Способ получения 3-(4-изобутил-2-метилфенил)пропаналя, используемого в парфюмерной промышленности - RU2708669C2
Код документа: RU2708669C2
Чертежи

Описание
Настоящее изобретение в целом относится к способам получения сырьевых материалов для парфюмерной промышленности и основных промежуточных соединений, применяемых в таких способах или полученных в процессе таких способов.
Соединения, имеющие характеристики запаха ландыша, очень востребованы на рынке в качестве парфюмерных ингредиентов. Такие соединения являются важными ингредиентами в композициях-базах с цветочным запахом и могут вести себя как гармонизирующие вещества во многих типах разработанных парфюмерных композиций. Соединения этого типа широко используют в средствах личной гигиены и средствах ухода за пожилыми, а также в парфюмерии с утонченным ароматом для образования приятных запахов или для маскирования неприятных запахов.
Прекрасным парфюмерным ингредиентом, который высоко ценится за свою ноту запаха ландыша, является Lilial™ или 3-(4-трет-бутилфенил)-2-метилпропаналь (CAS 80-54-6). Это соединение нашло широкое применение в парфюмерии с утонченным ароматом, а также в продуктах личной гигиены и бытовой химии. Однако целесообразность его применения является спорной с точки зрения последних исследований, поскольку оно демонстрирует токсическое воздействие на репродуктивные органы самцов крыс и собак. Никакого эффекта не было обнаружено в исследованиях на мышах, морских свинках и приматах, однако, тем не менее, по Всемирной гармонизированной системе (ВГС) классификации и маркировки химических веществ это соединение классифицировано как материал категории CMR 2. Для материалов категории CMR 2 необходимо установить, что количества, предложенные для применения, безопасны для потребителей. Из-за регулируемого статуса Lilial™ его заменяют другими парфюмерными ингредиентами.
В WO 2010105 873 решают проблему замены Lilial™, причем предложенное решение состоит в применении смесей известных ингредиентов, обычно присутствующих в палитре парфюмеров, для воссоздания характеристик, по существу аналогичных характеристикам Lilial™.
Аналогично, в WO 2009027957 предлагают решение, состоящее в смешивании сочетаний известных парфюмерных ингредиентов, имеющихся в палитре парфюмеров.
В WO 2013045301 также предлагают решение по замене Lilial™, которое состоит в подборе смесей ингредиентов, включая соединение Lilyflore™ и специфическое соединение инданилпропаналь в комбинации с другими вспомогательными ароматическими ингредиентами.
К настоящему моменту заявитель обнаружил новое соединение, которое можно применять в качестве парфюмерного ингредиента в парфюмерных композициях, духах с утонченным ароматом и потребительских продуктах для придания необходимого запаха ландыша указанным композициям, духам и продуктам. В частности, новое соединение обладает характеристиками запаха, который может восприниматься и признаваться парфюмерами как очень напоминающий запах Lilial™. Кроме того, к новому соединению не относятся какие-либо проблемы с токсичностью, которые концентрируются вокруг Lilial™. Раз так, данное новое соединение можно использовать в качестве простой замены Lilial™.
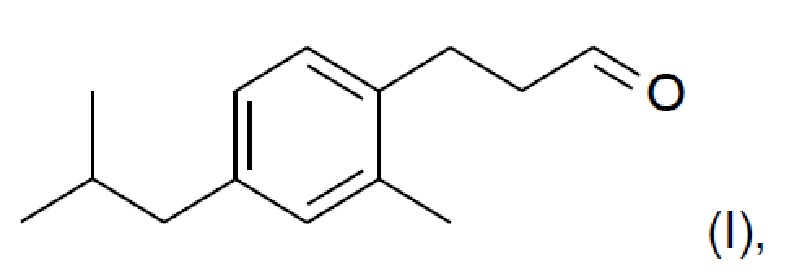
Новое соединение, которое описано в параллельно поданной патентной заявке РСТ/ЕР/2014059427 (включено в настоящую заявку во всей полноте посредством ссылки), характеризуется формулой (I):

Соединение формулы (I) обладает по существу аналогичными характеристиками запаха и свойствами при использовании, по меньшей мере настолько же хорошими, как Lilial™. Поэтому, и в противоположность предложениям уровня техники относительно замены Lilial™ на основе смесей известных ингредиентов, в настоящем изобретении предложена замена Lilial™ на основании одного соединения. Это имеет очевидное преимущество в том, что представлено эффективное с точки зрения стоимости решение по проблеме замены, которое, кроме того, упрощает процесс создания духов.
