Способ получения фенола - RU2430082C2
Код документа: RU2430082C2
Чертежи
Описание
Настоящее изобретение относится к способу получения фенола, точнее к способу, в котором продукт, полученный катализируемым кислотой расщеплением гидропероксида кумола, с помощью перегонки разделяют на фракцию, содержащую фенол, и водную фракцию, содержащую гидроксиацетон, в котором указанную водную фракцию обрабатывают окислительным реагентом в присутствии основания и получают щелочную водную среду с уменьшенным содержанием гидроксиацетона.
Уровень техники
Способ получения фенола из кумола хорошо известен. В этом способе кумол сначала окисляют кислородом воздуха в гидропероксид кумола. Эту стадию обычно называют окислением. На второй стадии реакции, так называемом расщеплении, гидропероксид кумола расщепляют на фенол и ацетон с использованием в качестве катализатора сильной неорганической кислоты, например серной кислоты. Затем продукт этой второй стадии реакции, так называемый продукт расщепления, фракционируют с помощью перегонки.
Требования к чистоте выпускающегося на рынок фенола становятся все более строгими. Поэтому для экономически эффективной эксплуатации установки по производству фенола необходимо улучшить суммарный выход и селективность по искомому конечному продукту и примеси, образующиеся на описанных выше стадиях реакции, необходимо удалить как можно полнее при наименьших возможных потерях искомого конечного продукта, в особенности фенола и ацетона, при низких капитальных и переменных затратах, в особенности затратах энергии. Основными побочными продуктами, образующимися на стадиях окисления, являются диметилбензиловый спирт и ацетофенон. Ацетофенон удаляют из системы вместе с высококипящими веществами, оставшимися после перегонки. Диметилбензиловый спирт на стадии расщепления дегидратируется в альфа-метилстирол, который на стадии каталитического расщепления кислотой частично образует высококипящие димеры и кумилфенолы. Высококипящие вещества отделяют от фенола на стадии перегонки. Непрореагировавший альфа-метилстирол отделяют и гидрируют с получением кумола, который возвращают в реакцию. В зависимости от потребности рынка альфа-метилстирол также можно дополнительно очистить и продать, как ценный продукт. Поэтому в предшествующем уровне техники изучали, каким образом проводить стадию окисления, а также стадию расщепления, чтобы уменьшить образование этих высококипящих веществ, которое можно рассматривать, как прямые потери кумола. Например, эти методики расщепления описаны в US-A-4358618, US-A-5254751, WO 98/27039 и US-6555719.
Однако наряду с этими высококипящими веществами при расщеплении образуются другие компоненты, такие как, например, гидроксиацетон, 2-метилбензофуран и мезитилоксид. Эти так называемые микропримеси нелегко отделить от фенола с помощью перегонки. Гидроксиацетон является самым критически важным компонентом, поскольку его почти невозможно отделить от фенола с помощью обычной перегонки. Гидроксиацетон обычно также является микропримесью, концентрация которой в продукте, полученном на стадии расщепления, является самой высокой. Концентрация гидроксиацетона в продукте расщепления может составлять от 200 до 3000 мас.част./млн (массовых частей на миллион).
Поэтому в предшествующем уровне техники предпринимали значительные усилия для удаления и выделения гидроксиацетона из продукта, полученного на стадии расщепления (см., например, US 6066767, US 6630608, US 6576798 и US 6875898). Недостатком всех этих методик является то, что необходимо обработать большие объемы потока продукта расщепления. Кроме того, в US 6875898 большой объем потока продукта расщепления необходимо обработать окислительным реагентом, что может привести к большим затруднениям при обеспечении безопасности технологии.
До перегонки продукт расщепления нейтрализуют водным раствором основания, такого как фенолят натрия или каустическая сода. Затем продукт расщепления, который насыщен водой, обрабатывают с помощью перегонки. Хорошо известной методикой является отделение большей части гидроксиацетона с водной фазой, которую отделяют в первой дистилляционной колонне, в которой неочищенный фенол вместе с высококипящими веществами отбирают в качестве нижнего погона, как это описано в US 3405038 или в US 6657087.
По данным US 6657087 часть водной фазы, полученной после фазового разделения продукта, отбираемой из средней части первой дистилляционной колонны, отбрасывают, а остальную часть возвращают в первую дистилляционную колонну. В соответствии с нормами техники безопасности и охраны окружающей среды отброшенную часть водной фазы необходимо обработать, как сточные воды. Это значительно увеличивает технологические затраты. Кроме того, порцию воды, удаленной из системы, необходимо возместить чистой водой, что приводит к дополнительному расходу ресурсов. Таким образом, задачей настоящего изобретения является разработка способа получения фенола, в котором устранены недостатки предшествующего уровня техники; рассмотренные выше, и который обеспечивает эффективное удаление гидроксиацетона из потока неочищенного фенола при низких капитальных и переменных затратах.
