Способ получения высокочистого, свободного от галогенов о-фталальдегида - RU2423343C2
Код документа: RU2423343C2
Описание
Фталальдегиды, такие как о-фталальдегид (ОФА), используют во многих областях в качестве промежуточных соединений для получения красок, оптических осветлителей или особых полимеров, в биоциде или фотографической промышленности, и для синтеза фармахимических препаратов. Некоторые варианты способа описаны. Таким образом, о-фталальдегид (ОФА) можно, например, согласно ЕР-В-0147593, получать озонолизом нафталина в метаноле и каталитическим восстановлением получаемых при этом пероксидов с их последующей экстракцией или кристаллизацией. Недостаток указанного способа заключается в том, что сложный эфир, который образуется в качестве побочного продукта, трудно отделяется от ОФА.
Кроме того, ОФА представляет собой реакционно-способное соединение, которое нестабильно термически и к окислению, и которое при длительном хранении подвержено комкованию, таким образом, делая необходимыми проведение длительных процессов растворения, которые могут привести к обесцвечиванию ОФА. Возможность, описанная в ЕР-А1-0522312 для защиты альдегида от нежелательных реакций, заключается в применении тетраалкилацеталей о-фталальдегида, полученных электрохимическим окислением, в качестве соединений, способных к длительному хранению.
ЕР-В-0839789 дополнительно описывает превращение ОФА с образованием ацеталя при катализе кислотой, с последующей дистилляцией в пригодное для хранения соединение, такое как, например, диалкоксифталан или тетраалкилацеталь, из которого, при необходимости, получают сырой ОФА с чистотой более чем 99,5% после полного разложения ацеталя кислотным гидролизом.
Однако сырой ОФА, например, полученный согласно ЕР-В-0 839 789, имеет красновато-оранжевый цвет, и необходима последующая перекристаллизация до обесцвечивания, например, активированным углем или Tonsil.
Мелкий порошок ОФА с температурой плавления 57°С, который получают кристаллизацией, также склонен к комкованию. Кроме того, разные партии не показывают постоянного цвета и качества.
Патент США 5107032 описывает получение ОФА с помощью тетрагало-о-ксилола с последующим гидролизом при от 90 до 146°С. Это осуществляют, например, гидролизом тетрагало-о-ксилола ацетатом натрия в водном растворе уксусной кислоты при максимальной температуре 146°С и 3,5 бар, после чего 5-кратная экстракция толуолом и последующая отгонка объединенной органической фазы дает выход ОФА 87%. Однако недостаток указанного способа заключается в том, что полученный продукт не свободен от галогенов.
Поэтому целью настоящего изобретения было найти улучшенный способ получения высокочистого, свободного от галогенов ОФА.
Неожиданно оказалось возможным достичь указанной гидролизом тетрагало-о-ксилола при температуре свыше 155°С, последующей ацетализации в соответствующий ацеталь о-фталальдегида, очистки ацеталя перегонкой и разложением ацеталя при рН >1,5.
Изобретение, следовательно, касается способа получения высокочистого, свободного от галогенов о-фталальдегида, который включает:
а) гидролиз тетрагало-о-ксилола при температуре от 155°С до 160°С и давлении от 2 до 5 бар, если подходит, в присутствии катализатора межфазного переноса, в о-фталальдегид,
b) превращение о-фталальдегида в кислом спиртовом растворе при температуре от 0 до температуры кипения с обратным холодильником в соответствующий диалкоксифталан и, потом
с) разложение ацеталя кислотным гидролизом при рН от >1,5 до рН 7, с получением высокочистотного, свободного от галогенов о-фталальдегид.
Высокочистый, свободный от галогенов о-фталальдегид (ОФА) получают по способу изобретения.
Исходное вещество представляет собой тетрагало-о-ксилол. Подходящие тетрагало-о-ксилолы представляют собой, например, соединения формулы
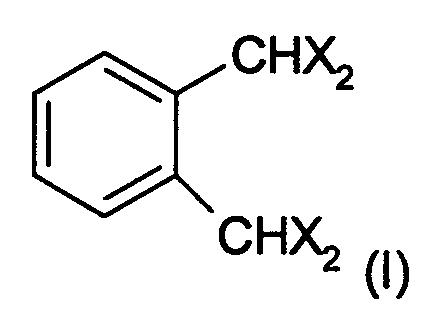
в которой Х может быть Cl, Br или I.
Тетрагало-о-ксилол (ТГК), кроме того, может быть необязательно замещен одними или более подходящими радикалами. Подходящие радикалы представляют собой С1-С4 алкил, С5-С20 арил, ОН, NO2, CN, Cl, Br или СО2Н.
Предпочтительно, если используют незамещенные ТГК соединения, и Х, предпочтительно, представляет собой хлор.
