Способ получения 4-гидроксибензальдегида и его производных - RU2194032C2
Код документа: RU2194032C2
Чертежи

Описание
Предметом настоящего изобретения является способ получения 4-гидроксибензальдегида и его производных.
В частности, изобретение касается получения 3-метокси-4-гидроксибензальдегида и 3-этокси-4-гидроксибензальдегида, называемых соответственно "ванилин" и "этилванилин".
В заявке на патент Франции 95/06186 описан способ получения 4-гидроксибензальдегида и, в частности, ванилина и этилванилина.
Описанный способ заключается в получении 3-карбокси-4-гидроксибензальдегида, затем в проведении декарбоксилирования названного соединения с получением таким образом 4-гидроксибензальдегида.
3-Карбокси-4-гидроксибензальдегид получают согласно патенту Франции
95/06186 из одного из соединений и их смесей, отвечающих, в частности, следующим формулам (IIа), (IIb), (IIc) и (IId), приводимым ниже:


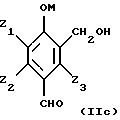

в которых М представляет собой атом водорода и/или катион металла группы (Iа) или (IIа) или катион аммония;
- Z1, Z2 и Z3, одинаковые или различные, представляют собой атом водорода, радикал алкил, алкенил, алкокси, гидроксиалкил, алкоксиалкил, циклоалкил, арил, гидроксильную группу, нитрогруппу, атом галогена, трифторметильную группу.
По запатентованному способу исходят из двухфункционального фенольного соединения, имеющего на ароматическом ядре две функциональные орто- и пара-группы, которые могут быть группой -СНО и/или группой -СН2ОН.
Вначале проводят селективное окисление группы, расположенной в орто-положении до карбоксильной группы; при этом группа, расположенная в пара-положении, окисляется самое большое до формильной группы. После удаления карбоксильной группы, расположенной в орто-положении, получают 4-гидроксибензальдегид.
Таким образом, получают преимущественно ванилин и этилванилин по селективному, а также очень конкурентоспособному с промышленной точки зрения способу, так как в нем используются недорогие реагенты.
Однако при этом способе трудно получить выход реакции (выражаемый по отношению к исходному фенолу) выше 70%, так как получение двухфункционального фенольного соединения с высоким выходом сопровождается высоким выходом побочного продукта, а именно бис-арилметана.
Продолжая свои поиски, заявитель открыл в заявке на патент Франции 96/12479, что возможно получить 4-гидроксибензальдегид из смеси однозамещенных фенольных соединений, одно из которых (А) является носителем формильной или гидроксиметильной группы в положении 2, а другое (В) - носителем формильной или гидроксиметильной группы в положении 4, путем избирательного окисления формильной или гидроксиметильной группы в положении 2 соединения (А) в карбоксильную группу, и при необходимости гидроксиметильной группы соединения (В) в положении 4 в формильную группу с получением таким образом смеси 2-гидроксибензойной кислоты и 4-гидроксибензальдегида, от которой этот последний отделяется.
Таким образом, используемая смесь фенольных соединений соответствует, в частности,
общей формуле (II):


в названных формулах (IIА) и (IIB):
- Y1 и Y2, одинаковые или различные, представляют собой одну из следующих групп:
группу -СНО,
группу -СН20Н,
- Z1, Z2 и Z3, одинаковые или различные, представляют собой атом водорода, радикал алкил, алкенил, алкокси, гидроксиалкил, алкоксиалкил, циклоалкил, арил, гидроксильную группу, нитрогруппу, атом галогена, трифторметильную группу.
Недостаток данного способа заключается в том, что для получения соединений формулы (IIА) или (IIB) гидроксиметилированием фенола важно действовать при низкой степени конверсии исходного фенола, что приводит к низкой производительности.
Для того чтобы иметь способ с высокой экономической эффективностью, который дает минимальное количество побочных продуктов и позволяет добиться высокой производительности, желательно внести усовершенствования в существующие способы.
Таким образом, был найден, и именно это является предметом настоящего изобретения, способ получения 4-гидроксибензальдегида и его производных,
отличающийся тем, что он заключается в избирательном окислении группы в положении 2 по отношению к гидроксильной группе в карбоксильную группу, присутствующую в фенольных соединениях смеси,
содержащей, по меньшей мере:
- одно фенольное соединение (А) - носитель формильной и/или гидроксиметильной групп в положении 2 и 4,
- одно фенольное соединение (В) - носитель
формильной или гидроксиметильной группы в положении 4,
- одно фенольное соединение (С) - носитель формильной или гидроксиметильной группы в положении 2,
при этом получают смесь,
содержащую 3-карбокси-4-гидроксибензальдегид, 4-гидроксибензальдегид, 2-гидроксибензойную кислоту, которую подвергают затем операции декарбоксилирования, позволяющей таким образом получить
4-гидроксибензальдегид и фенол, который может быть при необходимости рециркулирован.
Другим предметом изобретения является заявленная исходная смесь фенольных соединений как таковая, и смесь, получаемая после окисления.
Наконец, прочими предметами изобретения являются способы получения названных смесей.
В соответствии со способом по изобретению было открыто, что, исходя из смеси определенных выше исходных соединений, возможно провести одновременно внутримолекулярное (А) и межмолекулярное окисление (В+С), поскольку окисление до карбоксильной группы происходит предпочтительно на замещенной гидроксиметильной или формильной группе в орто-положении.
Способ по изобретению включает таким образом стадию окисления и стадию декарбоксилирования 3-карбокси-4-гидроксибензальдегида до 4-гидроксибензальдегида и 2-гидроксибензойной кислоты, позволяющие получить исходное фенольное соединение, которое может быть при этом рециркулировано; 4-гидроксибензальдегид при этом извлекается обычным способом.
Исходными субстратами, используемыми в способе по изобретению, являются смеси фенольных соединений, одно (А) - носитель формильной и/или гидроксиметильной групп в положении 2 и 4, второе (В) - носитель формильной или гидроксиметильной группы в положении 4 и последнее (С) - в положении 2.
Под "фенольным соединением" подразумевают любое ароматическое соединение, ароматическое ядро которого является носителем гидроксильной группы.
В нижеследующем описании настоящего изобретения под "ароматическим" подразумевают обычное понятие "ароматичности", такое как определенное в литературе, в частности Jerry MARCH, Advanced Organic Chemistry, 4-е издание, John Wiley and Sons, 1992, стр. 40 и последующие.
Таким образом, используют смесь (II) фенольных соединений, отвечающих, в частности, следующим формулам:



в этих формулах (IIА)-(IIС):
- Y1 и Y2, одинаковые или различные, представляют собой одну из следующих групп:
- группу -СНО,
- группу -СН2ОН,
- Z1, Z2 и Z3, одинаковые или различные, представляют собой атом водорода, радикал алкил, алкенил, алкокси, гидроксиалкил, алкоксиалкил, циклоалкил, арил, гидроксильную группу, нитрогруппу, атом галогена, трифторметильную группу.
Соединения, которые особенно подходят для
применения в способе по изобретению, отвечают формулам (IIА) - (IIС), в которых Z1, Z2 и Z3, одинаковые или различные, представляют собой один из следующих атомов или
групп:
- атом водорода,
- радикал алкил, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, предпочтительно от 1 до 4 атомов углерода, такой как метил, этил, пропил,
изопропил, бутил, изобутил, вторичный бутил, третичный бутил,
- радикал алкенил, линейный или разветвленный, имеющий от 2 до 12 атомов углерода, предпочтительно от 2 до 4 атомов углерода,
такой как винил, аллил,
- радикал алкокси, линейный или разветвленный, имеющий от 1 до 12 атомов углерода, предпочтительно от 1 до 4 атомов углерода, такой как радикалы метокси, этокси,
пропокси, изопропокси, бутокси, изобутокси, вторичный бутокси, третичный бутокси,
- радикал фенил,
- атом галогена, предпочтительно атом фтора, хлора или брома.
Настоящее изобретение не исключает наличие на ароматическом цикле заместителей другого типа, если только они не противодействуют реакциям способа по изобретению.
Настоящее изобретение применяется предпочтительно к соединениям формулы (IIА)-(IIС), в которых Z1 представляет собой атом водорода или радикал алкил или алкокси, линейный или разветвленный, имеющий от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода; при этом Z2 и Z3 представляют собой атом водорода; Y1 и Y2, одинаковые или различные, представляют собой формильную группу или гидроксиметильную группу.
В качестве предпочтительных примеров смесей фенольных соединений, которые могут быть использованы в способе по
изобретению, можно привести, в частности:
- о-гидроксиметилгваякол, р-гидроксиметилгваякол и 4,6-ди(гидроксиметил)гваякол,
- о-формилгваякол, р-формилгваякол и 4,6-диформилгваякол,
- о-гидроксиметилгветол, р-гидроксиметилгветол и 4,6-ди(гидроксиметил)гветол,
- о-формилгветол, р-формилгветол и 4,6-диформилгветол.
В соответствии со способом по изобретению исходят из смеси фенольных соединений, отвечающих предпочтительно формуле (II).
Соотношение в смеси каждого фенольного соединения зависит от способа их получения.
Следует уточнить, что предпочтительные смеси содержат:
- от 30 до 70 вес. % и более предпочтительно от 50 до 70 вес. % фенольного соединения (А),
- от 30 до 70 весовых % и
более предпочтительно от 50 до 70 весовых % фенольных соединений (В+С).
Для сведения следует отметить, что количества изомеров В и С находятся приблизительно в равных молярных количествах в их смеси.
Схема реакции способа по изобретению для облегчения понимания описания изобретения, без ограничения тем не менее объема изобретения этой схемой приводится в конце описания.
В указанных формулах (I) - (VI) схемы:
- Y1 и Y2, одинаковые или различные, представляют собой одну из следующих групп:
- группа
-СНО,
- группа -СН2ОН,
- М представляет собой атом водорода и/или катион металла группы (Iа) или (IIа) Периодической системы элементов или катион аммония,
- Z1, Z2, Z3 имеют приведенные выше значения.
В настоящем тексте ниже использованы данные Периодической системы элементов, напечатанной в Бюллетене Химического Общества Франции, 1 (1966 г.).
В соответствии с изобретением проводят избирательное окисление группы Y1 в положении 2 до карбоксильной группы фенольных соединений (А) и (С), отвечающих предпочтительно формулам (IIА) и (IIС), и гидроксиметильной группы, если такая есть, в положении 4 до формильной группы фенольных соединений (А) и (В), отвечающих предпочтительно формулам (IIА) и (IIB).
Окисление осуществляется молекулярным кислородом или содержащим его газом обычно в присутствии катализатора.
Предпочтительный способ окисления заключается в окислении смеси фенольных соединений формулы (II) в жидкой фазе с помощью молекулярного кислорода или содержащего его газа в водной среде, содержащей основный агент, в присутствии катализатора на основе металла M1, выбираемого среди металлов группы 1b и 8 Периодической системы элементов, содержащего при необходимости в качестве активаторов металлы, такие как кадмий, церий, висмут, свинец, серебро, теллур или олово.
В соответствии с изобретением было совершенно неожиданно установлено, что при повышении температуры и проведении реакции предпочтительно под давлением или при увеличении количества основания, присутствующего при окислении, проводится избирательное окисление группы Y1 в положении 2 до карбоксильной группы фенольных соединений (А) и (С), отвечающих предпочтительно формулам (IIА) и (IIС), а группа, расположенная в положении 4 в фенольных соединениях (А) и (В), отвечающих предпочтительно формулам (IIА) и (IIВ), при этом самое большое окисляется до формильной группы.
В способе по изобретению используют катализаторы на основе металла группы 1b и 8 Периодической системы.
В качестве примеров катализаторов на основе металла группы 8 Периодической системы можно назвать никель, рутений, родий, палладий, осмий, иридий, платину и их смеси. Что касается металлов группы 1b, предпочитают использовать медь.
Предпочтительно используют платиновые и/или палладиевые катализаторы в любых имеющихся формах, например, таких как: платиновая сажа, палладиевая сажа, оксид платины, оксид палладия или сам благородный металл, осажденный на различных подложках, таких как углеродная сажа, карбонат кальция, активированные оксиды алюминия или кремния или подобные материалы. Особенно подходят каталитические массы на основе углеродной сажи.
