Способ удаления карбонильного соединения кобальта или родия из водного раствора 3-гидроксипропаналя - RU2203734C2
Код документа: RU2203734C2
Чертежи





Описание
Изобретение относится к селективному удалению металлического компонента из водного потока, содержащего термочувствительный компонент в растворе. Изобретение относится к производству 1,3-пропандиола. Один вариант данного изобретения относится к катализируемому кобальтом процессу получения 1,3-пропандиола, где кобальт эффективно удаляют из промежуточного водного потока.
1, 3-Пропандиол является важным промышленным химикатом, который может быть получен в двухстадийном процессе, где этиленоксид вначале гидроформилируют в органическом растворе в присутствии металлического катализатора, такого как карбонил кобальта или родия, получая 3-гидроксипропаналь. Промежуточное соединение 3-гидроксипропаналь экстрагируют водой под давлением и кобальтовый катализатор возвращают в цикл в реакцию гидроформилирования в органической фазе. Водный 3-гидроксипропаналь затем гидрируют до 1,3-пропандиола. 3-Гидроксипропаналь может быть направлен непосредственно в реактор гидрирования. Однако моноксид углерода, растворенный в воде, является ядом для большинства гетерогенных катализаторов гидрирования, когда небольшое количество металлического катализатора переходит в водную фазу во время экстракции 3-гидроксипропаналя. Для приемлемых выходов продукта катализатор должен быть удален из водного раствора 3-гидроксипропаналя в условиях, в которых не происходит разложения 3-гидроксипропаналя.
Поэтому задачей изобретения является создание способа, позволяющего эффективно удалять соединения кобальта и родия из водного раствора 3-гидроксипропаналя без существенного разложения 3-гидроксипропаналя. В одном из воплощений изобретения дополнительной целью является обеспечение водного потока 3-гидроксипропаналя для гидрирования, который по существу не содержит моноксида углерода и остаточных соединений металла.
Согласно изобретению карбонильные соединения кобальта или родия удаляют из водного раствора 3-гидроксипропаналя способом,
содержащим стадии:
(a) контактирования раствора 3-гидроксипропаналя с кислородом в кислотных условиях при температуре в интервале от 5 до 45oС с получением смеси продуктов
окисления, содержащей водный раствор 3-гидроксипропаналя, одно или несколько растворимых в воде соединений кобальта или родия и побочный продукт моноксид углерода;
(b) удаления образующегося
побочного продукта моноксида углерода из смеси продуктов окисления, и
(c) пропускания смеси продуктов окисления в контакте с кислотной ионообменной смолой, температуру которой поддерживают
менее 45oС, и удаления, по меньшей мере, части растворимых соединений металла из смеси продуктов окисления.
Такой способ полезен, например, при производстве 1,3-пропандиола из этиленоксида через промежуточный раствор 3-гидроксипропаналя, содержащий остаточный моноксид углерода и нерастворимые каталитические соединения кобальта или родия.
Подробное описание
изобретения
Способ согласно изобретению относится к способу удаления соединений кобальта и моноксида углерода из водного раствора 3-гидроксипропаналя в процессе получения 1,3-пропандиола
гидроформилированием этиленоксида до 3-гидроксипропаналя с последующим гидрированием 3-гидроксипропаналя до 1,3-пропандиола.
Раздельные или объединенные потоки ЕО (этиленоксид), СО и Н2 (синтез-газ) подают в резервуар гидроформилирования, который может быть реактором высокого давления, таким как колонна барботирования или резервуар с перемешиванием, работающий периодически или в непрерывном режиме. Подаваемые потоки контактируют в присутствии катализатора гидроформилирования, обычно карбонила металла, выбранного из карбонилов родия и кобальта. Катализатор гидроформилирования обычно присутствует в реакционной смеси в количестве в пределах от 0,01 до 1 мас.%, предпочтительно от 0,05 до 0,3 мас.%, от массы реакционной смеси гидроформилирования. Водород и моноксид углерода обычно должны быть введены в реактор в молярном отношении в пределах от 1:2 до 8:1, предпочтительно от 1:1 до 6:1.
Реакцию гидроформилирования проводят в условиях, эффективных для получения смеси продуктов реакции гидроформилирования, содержащей большую часть 3-гидроксипропаналя и меньшую часть ацетальдегида и 1,3-пропандиола, в то время как содержание 3-гидроксипропаналя поддерживают в реакционной смеси менее чем 15 мас. %, предпочтительно в пределах от 5 до 10 мас.%. (Чтобы обеспечить растворители, имеющие различные плотности, желательная концентрация 3-гидроксипропаналя в реакционной смеси может быть выражена в молярности, т. е. менее чем 1,5 М, предпочтительно в пределах от 0,5 до 1М). Как правило, катализируемую кобальтом реакцию гидроформилирования проводят при повышенной температуре менее чем 100oС, предпочтительно от 60 до 90oС, наиболее предпочтительно от 75 до 85oС, в случае катализируемых родием реакций гидроформилирования соответственно приблизительно на 10oС выше. Реакцию гидроформилирования обычно проводят при давлении в пределах от 0,69 до 34,47 МПа (7,0307-351,535 кг/см2), предпочтительно (для экономичности процесса) 6,89-24,13 МПа (70,307-246,074 кг/см2), более высокие давления предпочтительны для большей селективности.
Реакцию гидроформилирования проводят в жидком растворителе, инертном по отношению к реагентам. Под "инертным" подразумевается то, что растворитель не расходуется в течение реакции. Как правило,
идеальные растворители для процесса гидроформилирования должны солюбилизировать моноксид углерода, должны быть по существу не смешивающимися с водой и должны проявлять полярности от низкой до
умеренной, так как промежуточный продукт 3-гидроксипропаналь должен быть солюбилизирован до желательной концентрации, по меньшей мере, около 5 мас.% в условиях гидроформилирования, в то время как
значительное количество растворителя должно оставаться в виде отдельной фазы при экстракции водой. Под "по существу не смешивающийся с водой" подразумевается, что растворитель имеет растворимость в
воде при 25oС менее чем 25 мас. %, так что образует отдельную обогащенную углеводородом фазу при экстракции водой 3-гидроксипропаналя из реакционной смеси гидроформилирования.
