Способ получения алкоксидов алкилолова - RU2338749C2
Код документа: RU2338749C2
Чертежи
Описание
Настоящее изобретение относится к способу получения алкоксидов алкилолова, к способу получения эфира угольной кислоты (карбоната) или изоцианата с использованием в качестве катализатора алкоксида диалкилолова, полученного вышеуказанным способом, и к эфиру угольной кислоты или изоцианату, полученному указанным способом.
Алкоксиды алкилолова весьма полезны в качестве катализаторов синтеза или переэтерификации сложных эфиров или реакции вулканизации силиконового полимера или уретана.
Современными способами получения алкоксидов алкилолова являются, например, способ, в котором в качестве исходного материала используют диалкилдихлоролово (см., например, ссылку на патент [1]); и способ, в котором в качестве исходного материала используют оксид диалкилолова (см., например, ссылку на патент 2). В первом способе, в котором в качестве исходного материала используется диалкилдихлоролово, в качестве вторичного материала используется дорогостоящий алкоголят металла. Кроме того, в этом способе в качестве конечного продукта образуются два моля соли металла на один моль алкоксида диалкилолова, как показано в приведенном ниже уравнении реакции (8), в связи с чем возникает проблема утилизации отходов или т.п. Таким образом, применение первого способа для промышленного производства связано с такими проблемами, как высокая себестоимость и образование большого количества отходов.
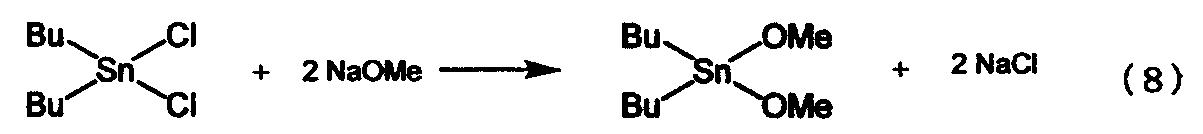
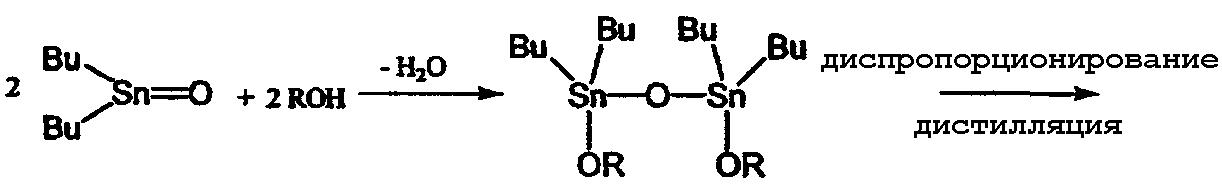
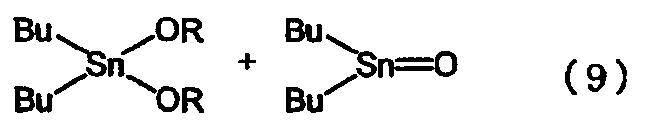
Второй способ, в котором в качестве исходного материала используется оксид диалкилолова, является предпочтительным, поскольку он не дает большого количества отходов. Поэтому были проведены исследования, направленные на получение алкоксидов диалкилолова с применением указанного второго способа. Одним из примеров такого способа производства является способ, в котором алкоксид диалкилолова получают из оксида дибутилолова и спирта путем проведения двухстадийной реакции, как показано в приведенном ниже уравнении реакции (9) (см. ссылку на патент [3]). На первой стадии оксид дибутилолова и спирт подвергают взаимодействию в бензоле или в толуоле при температуре от 80°С до 100°С с последующим удалением образовавшейся воды путем азеотропной перегонки и получением 1,1,3,3-тетрабутил-1,3-диалкоксидистанноксана. На второй стадии указанный дистанноксан подвергают реакции диспропорционирования при температуре от 180°С до 220°С с последующей перегонкой и получением диалкоксида дибутилолова. Указанный способ имеет то преимущество, что он является безотходным, однако реакция диспропорционирования на второй стадии данного способа требует перегонки высококипящего алкоксида диалкилалова при высокой температуре и поэтому потребляет большое количество энергии. Таким образом, применение второго способа для промышленного производства также связано с некоторыми проблемами, такими как, например, потребление большого количества энергии. Кроме того, этот способ имеет низкую производительность.
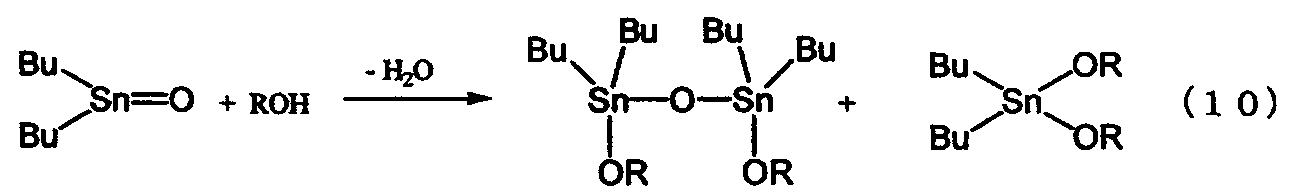
Был описан способ получения диалкоксида диалкилалова непосредственно из оксида диалкилолова и спирта, в котором используется высококипящий спирт, как показано в нижеследующем уравнении реакции (10) (см. ссылку на патент [3]). Поскольку в данном способе реакция протекает при температуре кипения спирта, используемого в качестве реагента, то указанную реакцию осуществляют при температуре, превышающей температуру реакции, проводимой в бензоле или толуоле, с последующим удалением образовавшейся воды в виде азеотропной смеси воды и спирта, используемого в качестве реагента. По сравнению с вышеописанным способом этот способ имеет то преимущество, что он не требует высокотемпературной перегонки высококипящего диалкоксида диалкилолова. Однако поскольку реакцию проводят при температуре кипения спирта, используемого в качестве реагента, то скорость реакции является низкой для спиртов с небольшим числом атомов углерода и даже для спиртов с большим числом атомов углерода. Поэтому указанный способ имеет низкую производительность.
Кроме того, поскольку в вышеописанном способе реакцию проводят в высококипящем спирте при высокой температуре, то фактически образуется большое количество соединения триалкилолова, которое, вероятно, образуется в соответствии с приведенным ниже уравнением реакции (11). Действительно, хорошо известно, что соединения триалкилолова получают путем пиролиза алкоксидов диалкилолова (см. ссылку 1 на не патентную работу), и полученное соединение триалкилолова может образовывать комплексную смесь побочных продуктов реакции, не являющихся диалкоксидом диалкилолова согласно изобретению. Поэтому указанный способ не является предпочтительным, особенно для промышленного производства.

Для увеличения производительности, которая является одной из главных проблем, связанных с применением вышеописанных способов, был разработан способ, где в качестве реагентов используется спирт и эфир угольной кислоты, как показано в уравнении реакции (12) (см. ссылку на патент 2). В этом способе в качестве реагента используется дорогостоящий эфир угольной кислоты, и хотя этот способ является более производительным, чем вышеописанные способы, однако проблема, связанная с применением указанного способа, а именно высокая себестоимость производства, пока остается нерешенной.

[Ссылка на патент 1] US-2700675
[Ссылка на патент 2] US-5545600
[Ссылка на патент 3] NL-6612421
[Ссылка на непатентную работу 1] Journal of Society of Chemical Industry, 72,7 (1969), 1543.
Как описано выше, в общепринятых способах получения алкоксидов алкилолова для повышения производительности всегда применяются дорогостоящие исходные материалы. Поэтому необходимо разработать удобный и высокоэффективный способ получения алкоксидов алкилолова.
В соответствии с этим настоящее изобретение относится к способу промышленного производства алкоксидов алкилолова и, в частности, к способу промышленного и непрерывного производства алкоксидов алкилолова.
После проведения многочисленных исследований, направленных на решение вышеописанных проблем, авторами настоящего изобретения было обнаружено, что если исходный материал, выбранный из группы, состоящей из оловоорганических соединений, каждое из которых имеет связь олово-кислород-олово, и гидроксисоединения, используемого в качестве реагента, непрерывно подается в реактор и низкокипящие компоненты, образующиеся в такой реакции, непрерывно удаляются из реактора, то реакционный раствор, содержащий алкоксид алкилолова, соответствующий исходному материалу и реагенту, может непрерывно выводиться из реактора как компонент, осаждающийся на дне реактора. Таким образом, авторами настоящей заявки было разработано настоящее изобретение.
Более конкретно, объектами настоящего изобретения являются следующие.
[1] Способ получения алкоксидов алкилолова, включающий реакцию дегидратации, по меньшей мере, одного соединения алкилолова, используемого в качестве исходного материала и выбранного из группы, состоящей из оловоорганических соединений, каждое из которых имеет связь олово-кислород-олово, и гидроксисоединения, используемого в качестве реагента, с получением алкоксида алкилолова, соответствующего исходному материалу и реагенту, в котором указанный исходный материал и реагент непрерывно подаются в реактор; низкокипящие компоненты, содержащие воду, удаляются из реактора, и реакционный раствор, который в виде компонента образуется на дне реактора и содержит алкоксид алкилолова, непрерывно выводится из реактора.
[2] Способ по вышеуказанному пункту [1], где вышеописанное, по меньшей мере, одно соединение алкилолова, используемое в качестве исходного материала, представляет собой тетраалкилдиалкокси-1,3-дистанноксан и/или оксид диалкилолова, обычно присутствующий в форме полимера, образующегося в результате полимеризации по связи олово-кислород-олово.
[3] Способ по вышеуказанному пункту [2], где указанный тетраалкилдиалкокси-1,3-дистанноксан представляет собой тетраалкилдиалкокси-1,3-дистанноксан, представленный химической формулой (1):
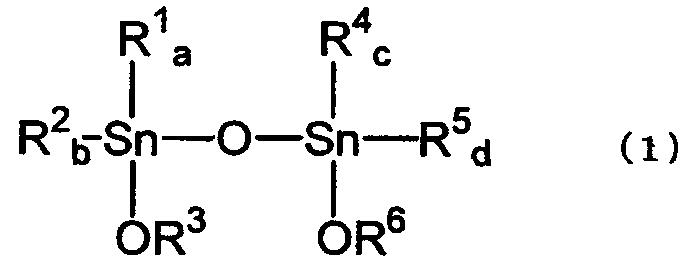
где R1, R2, R4 и R5, каждый независимо, представляет собой алкильную группу, аралкильную группу или арильную группу; R3 и R6, каждый представляет собой алкильную группу или аралкильную группу; а и b равны целому числу от 0 до 2; а+b равно 2; с и d равны целому числу от 0 до 2; и с+d равно 2.
[4] Способ по вышеуказанному пункту [2], где указанный оксид диалкилолова представляет собой полимер оксида диалкилолова, представленный химической формулой (2):
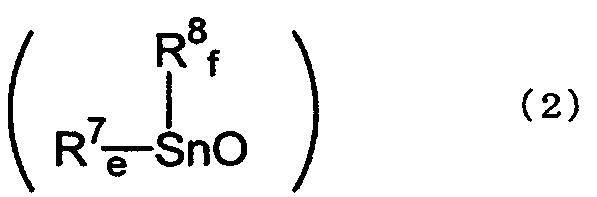
где R7 и R8, каждый независимо, представляет собой алкильную группу, аралкильную группу или арильную группу; е и f равны целому числу от 0 до 2; и е+f равно 2.
[5] Способ по вышеуказанному пункту [2], где указанный исходный материал представляет собой любой материал, выбранный из мономера, димера (агрегата, состоящего из одного и того же мономера, или агрегата, состоящего из различных мономеров), олигомера или полимера.
[6] Способ по вышеуказанному пункту [1], где описанное выше гидроксисоединение представляет собой спирт, представленный химической формулой (3):

где R9 представляет собой н-бутильную группу; 2-метилпропильную группу; алкильную группу с прямой или разветвленной цепью, имеющей 5-12 атомов углерода; циклоалкильную группу, имеющую 5-12 атомов углерода; алкенильную группу с прямой или разветвленной цепью, имеющей 2-12 атомов углерода; замещенный или незамещенный арил, имеющий 6-19 атомов углерода; или аралкильную группу, имеющую 7-20 атомов углерода и содержащую алкил, выбранный из группы, состоящей из алкила с прямой или разветвленной цепью, имеющей 1-14 атомов углерода, и циклоалкила, имеющего 5-14 атомов углерода.
[7] Способ по вышеуказанному пункту [6], где указанный спирт выбирают из группы, состоящей из 1-бутанола, 2-метил-1-пропанола и алкилового спирта, имеющего 5-8 атомов углерода.
[8] Способ по вышеуказанному пункту [1], включающий стадию непрерывной подачи исходного материала и реагента в реактор для осуществления реакции дегидратации в жидкой фазе или в газожидкой фазе в реакторе; и одновременного выведения высококипящей реакционной смеси в виде жидкости, содержащей полученный алкоксид алкилолова или смесь алкоксидов алкилолова, из нижней части реактора, при непрерывном удалении из реактора низкокипящей реакционной смеси, содержащей образуемую воду в виде газа, путем перегонки.
[9] Способ по вышеуказанному пункту [1] или [8], где указанный реактор включает линии подачи вышеописанного исходного материала и вышеописанного реагента соответственно или линию подачи смешанного раствора, состоящего из вышеописанного исходного материала и вышеописанного реагента; линию удаления низкокипящей реакционной смеси, содержащей воду; и линию выведения высококипящей реакционной смеси.
[10] Способ по вышеуказанному пункту [9], где указанная линия удаления низкокипящей реакционной смеси, содержащей воду, находится в месте удаления газофазных компонентов, и линия выведения высококипящей смеси находится в месте, расположенном ниже места выведения жидкофазного компонента.
[11] Способ по любому из вышеуказанных пунктов [1]-[10], где указанный реактор представляет собой резервуарный реактор или колонный реактор.
[12] Способ по любому из вышеуказанных пунктов [1]-[10], где указанный реактор представляет собой реактор, который включает смесительный резервуар, резервуар для многостадийного смешения, дистилляционную колонну, многоходовую дистилляционную колонну, многоходовую дистилляционную колонну для непрерывной перегонки, насадочную колонну, тонкопленочный испаритель, реактор с носителем, реактор с принудительной циркуляцией, испаритель с падающей пленкой, испаритель с падающей каплей, реактор, имеющий слой со струйным течением жидкости, или колонну-барботер.
[13] Способ по любому из вышеуказанных пунктов [1]-[12], где в указанный реактор подается инертный газ и/или газообразный реагент, и/или газообразное инертное органическое соединение, и/или органический растворитель, образующий азеотропную смесь с водой.
[14] Способ по вышеуказанному пункту [13], где указанный инертный газ выбирают из азота, диоксида углерода и аргона.
[15] Способ по вышеуказанному пункту [1], где вышеописанную реакцию дегидратации проводят при температуре в пределах от 60°С до 160°С.
[16] Способ по вышеуказанному пункту [1], где отношение общего числа молей атомов олова, имеющихся в исходном соединении, к числу молей реагента, то есть отношение исходного соединения к реагенту, находится в пределах от 3 до 100.
[17] Способ по любому из вышеуказанных пунктов [4]-[16], где реакцию дегидратации осуществляют со скоростью дегидратации, определяемой по уравнению (4):
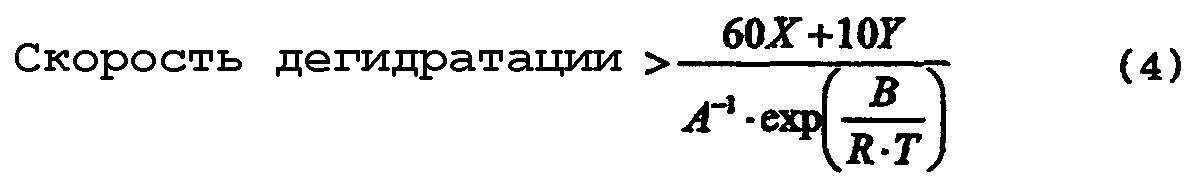
где термин "скорость дегидратации" означает количество воды, которое образуется в процессе реакции дегидратации и выводится из системы за единицу времени [моль·час-1]; Х означает общее число молей [моль] атомов олова, имеющихся в соединении алкилолова, представленном общей формулой (2) и содержащемся в исходном материале; Y означает общее число молей [моль] атомов олова, имеющихся в соединении алкилолова, представленном общей формулой (1) и содержащемся в исходном материале; Т означает температуру [К], при которой осуществляется реакция дегидратации; R представляет собой газовую постоянную, равную 8,314 Дж·моль-1·К-1; и А и В представляют собой коэффициенты, зависящие от типа соединения алкилолова, где коэффициенты А и В в вышеуказанном уравнении (4) зависят от типа соединения алкилолова, используемого в качестве исходного материала, и вычислены исходя из выбранного первичного стандартного вещества. Если исходный материал содержит соединение алкилолова, представленное химической формулой (1), то указанные коэффициенты А и В представляют собой фактор частоты и энергию активации пиролитической реакции первичного стандартного вещества, которым является соединение алкилолова, произвольно выбранное из соединений алкилолова, представленных химической формулой (1) и содержащихся в исходном материале, и эти коэффициенты вычисляют по уравнению (5). Если исходный материал не содержит соединения алкилолова, представленного химической формулой (1), но содержит соединение алкилолова, представленное химической формулой (2), то указанные коэффициенты А и В представляют собой фактор частоты и энергию активации пиролитической реакции первичного стандартного соединения, которым является алкоксид алкилолова, произвольно выбранный из алкоксидов алкилолова, представленных химической формулой (7) и образованных из соединений алкилолова, представленных химической формулой (2) и содержащихся в исходном материале и реагенте, где указанные коэффициенты вычисляют по уравнению (5)

где k означает константу скорости первого порядка [час-1]; А означает фактор частоты [час-1]; В означает энергию активации [Дж·моль-1]; R означает газовую постоянную=8,314 Дж·моль-1·К-1; и Т означает температуру [К], при которой осуществляется пиролитическая реакция. Вышеуказанная константа k представляет собой константу скорости первого порядка для пиролитической реакции, которую вычисляют по уравнению (6)

где k означает константу скорости первого порядка [час-1]; t означает время нагревания; и Х [час] означает коэффициент редукции [моль/моль] по отношению к начальной концентрации первичного стандартного вещества,
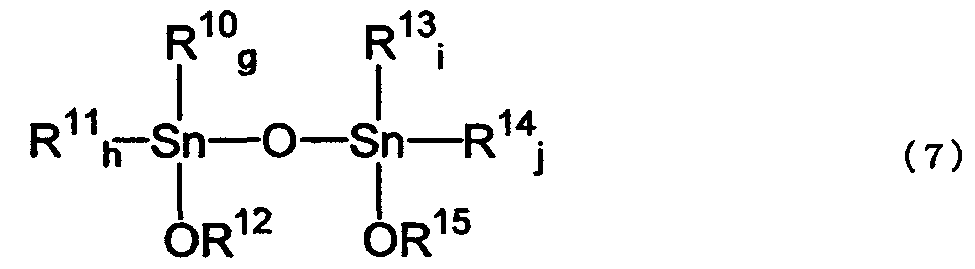
где R10, R11, R13 и R14 соответствуют R7 или R8 исходного соединения; g, h, i и j соответствуют е или f исходного соединения; по меньшей мере, один из R12 и R15 соответствует значению R9 указанного реагента; g и h равно целому числу от 0 до 2, g+h=2; i и j равны целому числу от 0 до 2, i+j=2.
[18] Способ получения эфиров угольной кислоты, где используют алкоксиды диалкилолова в качестве катализатора, полученные способом по любому из вышеуказанных п.п. [1] - [17].
[19] Эфир угольной кислоты, полученный с использованием алкоксида диалкилолова, полученного способом по любому одному из вышеуказанных пунктов [1] - [17], в качестве катализатора.
[20] Изоцианат, полученный с использованием эфира угольной кислоты по вышеуказанному пункту [19].
[21] Поликарбонат, полученный с использованием эфира угольной кислоты по вышеуказанному пункту [19].
В способе получения алкоксидов алкилолова согласно изобретению исходный материал и реагент подвергают непрерывной реакции дегидратации с одновременным удалением из системы образующейся воды и продуктов, в результате чего могут быть получены алкоксиды алкилолова с высоким выходом. Таким образом, указанный способ является в высокой степени полезен для его промышленного применения.
Настоящее изобретение отличается тем, что осуществляют непрерывную подачу в реактор исходного материала, содержащего оксид диалкилолова и/или тетраалкилдиалкоксидистанноксан и гидроксисоединение, используемое в качестве реагента, и удаление из реактора низкокипящих компонентов, содержащих воду, с непрерывным образованием реакционного раствора в виде компонента, образующегося в нижней части реактора и содержащего алкоксид алкилолова, соответствующий исходному материалу и реагенту.
Авторами настоящего изобретения было высказано предположение, что реакция взаимодействия оксида диалкилолова со спиртом, в результате которой образуется алкоксид диалкилолова, является равновесной реакцией, представленной уравнениями реакций (13) и (14).
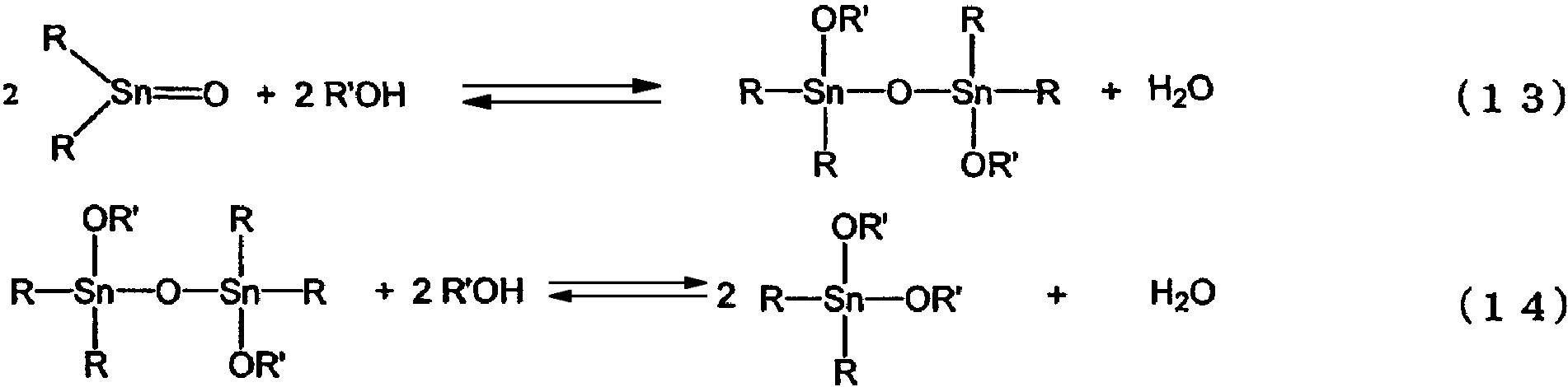
Обычно описанную выше реакцию осуществляют периодическим способом, где образующуюся воду отгоняют при атмосферном давлении или при пониженном давлении. Это вызвано тем, что поскольку равновесие в реакциях, представленных формулами (13) и (14), смещается влево (в сторону реагентов), то для установления равновесия необходимо смещение вправо (в сторону продуктов), и для дальнейшего прохождения реакции требуется выведение образующейся воды из системы. При этом для увеличения скорости реакции эту реакцию осуществляют при высоких температурах.
В результате, если в вышеописанной реакции в качестве реагента используется спирт с большим числом атомов углерода и высокой температурой кипения, то реакция протекает за несколько часов, и если в качестве реагента используется спирт с небольшим числом атомов углерода и низкой температурой кипения, то образуется алкоксид диалкилолова, соответствующий указанному спирту и производительность данной реакции является очень низкой. Известно, что такая реакция, проиллюстрированная реакционной формулой (13), то есть реакция дегидратации оксида диалкилолова спиртом с образованием тетраалкилдиалкоксидистанноксана с количественным выходом протекает относительно легко даже при ее проведении общепринятыми способами. Однако поскольку в реакции, проиллюстрированной реакционной формулой (14), равновесие значительно смещается влево (в сторону реагентов), то вода, образующаяся в этой реакции, быстро реагирует с образовавшимся диалкоксидом диалкилолова и поэтому диалкоксид диалкилолова не может быть получен с высоким выходом. Для получения большего количества нужного продукта в реакции, проиллюстрированной реакционной формулой (14), необходимо использовать большое количество гидроксисоединения. Очевидно, что использование большого количества гидроксисоединения позволяет повышать уровень превращения тетраалкилдиалкокси-1,3-дистанноксана в диалкоксид диалкилолова, однако для этого необходим огромный реактор, и, кроме того, необходимо удалять большое количество непрореагировавшего гидроксисоединения путем перегонки и, следовательно, производительность такой реакции не может быть увеличена.
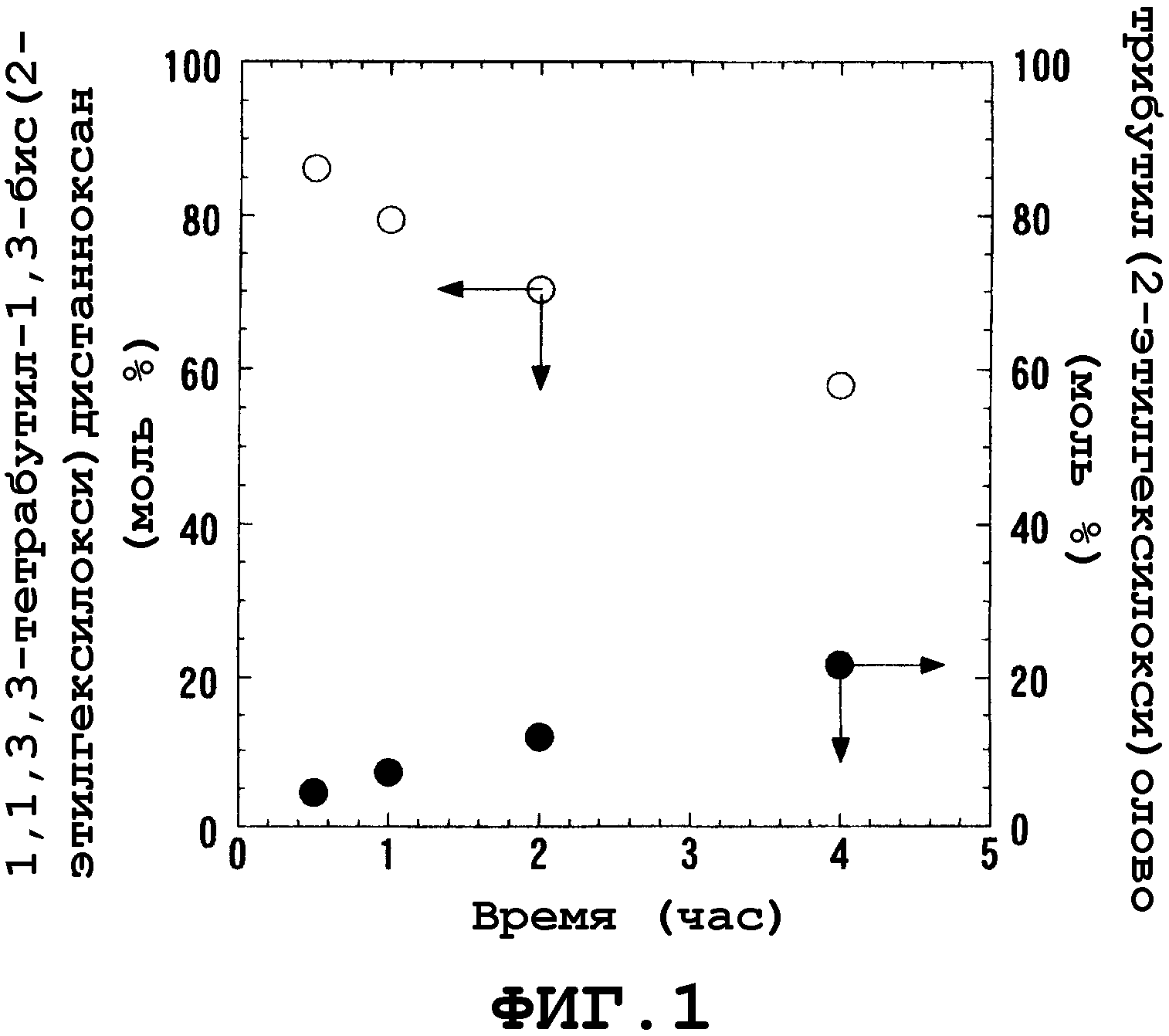
После интенсивного исследования общепринятых способов авторами настоящего изобретения было обнаружено, что в вышеописанных общепринятых способах образуется большое количество соединения трибутилолова, не являющегося целевым соединением, поскольку реакция, осуществляемая этими способами, протекает при высоких температурах и в течение длительного периода времени, хотя это явно не описано в документах. В частности, при осуществлении реакции общепринятым периодическим способом возникают серьезная проблема, связанная с образованием побочного продукта триалкилолова, описанного выше (на фиг.1 показана реакция пиролиза, в процессе которой происходит превращение 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана в соединение трибутилолова), поскольку из-за низкой скорости реакции и периодического способа ее осуществления, алкоксид алкилолова, как исходный материал или как продукт, остается в реакторе.
После многочисленных исследований, направленных на решение вышеописанной проблемы, авторами настоящего изобретения было неожиданно обнаружено, что в случае применения способа, который включает непрерывную подачу в реактор исходного материала, выбранного из группы, состоящей из оксидов диалкилолова, тетраалкилдиалкоксидистанноксанов и их смесей, а также реагента, то есть гидроксисоединения; удаление низкокипящих компонентов из реактора; и непрерывное выведение реакционного раствора, как компонента, накапливающегося на дне реактора, который содержит алкоксид алкилолова, соответствующий исходному материалу и реагенту; указанная реакция осуществляется за короткий промежуток времени и имеет высокую производительность по сравнению с реакциями, проводимыми общепринятыми периодическими способами, и более того в этой реакции ингибируется образование побочного продукта трибутилолова.
В частности, авторы настоящего изобретения пришли к выводу, что скорость образования алкоксидов алкилолова общепринятыми периодическими способами значительно ограничена скоростью удаления образующейся воды, и затем они обнаружили, что эта проблема может быть решена путем применения способа, который включает быстрое и непрерывное удаление образующейся воды из системы и одновременно с этим быстрое и непрерывное выведение из этой системы полученного продукта, а именно алкоксида алкилолова; и который позволяет получать алкоксид диалкилолова с высоким выходом. Авторами настоящего изобретения было также обнаружено, что способ согласно изобретению позволяет снижать уровень образования побочного соединения, а именно соединения трибутилолова.
Ниже, в первую очередь, будут описаны исходные материалы, используемые в настоящем изобретении.
Исходный материал представляет собой композицию, содержащую оксид диалкилолова и/или тетраалкилдиалкоксидистанноксан. Такая композиция может содержать только один тетраалкилдиалкоксидистанноксан или произвольное количество оксида диалкилолова, который является предшественником тетраалкилдиалкоксидистанноксана. В частности, как реакция, проиллюстрированная реакционной формулой (13), в которой тетраалкилдиалкоксидистанноксан получают из оксида диалкилолова, так и реакция, проиллюстрированная реакционной формулой (14), в которой диалкоксид диалкилолова получают из тетраалкилдиалкоксидистанноксана, представляют собой реакции дегидратации, и поэтому алкоксид диалкилолова может быть получен даже из исходного соединения, содержащего произвольное количество оксида диалкилолова.
