Синтез кислородзамещенных бензоциклогептенов в качестве ценных промежуточных продуктов для получения тканеселективных эстрогенов - RU2310643C2
Код документа: RU2310643C2
Описание
Настоящее изобретение относится к промежуточным продуктам и к новому способу получения бензоциклогептена С. Предлагаемый в изобретении способ получения его промежуточных продуктов основан на использовании недорогих исходных материалов, обеспечивает высокий выход и высокую степень чистоты промежуточных продуктов без операций по их хроматографической очистке и позволяет реализовать технологический процесс в промышленном масштабе.
Соединения общей формулы А (WO 00/03979) представляют собой соединения, обладающие сильным антиэстрогенным действием. Речь идет при этом о селективных эстрогенах, действие которых проявляется тканеселективно (избирательно по отношению к тканям). Это эстрогенное действие проявляется прежде всего по отношению к костной ткани. Преимущество данного класса соединений заключается в том, что на матку и печень они не оказывают никакого воздействия или оказывают его лишь в самой минимальной степени.
Указанные соединения могут вместе с тем обладать и антиэстрогенной эффективностью, что можно подтвердить, в частности, тестированием по предупреждению роста матки или на моделях опухолей.
Соединения с подобным профилем действия обозначают как селективные модуляторы эстрогенных рецепторов (SERM, от англ. "selective estrogene receptor modulator"). Наиболее известным представителем этого класса соединений является ралоксифен, допущенный для применения в качестве медикамента, предназначенного для предупреждения и лечения постменопаузального остеопороза.
Поскольку речь, как правило, идет о соединениях, применяемых в высокой дозировке, возникают значительные трудности, связанные с получением действующих веществ в больших количествах. Особенно актуальной поэтому для данного класса веществ остается потребность в разработке простого в осуществлении и недорогого синтеза.
Получение соединений общей формулы А
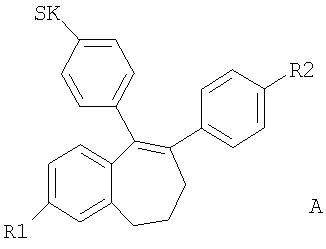
описано в заявке WO 00/03979. В приведенной формуле SK, R1 и R2 обозначают особые остатки боковых цепей, которые более подробно поясняются в указанной заявке.
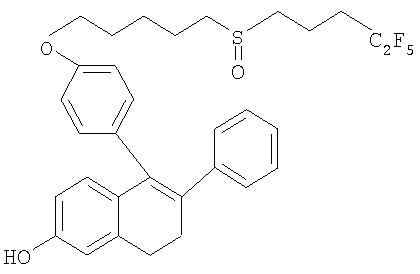
Исходя из вышеизложенного в основу настоящего изобретения была положена задача разработать более эффективный способ получения соединения формулы С (пример 9 в WO 00/03979).
Обменную реакцию с получением целевого соединения С осуществляют согласно WO 00/03979 следующим образом (см. примеры 8 и 9):

Описанный в WO 00/03979 синтез включает большое число стадий и предусматривает несколько операций по хроматографической очистке. Крупномасштабный промышленный синтез, когда требуется получать несколько сот килограммов соединения С, связан с исключительными трудностями. Таким образом, промышленное производство по получению тканеселективных эстрогенов общей формулы А, т.е. соединений, описанных в заявке WO 00/03979 и прежде всего в примере 9 (в данной заявке соединение С), диктует необходимость в разработке более эффективного и включающего меньшее число стадий синтеза.
В отличие от синтеза, представленного в WO 00/03979, предлагаемый в настоящем изобретении новый способ получения соединения С отличается тем, что путь к получению главного промежуточного продукта В удалось сократить за счет существенного снижения числа стадий. Вместе с тем дальнейшая последовательность проведения реакции с получением конечного продукта С осталась неизменной.
Благодаря упрощенной технологии (несколько методов получения продуктов в одном аппарате) для получения соединения С требуется выделять лишь 8 промежуточных продуктов.
Нижеприведенная таблица наглядно иллюстрирует преимущества нового способа по сравнению с уровнем техники.
Еще одним важным преимуществом предлагаемого в изобретении способа является практическая возможность получения продуктов без операций по хроматографической очистке и с высокой степенью чистоты.
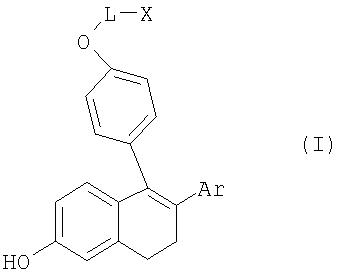
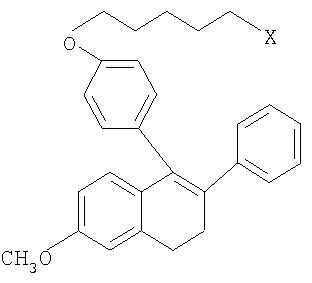
Предлагаемый в изобретении способ позволяет, кроме того, вводить еще и другие, замещенные заместителем R2 ароматические соединения благодаря применению предлагаемого промежуточного продукта общей формулы I
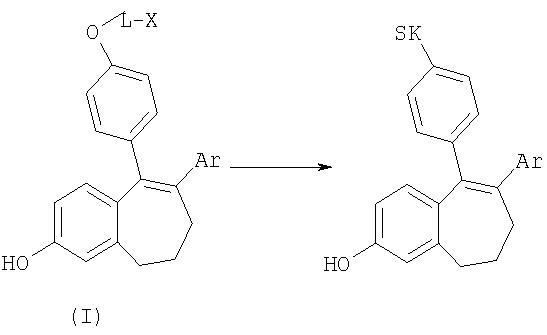
где
L обозначает С2-С10алкиленовую цепь, которая может быть неразветвленной или разветвленной,
Х обозначает Cl или Br,
Ar обозначает ароматический или гетероароматический остаток, который необязательно может содержать до 3-х заместителей.
