Бензопирановые и бензоконденсированные соединения, промежуточные соединения, фармацевтическая композиция и способ ингибирования - RU2128655C1
Код документа: RU2128655C1
Чертежи




Описание
Настоящее изобретение относится к новым бензопирановым и другим бензоконденсированным антагонистам лейкотриена B4 (ЛТВ4), к фармацевтически приемлемым солям указанных соединений, к фармацевтическим композициям, содержащим такие соединения, и к способу использования таких соединений в качестве антагонистов ЛТВ4.
Соединения настоящего изобретения ингибируют действие ЛТВ4 и поэтому являются полезными при использовании для лечения болезней, индуцированных ЛТВ4, таких как воспалительные заболевания, включая ревматоидный артрит, остеоартрит, воспалительные заболевание кишечника, псориаз и другие кожные заболевания, такие как экзема, эритема, чесотка и угревая сыпь, раздражение и другие формы реперфузионных заболеваний, отторжение трансплантата, аутоиммунные заболевания, астма и другие состояния, где происходит заметная инфильтрация нейтрофилов.
Антагонисты лейкотриена B4 описаны в Европейских патентных публикациях 276064 и 292977, которые упоминают дифениловые эфиры, бензофеноны и другие соединения, включающие две фенильных группы, и 7-(3-алкокси-4-алканоил-фенокси)алкоксибензопирановые производные соответственно.
Краткое описание изобретения
Настоящее изобретение относится к новым бензопиранам и другим
бензоконденсированным соединениям - антагонистам лейкотриена B4 (ЛТВ4) формулы

и к их фармацевтически приемлемым солям,
где A1 является O, CH2, S, NH или N(C1-C6)алкилом;
R3 является водородом или гидрокси;
A2 обозначает

R4 является водородом или гидрокси;
R5 выбран из группы, включающей -(CH2)nCHX9X10, -(CH2)nX10 и -CH(OH)X10; где n равно 0, 1, 2 или 3; X9 является водородом, (C1-C6)алкилом или необязательно замещенным фенилом; где необязательно замещенный фенил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил; X10 является водородом, (C1-C6)алкилом, (C3-C8)циклоалкилом или одним из следующих необязательно замещенных колец: фенилом, тиенилом, пиридилом, фурилом, нафтилом, хинолилом, изохинолилом, пиримидилом или пиразинилом; где необязательно замещенные кольца являются необязательно замещенными одним или двумя заместителями, независимо, выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)-алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил, фенилсульфонил и необязательно замещенный фенил; где необязательно замещенный фенил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси,
(C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил;
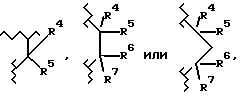
R6 и R7 являются, каждый, независимо водородом или (C1-C4)алкилом, или R6 и R7, вместе с атомом углерода, к которому они присоединены, образуют (C4-C7)циклоалкил;
R1 выбран из группы, включающей тетразолил, карбокси, цис- или транс (CH2)m-CX1= CX2-CO2H, -(CH2)m-CX3X4X5, -CO-NG1G2,
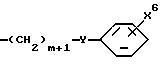
Y является О, CH2, S, NH или N(C1-C6 )алкилом;
X1 и X2 являются, каждый, независимо водородом или (C1-C4)алкилом;
X3 и X4 являются, каждый, независимо водородом или (C1-C4)алкилом, или X3 и X4 являются взятыми вместе с атомом углерода, к которому они присоединены, образуют (C4-C7 )циклоалкил;
X5 является гидрокси, карбокси, тетразолилом или -CO-NG3G4;
X6 является карбокси, тетразолилом, CH2ОН или -CO-NG5G6, G1, G2, G3 и G4, G5 и G6, каждый, независимо выбраны из группы, включающей водород, (C1 -C6)алкил, (C1-C4)-перфторалкил, (C1-C6)алкилсульфинил, фенилсульфинил,
(C1-C6)алкилсульфонил, фенилсульфонил, гидрокси, фенил и (Q1)a-замещенный фенил; где a равно 1 или 2; Q1 для каждого случая независимо выбран из фтора, хлора, (C1-C6)алкила, (C1-C6)алкокси, (C1-C4)перфторалкила, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6 )алкилсульфинила, фенилсульфинила, (C1-C6)алкилсульфонила и фенилсульфонила; замещенное пяти- или шестичленное ароматическое кольцо, замещено одним заместителем, выбранным из группы, включающей карбокси, тетразолил, -CO-N(H)(SO2-X7), -N(H)(SO2-X7), -N(H)(CO-X7) и -N(H)(CO-OX7), и одним или двумя заместителями, каждым, независимо, выбранным из группы, включающей фтор, хлор, (C1-C4)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио,
(C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил; где X7 является водородом, -CH2F, -CHF2, -CF3, (C1-C6)алкилом, (C3-C8)циклоалкилом или одним из следующих необязательно замещенных колец: фенилом, тиенилом, пиридилом, фурилом, нафтилом, хинолилом, изохинолилом, пиримидилом или пиразинилом; где необязательно замещенные кольца необязательно замещены одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4 )перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6 )алкилсульфонил, фенилсульфонил и необязательно замещенный фенил; где необязательно замещенный фенил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфонил, (C1-C4) алкилсульфонил и фенилсульфонил;
R2 представляет водород, фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6) алкилсульфонил или фенилсульфонил;
при условии, что
G1 и G2 не являются оба гидрокси в одно и то же время;
G3 и G4 не являются оба гидрокси в одно и то же время;
G5 и G6 не являются оба гидрокси в одно и тоже время; и
когда R3 является гидрокси и R4 является водородом, тогда R5 является -CH(ОН)X10.
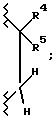
Предпочтительной группой
соединений являются такие соединения формулы I или их фармацевтически приемлемые соли,
где R3 является гидрокси; A2 является
Более предпочтительной группой соединений являются такие соединения формулы I или их фармацевтически приемлемые соли, где R3 является гидрокси; A1 является О или CH2; A2 является

Еще более предпочтительной группой соединений являются такие соединения формулы I или их фармацевтически приемлемые соли, где R3 является гидрокси; A1 является O или CH2; A2 является

Даже еще более предпочтительной группой соединений являются такие соединения формулы I или их фармацевтически приемлемые соли, где R3 является гидрокси; A1 является O или CH2; A2 является
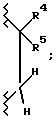
Наиболее предпочтительной группой соединений являются такие соединения формулы I или их фармацевтически приемлемые соли, где R3 является гидрокси; A1 является O или CH2; A2 является

(C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил; R4 является гидрокси, и R3 гидрокси является либо цис, либо транс с R4 гидрокси; и X7, R2 и R5 являются такими, как определено выше для формулы I. Из непосредственно указанной выше группы наиболее предпочтительных соединений, даже еще более предпочтительной группой соединений являются такие, где R3 и R4 гидроксигруппы являются цис по отношению друг к другу.
Еще одной наиболее предпочтительной группой соединений являются такие соединения формулы I или их фармацевтически приемлемые соли, где R3 является гидрокси; R4 является гидрокси; A1 является O или CH2; A2 является
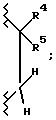
(C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6) алкилсульфонил и фенилсульфонил; R3 гидрокси и R4 гидрокси являются цис по отношению друг к другу; R5 является -(CH2)nCHX9X10, где X9 является водородом, и X10 является одним из необязательно замещенных колец, как определено выше для формулы I; и n, X7 и R2 являются такими, как определено выше для формулы I. Предпочтительной группой соединений из последней указанной выше группы соединений являются такие соединения, где n равно 1; и X10 является фенилом, замещенным в пара-положении фенилом. И следующая группа соединений представляет наиболее предпочтительной группой соединений, где R1 является замещенным фенилом, замещенным одним заместителем, выбранным из группы, включающей карбокси -N(H)(SO2-X7), и одним или двумя заместителями, каждый, независимо, выбранными из группы, включающей фтор, хлор и (C1-C4)перфторалкил.
Настоящее изобретение также относится к фармацевтической композиции для лечения заболеваний, вызванных ЛТВ4, которая содержит эффективное количество соединения формулы I или его фармацевтически приемлемой соли, как указано выше, и фармацевтически приемлемого носителя или разбавителя. Изобретение, кроме того, относится к фармацевтической композиции для лечения экземы, эритемы, чесотки, угревой сыпи, раздражения, отторжения трансплантата, аутоиммунных заболеваний и астмы, содержащей такое количество соединения формулы I, как определено выше, или его фармацевтически приемлемой соли, которое является достаточным для лечения указанных заболеваний, и фармацевтически приемлемый носитель или разбавитель. Предпочтительными композициями являются такие композиции, где соединение формулы I является предпочтительным соединением.
Настоящее изобретение, кроме того, включает способ ингибирования связывания рецепторов, ингибирования функциональной активности и ингибирования ЛТВ4 in vivo путем введения субъекту, нуждающемуся в таком ингибировании, соединения формулы I, как определено выше, или его фармацевтически приемлемой соли. Настоящее изобретение включает способ лечения воспалительных заболеваний, экземы, эритемы, чесотки, угревой сыпи, раздражения, отторжения трансплантанта, аутоиммунных заболеваний и астмы путем введения субъекту, нуждающемуся в таком лечении, соединения формулы I, как определено выше, или его фармацевтически приемлемой соли. Предпочтительные способы настоящего изобретения представляют собой такие способы, где соединение формулы I является предпочтительным соединением или его фармацевтически приемлемой солью.
Настоящее изобретение также относится к промежуточному
соединению формулы IA
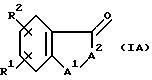
где A является O, CH2, S, NH или N(C1-C6)алкилом;
A2 обозначает

R4 является водородом или гидрокси;
R5 выбран из группы, включающей -(CH2)nCHX9X10, -(CH2)nX10 -CH(ОН)X10; где n равно 0, 1, 2 или 3;
X9 является водородом, (C1-C6)алкилом или необязательно замещенным фенилом; где необязательно замещенный фенил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил;
X10 является водородом, (C1-C6)алкилом, (C3-C8)циклоалкилом или одним из следующих необязательно замещенных колец: фенилом, тиенилом, пиридилом, фурилом, нафтилом, хинолилом, изохинолилом, пиримидилом или пиразинилом; где необязательно замещенные кольца являются необязательно замещенными одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4 )перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил, фенилсульфонил и необязательно замещенный фенил; где необязательно замещенный фенил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси,
(C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил;
R6 и R7 являются, каждый, независимо водородом или (C1 -C4)алкилом, или R6 и R7, взятые вместе с атомом углерода, к которому они присоединены, образуют (C4-C7)циклоалкил;
R1 выбран из группы, включающей тетразолил, карбокси, цис или транс -(CH2)m-CX1= CX2-CO2H, -(CH2)m-CX3X4X5, -CO-NG1G2,

