Фторэластомеры - RU2383555C2
Код документа: RU2383555C2
Описание
Данное изобретение относится к фторэластомерам, основанным на винилиденфториде VDF, имеющим Tg (температура стеклования) ниже -35°С и улучшенную комбинацию механических свойств и остаточной деформации при сжатии в широком пределе температур, как при высоких, так и при низких температурах.
Более конкретно данное изобретение относится к фторэластомерам, имеющим Tg ниже -35°С и существенно свободным от концевых -COF групп, причем эти группы не улавливаются рассмотренным ниже способом. Фторэластомеры в соответствии с данным изобретением имеют улучшенную молекулярную массу, что показывает улучшенная собственная вязкость, которые изготавливаются с помощью процесса полимеризации, имеющего повышенную производительность.
Хорошо известно, что фторэластомеры представляют собой полимеры, широко использующиеся в аэрокосмической, нефтяной, химической и электронной промышленностях благодаря комбинации их механических свойств и остаточной деформации при сжатии.
Однако требуется, чтобы эти полимеры имели комбинацию указанных выше свойств в широких пределах температур, как при высоких, так и при низких температурах.
В уровне техники предлагались различные фторэластомеры с комбинацией указанных выше свойств, которые особенно востребованы пользователями фторэластомеров. Однако значение концевых -COF групп во фторэластомерах не указывалось в уровне техники. Заявитель на основании глубоких исследований установил, что в том случае, когда в результате полимеризации получают полимеры, имеющие концевые -COF группы, фторэластомеры не имеют высоких механических свойств и не обладают высокой эластичностью.
Различные фторэластомеры, у которых температура стеклования указывается, известны из уровня техники. Однако с помощью известных из уровня техники решений не получают фторэластомеров с комбинацией низкой Tg и улучшенными механическими свойствами и эластичностью при высоких и низких температурах.
В патенте US 3132123 описывается получение перфторалкилвиниловых эфиров, их гомополимеров и сополимеров с TFE (тетрафторэтилен). Гомополимеры получают в экстремальных экспериментальных условиях, используя давление при полимеризации от 4000 до 18000 атм. Описываемые виниловые эфиры имеют следующую общую формулу:
CF2=CFOR0F,
где R0F - перфторалкильный радикал преимущественно с 1-5 атомами углерода. Исследования, проведенные заявителем, показали, что Tg гомополимера не очень низка и находится в пределе -6°С.
Патент US 3450684 относится к виниловым эфирам формулы:
CF2=CFO(CF2CFX0O)n'CF2CF2X0,
где Х0=F, Cl, CF3, H и n' может принимать значение от 1 до 20.
Сообщается также о гомополимерах, полученных УФ-поляризацией. Примеры сополимеров не характеризуются их механическими свойствами и эластичностью при низких температурах.
Патент US 3635926 относится к эмульсионной сополимеризации перфторвиниловых эфиров с TFE. Подчеркивается, что присутствие концевых -COF групп делает полимеры нестабильными. Об этом уже сообщалось в US 3085083 в отношении полимеризационных систем перфторвиниловых эфиров в растворителе.
Патент US 3817960 относится к получению и полимеризации перфторвиниловых эфиров формулы:
CF3O(CF2O)n"CF2CF2OCF=CF2,
где n" может изменяться от 1 до 5. Синтез винилового эфира сложен. Не приводится данных, характеризующих свойства полученного продукта.
Патент US 3896179 относится к выделению "первичных" изомеров фторвиниловых эфиров, например CF3CF2CF2OCF=CF2, из соответствующих "вторичных", менее устойчивых полимеров CF3(CF3)CFOCF=CF2. Последние являются нежелательными продуктами в отношении получения и низких характеристик получаемых полимеров.
Патент US 4487903 относится к получению фотоэластомерных сополимеров с использованием перфторвиниловых эфиров формулы:
CF2=CF(OCF2CFY0)n0OX2,
где n0 может принимать значение от 1 до 4; Y0=F, Cl, CF3, H; X2 может быть C1-С3 перфторалкил, C1-С3 ω-гидроперфторалкил, C1-С3 ω-хлорперфторалкил. Полимер содержит блок фторвиниловых эфиров в пределе от 15 до 50% молярных. Эти виниловые эфиры формируют сополимеры, имеющие при низких температурах свойства, которые выше, чем свойства указанных перфторэластомеров PVE (перфторпропилвиниловый эфир) и MVE. В этом патенте указывается на тот факт, что для достижения хороших свойств при низкой температуре требуется присутствие, по меньшей мере, двух эфирных связей в боковой цепи, примыкающих к двойной связи. Кроме того, в этом патенте для значений n0 более 4 трудно очистить мономеры, и эффект снижения Tg уменьшается. Кроме того, реактивность описанных виниловых эфиров очень низка, и трудно получать полимеры, имеющие большую молекулярную массу и способные обладать эластичностью для указанных назначений. Приводится пример сополимера TFE/перфторвиниловый эфир (n0=2), 31/69% по массе, с Tg -32°С. Однако этот полимер получают за очень долгий промежуток времени протекания реакции (96 часов полимеризации). В этом случае также не приводятся характеристические данные отвержденного эластомера.
В ЕР 130052 описывается полимеризация перфторвинилового полиэфира (PVPE), в результате которой получают аморфные перфторполимеры, имеющие Tg в пределах от -15°С до -100°С. В этом патенте описываются сополимеры и тройные полимеры TFE и MVE с виниловыми эфирами (PVPE) формулы:

где n′′′ находится в пределе от 3 до 30, a R0f' является перфторалкилом. В связи с трудностями очистки использующиеся виниловые эфиры являются смесью виниловых эфиров с различными значениями n′′′. В соответствии с этим патентом наибольший эффект снижения Tg достигается в тех случаях, когда n′′′ равен или более 3, преимущественно более 4. В соответствии с примерами полимеризации, описанными в указанном патенте, конечная масса полимера, кроме обработки нагревом и в вакууме, должна затем промываться фреоном ® TF для того, чтобы удалить весь непрореагировавший мономер (PVPE). Судя по этому примеру, реактивность всех описанных мономеров (PVPE) низкая.
Патент US 4766190 относится к полимеризации перфторвиниловых полиэфиров (PVPE), аналогичных тем, которые рассматриваются в US 4487903 с TFE и небольшим в процентном отношении содержании перфторпропена для улучшения механических свойств получаемых полимеров.
Патент US 5401818 относится к получению перфторвиниловых эфиров формулы:
R1f(OCF2CF2CF2)m'-OCF=CF2,
(где R1f является C1-С3 перфторалкильным радикалом и m' - целое число от 1 до 4) и соответствующих сополимеров, имеющих улучшенные свойства при низких температурах. Получение указанных перфторвиниловых эфиров проходит через 7 стадий, из которых некоторые имеют очень низкий выход, а также включают фторирование элементарным F2. Тем не менее реактивность указанных перфторвиниловых эфиров остается низкой.
Другие проблемы, отмеченные в документах из уровня техники, относятся к низкой реактивности перфторвиниловых эфиров, в связи с чем необходимо извлекать из полученных продуктов непрореагировавшие мономеры (английский патент GB 1514700), а также решать проблемы стабильности полимеров, имеющих концевые -C(O)F группы (US 3635926). Последние могут далее трансформироваться с помощью подходящих реагентов с целью повышения стабильности полимеров (ЕР 178935).
Перфтороксиалкилвиниловые эфиры используются также для придания резинам требующихся свойств при низких температурах и, в частности, для снижения температуры стеклования.
При увеличении перфтороксиалкиленовых блоков снижается Tg аморфных сополимеров, но в то же время реактивность винилового эфира значительно понижается, и получение полимеров с достаточно большой молекулярной массой и хорошими свойствами становится очень трудно или вообще невозможно. Отмеченные ранее проблемы удаления непрореагировавших мономеров из полученных полимеризацией продуктов или из самого полимера остаются (US 4487903 - ЕР 130052).
Аморфные TFE сополимеры с перфторметилвиниловым эфиром имеют Tg около 0°С или немного ниже (Maskornik, M. et al. "ECD-006 Fluoroelastomer - A high performance engineering material", Soc. Plast. Eng. Tech. Pao. (1974), 20, 675-7).
Экстраполированное значение Tg для гомополимера MVE составляет около -5°С (J.Macromol. Sci.-Phys., B1 (4), 815-830, Dec. 1967).
Известны другие патенты, описывающие виниловые эфиры для получения фторэластомеров. См. патенты US 6255536, ЕР 1117710, WO 99/48939 и US 6696216.
Более детально, известны фторэластомерные сополимеры, пригодные для изготовления уплотнительных колец, основанные на мономерных блоках, полученных из винилиденфторида (VDF), гексафторпропена (HFP), перфторалкилвиниловых эфиров (PAVE), как, например, метилвиниловый эфир, и, необязательно, тетрафторэтилен (TFE), который способен ионно отверждаться, имеют хорошие эластомерные свойства при низких и высоких температурах и хорошую способность к обработке, как, например, извлечение из формы после отверждения (см. US 5260393). Однако указанные фторэластомеры имеют температуру стеклования (Tg) выше -35°С. Поэтому они непригодны для использования при температурах ниже -35°С, так как теряют их эластомерные свойства.