Положенная в основу изобретения задача решается с помощью соединений общей формулы I
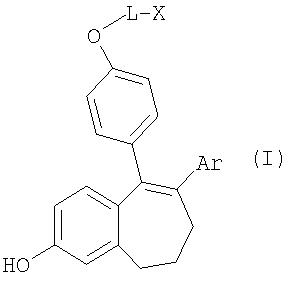
где
L обозначает С2-С10алкиленовую цепь, которая может быть неразветвленной или разветвленной,
Х обозначает Cl или Br,
Ar обозначает ароматический или гетероароматический остаток, который необязательно может содержать до 3-х заместителей.
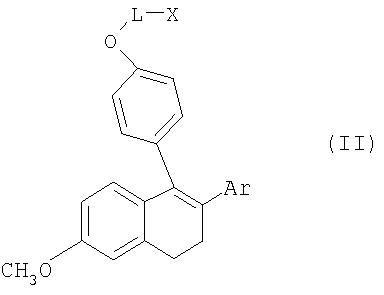
Соединения общей формулы I получают из метиловых эфиров общей формулы II
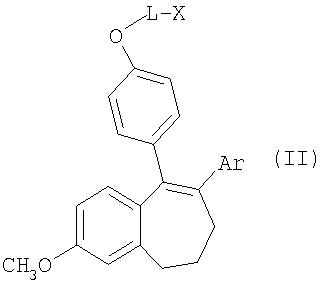
где L, Х и Ar имеют значения, указанные для общей формулы I, путем селективного отщепления метилового эфира с помощью реагента, состоящего из трибромида бора и 2,6-диметилпиридина (в соотношении 1:1-1:1,5).
Используемое количество этого реагента может составлять от 1 до 6 экв. (в пересчете на трибромид бора и отщепляемый ароматический метиловый эфир). Указанную реакцию проводят в апротонных растворителях, таких, например, как дихлорметан, хлороформ или 1,2-дихлорметан, предпочтительно, однако, в дихлорметане, при температурах в интервале от -30 до +50°С, предпочтительно от 10 до 30°С.
Исключительно высокая селективность отщепления ароматического метилового эфира в присутствии высших ароматических алкиловых эфиров является неожиданным фактором. Фенольные продукты при этом получают с высокой степенью чистоты и очень хорошим выходом.
Использование BBr3 для отщепления ароматических метиловых эфиров специалистам в принципе известно (см. Synthetic Communications 9 (5), 1979, cc.407-410). Без добавления лутидина селективное отщепление осуществлять не удается (происходит отщепление всех ароматических эфиров). Лишь комбинация с 2, 6-лутидином позволяет добиться неожиданного эффекта избирательности.
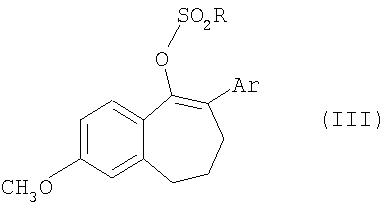
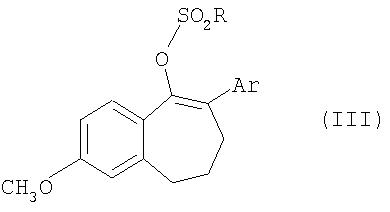
Соединения общей формулы II получают из соединений общей формулы III
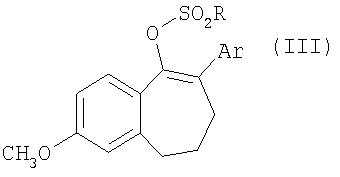
где
Ar обозначает ароматический или гетероароматический остаток, который необязательно может содержать до 3-х заместителей, а
R представляет собой перфторированную прямоцепочечную С1-С8алкильную группу, предпочтительно CF3, C4F9 или С8F17,
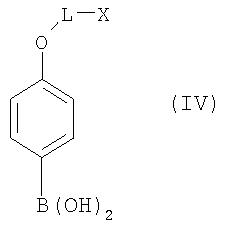
взаимодействием с фенолбороновыми кислотами общей формулы IV
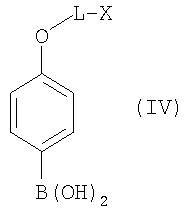
где L и Х имеют значения, указанные для формулы I, с помощью известных специалистам методов катализированной палладием реакции сочетания по Сузуки (Journ. Org. Chem. 64, 1999, cc.6797-6803; Chem. Rev. 95, 1995, cc.2457-2483; Pure Appl. Chem. 63, 1991, cc.419-422; Synlett, 1990, cc.221-223; Journ. Org. Chem. 58, 1993, cc.2201-2208). В этих целях могут применяться коммерчески доступные Pd-катализаторы, такие, например, как Pd(PPh3)4 илиPd(Cl2)(PPh3)2 (информацию о других катализаторах см., например, в каталоге STREM №18, 1999-2001, Chemicals for research, metals, inorganics and organometallics).