Y является О, CH2 , S, NH или N(C1-C6)алкилом;
X1 и X2 являются, каждый, независимо водородом или N(C1-C6)алкилом;
X3 и X4 являются, каждый, независимо водородом или (C1-C6)алкилом, или X3 и X4, взятые вместе с атомом углерода, к которому они присоединены, образуют (C3-C7) циклоалкил;
X5 является гидрокси, карбокси, тетразолилом или -CO-NG3G4;
X6 является карбокси, тетразолилом, CH2OH или -CO-NG5G6; G1, G2, G3, G4, G5 и G6, каждый, независимо, выбран из группы, включающей водород, (C1-C6)алкил, (C1-C4)перфторалкил, (C1-C6)алкилсульфинил, фенилсульфинил,
(C1-C6 )алкилсульфонил, фенилсульфонил, гидрокси, фенил и (Q1)a-замещенный фенил; где a равно 1 или 2; Q1 для каждого случая, независимо, выбран из фтора, хлора, (C1-C6)алкила, (C1-C6)алкокси, (C1-C4)перфторалкила, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинила, фенилсульфинила, (C1-C6)алкилсульфонила и фенилсульфонила; замещенное пяти- или шестичленное ароматическое кольцо, замещено одним заместителем, выбранным из группы, включающей карбокси, тетразолил, -CO-N(H)(SO2-X7), -N(H)(SO2-X7), -N(H)(CO-X7) и -N(H)(CO-OX7), и одним или двумя заместителями, каждый, независимо, выбранный из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4 )перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6 )алкилсульфонил и фенилсульфонил; где X7 является водородом, -CH2F, -CHF2, -CF3, (C1-C6)алкилом, (C3-C8 )циклоалкилом или одним из следующих необязательно замещенных колец: фенилом, тиенилом, пиридилом, фурилом, нафтилом, хинолилом, изохинолилом, пиримидилом или пиразинилом; где необязательно замещенные кольца являются необязательно замещенными одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6 )алкокси, (C1-C4)перфторалкил, (C1-C4)-перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6 )алкилсульфонил, фенилсульфонил и необязательно замещенный фенил; где необязательно замещенный фенил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, включающей фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4 )перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил и фенилсульфонил;
R2 представляет водород, фтор, хлор, (C1-C6)алкил, (C1-C6)алкокси, (C1-C4)перфторалкил, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинил, фенилсульфинил, (C1-C6)алкилсульфонил или фенилсульфонил;
при условии, что G1 и G2 не являются оба гидрокси в одно и то же время; G3 и G4 не являются оба гидрокси в одно и то же время; G5 и G6 не являются оба гидрокси в одно и тоже время.
Подробное описание настоящего изобретения
Термин "C1-C6алкил" всякий раз, когда он используется в настоящем описании,
обозначает насыщенные одновалентные алифатические углеводородные радикалы с прямой или разветвленной цепью, имеющие от одного до шести атомов углерода, такие как метил, этил, пропил, трет-бутил,
гексил и т.д. Также термины C3-C7циклоалкил и C3-C8циклоалкил обозначают циклоалкильные группы, имеющие от трех до семи или восьми атомов углерода
соответственно, такие как циклопропил, циклогексил, циклооктил и т.д.
Когда A1 является кислородом и A2 является

Соединения настоящего
изобретения, когда R3 является ОН, имеют два асимметричных атома углерода, обозначенных звездочкой в следующей формуле:

Стереоизомеры могут быть обозначены с указанием R и S вращения в соответствии со стандартной номенклатурой. Когда здесь делается отсылка к S, R или R, S, подразумевается единственное энантиомерно чистое соединение, причем S*, R* и R*, S* обозначают рацемическую смесь. Изобретение включает рацемические смеси и оптические изомеры формулы I.
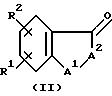


В соответствии с конкретным способом настоящего изобретения соединения указанной выше формулы II, которые являются промежуточными соединениями формулы I, где R1 является -(CH2)mX3X4 X5, где m равно 0 и X5, является карбокси или его сложными эфирами, получают путем взаимодействия соединений указанных выше формул III и IV с образованием соединения формулы V (не показана) с последующим восстановлением с образованием соединения формулы I.
Взаимодействие соединений III и IV обычно проводится в растворителе. Подходящими растворителями являются эфирные растворители, такие как тетрагидрофуран, диэтиловый эфир, этиленгликоль диметиловый эфир и 1,4-диоксан, диполярные апротонные растворители, такие как диметилформамид, N, N-диметилацетамид, ацетонитрил, диметилсульфоксид, гексаметилфосфорамид, N,N-диметилпропиленмочевина, неполярные ароматические растворители, такие как ксилол, бензол, хлорбензол и толуол, и галогенированные растворители, такие как метиленхлорид, хлороформ и дихлорэтан. Особенно подходящими растворителями являются ксилол или смесь равных объемов этиленгиколь диметилового эфира и диметилформамида. Температура реакции находится в диапазоне от -78oC до 200oC, в зависимости от температуры кипения используемого растворителя, и обычно находится в диапазоне от около 80oС до около 150oC.
Взаимодействие может проводиться в присутствии кислот Льюиса, таких как хлорид цинка, хлорид алюминия, бромид магния, хлорид олова и хлорид титана. Когда она присутствует, количество кислоты Льюиса находится в диапазоне от около 0,05 до около 2 эквивалентов на моль соединения III.
Взаимодействие обычно проводится с палладиевым катализатором. Подходящими палладиевыми катализаторами являются тетракистрифенилфосфин палладия, хлорид бис-бензонитрил палладия, димер аллилхлорид палладия, хлорид палладия, ацетат палладия, палладий на угле и хлорид бисацетонитрил палладия. Конкретный катализатор включает 5% весовых димера аллилхлорида палладия или 5% весовых бисбензонитрила хлорида палладия. Как правило, используют от около 0,001 эквивалента до одного эквивалента катализатора на моль субстрата.
Взаимодействие обычно проводится в присутствии фосфинового лиганда, такого как трифенилфосфин, три-о-толилфосфин или три-2-фурилфосфин в количестве от около 0,1 до около 5, предпочтительно от 1 до 2 молярных эквивалентов на моль используемого субстрата.
Восстановление соединения формулы V проводится обычным способом с боргидридом натрия в спиртовом растворителе при температуре окружающей среды с образованием соединения формулы I после омыления.
Соединения формулы III, где R5 является -(CH2)nX9X10 или -(CH2)mX10, могут быть получены следующим образом.
Соединение формулы VI

Группа R5 , когда она определена как -(CH2)nX9X10 или -(CH2)nX10, где n, X9 и X10 являются такими, как определено выше для формулы I, может вводиться в трифлатный аналог путем двухстадийного способа, включающего взаимодействие с альдегидом формулы X9X10CH(CH2)q-1CHO или X10(CH2)q-1CHO с образованием соответствующего алкенового аналога, где R5 представляет =CH(CH2)q-1CHX9 X10 или =CH(CH2)q-1X10, где q равно 1, 2, 3 или 4 соответственно, а затем гидрирование. Взаимодействие с альдегидом проводится в присутствии пирролидинового катализатора или с катализатором на основе хлористоводородной кислоты в уксусной кислоте. Гидрирование проводится обычным способом с водородом и палладиевым катализатором.
Соединения формулы VI обычно являются коммерчески доступными. В противном случае они могут быть получены с помощью способов, хорошо известных специалистам в данной области. Например, соединения формулы VI, где A1 является кислородом, и A2 является


Соединения формулы VI, где A2 является

Соединения формулы I, где R1 является -(CH2)m-CH2CX1=CX2-CO2H, могут быть синтезированы путем взаимодействия соединения формулы III с (CH3)3SnSn(CH3)3 и палладиевым катализатором, таким как тетракистрифенилфосфинпалладий (Pd(PPh3)4), в присутствии фосфинового лиганда, как описано выше для реакции соединений формулы III и IV, с получением соответствующего аналога триметилололова. Аналог триметилолова превращают в эфир-защищенное соединение формулы Z1O2CX2C=CX1-(CH2)m Z2, где Z1 является алкилом или циклоалкилом и Z2 является йодом, бромом или CF3SO3. Реакция связывания происходит в присутствии палладиевого катализатора, такого как бистрифенилфосфин хлорид палладия, как описано выше. Кетоновые эфиры сначала восстанавливают до соответствующих гидроксильных соединений, а затем гидролизуют до соответствующей кислоты формулы I. Восстановление осуществляют с боргидридом натрия. Как правило, восстановление проводят в растворителе. Подходящими растворителями являются низшие спирты, имеющие от одного до шести атомов углерода, смеси низших спиртов с органическими растворителями, такие как тетрагидрофуран или диоксан, и смеси из водорастворимых низших спиртов или других смешивающихся с водой органических растворителей и воды. Растворитель предпочтительно является низшим спиртом, таким как метанол или этанол. Диапазон значений температуры реакции главным образом находится в пределах от около -78oC до около 100oC, и обычно от около 0oC до около 25oC.
Стадия восстановления дает стереоизомерную смесь эфирных соединений формулы
I, имеющих следующие структуры:

Эти цис и транс изомеры могут быть разделены с помощью обычной колоночной хроматографии.
Разделение энантиомерной смеси, получаемой после разделения цис и транс изомеров, может быть достигнуто с помощью способов, известных в данной области. В одном из способов соединение формулы I, где R1 содержит карбоксильную группу (COOH), взаимодействует с хиральным основанием, таким как d-эфедрин, в полярном растворителе, таком как простой эфир, с образованием диастереомерных солей, которые разделяют, а затем преобразуют в оптически чистые кислоты путем обработки кислотой, такой как водный или метанольный раствор хлористого водорода. В другом способе соединение формулы I, где R1 содержит эфирную группу карбоновой кислоты, взаимодействует с оптически активной кислотой, такой как R-миндальная кислота или N-трет-бутоксикарбонил-D-триптофан с образованием диастереомерных эфиров с гидроксильной группой, которые после разделения преобразуют в оптически чистые кислоты путем обработки основанием, таким как гидроксид натрия, в метаноле или этаноле. Удаление разделяющей эфирной группы и гидролиз эфирной группы карбоновой кислоты в R1 удобно осуществляется водным раствором основания, такого как гидроксид щелочного металла, например гидроксид натрия, при температурах в диапазоне от около комнатной температуры до температуры дефлегмации или кипения используемого растворителя или смеси растворителей. Реакцию можно проводить в присутствии сорастворителя, такого как метанол, этанол или тетрагидрофуран.
Соединения формулы I, где R1 является карбокси и R2 является водородом, могут быть получены из промежуточного соединения формулы III сначала путем замещения CF3SO3-группы метоксикарбонилом, а затем гидролизом. Реакцию замещения осуществляют с моноокисью углерода в присутствии ацетата палладия, 1,1'-бис(дифенилфосфин)ферроцена (ДФФФ), метанола и триэтиламина. Гидролиз производят так, как описано ранее.
Соединения формулы I, где R1 является -(CH2)mCX3X4X5, где m, X3, X4 и X5 являются такими, как определено выше для формулы I, здесь и далее обозначены как соединения формулы XXI (не показана). Хотя последующее далее описание химических реакций описывает получение соединений формулы XVI, где R1 является -(CH2)mCX3X4CO2C2H5, понятно, что то же самое описание химических реакций относится и к соединениям формулы XVI, имеющим различные R1, как определено со ссылкой на формулу I, которые являются инертными в условиях реакции, описанных ниже.
Соединения формулы XXI, где X5 является тетразолилом, могут быть получены из соединений формулы XVI

Согласно этому способу, соединение XVI сначала подвергают взаимодействию с трет-бутилдиметилсилилхлоридом в присутствии имидазола и диметилформамида для защиты гидроксильной группы, как известно в данной области. Защищенное соединение взаимодействует с аммиаком и триэтилалюминием в ксилоле с замещением -CO2C2H5 группы на цианогруппу. Цианогруппу заменяют триметилстаннилтетразолилом путем взаимодействия с триметилстаннилазидом в толуоле. Преобразование в тетразолил и удаление силильной защитной группы достигается путем взаимодействия с тетрабутиламмонийфторидом в тетрагидрофуране.
Исходный продукт формулы XVI является идентичным соединению формулы II, указанной выше, где R1 является (CH2)mCX3X4X5 , где X5 является карбоксиэтиловым эфиром и m равно 0. Получение этого исходного продукта описано выше.