Регуляторные проблемы вокруг Lilial™ возникают из-за того, что это вещество подвергается ферментативному разложению в организме крыс и собак до трет-бутилбензойной кислоты (t-BBA). Известно, что трет-бутилбензойная кислота ингибирует синтез глюкозы и синтез жирных кислот in vitro (McCune et al, Arch Biochem Biophys (1982) 214 (1): 124-133).
Известно, что трет-бутилбензойная кислота является причиной нарушений работы яичек у самцов крыс (Hunter et al. Food Cosmet. Toxicol. 1965, 3: 289-298; Cagen et al. J. Am. Coll. Toxicol. 1989, 8 (5): 1027-1038).
К настоящему моменту заявитель обнаружил, что соединение формулы (I) не является восприимчивым к ферментативному разложению до соответствующего производного бензойной кислоты. Не связывая себя какой-то конкретной теорией, заявитель считает, что производное бензойной кислоты является основным промежуточным соединением, от которого происходит каскад метаболической активности, приводящей к репродуктивной токсичности у самцов и пониженному образованию спермы у самцов крыс. В частности, заявитель считает, что трет-бутилбензойная кислота и родственные разветвленные алкилзамещенные бензойные кислоты связываются с коферментом А в клетках крыс с образованием серосодержащего сложного эфира с этим кофактором. В свою очередь, считается, что данный серосодержащий сложный эфир ингибирует другие ферменты, которые ответственны за метаболизм жирных кислот в клетках крыс, и именно эта взаимосвязь с реакциями, зависимыми от СоА, приводит к наблюдаемой репродуктивной токсичности.
Неожиданное открытие, сделанное заявителем, о том, что арил-замещенные соединения-алканали, содержащие заместитель, например, метильный заместитель, в кольце в орто-положении к группе, содержащей альдегидную функциональную группу, не чувствительны к ферментативному разложению до их соответствующих производных бензойной кислоты, обеспечило прорыв в понимании сущности происходящих процессов, до сих пор не известных в уровне техники. Этот прорыв в понимании сущности происходящих процессов позволил заявителю разработать соединение формулы (I) и структурно родственные ему производные, тем самым расширив палитру парфюмерных ингредиентов, причем новые парфюмерные ингредиенты не только применимы сами по себе, но и подходят в качестве заменителей Lilial™.
Однако, несмотря на привлекательность соединения формулы (I) (и структурно родственных производных) в качестве парфюмерного материала и на его относительно простую химическую структуру, заявитель обнаружил, что его трудно и затратно получать, используя химические способы, которые нужно масштабировать до промышленного масштаба. Трудности происходят от того, что соединение представляет собой тризамещенное арильное соединение. С одной стороны, тризамещенные арильные исходные материалы дефицитны и дороги, но, с другой стороны, введение в ароматическое кольцо трех функциональных заместителей обычно требует длительных и сложных схем синтеза.
Следовательно, сохраняется необходимость в обеспечении экономичной и масштабируемой в промышленном масштабе схемы синтеза соединения формулы (I).
В процессе проведения своих исследований заявитель рассмотрел большое число возможных способов синтеза соединения формулы (I). Было обнаружено, что нельзя экономически эффективно ввести в кольцо метильный заместитель в процессе синтеза соединения (I). Метильный заместитель уже должен был присутствовать в легко доступном исходном материале. м-Ксилол представлял собой такой недорогой и легко доступный исходный материал, из которого легко можно было получить гомолог у одного из его метильных заместителей с получением основного промежуточного соединения (соединения II) в процессе образования соединения формулы (I). В это основное промежуточное соединение необходимо было ввести в кольцо функциональную группу, если нужно было осуществить способ синтеза, экономически пригодный в практическом плане и масштабируемый в промышленном масштабе.