Тетраалкилдиалкоксидистанноксанами, используемыми в настоящем изобретении, могут быть тетраалкилдиалкоксидистанноксаны, представленные нижеследующей химической формулой (1), а также их мономеры, агрегаты, олигомеры или полимеры, имеющие структуру, представленную химической формулой (1).
где R1, R2, R4 и R5, каждый независимо, представляет собой алкильную группу, аралкильную группу или арильную группу; R3 и R6, каждый представляет собой алкильную группу или аралкильную группу; а и b равны целому числу от 0 до 2; а+b равно 2; с и d равны целому числу от 0 до 2; с+d равно 2.
Примерами R1, R2, R4 и R5 в тетраалкилдиалкоксидистанноксанах, представленных химической формулой (1), являются алкильные группы, а именно алифатические углеводородные группы, имеющие 1-12 атомов углерода, и циклоалкильные группы, а именно алифатические углеводородные группы, имеющие 5-12 атомов углерода, такие как метил, этил, пропил, бутил (его изомеры), пентил (его изомеры), гексил (его изомеры), гептил (его изомеры), октил (его изомеры), нонил (его изомеры), децил (его изомеры), ундецил (его изомеры), додецил (его изомеры), 2-бутенил, циклобутенил, циклобутил, циклопентил, циклогексил, циклопентил, циклопентадиенил и циклогексенил; аралкильные группы, имеющие 7-20 атомов углерода, такие как бензильная и фенилэтильная группы; и арильные группы, имеющие 6-20 атомов углерода, такие как фенильная, толильная и нафтильная группы. Эти группы могут содержать простую эфирную связь, либо они могут быть галогенированными углеводородными группами, в которых все атомы водорода или часть атомов водорода каждой углеводородной группы заменены атомами галогена и которыми являются нонафторбутил и гептафторбутил (их изомеры), однако эти группы не ограничиваются указанными выше примерами. Предпочтительно, R1, R2, R4 и R5 представляют собой низшие алкильные группы. Более предпочтительно, они представляют собой алкильные группы с прямой или разветвленной цепью, имеющей 1-8 атомов углерода. Могут быть также использованы группы вышеописанных типов с еще большим числом атомов углерода, однако такие группы иногда негативно влияют на текучесть или выход алкоксидов диалкилолова. R1, R2, R4 и R5 в тетраалкилдиалкоксидистанноксанах, представленных химической формулой (1) могут быть одинаковыми или различными.
R3 и R6 представляют собой алкильную группу с прямой или разветвленной цепью, имеющей 1-12 атомов углерода; циклоалкильную группу, имеющую 5-12 атомов углерода; или алкенильную группу с прямой или разветвленной цепью, имеющей 2-12 атомов углерода; и аралкильные группы, имеющие 7-20 атомов углерода, которые включают замещенный или незамещенный арил, имеющий 6-19 атомов углерода; и алкильную группу, выбранную из группы, состоящей из алкилов с прямой или разветвленной цепью, имеющей 1-14 атомов углерода, и циклоалкилов, имеющих 5-14 атомов углерода. Эти группы могут содержать простую эфирную связь, либо они могут быть галогенированными углеводородными группами, в которых все атомы водорода или часть атомов водорода каждой углеводородной группы заменены атомами галогена, и которыми являются нонафторбутил и гептафторбутил (их изомеры), однако эти группы не ограничиваются указанными выше примерами. Предпочтительно, R3 и R6 представляют собой низшие алкильные группы. Более предпочтительно, они представляют собой н-бутильную группу, 2-метилпропильную группу или алкильные группы с прямой или разветвленной цепью, имеющей 5-9 атомов углерода. Могут быть также использованы группы вышеописанных типов с еще большим числом атомов углерода, однако такие группы иногда негативно влияют на текучесть или выход алкоксидов диалкилолова. R3 и R6 в тетраалкилдиалкоксидистанноксанах, представленных химической формулой (1), могут быть одинаковыми или различными.
Примерами тетраалкилдиалкоксидистанноксанов, представленных химической формулой (1), являются:
тетраалкилдиалкоксидистанноксаны и
тетраалкилдиаралкилоксидистанноксаны, такие как
1,1,3,3-тетраметил-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетраметил-1,3-бис(2-метилпропилокси)дистанноксан, 1,1,3,3-тетраметил-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетраметил-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетраметил-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетраметил-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетраметил-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетраметил-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетраметил-1,3-дибензилоксидистанноксан,
1,1,3,3-тетраметил-1,3-дифенилэтоксидистанноксан,
1,3-дибутил-1,3-диметил-1,3-ди(н-бутокси)дистанноксан,
1,3-дибутил-1,3-диметил-1,3-бис(2-метилпропил)дистанноксан,
1,3-дибутил-1,3-диметил-1,3-дипентилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диметил-1,3-дигексилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диметил-1,3-дигептилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диметил-1,3-диоктилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диметил-1,3-динонилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диметил-1,3-дидецилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диметил-1,3-дибензилоксидистанноксан,
1,3-дибутил-1,3-диметил-1,3-дифенилэтоксидистанноксан,
1,3-дибутил-1,3-диэтил-1,3-ди(н-бутокси)дистанноксан,
1,3-дибутил-1,3-диэтил-1,3-бис(2-метилпропил)дистанноксан,
1,3-дибутил-1,3-диэтил-1,3-дипентилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диэтил-1,3-дигексилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диэтил-1,3-дигептилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диэтил-1,3-диоктилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диэтил-1,3-динонилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диэтил-1,3-дидецилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-диэтил-1,3-дибензилоксидистанноксан,
1,3-дибутил-1,3-диэтил-1,3-дифенилэтоксидистанноксан,
1,3-дибутил-1,3-дипропил-1,3-ди(н-бутокси)дистанноксан,
1,3-дибутил-1,3-дипропил-1,3-бис(2-метилпропил)дистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-дипентилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-дигексилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-дигептилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-диоктилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-динонилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-дидецилоксидистанноксан (его изомеры),
1,3-дибутил-1,3-дипропил-1,3-дибензилоксидистанноксан,
1,3-дибутил-1,3-дипропил-1,3-дифенилэтоксидистанноксан,
1,1,3,3-тетрабутил-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетрабутил-1,3-бис(2-метилпропил)дистанноксан,
1,1,3,3-тетрабутил-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетрабутил-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетрабутил-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетрабутил-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетрабутил-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетрабутил-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетрабутил-1,3-дибензилоксидистанноксан,
1,1,3,3-тетрабутил-1,3-дифенилэтоксидистанноксан,
1,1,3,3-тетрафенил-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетрафенил-1,3-бис(2-метилпропил)дистанноксан,
1,1,3,3-тетрафенил-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетрафенил-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетрафенил-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетрафенил-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетрафенил-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетрафенил-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетрафенил-1,3-дибензилоксидистанноксан,
1,1,3,3-тетрафенил-1,3-дифенилэтоксидистанноксан,
1,1,3,3-тетра(трифторбутил)-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетра(трифторбутил)-1,3-бис(2-метилпропил)дистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетра(трифторбутил)-1,3-дибензилоксидистанноксан,
1,1,3,3-тетра(трифторбутил)-1,3-дифенилэтоксидистанноксан,
1,1,3,3-тетра(пентафторбутил)-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетра(пентафторбутил)-1,3-бис(2-метилпропил)дистанноксан,
1,1,3,3-тетра(пентафторбутил)-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетра(пентафторбутил)-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетра(пентафторбутил)-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетра(пентафторбутил)-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетра(пентафторбутил)-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетра(пентафторбутил)-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетра(пентафторбутил)-1,3-дибензилоксидистанноксан,
1,1,3,3-тетра(пентафторбутил)-1,3-дифенилэтоксидистанноксан,
1,1,3,3-тетра(гептафторбутил)-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетра(гептафторбутил)-1,3-бис(2-метилпропил)дистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетра(гептафторбутил)-1,3-дибензилоксидистанноксан,
1,1,3,3-тетра(гептафторбутил)-1,3-дифенилэтоксидистанноксан,
1,1,3,3-тетра(нонафторбутил)-1,3-ди(н-бутокси)дистанноксан,
1,1,3,3-тетра(нонафторбутил)-1,3-бис(2-метилпропил)дистанноксан,
1,1,3,3-тетра(нонафторбутил)-1,3-дипентилоксидистанноксан (его изомеры),
1,1,3,3-тетра(нонафторбутил)-1,3-дигексилоксидистанноксан (его изомеры),
1,1,3,3-тетра(нонафторбутил)-1,3-дигептилоксидистанноксан (его изомеры),
1,1,3,3-тетра(нонафторбутил)-1,3-диоктилоксидистанноксан (его изомеры),
1,1,3,3-тетра(нонафторбутил)-1,3-динонилоксидистанноксан (его изомеры),
1,1,3,3-тетра(нонафторбутил)-1,3-дидецилоксидистанноксан (его изомеры),
1,1,3,3-тетра(нонафторбутил)-1,3-дибензилоксидистанноксан и
1,1,3,3-тетра(нонафторбутил)-1,3-дифенилэтоксидистанноксан.
При этом может быть использовано одно соединение, выбранное из описанной выше группы тетраалкилдиалкоксидистанноксанов, либо могут быть использованы два или несколько соединений, выбранных из этой группы, в виде смеси.
Оксиды диалкилолова, используемые в настоящем изобретении, представлены нижеследующей формулой (2). Хотя эти соединения представлены структурной формулой (2), однако такими соединениями могут быть мономеры, агрегаты, олигомеры или полимеры. Известно, что оксиды диалкилолова не существуют в форме мономера, поскольку двойная связь, такая как Sn=O, обычно не может образовываться, и поэтому они существуют в форме полимера, образующегося в результате полимеризации по связи олово-кислород-олово, как показано в нижеследующей формуле (15).
где R7 и R8, каждый независимо, представляет собой алкильную группу, аралкильную группу или арильную группу; е и f равны целому числу от 0 до 2, и е+f равно 2.
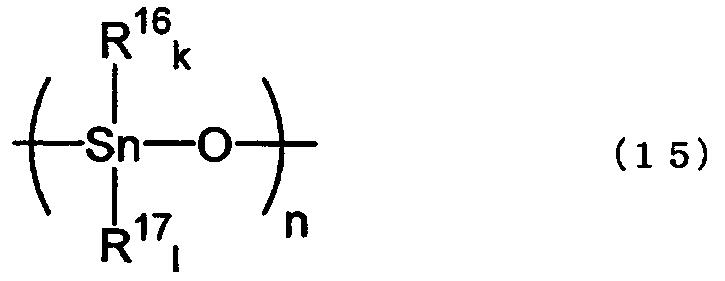
где R16 и R17, каждый представляет собой группы, как определено выше для R7 и R8, k и l имеют значения, как определено выше для е и f, и n равно целому числу 2 или более. Структура концевой группы не известна, и поэтому она не указана.
Примерами R7 и R8 в оксидах диалкилолова, представленных формулой (2), являются алкильные группы, а именно алифатические углеводородные группы, имеющие 1-12 атомов углерода, и циклоалкильные группы, а именно алифатические углеводородные группы, имеющие 5-12 атомов углерода, такие как метил, этил, пропил (его изомеры), бутил (его изомеры), пентил (его изомеры), гексил (его изомеры), гептил (его изомеры), октил (его изомеры), нонил (его изомеры), децил (его изомеры), ундецил (его изомеры), додецил (его изомеры), 2-бутенил, циклобутенил, циклобутил, циклопентил, циклогексил, циклопентил, циклопентадиенил и циклогексенил; аралкильные группы, имеющие 7-20 атомов углерода, такие как бензильная и фенилэтильная группы; и арильные группы, имеющие 6-20 атомов углерода, такие как фенильная, толильная и нафтильная группы. Эти группы могут содержать простую эфирную связь, либо они могут быть галогенированными углеводородными группами, в которых все атомы водорода или часть атомов водорода каждой углеводородной группы заменены атомами галогена и которыми являются нонафторбутил и гептафторбутил (их изомеры), однако эти группы не ограничиваются указанными выше примерами. Предпочтительно, R7 и R8 представляют собой низшие алкильные группы. Более предпочтительно, они представляют собой алкильные группы с прямой или разветвленной цепью, имеющей 1-8 атомов углерода. Могут быть также использованы группы вышеописанных типов с еще большим числом атомов углерода, однако такие группы иногда негативно влияют не текучесть или выход алкоксидов диалкилолова.
Примерами таких оксидов диалкилолова являются оксиды диалкилолова, такие как оксид диметилолова, оксид диэтилолова, оксид дипропилолова (его изомеры), оксид дибутилолова (его изомеры), оксид дипентилолова (его изомеры), оксид дигексилолова (его изомеры), оксид дигептилолова (его изомеры), оксид диоктилолова и оксид дициклогексилолова; оксиды диаралкилолова, такие как оксид дитолилолова и оксид дифенилэтилолова; и оксиды диарилолова, такие как оксид дифенилолова, оксид бис(2,6-диметилфенил)олова и оксид динафтилолова. При этом может быть использовано одно соединение, выбранное из описанной выше группы оксидов диалкилолова, либо могут быть использованы два или несколько соединений, выбранных из этой группы, в виде смеси.
В качестве исходного материала может быть использован агрегат или полимер тетраалкилдиалкоксидистанноксана, представленного формулой (1), и оксида диалкилолова, представленного формулой (2).
Тетраалкилдиалкоксидистанноксан, представленный формулой (1) и используемый в качестве исходного соединения, может быть получен любым известным способом. Указанный тетраалкилдиалкоксидистанноксан может быть также получен способом согласно изобретению с использованием в качестве исходного соединения оксида диалкилолова, представленного формулой (2), и с использованием в качестве реагента гидроксисоединения, представленного формулой (3).
Реагентом, используемым в настоящем изобретении, является гидроксисоединение, и предпочтительно, спирт, представленный нижеследующей формулой (3)
где R9 представляет собой н-бутильную группу; 2-метилпропильную группу; алкильную группу с прямой или разветвленной цепью, имеющей 5-12 атомов углерода; циклоалкильную группу, имеющую 5-12 атомов углерода; алкенильную группу с прямой или разветвленной цепью, имеющей 2-12 атомов углерода; замещенную или незамещенную арильную группу, имеющую 6-19 атомов углерода; или аралкильную группу, имеющую 7-20 атомов углерода, которая включает алкил, выбранный из группы, состоящей из алкила с прямой или разветвленной цепью, имеющей 1-14 атомов углерода, и циклоалкила, имеющего 5-12 атомов углерода.
Конкретными примерами вышеописанных гидроксисоединений являются алифатические спирты, имеющие 1-12 атомов углерода, и алициклические спирты, имеющие 5-12 атомов углерода, такие как 1-бутанол, 2-метил-1-пропанол, 2-метил-2-пропанол, циклобутанол,
1-пентанол, 2-пентанол (его изомеры), 3-пентанол,
3-метил-1-бутанол, 2-метил-1-бутанол,
2-метил-2-бутанол (его изомеры),
3-метил-2-бутанол (его изомеры), циклопентанол,
2-метил-1-циклобутанол (его изомеры),
3-метил-1-циклобутанол (его изомеры),
1-метил-1-циклобутанол (его изомеры),
циклобутилметанол (его изомеры), 1-гексанол,
2-гексанол (его изомеры), 3-гексанол (его изомеры),
4-метил-1-пентанол (его изомеры),
3-метил-1-пентанол (его изомеры),
2-метил-1-пентанол (его изомеры), 2-этил-1-бутанол,
3-метил-2-пентанол (его изомеры),
3-метил-3-пентанол (его изомеры), циклогексанол,
1-метил-1-циклопентанол (его изомеры),
2-метил-1-циклопентанол (его изомеры),
циклобутилметанол (его изомеры),
2-циклобутилэтанол (его изомеры),
1-циклобутилэтанол (его изомеры),
(1-метилциклобутил)метанол (его изомеры),
(2-метилциклобутил)метанол (его изомеры),
гептанол (его изомеры), циклогексилметанол (его изомеры),
(метилциклогексил)метанол (его изомеры),
циклогексилэтанол (его изомеры),
(этилциклобутил)метанол (его изомеры),
(метилциклопропил)этанол (его изомеры),
(этилциклопропил)метанол (его изомеры), октанол (его изомеры),
нонанол (его изомеры), деканол (его изомеры),
ундеканол (его изомеры), додеканол (его изомеры),
пропениловый спирт, бутениловый спирт (его изомеры),
пентениловый спирт (его изомеры), циклопентенол (его изомеры),
циклопентадиениловый спирт, гексенол (его изомеры) и
циклогексенол (его изомеры); и аралкиловые спирты, такие как бензиловый спирт и фенилэтиловый спирт.
Из этих гидроксисоединений предпочтительными являются первичные или вторичные одноатомные спирты, имеющие 1-8 атомов углерода, такие как 1-бутанол, 2-бутанол (его изомеры),
2-метил-1-пропанол, 2-метил-2-пропанол, циклобутанол,
1-пентанол, 2-пентанол (его изомеры), 3-пентанол,
3-метил-1-бутанол, 2-метил-1-бутанол,
2-метил-2-бутанол (его изомеры),
3-метил-2-бутанол (его изомеры), циклопентанол,
2-метил-1-циклобутанол (его изомеры),
3-метил-1-циклобутанол (его изомеры),
1-метил-1-циклобутанол (его изомеры),
циклобутилметанол (его изомеры), 1-гексанол,
2-гексанол (его изомеры), 3-гексанол (его изомеры),
4-метил-1-пентанол (его изомеры),
3-метил-1-пентанол (его изомеры),
2-метил-1-пентанол (его изомеры), 2-этил-1-бутанол,
3-метил-2-пентанол (его изомеры),
3-метил-3-пентанол (его изомеры), циклогексанол,
1-метил-1-циклопентанол (его изомеры),
2-метил-1-циклопентанол (его изомеры),
циклобутилметанол (его изомеры),
2-циклобутилэтанол (его изомеры),
1-циклобутилэтанол (его изомеры),
(1-метилциклобутил)метанол (его изомеры),
(2-метилциклобутил)метанол (его изомеры),
гептанол (его изомеры), циклогексилметанол (его изомеры),
(метилциклогексил)метанол (его изомеры),
циклогексилэтанол (его изомеры),
(этилциклобутил)метанол (его изомеры),
(метилциклопропил)этанол (его изомеры),
(этилциклопропил)метанол (его изомеры), октанол (его изомеры) и
гексенол; и первичные или вторичные аралкиловые спирты, имеющие 7-8 атомов углерода, такие как бензиловый спирт.
Из группы описанных выше гидроксисоединений, более предпочтительными являются первичные алкиловые спирты и аралкиловые спирты, которые имеют температуру кипения при атмосферном давлении, превышающую температуру кипения воды, и в которых атом углерода с присоединенной к нему гидроксильной группой образует СН2-ОН. Наиболее предпочтительными спиртами являются 1-бутанол, 2-метил-1-пропанол и алкиловые спирты, имеющие 5-8 атомов углерода. Такие гидроксисоединения могут быть использованы отдельно или в виде смеси соединений, выбранных из вышеуказанной группы.
Помимо указанных исходных соединений и/или реагентов могут быть добавлены и другие металлорганические или неорганические металлсодержащие соединения, либо может быть также добавлен растворитель.
Ниже описаны алкоксиды диалкилолова, полученные способом согласно изобретению.
Алкоксидами диалкилолова, полученными способом согласно изобретению, являются алкоксиды диалкилолова, полученные посредством взаимодействия вышеописанного(ых) исходного(ых) соединения(ий) с реагентом(ами).
Алкоксидами диалкилолова, полученными способом согласно изобретению, являются тетраалкилдиалкоксидистанноксаны, имеющие структуру, представленную нижеследующей формулой (22), и диалкоксиды диалкилолова, имеющие структуру, представленную формулой (16). Такими соединениями могут быть мономеры, агрегаты, олигомеры или полимеры.
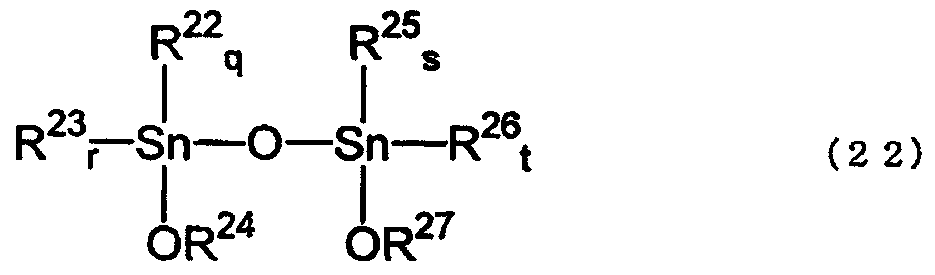
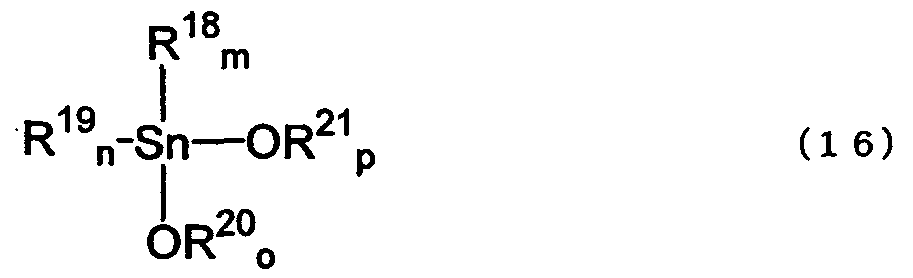
где R22, R23, R25, R26, R18 и R19, каждый соответствует любому одному из значений R1, R2, R4, R5, R7 и R8 в исходном(ых) соединении(ях); R24, R27, R20 и R21 выбраны из R3, R6 и R9 соответствующего исходного соединения и реагента (при условии, что, по меньшей мере, любой один из R24 и R27 является таким, как определено для R9); значения q, r, s, t, m и n зависят от исходного(ых) соединения(ий) и представляют собой целые числа от 0 до 2, q+r=2; s+t=2; и m+n=2; o и р представляют собой целые числа от 0 до 2, o+р=2.
Тетраалкилдиалкоксидистанноксанами, представленными вышеуказанной формулой (22), являются тетраалкилдиалкоксидистанноксаны, полученные из исходного(ых) соединения(ий) и реагента(ов). Если реагент(ы) содержит(ат) соединения, представленные формулой (1) и/или формулой (2), то R22, R23, R25 и R26, каждый соответствует любому одному из значений R1, R2, R4, R5, R7 и R8 в формуле (1) и/или в формуле (2), и R24 и R27, каждый соответствует любому одному из значений R3, R6 и R9 в формуле (1) и/или в формуле (3) (при условии, что, по меньшей мере, любой один из R24 и R27 является таким, как определено для R9). Примерами таких тетраалкилдиалкоксидистанноксанов являются тетраалкилдиалкоксидистанноксаны, представленные вышеуказанной формулой (1).
Диалкоксидами диалкилолова, представленными вышеуказанной формулой (16), являются диалкоксиды диалкилолова, полученные из исходного(ых) соединения(ий) и реагента(ов).
Если реагент(ы) содержит(ат) соединения, представленные формулой (1) и/или формулой (2), то R18 и R19 в оксидах диалкилолова, представленных формулой (16), соответствует любому одному из значений R1, R2, R4, R5, R7 и R8 в формуле (1) и/или в формуле (2), и R20 и R21, каждый соответствует любому одному из значений R3, R6 и R9 в формуле (1) и/или в формуле (3) (при условии, что, по меньшей мере, любой один из R20 и R21 является таким, как определено для R9).
Примерами таких диалкоксидов диалкилолова являются диметилди(н-бутокси)олово, диметил-бис(2-метилпропилокси)олово,
диметилдипентилоксиолово (его изомеры),
диметилдигексилоксиолово (его изомеры),
диметилдигептилоксиолово (его изомеры),
диметилдиоктилоксиолово (его изомеры),
диметилдинонилоксиолово (его изомеры),
диметилдидецилоксиолово (его изомеры),
бутилметилди(н-бутокси)олово,
бутилметил-бис(2-метилпропилокси)олово (его изомеры),
бутилметилдипентилоксиолово (его изомеры),
бутилметилдигексилоксиолово (его изомеры),
бутилметилдигептилоксиолово (его изомеры),
бутилметилдиоктилоксиолово (его изомеры),
этилбутилди(н-бутокси)олово,
этилбутил-бис(2-метилпропилокси)олово,
этилбутилдипентилоксиолово (его изомеры),
этилбутилдигексилоксиолово (его изомеры),
этилбутилдигептилоксиолово (его изомеры),
этилбутилдиоктилоксиолово (его изомеры),
бутилпропилди(н-бутокси)олово,
бутилпропил-бис(2-метилпропилокси)олово,
бутилпропилдипентилоксиолово (его изомеры),
бутилпропилдигексилоксиолово (его изомеры),
бутилпропилдигептилоксиолово (его изомеры),
бутилпропилдиоктилоксиолово (его изомеры),
дибутилди(н-бутокси)олово, дибутил-бис(2-метилпропилокси)олово,
дибутилдипентилоксиолово (его изомеры),
дибутилдигексилоксиолово (его изомеры),
дибутилдигептилоксиолово (его изомеры),
дибутилдиоктилоксиолово (его изомеры),
дибутилдинонилоксиолово (его изомеры),
дибутилдидецилоксиолово (его изомеры), дибутилдибензилоксиолово,
дибутилдифенилэтоксиолово, дифенилди(н-бутокси)олово,
дифенил-бис(2-метилпропилокси)олово,
дифенилдипентилоксиолово (его изомеры),
дифенилдигексилоксиолово (его изомеры),
дифенилдигептилоксиолово (его изомеры),
дифенилдиоктилоксиолово (его изомеры),
дифенилдинонилоксиолово (его изомеры),
дифенилдидецилоксиолово (его изомеры),
дифенилдибензилоксиолово, дифенилдифенилэтоксиолово,
ди(н-бутокси)дитрифторбутилолово,
бис(2-метилпропилокси)дитрифторбутилолово,
дипентилоксидитрифторбутилолово (его изомеры),
дигексилоксидитрифторбутилолово (его изомеры),
дигептилоксидитрифторбутилолово (его изомеры),
диоктилоксидитрифторбутилолово (его изомеры),
динонилоксидитрифторбутилолово (его изомеры),
дидецилоксидитрифторбутилолово (его изомеры),
дибензилоксидитрифторбутилолово,
дифенилэтоксидитрифторбутилолово,
ди(н-бутокси)дипентафторбутилолово,
бис(2-метилпропилокси)дипентафторбутилолово (его изомеры),
дипентилоксидипентафторбутилолово (его изомеры),
дигексилоксидипентафторбутилолово (его изомеры),
дигептилоксидипентафторбутилолово (его изомеры),
диоктилоксидипентафторбутилолово (его изомеры),
динонилоксидипентафторбутилолово (его изомеры),
дидецилоксидипентафторбутилолово (его изомеры),
дибензилоксидипентафторбутилолово (его изомеры),
дифенилэтоксидипентафторбутилолово,
ди(н-бутокси)дигептафторбутилолово,
бис(2-метилпропилокси)дигептафторбутилолово,
дипентилоксидигептафторбутилолово (его изомеры),
дигексилоксидигептафторбутилолово (его изомеры),
дигептилоксидигептафторбутилолово (его изомеры),
диоктилоксидигептафторбутилолово (его изомеры),
динонилоксидигептафторбутилолово (его изомеры),
дидецилоксидигептафторбутилолово (его изомеры),
дибензилоксидигептафторбутилолово,
дифенилэтоксидигептафторбутилолово,
ди(н-бутокси)динонафторбутилолово,
бис(2-метилпропилокси)динонафторбутилолово, дипентилоксидинонафторбутилолово (его изомеры),
дигексилоксидинонафторбутилолово (его изомеры),
дигептилоксидинонафторбутилолово (его изомеры),
диоктилоксидинонафторбутилолово (его изомеры),
динонилоксидинонафторбутилолово (его изомеры),
дидецилоксидинонафторбутилолово (его изомеры),
дибензилоксидинонафторбутилолово и
дифенилэтоксидинонафторбутилолово.
Ниже описан метод анализа исходных соединений, используемых в настоящем изобретении, и соединений, полученных посредством описанной реакции.
Для анализа алкоксидов алкилолова, представленных формулами (1), (7), (22) и (16), может быть применен119Sn-ЯМР-метод. Этот метод известен как метод анализа алкоксидов алкилолова (см., например, US-5545600). Однако величины119Sn-ЯМР-сдвигов для диалкоксидов диалкилолова, представленных формулой (16), широко варьируются в зависимости от концентрации металлорганических соединений, представленных формулой (16), или от присутствия спирта в образце и поэтому указанный анализ предпочтительно проводить с применением1Н-ЯМР вместе с13С-ЯМР.
В качестве примера в таблице показаны величины119Sn-ЯМР-сдвигов, которые соответствуют структуре алкоксида алкилолова формулы (16), синтезированного с использованием 2-этил-1-гексанола в качестве реагента и оксида дибутилолова в качестве исходного соединения.

Ниже приводится подробное описание стадий способа получения соединений согласно изобретению.
Настоящее изобретение относится к способу получения алкоксидов алкилолова, включающему проведение реакции дегидратации исходного соединения, выбранного из группы, состоящей из тетраалкилдиалкоксидистанноксанов, представленных химической формулой (1); оксидов диалкилолова, представленных химической формулой (2), и их смеси; агрегатов или полимеров, с реагентом, представленным химической формулой (3), а именно с гидроксисоединением, с образованием алкоксида алкилолова, представленного химической формулой (22) и/или химической формулой (16), который соответствует исходному соединению и реагенту, где указанный способ отличается тем, что исходное соединение и реагент непрерывно подаются в реактор; низкокипящие компоненты, включающие воду, удаляются из реактора; и реакционный раствор, как компонент, накапливающийся в нижней части реактора и содержащий алкоксид алкилолова, представленный химической формулой (22) и/или химической формулой (16), непрерывно выводится из реактора.
Исходный материал и реагент могут подаваться в реактор отдельно либо перед подачей в реактор они могут быть смешаны друг с другом. Если исходным материалом является твердый материал, то он может быть доведен до жидкого состояния путем нагревания, либо он может быть доведен до жидкого состояния или состояния взвеси с помощью реагента и/или растворителя и затем подан в реактор. Исходный материал и реагент могут подаваться в реактор непрерывно или периодически.