Соединение формулы XVII преобразуют путем последовательных взаимодействий с (1) акрилонитрилом, (2) гидролиза с концентрированным хлористым водородом и (3) циклизации с полифосфорной кислотой с образованием соединения формулы XVIII. Введение группы R5 с получением соединения формулы XIX происходит так, как описано для соединений формулы VI. Гидрирование и гидролиз соединения формулы XIX происходит так, как описано выше.
Соединение формулы XVII может быть получено из 3-гидроксифенилуксусной кислоты путем введения групп X3 и X4 с помощью известных способов.
Исходный продукт XVI, когда m равно 0, 1 или 2, A2 является

Исходный продукт XVI, где A1 является CH2, m равно 0, 1 или 2, и A2 является
Бензол, замещенный -(CH2)m-CX3X4CO2C2H5, взаимодействует с моноэфиром монохлорангидрида малоновой, янтарной или глутаровой кислоты в присутствии катализатора Фриделя-Крафтса, такого как хлорид алюминия. Полученный кетон преобразуют в соответствующий пропилендитиол с пропилендитиоловым и бортрифторидным катализатором. Образовавшееся соединение восстанавливают с помощью никеля Рани, а затем омыляют. Образуется кольцо с полифосфорной кислотой с образованием бициклического соединения XIX. Введение группы R5 происходит так, как описано выше.
Соединения формулы XXI, где X5 является CO2H, могут быть получены путем омыления соединения формулы I, где R1 является -(CH2)mCO2CH3, получение которого описано выше.
Соединения формулы XXI, где X5 является ОН, m равно 0, 1 или 2, и X3 и X4 являются, каждый, водородом, могут быть получены с помощью обычного гидрирования соединения формулы I, где R1 является -(CH2)mCO2 CH3, где m равно 0, 1 или 2, с помощью литийалюминийхлорида.
Соединения формулы XXI, где X5 является ОН, m равно 0, 1 или 2, и X3 и X4 являются, каждый, алкилом, могут быть получены путем взаимодействия соответствующих соединений, где X3 и X4 являются водородом, с одним эквивалентом реагента Гриньяра, содержащего группу X3, например, X3MgCl, а затем с одним эквивалентом реагента Гриньяра, содержащего группу X4, например X4MgCl.
Соединения формулы XXI, где X5 является ОН, m равно 0, 1 или 2, и X3 и X4, взятые вместе с образованием C3-C7циклоалкила, получают подобным же образом путем взаимодействия соответствующих соединений, где X3 и X4 являются водородом, с реагентом Гриньяра, производным C3-C7дигалогеналкана, например, ClMg(C3-C7алканил)MgCl.
Соединения формулы I, где R1 является
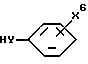
с трифлатным производным формулы I, где R1 является CF3SO3(CH2)m в присутствии основания, такого как триэтиламин или гидрид натрия, в реакционно-инертном растворителе.
Трифлаты могут получены путем взаимодействия трифлинового ангидрида с соединением формулы XXI, где m равно 0, 1 или 2; X3 и X4 являются водородом; и X5 является гидроксилом, синтез которого описан выше.
Соединения формулы I, где R1 является -CONG1G2, могут быть получены из соответствующего соединения, где R1 является карбокси, путем взаимодействия с амином формулы NHG1G2.
Согласно конкретному способу настоящего изобретения промежуточные соединения указанной выше формулы II, где R1 является CF3-SO2-O-, получают путем взаимодействия соединения формулы III, как определено выше, с соединением формулы
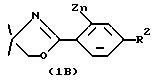
Соединения формулы IB получают in situ из соединения формулы

Кетоны формулы II, где A1, A2, R1 и R2 являются такими, как определено с отсылкой к формуле I, могут быть восстановлены до соответствующих гидроксильных производных формулы I путем взаимодействия с боргидридом натрия. Обычно восстановление проводят в растворителе. Подходящими растворителями являются низшие спирты, имеющие от одного до шести атомов углерода, смеси низших спиртов с органическими растворителями, такими как тетрагидрофуран или диоксан, и смеси, состоящие из смешивающихся с водой низших спиртов или других смешивающихся с водой органических растворителей воды. Растворителем является предпочтительно низший спирт, такой как метанол или этанол. Температура реакции обычно находится в области от около -78oC до около 100oC, и, как правило, от около 0oC до около 25oC.

Соединение формулы XIV образуется путем взаимодействия соединения формулы III с (CH3)3SnSn(CH3)3 и палладиевым катализатором, таким как тетракистрифенилфосфин палладия (Pd(PPh3 )4), или хлорида бис-бензонитрилпалладия, в присутствии фосфинового лиганда, такого как трифенилфосфин, в количестве от около 0,1 до около 5 молярных эквивалента на моль используемого субстрата. Соединение формулы XIV преобразуют в соединение формулы XV путем взаимодействия с эфир-защищенным соединением формулы

где X является С, CH, N, О или S;
K1 является карбокси, защищенным в виде сложного эфира, тетразолилом, -CO-N(H)(SO2-X7 ), -N(H)(SO2-X7), -N(H)(CO-X7) или -N(H)(CO-OX7) (эту группу получают в виде кислоты перед преобразованием в сложный эфир);
K2 является независимо F, Cl, (C1-C6)алкилом, (C1-C6)алкокси, (C1-C4)перфторалкилом, (C1-C4)перфторалкокси, (C1 -C6)алкилтио, (C1-C6)алкилсульфинилом, фенилсульфинилом, (C1-C6)алкилсульфонилом или фенилсульфонилом;
l равно 1 или 2;
Z является йодом, бромом или CF3SO3.
Реакцию связывания проводят в присутствии палладиевого катализатора, такого как тетракистрифенилфосфинпалладия или хлорид бис-трифенилфосфинпалладия.
Кетоновые эфиры формулы XV сначала восстанавливают до соответствующего гидроксила, а затем гидролизуют до соответствующей кислоты формулы I. Восстановление проводят с боргидридом натрия, как описано выше с отсылкой к восстановлению кетонов формулы II. Гидролиз до кислоты может проводиться с водным раствором основания, такого как гидроксид щелочного металла, например гидроксид натрия, необязательно в присутствии сорастворителя, такого как метанол или этанол, при температурах в диапазоне от комнатной температуры до температуры дефлегмации или кипения используемого растворителя.
Соединения формулы I, где R3 и R4 являются гидрокси, могут быть получены из эфиров формулы I путем отщепления гидроксильной группы с образованием олефина. Это осуществляют способом, хорошо известным специалистам в данной области, путем обработки кислотой, такой как хлористоводородная, серная, трифлиновая или предпочтительно толуолсульфоновая кислота в растворителе, таком как бензол, уксусная кислота, диоксан или предпочтительно толуол, при температуре около 25oC до температуры кипения с обратным холодильником в течение от около 0,5 до 5 часов.
Полученный алкен может быть гидроксилирован с использованием каталитического тетраоксида осмия и окислителя, такого как морфолин-N-оксид или тому подобное, в растворителе, таком как эфир, ТГФ или предпочтительно ацетон, смешанный с водой. Смесь перемешивают при комнатной температуре до тех пор, пока весь исходный продукт не расходуется, что отнимает всегда от около 1 до 5 часов.
Это дает исключительно цис-дигидрокси соединения. Трансдигидрокси аналоги могут быть получены путем более длительного окисления, от около 10 до 24 часов, в таких же реакционных условиях, с последующим восстановлением 3-гидрокси-4-кето продукта с гидридным восстанавливающим агентом, таким как LiAlH4 или LiBH4, или предпочтительно NaBH4, в растворителе, таком как ТГФ, но предпочтительно MeOH.
Гидроксилированные сложные эфиры омыляют, используя основания щелочного металла с сорастворителем, таким как кипящие низшие спирты, предпочтительно этанол, с получением кислотных продуктов.
Алкен может быть также обработан надкислотой, предпочтительно метахлорнадбензойной кислотой, в растворителе, таком как ТГФ или предпочтительно дихлорэтан, при температуре от около 0oC до комнатной температуры, предпочтительно при температуре 0oC, в течение 1-5 часов с получением эпоксида. Эпоксид гидролизуют с помощью H2 и 10% Pd/C в инертном растворителе, таком как этилацетат, с получением 3-гидрокси эфира (R3 = H, R4 = ОН).
Соединения формулы I, где R1 является

где R10 является независимо фтором, хлором, (C1-C6)алкилом, (C1-C6)алкокси, (C1-C4)перфторалкилом, (C1-C4)перфторалкокси, (C1-C6)алкилтио, (C1-C6)алкилсульфинилом, фенилсульфинилом, (C1-C6)алкилсульфонилом или фенилсульфонилом;
r равно 1 или 2,
могут быть получены путем взаимодействия соединений формулы I, где R1 является