Используемое количество этого катализатора, выраженное по весу металла M1 по отношению к весу смеси фенольных соединений формулы (II), может колебаться от 0,01 до 10%, предпочтительно от 0,04 до 2%.
Для получения более детальных сведений о катализаторах можно обратиться к патентам США А-3 673 257, Франции А-2 305 420, Франции А-2 350 323.
Активатор можно выбрать среди всех тех, которые были упомянуты в приведенных выше патентах. Предпочтительно используют висмут, свинец и кадмий в виде свободных металлов или катионов. В последнем случае природа присоединенного аниона не имеет принципиального значения и можно использовать любые производные этих металлов. Предпочтительно используют металлический висмут или его производные.
Можно использовать органическое или неорганическое производное висмута, в котором атом висмута находится в степени окисления выше нуля, например равной 2, 3, 4 или 5. Остаток, связанный с висмутом, не имеет решающего значения при условии, что он отвечает этому требованию. Активатор может быть растворимым или нерастворимым в реакционной среде.
Примерами соединений активаторов, которые могут использоваться в способе по настоящему изобретению, являются: оксиды висмута; гидроксиды висмута; соли неорганических водородных кислот, такие как хлорид, бромид, иодид, сульфид, селенид, теллурид висмута; соли неорганических оксикислот, такие как сульфит, сульфат, нитрит, нитрат, фосфит, фосфат, пирофосфат, карбонат, перхлорат, антимонат, арсенат, селенит, селенат висмута; производные соли оксикислот переходных металлов, такие как ванадат, ниобат, танталат, хромат, молибдат, вольфрамат, перманганат висмута.
Другие подходящие соединения также являются солями органических алифатических или ароматических кислот, таких как ацетат, пропионат, бензоат, салицилат, оксалат, тартрат, лактат, цитрат висмута; фенаты, такие как галлат и пирогаллат висмута. Эти соли и фенаты также могут быть солями висмутила.В качестве других органических или неорганических соединений можно использовать бинарные комбинации висмута с такими элементами, как фосфор и мышьяк; гетерополикислоты, содержащие висмут, а также их соли; подходят также алифатические и ароматические висмутины.
В качестве особых примеров можно привести:
- в качестве оксидов: BiO, Bi2O3, Bi204, Bi2O5;
- в качестве гидроксидов: Вi(ОН)3;
- в качестве неорганических солей водородных кислот: хлорид висмута ВiС13, бромид висмута ВiВr3, иодид
висмута ВiI3, сульфид висмута Вi2S3, селенид висмута Bi2Se3, теллурид висмута Вi2Te3;
- в качестве неорганических
солей оксикислот: основный сульфит висмута Вi2(S03)3, Вi2O3, 5H2O; нейтральный сульфат висмута Bi2(S04)3; сульфат висмутила (BiO)HSO4; нитрит висмутила (BiO)NO2, 0,5H2O; нейтральный нитрат висмута Вi(NО3)3, 5H2O; двойной нитрат
висмута и магния 2Вi(NO3)3, 3Mg(NO3)2, 24H2O; нитрат висмутила (ВiO)NO3; фосфит висмута Вi2(РО3Н)3,
3Н2О; нейтральный фосфат висмута BiPO4; пирофосфат висмута Вi4(Р207)3, карбонат висмутила (Вi0)2СО3, 0,5H2O; нейтральный перхлорат висмута Вi(С104)3, 5H2O; перхлорат висмутила (BiO)C104; антимонат висмута BiSb04; нейтральный арсенат висмута
Вi(Аs04)3; арсенат висмутила (BiO)As04, 5H2O; селенит висмута Вi2(Sе03)3;
- в качестве производных солей
оксикислот переходных металлов: ванадат висмута BiV04, ниобат висмута BiNbO4, танталат висмута BiTaO4, нейтральный хромат висмута Вi2(Сr04),
бихромат висмутила [(BiO)2]2Cr2O7, кислый хромат висмутила H(BiO)Cr04, двойной хромат висмутила и калия K(BiO)Cr04, молибдат висмута
Вi2(Мо04)3, вольфрамат висмута Bi2(W04)3, двойной молибдат висмута и натрия NaBi(МоО4)2, основный перманганат
висмута Bi202(OH)Mn04;
- в качестве солей органических алифатических и ароматических кислот: ацетат висмута Вi(С2Н3О2)3; пропионат висмутила (BiO)C3H5О2; основный бензоат висмута C6H5CO2Bi(ОН)2; салицилат висмутила C6H4CО2(BiO)(ОН); оксалат висмута (С2О4)2Вi2; тартрат висмута Вi2(С4Н4О6)3, 6Н2О; лактат висмута (С6Н9О5)OBi, 7Н20; цитрат висмута C6H5О7Bi;
- в качестве фенатов: основный галлат висмута
C7H7О7Bi, основный пирогаллат висмута С6Н3(ОН)2(ОВi)(ОН).
В качестве других неорганических или органических соединений подходят также: фосфид висмута BiP; арсенид висмута Вi3As4; висмутат натрия NаВiO3; висмут-тиоциановые кислоты Н2[Bi(BNS)5], Н3 [Вi(СNS)6] и их соли натрия и калия; триметилвисмутин Вi(СН3)3, трифенилвисмутин Вi(С6Н5)3.
Производные висмута, которые предпочтительно используются для проведения способа по изобретению, таковы: оксиды висмута; гидроксиды висмута; соли висмута или висмутила неорганических водородных кислот; соли висмута или висмутила неорганических оксикислот; соли висмута или висмутила органических алифатических или ароматических кислот; и феналаты висмута или висмутила.
В группу активаторов, которые особенно подходят для осуществления изобретения, входят: оксиды висмута Вi2О3 и Bi2O4; гидроксид висмута Вi(ОН)3, нейтральный сульфат висмута Вi2(SО4)3; хлорид висмута ВiСl3; бромид висмута ВiВr3; йодид висмута ВiI3; нейтральный нитрат висмута Bi(NО3)3, 5Н2О; нитрат висмутила ВiO(NО3); карбонат висмутила (ВiO)2СО3, 0,5Н2О; ацетат висмута Вi(С2Н3О2)3; салицилат висмутила С6Н4СО2(BiO)(ОН).
Используемое количество активатора, выражаемое количеством металла, содержащегося в активаторе по отношению к весу задействованного металла M1, может колебаться в широких пределах. Например, это количество может составлять всего 0,1% и может достигать веса привлеченного металла M1 и даже превосходить его без всяких проблем.