Соответственно, в первом аспекте изобретения предложен способ региоселективного введения функциональной группы в положение (а) кольца алкилтолуола - соединения (II)

где заместитель R представляет собой изобутильную группу.
В другом аспекте настоящего изобретения предложен способ образования соединения формулы (I)

включающий стадию региоселективного введения функциональной группы в положение (а) в кольце 3-метил-1-изобутилбензола (соединения II):
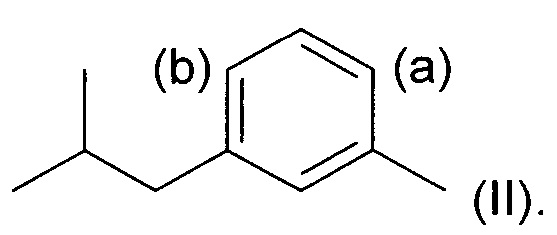
Под "региоселективностью" понимают, что процесс введения функциональной группы преимущественно направлен в положение (а) в кольце, а не в положение (b). Более конкретно, "региоселективность" означает, что соотношение (а):(b) составляет по меньшей мере 70:30, более предпочтительно по меньшей мере 80:20, еще более предпочтительно по меньшей мере 85:15, и еще более предпочтительно по меньшей мере 90:10.
В еще одном аспекте настоящего изобретения предложено применение 3-метил-1-изобутилбензола (соединение II) в качестве промежуточного соединения в синтезе соединения (I).
Как указано выше, заявитель обнаружил, что существенным для экономически пригодного в практическом плане и масштабируемого в промышленном масштабе способа синтеза соединения (I) является то, что метильная группа уже расположена в доступном исходном материале. Этот факт определяет то, что соединение (II) (3-метил-1-изобутилбензол) является основным промежуточным соединением в любом способе, и что введение функциональной группы в это соединение должно протекать с такого рода высокой региоселективностью.
Кроме того, идея получения соединения формулы (I) через основное промежуточное соединение, соединение (II), является неочевидной, так как в уровне техники озвучено предубеждение против ее осуществления. В частности, Rao et al. в Indian Journal of Chemistry vol. 16B, May 1978 вводит альдегидную функциональную группу в 3-метил-1-изобутилбензол (III е в указанной статье). Полученное соединение представляло собой 2-изобутил-4-метилбензальдегид (III g), демонстрируя, что изобутильная группа направляет альдегидную функциональную группу в орто-положение, а не в пара-положение к изобутильной группе. Другими словами, изобутильная группа не способствует высокой региоселективности в пара-положение относительно нее.
По собственным наблюдениям заявителя, которые суммированы в таблице 1 ниже, неожиданно и в противоположность Rao et al., при карбонилировании соединений 1-бутил-3-метилбензола в присутствии монооксида углерода (примерно 4 МПа (40 бар)) и трифлатной кислоты получили, что третичный бутильный и втор-бутильный заместители в кольце вообще не являются эффективными для селективного направления карбонилирования в пара-положение кольца относительно бутильного заместителя. В то же время н-бутильный заместитель, который по существу более эффективен, чем третичный бутильный и втор-бутильный заместители, является менее эффективным по сравнению с изобутильным заместителем.

Согласно настоящему изобретению, 3-метил-1-изобутилбензол (соединение II) можно получить из 3-изобутил-1-метилциклогекс-1-ена (соединение (Ia)) в соответствии с известными способами. В частности, (Ia) можно дегидрировать при пониженном давлении при использовании палладиевого катализатора, закрепленного на оксиде алюминия или углероде. Дегидрирование можно проводить при комнатной температуре или повышенных температурах, предпочтительно при 200°С.

Выбор функциональной группы, которую можно ввести в пара-положение (относительно изобутильной группы) 3-метил-1-изобутилбензола (соединение II) может существенно различаться.
Региоселективное введение бензальдегидной функциональной группы является предпочтительным воплощением изобретения. Введение этой функциональной группы может быть достигнуто несколькими способами. Один способ состоит в том, чтобы сначала хлорметилировать соединение (II), перед тем, как превратить 1-хлорметил-2-метил-4-изобутилбензол в 2-метил-4-изобутилбензальдегид. Однако по причинам экономической эффективности способа хлорметилирование соединения (II) не является предпочтительным воплощением настоящего изобретения.