Предпочтительным классом растворителей являются спирты и простые эфиры, которые могут быть описаны формулой
R2-O-R1 (1),
где R1 означает водород или
C1-20 линейный, разветвленный, циклический или ароматический углеводород или моно- или полиалкиленоксид, и R2 означает C1-20 линейный, разветвленный, циклический или
ароматический углеводород, алкокси или моно- или полиалкиленоксид. Наиболее предпочтительными растворителями гидроформилирования являются простые эфиры, такие как метил-трет-бутиловый эфир,
этил-трет-бутиловый эфир, диэтиловый эфир, фенил-изобутиловый эфир, этоксиэтиловый эфир, дифениловый эфир и диизопропиловый эфир. Смеси растворителей, такие как тетрагидрофуран/толуол,
тетрагидрофуран/гептан и третбутиловый спирт/гексан, также могут быть использованы для достижения желательных свойств растворителя. В настоящее время предпочтительным растворителем благодаря высоким
выходам 3-гидроксипропаналя, которые могут быть достигнуты при умеренных условиях реакции, является метил-трет-бутиловый эфир.
Для дальнейшего увеличения выходов при умеренных условиях реакции реакционная смесь гидроформилирования предпочтительно будет содержать промотор катализатора, чтобы ускорить скорость реакции. Предпочтительные промоторы содержат липофильные соли фосфония и липофильные амины, которые ускоряют скорость гидроформилирования без придания гидрофильности (растворимости в воде) активному катализатору. Как используется здесь, "липофильный" означает, что промотор имеет тенденцию оставаться в органической фазе после экстракции 3-гидроксипропаналя водой. Промотор будет обычно присутствовать в количестве в пределах от 0,01 до 1,0 моль на моль кобальта. Предпочтительными в настоящее время липофильными промоторами являются ацетат тетрабутилфосфония и диметилдодециламин.
При низких концентрациях вода служит в качестве промотора образования частиц желательного карбонильного катализатора. Оптимальное содержание воды для гидроформилирования в растворителе простом метил-трет-бутиловом эфире находится в пределах от 1 до 2,5 мас.%. Избыточные количества воды, однако, снижают селективность (3-гидроксипропаналь + 1,3-пропандиол) ниже приемлемых уровней и могут вызывать образование второй жидкой фазы.
Вслед за реакцией гидроформилирования смесь продуктов реакции гидроформилирования, содержащую 3-гидроксипропаналь, растворитель реакции, 1,3-пропандиол, катализатор, остаточный синтез-газ и небольшое количество побочных продуктов реакции, охлаждают и пропускают в резервуар экстракции, где водную жидкость, обычно воду и необязательный улучшающий смешиваемость растворитель добавляют для экстракции и концентрирования 3-гидроксипропаналя для последующей стадии гидрирования.
Жидкостная экстракция 3-гидроксипропаналя в воду может быть осуществлена какими-либо подходящими средствами, такими как смесители-отстойники, экстракционные колонны с насадкой или тарелками или контактные аппараты с вращающимися дисками. Количество воды, добавленной к смеси продуктов реакции гидроформилирования, как правило, должно быть таким, чтобы обеспечивать отношение вода-смесь в пределах от 1:1 до 1:20, предпочтительно от 1:5 до 1: 15. Экстракцию водой предпочтительно проводят при температуре в пределах от 25 до 55oС, причем предпочтительны более низкие температуры. Экстракция водой при 0,34-1,38 МПа (3,5154-14,0614 кг/см2) моноксида углерода при 25-55oС приводит к максимальному извлечению катализатора в органическую фазу.
Органическая фаза, содержащая реакционный растворитель и большую часть кобальтового катализатора, может быть рециркулирована с необязательной очисткой тяжелых концевых фракций из экстракционного резервуара в зону реакции гидроформилирования. Водный экстракт пропускают в зону гидрирования через колонну однократной равновесной перегонки и через ионообменную смолу для удаления остаточного кобальтового или родиевого катализатора. Большую часть остаточного синтез-газа удаляют из водного раствора однократной равновесной перегонкой. Обнаружено, однако, что даже очень малые количества моноксида углерода, остающиеся в растворе, могут влиять на эксплуатационные характеристики катализатора гидрирования, и предпочтительное воплощение способа данного изобретения обеспечивает удаление указанного остаточного моноксида углерода, как описано ниже, перед пропусканием водного раствора 3-гидроксипропаналя на гидрирование.
Водный раствор 3-гидроксипропаналя, переработанный способом согласно изобретению, как правило, будет содержать от 4 до 60 мас.% 3-гидроксипропаналя, обычно от 20 до 40 мас.% 3-гидроксипропаналя, и от 10 до 400 м.д. растворимых в воде и не растворимых в воде соединений кобальта или родия, таких как Co[Co(CO)4]2, СО2 (СО)8 и Rh6(CO)16.
В способе согласно изобретению слабокислый содержащий кобальт водный раствор 3-гидроксипропаналя приводят в контакт с кислородом в условиях, эффективных для окисления нерастворимых соединений кобальта до растворимых в воде соединений кобальта. Водный раствор 3-гидроксипропаналя может быть приготовлен достаточно кислым путем добавления органической или неорганической кислоты в количестве, эффективном для получения раствора, имеющего рН в пределах от 3 до 6, предпочтительно от 3 до 4. К подходящим кислотам относятся C1-4 органические кислоты. Альтернативно водная кислота может быть получена в качестве побочного продукта гидроформилирования этиленоксида в условиях, благоприятствующих образованию 3-гидроксипропионовой кислоты.
