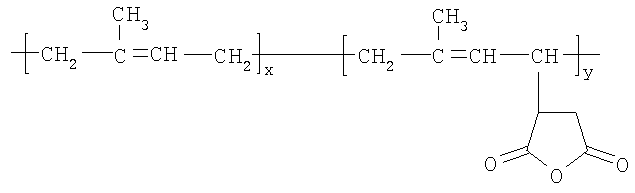
имеет CAS №139948-75-7 и имеет среднюю Мw (молекулярную массу) приблизительно 25000, вязкость по Брукфилду 10-50 сП (в виде 20 мас.% раствора в толуоле) при 30°С и плотность 0,92 г/мл (при 25°С). Соотношение изопреновых мономерных звеньев к привитым мономерным звеньям малеинового ангидрида составляет 98:2.
10 г полиизопрен-графт-малеинового ангидрида (PIP-g-MA) (Aldrich) растворяли в 50 мл толуола в реакционной колбе, добавляли 2 мл катализатора триэтиламина и включали магнитную мешалку. Затем добавляли 6 г полиэтиленгликоля и дополнительно добавляли 50 мл толуола для его растворения. После того как все растворилось, реакционную смесь нагревали до 85°С и оставляли в течение 7 дней при этой температуре.
Реакционную смесь затем охлаждали до комнатной температуры и затем добавляли к 800 мл пентана в химическом стакане. Образовавшуюся белую суспензию и реакционную смесь оставляли в холодильнике в течение ночи (охлаждали до 8°С в течение 24 часов). Происходило разделение фаз, и белый осадок удерживался на дне химического стакана, тогда как полупрозрачная верхняя фаза располагалась выше. Понятно, что некоторая часть продукта была растворима в пентане, в то время как другая часть продукта - нет. (Предполагают, что когда эффективность прививки увеличивается, тогда увеличение количества боковых цепей, присоединенных к полимерному скелету, делает полученный полимер менее растворимым в пентане, создавая таким образом спектр продуктов, некоторые из которых растворимы в пентане, а некоторые не растворимы в пентане и поэтому осаждаются из раствора и оседают на дне.)
Фазы осторожно разделяли и концентрировали для анализа. Концентрирование происходило путем выпаривания избытка пентана из обоих образцов под высоким вакуумом.
Пример 3
Взаимодействие полиизопрен-графт-малеинового ангидрида с попи(этиленгликоль)метиловым эфиром с использованием азеотропной перегонки
20 г полиизопрен-графт-малеинового ангидрида (PIP-g-MA), который использовали в примере 2, и 12 г поли(этиленгликоль)метилового эфира (PEGME) (также приобретенного у Aldrich), имеющего среднюю молекулярную массу 2000, взвешивали и добавляли в круглодонную колбу. Затем в эту колбу добавляли 200 мл толуола и смесь перемешивали с помощью магнитной мешалки.
Используя ловушку Дина-Старка (Dean Stark), реакционную колбу нагревали при 120°С в течение двух часов для того, чтобы высушить поли(этиленгликоль)метиловый эфир посредством азеотропной перегонки. Температуру реакции затем увеличивали до 130°С и нагревание продолжали при этой температуре в течение двух часов. После стадии нагревания 20 мл отогнанной жидкости мутнело, что указывает на азеотропную смесь толуола и воды. Еще 40 мл жидкости отогоняли, но она была прозрачной, что указывает на то, что всю воду удалили.
Реакционную смесь затем продували азотом и оставляли на 24 часа для развития реакции.
Реакционную смесь охлаждали до комнатной температуры и затем осаждали в 1200 мл пентана, превращая смесь в мутную эмульсию. Эту эмульсию помещали в холодильник (8°С в течение 24 часов), позволяя нерастворимому веществу осаждаться.
Полученные две фазы, растворимый верхний слой и нерастворимый осадок, разделяли. Растворимую пентановую фракцию концентрировали в роторном испарителе. Нерастворимый осадок сушили в вакууме, растворяли в хлороформе и осаждали в метаноле (800 мл) для того, чтобы растворить избыточное количество PEGME. Снова получали две фазы, но разделение было затруднено, поскольку новый модифицированный гидрофильный полимер образовал коллоид в метаноле. Для отделения коллоидного полимера от растворителя с избытком PEGME необходимо было трижды центрифугировать смесь в течение 45 мин при 15000 об/мин. Белый продукт, собранный со дна центрифужных пробирок, затем сушили.
Как ожидалось, метанольный элюент содержал некий полимер, который также концентрировали в роторном испарителе и растворяли в хлороформе. Для удаления избытка PEGME раствор осаждали в диэтиловом эфире и сушили.
Пример 4
Взаимодействие полиизопрен-графт-малеинового ангидрида с поли(этиленгликоль)метиловым эфиром посредством азеотропной перегонки
262 г полиизопрен-зрафт-малеинового ангидрида (PIP-g-MA), который использовали в примере 2, и 200 г поли(этиленгликоль)метилового эфира (PEGME) (также приобретенного у Aldrich), имеющего среднюю молекулярную массу 2000, взвешивали и добавляли в трехлитровую круглодонную колбу, снабженную магнитной мешалкой. Перед этим к толуолу добавляли гидрид кальция для удаления воды из толуола и после фильтрации 700 мл этого слегка подсушенного толуола добавляли в круглодонную колбу для растворения исходных материалов.
Используя ловушку Дина-Старка, реакционную колбу нагревали до 120°С для того, чтобы удалить воду из поли(этиленгликоль)метилового эфира и из толуола посредством азеотропной перегонки. Температуру реакции увеличивали до 130°С и нагревали при этой температуре в течение двух часов (удаление 50 мл жидкости). Как только достигали ее определенной постоянной точки кипения, азеотропную смесь (толуола и воды) упаривали и конденсировали в виде мутной жидкости. Как только жидкость, которую конденсировали, становилась прозрачной, вся вода была удалена и выпаривали только толуол.