Краткое описание изобретения
Эта задача решена с помощью способа получения фенола, включающего:
а) окисление кумола с образованием продукта окисления, содержащего гидропероксид кумола;
b) расщепление указанного продукта окисления с помощью кислотного катализатора с образованием продукта расщепления, содержащего фенол, ацетон, гидроксиацетон и примеси;
c) нейтрализацию и промывку указанного продукта расщепления водной щелочной средой с образованием нейтрализованного продукта расщепления;
d) разделение указанного нейтрализованного продукта расщепления с помощью, по меньшей мере, одной стадии перегонки с получением, по меньшей мере, фракции, содержащей фенол, и водной фракции, содержащей гидроксиацетон;
e) обработку указанной водной фракции окислительным реагентом в присутствии основания с получением щелочной водной среды с пониженным содержанием гидроксиацетона;
f) рециркуляцию, по меньшей мере, части указанной щелочной водной среды на стадии нейтрализации и промывки с); и
g) извлечение фенола из указанной фракции, содержащей фенол, полученной на стадии d).
По сравнению с описанием в US 6576798, обработать окислительным реагентом необходимо только небольшой объем водного потока, содержащего гидроксиацетон, полученного после разделения продукта расщепления, проведенного с помощью перегонки. Кроме того, по сравнению с экспериментальными данными, приведенными в US 6576798, остаточное количество гидроксиацетона в потоке неочищенного фенола при использовании способа, соответствующего настоящему изобретению, значительно снижено. Кроме того, по сравнению с описанием в US 6657087, без ухудшения качества неочищенного фенола с точки зрения содержания остаточного гидроксиацетона не отбрасывают содержащий гидроксиацетон водный поток, полученный при перегонке продуктов расщепления, который необходимо обработать, как сточные воды.
Подробное описание изобретения
В соответствии с настоящим изобретением большую часть гидроксиацетона, содержащегося в продукте расщепления, полученном катализируемым кислотой расщеплением гидропероксида кумола, предпочтительно более 90%, удаляют с водной фракцией, полученной при разделении нейтрализованного продукта расщепления с помощью, по меньшей мере, одной стадии перегонки. Водную фазу, содержащую гидроксиацетон, удаленную из продукта расщепления, обрабатывают окислительным реагентом в присутствии основания. При этом гидроксиацетон превращается в нейтрализованные продукты окисления, например соли соответствующего вещества, содержащего карбоксигруппы, что приводит к щелочной водной среде, обладающей сниженным содержанием гидроксиацетона. По меньшей мере, часть указанной щелочной водной среды используют для нейтрализации продукта расщепления.
В предпочтительном варианте осуществления водную фракцию, обработанную окислительным реагентом в присутствии основания, полностью рециркулируют на стадию нейтрализации и промывки продукта расщепления. Тем самым исключается какой-либо дополнительный поток сточных вод, образующийся при разделении нейтрализованного продукта расщепления путем перегонки.
В предпочтительном варианте осуществления на стадии нейтрализации и промывки с) смесь продукта расщепления и водной среды является гетерогенной и после стадии нейтрализации и промывки с) и до стадии разделения d) гетерогенную смесь подвергают фазовому разделению на водную фазу, содержащую, по меньшей мере, часть нейтрализованных продуктов окисления гидроксиацетона и любые дополнительные соли, образовавшиеся при нейтрализации продукта расщепления, и насыщенную водой органическую фазу, которую подают на стадию разделения d).
Затем все соли, включая нейтрализованные продукты окисления гидроксиацетона, удаляют из системы в виде единого потока сточных вод, образующегося после нейтрализации продукта расщепления.
В предпочтительном варианте осуществления основание прибавляют к водной фракции, полученной после обработки нейтрализованного продукта расщепления до окислительной обработки. Основание предпочтительно прибавлять в количестве, необходимом для доведения значения рН до более 8, предпочтительно от 10 до 12. В настоящем изобретении можно использовать любое растворимое в воде основание, но, предпочтительно, если основание выбрано из группы, включающей водный раствор NaOH и водные растворы феноксида. Особенно предпочтительно использовать водный раствор фенолята натрия, в котором концентрация фенолята натрия предпочтительно составляет от 5 до 50, более предпочтительно от 30 до 45 и наиболее предпочтительно от 40 до 45 мас.%. Такие растворы фенолята натрия обычно получают в виде технологического потока стандартной установки производства фенола, как результат одной или множества последующих стадий обработки. Применение технологического потока, полученного на каком-либо участке установки производства фенола, обладает тем преимуществом, что на стадию способа, соответствующего настоящему изобретению, не требуется вводить ни чистую воду, ни свежую щелочь. Кроме того, фенол, теряющийся в виде фенолята натрия на стадиях обработки стандартной установки производства фенола, тем самым рециркулируется в систему, что сводит к минимуму потери ценного вещества.