Прямое введение бензальдегидной функциональной группы в кольцо путем карбонилирования является предпочтительным воплощением изобретения.

В частном воплощении изобретения, эту реакцию можно осуществлять в 6,8 молярных эквивалентах трифлатной кислоты в автоклаве при примерно 4-6 кПа (примерно 40-60 атмосферах) монооксида углерода. Карбонилирование обеспечит смесь региоизомеров, которая может содержать 70%, и более предпочтительно 80% (и даже более высокие количества) соединения (III).
Альтернативно, реакцию можно проводить в HF/BF3 в условиях, обычно известных в уровне техники. Эту реакцию часто называют химической реакцией Мицубиси.
Пример, иллюстрирующий химическую реакцию Мицубиси, описан в US 3962343, который включен в настоящую заявку посредством ссылки.
В другом воплощении по изобретению, соединение формулы (II) можно бромировать в условиях реакции, обычно известных в уровне техники

Рабочими условиями реакции являются обработка чистого соединения (II) эквимолярным количеством брома в присутствии 0,05 молярного эквивалента порошка железа при 10°С.
В еще одном воплощении изобретения соединение формулы (II) можно напрямую превратить в соединение формулы (I)

Условия реакции этого превращения в целом известны в уровне техники, которое может проходить по реакции соединения (II) с тетрахлоридом титана и соединением формулы (V) в дихлорметане при -70°С. Гидролиз енол-ацетатного интермедиата разбавленной серной кислотой дает до 40% соединения (VI) в виде смеси региоизомеров.

Соединение формулы (III) можно превратить в соединение (VI) в условиях реакции Мюллера-Конради-Пэро (Muller Conradi-Pieroh), в целом известных в уровне техники.
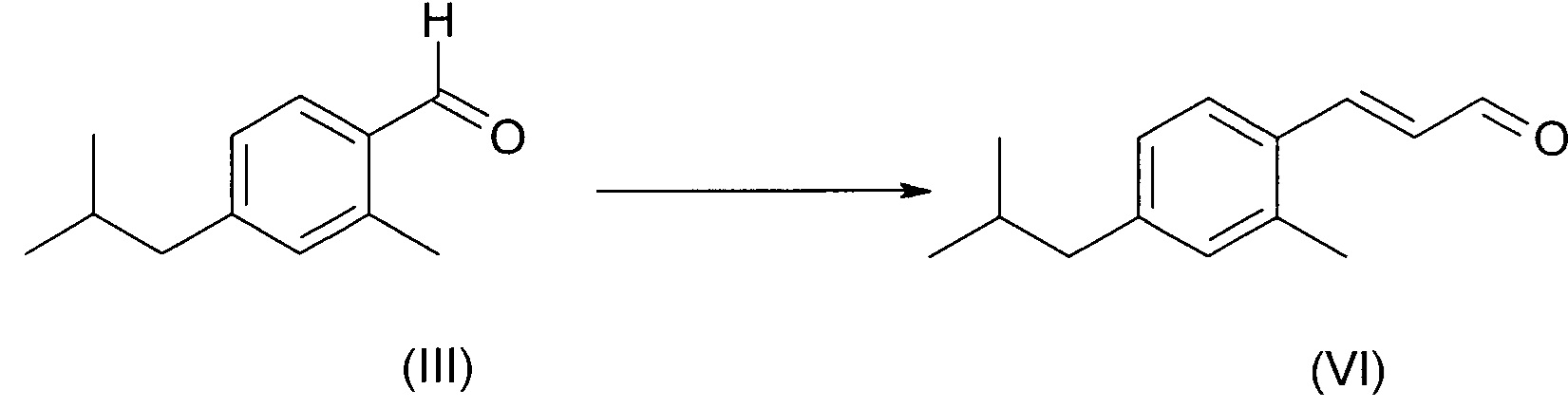
Специфические условия реакции включают превращение бензальдегида (III) в диметилацеталь посредством обработки триметил ортоформиатом с последующей реакцией с этилвиниловым эфиром в присутствии каталитических количеств эфирата трифторида бора при комнатной температуре. Промежуточные этоксиметоксиацетали подвергали гидролизу в присутствии 5%-ной HCl с получением соединения (VI).