Патентная заявка ЕР 1308467 описывает перфторэластомеры, содержащие фторалкоксивиниловые эфиры формулы CFXA=CXAOCF2ORA, где XA=F, H; RA - С2-С6 перфторалкил, перфтороксиалкил или C5-C6 циклический (пер)фторалкил. В частности, описываются следующие перфторалкоксивиниловые эфиры: CF2=CFOCF2OCF2CF3 (MOVE 1) и CF2=CFOCF2OCF2CF2OCF3 (MOVE 2). В Примерах описываются перфторэластомеры, содержащие не более 40% указанных перфторалкоксивиниловых эфиров. MOVE перфторэластомеры с TFE содержат концевые -COF группы. См. сравнительные Примеры. Для приготовления полимерных композиций, содержащих TFE в количестве, ниже или равном 60% в молярном отношении, необходимо проводить полимеризацию в течение длительного времени. С промышленной точки зрения это представляет собой недостаток, так как производительность ухудшается.
В ЕР 1304341 описываются фторэластомеры, содержащие фторалкоксивиниловые эфиры формулы CFXA=-CXAOCF2ORA, где ХА=F, H; RA - С2-С6 перфторалкил, перфтороксиалкил или C5-C6 циклический (пер)фторалкил. В частности, описаны следующие перфторалкоксивиниловые эфиры: CF2=CFOCF2OCF2CF3 (MOVE 1) и CF2=CFOCF2OCF2CF2OCF3 (MOVE 2). В Примерах приводятся фторэластомеры, содержащие не более 19% указанных перфторалкоксивиниловых эфиров. Тесты, проведенные заявителем, показали, что указанные фторэластомеры имеют концевые -COF группы, которые, как указывалось, ухудшают механические свойства при высоких температурах и термостойкость указанных полимеров.
Ощущается потребность в получении основанных на VDF фторэластомеров, имеющих следующую комбинацию свойств:
- Tg ниже -35°С, преимущественно ниже -40°С, более преимущественно ниже -45°С;
- существенно свободные от концевых -COF групп, причем указанные группы не улавливаются способом, указанным ниже;
- улучшенная молекулярная масса, определяемая по более высокой внутренней вязкости;
- улучшенные механические свойства и остаточная деформация при сжатии в широких пределах как высоких, так и низких температур;
- улучшенная производительность процесса получения фторэластомеров, выраженная через величину (масса полимера в кг/час × литр воды).
Заявитель неожиданно и к его удивлению открыл фторэластомеры, имеющие удивительно улучшенную комбинацию рассмотренных выше качеств.
Целью данного изобретения являются отверждаемые VDF-base фторэластомеры, имеющие температуру стеклования ниже -35°С, преимущественно ниже -40°С, более преимущественно ниже -45°С, и имеющие количество концевых -COF групп ниже, чем уровень чувствительности описанного ниже метода; в конце полимеризации полимер изолируется коагуляцией с помощью замораживания и последующего размораживания; он промывается дважды деминерализованной водой и высушивается в печи до достижения постоянного веса; концевые -COF группы определяются ФТ-ИК-спектроскопией, когда на полимерной пленке, имеющей толщину от 50 до 300 микрон, проводится сканирование между 4000 см-1 и 400 см-1, пленка выдерживается в течение 12 часов в атмосфере, насыщенной парами аммиака, и после этого ведется запись ИК-спектра в тех же условиях, в которых получался первоначальный ИК-спектр; обрабатываются оба спектра, вычитая из сигналов спектра необработанного образца (первоначальный спектр) соответствующие сигналы спектра образца после обработки парами аммиака, получают спектр "различия", нормализованный следующим уравнением:

измеряются оптические плотности, относящиеся к концевым -COF группам, прореагировавшим с парами аммиака; оптические плотности преобразовываются в ммоль/кг полимера, используя коэффициент удаления, указанный в таблице страница 73 отчета М.Pianca и др. "End groups in fluoropolymers", J. Fluorine Chem. 95 (1999), 71-84 (использован здесь в качестве ссылки); полученные значения выражают концентрацию остаточных концевых -COF групп в ммолях концевых -COF групп на 1 кг полимера: в спектре фторэластомера не отслеживается линий, относящихся к концевым -COF группам (1900-1830 см-1) при пределе чувствительности метода 0.05 ммоль/кг.
Более точно, количество концевых -COF групп в полимере определяется на ФТ-ИК оборудовании Nicolet® Nexus (256 сканирований, разрешение 2 см-1).
Фторэластомеры в соответствии с данным изобретением содержат также блоки, извлекаемые из бис-олефинов общей формулы:
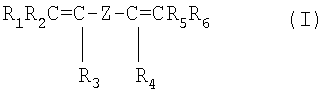
где R1, R2, R3, R4, R5, R6, равные или отличающиеся друг от друга, представляют Н или C1-C5 алкилы;
Z представляет С1-С58 линейный или разветвленный алкилен или циклоалкиленный радикал, необязательно содержащий атомы кислорода, преимущественно, по меньшей мере, частично фторированный, или (пер)фторполиоксиалкиленный радикал, как тот, который описан в патенте ЕР 661304, выданном на имя заявителя.
Количество бис-олефинов обычно составляет от 0.01 до 1.0% молярных, преимущественно от 0.03 до 0.5% молярных и еще более преимущественно от 0.05 до 0.2 молей на 100 молей приведенных выше мономерных блоков, составляющих основную структуру фторэластомера; полная сумма мономеров составляет 100%.
В формуле (I) Z преимущественно является С4-С12, более преимущественно С4-С8, перфторалкиленовый радикал, в то время как R1, R2, R3, R4, R5, R6 являются преимущественно водородом; когда Z является (пер)фторполиоксиалкиленом, он может включать блоки, выбранные из следующих:
-CF2CF2O-, -CF2CF(CF3)O-, -CFX1O-, где X1=F, CF3, -CF2CF2CF2O-, -CF2-CH2CH2O-, -C3F6O-.
Преимущественно Z выражается формулой:
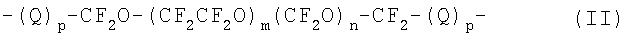
где Q является C1-С10 алкиленовым или оксиалкиленовым радикалом; p=0 или 1; m и n - такие числа, что отношение m/n находится в пределе между 0.2 и 5, и молекулярная масса указанного (пер)фторполиоксиалкиленового радикала находится в пределах 500-10000, преимущественно 700-2000.
Преимущественно Q выбирается из
-CH2OCH2-; -CH2O(CH2CH2O)sCH2-, где s=1-3.
Бис-олефины формулы (I), где Z является алкиленом или циклоалкиленом, могут получаться в соответствии с описанием в литературе, например, у И.Л.Кнуньянц и др. в Изв. Акад. наук СССР, сер. Химия, 1964(2), стр.384-6. Бис-олефины, содержащие структуры (пер)фторполиоксиалкилена, описаны в патенте US 3810874.
Более преимущественно бис-олефины имеют формулу:
CH2=CH-(CF2)t0-CH=CH2
где t0 - целое число от 6 до 10.
Бис-олефины формулы:
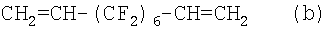
являются особенно преимущественными.
Фторэластомеры в соответствии с данным изобретением отверждаются пероксидным путем. Когда фторэластомеры по данному изобретению отверждаются пероксидным путем, они содержат преимущественно йод и/или бром, более преимущественно йод, в количествах, обычно находящихся в пределах от 0.001% до 5% по массе, преимущественно в пределах от 0.01% до 2.5% по массе в отношении к полной массе полимера. Атомы йода могут находиться внутри цепи и/или в концевом положении.
Для введения атомов йода и/или брома в цепь проводится сополимеризация основных мономеров фторэластомера с подходящими фторированными сомономерами, содержащими йод и/или бром (мономеры при отверждении), см., например, патенты US 4745165, US 4831085, US 4214060, EP 683149. Указанный фторированный сомономер, содержащий йод, может выбираться, например, из следующих соединений:
(a) йодо(пер)фторалкил-перфторвиниловый эфир формулы:

где Rf-C1-C12 (пер)фторалкилен, необязательно содержащий хлор и/или атомы кислорода эфира;
например: ICF2-O-CF=CF2, ICF2CF2-O-CF=CF2, ICF2CF2CF-O-CF=CF2, CF3CFICF2-O-CF=CF2 и подобные им;
(b) йод-(пер)фторолефины формулы:
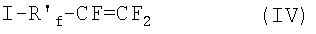
где R′f - C1-C12 (пер)фторалкилен, необязательно содержащий атомы хлора; например: йодтрифторэтилен, 1-йод-2,2-дифторэтилен, йод-3,3,4,4-тетрафторбутан-1, 4-йодперфторбутан-1 и подобные им;
(c) йод-(пер)фторолефины формулы:
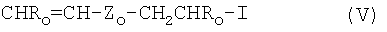
где Ro=Н или -СН3; Zo-C1-C-18 линейный или разветвленный (пер)фторалкилен, необязательно содержащий один или более атомов кислорода, или (пер)фторполиоксиалкилен, определенный выше.
Другими йодированными сополимерами при отверждении являются йодфторалкилвиниловые эфиры, см. патенты US 4745165 и US 4564662.
Кроме или в дополнение к йодированным сомономерам, фторэластомер может содержать атомы йода в концевой позиции, получаемые из подходящего йодированного цепного трансферного агента, вводимого в реакционную среду во время приготовления полимера, как описано в патенте US 4501869. Указанные трансферные агенты имеют формулу RAf(I)x, где RAf - радикал C1-C-12 (пер)фторалкил, необязательно содержащий атомы хлора, а х=1 или 2.
Указанный трансферный агент может выбираться, например, из CF2I2, I(CF2)6I,
I(CF2)4I, CF2ClI, CF3CFICF2I и им подобных.