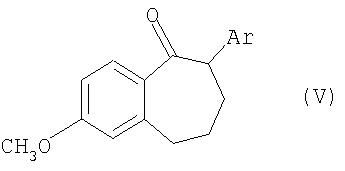
Соединения общей формулы III получают из кетонов общей формулы V
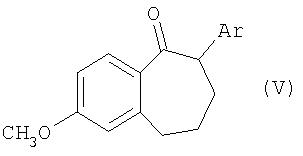
где Ar имеет значение, указанное для общей формулы I, взаимодействием, осуществляемым по известным специалистам методам получения енолтрифлатов (см. Journ. Amer. Soc., EN, 96, 1974, cc.1100-1110; Chem. Ber., GE, 110, 1977; Tetrahedron Lett, EN, 23, 1, 1982, cc.117-120; Synthesis, EN, 1, 1981, cc.29-30; Tetrahedron Lett, EN, 40, 29, 1999, cc.5337-5340), с реагентом общей формулы VI
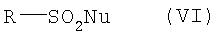
где
R имеет значение, указанное для общей формулы III, а
Nu обозначает уходящую группу, такую, например, как F, Cl, J или R-SO3-.
Особенно успешной зарекомендовала себя комбинация нонафторбутилсульфонилфторида и ДБУ (диазабициклоундецена) в ТГФ при 0°С. Полученные таким путем нонафлаты неожиданно отличаются высокой устойчивостью, и при необходимости их можно выделять в твердой форме. Как правило, однако, растворы сырых продуктов непосредственно используют в последующих реакциях. Соединения общей формулы VI являются коммерчески доступными продуктами (поставщики: фирмы Aldrich, Fluorochem и др.).
Соединения общей формулы IV можно получать из соединений общей формулы VII
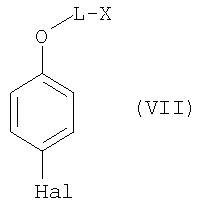
где L и Х имеют значения, указанные для общей формулы I, а Hal обозначает атом галогена, такого как Cl, Br или J, по известным специалистам методам получения фенилбороновых кислот из галоароматических соединений (см. Houben-Weyl, "Methoden der organiscen Chemie", изд-во Georg Thieme Verlag Stuttgart, New York, 1982, том 13/3а, cc.637 и далее).
Особенно целесообразным оказалось использовать н-бутиллитий для обмена галогена на металл, после чего литиевое соединение подвергают взаимодействию с В(ОМе)3 и затем гидролизуют кислотой с образованием требуемого производного фенилбороновой кислоты.
Процесс получения соединений общей формулы VII осуществляют по известным специалистам методам получения простых феноловых эфиров (см. Mol. Cryst. Liq. Cryst, EN, 158, 1988, cc.209-240; Synth. Commun., EN 28, 16, 1998, cc.3029-3040; Journ. Chem. Soc. Perkin Trans., EN, 2, 1989, cc.2041-2054) из соответствующих галофенолов и симметричных или несимметричных дигалоалканов (например 5-бром-1-хлорпентана). Соответствующие фенолы и дигалогениды являются коммерчески доступными продуктами.
Получение кетонов общей формулы V в тех случаях, когда Ar обозначает фенильный остаток, описан в Indian Journ. Chem., август 1996, том 25В, cc.832-837. Описанная в данной публикации многостадийная последовательность получения этого промежуточного продукта, во-первых, требует значительного времени, а во-вторых, крайне трудна для реализации в промышленном масштабе.
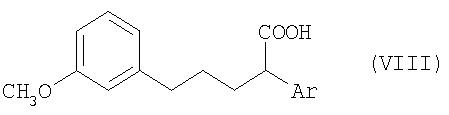
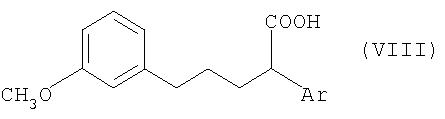
В отличие от этой технологии, особенно в условиях крупномасштабного промышленного производства, наиболее целесообразно получать соединения общей формулы V из соединений общей формулы VIII, где Ar имеет значение, указанное для общей формулы I, с помощью известных специалистам методов циклизации (замыкания цикла) по реакции Фриделя-Крафтса (Chem. Rev. 70, 1970, с.553).
Как особенно предпочтительное можно назвать применение полифосфорной кислоты при температурах в интервале от 80 до 120°С.
Полифосфорную кислоту можно приобрести на рынке или же использовать в свежеприготовленном виде.