Окисление обычно может быть проведено путем введения содержащего кислород газа, такого как воздух, в водный раствор 3-гидроксипропаналя. Предпочтительная технология окисления предусматривает барботирование воздуха в направлении вверх через тарельчатую колонну, когда раствор 3-гидроксипропаналя, который должен быть обработан, течет в направлении вниз через колонну. Процесс проводят при температуре в пределах от 5 до 45oС и при атмосферном давлении. Длительности пребывания зависят от других переменных, но обычно находятся в пределах от 1 до 15 минут.
Использование приема барботирования для окисления нерастворимых соединений металла создает дополнительный эффект вытеснения моноксида углерода из водного раствора, особенно если в состав окислительного газа вводят инертный газ, такой как азот или диоксид углерода, для предотвращения образования воспламеняющихся смесей.
Некоторые типы смол эффективны для удаления кобальта из водного потока, включая соли щелочных металлов сильнокислых смол (например натриевые соли сульфированных полистиролов), соли щелочных металлов слабокислых смол и кислотные формы как сильно-, так и слабокислых смол. Оптимальных результатов в коммерческих процессах достигают, когда смола, выбранная для удаления кобальта или родия, имеет низкий потенциал разложения 3-гидроксипропаналя, может быть регенерирована одностадийным процессом и сильно адсорбирует частицы целевого металла. Этим требованиям наилучшим образом отвечает кислотная форма сильнокислой смолы, которая сильно адсорбирует окисленные частицы кобальта и легко регенерируется серной кислотой за одну стадию. Применение такой смолы в слое с коротким временем контакта в настоящее время предпочтительно для удаления металла. Подходящие смолы для удаления металла коммерчески доступны как смолы IR120, А1200 или А-15 от Rohm & Haas и смола М-31 от Dow Chemical.
Для того чтобы минимизировать разложение 3-гидроксипропаналя, температуру ионообменной смолы следует поддерживать ниже около 45oС и длительности пребывания следует сводить к минимуму, например, за счет применения укороченных слоев ионообменной смолы. Такие слои устроены так, чтобы заострять профиль зоны абсорбции/ионообмена к пункту, где пропускание по каналу не будет лимитировать эксплуатационные характеристики слоя.
Обнаружено, что ионообменная смола подвергается загрязнению остаточным ЕО в водном потоке. В соответствии с одним аспектом изобретения контактирование смолы с кислотой, такой как 10% серная кислота, очищает смолу и восстанавливает стабильный ионообмен. Кислота находится предпочтительно при повышенной температуре в пределах от 70 до 110oС. Длительности обработки от 0,5 до 2 часов являются обычно достаточными. Для экономичности способа желательно извлечение концентрированного кобальта или родия из смолы для превращения снова в каталитическую карбонильную форму.
Обработанный водный поток 3-гидроксипропаналя пропускают в зону гидрирования и подвергают реакции с водородом в присутствии катализатора гидрирования, чтобы получить смесь продуктов гидрирования, содержащую 1,3-пропандиол. Катализатором гидрирования предпочтительно служит никелевый катализатор на носителе в стационарном слое, такой как коммерчески доступный как Calsicat E-475SR и R-3142 от W.R. Grace.
Способ гидрирования данного изобретения может быть проведен в одну стадию, или в две, или несколько последовательных температурных стадий. В предпочтительном воплощении гидрирование проводят, как описано выше, при температуре в пределах от 50 до 130oС с последующей второй стадией, проводимой при температуре выше, чем температура первой стадии и в пределах от 70 до 155oС и затем, необязательно, третьей стадией при температуре выше чем 120oС для обратного превращения тяжелых концевых фракций в 1,3-пропандиол. В таком процессе зона гидрирования содержит ряды из двух или более отдельных реакторов.
Остаточный растворитель и вода экстрагента могут быть извлечены перегонкой в колонне и рециркулированы в процесс водной экстракции посредством дополнительной перегонки для отделения и устранения легких концевых фракций. Поток продукта, содержащий 1,3-пропандиол, может быть направлен в перегонную колонну для извлечения 1,3-пропандиола из тяжелых концевых фракций.
Пример 1
Отравление кобальтом
никелевого катализатора гидрирования.
Водные растворы промежуточного соединения 3-гидроксипропаналя (3-гидроксипропаналь) с добавленным кобальтом или без него гидрируют до 1, 3-пропандиола над никелевым катализатором на носителе (50% никель на диоксиде кремния - оксиде алюминия, 8•14 меш). В каждом опыте используют 28 г свежего катализатора, удерживаемого в кольцевой корзине для катализатора, размещенной в 500 мл реакторе с перемешиванием, снабженном вытяжным трубчатым импеллером для повторного диспергирования водорода из верхнего пространства в жидкость. В реактор загружают 320-340 г водного промежуточного продукта, богатого 3-гидроксипропаналем, затем в реакторе создают давление до 6,89 МПа (70,307 кг/см2) газообразным водородом. Потом реактор нагревают до желательной температуры реакции, 1-2 мл пробы периодически отбирают для анализа компонентов путем газовой хроматографии.
В опытах 1 и 2 водный раствор 3-гидроксипропаналя окисляют воздухом, содержащим кислород, путем барботирования через погруженную трубку в резервуарах с последующим ионообменом с сильнокислой смолой (сульфированным полистиролом). Газовая хроматография показывает, что 3-гидроксипропаналь быстро гидрируется до 1,3-пропандиола.
В опыте 3 водный раствор 3-гидроксипропаналя не барботируют воздухом и не обрабатывают в контакте с ионообменной смолой. Как результат 92 м.д. кобальта и остаточный моноксид углерода остаются в растворе. Скорость гидрирования 3-гидроксипропаналя до 1,3-пропандиола значительно снижена по сравнению со скоростью в опытах 1 и 2.