Для йода, вводимого в качестве цепной концевой группы путем добавления йодированного цепного трансферного агента, как упомянутый выше, см., например, US 4243770 и US 4943622.
Возможно также использовать в качестве цепных трансферных агентов йодиды щелочных или щелочноземельных металлов в соответствии с патентной заявкой ЕР 407937.
В комбинации с цепными трансферными агентами, содержащими йод, могут использоваться другие известные в уровне техники цепные трансферные агенты, как этилацетат, диэтилмалонат и т.п.
Количество йода в концевой позиции фторэластомера обычно составляет между 0.001% и 3%, преимущественно между 0.01% и 1% по массе в отношении к массе фторэластомера. См. US 4035565 и US 4694045.
Кроме того, отвержденные фторэластомеры могут также содержать, кроме йода или в комбинации с ним, бром в цепи или в концевой позиции. Бром может вводиться в цепь с помощью известной технологии, см., например, US 4035565, US 4745165, EP 199138; или как концевой бром, как описывается в US 4501869.
Преимущественно перфторэластомер содержит атомы йода в цепи и/или в концевой позиции.
Необязательно перфторэластомеры по данному изобретению содержат в смеси полукристаллический (пер)фторполимер в количестве в процентах по массе, соотнесенных с полной сухой массой смеси фторэластомер + полукристаллический (пер)фторполимер, от 0% до 70%, преимущественно от 0% до 50%, еще более преимущественно от 2% до 30% по массе.
Под понятием полукристаллический (пер)фторполимер имеется в виду (пер)фторполимер, который имеет не только температуру стеклования Tg, но и, по меньшей мере, одну температуру плавления кристаллов.
Полукристаллический (пер)фторполимер составляется из тетрафторэтиленовых (TFE) гомополимеров или TFE сополимеров с одним или более мономерами, содержащими, по меньшей мере, одну ненасыщенность этиленового типа, в количестве от 0.01% до 10% молярных, преимущественно от 0.05% до 7% молярных.
Указанные сомономеры, имеющие этиленовую ненасыщенность, могут быть как гидрированного, так и фторированного типа. Среди гидрированных можно указать этилен, пропилен, акриловые мономеры, например метилметакрилат, (мет)акриловая кислота, бутилакрилат, гидроксиэтилгексилакрилат, стироловые мономеры.
Среди фторированных сомономеров можно назвать:
- С3-С8 перфторолефины, как гексафторпропен (HFP), гексафторизобутан;
- С2-С8 гидрированные фторолефины, как винилфторид (VF), винилидинфторид (VDF), трифторэтилен, перфторалкилэтилен CH2=CH-Rf, где Rf - C1-C6 перфторалкил;
- С2-С8 хлор- и/или бром- и/или йод-фторолефины, как хлортрифторэтилен (CTFE);
- CF2=CFORf (пер)фторалкилвиниловые эфиры (PAVE), где Rf - C1-C6 (пер) фторалкил, например CF3, C2F5, C3F7;
- CF2=CFOX (пер)фтороксиалкилвиниловые эфиры, где Х=C1-C12 алкил или C1-C12 оксиалкил или C1-C12 (пер)фтороксиалкил, имеющий одну или более эфирных групп, например перфтор-2-пропоксипропил; фтордиоксолы, преимущественно перфтордиоксолы.
PAVEы, в частности перфторметил-, этил-, пропилвиниловый эфир и фтордиоксолы, преимущественно перфтордиоксолы, являются преимущественными сомономерами.
Когда фторэластомеры по данному изобретению содержат полукристаллические (пер)фторполимеры, проводится смешивание в желательном соотношении латекса перфторэластомера с латексом полукристаллического перфторполимера, а затем проводится сокоагуляция полученной смеси так, как описано в US 6395834 и US 6310142.
В другом варианте полукристаллический (пер)фторполимер может полимеризироваться, а затем фторэластомер полимеризируется на частицах (пер)фторполимера. Таким образом получают структуру, состоящую из ядер и оболочек.
Заявитель обнаружил, что когда концевые -COF группы во фторполимере после полимеризации существенно отсутствуют при обнаружении изложенным выше методом анализа, получается наилучшая комбинация механических свойств и остаточной деформации при сжатии в широком диапазоне температур, как при высоких, так и при низких температурах.
Другим объектом данного изобретения являются композиции, которые включают:
- фторэластомеры по данному изобретению, имеющие концевые -COF группы в количестве менее чем 0.05 ммоль/кг и Tg, как она была определена выше,
и
- фторэластомеры, получаемые из полимеров, содержащих концевые -COF группы в количестве более 0.05 ммоль/кг при условии, что количество перфторэластомера по данному изобретению составляет, по меньшей мере, 5-10% по массе, преимущественно 20-40% по массе, более преимущественно 50% по массе в отношении полной массы фторэластомеров в композиции.
Эти композиции могут быть получены различными путями. Например, когда мономеры, привносящие концевые -COF группы, используются в полимеризации для получения улучшенных свойств в соответствии с данным изобретением, проводится частичная полимеризация мономеров в отсутствие тех мономеров, которые привносят концевые -COF группы, чтобы получить определенное количество полимера, существенно свободного от концевых -COF групп, что позволяет получить комбинацию с указанными выше свойствами. Например, полимер, полученный при такой полимеризации части исходного материала, проведенной в отсутствие мономеров, дающих концевые -COF группы, должен составлять, по меньшей мере, 5-10% по массе, преимущественно 20-40% по массе, более преимущественно 50% по массе в отношении окончательной массы полимера. В альтернативном процессе полимеры по данному изобретению, существенно свободные от концевых -COF групп, смешиваются с полимерами, содержащими концевые -COF группы в указанных выше отношениях.
Фторэластомеры, содержащие концевые -COF группы в количестве, более 0.05 ммоль/кг, содержат сомономеры, выбранные из следующих соединений:
- перфтордиоксолы, преимущественно имеющие следующую формулу:

где Y=F, ORf1, Rf1, являющийся C1-C5 перфторалкилом, преимущественно Rf1=CF3;
X1 и Х2, одинаковые или отличающиеся друг от друга, выбираются из F и CF3, преимущественно F;
Z1 выбирается из F, Cl, преимущественно F;
- перфторалкилвиниловые эфиры формулы CF2=CFORf, где Rf является С3 перфторалкилом;
- CF2=CFOXa перфтороксиалкилвиниловые эфиры, где Ха является С3-С12 перфтороксиалкилом, имеющим одну или более эфирных групп, например перфтор-2-пропоксипропил;
- перфторвиниловые эфиры (MOVE) общей формулы
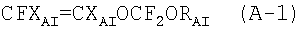
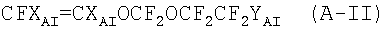
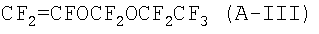
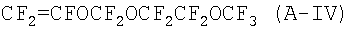
Отверждаемые фторэластомеры в соответствии с данным изобретением преимущественно содержат следующие мономеры (в молярных процентах):
A) от 1% до 99%, преимущественно от 5% до 99%, мономера формулы:

B) один или более перфорированных сомономеров, имеющих, по меньшей мере, одну ненасыщенность этиленового типа, от 1% до 99%, преимущественно от 1 до 95%;
указанные один или более сомономеров содержат VDF (VDF) в количестве от 1% до 85% по отношению к полной молярной массе мономеров для того, чтобы полимер был фторэластомерным; сумма молярных процентов мономеров равна 100%; количество концевых -COF групп является таким, как указано выше.
Когда полимер не содержит других мономеров В), кроме VDF, количество мономера формулы (а) должно быть не менее 15% молярных для того, чтобы получать фторэластомерные полимеры.
Указанные фторэластомеры преимущественно содержат один бис-олефин.
Более точно количество концевых -COF групп в полимере определяется с использованием ФТ-ИК прибора Nicolet® Nexus (256 сканирований, разрешение 2 см-1).
Эластомерные полимеры в соответствии с данным изобретением представляют собой полимеры, которые при использовании DSC (дифференциальная сканирующая калориметрия) не показывают пиков плавления, так как кристаллическая часть существенно отсутствует.
Когда кроме VDF присутствуют другие сомономеры (В), они выбираются из следующих соединений:
- С2-С8 перфторолефины, например TFE, гексафторпропен, гексафторизобутен;
- перфторалкилвиниловые эфиры формулы CF2=CFORf, где Rf является C1-C2 перфторалкилом, преимуществонно Rf=CF3.
Тетрафторэтилен (TFE) и/или перфторметилвиниловый эфир (MVE) являются преимущественными сомономерами В).
Преимущественными композициями (в молярных %) являются следующие, причем сумма молярных процентов мономеров равна 100%; более преимущественно указанные композиции содержат бис-олефин:
мономер формулы (а): 15-40%, VDF: 60-85%; преимущественно мономер формулы (а): 15-40% VDF: 60-85%, бис-олефин формулы (b): 0.01%-1%;
мономер формулы (а): 15-40%, VDF: 60-85%; TFE: 5-40%; преимущественно мономер формулы (а): 15-40%, VDF: 60-85%; TFE: 5-40%, бис-олефин формулы (b): 0.01%-1%;
мономер формулы (а): 5-40%, MVE: 5-30%; VDF: 50-85%; преимущественно мономер формулы (а): 5-40%, MVE: 5-30%; VDF: 50-85%; бис-олефин формулы (b): 0.01%-1%;
мономер формулы (а): 5-40%, MVE: 5-30%, VDF: 50-85%; TFE: 5-40%; преимущественно мономер формулы (а): 5-40%, MVE: 5-30%, VDF: 50-85%; TFE: 5-40%, бис-олефин формулы (b): 0.01%-1%;
мономер формулы (а): 40-99%; VDF: 1-60%; преимущественно мономер формулы (а): 40-99%, VDF: 1-60%; бис-олефин формулы (b): 0.01%-1%;
мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; преимущественно мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; бис-олефин формулы (b): 0.01%-1%;
мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; TFE: 5-40%; преимущественно мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; TFE: 5-40%; бис-олефин формулы (b): 0.01%-1%.