где R10 и r являются такими, как определено выше,
с сульфонамидом формулы X7SO2NH2 в присутствии связывающего агента, такого как 1,3-дициклогексилкарбодиимид или 1-[3-(диметиламино)пропил]-3-этилкарбодиимид, и в присутствии органического основания, такого как пиридин, диметиламинопиридин, триэтиламин, диизопропилэтиламин или диазобицикло[5,4,0] ундец-7-ен. Реакцию проводят в растворителе, таком как тетрагидрофуран, диэтиловый эфир, толуол и хлорбензол, при температуре в диапазоне от около комнатной температуры до около температуры кипения используемого реакционного растворителя.
Соединения формулы I, где X6 из R1 или ароматического замещения равен NHCO-X7, NHSO2-X7 или NHCO-OX7, могут быть получены взаимодействием соединений формулы I, где X5 или X6 на R1 является карбокси или замещенной ароматической или гетероароматической кислотой, с дифенилфосфорилазидом в растворителе, таком как толуол, ДМЭ, ТГФ или дихлорэтан, в присутствии бензилового спирта и основания амина, такого как пиридин, диизопропилэтиламин, пирролидин или предпочтительно триэтиламид, при температуре кипения используемого растворителя в течение 5-48 часов, предпочтительно 16 часов. Продукт от этой реакции гидрируют в низшем спирте в присутствии палладиевого катализатора, предпочтительно Pd(OH)2/C, с последующим ацилированием с помощью соответствующего хлорангидрида кислоты, карбомоилхлорида или сульфонилхлорида.
Способы синтеза, указанные выше, вместе с последующими примерами описывают способы, которые используются и могут быть использованы для получения соединений настоящего изобретения.
Там, где это возможно, как может определить специалист в данной области с помощью настоящего описания, фармацевтически приемлемые катионные соли некоторых соединений настоящего изобретения включают, но не ограничиваются ими, соли натрия, калия, кальция, магния, аммония, N,N'-дибензилэтилендиамина, N-метилглюкамина, этаноламина и диэтаноламина. Фармацевтически приемлемые катионные соли соединений формулы I могут быть получены путем смешивания соединения формулы I с одним эквивалентом аминового основания или основания щелочного металла.
Соединения настоящего изобретения могут вводится млекопитающим, включая человека, для лечения заболеваний, индуцированных ЛТВ4, различными путями, включая
пероральный, парентеральный и местный, включая использование суппозитариев и клизм. При пероральном введении уровни доз от около 0,5 до 1000 мг/день, более предпочтительно около 5-500 мг/день, могут
назначаться в виде одной дозы или в разделенных дозах, до трех доз. Для внутривенного введения уровни доз составляют 0,1-500 мг/день, более предпочтительно, около 0,1-100 мг/день. Внутривенное
введение может включать непрерывное введение с помощью капельницы. Изменения будут необходимы в зависимости от возраста, веса и состояния субъекта, подвергающегося лечению, и конкретного выбранного
пути введения, что понятно из данного описания, специалисту в данной области
Соединения настоящего изобретения могут вводится сами по себе, но обычно будут вводиться в смеси с
фармацевтическим носителем или разбавителем, выбранным с учетом предполагаемого пути введения и
обычной фармацевтической практики, хорошо известной специалисту в данной области. Например, они могут
приниматься во внутрь в виде таблеток, содержащих такие наполнители, как крахмал или лактоза, или в капсулах, либо сами по себе, либо в смеси с наполнителями, или в виде эликсиров или суспензий,
содержащих ароматизирующие или подкрашивающие агенты. Они могут вводиться парентерально, например внутримышечно, внутривенно или подкожно. Для парентерального приема их лучше всего использовать в виде
стерильного водного раствора, который может содержать другие растворенные вещества, например достаточное количество соли или глюкозы, чтобы сделать раствор изотоническим.
Активность соединений настоящего изобретения по отношению к ЛТВ4 может быть определена путем сравнения способности соединений настоящего изобретения к конкуренции с радиоактивно-меченным ЛТВ4 за специфические сайты ЛТВ4 рецепторов на мембранах клеток селезенки морской свинки. Мембраны клеток селезенки морской свинки получают, как описано Cheng et al. (J. Pharmacology and Experimental Therapeutics 232:80, 1985). Анализ связывания3H-ЛТВ4 производят в 150 мкл, содержащих 50 мМ Трис pH 7,3, 10 мМ MgCl2, 9% метанола, 0,7 нМ3 H-ЛТВ4 (NEN, примерно 200 Ci/ммоль) и 0,33 мг/мл мембран клеток селезенки морской свинки. Немеченный ЛТВ4 добавляют в концентрации 5 мкМ для определения неспецифического связывания. Соединения добавляют в различных концентрациях для оценки их влияния на связывание3H-ЛТВ4. Реакционные смеси инкубируют при температуре 4oC в течение 30 минут. Мембраны, связывающие3H-ЛТВ4, собирают с помощью фильтрования через стекловолоконные фильтры, и связанное количество определяют путем счета сцинцилляций. Величина ИК50 для соединения представляет собой концентрацию, при которой ингибируется 50% специфического связывания3H-ЛТВ4.
Функциональная активность соединений настоящего изобретения может быть определена несколькими способами, используя биологические испытания. Описаны как высоко-, так и низкоафинные формы ЛТВ4 рецептора, которые по-разному связаны с хемотаксисом лейкоцитов и регуляцией адгезии молекул (Sterman, J. W. ; Groetzl, E. J. et al., J. Immun., 1988, 140, 3900-3904). Xемотаксис нейтрофилов человека измеряют, как описано в Horvath, L. et al., J. Immunol. , 1987, 139, 3055. Регуляцию нейтрофилов человека CDIIb измеряют, как описано Marder, P. et al., Prostaglandins, Leukotriene Essent. Fatty Acids, 1991, 46, 265-278.
Кроме того, соединения формулы I могут быть исследованы in vivo в соответствии со способом, аналогичным способу, описанному Pettipler, E.R. et al. , Brit. J. Pharmacology, 1993, 423-427, путем инъекции ЛТВ4 в дермис морских свинок и измерения блокады миграции нейтрофилов в коже с помощью принимаемых перорально соединений формулы I.
Следующие примеры иллюстрируют получение соединений настоящего изобретения и не могут быть использованы для ограничения рамок настоящего изобретения каким-либо образом.
Пример 1
(5S*,
6S*)2-(6-Бензил-5,6-дигидрокси-5,6,7,8-тетрагидро- нафталин-2-ил)-4-фторбензойная кислота
A. 2-(4-Фторфенил)-4,4-диметил-4,5-дигидрооксазол
К перемешиваемому раствору
2-амино-метилпропанола (0,378 моль, 33,64 г) в 300 мл метиленхлорида при температуре около 0oC прибавляют раствор 4-фторбензоилхлорида (0,189 моль, 22,36 мл) в 100 мл метиленхлорида в
течение около 0,5 часа. Смеси позволяют нагреться до комнатной температуры и перемешивают около 3 часов. Смесь затем выливают в воду и слои разделяют. Органическую фазу промывают двумя частями 10% HCl,
одной частью раствора насыщенного хлорида натрия и сушат над безводным MgSO4. Удаление растворителя в вакууме дает бесцветный твердый продукт, который перемешивают при добавлении по каплям
SOCl2 (0,567 моль, 41 мл) в течение около 30 мин. Полученный раствор перемешивают около 0,5 часа, в это время добавляют диэтиловый эфир, при этом раствор быстро перемешивают. В продолжение
этой процедуры получается бесцветный преципитат. Суспензию фильтруют и твердый продукт промывают тремя частями по 250 мл диэтилового эфира. Твердый продукт затем растворяют в 300 мл 3н. KOH и
полученный раствор экстрагируют этилацетатом. Органические экстракты промывают раствором насыщенного NaCl и сушат над MgSO4. Удаление растворителя в вакууме дает 32 г указанного в заглавии
этого примера 1A соединения:1H-ЯМР (250 МГц, CDCl3) δ: 8,00-7,91 (м, 2H), 7,10-7,02 (м, 2H), 4,11 (с, 2H), 1,39 (с, 6H).
B. 2-Бензилиден-6-метокси-3,
4-дигидро-2H-нафталин-1-он
К перемешиваемому раствору 6-метокси-1-тетралона (0,227 моль, 40 г) и бензальдегида (0,272 моль, 27,5 мл) в 450 мл метанола прибавляют пирролидин (0,272 моль, 23,6
мл). Смесь перемешивают при комнатной температуре в течение около 4 дней до тех пор, пока ТСX не покажет, что исходный тетралон отсутствует. Смесь концентрируют в вакууме, затем растворяют в EtOAc,
промывают четырьмя частями 10% HCl, двумя частями насыщенного раствора NaHCO3 и одной частью солевого раствора. Растворитель удаляют в вакууме и сырое масло растирают с диэтиловым эфиром с
получением 38 г указанного в заглавии этого примера 1B соединения, т.пл. 100-102oC. Рассчитано для C18H16O2: 264, 1146. Найдено: 264, 1149.
C. 2-Бензил-6-метокси-3,4-дигидро-2H-нафталин-1-он
Аппарат Парра® загружают хроменоном (15 г), этилацетатом (150 мл) и 1 г 10% палладия на древесном угле. Смесь гидрируют в
аппарате Парра® со встряхиванием в течение около 15 часов при давлении водорода 20 фунтов на квадратный дюйм (1406 г/см2). Полученную смесь фильтруют через слой Целита и
концентрируют в вакууме с получением красного масла, которой очищают путем флэш-хроматографии (3:1 гексан/диэтиловый эфир) с получением 14,1 г соединения этого примера 1C, т.пл. 50-51oC.
Рассчитано для C18H18O2: 266, 1302. Найдено: 266, 1308.
D. 2-Бензил-6-гидрокси-3,4-дигидро-2H-нафталин-1-он
К перемешиваемому раствору
соединения примера 1C (бензилтетралон) (5 г, 19 ммоль) в метиленхлориде (40 мл) при температуре около -78oC прибавляют тиобромид бора (1,95 мл, 21 ммоль). Охлаждающую баню удаляют и
реакционную смесь перемешивают в течение ночи при комнатной температуре, по истечении этого времени добавляют дополнительные 1,5 мл трибромида бора. Перемешивание продолжают при комнатной температуре
дополнительно в течение еще около 4 часов, в это время смесь выливают в воду, охлажденную льдом, и перемешивают около 0,5 часа. Водную смесь насыщают хлоридом натрия и экстрагируют четырьмя частями
метиленхлорида. Слои разделяют и органическую фазу промывают водой и сушат над безводным сульфатом натрия. Фильтрование и удаление растворителя в вакууме дает коричневый твердый продукт, который
очищают путем флэш-хроматографии (3:2 гексан/эфир) с получением 3 г соединения этого примера 1D, т.пл. 160-162oC. Рассчитано для C17H16O2: 252, 1146.
Найдено: 252, 1144.
E. 6-Бензил-5-оксо-5,6,7,8-тетрагидронафталин-2-иловый эфир трифтрометансульфоновой кислоты
К перемешиваемому раствору соединения примера 1D (2,75 г, 11
ммоль), триэтиламина (4,56 мл, 33 ммоль) и ДМАФ (0,05 г) в метиленхлориде (100 мл) при температуре около -78oC прибавляют трифтрометансульфоновый ангидрид (2 мл, 12 ммоль). Охлаждающую баню
удаляют и реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Смесь выливают в воду со льдом и экстрагируют этилацетатом. Полученный органический слой промывают водой,
сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Сырой продукт очищают путем флэш-хроматографии с получением 3,9 г соединения этого примера 1E, т.пл. 52-53,7oC. Рассчитано для C18H15SF3: 384, 0638. Найдено: 384, 0602.
F. 2-Бензил-6-[2-(4,4-диметил-4,5-дигидрооксазол-2-ил)- 5-фторфенил]-3,
4-дигидро-2H-нафталин-1-он
К перемешиваемому раствору н-бутиллития (3,6 мл, 2,5 М раствор в гексане, 9 ммоль) в толуоле (10 мл) при температуре около -40oC добавляют раствор
арилоксазолина (1,76 г, 9 ммоль) в толуоле (5 мл) по каплям с помощью пипетки. Смесь перемешивают при температуре около -40oC в течение около 0,5 часа, затем нагревают до температуры около
-25oC и перемешивают еще в течение около 1 часа. К этой смеси добавляют хлорид цинка (9 мл, 1 М раствор в диэтиловом эфире, 0,009 моль). Охлаждающую баню удаляют и смесь нагревают до
комнатной температуры и перемешивают около 1 часа. Полученную смесь добавляют с помощью пипетки к раствору тетралонтрифлата (3,5 г, 9 ммоль) и тетракисфенилфосфинапалладия (0,5 ммоль, 0,63 г) в
тетрагидрофуране (15 мл). Реакционную смесь нагревают с обратным холодильником около 2 часов, охлаждают до комнатной температуры и выливают в насыщенный водный раствор хлорида аммония. Водную смесь
экстрагируют тремя порциями этилацетата. Органическую фазу промывают тремя порциями 1М HCl, насыщенным водным раствором бикарбоната натрия и солевым раствором. Органическую фазу затем сушат над
безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Сырой продукт очищают путем флэш-хроматографии (2:1 диэтиловый эфир/гексан) с получением 2,07 г соединения этого примера 1F, т.
пл. 114-115oC. Рассчитано для C28H26NO2F: 427, 1948. Найдено: 427, 1956.
G. 2-Бензил-6-[2-(4,4-диметил-4,
5-дигидрооксазол-2-ил)- 5-фторфенил]-1,2,3,4-тетрагидро-2H-нафталин-1-ол
К перемешиваемому раствору соединения примера 1F (1,5 г, 3,5 ммоль) в метаноле (35 мл) добавляют боргидрид натрия (0,
20 г, 5,25 ммоль). Полученную коричневую смесь перемешивают при комнатной температуре около 1 часа, затем выливают в солевой раствор и экстрагируют тремя порциями этилацетата. Органическую фазу сушат
над безводным сульфатом натрия и растворитель удаляют в вакууме с получением 1,20 г 1:1 смеси цис и транс спиртов, т.пл. 88-89oC. Рассчитано для C28H28NO2F:
429, 2087. Найдено: 429, 2067.
H. 2-(6-Бензил-5-гидрокси-5,6,7,8-тетрагидронафталин-2-ил)- 4-фторбензойная кислота
Соединение примера 1G (1,0 г, 2,34 ммоль) растворяют в 5 мл
метилйодида и перемешивают при комнатной температуре около 2 дней, за это время метилйодид удаляют в вакууме. Остаток извлекают метиленхлоридом и концентрируют в порядке удаления следов остатка
метилйодида. Темно-красный остаток растворяют в метаноле (5 мл) и добавляют 2н. NaOH (5 мл). Полученную смесь нагревают с обратным холодильником при перемешивании в течение около 5 часов. Смесь затем
охлаждают до комнатной температуры и подкисляют 3н. HCl. Полученную суспензию экстрагируют тремя порциями этилацетата и объединенную органическую фазу промывают солевым раствором. Органическую фазу
сушат над безводным сульфатом натрия и растворитель удаляют в вакууме с получением 0,80 г соединения этого примера 1H.1H-ЯМР (250 МГц, метанол-d4) δ: 7,83 (дд, 1H, J= 7,0,