В опыте 4 водный раствор 3-гидроксипропаналя вначале окисляют путем барботирования воздуха с последующей ионообменной обработкой для удаления остаточного кобальта. Кобальт затем повторно добавляют в виде ацетата кобальта, чтобы получить раствор 3-гидроксипропаналя, содержащий 533 м.д. кобальта. Скорость гидрирования этого раствора также значительно ниже, чем для растворов 3-гидроксипропаналя, которые были обработаны для удаления кобальта.
Опыты 1-4 демонстрируют, что и моноксид углерода, и кобальт являются ядами для катализатора гидрирования и что окислительное десорбирование раствора 3-гидроксипропаналя (для удаления моноксида углерода) является недостаточным для предотвращения отравления катализатора гидрирования остаточным кобальтом.
Пример 2
Влияние окисления на удаление кобальта катионообменной смолой
Для последующих опытов водные растворы 3-гидроксипропаналя
получают в опытной установке непрерывного действия с небольшой производительностью, состоящей из двух последовательно соединенных 2 л реакторов гидроформилирования, работающих при 80oС и 10,
34 МПа (108,976 кг/см2) 4:1 Н2/СО (синтез-газ), через которые осуществляют циркуляцию растворителя МТВЕ при 80-100 мл/мин и реагент ЕО подают на первую стадию реакции при
скорости подачи 1,8-3,0 мл/мин. Растворимый катализатор гидроформилирования дикобальтоктакарбонил подают при 1200-2000 м.д. Непрореагировавший ЕО, промежуточное соединение 3-гидроксипропаналь и
катализатор выводят со второй реакторной стадии и диспергируют в донной части экстракционной колонны диаметром 5,08 см (2 дюйма), содержащей 7 решетчатых тарелок, размещенных с промежутками 5,08 см (2
дюйма). Воду (45oС) подают при 4,5-7 мл/мин как экстракционный растворитель непрерывной фазы. Экстракционную колонну эксплуатируют при давлении синтез-газа 8,27-9,65 МПа (84,368-98,430
кг/см2). Водный поток 3-гидроксипропаналя, выходящий со дна указанной колонны, обычно содержит 25-35 мас.% 3-гидроксипропаналя, 0,2-0,4 мас.% ЕО и 30-200 м.д. кобальта.
Указанный водный поток 3-гидроксипропаналя направляют в смотровое стекло диаметром 5,08 см (2 дюйма) и высотой 20,32 см (8 дюймов), обычно работающее при 1/2 полной метки (200 мл), где происходит мгновенное испарение жидкости за счет снижения давления приблизительно до атмосферного. Значительная часть синтез-газа, растворенного в растворе 3-гидроксипропакаля, высвобождается таким образом из раствора. Водный раствор, извлеченный со дна сосуда на контроле уровня, содержит небольшое количество остаточного синтез-газа.
Для опытов 5 и 6 из указанного дегазированного водного промежуточного потока 3-гидроксипропаналя, содержащего 69 м.д. кобальта, отбирают две пробы в склянки под атмосферой азота. Каждая склянка содержит 1 часть по объему сильнокислой (катионной) ионообменной смолы на основе сульфированного полистирола и 3 части по объему жидкой пробы. В опыте 6 склянку барботируют воздухом в течение 5 минут, а другую склянку держат закрытой, чтобы исключить воздух. Обе склянки вращают в течение 3 часов для перемешивания с последующим анализом колориметрическим методом (тиоцианатная дериватизация), чтобы определить кобальт. Обнаружено, что 5 м.д. кобальта остается в неокисленной пробе, в то время как содержание кобальта окисленной пробы снижается до 1 м.д.
Пример 3
Одновременное удаление синтез-газа и окисление кобальта
Для исследования непрерывного окисления и удаления кобальта после стадии дегазации, описанной в примере 2 выше, добавляют 10-тарельчатую стеклянную перегонную колонну Oldershaw диаметром 5,08 см (2
дюйма). Водный продукт течет вниз на тарелки колонны при 6-12 мл/мин с максимальной загрузкой тарелки 0,48 см (3/16 дюйма) и обычно 0,24 см (3/32 дюйма). Окисляющий и десорбирующий газ добавляют
восходящим потоком через колонну путем смешивания воздуха и азота в двух тотаметрах, производящих общий поток 0,0057-0,028 м3/ч при нормальных условиях при концентрациях кислорода 2-10
мол.%. Разбавление кислорода ниже его концентрации в воздухе желательно, чтобы обеспечить работу вне области воспламенения. В зависимости от условий работы может быть предусмотрено изменяющееся число
тарелок для жидкости.
Вторую колонну диаметром 5,06 см (2 дюйма) с насадкой высотой 15,24 см (6 дюймов) или сечением 60,96 см (24 дюйма) из 0,64 см (1/4 дюйма) перфорированной нержавеющей стали эксплуатируют вместо тарельчатой колонны Oldershaw для таких же опытов, что позволяет исследовать влияние удерживающей способности тарелок и времени пребывания на характеристики окисления и десорбции.
Слой 350 мл сильнокислой катионообменной смолы размещают ниже по потоку от десорбирующих колонн. Неполное окисление кобальта в материале, подаваемом к этому слою, может быть определено по появлению кобальта на выходе из ионообменного слоя. Пробы водного промежуточного материала, подаваемого в десорбер, и пробы, взятые на выходе из ионообменного слоя, анализируют на кобальт колориметрическим методом, описанным выше. Ионообменный слой набивают свежей смолой перед экспериментами для гарантии того, что прорыв кобальта из слоя не может быть приписан загрязнению смолы этиленоксидом.