Как уже было сказано, фторэластомеры по данному изобретению имеют улучшенную комбинацию указанных выше свойств.
Фторэластомеры по данному изобретению показывают высокую эластичность при низких температурах, как, например, определяется значениями TR10 и TR70 (ASTM D 1329).
Фторэластомеры по данному изобретению по сравнению с фторэластомерами, известными в уровне техники, имеют Tg ниже -35°С, при сравнении, проводящемся с известными фторэластомерами, имеющими такую же Tg, показывают улучшенные механические свойства и остаточную деформацию при сжатии и более высокую устойчивость при высоких температурах. При сравнении, проведенном с фторэластомерами, основанными на перфторметилвиниловых эфирах, представляющими наиболее широко продающиеся фторэластомеры для использования при низких температурах, фторэластомеры по данному изобретению показали более низкую Tg и улучшенные свойства при низких температурах, что подтверждается значениями TR.
Заявитель обнаружил, что фторэластомеры по данному изобретению получаются с высокой полимеризационной кинетикой, и потому возможно получать гомополимеры и сополимеры, имеющие большую молекулярную массу. Фторэластомеры по данному изобретению могут производится с большим выходом, и потому извлечение непрореагировавших мономеров в конце полимеризации не имеет смысла. Это позволяет упростить конструкцию производящей установки, т.к. нет необходимости применять дорогие способы извлечения непрореагировавших мономеров.
Получение фторэластомеров по данному изобретению осуществляется полимеризацией мономеров в водной эмульсии в присутствии эмульсии, дисперсии или микроэмульсии перфторполиоксиалкиленов в соответствии с патентами US 4789717 и US 4864006. Преимущественно синтез проводится в присутствии микроэмульсии перфторполиоксиалкиленов.
В соответствии с хорошо известными в уровне техники способами используются радикалы-инициаторы, например персульфаты, перфосфаты, пербораты или перкарбонаты щелочи или аммония, необязательно в комбинации с солями железа, меди или серебра или других легко окисляющихся металлов. В реакционной среде необязательно также присутствуют поверхностно-активные вещества различных типов, среди которых особым преимуществом обладают фторированные поверхностно-активные вещества формулы:

где R3f - цепь C5-C16 (пер)фторалкила или (пер)фторполиоксиалкила, Xk- представляет - СОСТ- или -SO3-, M+ выбирается из Н+, NH4+ или иона щелочного металла. Среди наиболее обычно применяемых мы можем привести перфтороктаноат аммония, (пер)фторполиоксиалкилены, заканчивающиеся одной или более карбоксильными группами, и т.п. См. патенты US 4990283 и US 4864006.
Реакция полимеризации обычно проводится при температурах между 25°С и 150°С и при давлении между атмосферным и до 10 МПа.
В замену или в комбинации с цепными трансферными агентами, содержащими йод и/или бром, могут использоваться другие цепные трансферные агенты, известные из уровня техники, такие как этилацетат, диэтилмалонат, этан и т.п.
Как сообщалось ранее, фторэластомеры по данному изобретению отверждаются пероксидным или ионным путем.
При пероксидном отверждении преимущественно фторэластомер содержит в цепи и/или в концевой позиции макромолекулу йода и/или атомы брома.
К отверждающей смеси добавляются следующие компоненты:
- пероксиды, способные выделять радикалы при нагревании, например диалкилпероксиды, в частности дитербутилпероксид и 2,5-диметил1-2,5-ди(тербутилперокси)гексан; диалкиларилпероксиды, как, например, дикумилпероксид; дибензоилпероксид; дитербутилпербензоат; ди[1,3-диметил-3-(тербутилперокси)бутил]карбонат. Другие пероксидные системы описаны, например, в Европейских патентных заявках ЕР 136596 и ЕР 410351.
Количество пероксида обычно составляет от 0.5% до 10% по массе в отношении к массе полимера, преимущественно 0.6%-4% по массе;
- отверждающие соагенты, применяющиеся обычно в количестве между 0.5 и 10%, преимущественно между 1 и 7% по массе в отношении к массе полимера; среди них обычно используются бис-олефины формулы (I); триаллилцианурат, триаллилизоцианурат (TAIC), трис-(диалиламин)-s-триазин; триаллилфосфит; N,N-диаллилакриламид; N,N,N′,N′-тетраллилмалонамид; тривинилизоцианурат; 4,6-тривинилметилтрисилоксан и т.п.; особенно преимущественны TAIC и бис-олефин формулы:
CH2=CH-(CF2)6-СН=СН2;
необязательно
- соединение металла в количестве от 1 до 15%, преимущественно от 2 до 10% по массе в отношении к массе полимера, которое выбирается из оксидов или гидроксидов двухвалентных металлов, например, Mg, Zn, Са или Pb, необязательно комбинируется с солью слабой кислоты, как стеараты, бензоаты, карбонаты, оксалаты или фосфиты Ва, Na, K, Pb, Са;
- другие обычные добавки, как минеральные наполнители, полукристаллические фторполимеры в порошке, пигменты, антиоксиданты, стабилизаторы и т.п.
Когда отверждение осуществляется ионным путем, добавляются отверждающие и ускоряющие процесс агенты, известные из уровня техники. В этом случае фторэластомер преимущественно содержит блоки, получаемые из HFP.
Количество ускоряющего агента находится в пределах 0.05-5 ч/100 ч (частей на 100 частей полимера), отверждающего агента в пределах 0.5-15 ч/100 ч, преимущественно 1-6 ч/100 ч.
В качестве отверждающих агентов могут применяться полигидрооксилированные, ароматические или алифатические соединения или их производные, как излагается в патентах ЕР 335705 и US 4233427. Среди них мы, в частности, отмечаем следующие соединения: ди-, три-, тетра-гидроксибензолы, нафталины или антрацены; бисфенолы, у которых два ароматических кольца соединены друг с другом алифатическим, циклоалифатическим или ароматическим бивалентным радикалом или одним атомом кислорода или серы, или также карбонильной группой. Ароматические кольца могут замещаться одним или более атомами хлора, фтора, брома или карбонилами, алкилами, ацилами. В частности, преимущественным является бисфенол AF.
Ускоряющими агентами могут быть, например, четвертичные аммониевые или фосфоновые соли (см., например, ЕР 335705 и US 3876654); аминофосфоновые соли (см., например, US 4259463); фосфораны (см., например, US 3752787); иминовые соединения, описанные в ЕР 182299 и ЕР 120462 и т.д.
Преимущественными являются четвертичные фосфоновые соли и аминофосфоновые соли.
Взамен использования ускоряющего и отверждающего агентов можно использовать от 1 до 5 ч/100 ч (преимущественно 2-4.5) объединенного продукта ускоряющего агента и отверждающего агента в молярном отношении 1:2-1:5, преимущественно 1:3-1:5, причем ускоряющий агент является одним из ониум-органических соединений, имеющих положительный заряд, как указано выше, отверждающий агент, выбирающийся из упомянутых выше соединений, в частности ди- или полигидроксил или ди- или политиол; объединенный продукт получается плавлением реакционного продукта с ускоряющим агентом и отверждающим продуктом в указанном молярном отношении или плавлением смеси объединенного продукта 1:1 с добавлением отверждающего агента в указанных количествах. Необязательно ускоряющий агент может присутствовать в избытке по отношению к тому количеству, которое содержится в объединенном продукте, обычно от 0.05 до 0.5 ч/100 ч.
Для получения объединенного продукта особенно преимущественными являются катионы: 1,1-дифенил-1-бензил-N-диэтилфосфоранамин и тетрабутилфосфониум; из анионов: бисфенольные соединения, где два ароматических кольца соединены двухвалентным радикалом, выбирающимся из перфторалкильных групп с 3-7 атомами углерода, и ОН в двойных положениях особенно преимущественны.
Получение объединенного продукта описывается в европейской патентной заявке на имя данного заявителя ЕР 684277, использующейся здесь в качестве ссылки.
Смесь для ионного отверждения дополнительно содержит:
i) один или более акцепторов неорганических кислот, которые выбираются из тех, которые известны из ионного отверждения сополимеров винилиденфторида, в количестве 1-40 частей на 100 частей фторэластомерного полимера;
ii) один или более основных соединений из тех, которые известны из ионного отверждения сополимеров винилиденфторида в количестве 0.5-10 частей на 100 частей фторэластомерного полимера.
Основные соединения раздела ii) обычно выбираются из группы, состоящей из Са(ОН)2, Sr(OH)2, Ba(OH)2 соли слабых кислот и металлов, такие как, например, карбонаты, бензоаты, оксалаты и фосфиты Са, Sr, Ba, Na и K и смеси указанных выше гидроксидов с указанными выше солями металлов; среди соединений типа i) можно упомянуть MgO.