Авторами настоящего изобретения было высказано предположение, что взаимодействие исходного материала с реагентом, осуществляемое способом согласно изобретению, является равновесной реакцией, представленной следующими уравнениями (17) и (18)
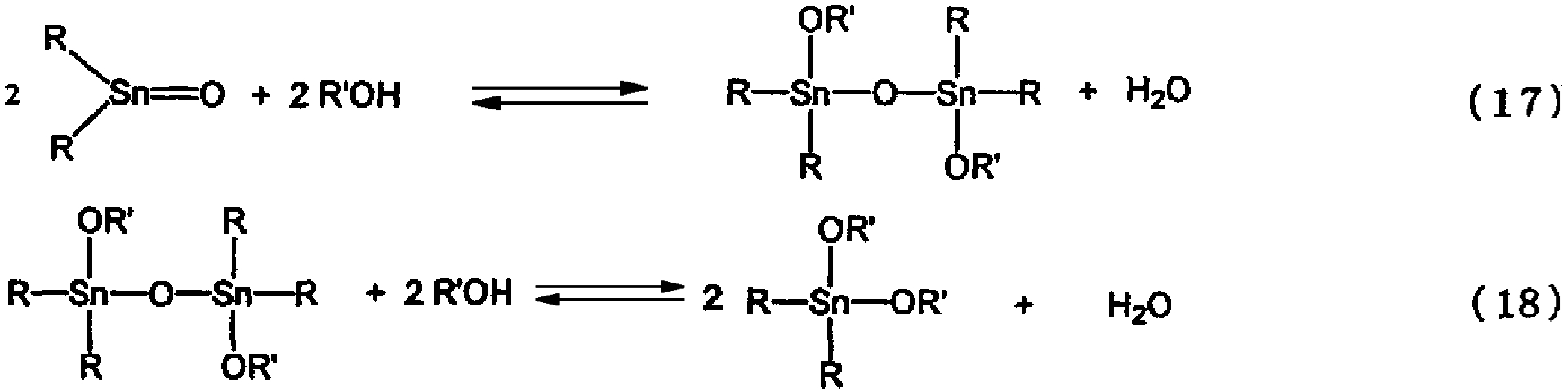
В соответствии с настоящим изобретением исходный материал и реагент подвергают реакции дегидратации в реакторе в соответствии с вышеуказанной схемой реакции (17) и/или (18) и низкокипящие компоненты, содержащие воду, удаляют из реактора, в результате чего тетраалкилдиалкоксидистанноксан и/или диалкоксид диалкилолова могут непрерывно выводиться из нижней части реактора.
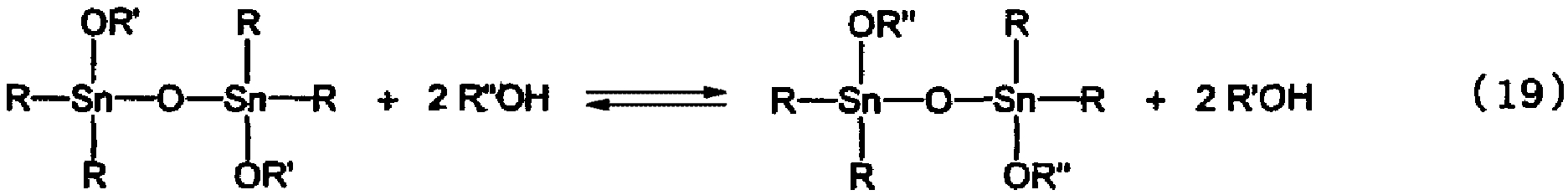
При использовании тетраалкилдиалкоксидистанноксана в качестве исходного материала и гидроксисоединения в качестве реагента, отличающегося от вышеуказанного дистанноксана отсутствием алкоксигруппы, иногда образуется соединение, являющееся продуктом переноса алкоксигруппы посредством обменной реакции, вероятно протекающей в соответствии с нижеследующей реакционной формулой (19).
В настоящем изобретении тип реактора, используемого для реакции дегидратации, не ограничен каким-либо одним конкретным видом. При этом может быть использован любой известный реактор резервуарного типа или колонного типа при условии, что образующиеся низкокипящие компоненты, содержащие воду, могут быть удалены из реактора путем перегонки, и образующаяся высококипящая реакционная смесь, содержащая образовавшиеся алкоксид алкилолова или смесь алкоксидов алкилолова, может быть выведена в жидком состоянии из нижней части реактора. Для этого могут быть применены известные способы, в которых может быть использован реактор, включающий смесительный резервуар, резервуар для многостадийного смешения, дистилляционную колонну, многоходовую дистилляционную колонну, многотрубчатый реактор, многоходовую дистилляционную колонну для непрерывной перегонки, насадочную колонну, тонкопленочный испаритель, реактор с носителем, реактор с принудительной циркуляцией, испаритель с падающей пленкой, испаритель с падающей каплей, реактор, имеющий слой со струйным течением жидкости или колонну-барботер, или в которых может быть использована комбинация реакторов вышеописанных типов. Для достижения эффективного смещения равновесия реакции дегидратации в сторону продуктов предпочтительно использовать колонный реактор. Такой реактор, предпочтительно реактор колонного типа, имеет большую площадь контакта газа с жидкостью, что позволяет образовавшейся воде быстро переходить в газовую фазу. Особенно предпочтительным является непрерывный способ, в котором используются многотрубчатый реактор, многоходовая дистилляционная колонна и насадочная колонна.
В качестве многоходовой дистилляционной колонны может быть использована любая многоходовая дистилляционная колонна, при условии, что ее теоретическое число тарелок равно двум или более и что она пригодна для непрерывной перегонки. При этом может быть использована многоходовая дистилляционная колонна любого типа, при условии, что она обычно используется как многоходовая дистилляционная колонна, например колонна тарельчатого типа, в которой используются тарелки, такие как колпачковые тарелки, перфорированные тарелки, клапанные тарелки или поперечно-точные тарелки; или колонна насадочного типа, в которой используются насадки различных типов, такие как кольцо Рашига, кольцо Лессинга, кольцо Палля, седло Берля, блокирующее седло, насадка Диксона, насадка Мак Магона, спиральная насадка, насадка Зульцера или насадка Меллапак. При этом может быть использована любая насадочная колонна, при условии, что она упакована вышеуказанным известным насадочным материалом. Могут быть также использованы насадки, эффективные для дегидратации.
Так, например, предпочтительно, использовать такие насадки, как молекулярные сита. В колонных реакторах также предпочтительно использовать колонны смешанного типа, то есть тарельчатые и насадочные колонны. Такие колонные реакторы предпочтительно имеют линии подачи вышеописанного исходного материала и вышеописанного реагента, соответственно, или линию подачи смешанного раствора, состоящего из вышеописанного исходного материала и вышеописанного реагента; линию удаления из реактора низкокипящей реакционной смеси, содержащей воду; и линию выведения из реактора высококипящей реакционной смеси. Особенно предпочтительно, чтобы в вышеописанных колонных реакторах, линия удаления из реактора низкокипящих компонентов, содержащих воду, была расположена таким образом, чтобы она обеспечивала удаление из реактора газофазных компонентов, и линия выведения из реактора полученной высококипящей реакционной смеси была расположена в нижней части реактора. В непрерывном способе в реактор непрерывно или периодически подается исходный материал и реагент, где указанные исходный материал и реагент подвергают реакции дегидратации в жидкой фазе или газожидкой фазе; полученную высококипящую реакционную смесь, включающую полученный алкоксид алкилолова, выводят в жидком состоянии из нижней части реактора; и полученные низкокипящие компоненты, содержащие воду, непрерывно выводят из реактора в виде газа. В результате получают алкоксиды алкилолова.
Вышеописанные колонные реакторы могут быть также снабжены отдельными линиями подачи инертного газа и/или газообразного, и/или жидкого реагента в реакторы из их нижней части, или линией подачи части или всей полученной высококипящей реакционной смеси обратно в реактор для рециркуляции. Низкокипящие компоненты, содержащие воду, которые были удалены из реакторов, могут быть очищены с использованием стандартного оборудования, такого как дистилляционная колонна, где азеотроп и/или сопутствующий реагент подвергаются рециркуляции. В некоторых случаях используемые исходные материалы принимают форму суспензии или твердого вещества при комнатной температуре (20°С), либо они имеют высокую вязкость, и поэтому указанные линии конструируют с учетом возможного засорения или т.п., либо они могут быть снабжены изоляцией и холодильным или нагревательным оборудованием.
При получении алкоксидов алкилолова способом согласно изобретению может быть использован только тот реактор, который удовлетворяет требованиям настоящего изобретения, либо может быть использована комбинация из двух или нескольких таких реакторов. Могут быть также использованы реактор, который удовлетворяет требованиям настоящего изобретения, и комбинация других реакторов, используемых для получения алкоксидов алкилолова. Так, например, некоторыми из вариантов настоящего изобретения также являются способы, в которых для осуществления взаимодействия оксида диалкилолова и спирта проводят реакцию с получением первой партии, содержащей лишь некоторое количество алкоксидов алкилолова, и затем для инициации реакции в реакционном растворе используют реактор, удовлетворяющий требованиям настоящего изобретения.
Конкретные примеры реакторов, которые могут быть использованы в соответствии с настоящим изобретением, описаны в прилагаемом графическом материале, однако при этом следует отметить, что реакторы, которые могут быть использованы в соответствии с настоящим изобретением, не ограничиваются этими примерами. Реакторы могут быть также снабжены системами приборов, такими как расходомер и термометр, и известными технологическими системами, такими как кипятильник, насос, холодильник и дистилляционная колонна, в зависимости от конкретной ситуации. Нагревание может быть осуществлено с использованием известного оборудования, такого как аппарат для обработки паром и нагреватель, и охлаждение также может быть осуществлено известными способами, такими как естественное охлаждение, охлаждение водой, и солевым раствором.
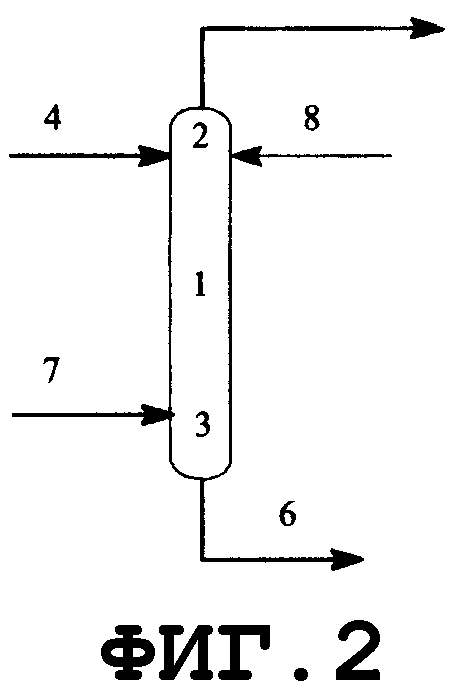
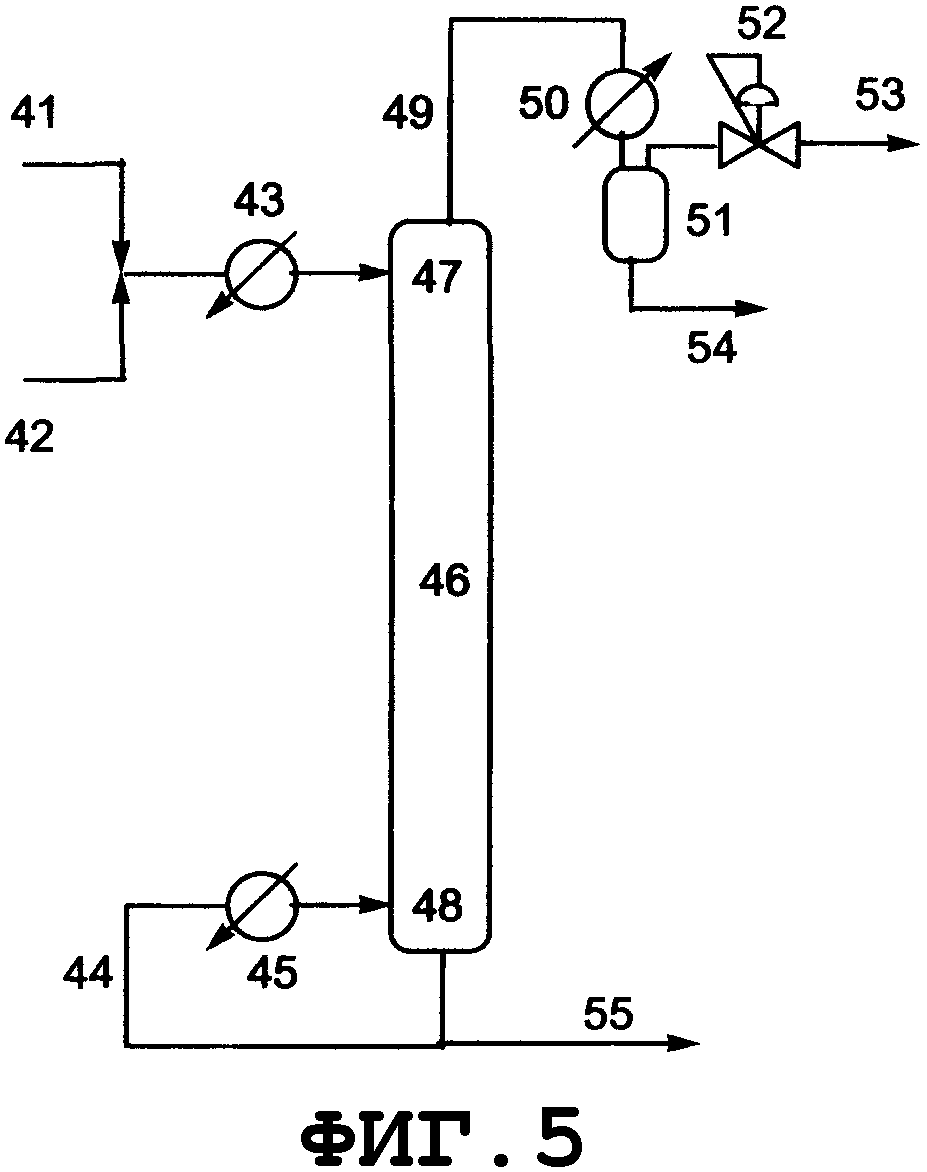
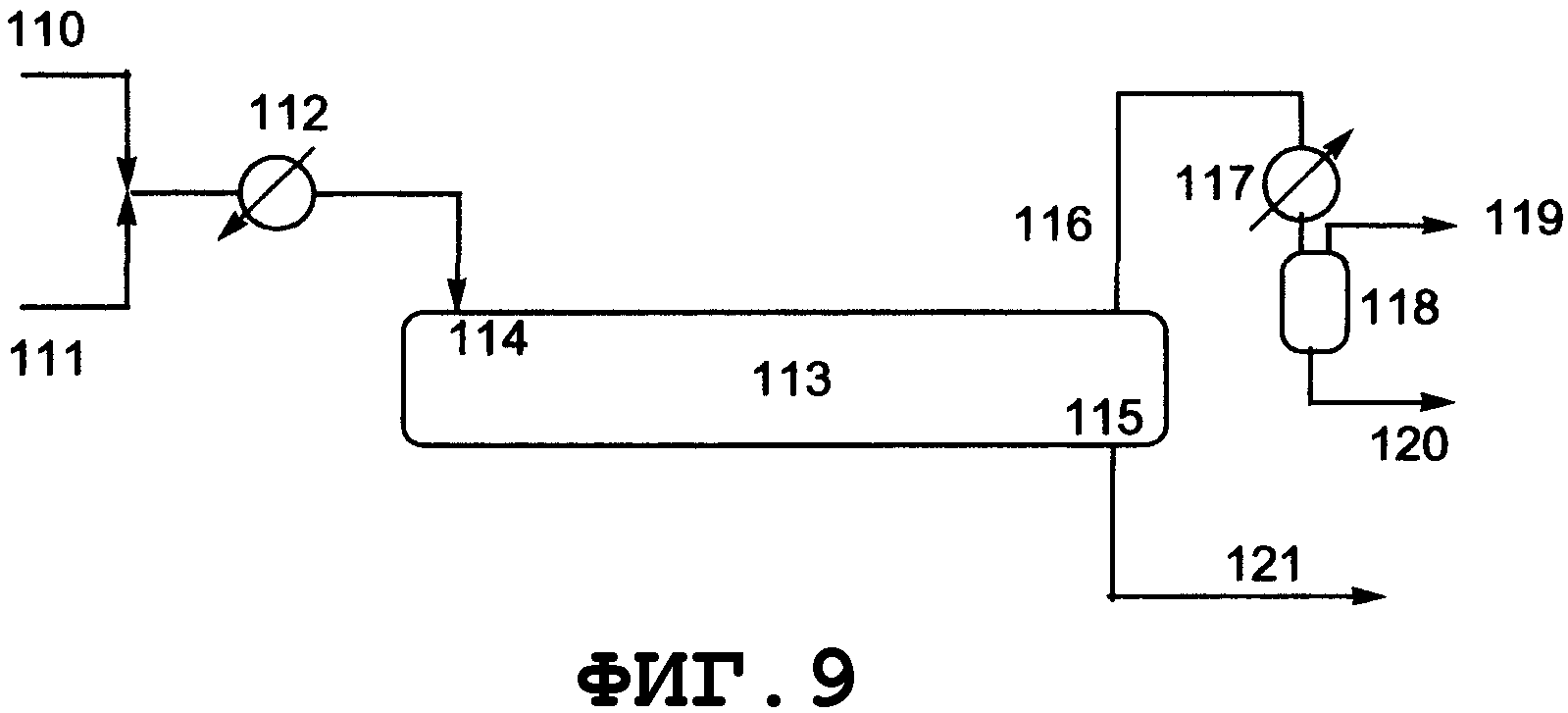
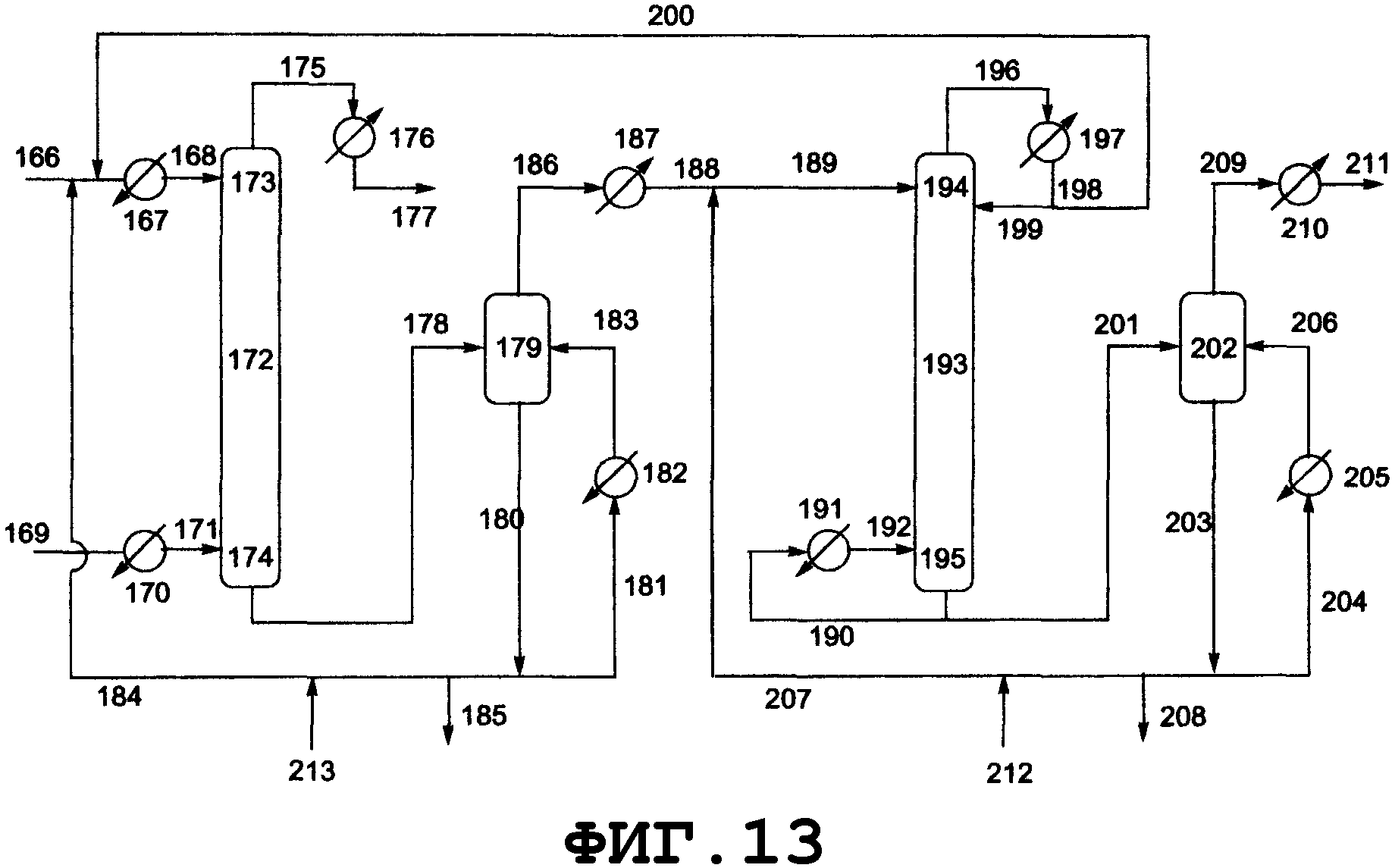
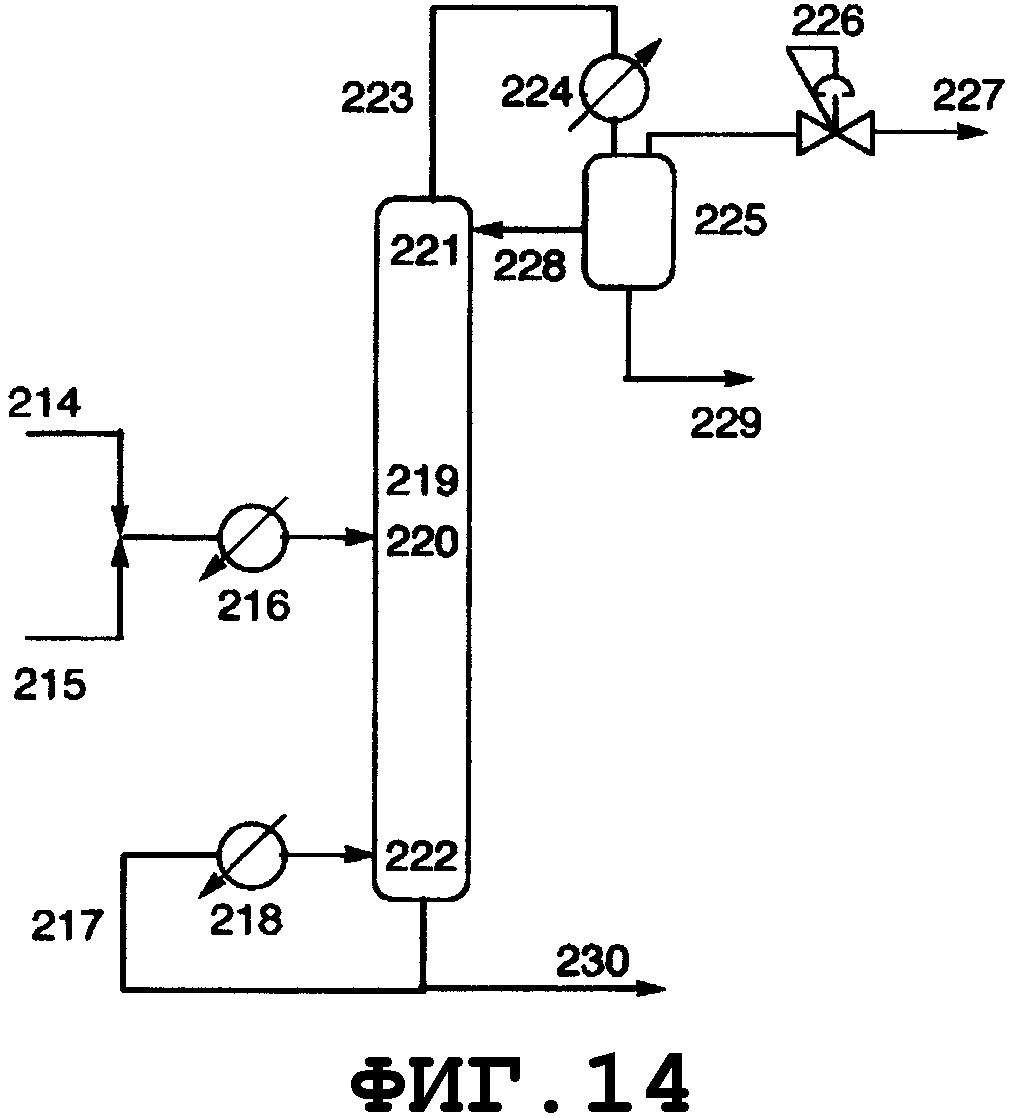
На фиг.2 показано поперечное сечение колонного реактора, вид спереди. Таким колонным реактором может быть реактор, который включает колонну с насадкой, многоходовую дистилляционную колонну или колонну любого другого типа. Описанным в данном описании реактором колонного типа является колонный реактор, включающий насадочную колонну. Смешанный раствор исходного материала и реагента вводят из питающего трубопровода 4 в реактор 1, либо исходный материал вводят из питающего трубопровода 4, и реагент вводят из питающего трубопровода 8 в реактор 1. Инертный газ вводят из питающего газового трубопровода 7 в реактор 1. Вводимый исходный материал и реагент диспергируются внутри реактора. Вышеуказанный смешанный раствор поступает вниз по насадке, упакованной в реакторе, и вода испаряется. Процесс внутри реактора может регулироваться при пониженном давлении, атмосферном давлении или при повышенном давлении, и инертный газ поступает из питающего газового трубопровода 7, в зависимости от ситуации, и/или низкокипящие компоненты, включающие газообразный реагент и воду, образующиеся в процессе реакции, поступают вниз из верхней части 2 реактора и выгружаются из спускного трубопровода 5. Реакционный раствор, образующийся внутри реактора и имеющий повышенную концентрацию алкоксида алкилолова в качестве продукта, выводится из нижней части 3 реактора и выгружается из отводного трубопровода 6. Указанные насадочная колонна и трубопроводы нагреваются или охлаждаются, в зависимости от ситуации, известными средствами, такими как рубашка или нагреватель.