Реакцию оставляли в течение 24 часов при 120°С и затем нагревали при 130°С в течение двух часов перед удалением еще большего количества растворителя азеотропной перегонкой. Реакцию оставляли на 48 часов.
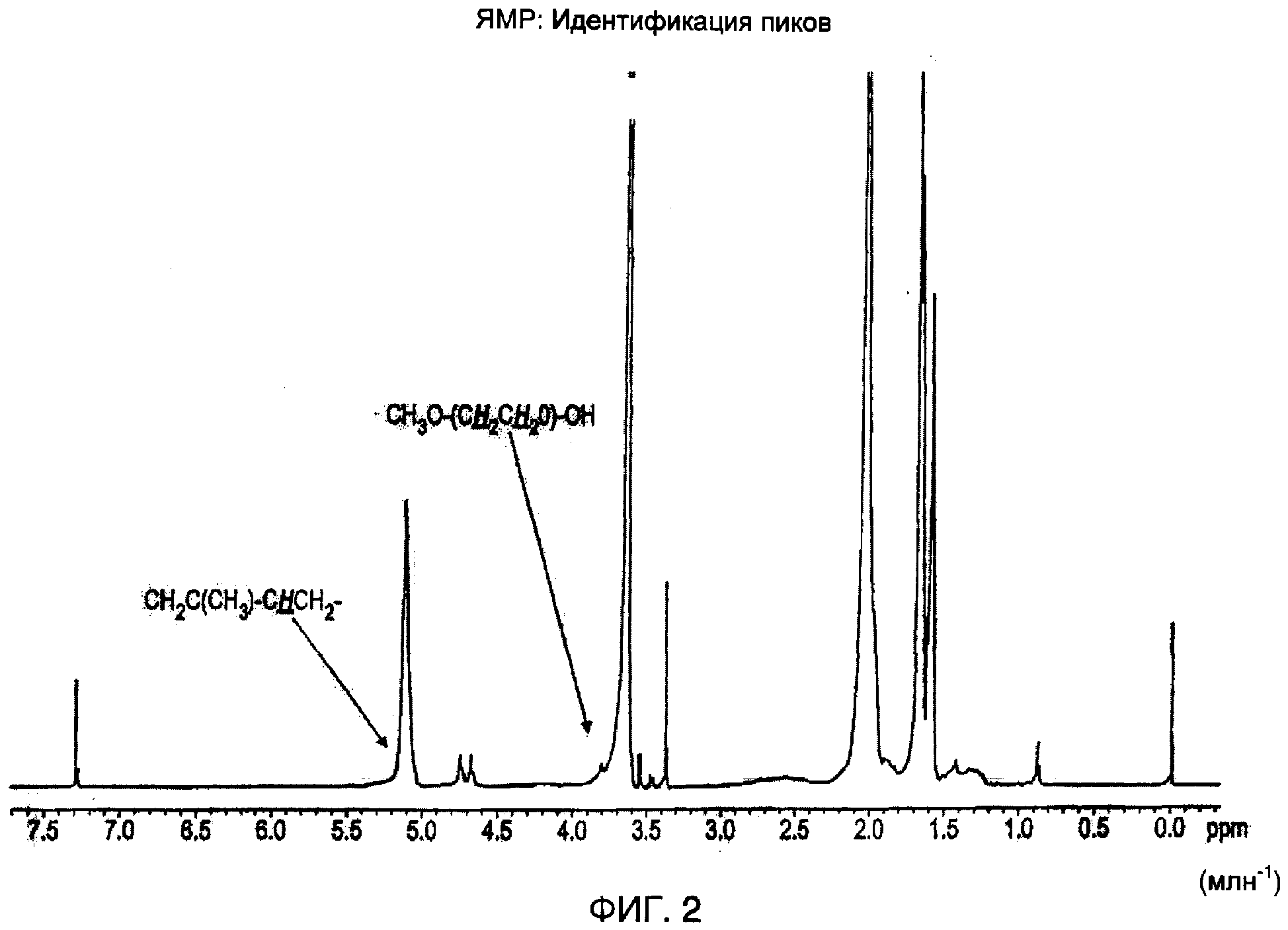
Затем реакционную смесь охлаждали до комнатной температуры и осаждали в метаноле (4 л). Продукт затем сушили в сушильном шкафу под высоким вакуумом в течение 72 часов для удаления метанола, после чего его промывали избытком воды (4 л) и сушили в сушильном шкафу под вакуумом в течение 72 часов.1H-ЯМР спектр этого продукта показан на фиг.2.1H-ЯМР спектр получали с использованием ЯМР спектрофотометра Delta/GX 400, работающего при 400 МГц, в CDCl3 (дейтерированном хлороформе). Определили, что температура стеклования (Tg) продукта составляет приблизительно 50°С при использовании синхронного термического анализа Netzsch STA-409 ЕР.
Пример 5
Взаимодействие жидкого изопренового каучука (LIR-410) с поли(этиленгликоль)метиловым эфиром с использованием азеотропной перегонки
В этом примере поли(этиленгликоль)метиловый эфир подвергали взаимодействию с карбоксилированным жидким полиизопреном, доступным от компании Kuraray Co. Ltd. в виде LIR-410. Этот карбоксилированный полиизопрен имеет общую формулу
и имеет функциональность 10 (т.е. 10 групп карбоновой кислоты на молекулу), молекулярную массу приблизительно 25000, вязкость расплава 180 Па·с при 38°С, удельную массу 0,92 г/см3 и температуру стеклования -59°С.