В частности, это количество выбирается таким образом, чтобы оно вносило в среду окисления от 10 до 900 частей на миллион от веса металла активатора по отношению к смеси фенольных соединений формулы (II). С этой целью высокие количества активатора порядка 900-1500 частей на миллион могут естественно использоваться, но без значительного дополнительного преимущества.
Согласно способу по изобретению окисление проводят в водной среде, содержащей в растворе основный агент, и в частности гидроксид аммония, основания щелочных или щелочно-земельных металлов, среди которых можно назвать такие гидроксиды, как гидроксид натрия, калия, лития и гидроксид бария; щелочные алканолаты, такие как метилат, этилат, изопропилат и третичный бутилат натрия или калия, карбонаты или бикарбонаты натрия или калия, и вообще соли щелочных или щелочно-земельных оснований и слабых кислот.
Таким образом, соединения формулы (III) - (V) могут быть полностью или частично преобразованы в соль в зависимости от используемого количества основного соединения. Отсюда следует, что в указанных формулах М - остаток основания - обычно обозначает атом водорода и/или катион металла группы (Iа) или (IIа) или катион аммония.
По экономическим соображениям используют гидроксид натрия или калия. Соотношение используемого неорганического основания может составлять от 0,5 до 10 моль, предпочтительно от 1 до 4 моль и еще более предпочтительно от 2 до 4 моль неорганического основания на моль фенольных соединений формулы (II).
Весовая концентрация смеси фенольных соединений формулы (II) в жидкой фазе обычно составляет от 1 до 60%, предпочтительно от 2 до 30%.
Практически, осуществление способа заключается в приведении в контакт с молекулярным кислородом или содержащим его газом, например воздухом, раствора, содержащего смесь фенольных соединений формулы (II), основного агента, катализатора на основе металла M1, при необходимости активатора в указанных выше соотношениях.
Можно работать при атмосферном давлении, но предпочитают работать при давлении 1-20 бар.
Затем смесь перемешивается при требуемой температуре до потребления количества кислорода, соответствующего количеству, необходимому для преобразования гидроксиметильной или формильной группы соединений (А) и (С) в карбоксильную группу, а гидроксиметильной группы, если таковая имеется, соединений (А) и (В) - в формильную группу.
Используемая температура реакции выбирается в зависимости от термостойкости получаемых соединений.
В соответствии с изобретением температура выбирается предпочтительно в интервале температур от 30 до 200oС, предпочтительно от 40 до 160oС.
Специалист выбирает температуру в зависимости от условий реакции (в частности, количества основания, типа металла M1, давления и перемешивания). В частности, было обнаружено, что чем ниже температура, тем большее количество основного агента выбирается для использования.
Можно уточнить в качестве примера предпочтительные условия в случае использования предпочтительных металлов, платины и палладия. Для платины, так как температура выбирается от 60 до 160oС, используемое количество основания преимущественно составляет от 1 до 3 моль на моль фенольных соединений формулы (II). В случае палладия температура может выбираться в интервале от 30 до 200oС, предпочтительно от 30 до 150oС, и для этого последнего интервала количество основания предпочтительно составляет 2-4 моль на моль фенольных соединений.
Таким образом, количество основания должно быть достаточным для окисления группы Y1 в орто-положении в карбоксильную группу. Оно определяется специалистом в зависимости от температуры и выбранного металла.
В конце реакции, которая продолжается предпочтительно от 30 мин до 6 ч, извлекают смесь, содержащую 3-карбокси-4-гидроксибензальдегид, отвечающий предпочтительно формуле (III), 4-гидроксибензальдегид, отвечающий предпочтительно формуле (V), и 2-гидроксибензойную кислоту, отвечающую предпочтительно формуле (IV); указанные соединения могут быть частично или полностью преобразованы в соль.
Затем после охлаждения, если оно имеет место, отделяют каталитическую массу от реакционной среды, например, фильтрованием.
На второй стадии способа по изобретению в конце реакции реакционную среду подвергают реакции декарбоксилирования.
Для этого подкисляют получаемую среду добавкой протоновой кислоты неорганического происхождения, предпочтительно соляной или серной кислотой или какой-либо органической кислотой, такой как, например, трифторметансульфокислота или метансульфокислота, до достижения рН ниже или равного 3.
Концентрация кислоты не имеет значения, и предпочтительно используют технические формы.
Реакционную среду нагревают до температуры, например, от 120 до 350oС, предпочтительно от 150 до 220oС.
Способ осуществляют предпочтительно при автогенном давлении реагентов.
В конце реакции реакционную смесь охлаждают до 20 - 80oС.
Получают двухфазную среду, состоящую из органической фазы, содержащей, с одной стороны, 4-гидроксибензальдегид, отвечающей предпочтительно формуле (VI), и исходное фенольное соединение формулы (I), а с другой стороны, водно-солевую фазу.
Разделяют органическую и водную фазы и извлекают 4-гидроксибензальдегид из органической фазы обычными методами отделения, например, экстракцией с помощью соответствующего растворителя (например, метилизобутилкетона или простого изопролилового эфира) или перегонкой.
В соответствии с усовершенствованным способом по изобретению исходят из смеси двух фенольных соединений, одно - с формильной или гидроксиметильной группой в положении 2 и 4, а два других - с формильной или гидроксиметильной группой в положении 2 или 4.
Таким образом, исходные смеси отвечают, в частности, приводимым ниже формулам:
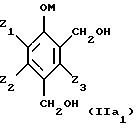
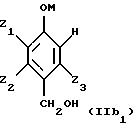