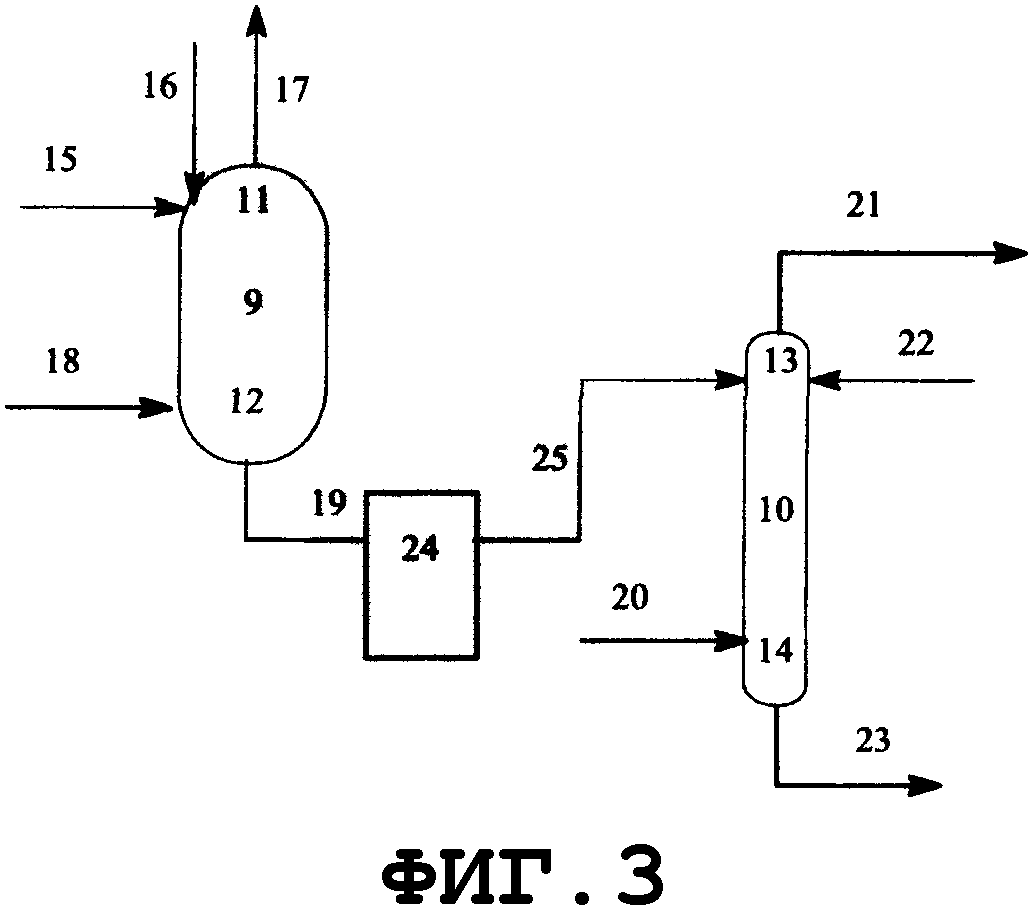
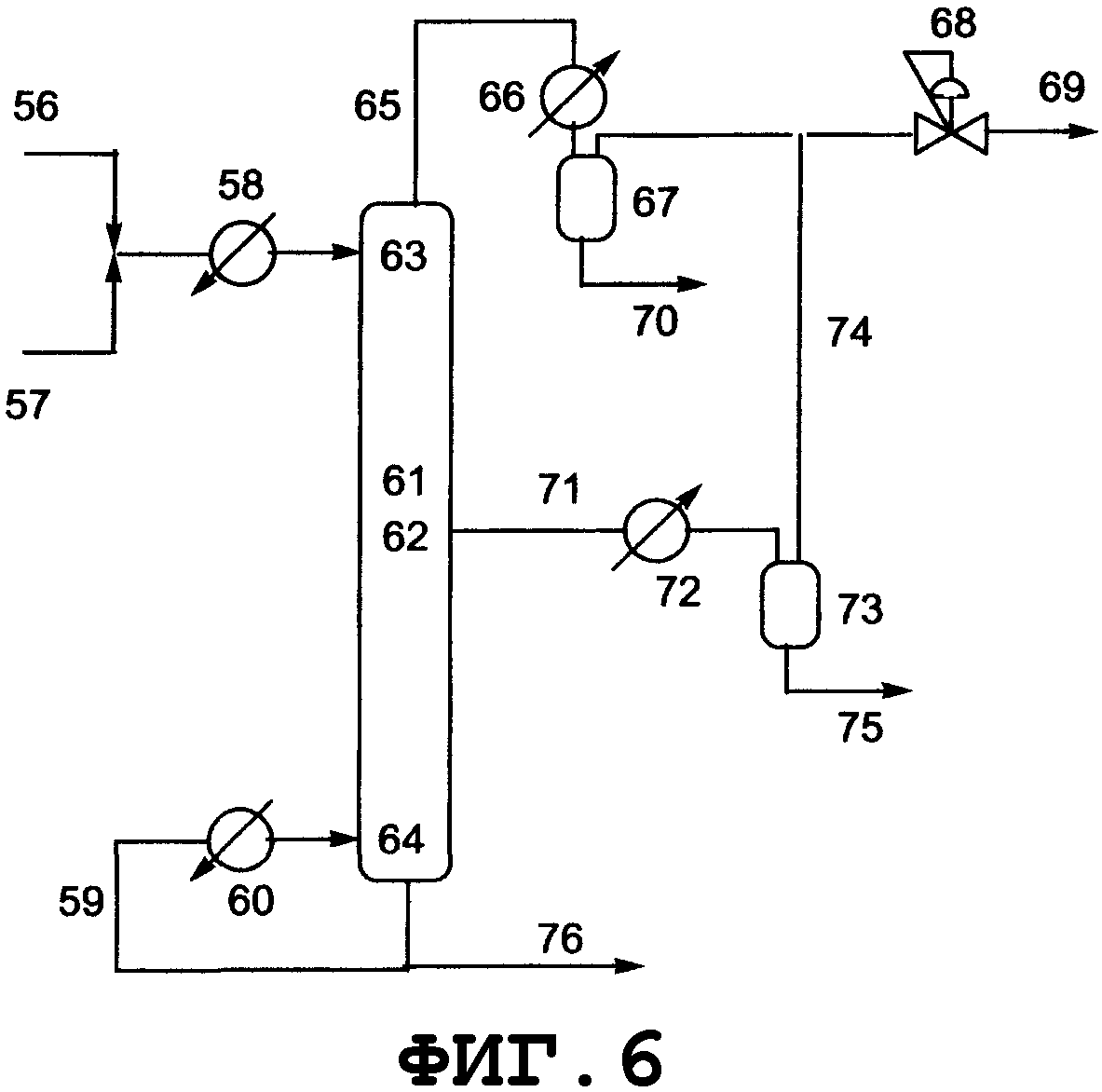
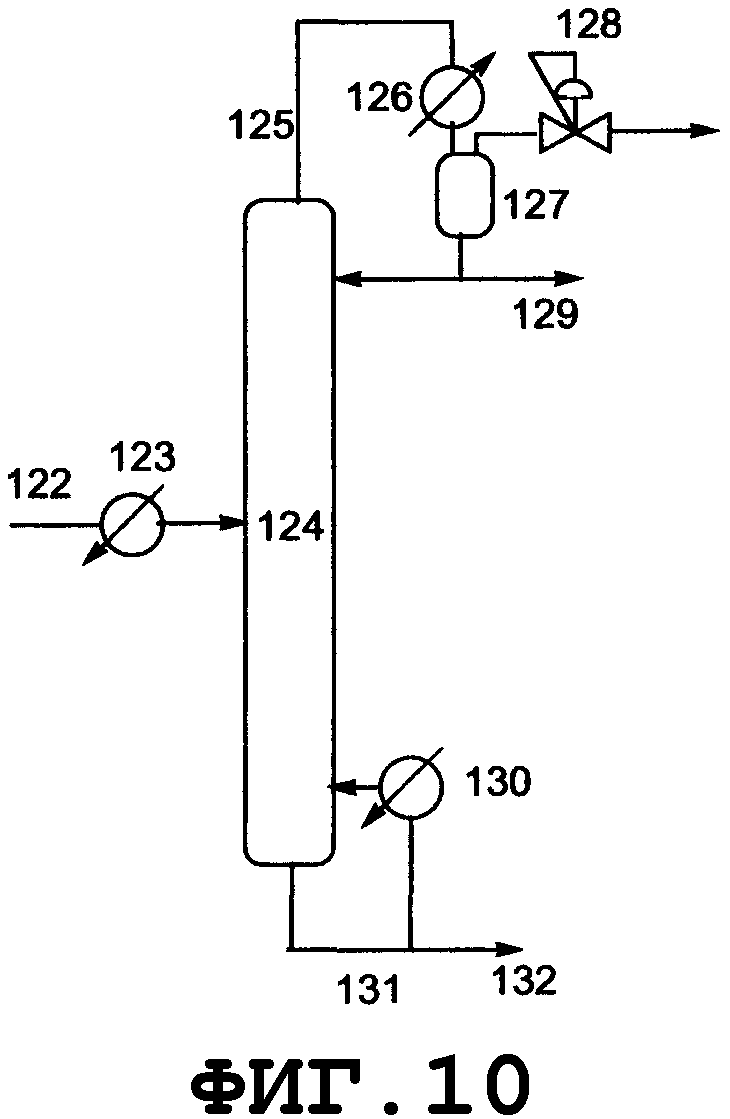
На фиг.3 показано поперечное сечение реактора смешанного типа, включающего резервуарный и колонный реактор, вид спереди. Резервуарный реактор может включать смесительный резервуар, циркуляционный резервуар или резервуар любого другого типа. В настоящем изобретении будет описан реактор резервуарного типа, включающий смесительный резервуар. Колонным реактором может быть насадочная колонна с имеющейся внутри нее насадкой, многоходовая дистилляционная колонна или колонный реактор любого другого типа. В настоящем изобретении будет описан колонный реактор, включающий насадочную колонну с имеющейся в ней насадкой. Реагент вводят из питающего трубопровода 15 в смесительный резервуар 9 и исходное вещество вводят из питающего трубопровода 16 в смесительный резервуар 9. Вводимый исходный материал и реагент диспергируются в смесительном резервуаре. Вышеуказанный смешанный раствор нагревается, в результате чего вода испаряется. Процесс внутри смесительного резервуара может регулироваться при пониженном давлении, при атмосферном давлении или при повышенном давлении; инертный газ поступает из питающего газового трубопровода 18, в зависимости от ситуации, и/или низкокипящие компоненты, включающие газообразный реагент и воду, образующиеся в результате реакции, поступают вниз из верхней части 11 смесительного резервуара и выгружаются из спускного трубопровода 17.
Реакционный раствор, образующийся внутри смесительного резервуара и имеющий повышенную концентрацию алкоксида алкилолова в качестве продукта, поступает из нижней части 12 смесительного резервуара в буферный резервуар 24 через соединительный трубопровод 19 и затем из буферного резервуара в колонный реактор через соединительный трубопровод 25. Раствор, введенный в реактор 10 через соединительный трубопровод 25 и содержащий алкоксид диалкилолова, диспергируется благодаря насадке, присутствующей внутри реактора. Вышеуказанный смешанный раствор стекает вниз по насадке, присутствующей в реакторе, и вода испаряется. Процесс внутри реактора может регулироваться при пониженном давлении, атмосферном давлении или при повышенном давлении; инертный газ поступает из питающего газового трубопровода 20 в зависимости от ситуации; и/или низкокипящие компоненты, включающие газообразный реагент и воду, образующиеся в процессе реактора, поступают вниз из верхней части 13 реактора и выгружаются из спускного трубопровода 21. Реакционный раствор, образующийся внутри реактора и имеющий повышенную концентрацию алкоксидов диалкилолова в качестве продуктов, выводится из нижней части 14 реактора и выгружается из отводного трубопровода 23. Реагент может пополняться из питающего трубопровода 22, в зависимости от ситуации. Смесительный резервуар, насадочная колонна и указанные трубопроводы нагреваются или охлаждаются в зависимости от ситуации известными средствами, такими как рубашка или нагреватель.
В реакторах или в трубопроводах могут быть использованы любые материалы, при условии, что они не оказывают негативного влияния на используемый исходный материал или реагент. Предпочтительными и недорогостоящими материалами являются SUS 304, SUS 316 или SUS 316L.
Время прохождения реакции дегидратации, осуществляемой в соответствии с настоящим изобретением (в случае применения непрерывного способа, время пребывания алкоксида алкилолова в реакторе), не имеет конкретных ограничений и обычно составляет от 0,001 до 50 часов, предпочтительно от 0,01 до 10 часов и более предпочтительно от 0,1 до 2 часов.
Температура реакции варьируется в зависимости от типа используемых исходных соединений и обычно, она находится в пределах от 50°С до 350°С, предпочтительно от 60°С до 160°С. Для поддержания в реакторе постоянной температуры вышеописанные реакторы могут быть снабжены стандартным холодильным оборудованием или нагревательным оборудованием. Давление, при котором осуществляется реакция, варьируется в зависимости от типа используемых исходных соединений или температуры реакции, и такая реакция может быть осуществлена при пониженном давлении, атмосферном давлении или при повышенном давлении. Обычно давление находится в пределах от 0,1 до 2,0×107 Па. В настоящем изобретении необязательно использовать реакционный растворитель, однако для облегчения прохождения реакции в качестве реакционных растворителей может быть использован соответствующий инертный растворитель, такой как эфир, алифатический углеводород или ароматический углеводород.
Время реакции и температура реакции будут более подробно описаны ниже.
Настоящее изобретение отличается тем, что предусматривает непрерывную подачу в реактор исходного материала, выбранного из группы, состоящей из оксида диалкилолова, тетраалкилдиалкоксидистанноксана и их смесей, и гидроксисоединения, используемого в качестве реагента, и выведение низкокипящих компонентов из реактора с непрерывным сбором реакционного раствора в виде компонента, аккумулирующегося в нижней части реактора и содержащего алкоксид алкилолова, соответствующий исходному материалу и реагенту. В соответствии с настоящим изобретением, в отличие от общепринятых способов алкоксиды алкилолова, являющиеся целевыми продуктами, могут быть получены с очень высокой эффективностью. Кроме того, неожиданным является тот факт, что настоящее изобретение позволяет осуществлять реакцию дегидратации, которая представляет собой равновесную реакцию, и помимо этого, оно позволяет значительно снизить уровень образования соединения триалкилолова благодаря реакции пиролиза алкоксидов алкилолова.
Если реакцию дегидратации осуществляют способом согласно изобретению в описанных ниже условиях, то количество соединения трибутилолова, образующегося как побочный продукт в процессе реакции дегидратации, может составлять 1% моль или менее на 100% атомов олова, содержащихся в исходном соединении. Разумеется, что если исходный материал содержит соединение трибутилолова, то его количество иногда может выходить за пределы вышеуказанного интервала, и поэтому такое соединение трибутилолова необходимо удалять заранее, либо количество соединения трибутилолова, содержащегося в исходном материале, необходимо регулировать таким образом, чтобы оно не выходило за пределы допустимого интервала. Кроме того, поскольку образование соединения трибутилолова стимулируется не только в процессе реакции дегидратации, но и при пиролизе алкоксида алкилолова, то для ингибирования образования этого побочного продукта, то есть соединения трибутилолова, предпочтительно, снижать время пребывания алкоксида алкилолова в трубопроводах, а также снижать температуру в этих трубопроводах. Для снижения количества образующегося соединения трибутилолова вместо реактора для дегидратации может быть использовано и другое оборудование.
При образовании алкоксида алкилолова из тетраалкилдиалкоксидистанноксана, представленного химической формулой (1), и/или оксида диалкилолова, представленного химической формулой (2) и используемого в качестве исходного соединения, и спирта, представленного химической формулой (3) и используемого в качестве реагента, алкоксид алкилолова, представленный химической формулой (22) и/или химической формулой (16) и содержащий лишь очень небольшое количество соединения трибутилолова, может быть получен путем проведения реакции дегидратации при скорости дегидратации, определяемой по уравнению (4). При этом может быть также использован смешанный раствор, состоящий из оксида диалкилолова, представленного химической формулой (2), и спирта, представленного химической формулой (3), однако с точки зрения растворимости или подачи этого смешанного раствора предпочтительно использовать тетраалкилдиалкоксидистанноксан, представленный химической формулой (1), и спирт, представленный химической формулой (3)
где "скорость дегидратации" означает количество воды, которое образуется в процессе реакции дегидратации и выводится из системы за единицу времени [моль·час-1]; Х означает общее число молей атомов олова, имеющихся в соединении алкилолова, представленном общей формулы (2) и содержащемся в исходном материале; Y означает общее число молей [моль] атомов олова, имеющихся в соединении алкилолова, представленном общей формулы (1) и содержащемся в исходном материале; Т означает температуру [К], при которой осуществляется реакция дегидратации; R означает газовую постоянную, равную 8,314 Дж·моль-1·К-1; и А и В представляют собой коэффициенты, зависящие от типа соединения алкилолова, где коэффициенты А и В в вышеуказанном уравнении (4) зависят от типа соединения алкилолова, используемого в качестве исходного материала, и вычислены исходя из выбранного первичного стандартного вещества. Если исходный материал содержит соединения алкилолова, представленные химической формулой (1), то вышеуказанные коэффициенты А и В представляют собой фактор частоты и энергию активации пиролитической реакции исходного стандартного вещества, которым является соединение алкилолова, произвольно выбранное из соединений алкилолова, представленных химической формулой (1) и содержащихся в исходном материале, и эти коэффициенты вычисляют по уравнению (5). Если исходный материал не содержит соединений алкилолова, представленного химической формулой (1), но содержит соединения алкилолова, представленные химической формулой (2), то указанные коэффициенты А и В представляют собой фактор частоты и энергию активации пиролитической реакции первичного стандартного вещества, которым является алкоксид алкилолова, произвольно выбранный из алкоксидов алкилолова, представленных химической формулой (7) и образованных из соединений алкилолова, представленных химической формулой (2) и содержащихся в исходном материале и реагенте, где указанные коэффициенты вычисляют по уравнению (5)
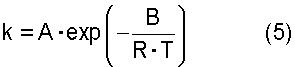
где k представляет собой константу скорости первого порядка [час-1]; А означает фактор частоты [час-1]; В означает энергию активации [Дж·моль-1]; R означает газовую постоянную, равную 8,314 Дж·моль-1·К-1; и Т означает температуру [К], при которой осуществляется пиролитическая реакция. Вышеуказанная константа k представляет собой константу скорости первого порядка для пиролитической реакции, которую вычисляют по уравнению (6):
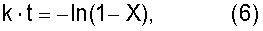
где k означает константу скорости первого порядка [час-1]; t означает время нагревания; и Х [час] означает коэффициент редукции [моль/моль] по отношению к начальной концентрации исходного стандартного вещества.
где R10, R11, R13 и R14 соответствуют значениям R7 или R8 исходного соединения; g, h, i и j соответствуют значениям е или f исходного соединения; и, по меньшей мере, один из R12 и R15 соответствует значениям R9 указанного реагента.
Если указанную реакцию дегидратации осуществляют со скоростью дегидратации, превышающей скорость реакции, описанную уравнением (4), то может быть получен алкоксид алкилолова, включающий меньшее количество соединения триалкилолова. Однако для получения значительно большего количества алкоксида алкилолова, представленного химической формулой (16), реакцию дегидратации предпочтительно проводить со скоростью, превышающей скорость реакции, описанную уравнением (20):
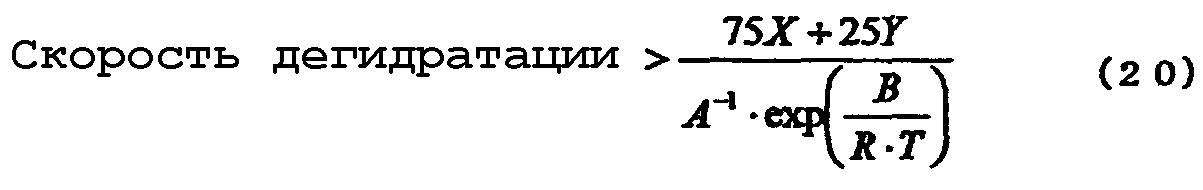
где Х, Y, А, В, R и Т, каждый имеет значения, как указано выше для уравнения (4), и А и В получены из уравнений (5) и (6), аналогично, как вычислено из уравнения (4).
Вышеописанная пиролитическая реакция представляет собой реакцию, описанную формулой (21) и представленную как репрезентативная реакция, в результате которой количество тетраалкилдиалкоксидистанноксанов, представленных химической формулой (1) и/или химической формулой (7), снижается. Более конкретно, изменение снижения количества тетраалкилдиалкоксидистанноксанов, представленных химической формулой (1) и/или химической формулой (7), в зависимости от времени, измеряют с помощью119Sn-ЯМР-спектроскопии при перемешивании раствора, содержащего тетраалкилдиалкоксидистанноксаны, представленные химической формулой (1) и/или химической формулой (7), в атмосфере азота и при постоянной температуре раствора, и затем скорость реакции анализируют в соответствии с вышеуказанными уравнениями (6) и (5). В уравнении (21) тетраалкилдиалкоксидистанноксаны описаны как мономеры, однако совершенно очевидно, что они могут быть димерами, агрегатами, олигомерами или полимерами.
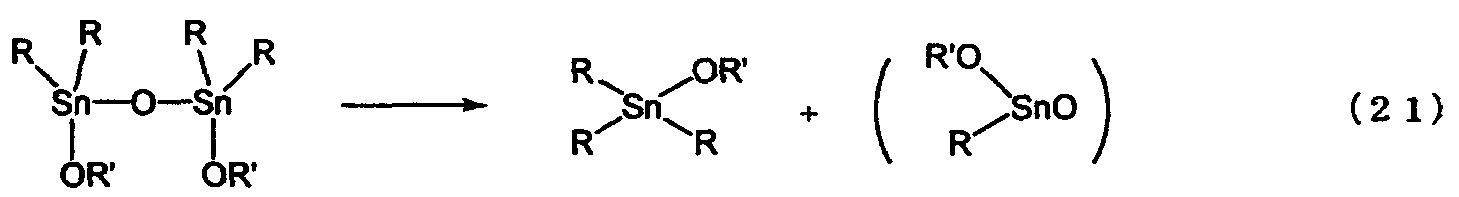
В вышеописанной пиролитической реакции температурой нагревания может быть любая температура в пределах от 100°С до 200°С (например, 120°С, 140°С и 160°С). В вышеописанной пиролитической реакционной системе содержание соединений, представленных химической формулой (1) и/или химической формулой (7), составляет 95% или более. Вышеописанную пиролитическую реакцию осуществляют при нагревании в условиях, которые обеспечивают защиту реакции от воздействия веществ (например, кислорода или воды), способных стимулировать разложение соединений, представленных химической формулой (1) и/или химической формулой (7). Количество соединений, представленных химической формулой (1) и/или химической формулой (7), снижающееся при нагревании в процессе реакции, представленной химической формулой (21), измеряют в зависимости от времени с помощью119Sn-ЯМР-спектроскопии. Продукты пиролитической реакции не могут быть точно определены, но можно с уверенностью сказать, что они включают алкоксид триалкилолова.
Алкоксиды алкилолова, полученные в соответствии с настоящим изобретением и при скорости дегидратации, определенной по уравнению (4) и превышающей эту скорость, имеют очень низкое содержание соединений триалкилолова и хлорсодержащих соединений, поскольку в способе согласно изобретению в качестве исходных материалов не используются хлорсодержащие соединения. Исходные соединения иногда могут включать хлорсодержащее соединение, однако в соответствии с настоящим изобретением, в такой реакции количество такого хлорсодержащего соединения, в принципе, не превышает количество хлорсодержащего соединения в вышеуказанном исходном соединении, и, таким образом, могут быть получены алкоксиды алкилолова с высокой степенью чистоты.
Если количество используемого реагента превышает количество исходного соединения, то химическое равновесие смещается преимущественно в сторону продуктов, однако для увеличения концентрации алкоксида алкилолова в выходящем из реактора растворе, содержащем алкоксид алкилолова, избыток непрореагировавшего гидроксисоединения должен быть удален путем перегонки. При этом потребление энергии значительно снижается. В противоположность этому, если количество используемого реагента является слишком низким, то должно быть собрано большее количество непрореагировавшего исходного соединения. В соответствии с этим, отношение исходного соединения к реагенту находится в пределах от 3 до 200 и означает отношение общего количества молей атомов олова, содержащихся в исходном соединении, к количеству молей указанного реагента. Для увеличения концентрации алкоксида диалкилолова, экстрагируемого из нижней части реактора, такое отношение предпочтительно должно находиться в пределах от 3 до 100 и более предпочтительно в пределах от 3 до 10.
Настоящее изобретение отличается тем, что предусматривает быстрое выведение из системы воды и алкоксида алкилолова, образующихся в такой реакции. Как указывалось выше, авторами настоящего изобретения было высказано предположение, что в общепринятых периодических способах образующаяся вода быстро подвергается обратимой реакции с продуцированным алкоксидом алкилолова, что негативно влияет на производительность реакции. Настоящее изобретение относится к способу, гарантирующему повышение производительности реакции посредством быстрого превращения свободной воды, образующейся в реакционном растворе, в газовую фазу, с ее удалением из реактора и одновременным выведением образовавшегося алкоксида алкилолова из системы. Авторами настоящего изобретения было высказано предположение, что свободная вода, образующаяся в вышеописанной реакции из реакционного раствора, превращается в газовую фазу, что обусловлено равновесием "газ-жидкость" в данной системе.
Целью осуществления способа получения соединений настоящего изобретения является ингибирование обратной реакции, где в указанном способе равновесные реакции, представленные формулами (13) и/или (14), смещаются влево (в сторону продуктов) посредством увеличения конкретной площади поверхности реакционного раствора, что ускоряет превращение образовавшейся воды в газ, которое зависит от равновесия "газ-жидкость", при одновременном выведении полученного алкоксида алкилолова из системы. В соответствии с этим, в вышеописанном резервуарном и/или колонном реакторе для быстрого превращения образовавшейся воды в газовую фазу предпочтительно, чтобы объем жидких компонентов составлял 2/3 или менее, и более предпочтительно 1/3 или менее от общей емкости реактора.
Используемый в данном описании термин "в системе" означает присутствие соединений внутри реакторов, трубопроводов или в периферическом оборудовании реакторов, или в оборудовании или трубопроводах системы экстракции. Термин "высококипящая реакционная смесь" означает подаваемый в реакторы раствор, содержащий вещество с высокой температурой кипения; находящийся в реакторах реакционный раствор, содержащий вещество с высокой температурой кипения; выгружаемый из реакторов раствор, содержащий вещество с высокой температурой кипения; или концентрированный раствор, содержащий повышенную концентрацию вещества с высокой температурой кипения благодаря испарению части реагента. Некоторые высококипящие реакционные смеси содержат растворенные высококипящие вещества, либо некоторые высококипящие реакционный смеси присутствуют в форме взвеси высококипящих веществ. Если высококипящая реакционная смесь присутствует в форме взвеси, то термин "высококипящая реакционная смесь" также включает и нерастворимую часть данной взвеси.
Используемый в данном описании термин "высококипящие вещества" означает органические вещества, имеющие температуру кипения, равную температуре кипения алкоксидов алкилолова или превышающую эту температуру. Так, например, термин "высококипящие вещества" также включает высокомолекулярные побочные продукты, полученные в указанной реакции.
Используемый в данном описании термин "низкокипящие компоненты, содержащие воду" означает воду, образующуюся в данной реакции, и часть реагентов, используемых в качестве органических веществ, имеющих температуру кипения ниже температуры кипения алкоксидов алкилолова, полученных в соответствии с настоящим изобретением. Так, например, термин "низкокипящие вещества" также включает низкомолекулярные побочные продукты, полученные в указанной реакции. При использовании инертных газов или органических растворителей термин "низкокипящее вещество" также включает часть таких органических растворителей.
При получении алкоксида алкилолова путем проведения взаимодействия исходного соединения и реагента способом согласно изобретению скорость реакции увеличивается в результате удаления из реакционной смеси воды, образующейся в процессе реакции. В этом способе для эффективного снижения парциального давления полученных низкокипящих компонентов, содержащих воду, реакцию стимулируют путем подачи инертного газа в систему. В соответствии с этим, предпочтительным способом является способ, при котором в данную систему подают инертный газ, не оказывающий негативного влияния на реакцию, такой как азот, аргон, гелий, диоксид углерода или низший углеводород, в результате чего этот газ захватывает образовавшиеся низкокипящие компоненты, включающие воду, с их удалением из системы, или способ, в котором внутренняя часть системы поддерживается под давлением, подходящим для удаления воды, образующейся в данной системе, то есть таком, чтобы образовавшаяся вода или азеотропные компоненты, включающие воду, находились под давлением пара при реакционной температуре, другими словами, чтобы образовавшаяся вода или азеотропные компоненты, включающие воду, могли переходить из жидкой фазы в газовую фазу.
Из вышеуказанных инертных газов диоксид углерода иногда может взаимодействовать с полученным алкоксидом алкилолова, в результате чего образуется алкоксид алкилолова, содержащий оксид углерода, или дополнительно образуется небольшое количество эфира угольной кислоты из указанного алкоксида алкилолова, содержащего оксид углерода. Однако поскольку диоксид углерода не оказывает значительного негативного влияния на реакцию, то он также входит в число инертных газов. Как показано в реакционных формулах (13) и/или (14), при увеличении концентрации гидроксисоединения, используемого в качестве реагента, химическое равновесие преимущественно смещается в сторону продуктов.
В частности, внутри вышеописанного реактора, по мере прохождения реакции, гидроксисоединение, используемое в качестве реагента, потребляется в качестве алкоксигруппы для алкоксида алкилолова, и концентрация гидроксисоединения постепенно снижается. Таким образом, эффект удаления образовавшихся низкокипящих компонентов, включающих воду, иногда может быть достигнут путем подачи реагента или реагента в газообразном состоянии даже из нижней части реактора для увеличения концентрации гидроксисоединения, используемого в качестве реагента, или путем подачи гидроксисоединения в газообразном состоянии из нижней части реактора, таким образом, чтобы газ захватывал низкокипящие компоненты, содержащие воду с их выведением из системы, аналогично случаям использования инертных газов.
Совершенно очевидно, что указанное гидроксисоединение или гидроксисоединение в газообразном состоянии, органическое соединение в виде инертного газа и/или органический растворитель, которые образуют азеотропную смесь с водой или органическим растворителем в газообразном состоянии, могут подаваться вместе с инертным газом из нижней части реактора. В качестве циркулирующего инертного газа или газа-реагента предпочтительно использовать газ, содержащий, по возможности, минимальное количество кислорода. В этом случае, такой газ может циркулировать через молекулярные сита или слой, упакованный ионообменной смолой или восстановителем, и может быть также использован газ, охлажденный до крайне низкой температуры и находящийся в дегидратированном состоянии. Содержание воды в циркулирующем газе определяется "точкой росы", которая, предпочтительно, составляет -10°С или менее, и более предпочтительно, -40°С или менее. При подаче инертного газа из нижней части реактора количество подаваемого инертного газа не имеет конкретных ограничений. Это количество зависит от типа, структуры или размера реактора. При использовании дистилляционной колонны в качестве реактора необходимо соблюдать меры предосторожности, чтобы не допустить сильного затопления колонны.
Алкоксиды алкилолова, полученные способом согласно изобретению, могут быть использованы как таковые, либо они могут быть использованы в разбавленной или концентрированной форме, либо они могут быть использованы вместе с другими компонентами.
Алкоксиды алкилолова известны как катализаторы, используемые для получения эфиров угольной кислоты, таких как диалкиловые эфиры угольной кислоты, алкилариловые эфиры угольной кислоты и диариловые эфиры угольной кислоты; изоцианатов; и поликарбонатов. Алкоксиды алкилолова, полученные способом согласно изобретению, имеют высокую чистоту и являются не дорогостоящими, и поэтому они могут быть использованы для промышленного производства эфиров угольной кислоты, таких как диалкиловые эфиры угольной кислоты, алкилариловые эфиры угольной кислоты и диариловые эфиры угольной кислоты; изоцианатов; и более предпочтительно, для получения поликарбонатов.
В частности, алкоксиды алкилолова, полученные способом согласно изобретению, отличаются тем, что они включают лишь очень небольшое количество соединений трибутилолова и соединений хлора. Такие алкоксиды диалкилолова весьма полезны в качестве катализаторов, используемых для получения эфиров угольной кислоты, таких как диалкиловые эфиры угольной кислоты, алкилариловые эфиры угольной кислоты и диариловые эфиры угольной кислоты; изоцианатов; и поликарбонатов, а также для синтеза эфиров, для переэтерификации и для вулканизации силиконовых полимеров или уретанов.
Использование некоторых соединений триалкилолова очень ограничено из-за их токсичности. Известно также, что присутствие хлорсодержащих соединений вызывает коррозию металла или разрушение полимеров. Если в качестве вышеописанных катализаторов используются традиционные алкоксиды алкилолова, то продукты, полученные с применением таких катализаторов, имеют примеси вышеописанных вредных соединений триалакилолова или соединений хлора. Однако до сих пор было неизвестно, на какой именно стадии происходит загрязнение продуктов указанными соединениями и от каких именно соединений образуются такие примеси.
После многочисленных исследований, направленных на решение вышеописанных проблем, авторами настоящего изобретения было обнаружено, что большинство соединений триалакилолова или хлорсодержащих соединений, которые загрязняют полученные продукты, содержатся в алкоксидах диалкилолова, используемых в самом начале процесса. Алкоксиды алкилолова, полученные способом согласно изобретению, имеют высокую чистоту, и поэтому они содержат лишь очень небольшое количество соединений триалакилолова или хлорсодержащих соединений, что позволяет решить проблемы, связанные с использованием стандартных алкоксидов алкилолова.
Так, например, известными способами получения эфиров угольной кислоты являются фосгеновый способ, осуществляемый с использованием фосгена, и способ окислительного карбонилирования, осуществляемый с использованием монооксида углерода. Известно также, что в этих способах, в качестве исходных материалов или катализаторов используются хлорсодержащие соединения, и эфиры угольной кислоты, полученные этими способами, содержат хлорсодержащие соединения, оказывающие значительное негативное воздействие на процесс образования поликарбонатов, в котором указанные эфиры угольной кислоты используются в качестве исходных материалов (например, таким воздействием является дезактивация катализаторов полимеризации, окрашивание или разрушение поликарбонатов). Кроме того, использование таких эфиров угольной кислоты в качестве добавок к бензину или к дизельному топливу может приводить к коррозии двигателей или систем трубопроводов.
Авторами настоящего изобретения в WO 03/055840 и WO 04/014840 уже был описан способ получения эфиров угольной кислоты и воды отдельно от диоксида углерода и спиртов с использованием алкоксида диалкилолова (в этих патентах термин "алкоксид диалкилолова" используется в более широком смысле и включает алкоксид диалкилолова и тетраалкилдиалкоксидистанноксан). Эти предшествующие изобретения были усовершенствованы благодаря настоящему изобретению. Кроме того, применение способа согласно изобретению позволяет эффективно получать алкоксиды диалкилолова при очень высокой скорости и с высокой чистотой и, следовательно, получать эфиры угольной кислоты, содержащие очень небольшое количество соединения трибутилолова или соединения хлора. Полученные эфиры угольной кислоты могут быть легко превращены посредством переэтерификации или диспропорционирования в диариловые эфиры угольной кислоты с очень небольшим содержанием хлора.
Предпочтительными способами производства эфиров угольной кислоты с использованием алкоксида алкилолова, полученного способом согласно изобретению, являются способы, описанные в вышеуказанных WO 03/055840 и WO 04/014840. Реакционный раствор, включающий диалкиловый эфир угольной кислоты, может быть получен посредством взаимодействия смеси, содержащей алкоксид алкилолова, с диоксидом углерода при температуре в пределах от 60°С до 200°С в течение 0,1-10 часов, под давлением в пределах от 0,1 до 20 МПа. Реакционный раствор, включающий диалкиловый эфир угольной кислоты, обрабатывают известным способом, таким как перегонка, в результате чего указанный раствор разделяется на компоненты, включающие диалкиловый эфир угольной кислоты и оловосодержащий остаток. Такой оловосодержащий остаток включает соединения, представленные химической формулой (1) или химической формулой (2), которые используются в качестве реагентов в способе согласно изобретению; и оловосодержащий компонент, структура которого не может быть точно не определена современным аналитическим методом. Неожиданно было обнаружено, что применение способа согласно изобретению также позволяет получать алкоксиды алкилолова, которые являются целевыми продуктами согласно изобретению, из оловосодержащего компонента, структура которого еще точно не определена.