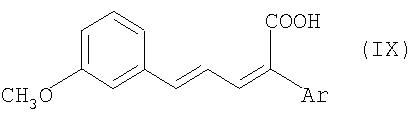
Соединения общей формулы VIII получают в свою очередь известным образом из соединений общей формулы IX
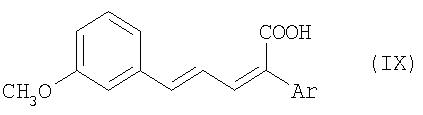
путем каталитического гидрирования (см. Houben-Weyl, "Methoden der organischen Chemie", изд-во Georg Thieme Verlag Stuttgart, New York 1980, том 4/1 с, часть 1, cc.14 и далее).
Соединения общей формулы VIII можно получать по реакции получения продукта в одном аппарате исходя из 3-метоксибензальдегида
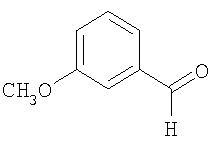
который катализируемым основанием взаимодействием с ацетальдегидом превращают в 3-метоксикоричный альдегид (см. Organic Reactions, том 16, 1968, cc.1 и далее; Justus Liebigs Ann. Chem. 412, 1917, с.322)
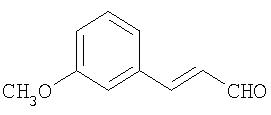
и без выделения подвергают затем по реакции Кневеннагеля взаимодействию с соединением общей формулы Х
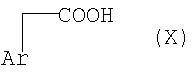
где Ar имеет значение, указанное для общей формулы I (получение соединений VIII, в том числе условия проведения реакции Кневеннагеля, см. в Organic Reactions, том 15, 1967, сс. 204 и далее).
Соединения общей формулы Х являются коммерчески доступными продуктами (поставщики, например, фирмы Aldrich, Fluka и др.) или же их можно без проблем получать по известным специальным методам, используемым для получения арилуксусных кислот (см. Journ. Amer. Chem. Soc. 78, 1956, с.6037; Can. Journ. Chem., EN 7031992, сс. 992-999; Journ. Amer. Chem. Soc., EN 11251990, сс. 1894-1896; Recl. Trav. Chim. Pays-Bas, 70, 1951, сс. 977, 983; Journ. Amer. Chem. Soc. 69, 1947, с.1797).
При получении коричного альдегида целесообразно, как было установлено, использовать неорганическое основание, такое как NaOH, КОН, предпочтительно КОН. Реакцию проводят в воде при температурах в интервале от 1 до 30°С. При этом ацетальдегид можно применять в количестве до 5 экв. Особенно предпочтительным зарекомендовал себя вариант, в котором ацетальдегид и основание добавляют небольшими порциями с интервалами времени перед каждым последующим добавлением по 10-30 мин.
Для реакции Кневеннагеля предпочтительно использовать в качестве основания ацетангидрид, а также триэтиламин. Реакция протекает при температуре в интервале от 60°С до температуры перегонки.
Предлагаемый в изобретении способ может применяться также для получения соединений общей формулы XI
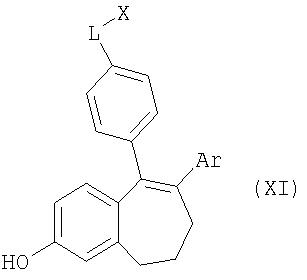
где Ar, L и X имеют значения, указанные для общей формулы I, при этом вместо галофенолов общей формулы VII в качестве исходных веществ используют соответствующие галоалканзамещенные галоароматические соединения общей формулы XII и далее работают аналогичным образом:
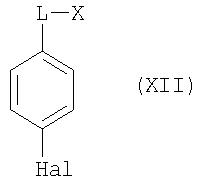
Соединения общей формулы XII известны из литературы и часть из них являются коммерчески доступными продуктами.
В качестве предпочтительных остатков L можно назвать, например, -С2Н4-, -С3Н6-, СН2-СН(СН3)-СН2, -С4Н8-, -С5Н10-, -С6Н12-, -С7Н14-, -С8Н16-. Наиболее предпочтителен из них остаток -C5Н10-.
Х обозначает предпочтительно атом хлора.
В качестве остатка Ar можно назвать, например, несколько остатков следующих формул:
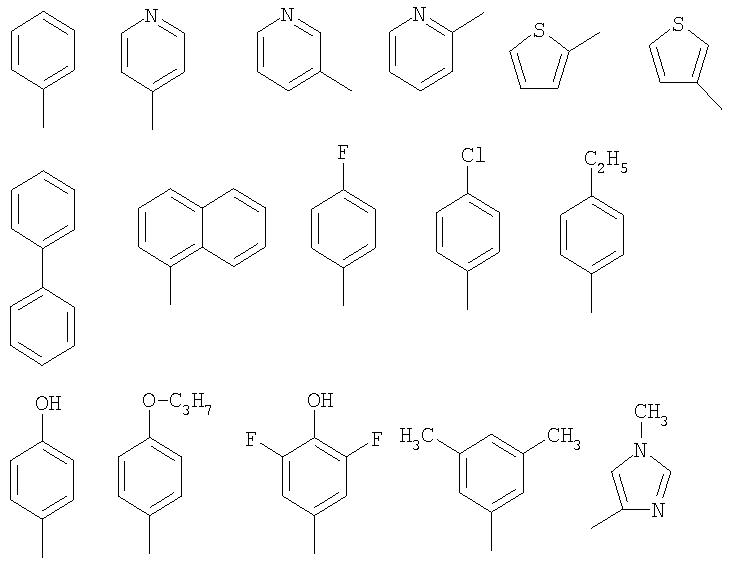
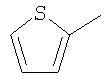
Особенно предпочтительными являются при этом фенильный, пиридильный и тиофеновый остаток.