7,5), 7,50 (д, 1H, J=7,0), 7,30-7,00 (м, 9Hx2), 4,50 (д, 1H, J=2,0), 4,41 (д, 1H, J= 8,0), 3,15 (дд, 1H, J=5,4, 13,9), 3,00-2,57 (м, 4H), 2,42 (дд, 1Н, J=11,4 13,5), 2,09-1,35 (м, 5Hx2).
I. Этиловый эфир 2-(6-бензил-5-гидрокси-5,6,7,8-тетрагидронафталин-2-ил) -4-фторбензойной кислоты
К перемешиваемому раствору соединения примера 1H (0,80г, 2,13 ммоль) и этилйодида
(0,34 мл, 4,26 ммоль) в ацетонитриле (20 мл) добавляют карбонат калия (1,03 г, 1,45 ммоль). Полученную суспензию нагревают до температуры около 60oC в течение около 24 часов. Смесь
охлаждают до комнатной температуры, разбавляют диизопропиловым эфиром и фильтруют через целит. Концентрирование в вакууме дает 0,72 г соединения этого примера в виде 1:1 смеси цис/транс. Данные для
смеси диастереомеров являются следующими:1H-ЯМР (250 МГц, хлороформ-d) δ: 7,88 (дд, 1H, J=7,0, 7,5), 7,53 (д, 1H, J=7,0), 7,39-7,70 (м, 9Hх2), 4,58-4,52 (м, 1Hх2), 4,12 (кв, 2H, J=7,
0), 4,11 (кв, 2H, J= 7,0), 3,13 (дд, 1H, J=6,1, 14,2), 3,00 (8Hх1, 14Hх2), 2,92-2,70 (м, 2H), 2,55 (дд, 1H, J=9,4, 14,2), 2,11-1,40 (м, 5Hх2), 1,08 (т, 3H, J=7,0), 1,07 (т, 3H, J=7,0).
J. Этиловый эфир 2-(6-бензил-7,8-дигидронафталин-2-ил)- 4-фторбензойной кислоты
В круглодонную колбу, содержащую соединение примера 1I (0,70 г) в бензоле (50 мл), добавляют
п-толуолсульфоновую кислоту (0,09 г). Колбу снабжают ловушкой Дина-Старка для удаления воды во время протекания реакции. Смесь нагревают с обратным холодильником в течение около 16 часов, затем
охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток извлекают хлороформом и промывают насыщенным водным раствором бикарбоната натрия. Xлороформовые экстракты сушат над
безводным сульфатом натрия и растворитель удаляют в вакууме с получением желтого масла, которое очищают путем флэш-хроматографии (6:1 гексан/этилацетат) с получением 0,56 г соединения этого примера
1J. Рассчитано для C26H23O2F: 386, 1676. Найдено: 386, 1713.
K. Этиловый эфир цис-(5S*, 6S*)-2-(6-бензил-5,6- дигидрокси-5,6,7,
8-тетрагидронафталин-2-ил)-4-фторбензойной кислоты
К перемешиваемому раствору соединения примера 1J (0,50 г, 1,3 ммоль) в 3:1 смеси ацетон/вода (6 мл) добавляют N-метилморфолин-N-оксид (0,168
г, 1,4 ммоль), после чего добавляют тетроксид осмия (0,79 мл, 4% раствор в воде, 0,1 ммоль). Реакционную смесь перемешивают при комнатной температуре и контролируют с помощью ТСX на исчезновение
исходного продукта. Смесь затем разбавляют хлороформом и промывают 10% водным раствором NaHSO3. Хлороформенные экстракты сушат над безводным сульфатом натрия и удаляют растворитель при
пониженном давлении. Остаток очищают путем флэш-хроматографии (1:1 гексан/этилацетат) с получением 0,34 г соединения этого примера 1K, т.пл. 59-60oC. Рассчитано для C26H25O4F: 420, 1736. Найдено: 420, 1722.
L. (5S*, 6S*)-2-(6-Бензил-5,6-дигидрокси-5,6,7,8- тетрагидронафталин-2-ил)-4-фторбензойная кислота
К перемешиваемому раствору соединения примера 1K (0,30 г, 0,71 ммоль) в смеси 3:1 метанол/вода (12 мл) добавляют гидроксидмоногидрат лития (0,15 г, 3,6 ммоль). Полученную смесь нагревают с обратным
холодильником в течение около 4 часов. Смесь охлаждают до комнатной температуры и подкисляют 1н. HCl. Водную суспензию экстрагируют этилацетатом. Органическую фазу промывают солевым раствором, сушат
над безводным сульфатом натрия и удаляют в вакууме растворитель с получением 0,24 г соединения этого примера 1L (указанный в заглавии продукт), т. пл. 127-130oC. Рассчитано для C24H21O4F: 392, 1423. Найдено: 392, 1948.
Пример 2
7-(2-Карбокси-5-трифторметилфенил)-З-фенилметил-З-гидрокси-2,3,4- дигидробензопиран
A.
2', 4' -Дигидрокси-3-хлорпропиофенон
К перемешиваемой смеси резорцинола (200 г, 1,82 моль) и 3-хлорпропионовой кислоты (200 г, 1,84 моль) за один раз добавляют трифторметансульфоновую кислоту
(1 кг). Раствор медленно нагревают в течение около 45 минут до температуры 80oC, затем охлаждают до комнатной температуры в течение около 15 минут и выливают в хлороформ (4,0 л).
Органическую часть медленно выливают в воду (4,0 л) и слои разделяют. Водный слой экстрагируют хлороформом (2 x 2,0 л). Объединенные органические слои промывают солевым раствором, сушат над сульфатом
натрия и фильтруют. Концентрирование в вакууме дает оранжевый полутвердый продукт (244,1 г), который используют сырым на следующей стадии.1 H-ЯМР (300 МГц, CDCl3 δ: 12,56
(1H, с), 7,63 (1H, д, J=7,6), 6,37-6,46 (2H, м), 3,92 (2H, т, J=6,3), 3,41 (2H, т, J=6,3).
B. 7-Гидроксибензопиран-4-он
К охлажденному (около 5oC) раствору 2 н.
гидроксида натрия (10,0 л) за один раз добавляют соединение примера 2A (244,1 г). Раствор нагревают до комнатной температуры в течение около 2 часов, используя баню с нагретой водой, затем повторно
охлаждают до около 5oC и устанавливают pH 2 с помощью 6 М серной кислоты (1,2 л). Смесь экстрагируют 3 x 3,0 л этилацетатом, промывают солевым раствором (1 x 2,0 л), сушат над сульфатом
натрия и фильтруют. Концентрирование в вакууме дает желтовато-коричневый твердый продукт. Растирание с гексаном и фильтрование дает 173,7 г (выход 58%) соединения этого примера 2B. Т.пл. 136-137oC.
C. 7-[Трифторметилсульфонилокси]-бензопиран-4-он
К перемешиваемому раствору соединения примера 2B (173,7г, 1,05 моль) в метиленхлориде (3,0 л) при температуре около
-78oC добавляют триэтиламин (320 г, 3,16 моль) и диметиламинопиридин (2,5 г). После полного растворения по каплям добавляют трифторметансульфоновый ангидрид (327 г, 1,16 моль) в течение
около 20 минут, продукт перемешивают в течение около 30 минут при температуре около -78oC и затем нагревают до комнатной температуры в течение около 2 часов. Реакционную смесь выливают в
насыщенный раствор хлорида аммония (2,5 л) и слои разделяют. Водный слой экстрагируют 2 x 2,0 л метиленхлорида. Объединенные органические фракции промывают водой (1 x 1,0 л), сушат над сульфатом
магния и фильтруют. Концентрирование в вакууме дает красное масло. Хроматография на силикагеле (1 кг) с элюированием (8:1) гексан: этилацетат дает после удаления растворителя 211,1 г (выход 69%)
указанного в заглавии этого примера 2C продукта. Т.пл. 43-44oC.
D. 7-[(Трифторметилсульфонил)окси]-З-фенилметилен-бензопиран-4-он
К перемешиваемому раствору
продукта примера 2C (27 г, 91,2 ммоль) в 183 мл метанола добавляют бензальдегид (11,1 мл, 109 ммоль), затем добавляют пирролидин (9,1 мл, 109 ммоль). Смесь перемешивают при комнатной температуре в
течение ночи, охлаждают до около 0oC и фильтруют. Твердый продукт промывают один раз 50 мл метанола со льдом и затем сушат в вакууме; выделяют 35,2 г (выход 75%) указанного в заглавии этого
примера 2D продукта. Т.пл. 133-135oC.1H-ЯМР (300 МГц, CDCl3) δ: 8,11 (1H, д, J=8,7), 7,91 (1H, ушир. с), 7,40-7,51 (2H, м), 7,24-7,38 (3H, м), 6,97 (1H, дд,
J=8,7, 2,4), 6,91 (1H, д, J=2,4), 5,40 (1H, ушир. с).
E. 7-[(Трифторметилсульфонил)окси]-З-фенилметил-бензопиран-4-он
К перемешиваемому раствору продукта примера 2D (26,6 г,
69,2 ммоль) в 250 мл этилацетата в 500 мл аппарате Парра® со встряхиванием добавляют катализатор из 10% палладия на угле (1,3 г). Смесь гидрируют при 40 фунтах на квадратный дюйм
(2812 г/см2) до тех пор, пока через примерно 3 часа не прекратится выделение водорода. Смесь фильтруют через Целит® (торговая марка диатомовой земли) для удаления
палладиевого катализатора и подвергают хроматографии на силикагеле (гексан-эфир); получают 25,1 г (выход 94%) указанного в заглавии этого примера 2E продукта. Т.пл. 56-58oC.1
H-ЯМР (300 МГц, CDCl3) δ: 8,01 (1H, д, J=8,5), 7,20-7,35 (5H, м), 6,81-6,96 (2H, м), 4,42 (1H, дд, J= 11,6, 4,4), 4,22 (1H, дд, J=11,6, 8,7), 3,26 (1H, дд, J= 14,0, 4,4), 2,90-3,05
(1H, м), 2,70 (1H, дд, J=14,0, 8,7).
F. 7-(2-Карбоэтокси-5-трифторметилфенил)-З-фенилметил- 3,4-дигидробензопиран
Используя способ, описанный в примерах 1F-1J, но используя
2-(4-трифторметилфенил)-4,4-диметил-4,5-дигидрооксазол в качестве реагента, получают желаемый продукт этого примера 2F.1H-ЯМР (300 МГц, CDCl3) δ: 7,87 (1H, д, J=8,0), 7,
68-7,61 (2H, м), 7,41-7,22 (5H, м), 6,97 (1H, д, J=7,8), 6,85-6,73 (2H, м), 6,21 (1H, с), 4,71 (2H, с), 4,20 (2H, кв, J=7,2), 3,48 (2H, с), 1,15 (4H, т, J=7,2).
G.
7-(2-Карбоэтокси-5-трифторметилфенил)-3-фенилметил- 3,4-оксиранобензопиран
К перемешиваемому раствору олефина из примера 2F (230 мг, 0,53 ммоль) в метиленхлориде при температуре около 0oC добавляют m-CPDA (89 мг, 0,53 ммоль). Через 1 час смесь разбавляют эфиром и промывают 1 н. раствором NaOH и солевым раствором. Органический слой сушат (MgSO4), фильтруют и
концентрируют. Очистка флэш-хроматографией (элюирование смесью 3:1 гексан-этилацетат) дает соответствующий эпоксид этого примера 2G (209 мг, 87%). Продукт используют непосредственно для следующей
стадии.
H. 7-(2-Карбоэтокси-5-трифторметилфенил)-З-фенилметил-З- гидроксибензопиран
К раствору эпоксида примера 2G (150 мг, 0,33 ммоль) в этилацетате добавляют 10% палладий на
угле (100 мг). Систему соединяют с баллоном, содержащим H2, и колбу продувают несколько раз. После перемешивания в течение около 16 часов раствор фильтруют и концентрируют.
Флэш-хроматографическое элюирование дает желаемый спирт этого примера 2H (80 мг,53%).1H-ЯМР (300 МГц, CDCl3) δ: 7,89 (1H, д, J=8,0), 7,70-7,62 (2H, м), 7,42-7,25 (5H, м),
7,07 (1H, д, J= 7,8), 6,90-6,80 (2H, м), 3,95 (2H, с), 4,19 (2H, кв, J=7,l), 3,04-2,72 (4H, м).
I. 7-(2-Карбокси-5-трифторметилфенил)-3-фенилметил-3- гидроксибензопиран
Омыление соединения примера 2H по способу, описанному в примере 1L, дает желаемый продукт примера 2.1H-ЯМР (300 МГц, CDCl3) δ: 7,98 (1H, д, J=7,8), 7,67-7,64 (2H, м), 7,
38-7,26 (5H, м), 7,06 (1H, д, J=7,8), 6,88-6,85 (2H, м), 2,99-2,72 (4H, м).
Пример 3
(3S*, 4R*)-7-(2-Карбокси-5-трифторметилфенил-3,
4-дигидроксифенил)-З-фенилметил-3,4-дигидробензопиран
A. 7-(Карбоэтокси-5-трифторметилфенил)-3-фенилметил-3- гидроксибензопиран-4-он
К раствору олефина из примера 2F (3,7 г, 8,44
ммоль) в смеси ацетон-вода (3:1) добавляют N-метилморфолин-N-оксид (3,0 г, 25,3 ммоль), затем добавляют OsO4. После перемешивания в течение ночи раствор разбавляют водой и экстрагируют
этилацетатом. Объединенные экстракты сушат и концентрируют. Очистка остатка путем флэш-хроматографии дает соответствующий гидроксикетон этого примера 3A (3,0 г, 76%). Т.пл. 85-87oC.
В. (3S*, 4R*)-7-(2-Карбоэтокси-5-трифторметилфенил)-3, 4-дигидрокси-З-фенилметилбензопиран
К раствору кетона примера 3A (3,0 г, 6,4 ммоль) в метаноле (50 мл)
при комнатной температуре добавляют NaBH4 (250 мг, 6,4 ммоль). По истечении около 30 минут реакционную смесь гасят (NH4Cl раст.), экстрагируют (этилацетат), сушат (MgSO4), фильтруют и концентрируют. Флэш-хроматография (элюирование смесью 2: 1 гексан-этилацетат) дает желаемый транс-диол этого примера 3B (2,9 г, 97%).1H-ЯМР (300 МГц, CDCl3
) δ: 7,89 (1H, д, J=8,0), 7,69-7,55 (2H, м), 7,42-7,20 (5H, м), 6,90-6,82 (2H, м), 4,43 (1H, с), 4,15 (IH, д, J=12,l), 3,90 (1H, д, J=12,1), 3,79 (3H, с), 3,13 (1H, д, J=13,5), 2,85 (1H, д,
J=13,5).
С. (3S*, 4R*)-7-(2-Карбокси-5-трифторметил-3,4- дигидрокси)-3-фенилметилбензопиран
Омыление соединения примера 3B по способу, описанному в
примере 1L, дает желаемую кислоту этого примера 3. Т.пл. 100-102oC.
Пример 4
(3S*, 4R*)-7-(2-Трифторметансульфониламино-5-фторфенил) -3,
4-дигидрокси-3-гидрокси-3-фенилметил-2H-1-3,4-дигидробензопиран
A. 7-[(5-фтор-(2-(4,4-диметил-2-оксазолинил)фенил] -3-фенилметилен-1- бензопиран-4-он
К перемешиваемому раствору
2-(4-фторфенил)-4,4-диметил-2-оксазолина (1,0 экв в тетрагидрофуране, концентрация 0,5 М) при температуре около -78oC в атмосфере N2 добавляют н-бутиллитий в гексане (1,1 экв, 2,
5 М раствор). Смесь перемешивают при температуре около -78oC в течение около 1 часа, затем добавляют ZnCl2 (1М раствор в эфире, 1,1 экв). Смесь нагревают до около 10oC
в течение около 1 часа с получением 2-(4-фторфенил)-2-хлорцинк)-4,4-диэтил-2-оксазолина (не выделяют). К этому раствору добавляют 7-[((трифтор-метил)сульфонил)окси] -3-фенилметилен-1-бензопиран-4-он
(1,0 экв) и Pd(PPh3)4 (0,02 экв). Смесь нагревают с обратным холодильником (около 68oC) в течение около 3 часов, охлаждают до комнатной температуры и выливают в
раствор NH4Cl. Раствор экстрагируют трижды диэтиловым эфиром и объединенную органическую фракцию сушат над MgSO4. Фильтрование с последующим удалением растворителя в вакууме и
колоночная хроматография (силикагель - смесь 2:1 гексан: эфир) дают соединение этого примера 4A в виде желтого твердого продукта, выход 65%, т.пл. 110-112oC.1H-ЯМР (300 МГц,
CDCl3) δ: 8,04 (1H, д), 7,91 (1H, с), 7,78 (1H, дд), 7,41-7,52 (3H, м), 7,31 (2H, д), 7,06-7,18 (3H, м), 7,02 (1H, с), 5,40 (2H, с), 3,86 (2H, с), 1,31 (6H, с).
В.
(3S*, 4R*)-7-[5-фтор-(2-(4,4-диметил-2-оксазолинил)- фенил]-4-гидрокси-3-фенилметил-2H-1-бензопиран
К перемешиваемому раствору соединения из примера 4A в ТГФ (0,1 М) при
температуре около 0oC по каплям добавляют LiAlH4 (1М в эфире, 2,2 экв) в течение около 10 минут. Смесь нагревают до комнатной температуры и перемешивают в течение около 12 часов.
Смесь охлаждают до около 0oC, гасят солью Рошеля и фильтруют через диатомовую землю. Водный слой экстрагируют дважды этилацетатом и объединенные органические слои промывают солевым
раствором и сушат над MgSO4. Фильтрование и удаление растворителя дают желтое масло. Хроматография на силикагеле (этилацетат: гексан) дает 60% выход указанного в заглавии соединения этого
примера 4B в виде белого твердого продукта. Т.пл. 65-70oC (разл. ). Рассчитано для C27H26NO3F: C 75,15; H 6,07; N 3,25. Найдено: C 74,75; H 6,02; N 3,09.1H-ЯМР (300 МГц, CDCl3) δ: 7,70 (1H, дд), 7,02-7,37 (8H, м), 6,96 (1H, дд), 7,91 (1H, д), 4,51 (1H, д), 4,23 (1H, дд), 4,39 (1H, дд), 3,87 (2H, дд), 2,74 (1H, дд), 2,55
(1H, дд), 2,18-2,28 (1H, м), 1,31 (6H, д).
C. (3S*,4R*)-7-(2-Карбокси-5-фторфенил)-4-гидрокси-3- фенилметил-2H-1-бензопиран