Другое обладающее преимуществом сопутствующее действие, связанное с включением метильного заместителя в исходный материал, состоит в том, что, в отличие от, например, твердого 4-изобутилбензальдегида, применяемого в качестве исходного материала в синтезе хорошо известного парфюмерного ингредиента Bourgeonal®, присутствие метильного заместителя в соединении (III) переводит соединение в жидкость, что облегчает обращение с ним в промышленном масштабе.
Следовательно, соединение формулы (VI) можно гидрировать с получением соединения формулы (I). Условия гидрирования в целом хорошо известны в уровне техники и включают каталитическое гидрирование двойной связи в пропенальной боковой цепи над палладием на 5%-ном углероде при давлении 0,05 МПа (500 мбар).
Соединение (IV), описанное выше, можно подвергнуть дальнейшим превращениям в соответствии со схемами реакций:

Схема реакции (а) описывает превращение соединения (IV) в соединение (I). Эту реакцию можно осуществить посредством реакции Хека (Heck reaction) из соединения (IV) с аллиловым спиртом, катализируемой палладием в присутствии вторичного амина с последующим окислением полученной пропанольной боковой цепи в условиях, в целом известных в уровне техники, с получением соединения (I).
Схема реакции (b) описывает аналогичный способ взаимодействия аллилового простого эфира с соединением формулы (IV) в условиях реакции Хека с получением смеси соединений (VII) и (VIII). Специалисту будет понятно, что кислотный гидролиз смеси соединений (VII) и (VIII) приведет к смеси соединения (IX) и целевого соединения (I), и, таким образом, схема синтеза, проходящая через образование смеси, с этой точки зрения, не выглядит многообещающей.

Однако, к удивлению заявителя, оба соединения (VII) и (VIII) превращались в соединение (I) в условиях кислотного гидролиза, что предполагает, что соединение (VII) подверглось миграции двойной связи, вызванной кислой средой, с образованием соединения (VIII), перед превращением в соединение (I).
Соответственно, в другом аспекте настоящего изобретения предложен способ образования соединения формулы (I), включающий стадию образования смеси соединений (VII) и (VIII).
Если соединение (III) получают в виде смеси региоизомеров, в частности, в смеси с 2-изобутил-4-метилбензальдегидом (X), их трудно разделить путем перегонки. Однако отделение соединения (III) можно обеспечить путем перегонки соответствующих диалкилацеталей, предпочтительно соответствующих диметилацеталей (IIIa+Ха) или диэтилацеталей (IIIb+Xb). Это продемонстрировано путем сопоставления увеличенной разницы времен удерживания при разделении посредством газовой хроматографии, которые являются указанием на улучшенное разделение путем перегонки (Фиг. 1).
Следовательно, соответствующие диалкилацетальные соединения формулы (III), в частности, диэтилацетальное соединение формулы (III), которое представляет собой соединение (IIIb), является промежуточным соединением, пригодным для получения целевого соединения формулы (I) и является основой следующего аспекта настоящего изобретения.
Смесь региоизомеров (III) и (X) превращают на первой стадии а) в соответствующую смесь диэтилацеталей (IIIb+Xb) путем обработки триэтилортоформиатом и каталитическими количествами BF3-Et2O. На второй стадии b), после нейтрализации неочищенной реакционной смеси, осуществляют перегонку с получением диэтилацеталя (IIIb) в по существу чистой форме. Соединение формулы (IIIb) можно гидролизовать до соединения формулы (III) или превратить непосредственно в соединение формулы (VI) аналогичным способом, описанным выше.

Альтернативно, BF3-Et2O можно заменить п-толуолсульфоновой кислотой (p-TSA) и после удаления нежелательного изомера можно осуществить реакцию Мюллера-Конради, даже в присутствии p-TSA. Таким образом, образование ацеталя, перегонку и реакцию Мюллера-Конради можно осуществить способом в одном реакционном сосуде без выделения промежуточных соединений.
Соответственно, в еще одном аспекте настоящего изобретения предложен способ очистки и отделения соединения (III) и, тем самым, обеспечения чистоты продуктов, полученных в последующих реакциях, в частности, соединения формулы (I).