Указанные количества компонентов определяются по отношению к 100 частям фторэластомера. К отверждающей смеси могут затем добавляться другие обычные добавки, такие как загустители, пигменты, антиоксиданты, стабилизаторы и т.п.
Полукристаллические (пер)фторполимеры, необязательные компоненты настоящего изобретения, получаются при использовании методов полимеризации эмульсии или микроэмульсии, описанных выше для фторэластомеров по данному изобретению.
Мономер формулы (а) CF3OCF2OCF=CF2, использующийся в полимерах по данному изобретению, может получаться синтезом, включающим следующие этапы:

для получения фторгалогенового эфира формулы:
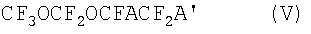
где А и А', одинаковые или различающиеся, являются Н, Cl или Br, при условии, что оба не являются Н; температура реакции фторирования может находиться в пределах от -120°С до -20°С, преимущественно от -100°С до -40°С; необязательно проходит в присутствии перфторированного растворителя, жидкого и инертного в условиях данной реакции; фтор может необязательно быть разбавлен неактивным газом, например азотом или гелием;
III удаление заместителей А и А' из фторгалогенового эфира (V) путем дегалогенизации, когда А или А' является водородом, а другой радикал - галогеном;
фторформу CF3OCOF этапа I можно получать с высокой конверсией и селективностью термической реакцией в газовой фазе CF3OF (фтороксиперфторметан) с СО в реакторе при температуре, поддерживаемой в пределах между 80°С и 250°С, преимущественно между 120°С и 230°С, более преимущественно между 150°С и 200°С.
Использующиеся реакции дегалогенизации или дегидрогалогенизации хорошо известны в уровне техники.
Молярное отношение CF3OF/CO находится в пределах между 0.1 и 10, преимущественно между 0.2 и 5, более преимущественно между 0.5 и 2.
Пергалогенизированный растворитель, необязательно использующийся на этапе II, преимущественно является органическим соединением, содержащим фтор и/или хлор, необязательно один или более атомов кислорода в цепи и/или аминные группы в качестве концевых групп.
Когда пергалогенированный растворитель перфторируется, он может, например, выбираться из перфторуглеродов, перфторэфиров, перфторполиэфиров, перфтораминов или соответствующих смесей.
Реакционная смесь, содержащая CF3OCOF этапа I, может подаваться непосредственно, без разделения компонентов смеси, в другой реактор для осуществления реакции этапа II. Процесс получения мономера этапа I, начиная с CF3OF. является особенно простым и эффективным. Как указывалось, преобразование CF3OF и селективность по отношению к CF3OCOP являются высокими (см. Примеры).
На этапе I за счет увеличения температуры в пределах 80°С-250°С преобразование увеличивается, и в то же время существенно поддерживается высокая селективность.
В другом варианте CF3OCOF этапа I может получаться фотохимическим способом, в жидкой фазе, вводом двух реагентов, как описано выше, в реактор, оборудованный ртутной УФ-лампой высокого давления, установленной в охлаждаемом кварцевом корпусе, погруженном в реакционную смесь при температуре между 0°С и 100°С, преимущественно между 20°С и 50°С.
Было установлено, что реакция формирования фторформы фотохимическим путем имеет высокую селективность, и получается более высокий выход по сравнению с такой же реакцией, проводимой в газовой фазе.
Реакция фотохимическим способом проводится в присутствии инертного перфторированного растворителя, находящегося в жидком состоянии в условиях проведения реакции.
Преимущественно перфторированный растворитель выбирается из перфторуглеродов, перфторполиэфиров, перфторированных третичных аминов, фторхлоруглеродов или их смесей.
Когда преобразование CF3OF неполное, газовый поток, выходящий из реактора, содержит смесь, сформированную из продукта реакции вместе с непреобразованными СО и CF3OF. Последний компонент может быть удален пропусканием газового потока через холодную ловушку, содержащую фторированный олефин, например, CFCl=CFCl, а затем отделением CF3OCOF фракционной дистилляцией.
В другом варианте реакционная газовая смесь, содержащая продукты реакции, сформированные на этапе I, охлаждается до получения конденсата фторформы, отделяя CF3OF и СО, которые могут возвращаться в реактор.
Преимущественно этап I проводится путем реакции фтороксиперфторметана и оксида углерода при температурах от 80°С до 250°С.
Преимущественно реактор, использующийся на этапе I, выполняется из стекла, инертных перфорированных пластиков, как, например, PTFE, PFA, металлических сплавов, например AISI 316, преимущественно покрытых стеклом или перфорированными пластиками. Более преимущественно в качестве материалов использовать стекло и фторированные пластики.
Фторэластомеры по данному изобретению, как указывалось, обладают улучшенной комбинацией механических свойств, включая модуль упругости, прочность при разрыве, деформацию при разрыве, эластомерных свойств, выраженных деформацией при сжатии и термостойкости; и одновременно они обладают указанными выше свойствами даже при низких температурах.
Фторэластомеры по данному изобретению используются для производства изделий, использующихся при температурах ниже -35°С и до 250°С, имеющих улучшенные механические и эластомерные свойства.
Приведенные далее Примеры иллюстрируют, но не ограничивают, настоящее изобретение.
ПРИМЕРЫ
Аналитические методы
Определение Tg полимера
Tg определялась DSC анализом в соответствии с методом ASTM D 3418. Значения Tg, которые приводятся в Примерах, представляют собой средние значения.
Определение внутренней вязкости
Внутренняя вязкость определялась в перфторгептане при температуре 30°С.
Определение полярных концевых -COF групп
В конце полимеризации полимер изолируется коагуляцией при заморозке при -20°С и последующем размораживании при комнатной температуре до получения суспензии, в которой полимер осаждается на дно; он промывается дважды в деминерализованной воде и высушивается в печи при 90°С до получения постоянного веса (около 12 часов); концевые -COF группы определяются ФТ-ИК спектроскопией с использованием прибора Nicolet® Nexus ФТ-ИК (256 сканирований, разрешение 2 см-1) на пленке полимера, имеющей толщину от 50 до 300 микрон, сначала проводится сканирование от 4000 см-1 до 400 см-1, пленка выдерживается в течение 12 часов в атмосфере, насыщенной парами аммиака, и в конце регистрируется ИК-спектр в тех же условиях, когда получают начальный ИК-спектр; обрабатывают два спектра путем вычитания из сигналов спектра, относящегося к необработанному образцу (начальный образец), соответствующих сигналов спектра образца после обработки парами аммиака и получают спектр "различия", нормализуемый следующим уравнением:

измеряется оптическая плотность, относящаяся к концевым -COF группам, реагирующим с парами аммиака; оптическая плотность преобразуется в ммоль/кг полимера с использованием молярного коэффициента удаления -COF групп при 1884 см-1, равного 215 литрам/(моль×см), как показано в таблице 1 на стр.77 сообщения М.Pianca и др. "End groups in fluoropolymers", J. Fluorine Chem. 95 (1999), 71-84 (используется здесь в качестве ссылки); найденные значения выражают концентрацию остаточных концевых -COF групп, как ммолей концевых -COF групп/кг полимера: в спектре перфторэластомеров не обнаруживается линий, относящихся к COF группам (1900-1830 см-1), предел чувствительности метода 0.05 ммоль/кг.
Определение вязкости по Муни
Вязкость по Муни (1+10' при 121°С) определяется по методу ASTM D 1646.
Определение остаточной деформации при сжатии
Остаточная деформация при сжатии определяется по методу ASTM D 395.
Определение TR
TR тест проводится по методу ASTM D 1329.
ПРИМЕР А
Получение CF3OCOF с помощью термической реакции при 170°С в реакторе из стекла.
Используется трубчатый стеклянный реактор, имеющий внутренний диаметр 55.6 мм и длину 510 мм, наполненный стеклянными 6х6 кольцами Рашига (свободный внутренний объем 842 мл), с поддержанием термостатического режима с помощью электрических сопротивлений.
В реактор, в котором поддерживается температура 170°С, в течение 5 часов вводится газовый поток CF3OF (1.5 литр/час), синтезированного, как описывается в патенте US 4400872, и одновременно подается поток СО (1.5 литр/час). Поток, выходящий из реактора, непрерывно анализируется с помощью газового хроматографа.
Поток, получаемый из реактора, конденсируется, за исключением СО, в ловушке, в которой поддерживается -110°С, содержащей 15 г CFCl=CFCl (A 1112), так что остаток CF3OF реагирует с олефином с получением CF3OCFClCF2Cl.
После фракционной дистилляции результирующей смеси получают 33.9 г CF3OCOF чистотой 99,8% (молярный выход на введенный CF3OF равен 76.5%); 12.3 г CF3OCFClCF2Cl; 3.4 г COF2.
Преобразование составляет 84.5%, и селективность равна 90%, рассчитанные по введенному CF3OF.
ПРИМЕР В
Получение CF3OCOF с помощью термической реакции при 170°С в реакторе PTFE.
Используется трубчатый термостатический реактор PTFE, имеющий внутренний диаметр 4 мм и длину 13.2 м.
В реактор вводится газовый поток CF3OF (1.5 литр/час), и одновременно подается поток СО (2.0 литр/час), причем реактор поддерживается при температуре 170°С.
Выходящий из реактора поток, анализирующийся с помощью газового хроматографа, имеет следующий молярный состав: 7.3% CF3OF, 54,2% CF3OCOF, 9.1% COF2 и 29.4% СО.
ПРИМЕР С
Получение CF3OCOF с помощью термической реакции при 120°С в реакторе PTFE.