Соединение из примера 4B
растворяют в метилйодиде (0,5 М) при комнатной температуре и перемешивают в течение около 24 часов. Метилйодид удаляют в вакууме, маслянистый твердый продукт растворяют в CH2Cl2
и растворитель удаляют в вакууме. Этот процесс повторяют до удаления следов метилйодида. Твердый продукт растворяют в метаноле (0,5 М) и добавляют 2 М NaOH (0,5 М). Смесь нагревают с обратным
холодильником в течение около 5 часов, охлаждают до комнатной температуры и подкисляют до pH 2 с помощью 1 М HCl. Смесь экстрагируют дважды этилацетатом, промывают солевым раствором, сушат над
MgSO4. Фильтрование и удаление растворителя в вакууме, а затем хроматография (силикагель, смесь 10:1 метиленхлорид:метанол) дают желаемую кислоту этого примера 4C, выход 93%.1
H-ЯМР (300 МГц, CD3COCD3) δ: 7,80 (1H, дд), 7,48 (1H, д), 7,18 (7H, м), 7,13 (1H, дд), 6,91 (1H, дд), 6,80 (1H, д), 4,52 (1H, д), 4,23 (1H, дд), 3,96 (1H, дд), 2,89 (1H,
дд), 2,54 (1H, дд), 2,19-2,30 (1H, м).
D. (3S*, 4R*)-7-(2-Карбокси-5-фторфенил)-3,4-дигидрокси- 3-фенилметил-2H-1-бензопиран
Используя способы примеров
от 1I до 1L, получают указанное в заглавии соединение этого примера 4D из соединения примера 4C.
Е. (3S*, 4R*)-7-(2-Карбобензилоксиамино-5-фтор)-3,
4- дигидрокси-3-фенилметил-2H-1-бензопиран
К раствору соединения, полученного в примере 4D (1 ммоль), в 10 мл 1,4-диоксана, добавляют 1,05 эквивалента дифенилфосфорилазида, 1,1 эквивалента
бензилового спирта и 2,2 эквивалента триэтиламина. Смесь нагревают с обратным холодильником в течение около 16 часов, растворитель удаляют в вакууме и остаток хроматографируют на силикагеле (1:1
- гексан:EtOAc) с получением N-CBZ продукта этого примера 4E.1H-ЯМР (300 МГц, CDCl3) δ: 8,02 (1H, с), 7,39-7,22 (1H, м), 7,03 (1H, дт, J=7,2), 6,95-6,87 (2H, м), 6,83 (1H,
с), 5,11 (2H, с), 4,48 (1H, с), 3,98 (1H, д, J=10,1), 3,80 (1H, д, J=10,1), 2,89 (2H, с).
F. (3S*, 4R*)-7-(2-Трифторметансульфониламино-5-фтор) - 3,
4-дигидрокси-3-гидрокси-3-фенилметил-2H-1-бензопиран
К раствору соединения, полученного в примере 4E, в 10 мл EtOH добавляют 0,05 экв весового Pd(OH)2 и суспензию гидрогенируют в
аппарате Парра® со встряхиванием при 1 атм в течение 3 часов. Смесь фильтруют через Целит® и фильтрат выпаривают. Желтое масло повторно растворяют в CH2
Cl2 (10 мл), охлаждают до температуры около 0oC и добавляют триэтиламин (2,2 экв), а затем ангидрид трифторметансульфоновой кислоты (1,1 экв). После перемешивания в течение около
2 часов добавляют 2 эквивалента твердого NaOMe, реакционную смесь перемешивают в течение около 15 минут и добавляют H2O (10 мл). Устанавливают pH смеси 2 с помощью 0,1 М HCl, затем
экстрагируют 3 x 10 мл EtOAc. Объединенные органические слои промывают солевым раствором, сушат над MgSO4, фильтруют и удаляют растворитель в вакууме с получением желтого полутвердого
продукта. Хроматография на силикагеле (1:1-10:1 EtOAc-гексан) дает желаемый полутвердый продукт этого примера 4F. Т.пл. 55-57oC.
Пример 5
1-(3S,
4S)-((3-(4-фенилфенилметил)-3-гидрокси)-4-гидрокси)хроман-7-ил) циклопентанкарбоновая кислота
A. Этил 1-(3-(4-фенилфенилметил)-4-хроманон-7-ил)циклопентанкарбоксилат
3-(4-Фенилбензил)-7-трифторметилсульфонилокси-4-хроманон (35,0 г, 91,0 ммоль), полученный из продукта примера 2C с бифенилальдегидом, используя способы примера 2D и 2E, растворяют в смеси
диметилформамида (230 мл) и диметоксиэтана (230 мл). К этому раствору добавляют в следующем порядке: трис(2-метилфенил)фосфин (7,48 г, 24,6 ммоль), хлорид бис(бензонитрил)палладия (II) (2,44 г, 6,37
ммоль), триметилсилилкетенацеталь этилциклопентанкарбоксилата (29,26 г, 136,5 ммоль) и 1,0 М эфирный раствор хлорида цинка (25 мл, 25 ммоль). Полученный прозрачный желтый раствор нагревают с обратным
холодильником в течение около 1 часа. В этот момент добавляют дополнительно триметилсилилкетенацеталь (9,75 г, 45,5 ммоль) и продолжают нагрев с обратным холодильником в течение еще 1 часа.
Охлажденную смесь разбавляют водой (1 л) и экстрагируют эфиром. Объединенные эфирные экстракты промывают водой (1 л) и затем сушат над сульфатом магния. Фильтрование и концентрирование экстракта дает
желтое масло, которое хроматографируют на силикагеле (8:92 этилацетат/гексан). Это дает 22,13 г (64%) указанного в заглавии этого примера 5A соединения в виде белого твердого продукта; т. пл.
50-56oC.1H-ЯМР (CDCl3) δ: 1,23 (3H, т, J=7,0), 1,75 (4H, м), 1,9 (2H, м), 2,6 (2H, м), 2,70 (1H, дд, J=10,5, 13,8), 2,9 (1H, м), 3,26 (1H, дд, J=4,3, 13,8), 4,
08 (2H, д, J=7,0), 4,15 (1H, дд, J=8,2, 11,5), 4,35 (дд, J= 4,3, 11,5), 6,95 (1H, д, J=1,7), 7,02 (1H, дд, J=1,7, 8,3), 7,2-7,4 (5H, м), 7,84 (1H, д, J=8,3).
B. Этил
2-(4-гидрокси-З-(4-фенилфенилметил)хроман-7-ил) циклопентанкарбоксилат
Соединение примера 5A (111,99 г, 295,9 ммоль) растворяют в этаноле (2,5 л) и осторожно нагревают с боргидридом натрия
(12,3 г, 325,5 ммоль) при комнатной температуре. По истечении около 3 часов реакционную смесь концентрируют до малого объема и разбавляют эфиром. Эфир промывают насыщенным раствором бикарбоната натрия
и сушат над сульфатом магния. Экстракт фильтруют и концентрируют до масла, которое хроматографируют на силикагеле (20:80 - этилацетат/гексан). Это дает 63,4 г (56%) высшего Rf продукта, то
есть 3R*, 4R*, в виде масла:1H-ЯМР (CDCl3 δ: 1,15 (т, J=7,1, 3H), 1,7 (м, 1,8-1,9 (м, 2H), 2,3 (м, 1H), 2,5-2,6 (м, 2H), 2,66 (дд, J=7,2, 13,6, 1H),
2,86 (дд, J= 8,4, 13,6, 1H), 4,0-4,1 (м, 4H), 4,47 (ушир. т, 1H), 6,85 (д, J=1,8, 1H,), 6,88 (дд, J=1,8, 7,9 1H,), 7,12 (д, J=7,9, 1H,), 7,1-7,35 (м, 5H); и 43,3 г (38%) низшего Rf продукта,
то есть 3S*, 4R*, в виде масла:1H-ЯМР (CDCl3) δ: 1,17 (т, J=7,0, 3H), 1,7 (м, 4H), 1,8-1,9 (м, 2H), 2,2 (м, 1H), 2,52 (дд, J= 9,3, 13,7, 1H), 2,6
(м, 2H), 2,70 (дд, J=6,3, 13,7, 1H), 3,95 (дд, J=3,7, 11,2, 1H), 4,07 (кв, J=7,0, 2H), 4,18 (дд, J=2,6, 11,2, 1H), 4,47 (ушир. т, 1H), 6,88 (д, J=1,8, 1H,), 6,94 (дд, J=1,8, 8,0 1H,), 7,15-7,3 (м,
6H).
C. 1-(3S, 4S)-((3-(4-Фенилфенилметил)-3-гидрокси)-4-гидрокси) хроман-7-ил)циклопентанкарбоновая кислота
Желаемый продукт получают из цис и транс смеси соединения примера
5B путем взаимодействия соединения примера 5B аналогично способам, описанным в Примерах 1J-1L, с получением указанного в заглавии соединения этого примера 5. Т.пл. 185-187oC.
Пример 6
(3S, 4S)-7-(Карбокси-5-трифторметилфенил)-3,4-дигидрокси-З-фенилметил- 3,4-дигидробензопиран
A. 7-(2-Карбометокси-5-трифторметилфенил)-4-L-трет-Boc-триптофановый
эфир-3-гидрокси-3-фенилметил)бензопиран
К раствору (3S, 4S)-7-(карбометокси-5-трифторметилфенил)-3,4-дигидрокси -3-фенилметилбензопирана (225 мг; 0,49 ммоль) в CH2Cl2 (6
мл) добавляют трет-BOC-L-триптофан (223 мг; 0,74 ммоль), ДМАФ (120 мг; 0,98 ммоль) и EDCI (165 мг; 0,86 ммоль). После перемешивания в течение около 16 часов раствор разбавляют этилацетатом и промывают
1н. HCl и солевым раствором, сушат (MgSO4), фильтруют и концентрируют. Очистка остатка путем флэш-хроматографии (элюирование смесью гексан-эфир-метиленхлорид 3:1:6) дает 137 мг, 38%,
выходящего быстрее диастереомера, а затем 135 мг, 37%, выходящего медленнее диастереомера этого примера 6A.1H-ЯМР (300 МГц, (CDCl3) δ: LP изомер: 8,98 (1H, с), 8,00 (1H,
д, J=8,0), 7,76-7,67 (2H, м), 7,68 (1H, д, J=7,5), 7,40 (1H, д, J= 7,6), 7,37-7,09 (9H, м), 6,92-6,86 (2H, м), 6,61 (1H, с), 5,80 (1H, с), 4,93 (1H, д, J=6,3), 4,57 (1H, дд, J=6,5, 3,0), 3,91 (2H, с),
3,82 (1H, д, J=10,2), 3,72 (1H, д, J=10,2), 3,50 (1H, дд, J=15,7, 5,0), 3,25 (1H, дд, J=15,0, 6,1), 2,84 (2H, с), 1,39 (9H, с). MP изомер: 8,72 (1H, с), 8,03 (1H, д, J=7,9), 7,78- 7,71 (2H, м), 7,59
(1H, д, J=8,0), 7,32-7,05 (9H, м), 6,93- 6,80 (2H, м), 6,41 (1H, м), 5,60 (1H, с), 5,17 (1H, д, J=6,0), 4,68-4,58 (1H, м), 3,95 (2H, с), 3,65 (1H, д, J=10,3), 3,43 (1H, д, J=10,2), 3,30 (1H, дд, J=15,
1, 5,3), 3,13 (1H, дд, J=15,0, 7,9), 2,72 (2H, с), 1,49 (9H, с).
B. (3S, 4S)-7-(2-Карбометокси-5-трифторметилфенил)-3,4-дигидроксил-3- фенилметилбензопиран
К раствору более
высокого Rf трет-BOC-триптофанового эфира примера 6A (130 мг; 0,17 ммоль) в метаноле-ТГФ (7 мл, например 5:2) добавляют 1н. NaOH (175 мл, 1н. ). По истечении около 1 часа раствор разбавляют
этилацетатом, промывают 1н HCl и солевым раствором, сушат (MgSO4), фильтруют и концентрируют. Очистка путем флэш-хроматографии (элюирование гексан-эфир-метиленхлорид 2: 1:6) дает
соответствующий спирт этого примера 6B (63 мг, 80%).1H-ЯМР (300 МГц, CDCl3) δ: 7,94 (1H, д, J=7,8), 7,72-7,60 (2H, м), 7,46 (1H, д, J=8,0), 7,40-7,22 (4H, м), 6,97-6,85
(3H, м), 4,52 (1H, с), 4,02 (1H, д, J= 12,1), 3,86 (IH, д, J=10,1), 3,72 (3H, с), 2,93 (2H, с), 2,48 (1H, ушир. с).
C. (3S, 4S)-7-(Карбокси-5-трифторметилфенил)-3,
4-дигидрокси-3- фенилметилбензопиран
К раствору эфира примера 6B (60 мг; 0,13 ммоль) в метаноле (3 мл) добавляют NaOH (3 мл; 3 н.). После нагревания при температуре около 60oC в
течение около 1 часа смесь охлаждают и подкисляют с помощью 1н. HCl. Раствор экстрагируют эфиром и объединенные экстракты промывают солевым раствором, сушат (MgSO4), фильтруют и
концентрируют с получением по существу чистой кислоты этого примера 6 (46 мг, 80%), которую в дальнейшем очищают перекристаллизацией из смеси гексан-этилацетат. Т.пл. 90-92oC.
Пример 7
7-(2-Карбоэтокси-5-фтор-фенил)-4-гидрокси-З-(4-фенил-фенилметил)-3,4- дигидробензопиран
A. 7-(Триметилстаннил)-3-(4-фенилфенилметил)бензопиран-4-он
К
перемешиваемому раствору соединения, полученного в примере 2E (10,95 г, 25,0 ммоль), в 200 мл диоксана добавляют хлорид лития (3,20 г, 75,0 ммоль), Pd(PPh3)4 (1,15 г, 1,0 ммоль),
3 кристалла бутилированного гидрокситолуола и гексаметилдиолова (9,0 г, 27,5 ммоль). Смесь нагревают с обратным холодильником в течение около 1,5 часа, охлаждают до комнатной температуры и выливают в
150 мл насыщенного водного раствора хлорида аммония. Смесь экстрагируют 3 x 150 мл диэтилового эфира и объединенные органические фракции промывают солевым раствором, сушат над сульфатом натрия и
фильтруют. Выпаривание в вакууме дает желтый полутвердый продукт, который хроматографируют на силикагеле (5: 1 гексан:эфир) с получением 9,8 г (выход 89%) указанного в заглавии продукта этого примера
7A.1H-ЯМР (300 МГц, CDCl3) δ: 7,85 (1H, д, J=8,7), 7,18-7,37 (9H, м), 7,14 (1H, д, J=8,7), 7,11 (1H, с), 4,38 (1H, дд, J= 11,6, 4,5), 4,17 (1H, дд, J=11,6, 8,4), 3,28 (1H,
дд, J= 14,0, 4,4), 2,84-2,95 (1H, м), 2,71 (1H, дд, J=14,11), 0,31 (9H, с).
B. 7-(2-Карбоэтокси-5-фторфенил)-3-(4-фенилфенилметил)бензопиран-4-он
К перемешиваемому раствору
соединения примера 7A (8,28 г, 17,5 ммоль) в диметилформамиде (ДМФ) (35 мл) добавляют Pd(PPh3)2Cl2 (490 мг, 0,7 ммоль), 3 кристалла ВНТ и этил-2-йод-5-фторбензоат (5,4
г, 19,1 ммоль). Смесь перемешивают при нагревании с обратным холодильником в течение около 1,5 часа, охлаждают до комнатной температуры и выливают в 150 мл насыщенного водного раствора хлорида
аммония. Смесь экстрагируют 3 x 150 мл диэтилового эфира и объединенные экстракты промывают 2 x 100 мл воды, а затем солевым раствором. Раствор сушат над сульфатом натрия, и выпаривают в вакууме с
получением желтого масла. Хроматография на силикагеле (элюирование 4:1 гексан:эфир) лает 6,51 г указанного в заглавии этого примера 7B соединения в виде вязкого масла.1H-ЯМР (300 МГц,
CDCl3) δ: 7,95 (2H, м), 7,28-7,65 (9H, м), 6,92-7,22 (4H, м), 4,49 (1H, дд, J=11,6, 4,5), 4,29 (1H, дд, J=11,6, 8,5), 4,15 (2H кв), 3,31 (1H, дд, J=14,0, 4,4), 2,91-2,99 (1H, м), 2,
73 (1H, дд, J=14,0, 11,1), 1,20 (3H, т).
C. 7-(2-Карбоэтокси-5-фтор-фенил)-4-гидрокси-З-( 4-фенил-фенилметил) бензопиран
К перемешиваемому раствору соединения примера 7B (6,
60 г, 17,5 ммоль) в 35 мл метанола при комнатной температуре добавляют боргидрид натрия (940 мг, 26,0 ммоль) за один раз. Темную смесь перемешивают при комнатной температуре в течение около 2 часов,
затем выливают в насыщенный водный раствор хлорида аммония (75 мл) и экстрагируют диэтиловым эфиром 3 x 75 мл. Объединенные экстракты промывают солевым раствором, сушат над сульфатом натрия, фильтруют
и концентрируют в вакууме с получением желтоватого масла. Хроматография на силикагеле (элюирование 4: 1 гексан:эфир) дает сначала 3,26 г цис кольцевого изомера указанного в заглавии этого примера 7
соединения, а затем 1,98 г транс-изомера указанного в заглавии этого примера 7 соединения в виде вязкого масла, общий выход 81%. Цис кольцевой изомер:1H-ЯМР (300 МГц, CDCl3)
δ: 7,95 (1H, дт), 6,8-7,61 (14H, м), 4,58 (1H, т, J=7,2), 4,28 (1H, дд, J=9,1, 2,5), 4,03 (1H, дд, J=9,1, 5,4), 4,15 (2H, кв), 2,78 (1H), 2,77 (1H, дд, J= 13,7, 6,2), 2,58 (1H, дд, J-13,7, 9,1),
2,20-2,29 (1H, м), 1,83 (1H, д, J= 7,2), 1,1 (3H, т). Транс кольцевой изомер:1H-ЯМР (300 МГц, CDCl3) δ: 7,95 (1H, дт), 6,8-7,60 (14H, м), 4,56 (1H, дт, J=4,7, 3,8), 4,
12-4,19 (2H, м), 4,10 (2H, кв), 2,90 (1H, дд, J=13,6, 8,4), 2,70 (1H, дд, J=13,6, 7,2), 2,36-2,39 (1H, м), 1,75 (1H, д, J=4,7), 1,12 (3H, т).
Ниже приведены результаты активности in
vitro соединения по примеру 10 настоящей заявки при анализе хемотаксиса (примеры 8 -16 см. в таблице). Для сравнения приведены также результаты исследований для соединений, которые имеют структуру,
подобную структуре соединения по примеру 10, в виде активности в том же самом анализе хемотаксиса. Анализ хемотаксиса является анализом влияния фармакологических соединений на нейтрофильный хемотаксис
человека за счет хемотаксических факторов, например, LTB4. Это дает представление о мере проникновения клеток в зону воспаления. После проникновения в зону воспаления, нейтрофилы подвергаются
окислительному распаду с выделением кислородных радикалов и матричных металлопротеаз, что влечет за собой поврежденные ткани. Таким образом, ингибирование LTB4 уменьшает повреждение ткани. Результаты
исследований:
Соединение по примеру 10:

Анализ хемотаксиса: IC50 = 0,003 мкМ.
В качестве соединений сравнения испытаны следующие соединения с аналогичной структурой:

IC50 = 39 мкМ
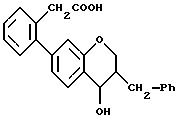
IC50 = 6 мкМ.
Вышеприведенные данные демонстрируют кардинальную разницу в активности между соединением по примеру 10 настоящей заявки и близкими по структуре соединениями.
Различие в активности настолько велико, что соединения сравнения, по существу, могут рассматриваться в качестве неактивных в отношении ингибирования LTB4. Существенно более низкие значения IC50 показывают, что соединения по настоящему изобретению являются намного более активными антагонистами LTB4, чем соединения сравнения.
Реферат
Бензопирановые и бензоконденсированные соединения формулы I и его фармацевтически приемлемые соли, где А' - О или СН2, R3, R4-Н или гидроксигруппа, R5 - -(СН2)nСНХ9Х10, -(СН2)nХ10, -СН(ОН)Х10; n=0 или 1; Х9 - Н или фенил; Х10 - Н, алкил, фенил, возможно замещенный R6 и R7, каждый Н или алкил, R1 - цис- или транс- (СН2)m-СХ3Х4Х5, и замещенное шестичленное ароматическое кольцо, где m = 0, и Х3 и Х4, взятые вместе с атомом углерода, к которому они присоединены, образуют С4-7-циклоалкил; Х5-карбоксигруппа, R2 - Н, являются антагонистами лейкотриена В4 (ЛТВ4). 4 с. и 8 з.п. ф-лы, 1 табл.
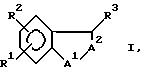