Далее приведен ряд примеров, которые служат для иллюстрации настоящего изобретения.
Пример 1: синтез 3-(4-изобутил-2-метилфенил)пропаналя
А) 3-изобутилтолуол (II)
Смесь свежеперегнанных 3- и 5-изобутил-1-метилциклогекс-1-енов (700 г, 4,6 моль) пропускали в вертикальном направлении через стеклянную трубку (2X50 см), заполненную 100 г палладия на пластинках из оксида алюминия (Aldrich, арт. 205745), и нагревали до 200°С. Циклогексен пропускали через колонку со скоростью 2 мл/мин при 3,2 кПа (32 мбар). Неочищенное соединение (II) конденсировали и собирали в приемник в нижней части колонки. Продукт, содержащий 90% соединения (II) и 10% 1-изобутил-3-метилциклогексана очищали путем перегонки (температура кипения 105°С, 8,8 кПа (88 мбар)) над колонкой со слоем 50 см с получением чистого соединения (II) (566 г, выход 83%).
Спектр ЯМР1Н (400 МГц, CDCl3): δ, м.д. (миллионные доли) = 7,24 (дд, J=7,58 Гц, 1Н), 7.05 (м, 3Н), 2,52 (д, J=7,07 Гц, 2Н), 2,41 (с, 3Н), 1,94 (м, 1Н), 0,99 (д, J=6,82 Гц, 6Н). Спектр ЯМР13С (400 МГц, CDCl3): δ, м.д.=141,7 (с), 137,6 (с), 130,0 (д), 128,0 (д), 126,4 (д), 126,2 (д), 45,5 (т), 30,3 (д), 22,5 (2 к), 21,5 (к). ГХ/МС (ЭИ) (газовая хроматография/масс-спектрометрия) (электронная ионизация): 148 (М+, 26), 106 (42), 105 (100), 103 (8), 91 (18), 79 (7), 77 (11), 43 (8), 41 (8), 39 (8).
В) 2-метил-4-изобутилбромид (IV)
Реактор продували азотом и добавляли соединение (II) (5440 г, 36,7 моль). Порошок железа (102 г, 1,8 моль) и иод (1 г) добавляли при перемешивании. Смесь охлаждали до 10°С и по каплям добавляли бром (5860 г, 36,7 моль) в течение периода 6 часов при 10°С. Во время добавления образовывался один молярный эквивалент бромоводородной кислоты, которую нужно было абсорбировать с помощью подходящих средств. По окончании добавления реакционную смесь перемешивали в течение 1 ч при комнатной температуре и затем промывали 10 л 2М-ного NaOH. Смесь дважды экстрагировали гексаном, затем органические фракции объединяли, промывали водой и насыщенным раствором соли и упаривали под вакуумом. Перегонка на короткой колонке (120°С, 0,8 кПа (8 мбар)) давала соединение (IV) (4580 г, выход 55%).
Спектр ЯМР1Н (400 МГц, CDCl3): δ, м.д.=7,46 (д, J=8,07 Гц, 1Н), 7.06 (с, 1Н), 6,87 (д, J=8,07 Гц, 1Н), 2,45 (д, J=7,09 Гц, 2Н), 2,42 (с, 3Н), 1,88 (м, 1Н), 0,95 (д, J=6,60 Гц, 6Н). Спектр ЯМР13С (400 МГц, CDCl3): δ, м.д.=141,0 (с), 137,2 (с), 132,0 (д), 131,7 (д), 128,3 (д), 122,0 (с), 44,7 (т), 30,1 (д), 22,9 (к), 22,3 (2к). ГХ/МС (ЭИ): 228 (М+, 20), 226 (М+, 20), 186 (21), 185 (97), 184 (23), 183 (100), 105 (19), 104 (14), 103 (17), 77 (13).