320 г карбоксилированного полиизопрена LIR-410 взвешивали в химическом стакане и растворяли в 850 мл толуола, который был предварительно слегка высушен с использованием гидрида кальция, как описано в примере 4. Толуольный раствор помещали в трехлитровую круглодонную колбу, снабженную магнитной мешалкой, и к этому добавляли 260 г поли(этиленгликоль)метилового эфира (PEGME) (Aldrich), имеющего среднюю молекулярную массу 2000, с перемешиванием до тех пор, пока не растворится.
Используя ловушку Дина-Старка, реакционную колбу нагревали до 130°С и нагревание продолжали в течение двух часов для удаления воды из PEGME и из толуола посредством азеотропной перегонки, после чего 80 мл растворителя удаляли. Реакционную смесь затем оставляли в течение 24 часов при 120°С. Цикл азеотропной перегонки (нагревание в течение двух часов при 130°C с последующим нагреванием в течение 24 часов при 120°С) повторяли трижды в течение следующих трех дней и затем реакционную смесь нагревали при 130°С для азеотропной перегонки в течение 5 часов, удаляя 120 мл растворителя. Поскольку цикл азеотропной перегонки продолжался, вязкость реакционной смеси увеличивалась, так что больше невозможно было вращать магнитную мешалку. Реакционную смесь затем охлаждали до комнатной температуры и промывали три раза водой, используя воронку Бюхнера. Промытый продукт затем сушили в сушильном шкафу под высоким вакуумом в течение 72 часов. Нашли, что температура стеклования (Tg) составляет приблизительно 50°С при использовании методики термического анализа, приведенной в примере 4.
Пример 6
Гидросилилирование полиизопрена с функционализированным поли(этиленгликоль)метиловым эфиром
Полиизопрен, используемый в этом примере, имел среднюю молекулярную массу 40000 (Aldrich). Поли(этиленгликоль)метиловый эфир (Aldrich) имел среднюю молекулярную массу 2000.
Поли(этиленгликоль)метиловый эфир (60 г) взвешивали в реакционную колбу и нагревали до 90°С под высоким вакуумом. К этому добавляли 1,75 г NaH, растворенного в тетрагидрофуране, и реакции позволяли продолжаться в течение 6-8 часов при 50°С. Затем в реакционную колбу добавляли аллилбромид (8 г) и оставляли реагировать в течение 20 часов. Реакционную смесь нагревали под вакуумом для удаления растворителя и продукт затем промывали бензолом. К 30 г продукта, высушенного под высоким вакуумом и растворенного в 40 мл сухого толуола в круглодонной колбе, добавляли H2PtCl6 (0,3 г), растворяли в 4 мл тетрагидрофурана и хлордиметилсилана (24 мл) и позволяли смеси реагировать в течение 24 часов. Реакционную смесь нагревали до 90°С под высоким вакуумом. Затем в реакционную колбу добавляли сухой толуол (45 мл) с последующим добавлением LiAlH4 (5 г) и смеси позволяли реагировать в течение 72 часов. После этого смесь фильтровали, используя бензол для промывки, и фильтрат собирали. Фильтрат упаривали в роторном испарителе, затем добавляли к пентану с получением осадка. Осадок собирали фильтрацией и сушили в роторном испарителе.