Формула
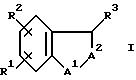
и его фармацевтически приемлемые соли,
где A1 является O, CH2;
R3 является атомом водорода или гидроксигруппой;
A2 обозначает

R4 является атомом водорода или гидроксигруппой;
R5 выбран из группы, включающей -(CH2)nCHX9X10, -(CH2)nX10 и -CH(OH)X10; где n равно 0 или 1;
X9 является атомом водорода или необязательно замещенным фенолом;
X10 является атомом водорода, C1-C6-алкилом, необязательно замещенным фенилом, который может быть замещен;
R6 и R7 являются каждый независимо атомом водорода или C1-C4-алкилом;
R1 выбран из группы, включающей цис- или транс- (CH2)m-CX3X4X5 и замещенное шестичленное ароматическое кольцо, где m равно 0, и X3 и X4, взятые вместе с атомом углерода, к которому они присоединены, образуют C4-C7-циклоалкил;
X5 является карбоксигруппой,
и замещенное шестичленное ароматическое кольцо замещено одним заместителем, выбранным из группы, включающей карбоксигруппу или -N(H)(SO2-X7) и одним или двумя заместителями, где каждый независимо выбран из группы, включающей атом фтора или хлора или C1-C4-перфторалкил, где X7 является -CF3;
R2 представляет атом водорода при условии, что, когда R3 является гидроксигруппой и R4 является атомом водорода, тогда R5 является -CH(OH)X10.
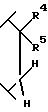
3. Соединение по п. 2 или его фармацевтически приемлемая соль, где R1 является -(CH2)mCX3X4X5 или замещенным шестичленным ароматическим кольцом, замещенным одним заместителем, выбранным из группы, включающей карбоксигруппу, или -CO-N(H)(SO2-X7), и одним или двумя заместителями, где каждый независимо выбран из группы, включающей атом фтора или хлора или C1-C4-перфторалкил.

где A1 является O, CH2;
A2 обозначает
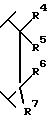
R4 является атом водорода или гидроксигруппой;
R5 выбран из группы, включающей -(CH2)nCHX9X10, -(CH2)nX10 и -CH(OH)X10, где n равно 0 или 1;
X9 является атомом водорода или необязательно замещенным фенилом;
X10 является атомом водорода, C1-C6-алкилом или необязательно замещенным фенилом;
R1 выбран из группы, включающей цис- или транс- -(CH2)m-CX3X4X5 и замещенное шестичленное ароматическое кольцо, где m равно 0;
X3 и X4, взятые вместе с атомом углерода, к которому они присоединены, образуют C3-C7-циклоалкил;
X5 является гидрокси- или карбоксигруппой, и замещенное шестичленное ароматическое кольцо замещено одним заместителем, выбранным из группы, включающей карбоксигруппу, -N(H)(SO2-X7), и одним или двумя заместителями, где каждый независимо выбран из группы, включающей атом фтора или хлора или C1-C4-перфторалкил, где X7 является CF3;
R2 представляет атом водорода,
при условии, что когда R3 является гидроксигруппой и R4 является атомом водорода, тогда R5 является -CH(OH)X10, и соединение формулы IA не может быть 2-гидрокси-1-тетралон-8-карбоновой кислотой.
Комментарии