В такой же реактор, который использовался в Примере В, вводится газовый поток CF3OF (1.5 литр/час) и одновременно подается поток СО (2.0 литр/час) в течение 6 часов, причем реактор поддерживается при температуре 120°С. Выходящий из реактора поток, анализирующийся с помощью газового хроматографа, имеет следующий молярный состав, без учета СО: 86.7% CF3OF, 13.3% CF3OCOF.
Выходящий из реактора поток конденсируется, за исключением СО, в ловушке, в которой поддерживается -110°С, содержащей 50 г А 1112, так что остаток CF3OF реагирует с олефином.
После фракционной дистилляции результирующей смеси получают 6.8 г CF3OCOF чистотой 99%.
Селективность равна 98%, рассчитанная по преобразованному CF3OF. Преобразование составляет 13.0%.
ПРИМЕР D
Получение CF3OCOF с помощью термической реакции при 170°С в реакторе AISI 316
Используется трубчатый термостатический реактор AISI 316, имеющий внутренний диаметр 4 мм и длину 11.3 м.
В реактор, в котором поддерживается температура 170°С, в течение 6 часов вводится газовый поток CF3OF (1.5 литр/час) и одновременно подается поток СО (1.5 литр/час). Поток, выходящий из реактора, конденсируется в ловушке, в которой поддерживается -110°С, содержащей 30 г А 1112.
После фракционной дистилляции содержания ловушки получают 31.2 г CF3OCOF чистотой 99%, 31.8 г фторгалогенэфира и 3.7 г COF2.
Преобразование составляет 66.6%, и селективность равна 86.5%.
ПРИМЕР Е
Получение CF3OCOF с помощью фотохимической реакции
500 г перфторполиэфира Galden®LS-165 вводятся в цилиндрический стеклянный реактор, оборудованный мешалкой и УФ лампой Hanau TQ 150 мощностью 150 Вт и оптическим расстоянием 1 см. Затем одновременно в течение 5 часов подается 2.0 литр/час CF3OF, разбавленное 3.0 литр/час Не, и 2.0 литр/час СО.
Газы, выходящие из реактора, конденсируется в ловушке, в которой поддерживается -110°С, содержащей 30 г А 1112. После фракционной дистилляции конденсированной смеси получают 22.9 г CF3OCOF чистотой 99%, 41.8 г фторгалогенэфира CF3OCFClCF2-Cl, 5.8 г COF2, 5.4 г трифторметилкарбоната.
Преобразование CF3OF составляет 60.5%. Селективность равна 63.6%.
ПРИМЕР F
Получение мономера формулы (а) реакцией CF3OCOF с элементарным фтором и фторолефином формулы CFCl=CFCl и последующей дегалогенизацией фторгалогенэфира.
20 г CFCl=CFCl (A 1112), 30 г CF3OCOF, полученных, как описано в Примере А, переносятся в 50 мл стеклянный реактор. Сформированный раствор поддерживается при температуре -100°С, а фтор, разбавленный азотом, барботируют с расходом 1 литр/час.
Баланс массы в конце реакции составляет 92%,19F-ЯМР анализ продукта реакции (52 г) показывает, что преобразование фторформы составляет 54%, и селективность получения фторгалогенэфира CF3OCFClCF2-Cl равна 93%. Непрореагировавшая фторформа удаляется из продукта реакции путем добавления воды при перемешивании. При достижении 25°С органическая фаза извлекается и высушивается с помощью MgSO4. Смесь фильтруется, и полученный остаток дистиллируется с выделением фракции, кипящей при 74°С, которая представляет собой 31.8 г фторгалогенэфира, имеющего чистоту 99%.
Дегалогенирование фторгалогенэфира проводится с использованием 1-литрового сосуда, снабженного механической мешалкой, термометром, капельницей, дистилляционной колонной и ловушкой, находящейся при температуре -78°С. 450 мл диметиформамида (DMF), 62 г цинка в порошке и 8.3 г ZnCl2 помещается в сосуд. Температура суспензии доводится до 80°С и добавляется 150 г фторгалогенэфира, полученного в предшествующей реакции. Когда добавка завершена, смесь оставляется реагировать в течение одного часа. В конце температура постепенно повышается до 120°С, и смесь снова оставляется реагировать в течение еще одного часа. Наконец, сосуд отсоединяется, и выделяется 106 г мономера формулы (а) CF3OCF2OCF=CF2, имеющего чистоту 99% (точка кипения 23°С).
ПРИМЕР 1
Получение микроэмульсии
Микроэмульсия получается смешиванием следующих ингредиентов в указанных ниже количествах, необходимых для получения одного литра микроэмульсии:
- 220.7 мл перфторполиоксиалкилена, имеющего концевую кислотную группу со средней молекулярной массой 600 формулы:
CF2ClO(CF2-CF(CF3)О)n(CF2O)mCF2COOH
где n/m=10;
- 220.7 мл водного 30% по объему раствора NH3;
- 427.6 мл деминерализованной воды;
- 131 мл Galden® D02, имеющего среднюю молекулярную массу 450, формулы:
CF3O(CF2-CF(CF3)О)n(CF2O)CF3
где n/m=20.
ПРИМЕР 2
Сополимер мономер формулы (a)/VDF 75/25 (% молярное отношение)
20 мл деминерализованной воды и 1 мл микроэмульсии, приготовленной в Примере 1, вводятся последовательно в 42 мл стальной автоклав, оборудованный магнитной мешалкой, после того, как устанавливается вакуум с помощью масляного насоса. Добавляются 9 г мономера формулы (а) и автоклав нагревается до 80°С. VDF вводится в реактор до тех пор, пока не установится давление 1.5 МПа. Затем вводится 4 мг персульфата аммония. Давление в реакторе поддерживается постоянным путем добавления VDF при каждом снижении давления на 0.01 МПа.
Реакция заканчивается после 6 ч. Полученный латекс дегазируется. Латекс коагулируется замораживанием и последующим размораживанием. Таким способом полимер отделяется от жидкой фазы, он дважды промывается деминерализованной водой и высушивается в печи при 100°С в течение 8 ч. Получают около 8.0 г полимера, что соответствует 90% введенного мономера формулы (а). Tg полимера равна -54.3°С. Внутренняя вязкость, измеренная при 30°С в перфторгептане (Galden® D80), равна 40.8 см3/г.
Состав полимера анализировался с помощью ЯМР, и анализ показал, что полимер содержит 75% молярных мономера формулы (а).
С помощью ИК-анализа определяется, что уровень концевых -COF групп в полимере ниже, чем чувствительность метода.
ПРИМЕР 3 (сравнительный)
Сополимеризация MOVE 1 с VDF в условиях Примера 2
Процесс осуществляется так же, как в Примере 2, но вводится 9 г MOVE 1 взамен мономера формулы (а). Полимеризация начинается через 5 мин после введения инициатора и заканчивается через 6 часов.
Получают полимер в количестве, равном 3 г.
Состав полимера анализировался с помощью ЯМР, и анализ показал, что полимер содержит 20% молярных MOVE 1, что соответствует 52% введенного мономера MOVE 1.
С помощью ИК-анализа определяется, что уровень концевых -COF групп устанавливается указанным способом, и потому он более 0.05 ммоль/кг.
Комментарии к примеру 2 в соответствии с изобретением и примеру 3 сравнительному
Выдающаяся реактивность мономера (а) при сравнении с MOVE 1 становится очевидной при сравнении % винилэфирных мономеров в сополимерах примера 2 и сравнительного примера 3. Эти примеры показывают, что в одинаковых условиях полимеризации при использовании мономера формулы (а) взамен MOVE 1, в полимере примера 2 содержится количество мономера формулы (а), равное 75% молярных, в то время как в сравнительном примере 3, в котором используется MOVE 1, в полимере находится 20% молярных MOVE 1. Кроме того, сополимер примера 2 свободен от концевых -COF групп, в то время как в полимере сравнительного примера 3 эти концевые группы присутствуют, хотя он содержит значительно меньшее количество винилового эфира.
Пример 2 подчеркивает удивительное и неожиданное поведение мономера формулы (а): Tg полученного в этом примере сополимера равна -54.3°С. Удивительно, насколько она ниже Tg гомополимера мономера формулы (а), т.е. -39.4°С (см. пример 9), а также гомополимера VDF, т.е. 47°С.
ПРИМЕР 4
Сополимер мономер (а)/VDF 21/79 (% молярного отношения)
В 5 л автоклаве, оборудованном мешалкой, работающей на 630 об/мин, вводится после откачки 3.0 л деминерализованной воды и 30 мл микроэмульсии перфторполиоксиалкиленов, предварительно полученных, как в Примере 1. Затем автоклав нагревался до 60°С и поддерживался при этой температуре в течение всей реакции. В него вводилась следующая смесь мономеров: винилиденфторид (VDF) 75% молярных, мономер (а) 25% молярных, и при этом давление поднималось до 1.1 МПа. Затем в автоклав вводились 3.75 г персульфата аммония (APS) в качестве инициатора, 5.48 г 1,4-дийодперфторбутана (C4F8I2) в качестве трансферного агента полимерной цепи, 2.26 г бис-олефина формулы СН2=СН-(CF2)6-СН=СН2; добавление бис-олефина проводилось в 20 аликвотных частей, каждая по 0.113 г, вводя с начала полимеризации и при каждом 5% увеличении конверсии мономера. Давление 1.1 МПа поддерживалось постоянным в течение всего процесса полимеризации путем подачи смеси, сформированной винилиденфторидом (VDF) 75% молярных, мономером (а) 25% молярных. После 189 минут протекания реакции, соответствующих 100% конверсии мономера, автоклав охлаждался и латекс извлекался. Полученный таким образом латекс коагулируется с раствором сульфата алюминия (6 г Al2(SO4)3 на каждый литр латекса), высушивается при 90°С в циркулирующем воздухе в течение 16 часов. Получается 580 г полимера.