Окислительным реагентом, использующимся в способе, соответствующем настоящему изобретению, может быть любой подходящий окислительный реагент, который может превратить гидропероксид в продукты окисления. Предпочтительно, если окислительный реагент выбран из группы, включающей пероксид водорода, кислород, воздух и любую смесь кислорода с азотом, из которых воздух является наиболее предпочтительным. Газ, содержащий кислород, например, воздух, можно растворить в водной фракции, полученной на стадии разделения d), как это указано выше, путем выдерживания системы под достаточно высоким давлением, или газ, содержащий кислород, диспергируют в жидкости в газожидкостном реакторе. Обе возможности взаимодействия водной фазы с газообразным окислительным реагентом, хорошо известны в предшествующем уровне техники.
Температура обработки указанной водной фракции окислительным реагентом может равняться от 20 до 150°С, предпочтительно от 80 до 120°С, и более предпочтительно от 90 до 110°С. Предпочтительно, если температуру и время пребывания на стадии обработки подбирают для превращения не менее 90% гидроксиацетона, содержащегося в водной фазе, полученной на стадии разделения, в нейтрализованные продукты окисления гидроксиацетона.
Водная фаза, полученная на стадии разделения d) в соответствии с настоящим изобретением, может содержать остаточные количества ацетона и фенола, но неожиданно было установлено, что во время окисления гидроксиацетона, ни один из этих компонентов не вступает в реакции с образованием нежелательных побочных продуктов. Вследствие рециркуляции водной фазы в систему производства фенола, несмотря на стадию окисления, исключены нежелательные потери ценных продуктов, таких как ацетон и фенол. В предпочтительном варианте осуществления настоящего изобретения содержание ацетона составляет менее 0,1 мас.%, предпочтительно, если оно находится в диапазоне мас.част./млн. Это можно обеспечить посредством достаточно высокой эффективности разделения на стадии d) или путем обработки водной фазы до окисления в устройстве удаления ацетона. При таком низком содержании ацетона не возникает затруднений при взаимодействии водной фазы с любым типом окислительного реагента, поскольку любая газовая фаза не является взрывоопасной. Это является одним из наиболее важных преимуществ по сравнению со способом, описанным в US 6576798 и в публикации Vasileva, I.I et al. 2000, Neftepereab. Neftekhim. Moscow, Russ. Fed., 12:34-38.
Другим важным преимуществом настоящего изобретения является то, что по сравнению со способами, описанными в US 6066767, US 6630608, US 6576798 и US 6875898, необходимо обработать лишь относительно небольшой объемный поток водной фазы, поэтому объем реактора, необходимый для стадии обработки е), невелик, что приводит к низким капитальным затратам.
Обычно в зависимости от содержания гидроксиацетона в продукте расщепления неочищенный фенол, полученный на стадии разделения d) в соответствии с настоящим изобретением, содержит от 50 до 400 мас.част./млн гидроксиацетона. Предпочтительно дополнительно обработать поток неочищенного фенола, полученного на стадии разделения d), чтобы дополнительно снизить содержание гидроксиацетона и других примесей.
Таким образом, в предпочтительном варианте осуществления настоящего изобретения поток неочищенного фенола, содержащий метилбензофуран и гидроксиацетон, обрабатывают в непрерывном режиме путем пропускания потока неочищенного фенола, по меньшей мере, через два последовательных реактора, содержащих кислую ионообменную смолу, при котором температура в последовательных реакторах снижается в направлении потока фенола, так что температура в первом реакторе в направлении потока фенола составляет от 100 до 200°С, и температура в последнем реакторе в направлении потока фенола составляет от 50 до 90°С, без стадии термического разделения между двумя последовательными реакторами.
Авторы настоящего изобретения установили, что при использовании множества последовательных реакторов, содержащих кислую ионообменную смолу, и, что важно, регулировании профиля температуры в последовательности реакторов, как указано выше, поток неочищенного фенола можно очистить, так что он будет обладать низким содержанием гидроксиацетона, а также метилбензофурана без предварительного удаления гидроксиацетона до взаимодействия с кислой ионообменной смолой и без требующей расхода энергии стадии перегонки, проводимой между двумя реакторами, содержащими кислую ионообменную смолу. Кроме того, неожиданно установлено, что, хотя необходимо использовать, по меньшей мере, два реактора, полная среднечасовая скорость подачи сырья в способе, соответствующем этому предпочтительному варианту осуществления, намного больше, чем в одностадийном способе, описанном в US 2005/0137429, и при этом полный объем реактора, требующийся в соответствии с настоящим изобретением, даже меньше, чем в одностадийном способе, описанном в US 2005/0137429.
Методику обработки потока неочищенного фенола можно легко включить в способ, соответствующий настоящему изобретению.