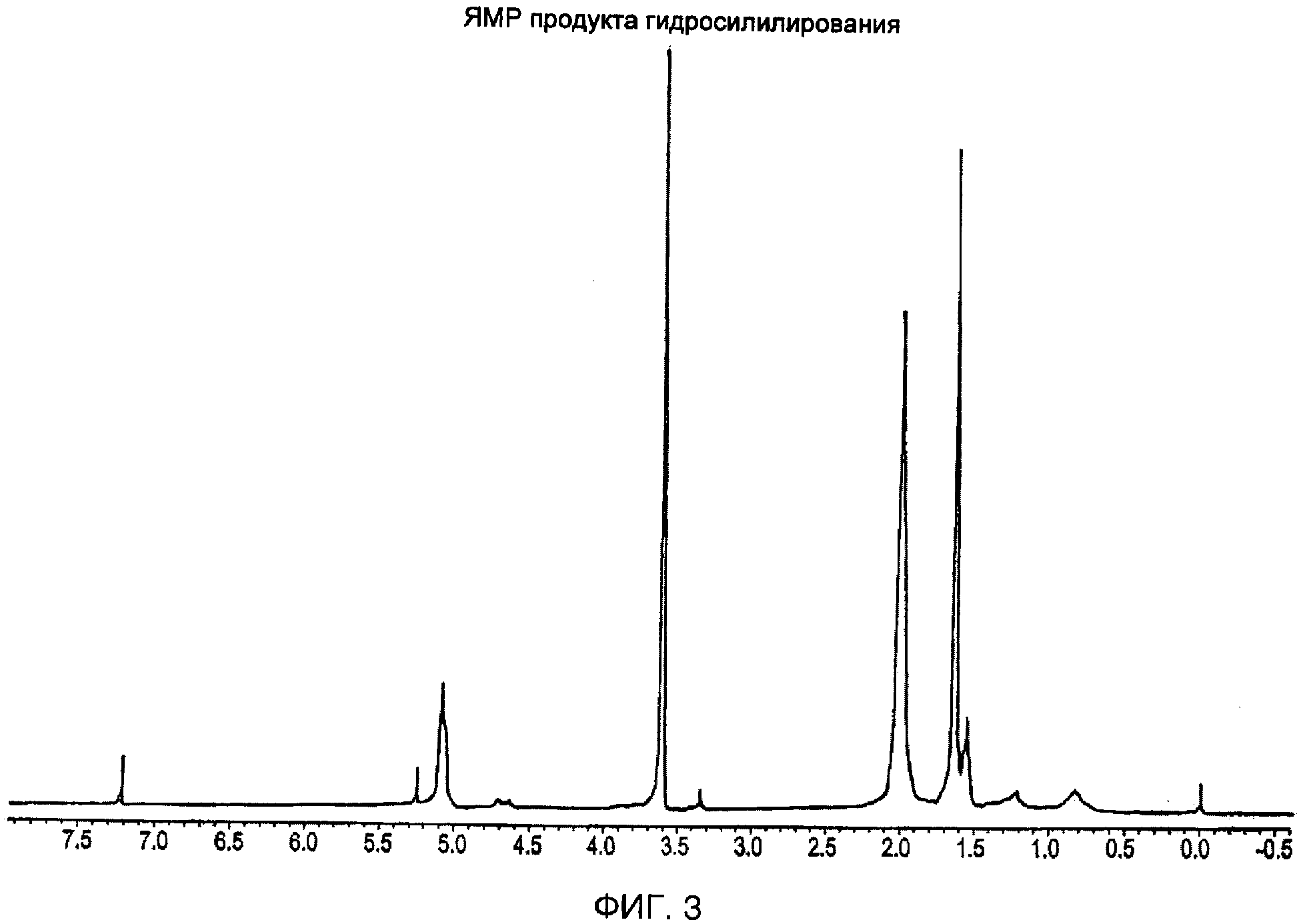
Сухой продукт (8 г) и полиизопрен (5 г) растворяли в бензоле (50 мл) в реакционной колбе. К этому затем добавляли H2PtCl6 (0,175 г), растворенную в 1 мл тетрагидрофурана, данную реакционному смесь нагревали до 50°С и реакции позволяли протекать при этой температуре в течение 48 часов, а затем оставляли охлаждаться до комнатной температуры. После этого реакционную смесь растворяли в дихлорметане (200 мл) и дважды фильтровали через колонку, содержащую 50 г Аl2О3, и дважды промывали дихлорметаном. Фильтрат собирали и затем упаривали в роторном испарителе и затем добавляли к пентану, вызывая образование осадка. Пентановую смесь, содержащую осадок, фильтровали и отфильтрованный осадок и элюент собирали. Собранный осадок сушили и промывали метанолом с образованием мутного раствора. Мутный раствор центрифугировали в течение 30 мин при 15000 об/мин и нерастворимый продукт собирали и сушили.1H-ЯМР спектр продукта показан на фиг.3.1H-ЯМР спектр получали, используя ЯМР спектрофотометр Delta/GX 400, работающий при 400 МГц, в CDCl3 (дейтерированный хлороформ). Рассчитали, что среднее количество боковых цепей, привитых на каждый полиизопреновый скелет, равно 145. Этот нерастворимый продукт состоял из изопрена, на который были привиты боковые цепи формулы

Количество привитых боковых цепей было достаточным для лишения продукта растворимости в органических растворителях. Элюент, собранный после вышеупомянутой фильтрации, концентрировали с помощью ротационного выпаривания. Считали, что продукт, который растворялся в пентане, состоит из изопрена, имеющего недостаточное количество привитых боковых цепей, так что растворимость в органических растворителях сохранялась.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
1. Тест на липкость
Образцы полимерных материалов, полученных в примере 4 (REV-7) и в примере 5 (REV-10), подвергали тестам на липкость. Для сравнения той же методике тестирования также подвергали образцы полиизопрена (PIP), имеющего Mw 40000, полиизопрен-g-малеинового ангидрида (исходного материала в примере 4) (PIP-g-MA) и LIR-410 (исходного материала в примере 5).
Образцы приготавливали для применения следующим образом. В каждом случае один грамм образца растворяли в хлороформе и раствор наносили равномерно на поверхность прямоугольного предметного стекла для микроскопа размером 2,5 см × 7,5 см. После выпаривания растворителя на поверхности предметного стекла оставалась пленка полимерного материала, имеющая одинаковую толщину примерно 0,5 мм.