Ниже изобретение более подробно поясняется на примерах
Пример 1
Пример 1а
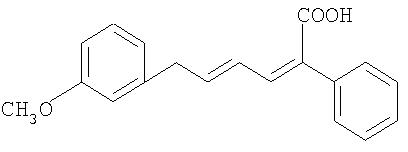
(2Z,4Е)-5-(3-метоксифенил)-2-фенилпентадиен-2,4-диеновая кислота
К 750г (5,5 моля) 3-метоксибензальдегида в 3750 мл воды добавляют 310 мл 20%-ного водного едкого кали. Затем в течение 30 мин по каплям добавляют 160 мл ацетальдегида, растворенных в 450 мл воды, таким образом, чтобы внутренняя температура не превышала 30°С. После этого данную операцию повторяют 7 раз (сначала 310 мл 20%-ного водного КОН, затем 160 мл ацетальдегида/450 мл воды/30 мин). После добавления последней порции перемешивают в течение 30 мин при комнатной температуре. Далее добавляют 3750 мл метил-трет-бутилового эфира и тщательно перемешивают в течение 15 мин. Органическую фазу отделяют и смешивают со смесью из 250 мл ледяной уксусной кислоты и 250 мл воды. После 5-минутного перемешивания добавляют 1500 мл воды и перемешивают еще в течение 5 мин. Органическую фазу отделяют, после чего концентрируют ее досуха в вакууме.
К остатку (масло оранжевого цвета) добавляют 748,80 г (5,5 моля) фенилуксусной кислоты, 1000 мл ангидрида уксусной кислоты и 1470 мл триэтиламина и в течение 4 ч нагревают до 100°С. Затем при давлении 15 мбар отгоняют растворитель и остатку дают нагреться до комнатной температуры. После этого остаток растворяют в 2500 мл метил-трет-бутилового эфира. Далее охлаждают до 0°С и по каплям медленно добавляют 600 мл концентрированной соляной кислоты. Затем добавляют 2000 мл воды и перемешивают в течение 5 мин. Органическую фазу отделяют и смешивают с 1100 мл 50%-ного водного едкого натра. Далее добавляют 830 мл воды. В течение 30 мин интенсивно перемешивают и водную фазу отделяют (продукт в водяной фазе). Органическую фазу повторно отделяют путем размешивания с 1100 мл едкого натра и 830 мл воды. Водную фазу отделяют и объединяют с первой водной фазой.
К обеим объединенным водным фазам добавляют 1700 мл метал-трет-бутилового эфира и интенсивно перемешивают в течение 5 мин. Затем водную фазу отделяют и с помощью концентрированной соляной кислоты значение рН устанавливают на 1,5. Выпавшую в осадок кислоту экстрагируют 4000 мл метил-трет-бутилового эфира. Органическую фазу концентрируют досуха в вакууме. Затем добавляют 2000 мл этанола и примерно 1000 мл этанола отгоняют в вакууме. Далее повторно добавляют 1000 мл этанола и выпавший осадок отделяют путем перемешивания в течение 1 ч при 0°С. После этого отфильтровывают от осадка и промывают 350 мл холодного этанола.
Светложелтое твердое вещество сушат в вакууме при 40°С.
Выход: 1094 г (71% от теории) твердого вещества светло-желтого цвета.
Элементный анализ:
Пример 1б
5-(3-метоксифенил)-2-фенилпентановая кислота
1000 г (3,567 моля) соединения, указанного в заголовке примера 1а, растворяют в 8 л тетрагидрофурана и добавляют 75 г палладиевого катализатора 30 (10%-ного Pd/С). Затем гидрируют при комнатной температуре при давлении 2 бара. После этого отфильтровывают от катализатора, промывают 500 мл тетрагидрофурана и раствор концентрируют досуха в вакууме.
Выход: 1015 г (100% от теории) бесцветного вязкого масла.
Элементный анализ:
Пример 1в
7-метокси-2-фенил-1-бензосуберон
К 560 г (1,97 моля) соединения, указанного в заголовке примера 1б, добавляют 5,6 кг 115%-ной полифосфорной кислоты и в течение 3 ч нагревают до 95°С. Затем раствору дают остыть до 50°С и этот все еще теплый раствор сливают в 9 л смеси воды со льдом. Далее добавляют 5000 мл метил-трет-бутилового эфира и интенсивно перемешивают в течение 10 мин. Органическую фазу отделяют, а водную фазу еще раз промывают 1500 мл метил-трет-бутилового эфира. Объединенные органические фазы промывают один раз 4000 мл воды, а затем 1200 мл 5%-ного водного едкого натра. Органическую фазу отделяют и смешивают с 280 г активированного угля. После 2-часового кипячения с обратным холодильником активированный уголь отфильтровывают, промывают небольшим количеством метил-трет-бутилового эфира и фильтрат досуха концентрируют в вакууме.