Неочищенный фенол, который можно эффективно очистить с помощью стадии обработки предпочтительного варианта осуществления настоящего изобретения, в качестве примесей содержит в основном гидроксиацетон, а также метилбензофуран. Концентрация гидроксиацетона может составлять до 1000 мас.част./млн и концентрация метилбензофурана может составлять до 200 мас.част./млн. Одним преимуществом настоящего изобретения является то, что гидроксиацетон, а также метилбензофуран можно эффективно удалить, даже если концентрация гидроксиацетона составляет более 260 мас.част./млн. Таким образом, можно успешно очистить поток неочищенного фенола, содержащий до 1000 мас.част./млн, предпочтительно от более 260 мас.част./млн до 1000 мас.част./млн гидроксиацетона и до 200 мас.част./млн, предпочтительно от 50 до 200 мас.част./млн метилбензофурана.
В дополнение к гидроксиацетону и метилбензофурану могут содержаться другие примеси:
мезитилоксид - до 1000 мас.част./млн,
2-фенилпропионовый альдегид - до 500 мас.част./млн,
метилизобутилкетон - до 500 мас.част./млн,
ацетофенон - до 500 мас.част./млн,
3-метилциклогексанон - до 500 мас.част./млн,
альфа-метил стирол - до 2000 мас.част./млн,
фенилбутены - до 1000 мас.част./млн.
Эти диапазоны концентрации охватывают встречающиеся концентрации этих компонентов в неочищенном феноле, который отделяют от ацетона, кумола и альфа-метилстирола, воды и высококипящих веществ путем перегонки до очистки на ионообменной смоле.
При взаимодействии потока неочищенного фенола с кислой ионообменной смолой гидроксиацетон и метилбензофуран взаимодействуют с высококипящими веществами. Мезитилоксид взаимодействует с фенолом с образованием высококипящих веществ и воды. В присутствии воды, которая также образуется по реакции между гидроксиацетоном и фенолом, часть мезитилоксида может разлагаться на ацетон на кислой ионообменной смоле. Ацетон может далее взаимодействовать с фенолом с образованием бисфенола А. Кроме гидроксиацетона и мезитилоксида существуют другие карбонилсодержащие компоненты, которые также могут содержаться в феноле в небольших количествах, такие как фенилпропионовый альдегид, метилизобутилкетон, ацетофенон и 3-метилциклогексанон. Кроме того, конечный фенол может содержать следы ненасыщенных углеводородов, таких как альфа-метил стирол и фенолбутены, которые являются нежелательными компонентами очищенного фенола. Как и содержащие карбонильные группы компоненты, ненасыщенные углеводороды образуют высококипящие вещества с фенолом при взаимодействии с кислыми ионообменными смолами. Обнаружено, что, даже если в неочищенном феноле содержатся эти дополнительные примеси, это не оказывает неблагоприятного влияния на превращение гидроксиацетона и метилбензофурана. Кроме того, превращение этих дополнительных примесных компонентов в высококипящие вещества всегда завершается, когда завершается превращение гидроксиацетона и метилбензофурана. Поэтому способ, соответствующий настоящему изобретению, обеспечивает превращение всех нежелательных примесей, содержащихся в неочищенном феноле, в высококипящие вещества, которые легко можно удалить из очищенного фенола на заключительной стадии перегонки после взаимодействия неочищенного фенола с кислой ионообменной смолой в соответствии со способом, соответствующим настоящему изобретению.
После взаимодействия неочищенного фенола с кислой ионообменной смолой можно обеспечить конечную концентрацию гидроксиацетона, составляющую менее 1 мас.част./млн, и концентрацию метилбензофурана, составляющую менее 20 мас.част./млн, предпочтительно менее 10 мас.част./млн. Как отмечено выше, все остальные примеси количественно превращаются в высококипящие вещества. Поэтому способ, соответствующий настоящему изобретению, применим для получения фенола высокой чистоты. Ряд реакторов, содержащих кислую ионообменную смолу, соединяют последовательно и поэтому количество различных значений температуры в соответствии с настоящим изобретением особо не ограничивается, но с учетом экономических соображений, т.е. капитальных затрат и переменных затрат, предпочтительно использовать 2-4 соединенных последовательно реактора и наиболее предпочтительно 2 соединенных последовательно реактора. Таким образом, в этом наиболее предпочтительном варианте осуществления, способ осуществляют при двух разных значениях температуры.
Кроме того, авторы настоящего изобретения установили, что дезактивация имеющейся в продаже ионообменной смолы очень хорошо коррелирует со степенью использования. Степень использования определяется как полное количество обработанного фенола, которое вступило во взаимодействие с ионообменной смолой за определенный промежуток времени. Для реактора идеального вытеснения непрерывного действия - это полное количество обработанного фенола в пересчете на площадь сечения реактора.