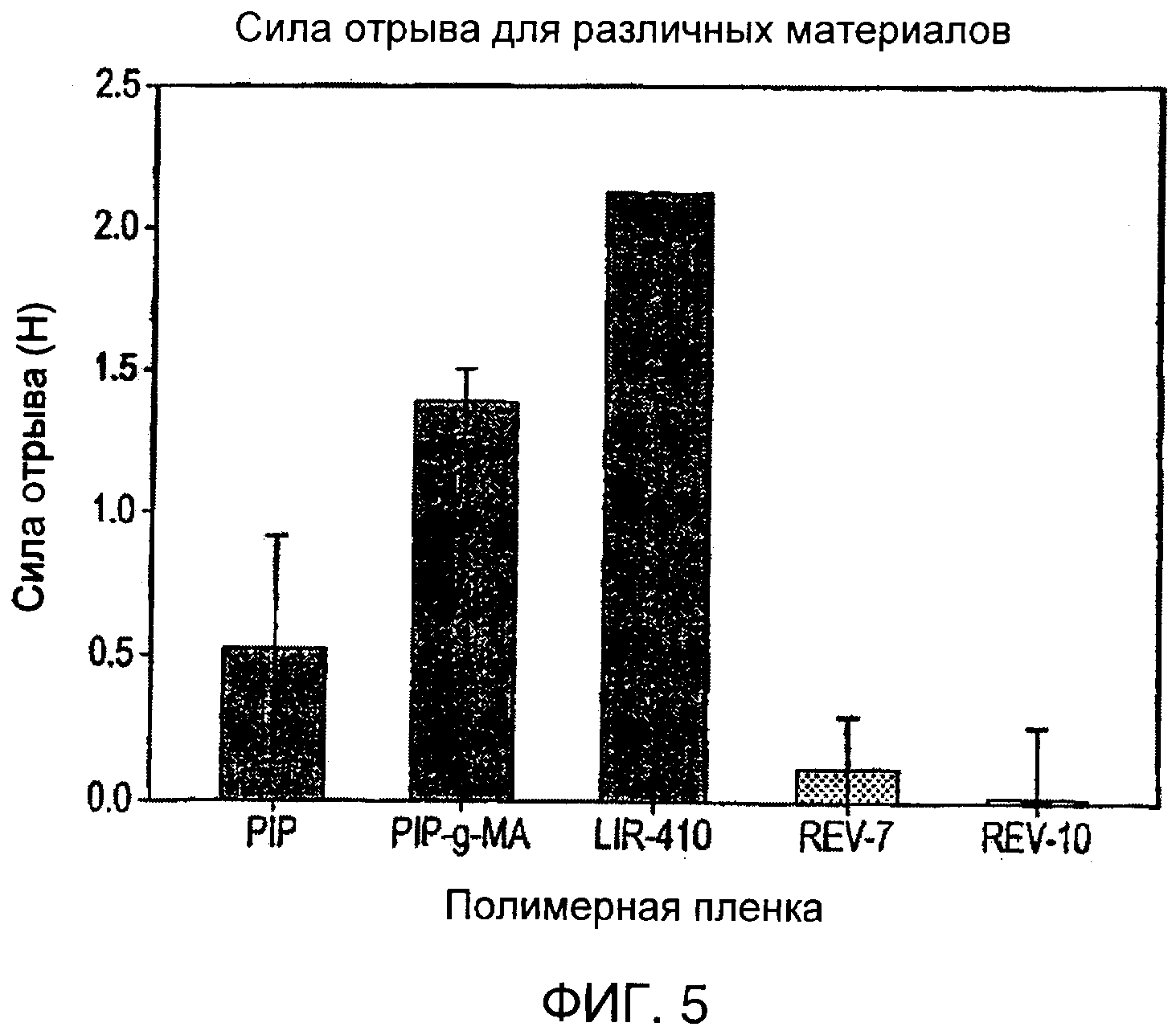
Тест на липкость представляет собой простую методику измерения липкости. В соответствии с процедурой тестирования, стандартный зонд из нержавеющей стали, имеющий круговую область контакта диаметром 5 мм, приводят в контакт с пленкой образца, нанесенного на предметное стекло (которое удерживается строго на месте), при низком усилии контакта (100 г/см2) в течение короткого времени контакта (10 с) и затем отрывают при постоянной скорости (1 см/с). Измеряют максимальное усилие на отрыв. Аппарат, используемый в этих экспериментах, представлял собой тестер липкости, модель №80-02 (Testing Machines, Inc.), и эксперименты осуществляли при комнатной температуре. Максимальное усилие на отрыв измеряли в граммах и затем превращали в Ньютоны. Полученные результаты приведены в следующей таблице.
Самой поразительной особенностью результатов, приведенных выше, является явное отсутствие липкости, демонстрируемое образцом REV-10. По существу это дает представление о его неадгезивном характере при сравнении с другими четырьмя тестируемыми материалами. Максимальное усилие, которое аппарат может измерить, составляет 2000 г (или 19,61 Н). Когда очень липкий материал превышает этот максимум, тогда значение, взятое в тесте на липкость, представляет собой это максимальное значение (как видно в случае LIR-410).
Общая тенденция изменения липкости для исходных тестируемых материалов выражается как LIR-410>PIP-g-MA>PIP. По сравнению с этим модифицированные полимеры (REV-7 и REV-10) демонстрируют значительное снижение липкости. Температуры стеклования модифицированных полимеров являются значительно более высокими, чем таковые для исходных материалов, как результат привитых боковых цепей. Все исходные материалы находились в жидкой фазе с Tg намного ниже комнатной температуры (около -60°С), обеспечивающей удобство при смачивании зонда. Твердые модифицированные полимеры (REV-7 и REV-10) также были менее липкими в результате их неспособности прилипать.
Среднее усилие на отрыв для каждого материала, указанного в таблице выше, показано на гистограмме на фиг.4. Максимальные погрешности также показаны.
Воспроизводимость этого эксперимента ограничивается неточностью некоторых параметров, таких как площадь контакта между зондом и тестируемыми пленками и фактическая температура образцов. Однако из качественного анализа полученных результатов ясно, что пониженная адгезия, продемонстрированная REV-7 и REV-10 при комнатной температуре, является следствием боковых цепей, присоединенных к скелету полимера.
2. Петлевой тест на липкость
Образцы полимерных материалов, полученных в примере 4 (REV-7) и в примере 5 (REV-10), подвергали петлевым тестам на липкость. Для сравнения той же методике тестирования также подвергали образцы полиизопрена (PIP), имеющего среднюю молекулярную массу 40000 (полученного от Aldrich), полиизопрен-g-малеинового ангидрида (исходного материала в примере 4) и LIR-410 (исходного материала в примере 5).
Образцы приготавливали для применения следующим образом. В каждом случае один грамм образца растворяли в трихлорметане и раствор наносили равномерно на поверхность прямоугольного предметного стекла для микроскопа размером 2,5 см × 7,5 см. После выпаривания растворителя на поверхности предметного стекла оставалась пленка полимерного материала, имеющая одинаковую толщину примерно 0,5 мм.