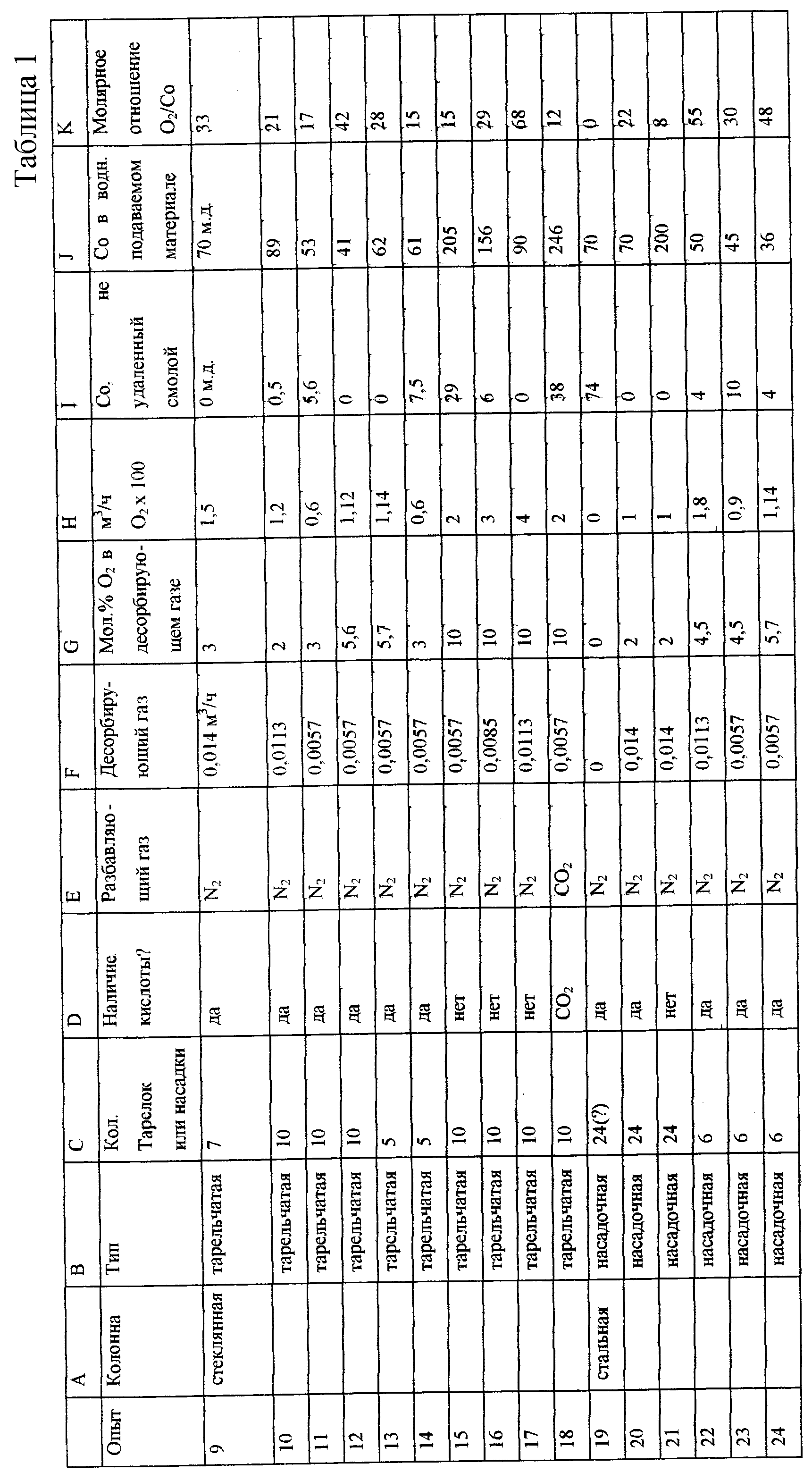
Результаты показаны в таблице 1. Столбец С описывает число тарелок стеклянной колонны, загруженных жидкостью во время испытания, или высоту насадочной колонны в дюймах для опытов, проводимых в насадочной колонне. Увеличение высоты насадочной зоны или увеличение числа тарелок, смоченных жидкостью, увеличивает площадь, доступную для контакта между жидкой и газовой фазами, и увеличивает время контакта между газовой и жидкой фазами.
Столбец D показывает, присутствует ли свободная кислота. Небольшое количество органической кислоты является побочным продуктом гидроформилирования ЕО. Указанная кислота соответствует по меньшей мере 10-кратному молярному избытку по отношению к присутствующему кобальту или 5-кратному избытку по отношению к количеству кислоты, необходимой для окисления кобальта до Со(ОАс)2. Если смесь растворителей гидроформилирования рециркулируют при температуре реакции без добавления ЕО, никакой кислоты не образуется. Более значительная фракция кобальта экстрагируется в водный поток в отсутствие кислоты. Данный случай соответствует отметке "нет" кислоты в столбце D. Столбец Е показывает разбавляющий газ, смешанный с воздухом, чтобы обеспечивать десорбирующую способность и производить работу вне области воспламеняемости. В большинстве случаев используют азот, хотя опыт 19 проводят с диоксидом углерода. Столбец F дает общий расход смешанного десорбирующего газа в м3/ч при стандартных условиях, в то время как столбец G дает мол.% кислорода в смешанном десорбирующем газе. Столбец Н дает их произведение, чтобы описать расход в м3/ч только кислорода. Столбец I показывает кобальт (м. д. ), выходящий из ионообменного слоя. Это неокисленный кобальт, который не был удален путем ионообмена. Столбец J показывает исходное количество кобальта в водном промежуточном продукте перед обработкой окислением и ионообменом. Столбец К показывает молярное отношение кислорода к кобальту в окислительном десорбере. Во всех случаях подают избыток кислорода.