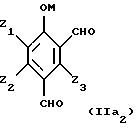

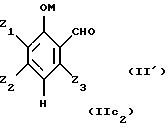
в этих формулах:
- М представляет собой атом водорода и/или катион металла группы (Iа) или (IIа) или катион аммония,
- Z1, Z2, Z3 имеют приведенные выше значения.
Смеси фенольных соединений, к которым можно применить способ по изобретению, получают по способу, который составляет другой предмет изобретения.
Так, смеси фенольных соединений формул (IIa1)-(IIc1) могут быть получены способом гидроксиметилирования фенола посредством его конденсации с формальдегидом или соединением генератором формальдегида в водной фазе в присутствии основания щелочного или щелочно-земельного металла таким образом, чтобы степень конверсии фенола составляла бы не более 95%, за которой следует при необходимости стадия окисления.
Точнее исходят из фенола, незамещенного в орто- и пара-положениях по отношению к гидроксильной группе, общей формулы (I):

в которой Z1, Z2, Z3 имеют приведенные выше значения.
Среди фенолов формулы (I), которые могут служить исходной точкой для синтеза соединений формулы (II), можно привести гваякол, гветол, 3-метоксифенол, 3-этоксифенол, 3-изопропоксифенол, 3-т-бутоксифенол, m-крезол, о-крезол.
Условия для проведения этой стадии гидроксиметилирования описаны в приводимом ниже уровне техники: см. , в частности, H.G.PEER Rec. Trav. Chim. Pays-Bas 19 825-835 (1960); патент Великобритании А-774 696; патент Великобритании А-751 845; ЕП-А-165; J. H.FREEMAN J. Am. Chem. Soc. 74 6275-6260 (1952) и 76 2080-2087 (1954); H.G.PEER Rec. Trav. Chim. Pays-Bas 78 851-863 (1959); H. EULER и др. Arkiv fur Chem. 13 1-7 (1939); P.CLAUS и др. Monath. Chem. 103 1178-11293 (1972).
Можно использовать формальдегид или любой генератор формальдегида, такой как, например, триоксан или параформальдегид, используемый в виде линейных полиформальдегидов с любой степенью полимеризации, с числом звеньев (СН20) предпочтительно от 8 до 100.
Формальдегид может использоваться в виде водного раствора, концентрация которого не имеет решающего значения. Она может изменяться от 20 до 50 вес. %; используют предпочтительно технические растворы, концентрация которых составляет около 30-40 вес. %.
Количество формальдегида, выраженное в числе моль формальдегида на моль фенола, может колебаться в широких пределах. Молярное отношение формальдегид/фенол может колебаться от 1,0 до 4,0 и предпочтительно от 1,0 до 2,5.
Количество основания, присутствующего в среде гидроксиметилирования, выражаемое числом моль основания на гидроксильную группу фенола, подлежащего гидроксиметилированию, может колебаться в широких пределах. В целом это отношение, изменяющееся в зависимости от типа основания, может колебаться от 1,0 до 4,0 и предпочтительно от 0,9 до 2,0. В качестве основания можно использовать те, которые были приведены выше для фазы окисления. Использование гидроксидов щелочных металлов в водном растворе является особенно удобным.
В целом стадия гидроксиметилирования проводится при температуре от 0 до 100oС и предпочтительно от 20 до 70oС.
Способ осуществляют предпочтительно при автогенном давлении реагентов во избежание возможных потерь параформальдегида, который может быть газообразным при рабочих температурах.
Предпочитают проводить реакцию в регулируемой атмосфере инертных газов, таких как азот или благородные газы, например аргон.
Продолжительность реакции легко определяется специалистом в зависимости от желаемой степени конверсии исходного фенола и учитывая необходимость свести к минимуму побочные продукты, такие как бис-арилметан. Чаще всего она составляет от 15 мин до 4 ч, предпочтительно от 1 до 3 ч.
Степень конверсии фенола контролируется посредством различных параметров (температура, продолжительность, количество реагентов). Преимущественно она составляет от 60 до 95%, предпочтительно от 80 до 95%.
С практической точки зрения реакцию легко осуществить, загружая в реактор фенол и формальдегид, при необходимости основание, затем нагревая реакционную смесь при перемешивании до требуемой температуры в течение периода времени, необходимого для завершения реакции.
Порядок загрузки реагентов не имеет решающего значения и может поэтому быть различным.
Получают смесь фенольных соединений формулы (IIa1)-(IIc1).
Соединения формулы (IIa2)-(IIc2) могут быть получены окислением гидроксиметилированных фенольных соединений формулы (IIa1)-(IIc1) посредством окисления молекулярным кислородом или содержащим его газом в щелочноводной фазе в присутствии катализатора на основании металла группы 8 Периодической системы элементов, предпочтительно платины и палладия, содержащего при необходимости в качестве активатора такие металлы, как кадмий, церий, висмут, свинец, серебро, теллур или олово. Такие способы были описаны в патентах США А-3 673 257, Франции А-2 305 420 и Франции А-2 350 323.
Если это необходимо, рН раствора доводится до значения, составляющего от 8 до 13 посредством возможной добавки основания щелочного или щелочно-земельного металла. Оптимальное значение рН зависит от типа гидроксиметилированных фенолов.
Температура реакции окисления составляет от 10 до 100oС и предпочтительно от 20 до 60oС.
В частности, способ по изобретению подходит для получения соединений формулы (IIa2) - (IIc2) из фенольных соединений формулы (IIa1) - (IIc1), получаемых на первой стадии, посредством молекулярного кислорода или содержащего его газа в присутствии катализатора на основе металла группы 8 Периодической системы элементов, содержащего при необходимости один из металлов, используемых в качестве активатора, без промежуточного разделения гидроксиметилированных фенольных соединений.