Во всех случаях аппарат настраивали следующим образом:
1) отрезок эластичной ленты, имеющей постоянную ширину 25 мм, образовывал петлю путем сближения его свободных концов, и затем свободные концы фиксировали в зажимах на установке для тестирования, которую соединяли с устройством для измерения нагрузки;
2) петлю, подвешенную вертикально под фиксированными концами ленты, совмещали, хотя и удерживали выше, с предметным стеклом (фиксированным в установке для тестирования), покрытым пленкой образца.
Тесты осуществляли при комнатной температуре следующим образом:
а) петлю опускают с постоянной скоростью 300 мм/мин, так что она контактирует с пленкой образца на подложке до тех пор, пока не будет достигнута максимальная площадь контакта между лентой, образующей петлю, и пленкой образца (25 мм × 25 мм);
б) после периода в одну секунду пребывания в контакте с максимальной площадью пленки образца петлю отрывают от пленки образца с той же постоянной скоростью (300 мм/мин) и максимальное усилие на отрыв (в Н) измеряют с помощью аппарата; и
в) петлю затем снова опускают с той же постоянной скоростью, как на стадии (а) выше, для повторного контактирования максимальной площади пленки образца в течение одной секунды и затем снова отрывают с той же постоянной скоростью, как на стадии (а) выше, для обеспечения второго измерения максимальной силы отрыва (в Н) между контактной поверхностью петли и образцом при тестировании.
Пленку образца и петлю затем заменяли. Для каждого образца эту процедуру тестирования проводили пять раз.
Таким образом, в соответствии с процедурой тестирования для каждой пленки образца и каждой петли осуществляли два измерения силы отрыва: первое - после того как петля и пленка образца были приведены в контакт в течение первого времени и второе - после второго контакта между петлей и пленкой образца.
Следующие результаты регистрировали для каждой из пяти пленок при тестировании. (Обратите внимание, что "1" в колонке "Контакт" означает измерение, осуществленное после первого контакта между петлей и пленкой образца, а "2" в колонке "Контакт" означает измерение, осуществленное после второго контакта петли и пленки образца.)
PIP
PIP-g-MA
LIR-410
REV-7
REV-10
Средние значения максимальной силы отрыва наносят на график для разных полимеров, и их стандартное отклонение показано в виде величины погрешности на фиг.5. Снова наиболее поразительной особенностью является отсутствие липкости у REV-10. Ее значения силы отрыва не превышают 0,02 Н, и это значение является таким же, как в случае, когда петлю приводят в контакт с неадгезивной поверхностью, т.е. данный материал не смачивает зонд, не дает какого-либо сопротивления, когда втягивается в противоположном направлении.
В петлевых тестах на липкость общая тенденция, показанная для исходных материалов, такая же, как предварительно показано в тестах на липкость, т.е. LIR-410>PIP-g-MA>PIP. Из результатов петлевого теста на липкость ясно, что введение боковых цепей (как определено выше), присоединенных к полимерному скелету полиизопрена, приводит к снижению липкости.
3. Тесты на растворимость
Определяли растворимость полимерных материалов, полученных в примере 4 (REV-7) и в примере 5 (REV-10), в различных растворителях (вода, толуол, хлороформ, пентан и метанол). Для сравнения растворимости в тех же растворителях образцы полиизопрена (PIP), имеющего Мw 40000, полиизопрен-g-малеинового ангидрида (исходного материала в примере 4) (PIP-g-MA), LIR-410 (исходного материала в примере 5) и поли(этиленгликоль)метилового эфира (исходного материала в примерах 4 и 5) (PEGME) также подвергали той же методике тестирования.
В каждом случае один грамм образца при тестировании взвешивали в 20 мл стеклянную колбу и добавляли растворитель. Смесь образца и растворителя энергично встряхивали и затем помещали в ультразвуковую баню на 30 мин. Затем смесь снова энергично встряхивали, наблюдали и регистрировали внешний вид содержимого колбы, т.е. определяли, получен прозрачный раствор или нет. Результаты приведены в следующей ниже таблице. В этой таблице "НЕТ" указывает на то, что растворения образца в растворителе не наблюдали, и "ДА" указывает на то, что образец растворялся в растворителе.
Из результатов, приведенных выше, можно видеть, что:
1. PIP, PIP-g-MA и LIR-410 все ведут себя одинаково в каждом растворителе.
2. PEGME ведет себя противоположным образом по сравнению с PIP, PIP-g-MA и LIR-410 в воде, пентане и метаноле.
3. Толуол и хлороформ растворяют все образцы (исходные материалы и продукты).
4. REV-7 и REV-10 проявляют повышенную гидрофильность (по сравнению с исходными материалами) в воде благодаря присутствию гидрофильных боковых цепей в этих молекулах.
5. REV-7 и REV-10 демонстрируют относительное затруднение при растворении в пентане, что указывает на то, что они ведут себя по-разному в зависимости от изопреновых полимеров, из которых они получены, и что они, возможно, образуют коллоидные дисперсии.
6. Способность REV-7 и REV-10 растворяться, по меньшей мере частично, или образовывать коллоиды в метаноле демонстрирует сродство этих модифицированных полимеров к полярным растворителям в результате присутствия гидрофильных боковых цепей в молекулах.
4. Малоугловое рассеяние нейтронов (SANS, от Small Angle Neutron Scattering)
Измерения SANS осуществляли на D22 в ILL (Гренобль, Франция), используя кварцевые кюветы Hellma с длиной пробега 2 мм, уравновешенные при 298 K. Использовали холодные нейтроны 8 Å. Расстояние от детектора составляло 17,5 м, и расстояние от коллиматора составляло 17,6 м. Q-разрешение с этой установкой составляет 10% (ΔQ/Q). Полученный диапазон Q составляет от 0,002 до 0,035 Å-1. Время измерения выбрали, чтобы получить 1 миллион импульсов для хорошей статистики.