Опыт 19 в таблице 1 поясняет, что в отсутствие десорбции и окисления очень малое количество кобальта удаляется последующим ионообменом. В опыте 20 по существу весь кобальт удаляют после десорбции и окисления водного промежуточного продукта в такой же колонне при описанных условиях. Опыты с 9 по 11 показывают влияние расхода десорбирующего газа при фиксированном молярном проценте кислорода для стеклянной тарельчатой колонны. Удаление кобальта увеличивается, когда возрастает интенсивность десорбции. Подобный результат наблюдается в отсутствие кислоты для опытов 15-17. Опыты 12 и 13 показывают влияние мол.% кислорода на эффективность окисления: способность окислять кобальт усиливается, когда концентрация (парциальное давление) кислорода в десорбирующем газе увеличивается. Сравнение опытов 11 и 14 показывает, что когда число тарелок ("ступеней") уменьшают, окисление кобальта является менее полным. К подобному заключению приходят при сравнении опытов 20 и 21 в насадочной колонне 60,96 см (24 дюйма) с характеристиками колонны 15,24 см (6 дюймов) в опытах 22-24. В меньшей колонне кобальт окисляется не полностью, несмотря на увеличенные концентрации кислорода по отношению к подобным опытам в более высокой 60,96 см (24 дюйма) насадочной колонне.
Опыты 12 и 15 показывают влияние кислоты. В присутствии кислоты окисление является полным, тогда как (опыт 15) окисление кобальта является неполным в отсутствие кислоты несмотря на более высокую концентрацию кислорода.
Твердые соединения кобальта осаждаются в отсутствие кислоты, побочного продукта гидроформилирования. В опыте 18 СО2 используют в качестве разбавляющего газа, образующего угольную кислоту при абсорбции водной фазой промежуточного продукта. Хотя, казалось бы, это не должно увеличивать степень окисления, образование твердых веществ исключается. Наблюдается, что добавление СО2 в стеклянную колонну солюбилизирует осадки кобальта, образующиеся во время работы в отсутствие кислоты с N2 в качестве разбавителя.
Результаты, суммированные в таблице 1, подтверждают, что и десорбция, и окисление содержащего кобальт водного потока необходимы для превращения кобальта в форму, удаляемую ионообменным слоем.
Пример 4
Влияние кислоты на окисление кобальта
Для исследования влияния кислоты на окисление кобальта проводят серию экспериментов, в которых продукт гидроформилирования экстрагируют
в присутствии ацетата натрия, результатом чего является экстракция кобальта, главным образом, как NaCo(CO)4. Это позволяет более высокой концентрации кобальта быть экстрагированной в водный
промежуточный продукт, так что его концентрация теперь подавляет концентрацию кислоты, образующейся в качестве побочного продукта гидроформилирования ЕО, и так что окисление аниона тетракарбонила
кобальта может быть отслежено инфракрасной спектроскопией (1890 см-1). Уксусную кислоту затем снова добавляют, чтобы довести общие эквиваленты карбоксильных кислотных частиц до эквивалентов
Со++ и Na+ с последующим окислением путем барботирования воздухом пробы конечной жидкости.
Таблица 2 показывает результаты окисления кобальта при 35oС с избытком карбоновой кислоты. Окисление продолжают по существу до полного завершения. Таблица 3 показывает результаты подобного исследования, в котором начальная концентрация кислоты не была избыточной. Окисление в этом случае, видимо, прекращается до полного завершения и продолжается только после добавления избытка кислоты. Эти результаты подтверждают то, что органическая кислота облегчает окисление кобальта.
Пример 5
Влияние остаточного моноксида углерода на окисление карбонила кобальта.
Дополнительные исследования окисления проводят в 50 мл реакторе с перемешиванием, снабженном ZnS (45oC) инфракрасным кристаллом для отслеживания на месте аниона тетракарбонила кобальта. Водный раствор диспропорционированного кобальтового катализатора для этого исследования готовят путем экстрагирования растворов МТВЕ Co2(CO)8 водой при повышенных температурах при низких парциальных давлениях моноксида углерода. Его разбавляют дистиллированной водой, чтобы получить исходный раствор с концентрацией кобальта 212 м.д. по массе без карбоновых кислот. В опыте 27 25 мл указанного раствора помещают в реактор, который снабжают стальной трубкой 0,08 см (1/32") для ввода газа, вставленной в дно реактора. Смесь нагревают до 40oС с перемешиванием и барботированием 100 мл/мин азота при давлении окружающей среды. Первоначальный спектр указанной смеси показывает анион тетракарбонила кобальта при 1908 см-1 и кластерный анион при 1979 см-1.
Окисление затем проводят путем переключения барботирующего газа на 3% кислород в азоте. Степень реакции может быть измерена по изменениям в инфракрасном спектре, которые происходят при окислении. Спектр показывает первоначальное прибавление аниона тетракарбонила кобальта вследствие расходования кластерного аниона. Затем анион расходуется, образуя карбонилы кобальта (0) (обнаружены и Со2(СО)8, и Co(CO)12). Эти карбонилы затем окисляются, образуя при этом "основной" карбонат кобальта. В этих условиях полное окисление кобальта достигается в течение 45 минут.
В опыте 28 25 мл исходного раствора помещают в 50 мл реактор (без приспособлений для барботирования газа). Раствор нагревают до 40oС в атмосфере азота с тщательным перемешиванием. Окисление начинают, создавая в сосуде давление до 5,273 кг/см2 2% кислородом в азоте. Чтобы смесь не обеднялась кислородом, атмосферу замещают, сбрасывая давление и повторно создавая давление в реакторе свежим 2% кислородом в азоте на 35 и 50 минутах от начала реакции. Изменения, происходящие во время указанного окисления, такие же, как в опыте 27, где выделяющийся моноксид углерода удаляют из реакционной смеси, за исключением того, что скорость реакции значительно меньше. Приблизительно через один час окисление усиливают, замещая смесь с 2% кислорода воздухом под давлением 0,52 МПа (5,273 кг/см2). Для полного окисления требуется дополнительно 25 минут. Указанные результаты демонстрируют, что свободный моноксид углерода, не десорбированный из потока водного промежуточного продукта, включающего моноксид углерода, связанный с кобальтом, как лиганд, будет подавлять окисление карбонила кобальта.