Выход: 456,5 г (87% от теории) бесцветного масла, которое при стоянии кристаллизуется.
Элементный анализ:
Пример 1г
4-[(5-хлорпентил)окси]фенилбороновая кислота
1000 г (5,78 моля) 4-бромфенола, 1125,7 г (6,069 моля) 1-бром-5-хлорпентана и 1118,3 г (8,092 моля) карбоната калия загружают в 4000 мл диметилформамида и в течение 5 ч перемешивают при 60°С. Смесь охлаждают до 20°С и добавляют 3500 мл толуола. Затем отфильтровывают от выпавших в осадок солей и соли трижды промывают толуолом порциями по 3500 мл. ДМФ/толуоловый фильтрат смешивают с 4000 мл воды и в течение 5 мин размешивают. Толуолсодержащую фазу отделяют, а водную фазу один раз промывают 3000 мл толуола. Затем толуолсодержащие фазы объединяют и в течение 20 мин размешивают с 4000 мл 5%-ного водного едкого натра. После этого толуолсодержащую фазу отделяют, промывают 4000 мл воды и упаривают досуха в вакууме. Остаток растворяют в 9 л тетрагидрофурана и охлаждают до внутренней температуры -65°С. Затем медленно по каплям добавляют 2860 мл раствора н-бутиллития (1,6-молярного в гексане) таким образом, чтобы температура не превышала -60°С. При этой температуре -65°С перемешивают в течение 30 мин. Далее по каплям добавляют 1291 г триметилового эфира борной кислоты и перемешивают в течение 2 ч при -65° С. Затем нагревают до -20°С. Для последующей переработки осторожно добавляют по каплям раствор из 2200 мл воды/метанола (соотношение 1:1) таким образом, чтобы температура не превышала -15°С. По завершении указанного добавления осторожно добавляют 11 л 2н. водной соляной кислоты и перемешивают в течение 1 ч при 0°С. Затем температуре дают подняться до комнатной, органическую фазу отделяют, а водную фазу экстрагируют один раз 5000 мл и один раз 2000 мл метил-трет-бутилового эфира.
Органические фазы объединяют и размешивают с 11 л 2н. едкого натра в течение 30 мин. Водную фазу отделяют (продукт) и дважды промывают метил-трет-бутиловым эфиром порциями по 3000 мл. Значение рН водной фазы добавлением 6н. соляной кислоты устанавливают на 1, затем добавляют 6000 мл метил-трет-бутилового эфира и в течение 2 ч перемешивают при комнатной температуре. Органическую фазу отделяют, один раз промывают 3000 мл воды и в завершение досуха упаривают в вакууме.
Выход: 1219,5 г (87% от теории) твердого вещества розового цвета.
Элементный анализ:
Пример 1д
5-{4-[(5-хлорпентил)окси]фенил}-2-метокси-6-фенил-8,9-дигидро-7Н-бензоциклогептен
81,67 г (306,64 ммоля) соединения, указанного в заголовке примера 1в, растворяют в 400 мл тетрагидрофурана/метил-трет-бутилового эфира и раствор охлаждают до 3°С. Затем к раствору добавляют 56,02 г (367,97 ммоля) диазабициклоундецена (ДБУ), поддерживая при этом температуру на уровне 3°С. Затем промывают 40 мл ТГФ. Далее добавляют 111,46 г (367,97 ммоля) фторангидрида перфторбутан-1-сульфоновой кислоты (при 3°С), промывают 40,86 мл метил-трет-бутилового эфира, причем температура в процессе добавления не должна превышать 8°С. После 12-часового перемешивания при 3°С к реакционному раствору добавляют при 10°С 290 мл 10%-ного раствора карбоната калия (в пересчете на 3,5-кратное количество эдукта) и перемешивают в течение 5 мин. Органическую фазу отделяют и концентрируют в вакууме до объема порядка 500 мл. Полученный таким путем раствор сырого нонафлата используют на последующей стадии.
78,08 г (321,97 ммоля) соединения, указанного в заголовке примера 1г, растворяют при комнатной температуре в 390 мл МТБ и к раствору добавляют 310 мл 2-молярного водного раствора К2СО3. Затем добавляют 1,076 г хлорида бис(трифенилфосфин)палладия(II) (1,533 ммоля, 0,005 мол. экв.), суспендированного в 10 мл метил-трет-бутилового эфира. После этого добавляют полученный выше раствор нонафлата и в течение 30 мин нагревают с обратным холодильником. Далее охлаждают до комнатной температуры и смешивают с 455 мл 2н. водного NaOH. Затем в течение 15 мин перемешивают при комнатной температуре. Органическую фазу отделяют и перемешивают в 20 течение 15 мин при комнатной температуре с 455 мл 2н. водного HCl.