Исследуемые образцы состояли из 5%-ных растворов графт-сополимеров в частично дейтерированном метаноле (СН3OD). Данные рассеяния анализировали с использованием модели Гинье Дебая (Guinier Debye) с помощью следующего уравнения (Debye P.J. Phys. Coll. Chem. (1947) 51, 18)
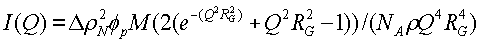
где ΔρN означает разность плотности длины рассеяния полимера и растворителя, ϕp означает объемную фракцию полимера, М означает молекулярную массу, NA означает число Авогадро, ρ означает физическую плотность, Q представляет собой вектор переноса импульса и RG означает радиус гирации.
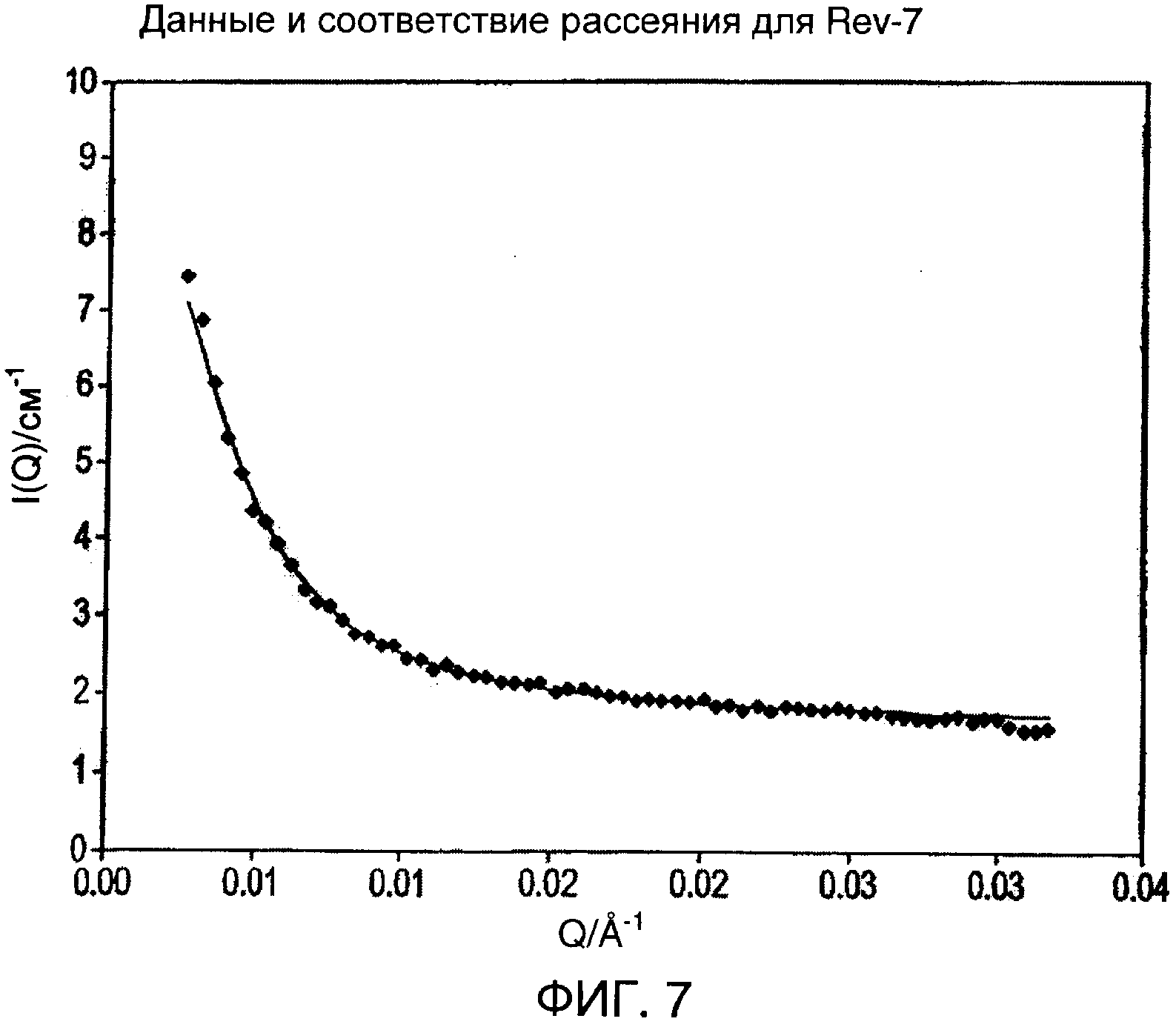
Эффективный радиус гирации для Rev-10 в метаноле из графика, показанного на фиг.6, составляет 673±15 Å. Аналогичный анализ для Rev-7 (фиг.7) давал 408+13 Å. Эти размеры указывают на коллоидную природу этих дисперсий.
Реферат
Изобретение относится к полимерным материалам, имеющим пониженную липкость. Предложен полимерный материал с низкой липкостью, который имеет углерод-углеродный полимерный скелет с прямой или разветвленной цепью и множество боковых цепей, присоединенных к этому скелету, где боковые цепи, которые присоединены непосредственно к атомам углерода полимерного скелета, имеют формулу (I) или имеют формулу (II) ! ! где R1 представляет собой Н, -C(O)OR4 или -C(O)Q и R2 представляет собой -C(O)OR4 или -C(O)Q, при условии, что по меньшей мере один из R1 и R2 представляет собой группу -C(O)Q; R3 представляет собой Н или -СН3; R4 представляет собой Н или алкильную группу, имеющую от 1 до 6 атомов углерода; Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, где каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода; а равно 3 или 4 и каждая из переменных b и с независимо равна 0 или целому числу от 1 до 125, при условии, что сумма b+с имеет значение в диапазоне от 10 до 250, предпочтительно от 10 до 120. Данный полимерный материал может быть использован для замены по меньшей мере части нерастворимой в воде гуммиосновы в композиции жевательной резинки с получением композиции, проявляющей пониженную адгезию к поверхностям. 6 н. и 41 з.п. ф-лы, 7 ил., 2 табл.