Оказывается очень выгодно с промышленной точки зрения для осуществления способа по настоящему изобретению
использовать соединения формулы (IIa2) - (IIc2), получаемые двухстадийным способом, включающим:
- гидроксиметилирование фенола в водной среде в присутствии основания
щелочного или щелочно-земельного металла посредством формальдегида или соединения генератора формальдегида, с получением смеси гидроксиметилированных фенольных соединений, одно гидроксиметилированное
- в положении 2 и 4, и два других гидроксиметилированных - в положении 2 или положении 4;
- и окисление без промежуточного разделения фенольных соединений, получаемых посредством
молекулярного кислорода или содержащего его газа в водно-щелочной фазе в присутствии катализатора на основе металла группы 8 Периодической системы элементов при необходимости в качестве активатора
одного из металлов, указанных выше.
Дополнительное значение способа по изобретению заключается в том, что он позволяет использовать смеси фенольных соединений, получаемых непосредственно на предыдущих стадиях гидроксиметилирования и, возможно, окисления.
Как указано выше, способ по изобретению особенно подходит для получения ванилина и этилванилина из смеси фенольных соединений, получаемых посредством гидроксиметилирования гваякола или гветола.
Так, ванилин можно получить, подвергая смесь фенольных соединений, 4, 6-ди(гидроксиметил)гваякола (А), р-гидроксиметилгваякола (В) и о-гидроксиметилгваякола (С), избирательному окислению гидроксиметильной группы в положении 2 соединений (А) и (С) до карбоксильной группы, а гидроксиметильной группы соединений (А) и (В) в положении 4 до формильной группы, с получением таким образом смеси 3-карбокси-4-гидрокси-5-метоксибензальдегида, ванилина и 2-гидрокси-3-метоксибензойной кислоты, из которой после декарбоксилирования получают ванилин и гваякол, который может быть рециркулирован.
Другой вариант заключается в том, что смесь фенольных соединений, 4,6-ди(формил)гваякола (А), р-формилгваякола (В) и о-формилгваякола (С), подвергают избирательному окислению формильной группы в положении 2 соединений (А) и (С) до карбоксильной группы, с получением таким образом смеси 3-карбокси-4-гидрокси-5-метоксибензальдегида, ванилина и 2-гидрокси-3-метоксибензойной кислоты, из которой после декарбоксилирования получают ванилин и гваякол, который может быть рециркулирован.
Что касается получения этилванилина по изобретению, то смесь фенольных соединений, 4,6-ди(гидроксиметил)гветола (А), р-гидроксиметилгветола (В) и о-гидроксиметилгветола (С) подвергают избирательному окислению гидроксиметильной группы в положении 2 соединений (А) и (С) до карбоксильной группы, а гидроксиметильной группы соединений (А) и (В) в положении 4 до формильной группы, с получением таким образом смеси 3-карбокси-4-гидрокси-5-этоксибензальдегида, этилванилина и 2-гидрокси-3-этоксибензойной кислоты, из которой после декарбоксилирования получают этилванилин и гветол, который может быть рециркулирован.
Другой вариант заключается в том, что смесь фенольных соединений, 4,6-ди(формил)гветола (А), р-формилгветола (В) и о-формилгветола (С), подвергают избирательному окислению формильной группы в положении 2 соединений (А) и (С) до карбоксильной группы, приводящему таким образом к получению смеси 3-карбокси-4-гидрокси-5-этоксибензальдегида, этилванилина и 2-гидрокси-3-этоксибензойной кислоты, из которой после декарбоксилирования получают этилванилин и гветол, который может быть рециркулирован.
Ниже приводят примеры осуществления изобретения. Эти примеры приводятся в качестве иллюстрации и не носят ограничительного характера.
В примерах определяют степень конверсии и получаемый выход.
Степень конверсии (ТТ) соответствует отношению числа преобразованного субстрата к числу задействованных моль субстрата.
Выход (RR) соответствует отношению между числом моль полученного продукта и числом моль задействованного субстрата.
Выход (RTванилин) соответствует отношению числа моль полученного ванилина к числу моль преобразованного гваякола на полимерной цепи.
В примерах использованы следующие сокращения:
- о-гидроксиметилгваякол = OMG
- р-гидроксиметилгваякол = PMG
- о-ванилин = 3-метокси-2-гидроксибензальдегид = OVA
- р-ванилин = 3-метокси-4-гидроксибензальдегид = PVA
- о-ванилиновая кислота =
2-гидрокси-3-метоксибензойная кислота = AOV
- р-ванилиновая кислота = 4-гидрокси-3-метоксибензойная кислота = APV
- 4,6-ди(гидроксиметил)гваякол = DMG
- 4,
6-(диформил)гваякол = DFG
- о-карбоксиванилин = OCVA
- 4,6-(дикарбокси)гваякол = DCG
ПРИМЕР 1
1 - Стадия конденсации
В реактор объемом 2 л, оборудованный
механическим перемешивателем и устройством регулирования температуры, загружают:
- 152 г гваякола,
- 246 г 30%-ного водного раствора формолина,
- 49,2 г гидроксида натрия
- и 672 г воды.
Реакционную среду выдерживают в течение 1 ч при температуре 45oС, затем охлаждают и подвергают анализу высокоточной жидкостной хроматографией.
Получают следующий баланс реакции:
- конверсия гваякола =90%
- выход о-гидроксиметилгваякола (OMG) =15%
- р-гидроксиметилгваякола (PMG) =18%
- 4,
6-ди(гидроксиметил)гваякола (DMG) =50%
- выходы (OMG+DMG+PMG) =83%
Сумма выходов полезных продуктов составляет 93%.
2 - Стадия окисления
При этом
реакционная среда разбавляется 1500 г воды и 148 г гидроксида натрия.
Реакционная среда затем подается в автоклав объемом 3,9 л, снабженный самовсасывающей турбиной.
Добавляют в эту реакционную среду 0,54 г триоксида висмута и 22 г палладиевого катализатора, осажденного на саже в количестве 3 вес. % металла.