C) 2-метил-4-изобутилбензальдегид (III)
Магниевые стружки (171 г, 7 моль) помещали в реактор и заливали ТГФ. Добавляли небольшое количество (6 мл) соединения (IV) и посредством при легком нагревании инициировали реакцию. Оставшееся соединение (IV) (1589 г, 7 моль) смешивали с ТГФ (3 л) и по каплям добавляли при неактивном кипении с обратным холодильником (70-85°С) без применения внешнего нагревания. По окончании добавления смесь перемешивали при кипячении с обратным холодильником в течение дополнительного часа. Реакционную смесь охлаждали до 10°С и по каплям в течение 1 часа добавляли диметилформамид (566 г, 7,7 моль), поддерживая температуру ниже 30°С. Реакционную смесь перемешивали в течение 1 часа и затем быстро охлаждали ледяной HCl (2М). Смесь экстрагировали гексаном, органические фракции объединяли и промывали водой и насыщенным раствором соли. Раствор сушили над MgSO4 и упаривали под вакуумом. Перегонка над колонкой слоем 100 см (т. кип. 105°С, 0,25 кПа (2,5 мбар)) давала чистое соединение (III) (592 г, выход 48%).
Спектр ЯМР1Н (400 МГц, CDCl3): δ, м.д.=10,22 (с, 1Н), 7,71 (д, J=7,82 Гц, 1Н), 7,14 (д, J=7,58 Гц, 1Н), 7,04 (с, 1Н), 2,65 (с, 3Н), 2,50 (д, J=7,34 Гц, 2Н), 1,91 (м, 1Н), 0,92 (д, J=6,85 Гц, 6Н). Спектр ЯМР13С (400 МГц, CDCl3): δ, м.д.=192,3 (д), 148,2 (с), 140,5 (с), 132,6 (д), 132,3 (д), 132,2 (с), 127,1 (д), 45,4 (т), 30,1 (д), 22,4 (2к), 19,6 (к) ppm. ГХ/МС (ЭИ): 176 (М+, 53), 134 (100), 133 (38), 106 (14), 105 (70), 103 (14), 91 (37), 77 (19), 43 (30), 41 (14).
D) 3-(4-изобутил-2-метилфенил)пропен-2-аль (VI)
В реактор загружали соединение (III) (1 кг, 5,68 моль), метанол (400 мл) и триметилортоформиат (900 г, 8,49 моль). Реакционную смесь охлаждали до -10°С и добавляли соляную кислоту (37%, 1 г). Реакция была экзотермическая, температуре позволяли повышаться до 25°С, смесь перемешивали в течение 30 минут. Реакцию быстро охлаждали ацетатом натрия (20 г) и летучие вещества удаляли путем упаривания под вакуумом. Оставшийся ацеталь загружали во второй реактор и добавляли эфират трифторида бора (1 г), а этилвиниловый эфир (538 г, 7,5 моль) добавляли по каплям в течение 4 часов при поддержании температуры 25-30°С. Реакционную массу быстро охлаждали насыщенным раствором карбоната натрия (500 мл). Оставшиеся неочищенные метоксиэтоксиацетали гидролизовали водой (500 мл), содержащей соляную кислоту (37%, 50 г), при 90°С в течение 5 часов. Промежуточное соединение (VI) отгоняли на короткой колонке при 120°С.
Спектр ЯМР1Н (400 МГц, CDCl3): δ, м.д.=9,87 (с, 1Н), 7,78 (д, J=15,89 Гц, 1Н), 7,55 (д, J=8,31, 1Н), 7,06 (м, 1Н), 6,96 (м, 1Н), 6,68 (м, 1Н), 2,50 (с, 2Н), 2,49 (с, 3Н), 1,88 (м, 1Н), 0,94 (д, J=6,60 Гц, 6Н). Спектр ЯМР13С (400 МГц, CDCl3): δ, м.д.=194,0 (д), 150,4 (д), 145,5 (с), 137,8 (с), 131,9 (д), 130,3 (с), 128,7 (д), 127,5 (д), 126,7 (д), 45,3 (т), 30,1 (д), 22,40 (2к), 19,8 (к). ГХ/МС (ЭИ): 202 (М+, 8), 187 (42), 159 (31), 145 (100), 141 (13), 131 (30), 129 (20), 128 (22), 116 (18), 115 (34).