Пример 6
Регенерация
ионообменной смолы
Слой 83 г сильнокислой смолы А-1200 в форме геля (Rohm and Haas) используют для обработки 7-12 мл/мин водного промежуточного продукта, экстрагированного из продукта
гидроформилирования ЕО в течение одного месяца. Водный промежуточный продукт, содержащий 22-30 мас.% 3-гидроксипропаналя, 0,1-0,5 мас. % остаточного этиленоксида и 40-120 м.д. кобальта, предварительно
подвергают окислению, данная стадия включает десорбирование смесью O2/N2 в условиях, эффективных для удаления остаточного моноксида углерода, и окисляют весь кобальт до катионной
формы. После проскока кобальта на выход из слоя слой регенерируют рециркуляцией 500 мл 10% серной кислоты в воде при температуре окружающей среды с последующим промыванием в течение 1 часа
деионизированной водой. Адсорбцию плюс регенерацию в этом случае считают "циклом" работы.
После одного месяца чередующихся операций наблюдается, что слой теряет эффективность в отношении удаления кобальта даже после попыток его регенерации. При окончательной кислотной регенерации слоя не обнаруживают, что кобальт выделяется из слоя с кислотой регенерации. Смола имеет скорее красноватый оттенок, чем коричневый цвет свежей смолы.
Пробу смолы удаляют из слоя и нагревают до 95oС в 10% серной кислоте в течение 3 часов. Некоторый розовый цвет, характерный для сульфата кобальта, наблюдается в супернатанте, подтверждая успешную регенерацию смолы. Более того, проба смолы приобретает коричневый цвет, характерный для свежей смолы. Обработанную пробу смолы тщательно промывают деионизированной водой и сушат воздухом до равномерной сухости. Часть замачивают с 75 частями 0,1н. NaOH в течение ночи с последующим обратным титрованием супернатанта 0,1н. НСl для определения количества Na+, обмененного смолой. Вторую часть сушат в вакууме в течение ночи при 90oС (общее давление около 6,89 кПа (0,0703 кг/см2)), чтобы определить содержание воды в смоле, используемой в эксперименте замачивания в течение ночи. Из этих определений равновесную обменную способность смолы определяют как 4,7 мэкв./г по отношению к теоретической максимальной способности 4,9-5,1 мэкв./г для свежей смолы.
Смолу, удаленную из рабочего слоя, но не подвергнутую регенерации горячей кислотой, также промывают, сушат воздухом и уравновешивают 0,1н. NaOH, чтобы определить способность. Наблюдаемая способность смолы менее чем 1 мэкв. центров обмена на грамм сухой смолы. Попытки регенерировать указанную смолу при температуре окружающей среды 20% серной кислотой также были сделаны, но по существу никакого кобальта не высвобождается и смола сохраняет свой красный цвет, характерный для загрязненной смолы. Обратное титрование смолы 0,1н. NaOH не показывает по существу никакого увеличения способности смолы (менее чем 1 мэкв./г).
Этот пример показывает, что сильнокислая катионообменная смола, подвергнутая воздействию водного потока промежуточного продукта со стадии гидроформилирования ЕО, теряет свою способность извлекать катионы, такие как кобальт, несмотря на регенерацию серной кислотой, как обычно практикуется, при окружающей температуре. На стадии регенерации кислотой требуется повышенная температура (95oС), чтобы восстановить способность смолы до близкой к ее исходной способности. В отсутствие регенерации горячей кислотой смола со временем теряет свою способность удалять кобальт.
Пример 7
Пробу макропористой сильнокислой (катионо)
обменной смолы А-15 открывают для доступа водного потока промежуточного продукта со стадии гидроформилирования ЕО приблизительно на один месяц, после чего способность к удалению кобальта после
регенерации при температуре окружающей среды 10% серной кислотой снижается по существу до ноля.
Пробу указанной смолы и пробу свежей смолы анализируют13С ЯМР. Загрязненная смола обнаруживает новые химические сдвиги при 70 и 60 м.д., показательные для простых эфирных связей -O-СН2-СН2- и концевой -СН2ОН соответственно. Высокотемпературная (80oС) регенерация 10% серной кислотой по существу устраняет указанные пики из спектра ЯМР. Этот результат говорит о том, что загрязнение смолы является результатом воздействия остаточного этиленоксида в водном потоке промежуточного продукта в противоположность 3-гидроксипропаналю, который давал бы -O-СН2СН2СН2- с соответствующим уникальным химическим сдвигом для центрального атома углерода из-за олигомеризации на смоле.
Пример 8
Ряд смол замачивают в водных растворах 3-гидроксипропаналя с добавлением изменяющихся
концентраций этиленоксида на 4-20 дней при окружающей температуре. После такого воздействия смолы тщательно промывают деионизированной водой, сушат воздухом и замачивают в 0,1н. NaOH для определения
ионообменной способности и оценки содержания твердых веществ смолы, как описано выше. Результаты показаны в таблице 4.
Сравнение опытов A, D и Е показывает, что степень загрязнения соотносится с концентрацией присутствующего ЕО при по существу фиксированной концентрации 3-гидроксипропаналя (25%), с 3-гидроксипропаналем, присутствующим по меньшей мере в 4:1 молярном избытке. Этот результат подтверждает заключение, выведенное из13С ЯМР, о загрязнении и потере ионообменной способности, являющихся результатом адсорбции и реакции ЕО на кислотных центрах смолы, а не 3-гидроксипропаналя.