19F-ЯМР анализ полимера, растворенного в горячем C6F6, показал, что молярный процент мономера (а) в полимере составляет 21% и VDF 79%. Tg, определенная с помощью DSC, составляет -47.9°С.
Вязкость по Муни (ML(1+10' при 121°С)) определялась в соответствии с методом ASTM D 1646 и составила 20 MU. Механические свойства показаны в таблице.
ПРИМЕР 5
Тройной сополимер мономер (a)/VDF/TFE 21/53/26 (% молярного отношения)
В 5 л автоклаве, оборудованном мешалкой, работающей на 630 об/мин, вводится после откачки 3.0 л деминерализованной воды и 30 мл микроэмульсии перфторполиоксиалкиленов, предварительно полученных, как в Примере 1. Затем автоклав нагревался до 70°С и поддерживался при этой температуре в течение всей реакции. Затем в него вводилась следующая смесь мономеров: винилиденфторид (VDF) 53% молярных, мономер (а) 21% молярных, тетрафторэтилен (TFE) 26% молярных, и при этом давление поднималось до 1.1 МПа. Затем в автоклав вводились 0.3 г персульфата аммония (APS) в качестве инициатора, 4.29 г 1,4-дийодперфторбутана (C4F8I2) в качестве трансферного агента полимерной цепи, 2.26 г бис-олефина формулы СН2=СН-(CF2)6-СН=СН2; добавление бис-олефина проводилось в 20 аликвотных частей, каждая по 0.113 г, вводя с начала полимеризации и при каждом 5% увеличении конверсии мономера. Давление 1.1 МПа поддерживалось постоянным в течение всего процесса полимеризации путем подачи смеси, сформированной винилиденфторидом (VDF) 49% молярных, мономером (а) 25% молярных, тетрафторэтиленом (TFE) 26% молярных. После 132 минут протекания реакции, соответствующих 100% конверсии мономера, автоклав охлаждался, и латекс извлекался. Полученный таким образом латекс коагулировался, как в Примере 4. Было получено 583 г полимера.
19F-ЯМР анализ полимера, растворенного в горячем C6F6, показал, что молярный процент мономера (а) в полимере составляет 21%, VDF 53% и TFE 26%. Tg, определенная с помощью DSC, составляет -43.6°С.
Вязкость по Муни (ML(1+10' при 121°С)) определялась в соответствии с методом ASTM D 1646 и составила 22 MU (единица вязкости по Муни).
С помощью ИК-анализа определяется, что уровень концевых -COF групп ниже, чем предел чувствительности метода определения. Механические свойства показаны в таблице.
ПРИМЕР 6
Четверной сополимер мономер (a)/VDF/TFE/HFP 15/62/16/7 (% молярного отношения)
В 5 л автоклаве, оборудованном мешалкой, работающей на 630 об/мин, вводится после откачки 3.0 л деминерализованной воды и 30 мл микроэмульсии перфторполиоксиалкиленов, предварительно полученных, как в Примере 1. Затем автоклав нагревался до 70°С и поддерживался при этой температуре в течение всей реакции. Затем в него вводилась следующая смесь мономеров: винилиденфторид (VDF) 53% молярных, мономер (а) 10% молярных, тетрафторэтилен (TFE) 20% молярных, гексафторпропен (HFP) 17% молярных, и при этом давление поднималось до 1.1 МПа. Затем в автоклав вводились 0.45 г персульфата аммония (APS) в качестве инициатора, 5.05 г 1,4-дийодперфторбутана (C4F8I2) в качестве трансферного агента полимерной цепи, 2.26 г бис-олефина формулы СН2=СН-(CF2)6- СН=СН2; добавление бис-олефина проводилось в 20 аликвотных частей, каждая по 0.113 г, вводя с начала полимеризации и при каждом 5% увеличении конверсии мономера. Давление 1.1 МПа поддерживалось постоянным в течение всего процесса полимеризации путем подачи смеси, сформированной винилиденфторидом (VDF) 64% молярных, мономером (а) 13% молярных, тетрафторэтиленом (TFE) 13% молярных, гаксафторпропеном (HFP) 10% молярных. После 118 минут протекания реакции, соответствующих 100% конверсии мономера, автоклав охлаждался, и латекс извлекался. Латекс коагулировался, как в Примере 4. Было получено 583 г полимера.
19F-ЯМР анализ полимера, растворенного в горячем ацетоне, показал, что молярный процент мономера (а) в полимере составляет 14.6%, VDF - 62.4% и TFE - 15.6% и HFP - 7.4%. Tg, определенная с помощью DSC, составляет -37.2°С.
Вязкость по Муни (ML(1+10' при 121°С)) определялась в соответствии с методом ASTM D 1646 и составила 9 MU.
С помощью ИК-анализа определяется, что уровень концевых -COF групп ниже, чем предел чувствительности метода определения (0.05 ммоль/кг). Механические свойства показаны в таблице.
ПРИМЕР 7
Четверной сополимер мономер (a)/VDF/TFE/MVE 15/57/22/6 (% молярного отношения)
В 5 л автоклаве, оборудованном мешалкой, работающей на 630 об/мин, вводится после откачки 3.5 л деминерализованной воды и 35 мл микроэмульсии перфторполиоксиалкиленов, предварительно полученных, как в Примере 1. Затем автоклав нагревался до 70°С и поддерживался при этой температуре в течение всей реакции. Затем в него вводилась следующая смесь мономеров: винилиденфторид (VDF) 44% молярных, мономер (а) 20% молярных, тетрафторэтилен (TFE) 23% молярных, метилвиниловый эфир (MVE) 13% молярных, и при этом давление поднималось до 1.6 МПа. Затем в автоклав вводились 0.35 г персульфата аммония (APS) в качестве инициатора, 5.05 г 1,4-дийодперфторбутана (C4F8I2) в качестве трансферного агента полимерной цепи, 2.26 г бис-олефина формулы СН2=СН-(CF2)6-СН=СН2; добавление бис-олефина проводилось в 20 аликвотных частей, каждая по 0.113 г, вводя с начала полимеризации и при каждом 5% увеличении конверсии мономера. Давление 1.6 МПа поддерживалось постоянным в течение всего процесса полимеризации путем подачи смеси, сформированной винилиденфторидом (VDF) 51% молярных, мономером (а) 15% молярных, тетрафторэтиленом (TFE) 26% молярных, метилвиниловым эфиром (MVE) 8% молярных. После 90 минут протекания реакции, соответствующих 100% конверсии мономера, автоклав охлаждался, и латекс извлекался. Латекс коагулировался, как в Примере 4. Было получено 603 г полимера.
19F-ЯМР анализ полимера, растворенного в горячем ацетоне, показал, что молярный процент мономера (а) в полимере составляет 14.6%, TFE 21.7%, VDF 57.5% и MVE 6.2%. Tg, определенная с помощью DSC, составляет -38.4°С.
Вязкость по Муни (ML(1+10' при 121°С)) определялась в соответствии с методом ASTM D 1646 и составила 20 MU.
Механические свойства показаны в таблице.
ПРИМЕР 8
Четверной сополимер мономер (a)/VDF/TFE/MVE 16/62/15/7 (% молярного отношения)
В 5 л автоклаве, оборудованном мешалкой, работающей на 630 об/мин, вводится после откачки 3.5 л деминерализованной воды и 35 мл микроэмульсии перфторполиоксиалкиленов, предварительно полученных, как в Примере 1. Затем автоклав нагревался до 70°С и поддерживался при этой температуре в течение всей реакции. Затем в него вводилась следующая смесь мономеров: винилиденфторид (VDF) 44% молярных, мономер (а) 20% молярных, тетрафторэтилен (TFE) 23% молярных, метилвиниловый эфир (MVE) 13% молярных, и при этом давление поднималось до 1.6 МПа. Затем в автоклав вводились 0.35 г персульфата аммония (APS) в качестве инициатора, 5.05 г 1,4-дийодперфторбутана (C4F8I2) в качестве трансферного агента полимерной цепи, 2.26 г бис-олефина формулы СН2=СН-(CF2)6-СН=СН2; добавление бис-олефина проводилось в 20 аликвотных частей, каждая по 0.113 г, вводя с начала полимеризации и при каждом 5% увеличении конверсии мономера. Давление 1.6 МПа поддерживалось постоянным в течение всего процесса полимеризации путем подачи смеси, сформированной винилиденфторидом (VDF) 61% молярных, мономером (а) 16% молярных, тетрафторэтиленом (TFE) 15% молярных, метилвиниловым эфиром (MVE) 8% молярных. После 97 минут протекания реакции, соответствующих 100% конверсии мономера, автоклав охлаждался, и латекс извлекался. Латекс коагулировался, как в Примере 4. Было получено 598 г полимера.
19F-ЯМР анализ полимера, растворенного в горячем ацетоне, показал, что молярный процент мономера (а) в полимере составляет 15.8%, TFE 15.4%, VDF 61.6% и MVE 7.2%. Tg, определенная с помощью DSC, составляет -40.9°С.
Вязкость по Муни (ML(1+10' при 121°С)) определялась в соответствии с методом ASTM D 1646 и составила 24 MU.