Е) 3-(4-Изобутил-2-метилфенил)пропаналь (I)
Отогнанное соединение (VI) загружали в автоклав и добавляли изопропанол (200 мл). Ненасыщенный альдегид гидрировали над палладием (5%) на углероде при давлении водорода 0,05 МПа (0,5 бар). Смесь отфильтровывали и упаривали под вакуумом. Неочищенный продукт очищали посредством перегонки над колонкой со слоем 50 см (т.кип. 116°С, 5 Па (0,05 мбар)) с получением продукта (I) (926 г, выход 80% в пересчете на соединение (III)).
Запах: цветочный, альдегидный, зеленый, нестойкий, лилиаль, влажный. Спектр ЯМР1Н (400 МГц, CDCl3): δ, м.д.=9,88 (т, J=1,5 Гц, 1Н), 7,07 (д, J=7,6 Гц, 1Н), 7,0-6,95 (м, 2Н), 2,98-2,93 (м, 2Н), 2,79-2,74 (м, 2Н), 2,46 (д, J=7,1 Гц, 2Н), 2,33 (с, 3Н), 1,95-1,82 (м, 1Н), 0,95 (д, J=6,6 Гц, 6Н). Спектр ЯМР13С (400 МГц, CDCl3): δ, м.д.=202,2 (д), 140,2 (с), 136 (с), 135,9 (с), 131,6 (д), 128,6 (д), 127,3 (д), 45,4 (т), 44,6 (т), 30,6 (д), 25,5 (т), 22,9 (к), 19,7 (к). ГХ/МС (ЭИ) (газовая хроматография/масс-спектрометрия) (электронная ионизация): 204 (М+, 23), 161 (100), 147 (26), 143 (49), 119 (84), 118 (34), 117 (33), 115 (33), 105 (59), 91 (36).
Пример 2: Синтез 1-(диэтоксиметил-4-изобутил-2-метилбензола (IIIb)
Смесь 85:15 соединений (III) и (X) (200 г, 1,13 моль) помещали в реактор и добавляли комплекс трифторида бора-ТГФ (1 г, 0,01 моль). Триэтилортоформиат (200 г, 1,35 моль) добавляли в течение 20 минут при 25-30°С при охлаждении ледяной баней. Темно-красную реакционную смесь перемешивали в течение 10 минут и затем добавляли триэтиламин (2 мл, 0,01 моль), и смесь, содержащую соединения (IIIb) и (Xb), перегоняли над колонкой размером 30 см, заполненной сетчатыми цилиндрами (2×3 мм) с получением чистого соединения (IIIb) (т.кип. 100°С, 0,26 кПа (2,6 мбар), 197 г, выход 69%).
Спектр ЯМР1Н (400 МГц, CDCl3): δ, м.д.=7,46 (д, J=7,83 Гц, 1Н), 6,96 (дд, J=7,58, 1,47 Гц, 1Н), 6,93 (с, 1Н), 5,54 (с, 1Н), 3,56 (м, 4Н), 2,42 (д, J=7,09 Гц, 2Н), 2,35 (с, 3Н), 1,84 (дт, J=13,39, 6,88х(2) Гц, 1Н), 1,22 (т J=7,09х(2) Гц, 6Н), 0,89 (д, J=6,60 Гц, 6Н). Спектр ЯМР13С (400 МГц, CDCl3): δ, м.д.=141,7 (с), 135,8 (с), 134,1 (с), 131,3 (д), 126,2 (2д), 100,2 (д), 61,3 (2 т), 45,1 (т), 30,2 (д), 22,4 (2к), 18,9 (к), 15,25 (2к). ГХ/МС (ЭИ): 250 (М+, 1), 206 (15), 205 (100), 177 (27), 162 (8), 134 (10), 105 (22), 103 (8), 91 (13), 57 (10), 29 (7).
Реферат
Изобретение относится к способу получения соединения формулы (I)включающему стадию региоселективного введения функциональной группы в диалкилбензольное соединениев котором соотношение соединения, содержащего соотношение соединения, содержащего функциональную группу в положении (а), к соединению, содержащему функциональную группу в положении (b), составляет по меньшей мере 70:30, и в котором заместитель R представляет собой изобутильную группу. 4 н. и 5 з.п. ф-лы, 1 ил., 1 табл., 2 пр.
Формула








Документы, цитированные в отчёте о поиске
Новые душистые соединения, метод их синтеза и применения
Комментарии