Органическую фазу отделяют и смешивают с 15 г активированного угля. После этого в течение короткого промежутка времени нагревают до температуры перегонки и еще горячий раствор фильтруют через кизельгур. Затем дважды промывают метил-трет-бутиловым эфиром порциями по 100 мл. Фильтрат концентрируют при 40°С в вакууме. К остатку добавляют 455 мл метанола и образовавшийся осадок размешивают в течение 6 ч при комнатной температуре. Суспензию охлаждают до 5°С, отфильтровывают и промывают 100 мл холодного метанола. В завершение сушат при 40°С в вакууме.
Выход: 112,4 г (82% от теории) бесцветного кристаллического твердого вещества.
Элементный анализ:
Пример 1е
5-{4-[(5-хлорпентил)окси]фенил}-6-фенил-8,9-дигидро-7Н-бензоциклогептен-2-ол
127,51 г (508,93 ммоля) трибромида бора растворяют при комнатной температуре в 650 мл дихлорметана. К этому раствору медленно при 0°С добавляют раствор из 57,26 г (534,37 ммоля) 2,6-диметилпиридина в 320 мл дихлорметана (следить за тем, чтобы температура оставалась на уровне 0°С). Затем раствор охлаждают до 0°С. Далее ему дают нагреться до 20°С и по каплям добавляют раствор из 65,00 г (145,4 ммоля) соединения, указанного в заголовке примера 1д (раствор в 300 мл дихлорметана), при этом внутренняя температура не должна превышать 20°С. После 4-часового перемешивания при 20°С раствор охлаждают до 0°С и при интенсивном перемешивании осторожно по каплям добавляют смесь из 10 мл воды и 65 мл тетрагидрофурана. Затем к реакционному раствору осторожно добавляют еще 1250 мл воды и в течение 30 мин перемешивают при 20°С. Органическую фазу отделяют, а водную фазу экстрагируют 325 мл дихлорметана. Органическую фазу отделяют. Обе органические фазы объединяют и смешивают с 13 г NaHCO3 и 312 мл воды. Органическую фазу в течение 30 мин перемешивают при 20°С вместе с раствором NaHCO3. Органическую фазу отделяют и концентрируют в вакууме до объема порядка 300 мл (кристаллизация начинается еще до достижения требуемого объема). Кристаллическую суспензию смешивают с 300 мл ацетона и при 40°С и давлении 300 мбар отгоняют примерно 320 мл растворителя. Затем в течение 1 ч перемешивают при 0°С. Кристаллы отфильтровывают и промывают небольшим количеством холодного ацетона. Из маточного раствора после концентрирования можно получить еще одну кристаллическую фракцию.
Выход: 51,6 г (82% от теории) бесцветного кристаллического порошка.
Элементный анализ:
Пример 1ж
5-(4-{5-[(4,4,5,5,5-пентафторпентил)сульфанил]пентилокси} фенил)-6-фенил-8, 9-дигидро-7Н-бензоциклогептен-2-ол
100 г (230,9 ммоля) соединения, указанного в заголовке примера 1е, и 132,1 г (881,1 ммоля) иодида натрия нагревают в 1000 мл метилэтилкетона (МЭК) в течение 16 ч с обратным холодильником. Затем при пониженном давлении отгоняют примерно 650 мл растворителя и смешивают с 1500 мл воды. Далее в течение 15 мин перемешивают при комнатной температуре, выпавший осадок отфильтровывают и промывают его смесью из 240 мл холодного этанола и 160 мл воды. Все еще немного влажный осадок растворяют в 1200 мл тетрагидрофурана. При нормальном давлении отгоняют 400 мл тетрагидрофурана и добавляют 400 мл метанола. К этому раствору добавляют другой раствор, приготовленный в отдельной колбе следующим образом: 64,8 г (274,2 ммоля) 4,4,5,5, 5-пентафторпентилтиоацетата растворяют в атмосфере азота в 300 мл метанола и при комнатной температуре добавляют 51 мл 30%-ного раствора метанолята натрия (в МеОН). Далее в течение 30 мин перемешивают при комнатной температуре. После объединения обоих растворов их оставляют на 1 ч для перемешивания.
Для последующей обработки концентрируют в вакууме до остаточного объема порядка 350 мл. Затем к остатку добавляют 800 мл воды и в течение 30 мин перемешивают при 10°С. После этого отфильтровывают и промывают смесью из 360 мл воды и 40 мл метанола. В завершение сушат в вакууме при 40°С.
Выход: 124,1 г (91% от теории) бесцветного мелкокристаллического порошка.
Элементный анализ:
Пример 1з
5-(4-{5-[(RS)-(4,4,5,5,5-пентафторпентил)сульфинил]пентилокси} фенил)-6-фенил-8,9-дигидро-7Н-бензоциклогептен-2-ол
100 г (169,3 ммоля) соединения, указанного в заголовке примера 1ж, растворяют в смеси из 800 мл ацетона и 500 мл метанола. Затем осторожно добавляют 175 мл воды. После этого добавляют 36,20 г (169,3 ммоля) периодата натрия и перемешивают в течение 16 ч при комнатной температуре. Далее добавляют 1000 мл дихлорметана и 1300 мл воды и в течение 30 мин перемешивают при комнатной температуре. Органическую фазу отделяют и концентрируют в вакууме до половины объема. К остатку добавляют 950 мл толуола и при нормальном давлении осторожно отгоняют низкокипящие компоненты (дихлорметан и метанол). Затем нагревают до температуры, обеспечивающей отгонку примерно 50 мл толуола. Далее температуре дают понизиться до комнатной, при этом продукт кристаллизуется. После этого перемешивают в течение 30 мин при 10°С и выпавший осадок отфильтровывают. Затем дважды промывают слегка холодным толуолом и в завершение сушат в вакууме при 50°С.