Формула
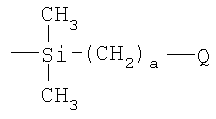
или имеют формулу
где R1 представляет собой Н, -C(O)OR4 или -C(O)Q, и R2 представляет собой
-C(O)OR4 или -C(O)Q,
при условии, что по меньшей мере один из R1 и R2 представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, имеющую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, где каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода;
а равно 3 или 4, и каждая из переменных b и с независимо равна 0 или целому числу от 1 до 125, при условии, что сумма b+с имеет значение в диапазоне от 10 до 250, предпочтительно от 10 до 120.
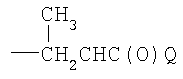
где Q является таким, как определено в п.1.
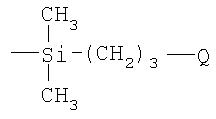
где Q является таким, как определено в п.1.
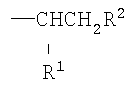
где один из R1 и R2 представляет собой -C(O)Q, а другой представляет собой -C(O)OR4, в которой Q и R4 являются такими, как определено в п.1.
где Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, где каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода;
и а равно 3 или 4, и каждая из переменных b и с независимо равна 0 или целому числу от 1 до 125, при условии, что сумма b+с имеет значение в диапазоне от 10 до 250, предпочтительно от 10 до 120.
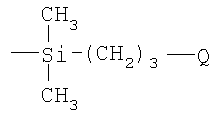
где Q является таким, как определено в п.16.
-СН3.
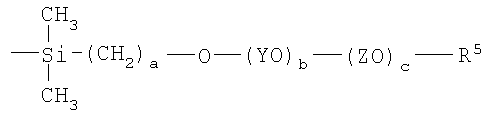
где Y, Z, R5, a, b и с являются такими, как определено в п.1, включающий:
(1) взаимодействие соединения формулы
HO-(YO)b-(ZO)c-R5
с гидридом натрия в сухом органическом растворителе в инертной атмосфере;
(2) взаимодействие продукта, полученного на стадии (1), с соединением
CH2=CH-(CH2)q-Br,
где q равно 1 или 2, с получением соединения II
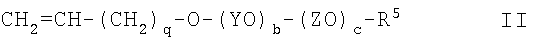
(3) взаимодействие соединения II с хлордиметилсиланом с получением соединения III
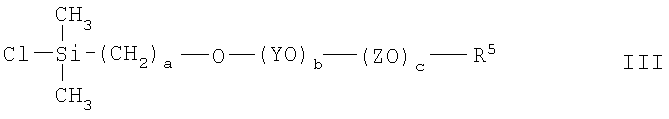
(4) восстановление соединения III и взаимодействие продукта d-гидродиметилсилилполиалкиленоксида с углеводородным полимером с прямой или разветвленной цепью, содержащим множество двойных углерод-углеродных связей в углеводородном полимерном скелете, в присутствии соли переходного металла.
где R1 представляет собой Н, -C(O)OR4 или -C(O)Q, и R2 представляет собой
-C(O)OR4 или -C(O)Q,
при условии, что по меньшей мере один из R1 и R2 представляет собой группу -C(O)Q;
R3 представляет собой Н или -СН3;
R4 представляет собой Н или алкильную группу, имеющую от 1 до 6 атомов углерода;
Q представляет собой группу, имеющую формулу -O-(YO)b-(ZO)c-R5, где каждый из Y и Z независимо представляет собой алкиленовую группу, имеющую от 2 до 4 атомов углерода, и R5 представляет собой Н или алкильную группу, имеющую от 1 до 4 атомов углерода;
и каждая из переменных b и с независимо равна 0 или целому числу от 1 до 125, при условии, что сумма b+с имеет значение в диапазоне от 10 до 250, предпочтительно от 10 до 120, и где углерод-углеродный полимерный скелет получен из гомополимера этилен-ненасыщенного полимеризующегося углеводородного мономера, содержащего 5 атомов углерода, или из гомополимера изобутилена, или из сополимера изобутилена и изопрена.
где Q является таким, как определено в п.32.
где один из R1 и R2 представляет собой -C(O)Q, а другой представляет собой -C(O)OR4, в которой Q и R4 являются такими, как определено в п.32.
-СН2СН(СН3)-С(O)-O-(YO)b-(ZO)с-R5,
в которой Y, Z, R5, b и с являются такими, как определено в п.32, включающий взаимодействие углеводородного полимера с прямой или разветвленной цепью в растворителе в инертной атмосфере с соединением
CH2=C(CH3)C(O)O-(YO)b-(ZO)c-R5,
в присутствии инициатора свободных радикалов.
в которой один из R1 и R2 представляет собой -C(O)Q, а другой представляет собой -C(O)OR4, где Q и R4 являются такими, как определено в п.1, включающий взаимодействие полиизопрен-графт-малеинового ангидрида или его моноэфирного производного в органическом растворителе с соединением
HO-(YO)b-(ZO)c-R5,
в котором Y, Z, R5, b и с являются такими, как определено в п.32.
Документы, цитированные в отчёте о поиске
Нелипкая жевательная резинка
Комментарии