Сравнение опытов В и А показывает, что Na-форма смолы менее склонна к загрязнению, чем сильнокислая смола. Анализ13С ЯМР показывает -О-СН2 -СН2- или происходящее от ЕО загрязнение для Na-формы смолы и для слабокислой смолы, хотя при более низких величинах, чем наблюдается для сильнокислой смолы. "Доля от способности свежей смолы" в таблице 4 также показывает более низкую долю способности (более обширное загрязнение), остающейся у сильнокислой смолы в кислотной форме по сравнению со слабой кислотой и особенно Na-формой сильной кислоты.
Пример 9
Степень регенерации загрязненной ЕО смолы.
Степень регенерации смолы исследуют как функцию температуры для загрязненной сильнокислой смолы посредством замачивания проб смолы в 10% серной кислоте для изменяющихся интервалов времени при изменяющихся температурах. Пробы смолы, обработанной таким образом, удаляют из нагревательной ванны и отделяют от кислотного супернатанта в фильтровальной воронке с тщательным промыванием деионизированной водой для удаления остаточного супернатанта. Пробы затем сушат воздухом и замачивают в 0, 1н. NaOH для обратного титрования, чтобы определить ионообменную способность, как описано выше.
Когда температуру увеличивают, степень регенерации серной кислотой активной ионообменной способности увеличивается, указывая на то, что возвращение загрязненной смолы к прежнему состоянию является зависящим от температуры кинетическим процессом.
Пример 10
Непрерывные исследования кислотной и Na-формы сильнокислой смолы проводят, чтобы исследовать характеристики в условиях промышленной эксплуатации. Условия испытаний непрерывного потока описаны в
примере 1. В ранних исследованиях используют 200 мл слой около 87 г сухой смолы. В более поздних исследованиях используют 12-13 г сухой смолы, набитой в 30 мл колонну, изготовленную из трубки
Hastelloy С с внутренним диаметром 12,7 мм. Кольцевая рубашка окружает трубку, чтобы обеспечить работу при контролируемой температуре выше окружающей, чтобы исследовать влияние температуры на степень
регенерации, что определяется по количеству кобальта, который может быть удален в следующем ионообменном цикле.
Результаты приведены в таблице 5. Столбец F показывает время на потоке для заданного цикла адсорбции, G - суммарное время всех циклов. Столбец Н дает количество обработанного подаваемого материала на единицу массы смолы для данного цикла, где "I" дает суммарное количество подаваемого материала, обработанного за все циклы, для данного типа смолы. Цикл адсорбции определяют как время течения в слое до тех пор, пока не происходит прорыв кобальта при 4 мас.м.д. в отходящий из слоя поток. Столбец J показывает мас.% кобальта, замененного на слое на время прорыва, в то время как К дает отношение количества удаленного кобальта к количеству, которое предположительно свежая смола могла бы удалить, если бы находилась в равновесии с количеством кобальта в подаваемом материале (са.70 мас.м.д.).
Кислотная форма смолы при регенерации при температуре окружающей среды 10% серной кислотой (серия А) обнаруживает загрязнение смолы, так что после шестого цикла смола становится далее неэффективной для удаления кобальта. Серия В исследований (проводимых в меньшем слое) демонстрирует, что поддерживаемое удаление кобальта может быть достигнуто посредством высокотемпературной (95oС) кислотной регенерации кислотной формы смолы. Достигают конфигурации устойчивого состояния, при которой способность удаления кобальта стабилизируют приблизительно на уровне 30% от количества, которое можно было бы ожидать в отсутствие загрязнения смолы. (Независимые исследования периодического процесса для определения изотермы адсорбции для удаления кобальта свежей смолой используют, чтобы определить равновесную способность свежей (незагрязненной) смолы для заданной концентрации кобальта в водном подаваемом промежуточном продукте).
Третье исследование (серия С) рассматривает натриевую форму сильнокислой смолы. Эта смола требует двух агентов регенерации: 10% серной кислоты, которая удаляет весь кобальт и большую часть обменного натрия на смоле, и 4% NaOH, который превращает смолу снова из кислотной формы в натриевую форму после регенерации кислотой. Требования к агентам регенерации, таким образом, значительно выше, чем для регенерации кислотной формы смолы, и регенерация может быть осуществлена в две стадии. Однако смола сохраняет свою по существу равновесную способность, которую она имеет в незагрязненном состоянии, в пределах ошибки эксперимента, на протяжении курса из одиннадцати циклов.
Конечное исследование (серия D) рассматривает кислотную форму слабокислой смолы с водным потоком промежуточного продукта с рН, доведенным до 5,5 добавлением щелочи, чтобы улучшить характеристики смолы. Требуется больший слой указанной смолы из-за ее более слабой адсорбции (более линейная изотерма адсорбции). Прорыв кобальта при 0-2 м.д. случается рано, до постепенного увеличения до существенного "прорыва", который отбирают как кобальт, вымываемый из слоя при более чем 4 м.д. Указанный слой регенерируют 10% серной кислотой при окружающей температуре. Как очевидно из столбцов J и К, слой в значительной степени поддерживает свою способность удалять кобальт в течение 9 циклов.
Реферат
Изобретение относится к производству 1,3-пропандиола гидроформилированием этиленоксида через промежуточный раствор 3-гидроксипропаналя, из которого удаляют остаточный диоксид углерода и нерастворимые каталитические соединения кобальта или родия. Процесс включает следующие стадии: (а) контактирование раствора 3-гидроксипропаналя с кислородом в кислотных условиях при температуре в пределах от около 5 до около 45oС с получением смеси продуктов окисления, содержащей водный раствор 3-гидроксипропаналя, одно или несколько растворимых в воде соединений кобальта или родия и побочный продукт моноксид углерода; (b) удаление побочного продукта моноксида углерода из смеси продуктов окисления и (с) удаление растворимых соединений металла из смеси продуктов окисления путем контакта с кислотной ионообменной смолой при температуре менее чем 45oС. Технический результат: эффективное удаление каталитических количеств кобальта и родия из продуктов реакции гидроформилирования. 6 з.п. ф-лы, 5 табл.
Комментарии