Алкилариловые эфиры угольной кислоты и диариловые эфиры угольной кислоты могут быть получены посредством взаимодействия вышеописанного диалкилового эфира угольной кислоты и ароматического гидроксисоединения в соответствии с известным способом.
Способы получения диариловых эфиров угольной кислоты, а именно такие, как фосгеновый способ, осуществляемый с использованием фосгена, и способ окислительного карбонилирования, осуществляемый с использованием монооксида углерода, являются известными. Однако в этих способах в качестве исходных материалов или катализаторов используются хлорсодержащие соединения, при этом известно, что диариловые эфиры угольной кислоты, полученные этими способами, содержат соединения хлора, и использование таких диариловых эфиров угольной кислоты приводит к значительному негативному воздействию на процесс производства поликарбонатов, в котором указанные эфиры угольной кислоты используются в качестве исходных материалов (например, таким воздействием является дезактивация катализаторов полимеризации, окрашивание или разрушение поликарбонатов). Кроме того, использование таких эфиров угольной кислоты в качестве добавок к бензину или дизельному топливу может приводить к коррозии двигателей или системы трубопроводов. Авторами настоящего изобретения в WO 03/055840 и WO 04/014840 уже был описан способ получения эфиров угольной кислоты и воды отдельно от диоксида углерода и спиртов с использованием алкоксида диалкилолова. Эти ранее описанные способы авторов настоящего изобретения были усовершенствованы благодаря настоящему изобретению. Кроме того, применение способа согласно настоящему изобретению позволяет легко и эффективно получать диариловые эфиры угольной кислоты с высокой чистотой и с очень низким содержанием соединения хлора.
Кроме того, использование диариловых эфиров угольной кислоты, полученных способом согласно изобретению, позволяет получать поликарбонаты, изоцианаты или диолы поликарбонатов. В этом случае, в качестве диарилового эфира угольной кислоты предпочтительно использовать дифениловый эфир угольной кислоты.
Ниже описаны поликарбонаты, изоцианаты или диолы поликарбонатов, которые могут быть получены вышеописанным способом.
Сначала будут описаны поликарбонаты. Диариловые эфиры угольной кислоты являются известными соединениями, которые обычно используются для производства поликарбонатов методом формования в расплаве. Однако стандартные диариловые эфиры угольной кислоты, которые получают с использованием хлорсодержащих соединений в качестве исходных материалов, имеют большое количество остаточных соединений хлора, и когда их подвергают реакции переэтерификации с бисфенолом А, то такие соединения хлора вызывают дезактивацию катализаторов. Использование большого количества катализатора как возможного способа предотвращения такой дезактивации, иногда может негативно влиять на стойкость поликарбонатов к атмосферным воздействиям, их цвет или физические свойства. В таких случаях необходимо проведение стадии удаления соединений хлора из диарилкарбоната.
Способы решения этой проблемы хорошо известны: так, например, в этих способах соединения хлора удаляют путем очистки диариловых эфиров угольной кислоты, содержащих соединение хлора, щелочью или путем перегонки. Однако применение таких методов все же связано с серьезными проблемами. Очистка щелочью может приводить к ингибированию реакции дегидратации диариловых эфиров угольной кислоты, поскольку температура плавления диариловых эфиров угольной кислоты является относительно высокой и эти эфиры подвергаются щелочной очистке в расплавленном состоянии. Очистка путем перегонки связана с другой серьезной проблемой, а именно она является дорогостоящей. Поскольку соединения хлора включают хлорсодержащие соединения различных типов, от низкокипящих компонентов до высококипящих компонентов, то высокая стоимость очистки при использовании таких диариловых эфиров угольной кислоты в промышленности является серьезной проблемой.
В способе производства дифенилкарбоната из этиленкарбоната, который получают с использованием диоксида углерода в качестве исходного материала, сначала получают диметилкарбонат из этиленкарбоната, затем получают метилфенилкарбонат и, в конечном счете, дифенилкарбонат. В данном способе диметилкарбонат является необязательным промежуточным соединением для получения дифенилкарбоната, что обусловлено ограничением, связанным с температурой кипения (в данной системе температура кипения метанола является наименьшей, и для смещения равновесия необходимо образование азеотропа с метанолом, имеющего минимальную температуру кипения). Очевидно, что метилфенилкарбонат, который всегда образуется как промежуточное соединение, вызывает вторичную реакцию, такую как карбоксилирование, и поэтому дифенилкарбонат, являющийся конечным продуктом, имеет небольшие примеси побочного продукта, такого как анизол, который включает метильную группу даже после проведения стадии очистки. Указанные примеси могут иногда приводить к снижению скорости полимеризации в процессе производства поликарбонатов с использованием дифенилкарбоната и вызывать изменение степени полимеризации или негативно влиять на цвет продукта.
В отличие от этого, в способе согласно изобретению не образуются побочные продукты. Идентификация вышеописанных побочных продуктов, имеющих метильную группу и происходящих от диметилкарбоната, представляет значительные трудности. Однако в способе получения диариловых эфиров угольной кислоты согласно изобретению промежуточным соединением является не диметилкарбонат, а диалкиловые эфиры угольной кислоты, имеющие алкильную группу с длинной цепью и полученные из спирта, представленного химической формулой (3), и поэтому может быть получен дифенилкарболнат, который не содержит побочных продуктов, имеющих метильную группу и оказывающих негативное воздействие на получение поликарбонатов.
Примерами диариловых эфиров угольной кислоты, предпочтительно используемых в качестве исходных материалов для получения поликарбонатов, являются диариловые эфиры угольной кислоты, содержащие вышеописанные органические соединения, имеющие метильную группу (побочные продукты) в концентрации 100 ч./млн или менее, и более предпочтительно 10 ч./млн или менее.
Ниже приводится описание изоцианатов. Изоцианаты могут быть получены сначала посредством реакции взаимодействия диарилового эфира угольной кислоты (в частности, дифенилкарбоната), полученного с использованием алкоксида алкилолова согласно изобретению, с полиаминовым соединением с образованием полиарилкарбамата, такого как гексаметилендиарилкарбамат; и затем посредством реакции пиролиза полиарилкарбамата. Обычно применяемый известный способ синтеза изоцианатов, в котором в качестве исходного материала используется фосген, является экономически невыгодным. Однако диариловые эфиры угольной кислоты, полученные способом согласно изобретению, имеют очень низкую себестоимость и содержат очень незначительное количество соединений хлора и поэтому этот способ является преимущественным способом получения изоцианатов. Стандартные изоцианаты получены из хлорсодержащих соединений, таких как фосгены, содержащие соединения хлора. Ранее, для получения уретана в качестве катализаторов использовали изоцианаты, но такие катализаторы, используемые для получения уретанов, являются чувствительными к дезактивации или денатурации хлором. В отличие от этого изоцианаты, полученные способом согласно изобретению, по существу, не содержат хлора, и поэтому при их применении не возникает каких-либо проблем.
Ниже приводится подробное описание поликарбонатдиолов. Поликарбонтатдиолы высокой чистоты могут быть получены с использованием диариловых эфиров угольной кислоты, которые получены с использованием алкоксидов алкилолова согласно изобретению.
Поликарбонаты, изоцианаты и поликарбонатдиолы, полученные с использованием диариловых эфиров угольной кислоты, которые получены с использованием алкоксидов алкилолова согласно изобретению, являются исключительно ценными соединениями с промышленной точки зрения, поскольку по сравнению с соединениями, полученными общепринятыми способами, они имеют высокую чистоту, могут быть легко получены (то есть с небольшими затратами) и не образуют побочных продуктов.
Изоцианаты могут быть получены с использованием вышеописанных диалкиловых эфиров угольной кислоты и/или вышеописанных диариловых эфиров угольной кислоты известным способом.
Диариловые эфиры угольной кислоты являются известными соединениями, которые обычно используются для производства поликарбонатов методом формования в расплаве. Однако стандартные диариловые эфиры угольной кислоты, которые получают с использованием хлорсодержащих соединений в качестве исходных материалов, содержат большое количество остаточных соединений хлора, и такие соединения хлора, когда их подвергают реакции переэтерификации с бисфенолом А, вызывают дезактивацию катализаторов. Использование большого количества катализатора как возможного способа предотвращения такой дезактивации может иногда негативно влиять на стойкость поликарбонатов к атмосферным воздействиям, их цвет или физические свойства. В этом случае, необходимо проведение стадии удаления соединений хлора из диарилкарбоната. Способы решения этой проблемы хорошо известны; так, например, в этих способах, соединения хлора удаляют путем щелочной очистки диариловых эфиров угольной кислоты, содержащих соединение хлора, или их очистки путем перегонки.
Однако применение таких способов все еще связано с серьезными проблемами. Очистка щелочью может приводить к ингибированию реакции дегидратации диариловых эфиров угольной кислоты, поскольку температура плавления диариловых эфиров угольной кислоты является относительно высокой и эти эфиры подвергаются щелочной очистке в расплавленном состоянии. Очистка путем перегонки также сталкивается с серьезной проблемой, то есть является дорогостоящей. Поскольку соединения хлора включают хлорсодержащие соединения различных типов, от низкокипящих компонентов до высококипящих компонентов, то высокая стоимость очистки при использовании таких диариловых эфиров угольной кислоты в промышленности является серьезной проблемой. Диариловые эфиры угольной кислоты, полученные с использованием алкоксидов диалкилолова, получаемых способом согласно изобретению, имеют низкую себестоимость и содержат очень небольшое количество соединений хлора и поэтому их использование для получения изоцианатов является преимущественным.
Таким образом, способ получения соединений, в котором используются алкоксиды алкилолова, полученные в соответствии с настоящим изобретением, позволяет осуществлять промышленное производство эфиров угольной кислоты, изоцианатов и поликарбонатов с низкой себестоимостью и высокой чистотой в отличие от соединений, получаемых общепринятыми способами.
Примеры
Более подробное описание настоящего изобретения приводится ниже в примерах, которые не должны рассматриваться как ограничение объема настоящего изобретения.
Аналитические методы:
1) ЯМР-анализ:
Оборудование: FT-ЯМР-система JNM-A400 (изготовленная JEOL Ltd., Japan).
(1) Получение образцов для1Н-ЯМР-,13С-ЯМР- и119Sn-ЯМР-анализов
Примерно 0,3 г соединения олова взвешивали и затем к нему добавляли примерно 0,7 г дейтерированного хлороформа (изготовленного Aldrich Chemical Co., 99,8%) и в качестве внутреннего стандарта для119Sn-ЯМР примерно 0,5 г тетраметилолова (изготовленного Wako Pure Chemical Industries Ltd., Wako, высокого качества). Полученный раствор смешивали до однородности для использования в качестве образца в ЯМР-анализе.
2) Анализ воды
Оборудование: Измеритель остаточной влажности СА-05 Trace Moisture Meter, изготовленный Mitsubishi Chemicals Corporation, Japan.
(1) Количественный анализ
Для отбора 0,12 мл образца для анализа использовали шприц, и этот шприц взвешивали. Затем образец в том же виде вводили в измеритель влажности для анализа воды. Затем шприц снова взвешивали и введенное количество образца оценивали для определения содержания влаги в данном образце.
(3) Анализ эфира угольной кислоты с помощью газовой хроматографии
Оборудование: система GC-2010 (изготовленная фирмой Shimadzu Corporation, Japan).
(1) Анализ препарата раствора образца
Примерно 0,5 мл дегидратированного диметилформамида или ацетонитрила добавляли к 0,4 г реакционного раствора. Затем в полученный раствор вводили примерно 0,4 г толуола или дифенилового эфира, которые используют в качестве внутреннего стандарта в растворе образца для анализа методом газовой хроматографией.
(2) Условия проведения анализа с помощью газовой хроматографии
Колонна: DB-1 (изготовленная J & W Scientific, USA)
Жидкая фаза: 100% диметилполисилоксан
Длина колонны: 30 м
Внутренний диаметр колонны: 0,25 мм
Толщина пленки: 1 мкм
Температура колонны: температуру повышали с 50 до 300°С со скоростью 10°С/мин
Температура инжекции: 300°С
Температура детектора: 300°С
Метод детекции: FID
(3) Количественный анализ
Количественный анализ проводили в растворах образца для анализа исходя из калибровочной кривой, построенной по данным анализа стандартного образца для каждого стандартного вещества.
4) Определение выхода алкоксида диалкилолова
Выход алкоксида диалкилолова вычисляли исходя из числа молей атомов олова (моль%) полученных соответствующих алкоксидов диалкилолова (соединений, представленных химической формулой (7) и/или химической формулой (16)) по отношению к числу молей атомов олова исходного материала (соединений, представленных химической формулой (1) и/или химической формулой (2)).
5) Определение выхода ароматического эфира угольной кислоты
Выход ароматического эфира угольной кислоты выражали в мас.% в реакционном растворе или определяли исходя из числа молей атомов олова (моль%) полученного алкиларилкарбоната и диарилкарбоната по отношению к числу молей подаваемого соединения (диалкилкарбоната).
6) Среднечисленная молекулярная масса ароматического поликарбоната
Среднечисленную молекулярную массу ароматического поликарбоната определяли с помощью гельпроникающей хроматографии (ГПХ).
Пример 1
(Получение смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксан и реагента 2-этил-1-бутанола)
В 1-литровую колбу-сборник загружали 24,9 г (0,1 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 208 г (2,0 моль) 2-этил-1-бутанола (изготовленного Aldrich Chemical Со., USA, 98%), в результате колба содержала образовавшуюся в ней смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), подсоединенному к масляной бане, снабженной терморегулятором (изготовленной Masuda Corporation, Japan, OBH-24), к вакуумному насосу (изготовленному ULVAC Inc., Japan, G-50A) и к вакуумному регулятору (изготовленному Okano Works Ltd., Japan, VC-10S). Температуру масляной бани устанавливали на 140°С. Затем колбу погружали на масляную баню, после чего начинали вращение испарителя. Вращение проводили в течение примерно 30 минут при перемешивании и нагревании при атмосферном давлении с открытым продувочным вентилем испарителя. Затем продувочный вентиль испарителя закрывали и давление в системе постепенно снижали примерно до 60 кПа с помощью вакуумного насоса и вакуумного регулятора. Этот режим поддерживали в течение 1 часа, после чего колбу вынимали из масляной бани. Реакционный раствор превращался в прозрачную жидкость. Затем продувочный вентиль постепенно открывали для установления в системе атмосферного давления. Дистиллят имел массу 9,9 г, был прозрачным и был разделен на 2 слоя. Анализ дистиллята показал, что содержание воды составляло примерно 1 г. Затем колбу вынимали из масляной бани и продувочный вентиль постепенно открывали для установления в системе атмосферного давления. Колба содержала 218 г реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализа показали, что при использовании оксида дибутилолова в качестве исходного соединения реакционный раствор содержал 11% дибутил-бис(2-этилбутилокси)олова и примерно 88% 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана.
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор, имеющий внутренний диаметр 15 мм и общую длину 850 мм (рабочую длину 750 мм) и изготовленный из SUS 316, к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5 и в его нижней части 3 - газовый питающий трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (Tokyo Tokushu Kanaami K.K., Japan). Температура нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с использованием нагревателя, установленного на 160°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с использованием нагревателя, установленного на 140°С.
Газообразный азот подавали из питающего трубопровода 7 со скоростью 0,04 л N/мин, при этом для подачи из питающего трубопровода 4 смешанного раствора, состоящего из исходного соединения и реагента, полученных, как описано выше, использовали питающий насос со скоростью подачи 20 г/час. Время пребывания в реакторе составляло примерно 16 минут. Низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из спускного трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6. Процесс непрерывной подачи и непрерывной экстракции продолжали в одном и том же режиме в течение 2 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения указанная жидкость содержала алкоксид дибутилолова, состоящий примерно на 94% из дибутил-бис(2-этилбутилокси)олова и примерно на 6% из 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана. Выход 2-этилбутилоксида трибутилолова составлял 0,2%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0033 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00025 моль/час.
Пример 2
(Получение исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана)
В 500-миллилитровую колбу-сборник загружали 59,8 г (0,15 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 122 г (1,2 моль) 2-этил-1-бутанола (изготовленного Aldrich Chemical Со., USA, 98%), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), подсоединенному к масляной бане, снабженной терморегулятором (изготовленной Masuda Corporation, Japan, OBH-24), к вакуумному насосу (изготовленному ULVAC Inc., Japan, G-50A) и к вакуумному регулятору (изготовленному Okano Works Ltd., Japan, VC-10S). Температуру масляной бани устанавливали на 140°С. Затем колбу погружали на масляную баню, после чего включали вращение испарителя. Вращение проводили в течение примерно 30 минут при перемешивании и нагревании при атмосферном давлении с открытым продувочным вентилем испарителя. Затем продувочный вентиль испарителя закрывали и давление в системе постепенно снижали примерно до 70 кПа с помощью вакуумного насоса и вакуумного регулятора. Такой режим поддерживали в течение 1 часа, после чего колбу вынимали из масляной бани. Реакционный раствор превращался в прозрачную жидкость. Затем продувочный вентиль постепенно открывали для установления в системе атмосферного давления. Дистиллят имел массу 3,6 г, был прозрачным и был разделен на 2 слоя. Анализ дистиллята показал, что содержание воды составляло примерно 2,2 г. Затем колбу вынимали из масляной бани и продувочный вентиль постепенно открывали для установления в системе атмосферного давления. Колба содержала 175 г реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что в случае использования оксида дибутилолова в качестве исходного соединения, реакционный раствор содержал примерно 99% 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана.
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор с внутренним диаметром 15 мм и общей длиной 1635 мм (рабочей длиной 1450 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединены питающий трубопровод 4 и спускной экстракционный трубопровод 5 и в его нижней части 3 - трубопровод для подачи газа 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с использованием нагревателя, установленного на 160°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с использованием нагревателя, установленного на 140°С.
Газообразный азот подавали из питающего трубопровода 7 со скоростью 0,04 л N/мин, и для подачи смешанного раствора, состоящего из исходного материала и реагента, полученных, как описано выше, через питающий трубопровод 4 использовали питающий насос со скоростью подачи 20 г/час. Время пребывания в реакторе составляло примерно 32 минуты. Затем низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6. Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 2 часов. Анализ полученного продукта, экстрагированного из экстракционного трубопровода 6, показал, что в случае использования оксида дибутилолова в качестве исходного соединения, полученный продукт содержал алкоксид дибутилолова, состоящий примерно на 94% из дибутил-бис(2-этилбутилокси)олова и примерно на 6% из 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана. Выход 2-этилбутилоксида трибутилолова составлял 0,4%. После охлаждения газовой фазы, выходящей из экстракционного спускного трубопровода 5, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0033 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,0005 моль/час.
Пример 3
(Синтез 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана)
В 2-литровую трехгорлую колбу, снабженную сборником влаги, подсоединенным к термометру, трехходовым запорным краном и холодильником Димрота, загружали 199,8 г (0,80 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%), 1045 г (8,0 моль) 2-этил-1-гексанола (изготовленного Aldrich Chemical Со., USA, 99,6%, дегидратированного) и 500 г толуола (изготовленного Wako Pure Chemical Industries Ltd., Japan, для органического синтеза), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу погружали на масляную баню (изготовленную Fine, Japan, FWB-240), установленную на 130°С. После 30-минутного нагревания при перемешивании температура смеси составляла 119°С, и затем вода и толуол начинали поступать в сборник влаги.
Этот режим поддерживали в течение 3 часов, после чего в сборнике влаги накапливалось примерно 7,2 мл воды. Затем температуру масляной бани снижали до 90°С и после снижения температуры смеси сборник влаги удаляли. Колбу подсоединяли к соединительному трубопроводу, снабженному трубопроводом с разветвлениями, холодильником Либиха, соединительным трубопроводом низкого давления и двумя сосудами-сборниками дистиллята. Давление в системе снижали до 29 кПа и после отгонки толуола из колбы давление в системе снижали до 0,6 кПа для отгонки избыточного 2-этилгексанола. Количество жидкости, собранной посредством перегонки, составляли 1420 г, и выход полученного продукта в колбе составлял 295,6 г. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что полученный продукт, а именно 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксан, имел чистоту, по меньшей мере, 95%.
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор, с внутренним диаметром 15 мм и общей длиной 850 мм (рабочей длиной 750 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 для инжекции исходного материала, питающий трубопровод 8 для инжекции реагента и спускной экстракционный трубопровод 5 и в его нижней части 3 - питающий трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с использованием нагревателя, установленного на 160°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с использованием нагревателя, установленного на 140°С.
Газообразный азот подавали из питающего газового трубопровода 7 со скоростью 0,04 л N/мин, при этом для введения исходного материала, полученного как описано выше и подаваемого через питающий трубопровод 4 со скоростью подачи примерно 3 г/час, и реагента 2-этил-1-гексанола (изготовленного Aldrich Chemical Со., USA, 99,6%, безводного), подаваемого из питающего трубопровода 8 со скоростью 17 г/час, использовали питающий насос. Время пребывания в реакторе составляло 15 минут. Затем низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного трубопровода 5, и высококипящие компоненты начинали выводить из экстракционного трубопровода 6. Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 2 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения, указанная жидкость содержала алкоксид дибутилолова, состоящий примерно на 45% из дибутил-бис(2-этилгексилокси)олова и примерно на 55% из 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана. Выход 2-этилбутилоксида трибутилолова составлял 0,3%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0018 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,000058 моль/час.
Пример 4
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор с внутренним диаметром 15 мм и общей длиной 850 мм (рабочей длиной 750 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5 и в его нижней части 3 - питающий газовый трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 160°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 140°С.
Газообразный азот подавали из питающего газового трубопровода 7 со скоростью 0,04 л N/мин, при этом для подачи из питающего трубопровода 4 взвеси, состоящей из 19,9 г (0,08 моль) исходного материала оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 817 г (8 моль) и реагента 2-этил-1-бутанола (изготовленного Aldrich Chemical Со., USA, 98%), со скоростью подачи 8 г/час использовали питающий насос. Время пребывания в реакторе составляло 35 минут. Низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного газового трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 5 часов. Анализ полученного продукта, экстрагированного из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученный продукт содержал алкоксид дибутилолова, состоящий примерно на 61% из дибутил-бис(2-этилбутилокси)олова и примерно на 38% из 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана. Выход 2-этилбутилоксида трибутилолова составлял 0,1%. После охлаждения газовой фазы, выходящей из спускного трубопровода 5, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,00062 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00005 моль/час.
Пример 5
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор с внутренним диаметром 15 мм и общей длиной 850 мм (рабочей длиной 750 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5 и в его нижней части 3 - питающий газовый трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 170°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 150°С.
Газообразный азот подавали из питающего газового трубопровода 7 со скоростью 0,04 л N/мин, при этом для подачи из питающего трубопровода 4 смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана и реагента 2-этил-1-бутанола, которые получены способом, описанным в примере 1, использовали питающий насос со скоростью подачи 8 г/час. Затем из питающего газового трубопровода 7 подавали 2-этил-1-бутанол (изготовленный Aldrich Chemical Со., USA, 98%) со скоростью 1 г/мин. Время пребывания в реакторе составляло 30 минут. Низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного газового трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 2 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученный продукт содержал алкоксид дибутилолова, состоящий примерно на 48% из дибутил-бис(2-этилбутилокси)олова и примерно на 52% из 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана. Выход 2-этилбутилоксида трибутилолова составлял 0,4%. После охлаждения газовой фазы, выходящей из спускного трубопровода 5, получали двухслойную прозрачную жидкость, в которой присутствовала влага.
Пример 6
(Получение диалкоксида дибутилолова с использованием комбинации резервуарного реактора и колонного реактора)
Алкоксид диалкилолова получали с использованием комбинации резервуарного реактора и колонного реактора, проиллюстрированной на фиг.3.
Использовали 1-литровый смесительный резервуар с перегородкой 9, изготовленный из SUS 304 и содержащий питающий трубопровод 15 для инжекции реагента, питающий трубопровод 16 для инжекции исходного материала и экстракционный спускной трубопровод 17, подсоединенный к дистилляционной колонне в верхней части 11 смесительного резервуара, питающий газовый трубопровод 18 и соединительный трубопровод 19, подсоединенный к нижней части 12 смесительного резервуара, мешалку, регулятор температуры и другое необходимое оборудование и вентили для регуляции работы каждого трубопровода; и колонный реактор 10, а именно трубчатый реактор со спиральной насадкой Helipack № 3 (изготовленной Tokyo Tokushu Kanaami K.K., Japan), имеющий внутренний диаметр 15 мм и общую длину 1635 мм (рабочую длину 1450 мм) и изготовленный из SUS 316, где указанный реактор снабжен регулятором температуры и необходимым оборудованием и вентилями для регуляции работы каждого трубопровода, соединенными с буферным резервуаром 24, промежуточным трубопроводом 25 для соединения буферного резервуара 24 с колонным реактором, питающим трубопроводом 22 для инжекции реагента и промежуточным трубопроводом 25, которые оба были подсоединены к реактору в его верхней части 13; экстракционным спускным трубопроводом 21, подсоединенным к дистилляционной колонне, питающим газовым трубопроводом 20 в его нижней части и экстракционным трубопроводом 23.
После продувки внутренней части смесительного резервуара 9 азотом в резервуар из питающего трубопровода 15 вводили 390 г (3,0) реагента 2-этил-1-гексанола (изготовленного Aldrich Chemical Со., USA, безводный, 99,6%) и из питающего трубопровода 16 вводили 199,8 г (0,80 моль) исходного соединения оксида дибутилолова (изготовленного Aldrich Chemical Co., USA, 98%). Газообразный азот подавали из питающего газового трубопровода 18 со скоростью 0,02 л N/мин. Реактор нагревали при перемешивании до тех пор, пока температура реакционного раствора не достигала 160°С, и из экстракционного спускного трубопровода 17 выпускали газ. Реакцию проводили в этом режиме в течение 20 минут, после чего реакционный раствор непрерывно экстрагировали из соединительного трубопровода 19 со скоростью примерно 40 мл/мин и одновременно подавали исходный материал и реагент из питающего трубопровода 16 и питающего трубопровода 15 таким образом, чтобы их молярное отношение составляло 1:3,75 и чтобы уровень поверхности жидкости внутри реактора оставался постоянным, в результате чего устанавливался стабильный процесс, при котором происходило непрерывное выведение газа из экстракционного спускного трубопровода 17.
Реакционный раствор поступал в буферный резервуар 24 через соединительный трубопровод 19. Анализ жидкости в буферном резервуаре 24, проведенный через 2 часа, показал, что при использовании оксида дибутилолова в качестве исходного соединения указанная жидкость содержала ранее полученный алкоксид дибутилолова, состоящий примерно на 5% из дибутил-бис(2-этилгексилокси)олова и примерно на 95% из 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана. Скорость дегидратации в резервуарном реакторе составляла 1,26 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,13 моль/час.
(Затем выход дибутил-бис(2-этилгексилокси)олова в реакторе 10 увеличивался в результате накопления в буферном резервуаре ранее полученного алкоксида дибутилолова в качестве исходного соединения).
Температуру нижней части фланца реактора 10 и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 160°С, и температуру части реактора, расположенной от нагревательного устройства и до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 140°С.
Газообразный азот подавали из питающего газового трубопровода 20 со скоростью 0,04 л N/мин и для введения алкоксида дибутилолова, полученного, как описано выше, и используемого в качестве исходного соединения, подаваемого из буферного резервуара 24 через промежуточный трубопровод 25 со скоростью 5 г/час, и реагента 2-этил-1-гексанол (изготовленный Aldrich Chemical Со., USA, 99,5%, безводный), подаваемого из питающего трубопровода 22 со скоростью 15 г/час, использовали питающий насос. Время пребывания в реакторе составляло 35 минут. Низкокипящие вещества, включающие воду, начинали выходить в газообразной форме из экстракционного спускного трубопровода 21, и высококипящие компоненты начинали выходить из экстракционного трубопровода 23.
Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 2 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 23, показал, что при использовании оксида дибутилолова в качестве исходного соединения, полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 75% из дибутил-бис(2-этилгексилокси)олова и примерно на 24% из 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана. Выход 2-этилгексилоксида трибутилолова составлял 0,9%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,021 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00025 моль/час.
Пример 7
(Получение диалкоксида дибутилолова с помощью колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор с внутренним диаметром 15 мм и общей длиной 1635 мм (рабочей длиной 1450 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5, а в его нижней части 3 - трубопровод 7 для подачи вспомогательного реагента и экстракционный трубопровод 6, вставляли насадку Диксона размером 3 мм (Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 170°С, и температуру части, расположенной от нагревательного устройства и до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 150°С.
Газообразный азот подавали из питающего газового трубопровода 7 со скоростью 0,04 л N/мин, при этом для подачи из питающего трубопровода 4 смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-этилбутлилокси)дистанноксана и реагента 2-этил-1-бутанола, полученных, как описано выше в примере 1, использовали питающий насос со скоростью подачи 15 г/час. Время пребывания в реакторе составляло 50 минут. Затем низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразном состоянии из экстракционного спускного трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 4 часов. Анализ полученной жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 63% из дибутил-бис(2-этилбутилокси)олова и примерно на 36% из 1,1,3,3-тетрабутил-1,3-бис(2-этилбутилокси)дистанноксана. Выход 2-этилбутилоксида трибутилолова составлял 0,3%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0016 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00026 моль/час.
Пример 8
(Получение исходного соединения 1,1,3,3-тетраоктил-1,3-ди(бутилокси)дистанноксана)
В 2-литровую колбу-сборник загружали 217 г (0,6 моль) оксида диоктилолова (изготовленного Wako Pure Chemical Industries Ltd., Japan, 95%) и 445 г (6 моль) 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленной Masuda Corporation, Japan, OBH-24).
Температуру масляной бани устанавливали на 127°С. Затем колбу погружали на масляную баню, после чего включали вращение испарителя. Вращение проводили в течение примерно 150 минут с перемешиванием и нагреванием при атмосферном давлении с открытым продувочным вентилем испарителя. Затем колбу вынимали из масляной бани и оставляли для охлаждения. Колба содержала 437 г вязкого реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида диоктилолова в качестве исходного соединения реакционный раствор содержал примерно 96% 1,1,3,3-тетраоктил-1,3-ди(2-бутилокси)дистанноксана и не содержал диоктилди(бутилокси)олова.
(Получение диалкоксида диалкилолова с использованием колонного реактора)
Алкоксид диоктилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор внутренним диаметром 15 мм и общей длиной 1850 мм (рабочей длиной 750 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5 и в его нижней части 3 - питающий газовый трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 150°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 140°С.