Механические свойства показаны в таблице.
ПРИМЕР 9
Гомополимер мономера формулы (а)
0.03 л деминерализованной воды, 1.5 мл микроэмульсии по Примеру 1 и 12 г мономера формулы (а) вводятся последовательно в 0.1 л (литр) стеклянный автоклав, оборудованный магнитной мешалкой, после того как устанавливается вакуум с помощью масляного насоса. Автоклав нагревается до 42°С. Затем вводится 0.1 г персульфата аммония. Реактор поддерживается при 42°С в течение 170 ч и затем охлаждается. Полученный латекс дегазируется. Латекс коагулируется замораживанием и последующим размораживанием. Таким способом полимер отделяется от жидкой фазы, он дважды промывается деминерализованной водой и высушивается в печи при 100°С в течение 8 ч.
Получают около 11 г полимера, что соответствует 92% преобразованию введенного мономера формулы (а). Tg полимера равна -39.4°С. Внутренняя вязкость, измеренная при 30°С в перфторгептане (Galden® D80), равна 30.5 см3/г. С помощью ИК-анализа определяется, что уровень концевых -COF групп в полимере ниже, чем чувствительность метода.
Преобразование 69% мономера формулы (а) получают при меньшем времени полимеризации.
Комментарии к примерам 4-9
Эти примеры показывают, что мономер (а) может сополимеризироваться с различными сомономерами, позволяя получать полимеры, у которых относительные количества сомономеров могут варьироваться в широких пределах композиций полимера. Это позволяет добиваться лучшей комбинации свойств полимера.
В качестве примера возможно понизить содержание водорода в полимере для того, чтобы увеличить его химическую стойкость, в то же время сохраняя хорошие качества при низких температурах. Смотрите, например, примеры 6 и 7, в которых описывается получение полимеров, имеющих одинаковые TR10, и показывающие различное содержание водорода.
Кроме того, было неожиданно и к удивлению изобретателей обнаружено, что значения Tg сополимеров ниже, чем значения у соответствующих гомополимеров мономера (а) и другого (других) сомономера (сомономеров) полимера.
Реферат
Отверждаемые фторэластомеры, основанные на винилиденфториде VDF, имеющие Tg ниже -35°С и колличество концевых -COF групп ниже чем 0,05 ммоль/кг. Фторэластомеры содержат (А) от 1% до 99% мономера формулы CF2=CFOCF2OCF3 и (В) от 1% до 99% одного или более перфторированных сомономеров, имеющих по меньшей мере одну ненасыщенность этиленового типа. Причем указанные один или более сомономеров (В) содержат винилиденфторид (VDF) в количестве от 1% до 85% по отношению к полным молям мономеров. Изделия из фторэластомеров обладают улучшенной комбинацией механических свойств и остаточной деформацией при сжатии в широком пределе как низких, так и высоких температур. 5 н.п. ф-лы, 1 табл.
Формула
A) от 1 до 99% мономера формулы
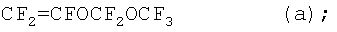
B) от 1 до 99% одного или более перфторированных сомономеров, имеющих, по крайней мере, одну ненасыщенность этиленового типа;
указанный один или более сомономеры содержат винилиденфторид (VDF) в количестве от 1 до 85% по отношению к полным молям мономеров для того, чтобы полимер был фторэластомерным;
сумма молярных процентов мономеров составляет 100%.
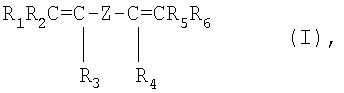
где R1, R2, R3, R4, R5, R6, равные или отличающиеся друг от друга, представляют Н или
C1-C5 алкилы;
Z представляет C1-C5 линейный или разветвленный алкилен или циклоалкилен, необязательно содержащий атомы кислорода, преимущественно, по меньшей мере, частично фторированный, или (пер)фторполиоксиалкилен.
когда Z является (пер)фторполиоксиалкиленом, он содержит блоки, выбранные из следующих:
-CF2CF2O-, -CF2CF(CF3)O-, -CFX1O-, где X1=F, CF3, -CF2CF2CF2O-, -CF2-CH2CH2O-,
-C3F6O-.
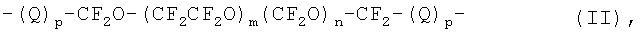
где Q является C1-С10 алкиленом или оксиалкиленом; р=0 или 1; m и n - такие числа, что отношение m/n находится в пределе между 0,2 и 5, и молекулярная масса указанного (пер)фторполиоксиалкилена находится в пределе 500-10,000, преимущественно 700-2,000.
-СН2ОСН2-; -CH2O(CH2CH2O)sCH2-, где s=1-3.
CH2=CH-(CF2)t0-CH=CH2
где t0 - целое число от 6 до 10.
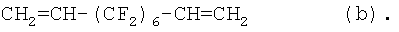
- С3-C8 перфторолефины;
- С2-С8 гидрированные фторолефины, перфторалкилэтилен CH2=CH-Rf, где Rf-С1-С6 перфторалкил;
- С2-С8 хлор-, и/или бром-, и/или йод- фторолефины;
- CF2=CFORf (пер)фторалкилвиниловые эфиры (PAVE), где Rf-С1-С6 (пер)фторалкил;
- CF2=CFOX (пер)фтороксиалкилвиниловые эфиры, где X=C1-C12 алкил, или С1-С12 оксиалкил, или С1-С12 (пер)фтороксиалкил, имеющий одну или более эфирных групп, например перфтор-2-пропоксипропил; фтордиоксолы, преимущественно перфтордиоксолы.
- фторэластомеры по любому из пп.1-16, и
- фторэластомеры, получаемые из полимеров, содержащих концевые -COF группы в количестве более 0,05 ммоль/кг;
количество фторэластомеров, содержащих концевые -COF группы в количестве менее 0,05 ммоль/кг и Tg, как определена в п.1, составляет, по меньшей мере, 5-10% по массе в отношении полной массы фторэластомеров.
- перфтордиоксолы, преимущественно имеющие следующую формулу:

где Y=F, ORf1, Rf1, являющийся С1-С5 перфторалкилом, преимущественно Rf1=CF3;
X1 и Х2, одинаковые или отличающиеся друг от друга, выбираются из F и CF3, преимущественно F;
Z1 выбирается из F, Н, CI, преимущественно F;
- перфторалкилвиниловые эфиры формулы CF2=CFORf, где Rf является С3 перфторалкилом;
- CF2=CFOXa перфтороксиалкилвиниловые эфиры, где Ха является C3-C12 перфтороксиалкилом, имеющим одну или более эфирных групп, например перфтор-2-пропоксипропил;
- перфторвиниловые эфиры (MOVE) общей формулы
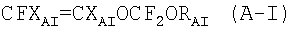

и (MOVE 2)
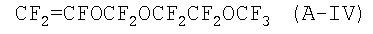
A) от 5 до 99% мономера формулы:

B) от 1 до 95% одного или более перфорированных сомономеров, имеющих, по меньшей мере, одну ненасыщенность этиленового типа;
указанный один или более сомономеры содержат винилиденфторид (VDF) в количестве от 1 до 85% по отношению к полным молям мономеров для того, чтобы полимер был фторэластомерным;
сумма молярных процентов мономеров составляет 100%.
- С2-С8 перфторолефины, например, TFE, гексафторпропен;
- перфторалкилвиниловые эфиры формулы CF2=CFORf, где Rf является С1-С2 перфторалкилом, преимущественно Rf=CF3.
мономер формулы (а): 15-40%, VDF: 60-85%; преимущественно мономер формулы (а): 15-40%, VDF: 60-85%, бис-олефин формулы (b): 0,01-1%;
мономер формулы (а): 15-40%, VDF: 60-85%; TFE: 5-40%; преимущественно мономер формулы (а): 15-40%, VDF: 60-85%; TFE: 5-40%, бис-олефин формулы (b): 0,01-1%;
мономер формулы (а): 5-40%, MVE: 5-30%; VDF: 50-85%; преимущественно мономер формулы (а): 5-40%, MVE: 5-30%; VDF: 50-85%; бис-олефин формулы (b): 0,01-1%;
мономер формулы (а): 5-40%, MVE: 5-30%, VDF: 50-85%; TFE: 5-40%; преимущественно мономер формулы (а): 5-40%, MVE: 5-30%, VDF: 50-85%; TFE: 5-40%, бис-олефин формулы (b): 0,01-1%;
мономер формулы (а): 40-99%; VDF: 1-60%; преимущественно мономер формулы (а): 40-99%, VDF: 1-60%; бис-олефин формулы (b): 0,01-1%;
мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; преимущественно мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; бис-олефин формулы (b): 0,01%-1%;
мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; TFE: 5-40%; преимущественно мономер формулы (а): 40-99%, MVE: 0-30%, VDF: 1-60%; TFE: 5-40%; бис-олефин формулы (b): 0,01-1%.
I получение фторформы CF3OCOF;
II реакция фторформы CF3OCOF с элементарным фтором и олефиновыми соединениями, имеющими формулу:

для получения фторгалогенового эфира формулы:

где А и А', одинаковые или различающиеся, являются Н, Cl или Br, при условии, что оба не являются Н; температура реакции находится в пределах от -120 до -20°С;
III удаление заместителей А и А' из фторгалогенового эфира формулы (V) мономера формулы (а) с помощью реакции, при которой:
на этапе I фторформа CF3OCOF получается реакцией CF3OF с СО при температуре между 80 и 250°С.
Комментарии