Подходящие ТГК соединения коммерчески доступны (с Х, равным Cl или Br) или могут быть получены известным способом, например, по реакции о-ксилола с элементарным хлором на свету или с использованием свободно-радикальных инициаторов, таких как АИБН, PCl3 и дибензоилпероксид и т.д.
ТГК гидролизуют по изобретению при температуре от 155°С до 160°С и давлении от 2 до 5 бар, предпочтительно, от 3 до 4 бар, в соответствующий о-фталальдегид.
Гидролиз проводят в водной системе, состоящей из С1-С4 карбоновой кислоты в присутствии основания, такого как, например, NaOH, LiOH, KOH и т.д. Предпочтительно применять уксусную кислоту в присутствии NaOH (от 40 до 50%).
Для указанной цели карбоновую кислоту или водный раствор карбоновой кислоты и ТГК, предпочтительно, смешивают и потом добавляют водный раствор основания и воду. Последовательность добавления можно менять.
Количество используемой карбоновой кислоты составляет от >4 до 20 мольных эквивалентов, исходя из ТГК. Предпочтительно от 8 до 10 мольных эквивалентов, исходя из ТГК.
Количество используемого основания составляет от 4,0 до 5,0 мольных эквивалентов, исходя из ТГК.
Можно добавлять в реакционную смесь катализаторы межфазного переноса для достижения более высокой скорости (МФК) (от 1 до 5 мол.%, исходя из ТГК). Подходящие МФК представляют собой обычные соединения, такие как, например, четвертичные аммониевые соли и фосфониевые соли, например соли тетраакиламмония и тетраалкилфосфония или соли арилалкилтриалиламмония. Предпочтительные соли в этой связи представляют собой галогениды, например хлорид или бромид тетрабутиламония, хлорид этилтриоктилфосфония, хлорид бензилтриэтиламмония и т.д.
После завершения гидролиза соответствующий сырой о-фталальдегид (ОФА) выделяют экстракцией обычными экстрагентами, такими как, например, метилтретбутиловый эфир, толуол, этилацетат и т.д., и последующей отгонкой растворителей и экстрагентов.
Затем на этапе b) сырой ОФА превращают в ацеталь, диалкоксифталан, с ацетализацией катализируемой кислотой со спиртом.
Предпочтительным спиртом в данном случае является С1-С4 спирт, особенно предпочтителен метанол или этанол.
Сырой ОФА с данной целью растворяют в спирте. Раствор затем доводят до рН от 0 до 3, предпочтительно, от 0,5 до 2, добавлением кислоты.
Подходящие кислоты представляют собой такие минеральные кислоты, как, например, HCl, H2SO4, H3PO4, такие органические кислоты, как, например, муравьиная кислота, уксусная кислота, п-толуолсульфоновая или метансульфоновая кислота или кислотные ионообменники.
Температура на данном этапе составляет от 0°С до температуры кипения с обратным холодильником, предпочтительно, до 50°С.
После завершения ацетализации водный раствор щелочи добавляют к раствору для нейтрализации кислоты или кислотных органических соединений. NaOH или КОН подходят в качестве щелочи.
Спирт, используемый в качестве растворителя, удаляют отгонкой потом или одновременно.
Диалкоксифталан, в свою очередь, выделяют экстракцией и последующей перегонкой. Выделенные ОФА ацетали в данном случае получаются с очень высоким, свободным от галогенов качеством (>99,5 ГХ % площади) и очень высоким выходом более чем 92%.
Разложение ацеталей на этапе с) также проводят обычным путем, известным из уровня техники кислотным гидролизом.
рН составляет от >1,5 до 7, предпочтительно, от 1,6 до 2,5, по изобретению.
Применяемые кислоты также представляют собой такие минеральные кислоты, как HCl, H2SO4, H3PO4, или такие органические кислоты, как уксусная кислота, муравьиная кислота и п-толуолсульфоновая или метансульфоновая кислота. Температура реакции, предпочтительно, от комнатной до 60°С. В то же время отгоняется удаляемый спирт и, где возможно, кислота. Полученный данным путем ОФА можно затем дополнительно очищать, где подходит, например, экстракцией, отмывкой или кристаллизацией.
ОФА получают с очень высоким, свободным от галогенов качеством (>99,5 ГХ % площади) и очень высоким выходом вплоть до свыше 92% очисткой по способу изобретения. Кроме того, ОФА, очищенный по изобретению, демонстрирует постоянный цвет и не нуждается в обесцвечивании с помощью Tonsil или угля. Кроме того, с помощью способа изобретения избегают потерь выхода ОФА.
Способ изобретения дополнительно отличается временем реакции, которое короче по сравнению с предшествующим уровнем техники, таким образом, делая возможным непрерывный процесс.