Газообразный диоксид углерода подавали из питающего газового трубопровода 7 со скоростью 80 мл/мин, при этом для подачи из питающего трубопровода 4 реакционного раствора, полученного, как описано выше (смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетраоктил-1,3-ди(бутилокси)дистанноксана и реагента 1-бутанола), использовали питающий насос со скоростью подачи 10 г/час. Время пребывания в реакторе составляло 37 минут. Внутреннее давление реактора, составляющее примерно 0,2 МПа-G, измеряли манометром. Низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 4 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида диоктилолова в качестве исходного соединения, полученная жидкость содержала алкоксид диоктилолова, состоящий примерно на 43% из диоктилди(бутилокси)олова и из 1,1,3,3-тетраоктил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида триоктилолова составлял 0,1%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0017 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00025 моль/час.
Пример 9
(Получение исходного соединения 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана)
В 1-литровую колбу-сборник загружали 50 г (0,2 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 178 г (2,4 моль) 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленной Masuda Corporation, Japan, OBH-24).
Температуру масляной бани устанавливали на 127°С. Затем колбу погружали на масляную баню, после чего включали вращение испарителя. Вращение проводили в течение примерно 2 часов с перемешиванием и нагреванием при атмосферном давлении с открытым продувочным вентилем испарителя. Затем колбу вынимали из масляной бани и оставляли для охлаждения. Колба содержала 212 г вязкого реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида дибутилолова в качестве исходного соединения реакционный раствор содержал 98% 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана и не содержал дибутилди(бутилокси)олова.
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор, имеющий внутренний диаметр 15 мм и общую длину 850 мм (рабочую длину 750 мм) и изготовленный из SUS 316, к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5, а в его нижней части 3 - питающий газовый трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 150°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 140°С.
Газообразный диоксид углерода подавали из питающего газового трубопровода 7 со скоростью 80 мл/мин, при этом для подачи из питающего трубопровода 4 реакционного раствора, полученного как описано выше (смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана и реагента 1-бутанола), использовали питающий насос со скоростью подачи 10 г/час. Время пребывания в реакторе составляло 37 минут. Внутреннее давление реактора, составляющее примерно 0,2 МПа-G, измеряли манометром. Низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции продолжали в том же режиме в течение 4 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 90% из дибутилди(бутилокси)олова и примерно на 9% 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,06%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали двухслойную прозрачную жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0033 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00025 моль/час.
Пример 10
(Получение исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана)
В 1-литровую колбу-сборник загружали 50 г (0,2 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 178 г (2,4 моль) 2-метил-1-пропанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., Japan, R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленным Masuda Corporation, Japan, OBH-24).
Температуру масляной бани устанавливали на 118°С. Затем колбу погружали на масляную баню, после чего включали вращение испарителя. Вращение проводили в течение примерно 2 часов с перемешиванием и нагреванием при атмосферном давлении с открытым продувочным вентилем испарителя. Затем колбу вынимали из масляной бани и оставляли для охлаждения. Колба содержала 212 г вязкого реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида дибутилолова в качестве исходного соединения реакционный раствор содержал 76% 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана и не содержал дибутил-бис(2-метил-1-пропилокси)олова.
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор с внутренним диаметром 15 мм и общей длиной 1635 мм (рабочей длиной 1450 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной экстракционный трубопровод 5, а в его нижней части 3 - питающий газовый трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 150°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 140°С.
Газообразный диоксид углерода подавали из питающего газового трубопровода 7 со скоростью 80 мл/мин, при этом для подачи через питающий трубопровод 4 реакционного раствора, полученного как описано выше (смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана и реагента 2-метил-1-пропанола), использовали питающий насос со скоростью подачи 10 г/час. Время пребывания в реакторе составляло 22 минуты. Внутреннее давление реактора, составляющее примерно 0,2 МПа-G, измеряли манометром. Низкокипящие вещества, включающие воду, начинали экстрагироваться в газообразной форме из экстракционного спускного трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции продолжали в этом режиме в течение 4 часов. Анализ жидкости, экстрагированной из экстракционного трубопровода 6, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 97% из дибутил-бис(2-метил-1-пропилокси)олова и примерно на 3% из 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана. Выход бутилоксида трибутилолова составлял 0,02%. После охлаждения газовой фазы, выходящей из спускного трубопровода, получали жидкость, в которой присутствовала влага. Скорость дегидратации в колонном реакторе составляла 0,0038 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00051 моль/час.
Пример 11
Стадия 1: (Получение исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана)
В 1-литровую колбу-сборник загружали 50 г (0,2 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 178 г (2,4 моль) 2-метил-1-пропанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленной Masuda Corporation, Japan, OBH-24).
Температуру масляной бани устанавливали на 118°С. Затем колбу погружали на масляную баню, после чего включали вращение испарителя. Вращение проводили в течение примерно 2 часов с перемешиванием и нагреванием при атмосферном давлении с открытым продувочным вентилем испарителя. Затем колбу вынимали из масляной бани и оставляли для охлаждения. Колба содержала 196 г вязкого реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида дибутилолова в качестве исходного соединения реакционный раствор содержал 76% 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана и не содержал дибутил-бис(2-метил-1-пропилокси)олова.
Стадия 2: (Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 1, таком как проиллюстрировано на фиг.2. В трубчатый реактор с внутренним диаметром 15 мм и общей длиной 1635 мм (рабочей длиной 1450 мм), который изготовлен из SUS 316 и к которому в его верхней части 2 подсоединен питающий трубопровод 4 и спускной трубопровод 5 для отгонки, а в его нижней части 3 - питающий газовый трубопровод 7 и экстракционный трубопровод 6, вставляли спиральную насадку Helipack № 3 (изготовленную Tokyo Tokushu Kanaami K.K., Japan). Температуру нижней части фланца трубчатого реактора и части, простирающейся примерно на 60 мм от указанного фланца, регулировали с помощью нагревателя, установленного на 150°С, и температуру части, расположенной выше от нагревательного устройства и простирающейся до верхнего фланца трубчатого реактора, регулировали с помощью нагревателя, установленного на 140°С.
Газообразный диоксид углерода подавали из питающего газового трубопровода 7 со скоростью 80 мл/мин, при этом для подачи через питающий трубопровод 4 реакционного раствора, полученного, как описано выше (смешанного раствора, состоящего из исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана и реагента 2-метил-1-пропанола), использовали питающий насос со скоростью подачи 10 г/час. Время пребывания в реакторе составляло 22 минуты. Внутреннее давление реактора, составляющее примерно 0,2 МПа-G, измеряли манометром. Низкокипящие вещества, включающие воду, начинали отгоняться в газообразной форме из спускного газового трубопровода 5, и высококипящие компоненты начинали выходить из экстракционного трубопровода 6.
Процесс непрерывной подачи и непрерывной экстракции жидкости, полученной, как описано в стадии 1, продолжали в этом режиме вплоть до удаления всей жидкости. Жидкость, экстрагированную из экстракционного трубопровода 6, собирали в 1-литровый промежуточный резервуар, изготовленный из SUS. Анализ собранной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения, полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 97% из дибутил-бис(2-метил-1-пропилокси)олова и примерно на 3% из 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана. Выход 2-метил-1-пропилоксида трибутилолова составлял 0,03%. После охлаждения газовой фазы, выходящей из спускного трубопровода, влагу удаляли с использованием многоходовой дистилляционной колонны для сбора 2-метил-1-пропанола. Скорость дегидратации в колонном реакторе составляла 0,0168 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,000648 моль/час.
Стадия 3: (Получение эфира угольной кислоты из алкоксида диалкилолова)
Реакционный раствор, собранный в промежуточном резервуаре, как описано в стадии 2, подавали со скоростью 3 г/мин в тонкопленочный перегонный аппарат (изготовленный Shibata Scientific Technology Ltd., Е-420), установленный на 130°С, и под давлением примерно 65 Па с помощью питающего насоса (изготовленного Shimadzu Corporation, Japan, LC-10AT) для удаления летучих компонентов. Нелетучие компоненты охлаждали и собирали, получая, таким образом, примерно 74 г собранной жидкости. Собранную жидкость загружали в 200-миллилитровый автоклав (изготовленный Toyo Koatsu Co., Ltd., Japan) и крышку закрывали. Внутреннюю часть автоклава продували газообразным азотом, и затем вторичное давление цилиндра с диоксидом углерода, соединенного с автоклавом посредством системы SUS-трубопроводов и вентиля, устанавливали на 4 МПа. Затем вентиль открывали и в автоклав подавали газообразный диоксид углерода.
После перемешивания в течение 10 минут вентиль закрывали. Затем температуру повышали до 120°С при перемешивании. На этой стадии внутреннее давление в автоклаве регулировали с использованием клапана обратного давления таким образом, чтобы давление поддерживалось при 4 МПа. В этом режиме реакцию проводили в течение 4 часов, после чего внутреннее давление в автоклаве снова доводили до атмосферного давления путем мягкой продувки диоксидом углерода из продувочного трубопровода. Жидкость в автоклаве быстро экстрагировали из экстракционного трубопровода, подсоединенного к нижней части автоклава, получая, таким образом, прозрачный реакционный раствор. Ди(2-метилпропил)карбонат получали с выходом 40% с использованием оксида дибутилолова в качестве исходного соединения. Прозрачный реакционный раствор подавали в перегонный аппарат с тонкой пленкой (изготовленный Shibata Scientific Technology Ltd., Japan, Е-420), установленный на 130°С, со скоростью 3 г/мин и под давлением примерно 65 Па с помощью питающего насоса (изготовленного Shimadzu Corporation, Japan, LC-10AT) для удаления летучих компонентов, включающих ди(2-метилпропил)карбонат. Нелетучие компоненты охлаждали и собирали, получая таким образом, примерно 62 г собранной жидкости. Летучие компоненты, включающие ди(2-метилпропил)карбонат, охлаждали и получали жидкость, в которой не обнаруживался 2-метил-1-пропилоксид трибутилолова. Результаты анализа на хлор также не выявили его присутствия.
119Sn-,1Н- и13С-ЯМР-анализы собранной жидкости показали, что жидкость содержала 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксан и не содержала дибутил-бис(2-метил-1-пропилокси)олова.
Стадия 4: (Получение алкоксида диалкилолова после образования эфира угольной кислоты с использованием собранной жидкости в качестве исходного материала)
В жидкость, собранную как описано в стадии 3, добавляли 2-метил-1-пропанол, полученный путем дегидратации в многоходовой дистилляционной колонне, как описано в стадии 1, в качестве реакционного раствора и, если его количество было недостаточным, добавляли еще некоторое количество 2-метил-1-пропанола (изготовленного Wako Pure Chemical Industries Ltd., специальной очистки) таким образом, чтобы в расчете на молярную концентрацию число молей пропанола примерно в десять раз превышало число молей атомов олова, содержащихся в оловоорганическом соединении, таком как 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксан, содержащийся в жидкости, полученной в стадии 3, получая таким образом смешанный раствор, состоящий из исходного соединения и реагента. Реакцию проводили способом, описанным в стадии 2, за исключением того, что вместо жидкости, подаваемой из питающего трубопровода 4, использовали вышеуказанный смешанный раствор. Непрерывную подачу и непрерывную экстракцию проводили до выведения смешанного раствора.
Жидкость, экстрагированную из экстракционного трубопровода 6, собирали в 1-литровый промежуточный резервуар, изготовленный из SUS. Анализ собранной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения, полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 97% из дибутил-бис(2-метил-1-пропилокси)олова и примерно на 3% из 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана. После охлаждения газовой фазы, выходящей из спускного трубопровода, влагу удаляли с использованием многоходовой дистилляционной колонны для сбора 2-метил-1-пропанола.
Затем стадию 3 и стадию 4 повторяли 3 раза, в результате чего образовавшийся раствор в промежуточном резервуаре после третьей операции на стадии 4 при использовании оксида дибутилолова в качестве исходного соединения содержал алкоксид дибутилолова, состоящий примерно на 95% из дибутил-бис(2-метил-1-пропилокси)олова и примерно на 2% из 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана. Скорость дегидратации в колонном реакторе превышала величину, вычисленную по уравнению (16). Выход 2-метил-1-пропилоксида трибутилолова составлял 0,05%.
Сравнительный пример 1
(Способ непрерывной экстракции только низкокипящих компонентов)
В 1-литровую колбу-сборник загружали 50 г (0,2 моль) исходного соединения оксида дибутилолова (изготовленного Aldrich Chemical Со., USA, 98%) и 178 г (2,4 моль) реагента 2-метил-1-пропанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленным Masuda Corporation, Japan, OBH-24). Температуру масляной бани устанавливали на 118°С. Затем колбу погружали на масляную баню, после чего включали вращение испарителя. Вращение проводили в течение примерно 8 часов с перемешиванием и нагреванием при атмосферном давлении с открытым продувочным вентилем испарителя. Затем колбу вынимали из масляной бани и оставляли для охлаждения. Колба содержала 139 г вязкого реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида дибутилолова в качестве исходного соединения реакционный раствор содержал 78% 1,1,3,3-тетрабутил-1,3-бис(2-метилпропилокси)дистанноксана и не содержал дибутил-бис(2-метил-1-пропилокси)олова.
Сравнительный пример 2
(Способ непрерывной перегонки только низкокипящих компонентов)
В 1-литровую колбу-сборник, снабженную холодильником и ловушкой Дина-Старка, загружали в качестве исходного материала 25 г (0,1 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA), 390 г (3,0 моль) реагента 2-этил-1-гексанола (изготовленного Aldrich Chemical Со., USA, 99,6%, безводного) и 300 мл толуола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки). Полученную смесь кипятили с обратным холодильником 12 часов на масляной бане и в течение этого времени температуру поддерживали при 120°С при перемешивании мешалкой. Ловушка Дина-Старка содержала 0,8 мл воды. При использовании оксида дибутилолова в качестве исходного соединения колба содержала примерно 95% 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана и не содержала дибутил-бис(2-этилгексилокси)олова.
Сравнительный пример 3
(Получение исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана)
Способом, описанным в сравнительном примере 1, из оксида дибутилолова, используемого в качестве исходного соединения, и 2-метил-1-пропанола получали 139 г жидкости, содержащей 78% исходного соединения 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана.
(Непрерывная экстракция только низкокипящих компонентов с использованием резервуарного реактора)
В 500-миллилитровый автоклав (от Yoyo Koatsu Co., Ltd., Japan), изготовленный из SUS и снабженный терморегулятором, мешалкой, трубопроводом для подачи азота и продувочным трубопроводом, загружали описанную выше жидкость и 74 г (1 моль) реагента 2-метил-1-пропанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, специальной очистки) и крышку закрывали. После продувки внутренней части автоклава газообразным азотом начинали перемешивание. Содержимое автоклава нагревали при температуре, установленной на 125°С, при перемешивании с открытым продувочным вентилем. Газообразные компоненты, образующиеся в результате повышения температуры, удаляли путем перегонки через продувочный трубопровод в течение 2 часов. Процесс перегонки низкокипящих компонентов контролировали для установления времени завершения реакции, после чего автоклав оставляли для охлаждения.119Sn-,1Н- и13С-ЯМР-анализы реакционного раствора во внутренней части автоклава показали, что при использовании оксида дибутилолова в качестве исходного соединения реакционный раствор содержал 79% 1,1,3,3-тетрабутил-1,3-бис(2-метил-1-пропилокси)дистанноксана, полученного из оксида дибутилолова и 2-метил-1-пропанола. Но при этом данный раствор не содержал дибутил-бис(2-метил-1-пропилокси)олова.
Сравнительный пример 4
(Способ непрерывной перегонки только низкокипящих компонентов)
В 1-литровую двухгорлую колбу, снабженную холодильником и ловушкой Дина-Старка, загружали 129 г (0,5 моль) исходного соединения оксида дибутилолова (изготовленного Aldrich Chemical Со., USA) и 510 г (5,0 моль) реагента 2-этил-1-бутанола (изготовленного Aldrich Chemical Со., USA, 98%). Перегонку осуществляли при перемешивании мешалкой путем нагревания в течение 6 часов на масляной бане, температуру которой поддерживали при 160°С, где образующуюся воду и 2-этил-1-бутанол подвергали взаимодействию с отгонкой. После охлаждения смеси с использованием оксида дибутилолова в качестве исходного соединения колба содержала примерно 8% дибутил-бис(2-этилбутилокси)олова, 80% 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана и 6% 2-этилбутилоксида трибутилолова. Скорость дегидратации в колонном реакторе составляла 0,045 моль/час и была ниже величины, вычисленной по уравнению (16), которая составляла 0,273 моль/час.
Сравнительный пример 5
(Способ непрерывной экстракции только низкокипящих компонентов, реагирующих при высокой температуре)
В 1-литровую двухгорлую колбу, снабженную холодильником Либиха и отверстием для взятия образцов, загружали 129 г (0,5 моль) исходного соединения оксида дибутилолова (изготовленного Aldrich Chemical Со., USA) и 651 г (5,0 моль) реагента 2-этил-1-гексанола (изготовленного Aldrich Chemical Со., USA, 99,6%, безводного). Перегонку осуществляли при перемешивании мешалкой путем нагревания в течение 2 часов на масляной бане, температуру которой поддерживали при 190°С, где образующуюся воду и 2-этил-1-гексанол подвергали взаимодействию с отгонкой. После охлаждения смеси с использованием оксида дибутилолова в качестве исходного соединения колба содержала примерно 40% дибутил-бис(2-этилгексилокси)олова, 1% 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана и 29% 2-этилгексилоксида трибутилолова. Скорость дегидратации в реакторе составляла 0,175 моль/час и была ниже величины, вычисленной по уравнению (16), которая составляла 3,44 моль/час.
Сравнительный пример 6
(Синтез эфира угольной кислоты из алкоксида дибутилолова, содержащего соединение трибутилолова и хлорсодержащее соединение)
Газообразный азот (0,3 л/мин) подавали через 100-миллилитровую колбу-сборник, снабженную трехходовым запорным краном. В колбу через газонепроницаемый шприц (изготовленный Hamilton Company, 1050TLL) вводили 23,80 г (0,063 моль) дибутоксида дибутилолова (изготовленного Azmax Co., содержание соединения трибутилолова составляло 1,5 моль%, содержание атомов хлора составляло 7600 ч./млн) и 26,44 г (0,30 моль) 3-метил-1-бутанола (изготовленного Aldrich Chemical Со., USA, ≥99%). Затем колбу встряхивали и получали однородную смесь раствора. Полученный смешанный раствор шприцем переносили в 150-миллилитровый сосуд под давлением, изготовленный из SUS 316L (Swagelok Company, 316L-50DF4-150) и снабженный вентилем. Вентиль закрывали, в результате чего сосуд становился герметично закрытым. 200-миллилитровый сосуд высокого давления (изготовленный Toyo Koatsu Co., Ltd., Japan, Система автоклавов серии FC), снабженный магнитной индукционной мешалкой, нагревательным кожухом, термометром, манометром, двумя вентилями для продувки газом и вентилем для взятия образцов жидкости, подсоединяли к цилиндру с газообразным азотом, снабженному регулятором давления, посредством системы трубопроводов, изготовленных из SUS316. Давление цилиндра с газообразным азотом устанавливали на 0,5 МПа с помощью регулятора давления. Вентиль для продувки газом сосуда высокого давления открывали и подавали азот до тех пор, пока давление в сосуде не достигало 0,5 МПа. Затем отдельный газовый вентиль открывали, снижая, таким образом, давление в сосуде до атмосферного давления. Эту процедуру повторяли 3 раза, в результате чего внутренняя часть реактора продувалась азотом. Сосуд под давлением, содержащий смешанный раствор, взвешивали. После подсоединения клапана для взятия образцов жидкости к сосуду высокого давления давление в сосуде повышали до 0,5 МПа путем подачи азота. Вентиль для взятия образцов жидкости медленно открывали и смешанный раствор вводили в сосуд высокого давления. Количество смешанного раствора, введенного в сосуд высокого давления, определяли по изменению массы сосуда под давлением. Затем автоклав начинали нагревать, включали мешалку при скорости вращения 450 об/мин и смешанный раствор нагревали при 120°С. Затем цилиндр с диоксидом углерода (изготовленный Showa Tansan Co., Ltd., чистота 99,99 об.%), снабженный регулятором давления, подсоединяли к вентилю для продувки газом сосуда высокого давления. Вторичное давление цилиндра с диоксидом углерода устанавливали на 4,5 МПа с помощью регулятора давления. Вентиль для продувки газом открывали и в сосуд высокого давления вводили газообразный диоксид углерода. Давление доводили до 4,0 МПа. После нагревания и перемешивания в течение 2 часов нагреватель вынимали из сосуда и сосуд оставляли для доведения внутренней температуры до комнатной температуры. Затем вентиль для продувки газом открывали, удаляя, таким образом газообразный диоксид углерода до тех пор, пока внутреннее давление сосуда не достигало 0,05 МПа. Вентиль для взятия образцов жидкости подсоединяли к 100-миллилитровой трехгорлой колбе, снабженной трехходовым запорным краном с использованием системы трубопроводов из тефлона (зарегистрированный торговый знак). После этого вентиль открывали и смешанный раствор переносили в колбу. Гравиметрический анализ содержимого колбы показал, что было собрано 22,07 г смешанного раствора. Затем колбу подсоединяли к соединительному трубопроводу, снабженному трубопроводом с разветвлениями, термометром, холодильником Либиха, соединительным трубопроводом низкого давления и двумя сосудами-сборниками дистиллята. Колбу погружали на масляную баню и смешанный раствор доводили до температуры 120°С. С помощью вакуумного клапана и вакуумного регулятора давление постепенно снижали примерно до 32 кПа. Этот режим поддерживали в течение примерно 1,5 часа, получая таким образом 11,51 г фракции 1, имеющей температуру парообразования 96°С. Затем давление в системе снова снижали до 0,15-0,06 кПа. Этот режим поддерживали в течение примерно 1 часа, получая таким образом 2,30 г фракции 2, имеющей температуру парообразования от 64°С до 80°С. Результаты GC-FID-анализа фракции 2 показали, что содержание ди-3-метилбутилкарбоната составляло 0,20 г. Кроме того, результаты анализа на содержание хлора показали, что оно составляло 70 ч./млн Результаты119Sn-ЯМР-анализа показали, что соединение трибутилолова имело выход 0,7 мас.%.
Сравнительный пример 7
(Получение эфира угольной кислоты из алкоксида дибутилолова, содержащего большое количество соединения трибутилолова и хлорсодержащего соединения)
В сосуд высокого давления загружали 23,56 г (0,079 моль) диметоксида дибутилолова (изготовленного Aldrich Chemical Со., USA, содержание соединения трибутилолова составляло 3,5 моль%) и 33,06 г (0,38 моль) 3-метил-1-бутанола (изготовленного Aldrich Chemical Со., USA, ≥99%), как описано в сравнительном примере 6. Нагревание и перемешивание также проводили способом, описанным выше. После снижения температуры смешанного раствора и удаления газообразного диоксида углерода аналогичным способом в 100-миллилитровой трехгорлой колбе, снабженной трехходовым запорным краном, собирали 37,51 г смешанного раствора. Затем колбу подсоединяли к соединительному трубопроводу, присоединенному к трубопроводу с разветвлениями, термометром, холодильнику Либиха, соединительному трубопроводу низкого давления и двум сосудам-сборникам дистиллята. Колбу погружали на масляную баню и смешанный раствор доводили до температуры 120°С. С помощью вакуумного насоса и вакуумного регулятора давление постепенно снижали примерно до 97-13 кПа. Этот режим поддерживали в течение примерно 1,5 часа, получая, таким образом, 16,85 г фракции 1, имеющей температуру парообразования 54-98°С. Затем давление в системе снова снижали до 0,06-0,2 кПа. Этот режим поддерживали в течение примерно 1 часа, получая таким образом 1,71 г фракции 2, имеющей температуру парообразования от 79°С до 81°С. Результаты GC-FID-анализа фракции 2 показали, что содержание ди-3-метилбутилкарбоната составляло 1,61 г. Кроме того, результаты119Sn-ЯМР-анализа показали, что содержание соединения трибутилолова в эфире угольной кислоты составляло 1,2 мас.%.
Пример 12
(Получение исходного соединения 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана)
В 2000-миллилитровую колбу-сборник загружали 542 г (2,18 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA) и 1400 г (18,9 моль) 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленной Masuda Corporation, Japan, OBH-24), к вакуумному насосу (изготовленному ULVAC Inc., Japan, G-50A) и к вакуумному регулятору (изготовленному Okano Works Ltd., Japan, VC-10S). Выходное отверстие продувочного вентиля испарителя подсоединяли к трубопроводу для подачи газообразного азота, подаваемого при атмосферном давлении. Затем продувочный вентиль испарителя закрывали, в результате чего давление в системе снижалось. После этого продувочный вентиль постепенно открывали и в систему поступал азот, создавая в системе снова атмосферное давление. Температуру масляной бани устанавливали на 126°С. Затем колбу погружали на масляную баню, после чего начинали вращение испарителя. Вращение проводили в течение примерно 30 минут при перемешивании и нагревании при атмосферном давлении с открытым продувочным вентилем испарителя, в результате чего реакционный раствор закипал, и затем начиналась перегонка низкокипящих компонентов. Этот режим поддерживали в течение 6 часов и затем продувочный вентиль закрывали, медленно снижая внутреннее давление системы. Остаточные низкокипящие компоненты отгоняли при поддержании внутреннего давления системы примерно от 76 до 54 кПа. После прекращения отгонки низкокипящих компонентов колбу вынимали из масляной бани. Реакционный раствор становился прозрачным. Дистиллятная жидкость, имеющая массу 1255 г, была прозрачной и была разделена на 2 слоя. Анализ дистиллятной жидкости показал, что содержание воды в этой жидкости составляло примерно 19,6 г. Затем колбу вынимали из масляной бани и продувочный вентиль постепенно закрывали для доведения внутреннего давления системы до атмосферного. Колба содержала 686 г реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида дибутилолова в качестве исходного соединения полученный продукт 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан имел выход 99%. Эту процедуру повторяли 6 раз и получали в итоге 4120 г 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана.
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 31, таком как проиллюстрировано на фиг.4. В колонный реактор с внутренним диаметром 15 мм и общей длиной 4000 мм, который изготовлен из SUS 316 и к которому в его верхней части 32 подсоединен питающий трубопровод 26, питающий трубопровод 27, теплообменник 28, трубопровод для сбора низкокипящих компонентов 34, холодильник 35, сепаратор для отделения газа от жидкости 36, клапан обратного давления 37, спускной трубопровод 38 и трубопровод для сбора жидкой фазы 39 и в его нижней части 33 - питающий газовый трубопровод 29, теплообменник 30 и экстракционный трубопровод 40, вставляли насадку Goodroll типа А (Tokyo Tokushu Kanaami K.K., Japan). Температуру реактора устанавливали на 140°С с помощью нагревателя.
Из питающего газового трубопровода 29 подавали 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 399 г/час и диоксид углерода со скоростью 3 л N/час. Весь 1-бутанол выпаривали с помощью теплообменника 30 и подавали в нижнюю часть реактора 33. Для подачи исходного соединения 1,1,3,3-тетрабутил-1,3-ди(2-бутилокси)дистанноксана из питающего трубопровода 26 со скоростью 210 г/час и для подачи реакционного раствора 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) из питающего трубопровод 27 со скоростью 951 г/час использовали питающий насос. Время пребывания в реакторе составляло 30 минут. Температура жидкости внутри реактора составляла 140°С и давление доводили до 0,096 МПа-G с помощью клапана обратного давления 37. Непрерывную подачу в этом режиме продолжали в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты выходили из верхней части реактора 32 и поступали через трубопровод для сбора низкокипящих компонентов в холодильник 35 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 39 со скоростью 753 г/час. Кроме того, из нижней части реактора 33 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 807 г/час из экстракционного трубопровода 40. Анализ полученной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения, полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 41,3% из дибутилди(бутилокси)олова и примерно на 58,7% из 1,1,3,3-тетрабутил-1,3-дис(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,04%. При этом жидкость, собранная из трубопровода для сбора жидкой фазы 39, была прозрачной и содержала 2500 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,144 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00015 моль/час.
Пример 13
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 31, таком как проиллюстрировано на фиг.4. В колонный реактор с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316 и к которому в его верхней части 32 подсоединен питающий трубопровод 26, питающий трубопровод 27, теплообменник 28, трубопровод для сбора низкокипящих компонентов 34, холодильник 35, сепаратор для отделения газа от жидкости 36, клапан обратного давления 37, спускной трубопровод 38 и трубопровод для сбора жидкой фазы 39 и в его нижней части 33 - питающий газовый трубопровод 29, теплообменник 30 и экстракционный трубопровод 40, вставляли металлическую насадку Gause CY (Sulzer Chemtech Ltd., Switzerland). Температуру реактора устанавливали на 140°С с помощью нагревателя.
Из питающего газового трубопровода 29 подавали 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 566 г/час и диоксид углерода со скоростью 3 л N/час. Весь 1-бутанол выпаривали с помощью теплообменника 30 и подавали в нижнюю часть реактора 33. Для подачи исходного соединения 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана, полученного способом, описанным в примере 12, из питающего трубопровода 26 со скоростью 280 г/час и реакционного раствора 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) из питающего трубопровода 27 со скоростью 1330 г/час использовали питающий насос. Время пребывания в реакторе составляло 13 минут. Температура жидкости внутри реактора составляла 140°С, и давление доводили до 0,096 МПа-G с помощью клапана обратного давления. Непрерывную подачу в этом режиме проводили в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты выходили из верхней части реактора 32 и поступали через трубопровод для сбора низкокипящих компонентов в холодильник 35 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 39 со скоростью 1006 г/час. Кроме того, из нижней части реактора 33 собирали компонент, содержащий алкоксид дибутилолова, который выходит со скоростью 1170 г/час из экстракционного трубопровода 40. Анализ жидкости, собранной из трубопровода для экстракции полученного продукта 40, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 37,5% из дибутилди(бутилокси)олова и примерно на 62,4% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутилоксида трибутилолова составлял 0,022%. При этом жидкость, собранная из трубопровода для сбора жидкой фазы 39, была прозрачной и содержала 2200 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,39 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,0015 моль/час.
Пример 14
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 46, таком как проиллюстрировано на фиг.5. Колонный реактор 46 в своей верхней части 47 снабжен питающим трубопроводом 41, питающим трубопроводом 42, теплообменником 43, трубопроводом для сбора низкокипящих компонентов 49, холодильником 50, резервуаром-сборником для низкокипящих компонентов 51, клапаном обратного давления 52, спускным трубопроводом 53 и трубопроводом для сбора жидкой фазы 54 и в его нижней части 48 - циркуляционным трубопроводом 44 для рециркуляции реакционного раствора, который аккумулируется в нижней части реактора, кипятильником 45 и экстракционным трубопроводом 55; при этом указанный реактор имел внутренний диаметр 50 мм и ситчатые тарелки общей длиной 3500 мм, снабженные сливными стаканами и изготовленные из SUS 316. Каждая ситчатая тарелка имела примерно 9 г части перегоняемой жидкости. Тарелки установлены с интервалами в 50 мм. Температуру колонного реактора устанавливали на 140°С с помощью нагревателя.