После достижения высокой степени использования активность катализатора составляет лишь несколько процентов от активности свежего катализатора. Неожиданно установлено, что температура, необходимая для компенсации дезактивации при постоянной среднечасовой скорости подачи сырья (ССПС), повышается пропорционально степени использования. По практическим соображениям максимальная температура равна 200°С, что необходимо для исключения термического разложения имеющихся в продаже ионообменных смол.
С другой стороны, установлено, что для потока фенола, содержащего метилбензофуран, а также значительные количества гидроксиацетона, например, до 200 мас.част./млн метилбензофурана и до 1000 мас.част./млн гидроксиацетона, в последнем реакторе температура должна быть ниже 90°С, чтобы обеспечить остаточное количество метилбензофурана, составляющее менее 20 мас.част./млн, предпочтительно менее 70°С, и чтобы обеспечить остаточное количество метилбензофурана, составляющее менее 10 мас.част./млн. По практическим соображениям температура должна быть не ниже 50°С, чтобы исключить слишком большой объем реактора даже при использовании свежего катализатора.
Одним преимуществом использования нескольких разных значений температуры для взаимодействия неочищенного фенола с кислой ионообменной смолой является то, что использованную или частично использованную кислую ионообменную смолу можно ввести во взаимодействие при относительно высокой температуре, так чтобы это, например, благоприятствовало превращению гидроксиацетона, но не превращению метилбензофурана, что приводит к тому, что даже в случае применения использованного или частично использованного катализатора вследствие высокой температуры можно поддерживать высокую активность уже отработанного катализатора. С другой стороны, при низком значении температуры свежий или частично использованный катализатор можно использовать при низких температурах, что благоприятствует превращению метилбензофурана, и, поскольку катализатор еще является относительно свежим, можно обеспечить высокую активность катализатора даже при низкой температуре. Поэтому можно обеспечить оптимальный баланс селективности взаимодействия с кислой ионообменной смолой и одновременно обеспечить оптимальную активность катализатора, что приведет к относительно высокой среднечасовой скорости подачи сырья и тем самым уменьшит объем катализатора, необходимого для обработки конкретного потока фенола.
Этот синергетический эффект оптимизации селективности катализатора по отношению к гидроксиацетону и метилбензофурану и активности катализатора в зависимости от степени дезактивации катализатора путем использования заявленного профиля температуры не описан в предшествующем уровне техники и не следует из него.
Другим преимуществом предпочтительного варианта осуществления настоящего изобретения является то, что если в непрерывной технологической схеме соединены последовательно несколько реакторов, включая, по меньшей мере, один запасной реактор, то полностью отработанный катализатор можно легко удалить из технологической линии. Реактор, содержащий наиболее отработанный катализатор, который нагрет до наивысшей температуры и находится в начале технологической линии, можно отсоединить от линии и присоединить реактор со свежим катализатором при наименьшем значении температуры, т.е. в конце линии. В реакторе, отсоединенном от линии, отработанный катализатор или заменяют свежим катализатором, или регенерируют на отдельной стадии способа для сохранения начальной активности свежего катализатора. Затем этот реактивированный реактор можно присоединить к линии при наименьшем значении температуры, когда от линии отсоединяют реактор, находящийся при наибольшем значении температуры, в котором катализатор дезактивировался до неприемлемого уровня. Это обеспечивает проведение непрерывной технологии, в которой эффективность очистки является приблизительно постоянной во времени, что обеспечивает получение продукта с почти постоянными характеристиками, что чрезвычайно важно для крупнотоннажного продукта, такого как фенол.
Предпочтительно использовать реакторы одинакового размера. Таким образом, в каждом положении на технологической линии ССПС для некоторого потока фенола одинаковы и не меняются при изменении положения реактора на линии. Можно без труда определить необходимую температуру в реакторах с ионообменными смолами разной активности.
Кроме того, для каждого значения температуры можно использовать множество реакторов, соединенных параллельно. Поэтому очень легко приспособить обработку к изменению пропускной способности. И в этом случае предпочтительно использовать реакторы одинакового размера и одинаковое количество реакторов для каждого значения температуры.
Кроме того, можно использовать теплообмен потоков фенола, проходящих через реакторы, для сведения расхода энергии к минимуму. Например, поток фенола можно пропускать через теплообменник, расположенный между первым реактором и последующим вторым реактором, и использовать более холодный выходной поток фенола из реактора, расположенного ниже по технологической линии от первого реактора, в качестве хладагента в теплообменнике. Этот вариант осуществления позволяет охлаждать поток фенола между двумя последовательными реакторами и одновременно поток фенола, выходящий из последнего реактора при самой низкой температуре, при его использовании в качестве хладагента для теплообменника нагревается, так что снижается потребление энергии на последующей стадии перегонки, предназначенной для удаления высококипящих веществ.
Кроме того, можно устанавливать дополнительные теплообменники между двумя последовательными реакторами с использованием обычных хладагентов, таких как вода, для установления необходимой температуры потока фенола.