Перемешивают со скоростью 1500 об/мин и повышают температуру реакционной среды до 45oС в азоте. Устанавливают давление 3 бар и подают в реакционную среду воздух с расходом 300 г/ч. В этих условиях реакционную среду выдерживают в течение 6 ч.
После этого реакционную среду охлаждают и доводят давление до атмосферного, затем отфильтровывают катализатор.
Затем реакционную среду подвергают анализу
высокоточной жидкостной хроматографией. Получают следующие выходы (для полной полимерной цепи):
- ТТ гваякол =92%
серия орто
- RR о-гидроксиметилгваякол (OMG)=0%
- RR ортованилин (OVA)=1%
- RR ортованилиновая кислота =14%
серия пара
- RR р-гидроксиметилгваякол (PMG) =0%
- RR ванилин (PVA) =16%
- RR параванилиновая
кислота (APV) =1%
серия ди
- RR 4,6-ди(гидроксиметил)гваякол (DMG) =0%
- RR 4,6-(диформил)гваякол (DFG) =1%
- RR ортокарбоксиванилин (OCVA) =47%
- RR 4,
6-(дикарбокси)гваякол (DCG) =10%
Сумма выходов "полезных" продуктов (гваякол+OAV+PVA+APV+OCVA+DCG) составляет 87%.
3 - Декарбоксилирование реакционной смеси
199,91 г
этой реакционной смеси нейтрализуют 16,69 г серной кислоты с концентрацией 92% и подают в автоклав объемом 300 мл, снабженный турбиной и системой регулирования температуры.
Реакционную среду нагревают в течение 3 ч до 175oС при автогенном давлении, после чего охлаждают и подвергают анализу методом жидкостной хроматографии.
Выход ванилина и
конверсия в гваякол составляют:
- ТТ гваякол =76%/исходный гваякол
- RR ванилин =61%/исходный гваякол, т.е. RT ванилин =80%
ПРИМЕР 2
1 - Стадия конденсации
В реактор объемом 2 л, снабженный механическим перемешиванием и устройством регулирования температуры, загружают:
- 133 г гваякола,
- 202 г водного раствора формалина с 30 вес. %,
- 145 г водного раствора гидроксида натрия с 30 весовыми %
- и 460 г воды.
Реакционную среду выдерживают в течение 0 ч 50 мин при 47oС, после чего охлаждают.
Добавляют 290 г водного раствора гидроксида натрия (30 вес. %).
Подвергают анализу высокоточной жидкостной хроматографией.
Получают
следующий баланс реакции:
- конверсия гваякола =97%
- выход о-гидроксиметилгваякола (OMG) =10%
- р-гидроксиметилгваякола (PMG) =12%
- 4,6-ди(гидроксиметил)гваякола
(DMG) =70%
- выходы (OMG+DMG+PMG) =92%
Сумма выходов RT полезных продуктов составляет 95%.
2 - Стадия окисления
Реакционную среду разбавляют 1230 г воды.
Затем реакционную среду подают в автоклав объемом 3,9 л, снабженный самовсасывающей турбиной.
Затем в эту реакционную среду добавляют 0,54 г триоксида висмута и 34,5 г катализатора, содержащего платину, осажденную на сажу в количестве 2 вес. % металла.
Перемешивают со скоростью 950 об/мин и повышают температуру реакционной среды до 70oС в атмосфере азота. Доводят давление до 4 бар и подают в реакционную среду воздух с расходом 200 г/ч. В этих условиях выдерживают реакционную среду в течение 5 ч. Затем охлаждают реакционную среду и доводят давление до атмосферного давления, после чего отфильтровывают катализатор.
После этого реакционную среду анализируют высокоточной жидкостной хроматографией.
Получают следующие выходы (для полной полимерной цепи):
- ТТ гваякол =97%
серия орто
- RR о-гидроксиметилгваякол (OMG) =0%
- RR ортованилин (OVA) =1%
- RR
ортованилиновая кислота =6%
серия пара
- RR р-гидроксиметилгваякол (PMG) =0%
RR ванилин (PVA) =9%- RR параванилиновая кислота (APV) =2%
серия ди
- RR 4,
6-ди(гидроксиметил)гваякол (DMG) =0%
- RR 4,6-(диформил)гваякол (DFG) =1%
- RR ортокарбоксиванилин (OCVA) =54%
- RR 4,6-(дикарбокси)гваякол (DCG) =6%
Сумма выходов
"полезных" продуктов (гваякол+OAV+PVA+APV+OCVA+DCG) составляет 80%.
3 - Декарбоксилирование реакционной смеси
150 г этой реакционной смеси нейтрализуют 15 мл серной кислоты с
10 моль/л и подают в автоклав объемом 300 мл, снабженный турбиной и системой регулирования температуры.
Реакционную среду нагревают в течение 2 ч до 175oС при автогенном давлении, после чего охлаждают и подвергают анализу жидкостной хроматографией.
Выход ванилина и конверсия в гваякол оставляют: ТТ гваякол =91%/исходный гваякол, RR ванилин =52%/исходный гваякол, т.е. RT ванилин =57%ы
Реферат
Изобретение относится к новому способу получения 4-гидроксибензальдегида и его производных, в частности касается получения 3-метокси-4-гидроксибензальдегида (ванилина) и 3-этокси-4-гидроксибензальдегида (этилванилина). Способ заключается в избирательном окислении, в присутствии основания и катализатора на основе палладия и/или платины, группы в положении 2 по отношению к гидроксильной группе до карбоксильной группы фенольных соединений смеси, содержащей, по меньшей мере, фенольное соединение (А) - носитель формильной и/или гидроксиметильной групп в положении 2 и 4, соответствующее формуле (IIA), фенольное соединение (В) - носитель формильной или гидроксиметильной группы в положении 4, соответствующее формуле (IIB), фенольное соединение (С) - носитель формильной или гидроксиметильной группы в положении 2, соответствующее формуле (IIС), причем в указанных формулах (IIA)-(IIС) Y1 и Y2, одинаковые или разные, представляют собой одну из следующих групп: группу -СHО, группу -СH2OH; Z1, Z2 и Z3, одинаковые или различные, представляют собой атом водорода, (С1-С4)- - алкил, (С1-С4)-алкенил или (С1-С4)-алкокси, прямые или разветвленные, фенил, гидроксильную группу, атом галогена, приводящем таким образом к получению смеси, содержащей 3-карбокси-4-гидроксибензальдегид, 4-гидроксибензальдегид, 2-гидроксибензойную кислоту, которая подвергается затем декарбоксилированию, позволяющему таким образом получить 4-гидроксибензальдегид и фенол, который может быть при необходимости рециркулирован. Экономически эффективный способ позволяет достичь высокой производительности при минимальном количестве побочных продуктов. 37 з.п.ф-лы.