С помощью питающего насоса из питающего трубопровода 41 подавали исходное соединение - 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 280 г/час, и из питающего трубопровода 42 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 1400 г/час. Время пребывания в реакторе составляло 24 минуты. Реакционный раствор, который аккумулировался в нижней части реактора, подвергали рециркуляции со скоростью 6000 г/час с использованием цикруляционного трубопровода 44 и кипятильника 45, при этом температура нагревания составляла примерно 140°С. Температура жидкости внутри реактора составляла 140°С, и давление в реакторе доводили до 0,096 МПа-G с помощью клапана обратного давления 52. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты выходили из верхней части реактора 47 и поступали через трубопровод для подачи низкокипящих компонентов в холодильник 50 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 54 со скоростью 1010 г/час. Кроме того, из нижней части реактора 48 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 670 г/час из экстракционного трубопровода 55. Анализ собранной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения, полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 44,4% из дибутилди(бутилокси)олова и примерно на 55,0% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,05%. При этом жидкость, собранная из трубопровода для жидкой фазы 54, была прозрачной и содержала 2300 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,20 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,0015 моль/час.
Пример 15
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 46, таком как проиллюстрировано на фиг.5. В колонный реактор с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316 и к которому в его верхней части 47 подсоединен питающий трубопровод 41, питающий трубопровод 42, теплообменник 43, трубопровод для сбора низкокипящих компонентов 49, холодильник 50, резервуар-сборник для низкокипящих компонентов 51, клапан обратного давления 52, спускной трубопровод 53 и трубопровод для сбора жидкой фазы 54, а в его нижней части 48 - циркуляционный трубопровод 44 для рециркуляции реакционного раствора, который аккумулируется в нижней части реактора, кипятильник 45 и экстракционный трубопровод 55; вставляли насадку Меллапак 750Y (Sulzer Chemtech Ltd., Switzerland). Температуру реактора доводили до 140°С с помощью нагревателя.
С помощью питающего насоса из питающего трубопровода 41 подавали исходное соединение - 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 280 г/час, и из питающего трубопровода 42 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 1330 г/час. Время пребывания в реакторе составляло 13 минут. Реакционный раствор, который аккумулировался в нижней части реактора, подвергали рециркуляции со скоростью 6000 г/час с использованием цикруляционного трубопровода 44 и кипятильника 45; при этом температура нагревания составляла примерно 140°С. Температура жидкости внутри реактора составляла 140°С, и давление в реакторе доводили до 0,096 МПа-G с помощью клапана обратного давления. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты выходили из верхней части реактора 47 и поступали через трубопровод для подачи низкокипящих компонентов в холодильник 50 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 54 со скоростью 1006 г/час. Кроме того, из нижней части реактора 48 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 604 г/час из экстракционного трубопровода 55. Анализ жидкости, собранной из экстракционного трубопровода 55, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 46,3% из дибутилди(бутилокси)олова и примерно на 53,6 % из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,022%. При этом жидкость, собранная из трубопровода для сбора жидкой фазы 54, была прозрачной и содержала 2200 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,21 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00081 моль/час.
Пример 16
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 61, таком как проиллюстрировано на фиг.6. В колонный реактор с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316 и к которому в его верхней части 63 подсоединен питающий трубопровод 56, питающий трубопровод 57, теплообменник 58, трубопровод для сбора низкокипящих компонентов 65, холодильник 66, резервуар-сборник для низкокипящих компонентов 67, клапан обратного давления 68, спускной трубопровод 69 и трубопровод для сбора жидкой фазы 70; в его средней части 62 - трубопровод для сбора низкокипящих компонентов 71, холодильник 72, резервуар-сборник для низкокипящих компонентов 73, газовый трубопровод 74 и трубопровод для сбора жидкой фазы 75, а в его нижней части 64 - циркуляционный трубопровод 59 для рециркуляции реакционного раствора, который аккумулируется в нижней части реактора, кипятильник 60 и экстракционный трубопровод 76, вставляли насадку Меллапак 750Y (Sulzer Chemtech Ltd., Switzerland). Температуру реактора доводили до 140°С с помощью нагревателя.
С помощью питающего насоса из питающего трубопровода 56 подавали исходное соединение - 1,1,3,3-тетрабутил-1,3-дис(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 280 г/час, и из питающего трубопровода 57 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 1332 г/час. Время пребывания в реакторе составляло 13 минут. Реакционный раствор, который аккумулировался в нижней части реактора, подвергали рециркуляции со скоростью 6000 г/час с использованием циркуляционного трубопровода 59 и кипятильника 60; при этом температура нагревания составляла примерно 140°С. Температура жидкости внутри реактора составляла 140°С, и давление в реакторе доводили до 0,096 МПа-G с помощью клапана обратного давления 68. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты поступали из верхней части реактора 63 через трубопровод для сбора низкокипящих компонентов 65 в холодильник 50 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 70 со скоростью 512 г/час. Низкокипящие компоненты также собирали из средней части реактора 62 с помощью трубопровода для сбора жидкой фазы 75 со скоростью 496 г/час. Кроме того, из нижней части реактора 64 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 603 г/час из экстракционного трубопровода 76. Анализ жидкости, собранной из экстракционного трубопровода 76, показал, что при использовании оксида дибутилолова в качестве исходного соединения полученная жидкость содержала алкоксид дибутилолова, состоящий примерно на 47,5% из дибутилди(бутилокси)олова и примерно на 52,4% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,021%. При этом жидкость, собранная из трубопроводов для сбора жидкой фазы 70 и 75, была прозрачной и содержала 2200 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,21 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00081 моль/час.
Пример 17
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 82, таком как проиллюстрировано на фиг.7. В колонный реактор 82 с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316, и к которому в его верхней части 84 подсоединен трубопровод для сбора низкокипящих компонентов 86, холодильник 87, резервуар-сборник для низкокипящих компонентов 88, клапан обратного давления 89, спускной трубопровод 90 и трубопровод для сбора жидкой фазы 91; в его средней части 83 - питающий трубопровод 77, питающий трубопровод 78 и теплообменник 79, и в его нижней части 85 - циркуляционный трубопровод 80 для рециркуляции реакционного раствора, который аккумулируется в нижней части реактора, кипятильник 81 и экстракционный трубопровод 92; вставляли насадку Меллапак 750Y (Sulzer Chemtech Ltd., Switzerland). Температуру реактора доводили до 140°С с помощью нагревателя.
С помощью питающего насоса, из питающего трубопровода 77 подавали исходное соединение - 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 280 г/час, и из питающего трубопровода 78 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 1330 г/час. Время пребывания в реакторе составляло 6 минут. Реакционный раствор, который аккумулировался в нижней части реактора, подвергали рециркуляции со скоростью 6000 г/час с использованием цикруляционного трубопровода 80 и кипятильника 81; при этом температура нагревания составляла примерно 140°С. Температура жидкости внутри реактора составляла 140°С, и давление в реакторе доводили до 0,096 МПа-G с помощью клапана обратного давления 89. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты поступали из верхней части реактора 84 через трубопровод-сборник для низкокипящих компонентов 86 в холодильник 87 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 91 со скоростью 1031 г/час. Кроме того, из нижней части реактора 85 также собирали низкокипящий компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 602 г/час из экстракционного трубопровода 92. Анализ полученной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения, жидкость содержала алкоксид дибутилолова, состоящий примерно на 47,0% из дибутилди(бутилокси)олова и примерно на 52,9% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,012%. При этом жидкость, собранная из трубопровода для сбора жидкой фазы 91 была прозрачной и содержала 2200 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,21 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00037 моль/час.
Пример 18
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 99, таком как проиллюстрировано на фиг.8. В колонный реактор 99 с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316, и к которому в его верхней части 101 подсоединен трубопровод для сбора низкокипящих компонентов 103, холодильник 104, резервуар-сборник для низкокипящих компонентов 105, клапан обратного давления 106, спускной трубопровод 107 и трубопровод для сбора жидкой фазы 108; в его средней части 100 - питающий трубопровод 93, питающий трубопровод 94 и теплообменник 95, и в его нижней части 102 - трубопровод для подачи органического растворителя 96, циркуляционный трубопровод 97 для рециркуляции аккумулированного реакционного раствора, кипятильник 98 и экстракционный трубопровод 109, вставляли насадку Меллапак 750Y (Sulzer Chemtech Ltd., Switzerland). Температуру реактора доводили до 140°С с помощью нагревателя.
С помощью питающего насоса из питающего трубопровода 93 подавали исходное соединение - 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 280 г/час, и из питающего трубопровода 94 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 1330 г/час. Время пребывания в реакторе составляло 6 минут. Для подачи гексана (изготовленного Wako Pure Chemical Industries Ltd., Japan, с чистотой, пригодной для дегидратации) из трубопровода для подачи органического растворителя 96, находящегося в нижней части реактора 102, использовали насос со скоростью подачи 300 г/час и аккумулированный реакционный раствор подвергали рециркуляции со скоростью 6000 г/час с использованием цикруляционного трубопровода 97 и кипятильника 98; при этом температура нагревания составляла примерно 140°С. Температура жидкости внутри реактора составляла 140°С, и давление в реакторе доводили до 0,12 МПа-G с помощью клапана обратного давления 106. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты поступали из верхней части реактора 101 через трубопровод для сбора низкокипящих компонентов 103 в холодильник 104 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 108 со скоростью 1088 г/час. Кроме того, из нижней части реактора 102 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 821 г/час из экстракционного трубопровода 109. Анализ полученной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения жидкость содержала алкоксид дибутилолова, состоящий примерно на 54,0% из дибутилди(бутилокси)олова и примерно на 46,0% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,013%. При этом жидкость, собранная из трубопроводов для сбора жидкой фазы 108 была прозрачной и содержала 2500 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,24 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00037 моль/час.
Пример 19
(Получение диалкоксида дибутилолова с использованием горизонтального перегонного аппарата с тонкой пленкой)
Алкоксид дибутилолова получали в горизонтальном перегонном аппарате с тонкой пленкой 113 (изготовленном Nitinan Engineering Co., Ltd., Japan, PFD1), таком как проиллюстрировано на фиг.9. Горизонтальный перегонный аппарат с тонкой пленкой 113, имеющий внутренний диаметр 50 мм и общую длину 1100 мм и изготовленный из SUS 316А, снабжен в его верхней части 114 питающим трубопроводом 110, питающим трубопроводом 111, теплообменником 112, трубопроводом для сбора низкокипящих компонентов 116, холодильником 117, резервуаром-сборником для низкокипящих компонентов 118, спускным трубопроводом 119 и трубопроводом для сбора жидкой фазы 120, и в его нижней части 115 - экстракционным трубопроводом 121. Температуру реактора доводили до 120°С с помощью нагревателя.
С помощью питающего насоса из питающего трубопровода 110 подавали исходное соединение 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 4600 г/час, и из трубопровода 111 для подачи спирта поступал реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 22000 г/час. Время пребывания в реакторе составляло 6 минут. Температуру жидкости внутри реактора доводили до 120°С. Непрерывную подачу проводили в этом режиме в течение 2 часов, после чего давление в системе достигало постоянного значения. Низкокипящие компоненты поступали из трубопровода для сбора низкокипящих компонентов 116 в холодильник 117 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 120 со скоростью 18000 г/час. Кроме того, из нижней части реактора 115 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 8600 г/час из экстракционного трубопровода 121. Анализ полученной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения, жидкость содержала алкоксид дибутилолова, состоящий примерно на 34,2% из дибутилди(бутилокси)олова и примерно на 65,7% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,015%. При этом жидкость, собранная из трубопроводов для сбора жидкой фазы 120, была прозрачной и содержала 2000 ч./млн влаги. Скорость дегидратации в реакторе составляла 2,5 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,0061 моль/час.
Пример 20
Стадия 1: (Получение исходного соединения 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана)
В 3000-миллилитровую колбу-сборник загружали 759 г (3,05 моль) оксида дибутилолова (изготовленного Aldrich Chemical Со., USA) и 1960 г (26,5 моль) 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan), в результате в колбе образовывалась смесь в виде белой взвеси. Колбу подсоединяли к испарителю (изготовленному Shibata Scientific Technology Ltd., R-144), соединенному с масляной баней, снабженной терморегулятором (изготовленным Masuda Corporation, Japan, OBH-24), к вакуумному насосу (изготовленному ULVAC Inc., Japan, G-50A) и к вакуумному регулятору (изготовленному Okano Works Ltd., Japan, VC-10S). Выходное отверстие продувочного вентиля испарителя подсоединяли к трубопроводу для подачи газообразного азота при атмосферном давлении. Затем продувочный вентиль испарителя закрывали, снижая давление в системе. После этого продувочный вентиль постепенно открывали и в систему поступал азот, создавая в системе снова атмосферное давление. Температуру масляной бани устанавливали на 127°С. Затем колбу погружали на масляную баню, после чего начинали вращение испарителя. Вращение проводили в течение примерно 40 минут при перемешивании и нагревании при атмосферном давлении с открытым продувочным вентилем испарителя, в результате чего реакционный раствор закипал, и затем начиналась перегонка низкокипящих компонентов. Этот режим поддерживали в течение 7 часов и затем продувочный вентиль закрывали, медленно снижая внутреннее давление системы. Остаточные низкокипящие компоненты отгоняли при поддержании внутреннего давления системы примерно от 76 до 54 кПа. После прекращения отгонки низкокипящих компонентов, колбу вынимали из масляной бани. Реакционный раствор становился прозрачным. Дистиллятная жидкость имела массу 1737 г, была прозрачной и была разделена на 2 слоя. Анализ дистиллятной жидкости показал, что содержание воды в этой жидкости составляло примерно 27,6 г. Затем колбу вынимали из масляной бани и продувочный вентиль медленно закрывали для доведения внутреннего давления системы до атмосферного. Колба содержала 958 г реакционного раствора. Результаты119Sn-,1Н- и13С-ЯМР-анализов показали, что при использовании оксида дибутилолова в качестве исходного соединения полученный продукт 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан имел выход 99%. Эти процедуры повторяли 6 раз и получали в итоге 5748 г 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана.
Стадия 2: (Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе, таком как проиллюстрировано на фиг.4. В колонный реактор 31 с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316 и к которому в его верхней части 32 подсоединен питающий трубопровод 26, питающий трубопровод 27, теплообменник 28, трубопровод для сбора низкокипящих компонентов 34, холодильник 35, сепаратор для отделения газа от жидкости 36, клапан обратного давления 37, спускной трубопровод 38 и трубопровод для сбора жидкой фазы 39, а в его нижней части 33 - питающий газовый трубопровод 29, теплообменник 30 и экстракционный трубопровод 40, вставляли насадку Goodroll типа А (Tokyo Tokushu Kanaami K.K., Japan). Температуру реактора устанавливали на 140°С с помощью нагревателя. Из питающего газового трубопровода 29 подавали 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 399 г/час и диоксид углерода со скоростью 3 л N/час. Весь 1-бутанол выпаривали с помощью теплообменника 30 и подавали в нижнюю часть реактора 33. Для подачи исходного соединения 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана из питающего трубопровода 26 со скоростью 210 г/час и для подачи реакционного раствора 1-бутанола (изготовленного Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) из питающего трубопровода 27 со скоростью 951 г/час использовали питающий насос. Время пребывания в реакторе составляло 30 минут.
Стадия 3: (Получение эфира угольной кислоты из алкоксида диалкилолова)
Реакционный раствор, полученный, как описано в стадии 2, подавали с помощью питающего насоса со скоростью 807 г/час в перегонный аппарат с тонкой пленкой (изготовленный Kobelco Eco-Solutions Co., Ltd., Japan), установленный на 80°С и примерно на 6,5 кПа, для удаления летучих компонентов. Нелетучие компоненты охлаждали и собирали и затем подавали в 990-миллилитровый автоклав со скоростью 241 г/час (изготовленный Toyo Koatsu Co., Ltd., Japan). После подсоединения к автоклаву с помощью SUS-трубопроводов цилиндра вторичного давления с диоксидом углерода и установления вентиля на 4 МПа вентиль открывали и в автоклав подавали диоксид углерода со скоростью 28 г/час с использованием регулятора потока массы вещества (изготовленного Oval Corporation, Japan). Температуру доводили до 120°С. Время пребывания в автоклаве составляло примерно 1 час. Жидкость, которая реагировала с диоксидом углерода, переносили в резервуар для удаления газообразного диоксида углерода и давление доводили до атмосферного давления. Подачу реакционного раствора в перегонный аппарат с тонкой пленкой (изготовленный Kobelco Eco-Solutions Co., Ltd., Japan), установленный на 130°С и примерно на 1,3 кПа, осуществляли с помощью питающего насоса со скоростью 267 г/час для удаления летучих компонентов, включая дибутилкарбонат, и нелетучие компоненты охлаждали и собирали. Летучий компонент, содержащий дибутилкарбонат, подавали со скоростью 202 г/час в средний ярус многоходовой колонны для непрерывной перегонки с внутренним диаметром 50 мм и длиной 2000 мм, снабженной насадкой Диксона (с внутренним диаметром 6 мм), и проводили разделение дистиллятов. Охлажденная жидкость представляла собой смесь 1-бутанола и дибутилкарбоната, в которой 98% составлял дибутилкарбонат. В полученном смешанном растворе не было обнаружено бутилоксида трибутилолова, при этом результаты анализа на хлор также не выявили присутствия хлора. Однако результаты119Sn-,1Н- и13С-ЯМР-анализов нелетучего компонента показали, что полученный компонент содержал 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан и не содержал дибутилди(бутилокси)олова.
1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, содержащийся в нелетучем компоненте, полученном в стадии 3, возвращали в колонный реактор 31 с помощью питающего насоса и стадию 2 повторяли.
Температуру жидкости внутри реактора на стадии 2 доводили до 140°С, и давление доводили до 0,096 МПа-G с помощью клапана обратного давления 37. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Низкокипящие компоненты поступали из верхней части реактора 32 через трубопровод для подачи низкокипящих компонентов 34 в холодильник 35 для разжижения, после чего они выходили из трубопровода для сбора жидкой фазы 39 со скоростью 753 г/час. Кроме того, из нижней части реактора 33 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 870 г/час из экстракционного трубопровода 40. Анализ полученной жидкости показал, что при использовании оксида дибутилолова в качестве исходного соединения жидкость содержала алкоксид дибутилолова, состоящий на 41,3% из дибутилди(бутилокси)олова и на 58,7% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,04%. При этом жидкость, собранная из трубопроводов для сбора жидкой фазы 39, была прозрачной и содержала 2500 ч./млн влаги. Скорость дегидратации в колонном реакторе составляла 0,144 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,0015 моль/час. Кроме того, выход дибутилкарбоната, полученного, как описано в стадии 3, составлял примерно 30 г/час. Полученный дибутилкарбонат не содержал ни соединений хлора, ни соединений трибутилолова.
Пример 21
(Получение дифенилкарбоната из дибутилкарбоната, полученного способом, описанным в примере 20)
Получение катализатора
Фенол (80 г) и 32 г моноксида свинца нагревали в течение 12 часов при 180°С и образовавшуюся воду отгоняли вместе с фенолом с получением катализатора А.
(Получение бутифенилкарбоната)
Бутифенилкарбонат получали с использованием дибутилкарбоната, полученного, как описано в примере 20, используя аппарат, проиллюстрированный на фиг.10. Реакцию проводили с использованием питающего насоса путем непрерывной подачи смешанного раствора дибутилкарбоната, фенола и катализатора А (чтобы массовое отношение в смеси дибутилкарбоната и фенола составляло 65/35, и концентрация свинца составляла примерно 1 мас.%) из питающего трубопровода 122 через теплообменник 123 со скоростью 270 г/час в средний ярус многоходовой колонны для непрерывной перегонки 124 с внутренним диаметром 50 мм и длиной 2000 мм, которая снабжена ситчатыми тарелками в количестве примерно 40 штук. Тепло, необходимое для проведения реакции и перегонки, подавали с помощью циркуляционного нагревания нижней части колонны, через циркуляционный трубопровод 131 и кипятильник 130. Температуру жидкости, находящейся в нижней части многоходовой дистилляционной колонны для непрерывной перегонки 124, доводили до 231°С, и давление в верхней части колонны доводили примерно до 200 кПа с помощью клапана обратного давления 128. Коэффициент орошения устанавливали примерно на 2. Низкокипящие компоненты, отогнанные из верхней части многоходовой дистилляционной колонны для непрерывной перегонки 124, поступали через трубопровод-сборник низкокипящих компонентов для их конденсации в холодильнике 126, затем они поступали через резервуар-сборник для низкокипящих компонентов 127, после чего их непрерывно выводили из трубопровода 129 со скоростью примерно 67 г/час. Из нижней части колонны низкокипящие компоненты поступали через трубопровод для низкокипящих компонентов 131 и затем непрерывно выводились из экстракционного трубопровода 132 со скоростью 203 г/час. Жидкость, экстрагированная из трубопровода для сбора жидкой фазы 129, содержала примерно 27 мас.% 1-бутанола, примерно 72 мас.% фенола и примерно 1 мас.% дибутилкарбоната. Жидкость, отогнанная из экстракционного трубопровода 132, содержала 330 ч./млн 1-бутанола, примерно 11 мас.% фенола, примерно 65 мас.% дибутилкарбоната, примерно 21 мас.% бутилфенилкарбоната и примерно 1 мас.% дифенилкарбоната, и имела концентрацию Pb примерно 1 мас.%.
(Получение дифенилкарбоната посредством реакции диспропорционирования бутилфенилкарбоната)
Дифенилкарбонат получали с использованием аппарата, проиллюстрированного на фиг.11. Реакцию проводили с использованием питающего насоса путем непрерывной подачи бутилфенилкарбоната из питающего трубопровода 133 через подогреватель 134 со скоростью примерно 203 г/час в средний ярус многоходовой колонны для непрерывной перегонки 135 с диаметром 50 см и длиной 2 м, которая снабжена ситчатыми тарелками в количестве примерно 40 штук. Тепло, необходимое для проведения реакции и перегонки, подавали посредством циркуляционного нагревания нижней части колонны, через циркуляционный трубопровод 142 и кипятильник 141. Температуру жидкости, находящейся в нижней части многоходовой дистилляционной колонны для непрерывной перегонки 135, доводили до 237°С, и давление в верхней части колонны доводили примерно до 27 кПа с помощью клапана обратного давления 139. Коэффициент орошения устанавливали примерно на 2. Низкокипящие компоненты, отогнанные из верхней части многоходовой дистилляционной колонны для непрерывной перегонки 135, поступали через трубопровод-сборник низкокипящих компонентов 136 в холодильник 137 для их конденсации, и затем они поступали через резервуар-сборник для низкокипящих компонентов 138, после чего их непрерывно выводили из трубопровода для сбора жидкой фазы 140 со скоростью примерно 172 г/час. Из нижней части колонны высококипящие компоненты непрерывно отгоняли через экстракционный трубопровод 143 со скоростью примерно 31 г/час. Жидкость, отогнанная из трубопровода для сбора жидкой фазы 140, содержала 390 ч./млн 1-бутанола, примерно 13 мас.% фенола, примерно 86 мас.% дибутилкарбоната и примерно 1 мас.% бутилфенилкарбоната. Жидкость, экстрагированная из экстракционного трубопровода 143, содержала 500 ч./млн дибутилкарбоната, примерно 26 мас.% бутилфенилкарбоната, и примерно 65 мас.% дифенилкарбоната, и имела концентрацию Pb примерно 8 мас.%.
Затем дифенилкарбонат очищали с использованием аппарата, проиллюстрированного на фиг.12. Жидкость непрерывно подавали со скоростью 315 г/час из экстракционного трубопровода 143 через питающий трубопровод 144 и теплообменник 145 в средний ярус многоходовой колонны для непрерывной перегонки 146 с диаметром 5 см и длиной 2 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм) и проводили разделение дистиллятов. Тепло, необходимое для проведения разделения дистиллятов, подавали путем циркуляционного нагревания нижней части колонны через циркуляционный трубопровод 153 и кипятильник 152. Температуру жидкости, находящейся в нижней части многоходовой дистилляционной колонны для непрерывной перегонки 146, доводили до 210°С, и давление в верхней части колонны доводили примерно до 1,5 кПа с помощью клапана обратного давления 150. Коэффициент орошения устанавливали примерно на 1. Низкокипящие компоненты, отогнанные из верхней части многоходовой дистилляционной колонны для непрерывной перегонки 146, поступали через трубопровод-сборник низкокипящих компонентов 147 в холодильник 148 для их конденсации, затем они поступали через резервуар-сборник для низкокипящих компонентов 149, после чего их непрерывно выводили из трубопровода для сбора жидкой фазы 151 со скоростью примерно 288 г/час. Из нижней части колонны, низкокипящие компоненты непрерывно выводились из системы через экстракционный трубопровод 154 со скоростью примерно 27 г/час. Жидкость, экстрагированная из трубопровода для сбора жидкой фазы 151, содержала 200 ч./млн дибутилкарбоната, примерно 29 мас.% бутилфенилкарбоната и примерно 71 мас.% дифенилкарбоната. Жидкость, непрерывно экстрагированную из трубопровода для сбора жидкой фазы 155, подавали со скоростью 288 г/час через соединительный трубопровод 155 и теплообменник 156 в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки 157 с диаметром 5 см и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), и проводили разделение дистиллятов. Тепло, необходимое для разделения дистиллятов, подавали путем циркуляционного нагревания нижней части колонны через циркуляционный трубопровод 164 и кипятильник 163. Температуру жидкости, находящейся в нижней части многоходовой дистилляционной колонны для непрерывной перегонки 157, доводили до 198°С, и давление в верхней части колонны доводили примерно до 6 кПа с помощью клапана обратного давления 161. Коэффициент орошения устанавливали примерно на 6. Низкокипящие компоненты, отогнанные из верхней части многоходовой дистилляционной колонны для непрерывной перегонки 157, поступали через трубопровод-сборник для низкокипящих компонентов 158 и конденсировались в холодильнике 159, и затем они поступали через резервуар-сборник для низкокипящих компонентов 160, после чего их непрерывно выводили из трубопровода для сбора жидкой фазы 162 со скоростью примерно 90 г/час. Из нижней части колонны, низкокипящие компоненты непрерывно выводились из системы через экстракционный трубопровод 165 со скоростью примерно 198 г/час. Жидкость, экстрагированная из трубопровода для сбора жидкой фазы 162, содержала примерно 700 ч./млн дибутилкарбоната, примерно 93 мас.% бутилфенилкарбоната и примерно 7 мас.% дифенилкарбоната. Жидкость, экстрагированная из экстракционного трубопровода 165, содержала бутилфенилкарбонат в количестве ниже измеримого предела и 99 мас.% дифенилкарбоната. Концентрация хлора в указанном реакционном растворе была ниже измеримых пределов.
Пример 22
(Получение гексаметилендиизоцианата из дифенилкарбоната, полученного, как описано в примере 21)
В 500-миллилитровую колбу, снабженную мешалкой, термометром и капельной воронкой, загружали 161 г (0,75 моль) дифенилкарбоната, полученного, как описано в примере 21, и 142 г (1,5 моль), фенола (изготовленного Aldrich Chemical Со., USA, предварительно отогнанного). После продувки сухим азотом колбу погружали на водяную баню при 50°С и начинали перемешивание. После подтверждения растворения твердого вещества в колбе температуру водяной бани устанавливали на 45°С. Капельная воронка содержала 35 г (0,3 моль) 1,6-гексаметилендиамина (изготовленного Aldrich Chemical Со., предварительно отогнанного), поддерживаемого при температуре 45-50°С. Затем из капельной воронки соединение начинали по каплям вводить в колбу. При этом скорость падения капель регулировали таким образом, чтобы температура жидкости в колбе составляла 50-60°С, и такое введение по каплям проводили в течение примерно 20 минут. После завершения введения по каплям температуру водяной бани корректировали таким образом, чтобы температура жидкости в колбе составляла 50°С, и перемешивание продолжали в течение 1 часа. Результаты анализа, проводимого с помощью высокоэффективной жидкостной хроматографии и гель-проникающей хроматографии, показали, что эффективность реакции 1,6-гексаметилендиамина составляла 100% с выходом полученного 1,6-гексаметиленфенилдикарбамата 99,6% и селективностью 99,6%. Выход соединений мочевины составлял примерно 0,4%.
Реакционный раствор, полученный описанным способом, подавали через подогреватель в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), где избыток фенола в газообразной форме удаляли из верхней части дистилляционной колонны, и высококипящие компоненты непрерывно удаляли из нижней части дистилляционной колонны. Нижнюю часть колонны подвергали циркуляционному нагреванию при 130°С с помощью кипятильника, и давление в верхней части колонны доводили примерно до 20 кПа. Тепловое разложение проводили путем подачи раствора, экстрагированного из нижней части колонны, через соединительный трубопровод и насос на участок, расположенный примерно на расстоянии 1 м от дна многоходовой дистилляционной колонны для непрерывной перегонки, имеющей диаметр 2 дюйма и длину 4 м и снабженной насадкой Диксона (с внутренним диаметром 6 мм). Нижнюю часть колонны подвергали циркуляционному нагреванию при 220°С с помощью кипятильника, и давление в верхней части колонны доводили примерно до 2,6 кПа. Компонент, содержащий гексаметилендиизоцианат в газообразной форме, экстрагировали из участка, расположенного на расстоянии 2 м от верха колонны, и фенол в газообразной форме экстрагировали из верхней части колонны. Компонент, содержащий гексаметилендиизоцианат, подавали в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), проводя тем самым очистку гексаметилендиизоцианата. Нижнюю часть колонны подвергали циркуляционному нагреванию при 120°С с помощью кипятильника, и давление в верхней части колонны доводили примерно до 0,13 кПа. В результате из верхней части колонны экстрагировались компоненты, содержащие гексаметилендиизоцианат с чистотой 99,9%. Кроме того, соединения, экстрагированные со дна колонны, содержали дифенилкарбонат в качестве главного компонента.
Сравнительный пример 8
(Получение гексаметилендиизоцианата из дифенилкарбоната, содержащего соединение хлора)
В 500-миллилитровую колбу, снабженную мешалкой, термометром и капельной воронкой, загружали 161 г (0,75 моль) дифенилкарбоната (изготовленного Bayer, Germany, гидролизуемое соединение с содержанием хлора 15 ч./млн) и 142 г (1,5 моль) фенола (изготовленного Aldrich Chemical Со., USA, предварительно отогнанного). После продувки сухим азотом колбу погружали на водяную баню при 50°С и начинали перемешивание.
После подтверждения растворения твердого вещества в колбе температуру водяной бани устанавливали на 45°С. Капельная воронка содержала 35 г (0,3 моль) 1,6-гексаметилендиамина (изготовленного Aldrich Chemical Со., предварительно отогнанного), поддерживаемого при температуре 45-50°С. Затем из этой капельной воронки начинали по каплям вводить в колбу указанное соединение. При этом скорость падения капель регулировали таким образом, чтобы температура жидкости в колбе составляла 50-60°С, и такое введение по каплям проводили в течение примерно 20 минут. После завершения введения по каплям температуру водяной бани регулировали таким образом, чтобы температура жидкости в колбе составляла 50°С, и перемешивание продолжали в течение 1 часа.