Пример 1
Этап а)
61 г (0,25 моль) тетрахлор-о-ксилола (ТХК) (чистота 99%) вводили в автоклав и добавляли 300 г (5 моль) уксусной кислоты. Затем медленно прибавляли раствор 44 г (1,1 моль) NaOH и 198 г деионизированной воды.
Реакционную смесь нагревали до 160°С и выдерживали при указанной температуре в течение 1 ч. Реакционное давление составляло 3,8-3,9 бар.
Затем смесь охлаждали до 40°С и выгружали из автоклава (606 г=525 мл реакционного раствора).
Образец 2 мл отбирали и анализировали.
Результат: 99,43 ГХ % площади сырого ОФА.
Экстракция сырого ОФА:
606 г раствора сырого ОФА сначала смешивали с 50 мл деионизированной воды и затем экстрагировали 4 раза с 93 г МТБЭ.
Органические фазы объединяли и концентрировали на роторном испарителе в вакууме 400-450 мбар и 34-72°С до прекращения отгонки.
После упаривания получали 54 г остатка.
Этап b)
Весь остаток после упаривания смешивали с 375 мл метанола и прибавляли 2 мл конц. H2SO4, чтобы довести pH до 0,5.
После 2 часов при 50°С прибавляли 50% раствор NaOH. Реакционную смесь затем концентрировали на роторном испарителе и затем смешивали с 175 мл деионизированной воды и экстрагировали 3×80 мл МТБЭ при 25°С.
Органические фазы объединяли и концентрировали на роторном испарителе.
Конечная масса 42,4 г.
Сырой ацеталь перегоняли при температуре бани 135-140°С, температура куба 113-114°С и температура верхнего погона 110-111°С. Давление составляло от 8 до 10 бар.
Конечная масса: 41,2 г ОФА диметоксиацеталя (91% от теории).
Этап с) разложение ОФА диметоксиацеталя:
200 г ОФА диметоксиацеталя прибавляли к 500 г деионизированной воды, которую доводили до pH 2,0 серной кислотой, и давление уменьшали до, примерно, 150 мбар. Затем начинали нагрев. Отбор дистиллята начинали при температуре куба 53°С и 155 мбар. Суммарно получали 270 мл дистиллята. IPC-ГХ анализ органической фазы после 4 ч показал 99,89% площади ОФА.
После добавления 500 мл DIPE ОФА экстрагировали при 50°С. После разделения фаз органическую фазу каждый раз промывали 2×100 мл деионизированной водой.
Промытый бледно-желтый окрашенный органический раствор кипятили с обратным холодильником и воду, оставшуюся в органической фазе, удаляли в виде азеотропа при атмосферном давлении. Суммарно получали 9,5 мл воды.
Затем начинали медленное охлаждение. Кристаллизация ОФА начиналась при 42°С. Дополнительное охлаждение продолжали до 15°С.
Кристаллический ОФА фильтровали через G-2 фритту, промывали 150 мл DIPE и сушили в вакууме при 40-45°С в течение ночи. Выделяли 91,5 г сухого ОФА с содержанием >99,8% и 450 мл маточного раствора. Выход составил 61,5% от теории.
Недостающий до 100% от теории ОФА присутствовал в маточном растворе, в воде после разложения и в воде после промывания, которые все могли быть повторно использованы в следующем цикле деацетализации, благодаря высокой чистоте и бледному цвету растворов. Количественная деацетализация была таким способом возможна.
Реферат
Настоящее изобретение относится к способу получения высокочистого о-фталальдегида, свободного от галогенов, имеющего чистоту >99,5 ГХ % площади, который может быть использован в качестве промежуточного соединения для получения красок, оптического осветлителя, в биоциде или фотографической промышленности. Предлагаемый способ включает следующие стадии: a) гидролиз тетрагало-о-ксилола при температуре 155-160°С и давлении от 2 до 5 бар до о-фталальдегида, который b) превращают в кислом спиртовом растворе при температуре от 0°С до температуры кипения с обратным холодильником в соответствующий диалкоксифталан и, далее c) ацеталь разлагают кислотным гидролизом при рН от >1,5 до рН 7, получая высокочистый, свободный от галогенов о-фталальдегид. 4 з.п. ф-лы.
Формула
a) гидролиз тетрагало-о-ксилола при температуре 155-160°С и давлении от 2 до 5 бар до о-фталальдегида, который
b) превращают в кислом спиртовом растворе при температуре от 0°С до температуры кипения с обратным холодильником в соответствующий диалкоксифталан, и далее
c) ацеталь разлагают кислотным гидролизом при рН от >1,5 до рН 7, получая высокочистый свободный от галогенов о-фталальдегид.
Комментарии