В одном варианте осуществления настоящего изобретения в качестве реакторов используют удлиненные емкости и их предпочтительно располагать вертикально с подачей фенола сверху на дно реактора. Однако в вертикальных емкостях можно использовать поток, идущий снизу вверх, или использовать горизонтальные емкости.
В предпочтительном варианте осуществления настоящего изобретения реакторы содержат кислую ионообменную смолу в неподвижном слое. Предпочтительно, если скорость поверхностной жидкости в неподвижном слое ионообменной смолы составляет от 0,5 до 5 мм/с, более предпочтительно от 1,0 до 3,0 мм/с и еще более предпочтительно от 1,5 до 2 мм/с.
В настоящем изобретении в качестве катализатора можно использовать любую кислую ионообменную смолу. При использовании в настоящем изобретении термин "кислая ионообменная смола" означает катионообменную смолу в водородной форме, в которой ионы водорода связаны с активными центрами, и их можно удалить или вследствие диссоциации в растворе, или путем замены другими катионами. Активные центры смол обладают разной способностью притягивать разные ионы, и это селективное притяжение обеспечивает ионный обмен. Неограничивающие примеры подходящих кислых ионообменных смол включают группу сульфированных сшитых сополимеров дивинилбензола со стиролом, такие как, например, Amberlyst 16, выпускающийся компанией Rohm & Haas, K2431, выпускающийся компанией Lanxess, CT-151, выпускающийся компанией Purolite.
Другие подходящие смолы можно приобрести у таких изготовителей, как Lanxess, Rohm and Haas Chemical Company и Dow Chemical Company.
В предпочтительном варианте осуществления настоящего изобретения температура в первом реакторе в направлении потока фенола составляет не менее 100°С и температура в последнем реакторе в направлении потока фенола составляет менее 90°С, предпочтительно менее 70°С.
Температура в первом реакторе в направлении потока фенола составляет не более 200°С, предпочтительно не более 150°С и наиболее предпочтительно не более 120°С. Температура в последнем реакторе в направлении потока фенола составляет не менее 50°С.
Настоящее изобретение дополнительно иллюстрируется предпочтительными вариантами осуществления и примерами.
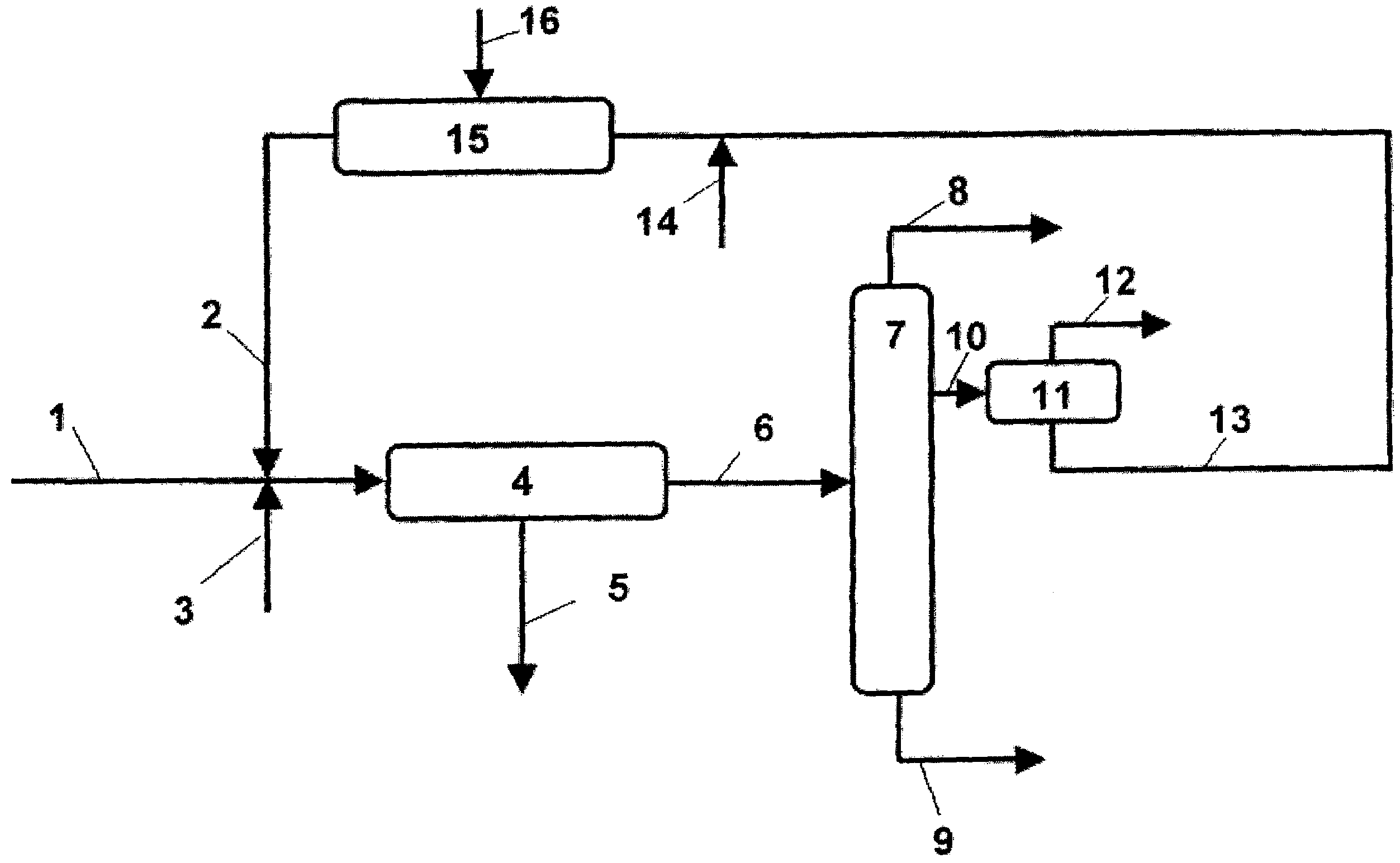
В соответствии с вариантом осуществления, представленным на чертеже, продукт расщепления, полученный катализируемым кислотой расщеплением гидропероксида кумола, подают по трубе 1 в отстойный барабан 4. До поступления продукта расщепления в отстойный барабан продукт расщепления смешивают с водной фазой, содержащей соли продуктов окисления гидроксиацетона и избыточное основание, предпочтительно фенолят натрия, который подают в систему по трубе 2. При необходимости для установки значения рН обычно в диапазоне от 4 до 8 по трубе 3 можно прибавить серную кислоту. Полученная смесь является гетерогенной и ее, обычно в отстойном барабане 4, разделяют на водную фазу 5, содержащую соли продуктов окисления гидроксиацетона, а также сульфат