Результаты анализа, проводимого с помощью высокоэффективной жидкостной хроматографии и гельпроникающей хроматографии, показали, что эффективность реакции 1,6-гексаметилендиамина составляла 99%, с выходом полученного 1,6-гексаметиленфенилдикарбамата 99% и селективностью 99,6%. Выход соединений мочевины составлял примерно 0,5%.
Реакционный раствор, полученный таким способом, подавали через котел-утилизатор в средний ярус многоходовой колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), где избыток фенола в газообразной форме удаляли из верхней части дистилляционной колонны, и высококипящие компоненты непрерывно удаляли из нижней части дистилляционной колонны. Нижнюю часть колонны подвергали циркуляционному нагреванию при 130°С с помощью кипятильника, и давление в верхней части колонны доводили примерно до 20 кПа. Тепловое разложение проводили путем подачи жидкости, экстрагированной из нижней части колонны, через соединительный трубопровод и насос, на участок, расположенный примерно на расстоянии 1 м от дна многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, снабженной насадкой Диксона (с внутренним диаметром 6 мм). Нижнюю часть колонны подвергали циркуляционному нагреванию при 220°С с помощью кипятильника, и давление в верхней части колонны доводили примерно до 2,6 кПа.
Компонент, содержащий гексаметилендиизоцианат в газообразной форме, экстрагировали из участка, расположенного на расстоянии 2 м от верха колонны, и фенол в газообразной форме экстрагировали из верхней части колонны. Компонент, содержащий гексаметилендиизоцианат, подавали в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), проводя тем самым очистку гексаметилендиизоцианата. Нижнюю часть колонны подвергали циркуляционному нагреванию при 120°С с помощью кипятильника, и давление в верхней части колонны доводили примерно до 130 Па.
В результате, из верхней части колонны экстрагировались компоненты, содержащие гексаметилендиизоцианат с чистотой 99,3%. Кроме того, соединения, отогнанные со дна колонны, содержали дифенилкарбонат в качестве главного компонента. Полученный гексаметилендиизоцианат содержал 5 ч./млн гидролизуемого соединения хлора.
Пример 23
(Получение поликарбоната из дифенилкарбоната, полученного, как описано в примере 21)
В вакуумный реактор, снабженный смесителем, загружали 23,5 г дифенилкарбоната (ЯМР-анализ не обнаруживал каких-либо примесей, включающих метильные группы (за исключением метильных групп, присутствующих на концах алкильных групп)) и 22,8 г бисфенола А. После продувки газообразным азотом полученную смесь полимеризовали под давлением 8 кПа в течение 30 минут и под давлением 4 кПа в течение 90 минут, после чего температуру повышали до 270°С и осуществляли полимеризацию при 0,07 кПа в течение 1 часа. В результате получали бесцветный и прозрачный ароматический поликарбонат хорошего качества, который имел среднечисленную молекулярную массу 105000.
Сравнительный пример 9
(Получение поликарбоната из дифенилкарбоната, содержащего соединение хлора)
В вакуумный реактор, снабженный смесителем, загружали 23,5 г дифенилкарбоната (изготовленного Bayer, Germany, с содержанием хлора 15 ч./млн) и 22,8 г бисфенола А. После продувки газообразным азотом полученную смесь полимеризовали под давлением 8 кПа в течение 30 минут и под давлением 4 кПа в течение 90 минут, после чего температуру повышали до 270°С и полимеризацию осуществляли при 0,07 кПа в течение 1 часа. Полимер не был получен. В результате был получен не полностью прореагировавший продукт, который содержал олигомер, имеющий среднечисленную молекулярную массу 800 или менее.
Сравнительный пример 10
(Синтез эфира угольной кислоты из алкоксида дибутилолова, содержащего большое количество соединения трибутилолова)
В сосуд высокого давления загружали 120 г 2-этилгексилоксида дибутилолова (с содержанием 2-этилгексилоксида трибутилолова 29 моль%), полученного, как описано в сравнительном примере 5, способом, описанным в сравнительном примере 6. Нагревание и перемешивали осуществляли аналогичным способом. После снижения температуры смешанного раствора и удаления газообразного диоксида углерода аналогичным образом в 200-миллилитровую трехгорлую колбу, снабженную трехходовым запорным краном, собирали 100 г смешанного раствора. Затем колбу подсоединяли к соединительному трубопроводу, присоединенному к трубопроводу с разветвлениями, термометру, холодильнику Либиха, соединительному трубопроводу низкого давления и двум сосудам-сборникам дистиллята. Колбу погружали на масляную баню и смешанный раствор доводили до температуры 130°С. С помощью вакуумного насоса и вакуумного регулятора давление постепенно снижали примерно до 0,13 кПа и получали 18 г фракции, имеющей температуру парообразования примерно 125°С. Результаты GC-FID-анализа данной фракции показали, что содержание в ней ди-2-этилгексилкарбоната составляло 55 мас.%. Кроме того, результаты119Sn-ЯМР-анализа показали, что в эфире угольной кислоты содержалось примерно 44 мас.% соединения трибутилолова.
Сравнительный пример 11
(Получение катализатора)
Фенол (40 г) и 8 г моноксида свинца нагревали в течение 10 часов при 180°С и образовавшуюся воду отгоняли вместе с фенолом с получением катализатора А.
(Получение дифенилкарбоната)
В 1000-миллилитровый автоклав (изготовленный Toyo Koatsu Co., Ltd., Japan) загружали 110 г (3-метил-1-бутил)карбоната, полученного способом описанным в сравнительном примере 7, 490 г предварительно отогнанного и очищенного фенола (изготовленного Aldrich Chemical Со., USA) и катализатор А (в количестве, при котором концентрация свинца в автоклаве составляла 0,4 мас.%), и автоклав закрывали крышкой.
Внутреннюю часть автоклава продували газообразным азотом, после чего вентиль закрывали и начинали перемешивание. Температуру внутри автоклава доводили до 230°С. В нижнюю часть автоклава подавали азот со скоростью 50 мл/мин. Вентиль в верхней части автоклава устанавливали таким образом, чтобы общее давление в автоклаве составляло в пределах от 100 до 200 кПа, и низкокипящие компоненты отгоняли в течение 4 часов. Через 4 часа введение азота прекращали и автоклав охлаждали.
Анализ содержимого автоклава показал, что было получено примерно 0,28 моль ди(3-метил-1-бутил)карбоната, 0,21 моль 3-метил-1-бутил(фенил)карбоната и 0,026 моль дифенилкарбоната.
Реакционный раствор переносили в 1000-миллилитровую трехгорлую колбу, снабженную холодильником, который был подсоединен к вакуумному регулятору и к вакуумному насосу, и ловушкой Дина-Старка. Для перемешивания подсоединяли мешалку. Затем колбу погружали на масляную баню, установленную на 150°С, и начинали перемешивание. Давление постепенно снижали примерно до 100 кПа. Непрореагировавший фенол и (3-метил-1-бутил)карбонат отгоняли в этом режиме и получали реакционный раствор, состоящий, главным образом, из 3-метил-1-бутил(фенил)карбоната и дифенилкарбоната.
Затем уровень вакуума доводили до 50 кПа и реакцию с перемешиванием проводили при температуре масляной бани 220°С. Реакцию продолжали в течение 6 часов с отгонкой ди(3-метил-1-бутил)карбоната и затем реакцию прекращали. Анализ реакционного раствора показал, что было получено примерно 0,26 моль (56 г) дифенилкарбоната.
Затем колбу подсоединяли к стеклянному трубчатому реактору с внутренним диаметром 25 мм и длиной колонны 500 мм, изготовленной из SUS 316 и снабженной спиральной насадкой Helipack № 2 и соединительным трубопроводом, к которому были подсоединены трубопровод с разветвлениями, термометр, холодильник Либиха, соединительный трубопровод низкого давления и два сосуда для сбора дистиллята. Колбу погружали на масляную баню и смешанный раствор доводили до температуры 185°С. С помощью вакуумного насоса и вакуумного регулятора давление постепенно снижали для отгонки низкокипящих компонентов, после чего давление в системе снижали примерно до 2 кПа. В результате получали примерно 50 г фракции, имеющей температуру парообразования 175°С. Результаты GC-FID-анализа данной фракции показали, что содержание в ней дифенилкарбоната составляло 98 мас.%. Кроме того, результаты119Sn-ЯМР-анализа подтвердили, что в дифенилкарбонате содержалось 1,5 мас.% соединения трибутилолова.
Сравнительный пример 12
(Получение гексаметилендиизоцианата из дифенилкарбоната, содержащего примеси, включающие соединение трибутилолова)
В 500-миллилитровую колбу, снабженную мешалкой, термометром и капельной воронкой, загружали 160 г (0,75 моль) дифенилкарбоната, полученного способом, описанным в сравнительном примере 11, и 142 г (1,5 моль) фенола (изготовленного Aldrich Chemical Со., USA, предварительно отогнанного). После продувки сухим азотом колбу погружали на водяную баню при 50°С и начинали перемешивание.
После подтверждения растворения твердого вещества в колбе температуру водяной бани устанавливали на 45°С. Капельная воронка содержала 35 г (0,3 моль) 1,6-гексаметилендиамина (изготовленного Aldrich Chemical Со., предварительно отогнанного), поддерживаемого при температуре 45-50°С. Затем из этой капельной воронки начинали по каплям вводить в колбу указанное соединение. При этом скорость падения капель регулировали таким образом, чтобы температура жидкости в колбе составляла 50-60°С, и такое введение по каплям проводили в течение примерно 20 минут. После завершения введения по каплям температуру водяной бани регулировали таким образом, чтобы температура раствора в колбе составляла 50°С, и перемешивание продолжали в течение 1 часа.
Результаты анализа, проводимого с помощью высокоэффективной жидкостной хроматографии и гельпроникающей хроматографии, показали, что эффективность реакции 1,6-гексаметилендиамина составляла 99%, с выходом полученного 1,6-гексаметиленфенилдикарбамата 99% и селективностью 99,6%. Выход соединений мочевины составлял примерно 0,5%.
Реакционный раствор, полученный описанным способом, подавали через котел-утилизатор в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), где избыток фенола в газообразной форме удаляли из верхней части дистилляционной колонны, и высококипящие компоненты непрерывно выводили из нижней части дистилляционной колонны. Нижнюю часть колонны подвергали циркуляционному нагреванию при 130°С с помощью кипятильника и давление в верхней части колонны доводили примерно до 20 кПа. Тепловое разложение проводили путем подачи раствора, отогнанного со дна колонны, через соединительный трубопровод и насос, на участок, расположенный примерно на расстоянии 1 м от дна многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, снабженной насадкой Диксона (с внутренним диаметром 6 мм). Нижнюю часть колонны подвергали циркуляционному нагреванию при 220°С с помощью кипятильника и давление в верхней части колонны доводили примерно до 2,6 кПа.
Компонент, содержащий гексаметилендиизоцианат в газообразной форме, экстрагировали из участка, расположенного на расстоянии 2 м от верха колонны, и из верхней части колонны экстрагировали фенол в газообразной форме. Компонент, содержащий гексаметилендиизоцианат, подавали в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки, имеющей диаметр 2 дюйма и длину 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), проводя тем самым очистку гексаметилендиизоцианата. Нижнюю часть колонны подвергали циркуляционному нагреванию при 120°С с помощью кипятильника и давление в верхней части колонны доводили примерно до 130 Па.
Компонент, экстрагированный из верхней части колонны, имел коричневую окраску и содержал гексаметилендиизоцианат с чистотой 95%. Кроме того, компонент, экстрагированный со дна колонны, содержал дифенилкарбонат в качестве главного компонента.
Сравнительный пример 13
(Получение поликарбоната из дифенилкарбоната, содержащего примеси, включающие соединение трибутилолова)
В вакуумный реактор, снабженный мешалкой, загружали 24,5 г дифенилкарбоната (содержащего примерно 1,5 мас.% соединения трибутилолова), полученного как описано в сравнительном примере 11, и 22,2 г бисфенола А. После продувки газообразным азотом полученную смесь полимеризовали под давлением 8 кПа в течение 30 минут и под давлением 4 кПа в течение 90 минут, после чего температуру повышали до 270°С и полимеризацию осуществляли при 0,07 кПа в течение 1 часа. В результате получали коричневый ароматический поликарбонат, который имел среднечисленную молекулярную массу 9000.
Сравнительный пример 14
(Получение дифенилкарбоната из диметилкарбоната)
(Получение катализатора)
Тетрабутоксид титана (340 г, 1 моль) (изготовленный Toyo Kasei Kogyo Co., Ltd., Japan) и 1882 г (20 моль) фенола нагревали в течение 6 часов при 180°С и образовавшийся 1-бутанол отгоняли вместе с фенолом, получая таким образом катализатор В.
(Получение дифенилкарбоната)
Дифенилкарбонат получали с использованием аппарата, проиллюстрированного на фиг.13. Реакцию проводили путем непрерывной подачи смешанного раствора диметилкарбоната, фенола и метилфенилкарбоната в жидкой форме со скоростью 312 г/час из питающего трубопровода 166 через теплообменника 167 и питающий трубопровод 168 в верхнюю часть 173 многоходовой дистилляционной колонны для непрерывной перегонки 172 с внутренним диаметром 50 мм и длиной 2000 мм, которая снабжена ситчатыми тарелками в количестве примерно 40 штук. При этом в вышеописанной смеси использовали соответствующие компоненты в таком количестве, чтобы жидкость в питающем трубопроводе 168 в процессе реакции имела состав: 50,1 мас.% диметилкарбоната, 44,6 мас.% фенола и 5,0 мас.% метилфенилкарбоната. В нижнюю часть 174 многоходовой дистилляционной колонны для непрерывной перегонки вводили диметилкарбонат из питающего газового трубопровода 169 через теплообменник 170 и диметилкарбонат, который превращался в газообразную форму, подавали со скоростью 550 г/час через питающий газовый трубопровод 171. Катализатор В подавали из трубопровода для подачи катализатора 213 в таком количестве, чтобы концентрация Ti в соединительном трубопроводе 178 составляла 0,046 мас.%. Температура в нижней части многоходовой дистилляционной колонны для непрерывной перегонки 172 составляла 203°С, и давление в верхней части колонны составляло 0,65 МПа-G. Газ, отогнанный из верхней части колонны 173, поступал через трубопровод-сборник низкокипящих компонентов в холодильник 176 для разжижения, после чего он выходил из трубопровода для сбора жидкой фазы 177 со скоростью 551 г/час. Реакционную смесь, экстрагированную из нижней части колонны 174 со скоростью 311 г/час, вводили в испаритель 179 через соединительный трубопровод 178. На этой стадии образовывался концентрат, содержащий катализатор и ароматический эфир угольной кислоты. Часть концентрата подавали на рециклизацию в испаритель 179 из соединительного трубопровода 180 и циркуляционного трубопровода 181 через кипятильник 182 и циркуляционный трубопровод 183. Остаточный концентрат снова подавали в многоходовую дистилляционную колонну 172 из испарителя 179 через соединительный трубопровод 180, соединительный трубопровод 184 и питающий трубопровод 166 со скоростью примерно 10 г/час. Часть концентрата, образовавшегося в испарителе 179, экстрагировали из системы через экстракционный трубопровод 185 со скоростью 0,5 г/час. Катализатор В поступал из трубопровода для подачи катализатора 213 в таком количестве, чтобы концентрация Ti в соединительном трубопроводе 178 поддерживалась при 0,046 мас.%. При этом дистиллят из испарителя 179 поступал через трубопровод-сборник для низкокипящих компонентов в холодильник 187 для разжижения. Для проведения реакции полученную жидкость подавали через соединительный трубопровод 188 и соединительный трубопровод 189 в многоходовую дистилляционную колонну 193 с внутренним диаметром 50 мм и длиной 1000 мм, которая снабжена ситчатыми тарелками в количестве примерно 20 штук. Жидкость в соединительном трубопроводе 189 имела следующий состав: 42,1 мас.% диметилкарбоната, 24,5 мас.% фенола, 28,1 мас.% метилфенилкарбоната и 4,5 мас.% дифенилкарбоната. Катализатор подавали из трубопровода для подачи катализатора 212 в таком количестве, чтобы концентрация Ti в соединительном трубопроводе 201 составляла 0,046 мас.%. Температура в нижней части многоходовой дистилляционной колонны для непрерывной перегонки 193 составляла 198°С, и давление в верхней части колонны составляло 38 кПа. Газ, отогнанный из верхней части колонны 194, поступал через трубопровод-сборник для низкокипящих компонентов 196 в холодильник 197 для разжижения. Часть полученного концентрата возвращали в верхнюю часть колонны 174 из соединительного трубопровода 199. Остаточный концентрат подавали на рециклизацию в многоходовую дистилляционную колонну 173 из соединительного трубопровод 198 и соединительного трубопровода 200 через теплообменник 167 и питающий трубопровод 168. После начала проведения рециркуляции фенол снова подавали из питающего трубопровода 166 таким образом, чтобы соединительный трубопровод 168 содержал смесь того же состава. Часть реакционной смеси, находящейся в нижней части многоходовой дистилляционной колонны 195, подавали на рециклизацию в нижнюю часть колонны 195 из циркуляционного трубопровода 190 через кипятильник 191 и циркуляционный трубопровод 192. Остаточную реакционную смесь подавали в испаритель 202 из соединительного трубопровода 201 со скоростью 690 г/час. В испарителе 202 образовывался концентрат, содержащий катализатор и ароматический эфир угольной кислоты. Часть этого концентрата подавали на рециклизацию в испаритель 202 из соединительного трубопровода 203 и циркуляционного трубопровода 204 через кипятильник 205 и циркуляционный трубопровод 206. Остаточный концентрат снова подавали в многоходовую дистилляционную колонну 193 из испарителя 202 через соединительный трубопровод 203, соединительный трубопровод 207 и соединительный трубопровод 189 со скоростью 20 г/час. Часть парового концентрата, образовавшегося в испарителе 202, экстрагировали из системы через экстракционный трубопровод 208 со скоростью 1 г/час. Катализатор В подавали из трубопровода для подачи катализатора 213 в таком количестве, чтобы концентрация Ti в соединительном трубопроводе 201 поддерживалась при 0,046 мас.%. При этом газ, отогнанный из испарителя 202, экстрагировали из трубопровода для сбора жидкой фазы 211 со скоростью 682 г/час через трубопровод для сбора низкокипящих компонентов 209 и холодильник 210. Полученная жидкость содержала 98% дифенилкарбоната.1Н-ЯМР-анализ показал, что примеси в виде метильных групп составляли 90 ч./млн.
Сравнительный пример 15
(Получение гексаметилендиизоцианата из дифенилкарбоната, содержащего примеси в виде метильных групп)
В 500-миллилитровую колбу, снабженную мешалкой, термометром и капельной воронкой, загружали 161 г (0,75 моль) дифенилкарбоната (содержащего 90 ч./млн метилфенилкарбоната), полученного способом, описанным в сравнительном примере 14, и 142 г (1,5 моль) фенола (изготовленного Aldrich Chemical Со., USA, предварительно отогнанного). После продувки сухим азотом колбу погружали на водяную баню при 50°С и начинали перемешивание.
После подтверждения растворения твердого вещества в колбе температуру водяной бани устанавливали на 45°С. Капельная воронка содержала 35 г (0,3 моль) 1,6-гексаметилендиамина (изготовленного Aldrich Chemical Со., предварительно отогнанного), поддерживаемого при температуре 45-50°С. Затем из капельной воронки начинали по каплям вводить в колбу указанное соединение. При этом скорость падения капель регулировали таким образом, чтобы температура жидкости в колбе составляла 50-60°С, и такое введение по каплям проводили в течение примерно 20 минут. После завершения введения по каплям, температуру водяной бани регулировали таким образом, чтобы температура жидкости в колбе составляла 50°С, и перемешивание продолжали в течение 1 часа.
Результаты анализа, проводимого с помощью высокоэффективной жидкостной хроматографии и гельпроникающей хроматографии, показали, что эффективность реакции 1,6-гексаметилендиамина составляла 99% с выходом полученного 1,6-гексаметиленфенилдикарбамата 99% и селективностью 99,6%. Выход соединений мочевины составлял примерно 0,5%.
Реакционный раствор, полученный описанным способом, подавали через котел-утилизатор в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), где избыток фенола в газообразной форме удаляли из верхней части дистилляционной колонны, и высококипящие компоненты непрерывно экстрагировали из нижней части дистилляционной колонны. Нижнюю часть колонны подвергали циркуляционному нагреванию при 130°С с помощью кипятильника и давление в верхней части колонны доводили примерно до 20 кПа. Тепловое разложение проводили путем подачи жидкости, отогнанной со дна колонны, через соединительный трубопровод и насос, на участок, расположенный примерно на расстоянии 1 м от дна многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, снабженной насадкой Диксона (с внутренним диаметром 6 мм). Нижнюю часть колонны подвергали циркуляционному нагреванию при 220°С с помощью кипятильника и давление в верхней части колонны доводили примерно до 2,6 кПа.
Компонент, содержащий гексаметилендиизоцианат, в газообразной форме экстрагировали из участка, расположенного на расстоянии 2 м от верха колонны, и из верхней части колонны экстрагировали фенол в газообразной форме. Компонент, содержащий гексаметилендиизоцианат, подавали в средний ярус многоходовой дистилляционной колонны для непрерывной перегонки диаметром 2 дюйма и длиной 4 м, которая снабжена насадкой Диксона (с внутренним диаметром 6 мм), проводя таким образом очистку гексаметилендиизоцианата. Нижнюю часть колонны подвергали циркуляционному нагреванию при 120°С с помощью кипятильника и давление в верхней части колонны доводили примерно до 130 Па.
Компонент, экстрагированный из верхней части колонны, имел коричневую окраску и содержал гексаметилендиизоцианат с чистотой 98%. Кроме того, компонент, экстрагированный со дна колонны, содержал дифенилкарбонат в качестве главного компонента.
Сравнительный пример 16
(Получение поликарбоната из дифенилкарбоната, содержащего примеси в виде метильных групп)
В вакуумный реактор, снабженный мешалкой, загружали 23,5 г дифенилкарбоната (содержащего 90 ч./млн метилфенилкарбоната) и 22,8 г бисфенола А. После продувки газообразным азотом полученную смесь полимеризовали под давлением 8 кПа в течение 30 минут и под давлением 4 кПа в течение 90 минут, после чего температуру повышали до 270°С и полимеризацию осуществляли при 0,07 кПа в течение 1 часа. Полученный бесцветный и прозрачный ароматический поликарбонат имел хорошее качество и среднечисленную молекулярную массу 9500.
Сравнительный пример 17
(Получение диалкоксида дибутилолова с использованием горизонтального перегонного аппарата с тонкой пленкой)
Алкоксид дибутилолова получали в горизонтальном перегонном аппарате с тонкой пленкой (изготовленном Nitinan Engineering Co., Ltd., Japan, PFD1), таком как аппарат, проиллюстрированный на фиг.9. Горизонтальный перегонный аппарат с тонкой пленкой 113, имеющий внутренний диаметр 50 мм и общую длину 1100 мм и изготовленный из SUS 316, снабжен в его верхней части 114 трубопроводом для подачи алкоксида олова 110, трубопроводом для подачи спирта 111, теплообменником 112, трубопроводом для сбора низкокипящих компонентов 116, холодильником 117, резервуаром для сбора низкокипящих компонентов 118, спускным трубопроводом 119 и трубопроводом для сбора жидкой фазы 120, и в его нижней части 115 - трубопроводом для экстракции полученного продукта 121. Температуру реактора доводили до 120°С с помощью нагревателя и внутреннее давление доводили до 54 кПа с использованием вакуумного насоса и вакуумного регулятора.
С использованием питающего насоса из питающего трубопровода 110 подавали исходное соединение 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный, как описано в примере 12, со скоростью 4600 г/час, и из трубопровода для подачи спирта 111 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, с чистотой, подходящей для дегидратации, с содержанием воды 50 ч./млн) со скоростью 22000 г/час. Время пребывания в реакторе составляло 10 минут. Температуру жидкости внутри реактора доводили до 100°С. Непрерывную подачу проводили в этом режиме в течение 1 часа, после чего давление в системе оставалось постоянным. Низкокипящие компоненты выходили из верхней части реактора 114 со скоростью 21000 г/час. Кроме того, из нижней части реактора 115 собирали компонент, содержащий алкоксид дибутилолова, который выходил со скоростью 5600 г/час. Анализ жидкости, собранной из экстракционного трубопровода 121, показал, что указанная жидкость не содержала дибутилди(бутилокси)олова, и весь непрореагировавший 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан выведен из реактора. Жидкость, выведенная из трубопровода для сбора жидкой фазы 120, была прозрачной и содержала 50 ч./млн влаги. Реакция дегидратации не происходила.
Пример 24
(Получение диалкоксида дибутилолова с использованием колонного реактора)
Алкоксид дибутилолова получали в колонном реакторе 219, таком как проиллюстрировано на фиг.14. В колонный реактор 219 с внутренним диаметром 50 мм и общей длиной 4000 мм, который изготовлен из SUS 316, и к которому в его верхней части 221 подсоединен трубопровод для сбора низкокипящих компонентов 223, холодильник 224, сепаратор 225, клапан обратного давления 226, спускной трубопровод 227, циркуляционный трубопровод для органического слоя 228 и трубопровод для сбора водного слоя 229; в его средней части 220 - питающий трубопровод 214, питающий трубопровод 215 и теплообменник 216, а в его нижней части 222 - циркуляционный трубопровод 217 для рециркуляции реакционного раствора, который аккумулировался в нижней части реактора, кипятильник 218 и экстракционный трубопровод 230, вставляли насадку Меллапак 750Y (Sulzer Chemtech Ltd., Switzerland). Температуру реактора доводили до 140°С с помощью нагревателя.
Из питающего трубопровода 214 подавали исходное соединение 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксан, полученный как описано в примере 12, с помощью питающего насоса со скоростью 280 г/час, и из питающего трубопровода 215 подавали реагент 1-бутанол (изготовленный Wako Pure Chemical Industries Ltd., Japan, промышленный продукт) со скоростью 1330 г/час. Время пребывания в реакторе составляло 6 минут. Реакционный раствор, аккумулированный в нижней части реактора, подавали на рециклизацию со скоростью 6000 г/час через циркуляционный трубопровод 217 и кипятильник 218 с нагреванием при температуре примерно 141°С. Низкокипящие компоненты поступали из верхней части реактора 221 через трубопровод для сбора низкокипящих компонентов 223 в холодильник 224 для разжижения, после чего они выходили со скоростью 2000 г/час. Затем органический слой медленно отделяли от воды в сепараторе 225. Органический слой в сепараторе 225, то есть 1-бутанол, возвращали в верхнюю часть реактора 221 со скоростью 1994 г/час через трубопровод для рециркуляции органического слоя 228, и нижний слой, который имел высокую концентрацию воды, собирали со скоростью 6 г/час из трубопровода для сбора водного слоя 229. Из нижней части реактора 222 собирали компонент, содержащий алкоксид дибутилолова со скоростью 1604 г/час через экстракционный трубопровод 230. Температура жидкости внутри реактора составляла 140°С и давление доводили до 0,96 МПа-G с помощью клапана обратного давления 226. Непрерывную подачу проводили в этом режиме в течение 10 часов, после чего система достигала стационарного состояния. Анализ жидкости, выделенной из экстракционного трубопровода 230, показал, что при использовании оксида дибутилолова в качестве исходного соединения, жидкость содержала алкоксид дибутилолова, состоящий примерно на 74,2% из дибутилди(бутилокси)олова и примерно на 25,7% из 1,1,3,3-тетрабутил-1,3-ди(бутилокси)дистанноксана. Выход бутоксида трибутилолова составлял 0,015%. При этом жидкость, собранная из трубопровода для сбора водного слоя 229, была прозрачной и содержала 90 мас.% влаги. Скорость дегидратации в колонном реакторе составляла 0,33 моль/час и превышала величину, вычисленную по уравнению (16), которая составляла 0,00037 моль/час.
Краткое описание графического материала
На фиг.1 схематически проиллюстрировано получение трибутил(2-этилгексилокси)олова и 1,1,3,3-тетрабутил-1,3-бис(2-этилгексилокси)дистанноксана с нагреванием при 180°С.
На фиг.2 схематически представлен пример одного из колонных реакторов в соответствии с настоящим изобретением.
На фиг.3 схематически представлен пример одного из комбинированных реакторов резервуарного и колонного типа в соответствии с настоящим изобретением.
На фиг.4 схематически представлен другой пример колонного реактора в соответствии с настоящим изобретением.
На фиг.5 схематически представлен еще один пример колонного реактора в соответствии с настоящим изобретением.
На фиг.6 схематически представлен еще один пример колонного реактора в соответствии с настоящим изобретением.
На фиг.7 схематически представлен другой пример колонного реактора в соответствии с настоящим изобретением.
На фиг.8 схематически представлен еще один пример колонного реактора в соответствии с настоящим изобретением.
На фиг.9 схематически представлен один пример горизонтального перегонного аппарата с тонкой пленкой в соответствии с настоящим изобретением.
На фиг.10 схематически представлен один пример многоходовой дистилляционной колонны для непрерывной перегонки в соответствии с настоящим изобретением.
На фиг.11 схематически представлен другой пример многоходовой дистилляционной колонны для непрерывной перегонки в соответствии с настоящим изобретением.
На фиг.12 схематически представлен еще один пример многоходовой дистилляционной колонны для непрерывной перегонки в соответствии с настоящим изобретением.
На фиг.13 схематически представлен еще один пример многоходовой дистилляционной колонны для непрерывной перегонки в соответствии с настоящим изобретением.
На фиг.14 схематически представлен другой пример колонного реактора в соответствии с настоящим изобретением.
Реферат
Описан способ получения алкоксидов алкилолова, включающий реакцию дегидратации, по меньшей мере, одного соединения алкилолова, используемого в качестве исходного соединения и выбранного из оловоорганических соединений, имеющих связи олово-кислород-олово, и гидроксисоединения, используемого в качестве реагента, с получением алкоксида алкилолова, соответствующего указанному исходному материалу и реагенту, где указанное исходное соединение и указанный реагент непрерывно подаются в реактор; и низкокипящие компоненты, содержащие воду, выводятся из указанного реактора при непрерывном удалении реакционной жидкости, содержащей алкоксид алкилолова, из нижней части реактора. 16 з.п. ф-лы, 1 табл., 14 ил.
Формула
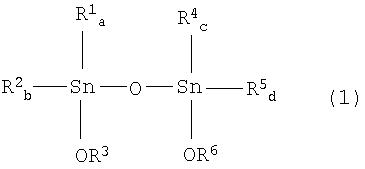
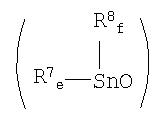

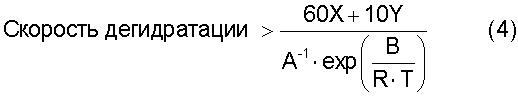
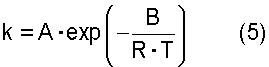
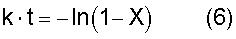
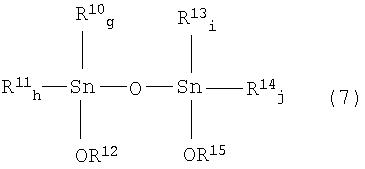
Комментарии