Выход: 99,6 г (97% от теории) бесцветного кристаллического порошка.
Элементный анализ:
Ниже рассмотрены другие примеры, иллюстрирующие получение универсального промежуточного продукта общей формулы I.
В нижеследующих таблицах указан выход и приведен элементный анализ других соединений. Эти соединения могут быть получены аналогичным описанному в примере 1 путем.
Аналогично примеру 1а получали следующие соединения, с тем, однако, отличием, что вместо фенилуксусной кислоты использовали другую арилуксусную кислоту:
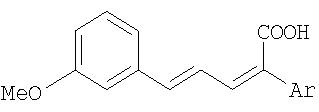
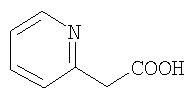
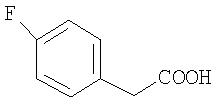
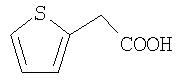
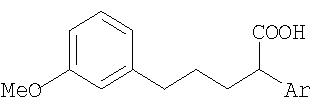
Аналогично примеру 1б осуществляли гидрирование системы с двойной связью:
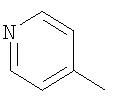
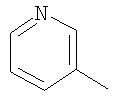
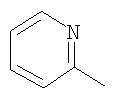
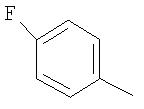
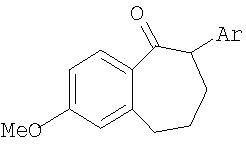
Аналогично примеру 1в осуществляли циклизацию с образованием бензосуберонов:
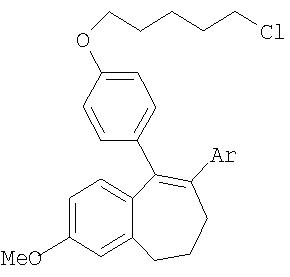
Аналогично примеру 1д через нонафлаты осуществляли реакцию сочетания по Сузуки:
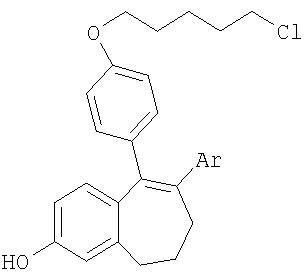
Аналогично примеру 1е отщепляли простые метиловые эфиры с помощью BBr3/2,6-лутидина:
Реферат
Изобретение относится к новым промежуточным продуктам и усовершенствованному способу получения соединения С:
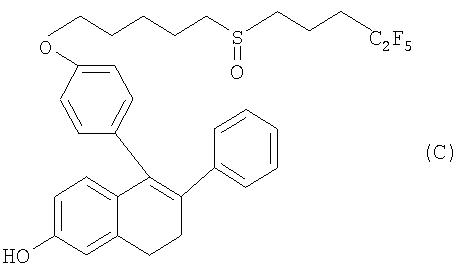
Предлагаемый в изобретении способ получения основан на использовании недорогих исходных материалов, позволяет получать промежуточные продукты с высоким выходом и высокой степенью чистоты без необходимости проводить операции по хроматографической очистке и может быть реализован в условиях крупномасштабного промышленного производства. Изобретение относится к усовершенствованным способам получения соединения формулы I:
соединения формулы II:
соединения формулы III:
соединения формулы VIII:
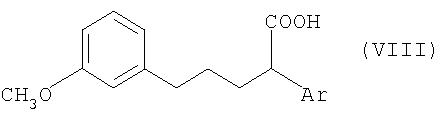
соединения формулы IX:
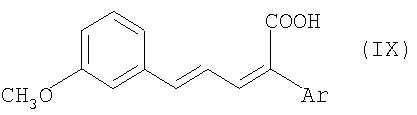
а также к реагенту, состоящему из трибромида бора и 2,6-диметилпиридина, для щадящего и селективного отщепления метильной группы в ароматических простых метиловых эфирах. 11 н. и 1 з.п. ф-лы.
Формула
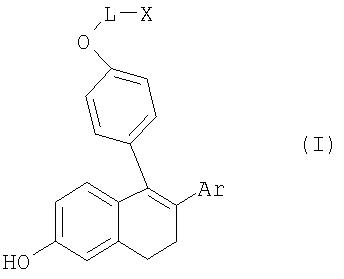
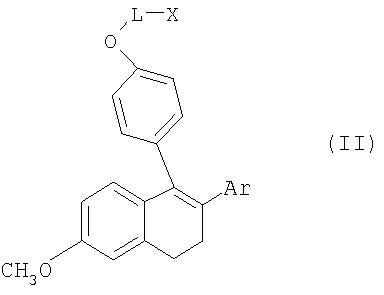
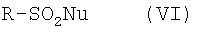
Комментарии