Формула



где Y1 и Y2, одинаковые или разные, представляют собой одну из следующих групп: группу -СНО, группу -СН2ОН;
Z1, Z2 и Z3, одинаковые или разные, представляют собой атом водорода, (С1-С4)-алкил, (C1-C4)-алкенил или (C1-C4)-алкокси, прямые или разветвленные, фенил, гидроксильную группу, атом галогена,
приводящем, таким образом, к получению смеси, содержащей 3-карбокси-4-гидроксибензальдегид, 4-гидроксибензальдегид, 2-гидроксибензойную кислоту, которая подвергается затем декарбоксилированию нагреванием, позволяющему, таким образом, получить 4-гидроксибензальдегид и фенол, который может быть при необходимости рециркулирован.
о-гидроксиметилгваякола, р-гидроксиметилгваякола и 4,6-ди(гидроксиметил)гваякола,
о-формилгваякола, р-формилгваякола и 4,6-диформилгваякола,
о-гидроксиметилгветола, р-гидроксиметилгветола и 4, 6-ди(гидроксиметил)гветола,
о-формилгветола, р-формилгветола и 4,6-диформилгветола.


где М представляет собой атом водорода и/или катион металла группы (Iа) или (IIа) или катион аммония;
Z1, Z2, Z3 имеют значения, приведенные в пп. 1 и 2.

где Z1, Z2, Z3 имеют значения, приведенные в пп. 1 и 2.



где М представляет собой атом водорода, и/или катион металла группы (Iа) или (IIа), или катион аммония;
Z1, Z2, Z3 имеют значения, приведенные в пп. 1 и 2.


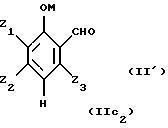
где М представляет собой атом водорода, и/или катион металла группы (Iа) или (IIа), или катион аммония,
Z1, Z2, Z3 имеют значения, приведенные в пп. 1 и 2.

в которой Z1, Z2, Z3 имеют приведенные выше значения.
Комментарии