натрия, формиат натрия и соли других органических кислот, и на насыщенную водой органическую фазу. Водную фазу, в дополнение к указанным выше солям содержащую небольшие количества гидроксиацетона, по трубе 5 отбирают для последующей обработки. Насыщенную водой органическую фазу подают в дистилляционную колонну 7 по трубе 6. В дистилляционной колонне 7 органическую фазу разделяют на фракцию неочищенного фенола, отбираемую со дна дистилляционной колонны 7 по трубе 9, фракцию неочищенного ацетона, отбираемую из верхней части колонны 7 по трубе 8, и фракцию, содержащую воду, кумол, альфа-метил стирол и большую часть гидроксиацетона, отбираемую из средней части дистилляционной колонны. Указанную водную фазу по трубе 10 подают в отстойный барабан 11, в котором фракцию разделяют на водную фазу, содержащую гидроксиацетон, и органическую фазу, содержащую кумол и альфа-метилстирол. Органическую фазу по трубе 12 направляют на последующие стадии обработки. Водная фаза кроме гидроксиацетона содержит небольшие количества ацетона, предпочтительно менее 0,1 мас.%, некоторое количество фенола, а также небольшие количества органических кислот, таких как муравьиная кислота или уксусная кислота, и ее смешивают с основанием, подаваемым по трубе 14, для установления значения рН, превышающего 8, предпочтительно равного от 10 до 12. В предпочтительном варианте осуществления используют водный раствор фенолята натрия в диапазонах концентрации, указанных выше. Предпочтительно, если соотношение смешивания водной фазы, полученной из дистилляционной колонны 7, и водного раствора фенолята натрия находится в диапазоне от 1:0,05 до 1:1, более предпочтительно от 1:0,1 до 1:0,3. Таким образом получают гомогенную смесь водной фазы и раствора фенолята натрия, которую подают в реактор 15. Окислительный реагент, предпочтительно воздух, вводят в реактор по трубе 16 и температуру и время пребывания на стадии обработки подбирают для превращения не менее 90% гидроксиацетона, содержащегося в водной фазе, полученной на стадии разделения, в соответствующие нейтрализованные продукты окисления. Содержание гидроксиацетона в водной фазе, полученной на стадии перегонки, обычно составляет от 0,5 до 2 мас.%. После этого выходной поток из реактора 15, содержащий соли продуктов окисления гидроксиацетона, избыток фенолята натрия и остаточные количества гидроксиацетона, как это показано выше, используют для нейтрализации продукта расщепления.
В качестве альтернативы способу, описанному на фиг.1, также можно использовать режим работы первой колонны без отбора из средней части колонны и получить головной продукт, содержащий ацетон, кумол, альфа-метилстирол, воду и большую часть гидроксиацетона. Затем этот головной продукт можно разделить на последующей колонне чистого ацетона, как это показано в US 5510543, и получить нижний погон, содержащий кумол, воду и гидроксиацетон. Затем этот нижний погон можно разделить в отстойном барабане и обработать, как это описано выше при рассмотрении чертежа.
Примеры
Сравнительный пример
Продукт расщепления содержит 42 мас.% фенола, 26 мас.% ацетона, 25 мас.% кумола, 3,1 мас.% альфа-метилстирола, 200 мас.част./млн растворенной серной кислоты и 1500 мас.част./млн гидроксиацетона, а также другие органические компоненты. Все концентрации приведены в пересчете на полное количество органических компонентов (безводных). Кроме того, содержится 1 мас.% растворенной воды. Для насыщения продукта расщепления водой и образования водной фазы, содержащей соли, к продукту расщепления необходимо дополнительно прибавить 10 мас.% пресной воды в пересчете на полное количество продукта расщепления. Соли образуются вследствие нейтрализации при добавлении количества сульфата натрия, достаточного для установления рН водной фазы, равного около 6. В первой дистилляционной колонне воду отбирают в виде бокового погона, содержащего 90% гидроксиацетона, что приводит к концентрации гидроксиацетона в воде, равной примерно 1,23 мас.%. Неочищенный фенол в виде нижнего погона содержит 340 мас.част./млн гидроксиацетона. Воду отбирают из системы и направляют на последующую обработку, в том числе на биологическую обработку.
Пример
Продукт расщепления, указанный в сравнительном примере, смешивают с водой из реактора окисления гидроксиацетона. Вода содержит 0,1 мас.% остаточного гидроксиацетона и фенолят натрия. Полученная концентрация гидроксиацетона в продукте расщепления равна около 1590 мас.част./млн. Для установления значения рН, равного около 6, прибавляют некоторое количество серной кислоты. В первой дистилляционной колонне воду отбирают в виде бокового погона, содержащего 90% гидроксиацетона, что приводит к концентрации гидроксиацетона в воде, равной примерно 1,30 мас.%. Неочищенный фенол в виде нижнего погона содержит 360 мас.част./млн гидроксиацетона. Воду смешивают с 40 мас.% водным раствором фенолята натрия в массовом соотношении 1:0,1 и вводят во взаимодействие с чистым кислородом при 95°С. Степень превращения гидроксиацетона равна 92%, что приводит к остаточной концентрации гидроксиацетона, равной 0,1 мас.%. Воду полностью рециркулируют на стадию нейтрализации продукта расщепления.
Из сопоставления данных сравнительного примера и примера, приведенных в настоящем изобретении, видно, что исключен дополнительный поток сточных вод без ухудшения качества потока неочищенного фенола, характеризующегося концентрацией гидроксиацетона.
Реферат
Настоящее изобретение относится к способу получения фенола, включающему: а) окисление кумола с образованием продукта окисления, содержащего гидропероксид кумола; b) расщепление указанного продукта окисления с помощью кислотного катализатора с образованием продукта расщепления, содержащего фенол, ацетон, гидроксиацетон и примеси; с) нейтрализацию и промывку указанного продукта расщепления водной щелочной средой с образованием нейтрализованного продукта расщепления; d) разделение указанного нейтрализованного продукта расщепления с помощью, по меньшей мере, одной стадии перегонки с получением, по меньшей мере, фракции, содержащей фенол, и водной фракции, содержащей гидроксиацетон; е) обработку указанной водной фракции окислительным реагентом в присутствии основания с получением щелочной водной среды с пониженным содержанием гидроксиацетона; f) рециркуляцию, по меньшей мере, части указанной щелочной водной среды на стадии нейтрализации и промывки с) и g) извлечение фенола из указанной фракции, содержащей фенол, полученной на стадии d). Предлагаемый способ позволяет получить целевой продукт при эффективном удалении побочного продукта - гидроксиацетона. 24 з.п. ф-лы, 1 ил.
Формула
a) окисление кумола с образованием продукта окисления, содержащего гидропероксид кумола;
b) расщепление указанного продукта окисления с помощью кислотного катализатора с образованием продукта расщепления, содержащего фенол, ацетон, гидроксиацетон и примеси;
c) нейтрализацию и промывку указанного продукта расщепления водной щелочной средой с образованием нейтрализованного продукта расщепления;
d) разделение указанного нейтрализованного продукта расщепления с помощью, по меньшей мере, одной стадии перегонки с получением, по меньшей мере, фракции, содержащей фенол, и водной фракции, содержащей гидроксиацетон;
e) обработку указанной водной фракции окислительным реагентом в присутствии основания с получением щелочной водной среды с пониженным содержанием гидроксиацетона;
f) рециркуляцию, по меньшей мере, части указанной щелочной водной среды на стадию нейтрализации и промывки с); и
g) извлечение фенола из указанной фракции, содержащей фенол, полученной на стадии d).
Комментарии