Отбеливающий состав и способы для его использования - RU2193050C2
Код документа: RU2193050C2
Чертежи






Описание
Изобретение относится к применению макроциклических комплексов металл-лиганд в качестве активаторов отбеливания и, более конкретно, к комплексам переходных металлов с макроциклическими тетраамидными лигандами в качестве катализаторов для ускорения реакций окислительного отбеливания.
Краткое описание уровня техники
Перекись водорода и другие перекисные соединения, образующие перекись водорода в
водных растворах, давно используются для отбеливания материалов и поверхностей. Однако такие перекисные соединения, как перборат натрия (моногидрат или тетрагидрат), перкарбонат натрия и т.п., имеют
относительно низкую эффективность отбеливания при низких температурах (например, ниже 38,8oС/100oF). Органические перкислоты или надкислоты, такие, как пербензойная кислота,
являются более сильными окислителями, но в большинстве случаев они неустойчивы, если их не стабилизировать с помощью дорогостоящих и обременительных методов. Кроме того, производство таких
предварительно обработанных перкислот часто является затратным. Активаторы отбеливания или предшественники перкислот, такие, как эфиры, кетоны, нитрилы и тому подобное, почти всегда эффективны при
повышении эффективности перекисных соединений. Однако активаторы отбеливания обычно присутствуют в стехиометрических или в более высоких количествах, и поэтому их производство может быть
невыгодным.
Известно применение хелатов переходных металлов, особенно содержащих марганец и железо, в качестве катализаторов отбеливания с помощью перекисных соединений. Они представлены, например, Favre и др., патент США. 5,246,621, Bragg и др. , патент США 5,002,682, Postlethwaite, патент США, 4,119,557 и Ellis, Jr. и др., патент США 4,900,871. Эти хелаты переходных металлов могут применяться, например, при стирке тканей с соответствующими перекисными соединенями, например, с моногидратом пербората натрия.
Несмотря на имеющиеся доказательства усиления окисляющей способности перекисных соединений с помощью таких хелатов переходных металлов, в некоторых случаях при использовании в качестве активаторов отбеливания они могут способствовать сходу краски и даже повреждению ткани.
Изучалось применение хелатов переходных металлов в областях, не связанных с изобретением. Например, известно, что комплексы переходных металлов с высокой степенью окисления функционируют в качестве окислителей в многочисленных биологических реакциях под воздействием белковой матрицы, и в последние годы проявляется огромный интерес к пониманию механизма действия и реакционной способности некоторых монооксигеназ.
Примером служит программа, описанная Collins T.J., "Создание лигандов для окисляющих комплексов", Accounts of Chemical Research, 279, 27 (9), 1994. В этой статье планируется создание направленного подхода к получению лигандов, которые устойчивы к окислительной деградации при координации с металлами высокой степени окисления. В статье Collins в Accounts of Chemical Research описаны некоторые ациклические диамидо-N-дифеноксидные и диамидо-N-алкоксидные хелатные соединения и макроциклические тетраамидные N-хелатные соединения.
Синтетический путь к макроциклическим тетраамидным лигандам описан Uffelman E.S., Ph.D. Thesis, California Institute of Technology (1992). Кроме того, синтез тетраамино-лиганда с арильным мостиком по азидному методу может быть осуществлен с использованием в качестве исходного вещества ароматического диамина.
Однако до сих пор оставалось неизвестным, что на основе некоторых макроциклических тетраамидныех лигандов можно получить новые и необычайно эффективные активаторы отбеливания с помощью перекисных соединений. Кроме того, не было сообщено, раскрыто в изобретении или предположено, что соединения этих типов будут иметь необычайные преимущества в областях, связанных с подавлением процесса переноса краски с одной ткани на другую, с подавлением процесса повторного отложения загрязнения и удалением пятен.
Краткое содержание изобретения
Изобретение включает отбеливающий состав, содержащий:
а)
устойчивый к окислению активатор отбеливания, имеющий структуру

где Y1, Y3 и Y4 по отдельности означают мостиковую группу, имеющую 1-3 углеродсодержащих узла для замещения, или одинарную связь,
Y2 означает мостиковую группу, имеющую, по меньшей мере, один углеродсодержащий узел для замещения, причем упомянутый узел содержит группы CR, CR1R2 или C(R)2, и каждый заместитель R означает то же самое или отличается от R и выбирается из группы, состоящей из атома водорода, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, атома галоида, алкоксила, феноксигруппы, трифторметильной группы, трифторэтильной группы или их комбинации, или образует замещенное либо незамещенное бензольное кольцо, в котором два кольцевых атома углерода образуют узел в ячейке Y, или вместе со спаренными заместителями R, присоединенными к одному атому углерода, образует циклоалкильное кольцо, например, циклопентильное или циклогексильное, которое может включать атом, отличный от атома углерода,
М означает переходный металл со степенью окисления I, II, III, IV, V или VI или выбирается из групп 6, 7, 8, 9, 10 и 11 Периодической системы элементов,
Q означает любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе,
L означает любой лабильный лиганд;
б) эффективное количество источника окисляющего соединения.
К составу могут добавляться поверхностно-активные вещества, наполнители, связывающие вещества, усилители экскреции, антиоксиданты, ферменты, флуоресцентные отбеливающие агенты, красители, оттеночные вещества, пигменты и другие стандартные вспомогательные вещества для чистки и/или стирки.
Предпочтительными активаторами отбеливания являются макроциклические тетраамидопроизводные. Из них особо предпочтительными являются те, которые содержат замещенный ароматичекий фрагмент, сочлененный непосредственно с циклическим лигандом.
Например, предпочтительное соединение имеет следующую структуру:

где Х и Z означают атом водорода, электронодонорную или электроноакцепторную группу,
R' и R" означают любую комбинацию атома водорода, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, атома галоида, алкоксила, феноксигруппы, или при соединении образуют циклоалкильное или циклоалкенильное кольцо, которое может содержать, по меньшей мере, один атом, отличный от атома углерода,
М означает переходный металл со степенью окисления I, II, III, IV, V или VI, или выбирается из 6, 7, 8, 9, 10 или 11 группы Периодической таблицы элементов,
Q означает любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе,
L означает любой лабильный лиганд.
Существует необходимость в создании перекисного активатора отбеливания с улучшенной способностью подавлять процесс переноса красителя на другую ткань. Существует необходимость в создании активатора отбеливания с улучшенными грязеотталкивающими свойствами. Существует также необходимость в создании активатора отбеливания, обладающего уникальными пятновыводящими свойствами. Кроме того, существует необходимость в создании активатора отбеливания с длительно сохраняющейся каталитической стабильностью в забуференном растворе. Помимо этого, существует необходимость в создании активатора отбеливания, который может применяться в субстехиометрических количествах по отношению к окисляющему соединению.
Краткое описание чертежей
Фиг. 1 представляет синтетический способ получения макроциклических тетраамидных лигандов по
изобретению азидным методом.
Фиг. 2 представляет синтетический способ получения макроциклических тетраамидных лигандов по изобретению азидным методом с использованием ароматического диамина в качестве исходного вещества.
Фиг. 3 отображает в графическом виде сопоставление с контролем сохраняющейся длительное время каталитической стабильности в одном из предпочтительных вариантов осуществления данного изобретения.
Подробное описание предпочтительных вариантов осуществления изобретения.
Изобретение включает отбеливающий
состав, содержащий:
а) устойчивый к окислению активатор отбеливания, имеющий структуру:
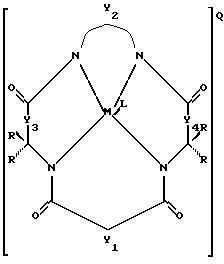
где Y1, Y3 и Y4 по отдельности означают мостиковую группу, имеющую 1-3 углеродсодержащих узла для замещения, или одинарную связь,
Y2 означает мостиковую группу, имеющую, по меньшей мере, один углеродсодержащий узел для замещения, причем упомянутый узел содержит группы CR, CR1R2 или С(R)2, и каждый заместитель R означает то же самое или отличается от R и выбирается из группы, состоящей из атома водорода, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, атома галоида, алкоксила, феноксигруппы, трифторметильной группы, трифторэтильной группы или их комбинации, или образует замещенное либо незамещенное бензольное кольцо, в котором два кольцевых атома углерода образуют узел в ячейке Y, или вместе со спаренными заместителями R, присоединенными к одному атому углерода, образует циклоалкильное или циклоалкенильное кольцо, например, циклопентильное или циклогексильное, которое может включать атом, отличный от атома углерода,
М означает переходный металл со степенью окисления I, II, III, IV, V или VI или выбирается из групп 6, 7, 8, 9, 10 и 11 Периодической системы элементов,
Q означает любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе,
L означает любой лабильный лиганд;
б) эффективное количество источника окисляющего соединения.
Предпочтительные макроциклические тетраамидные лиганды по изобретению проявили чрезвычайную эффективность при оценке с помощью различных рабочих характеристик для активаторов отбеливания.
Эти лиганды получают в соответствии с методами, описанными в одновременно внесенных в реестр Соединенных Штатов заявке на изобретение Gordon-Wylie и др., озаглавленной "Синтез макроциклических тетраамидо-N-лигандов", регистрационный номер 08/681,187 и международной заявке 98/03263, Collins и др., озаглавленной "Долгоживущие гомогенные катализаторы окисления", которые включены в данную заявку в виде ссылок.
Особо предпочтительный вариант соединений по данному изобретению представлен
структурой макроциклических тетраамидных производных:

где Х и Z означают атом водорода, электронодонорную или электроноакцепторную группу,
R' и R" означают любую комбинацию атома водорода, алкила, циклоалкила, циклоалкенила, алкенила, арила, алкинила, алкиларила, атома галоида, алкоксила, феноксигруппы, или при соединении образуют циклоалкильное или циклоалкенильное кольцо, которое может содержать, по меньшей мере, один атом, отличный от атома углерода,
М означает переходный металл со степенью окисления I, II, III, IV, V или VI, или выбирается из 6, 7, 8, 9, 10 или 11 группы Периодической таблицы элементов,
Q означает любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе,
L, если присутствует, означает лабильный лиганд.
Х и Z могут быть атомами водорода или электронодонорными, либо электроноакцепторными группами. Электроноакцепторные группы включают атом галоида, такой, как атом брома, атом йода и, предпочтительнее, атом хлора. Кроме того, подходящими группами являются SО3-, ОSО3-, OSO3R (причем R без ограничений означает атом водорода, алкил, арил, алкиларил) и группа NО2- . Электронодонорные группы включают алкоксигруппу (без ограничений метокси, этокси, пропокси и бутокси), алкил (без ограничений метил, этил, пропил, н-бутил и трет. -бутил) и атом водорода. Эти группы изменяют электронную плотность комплекса металл-лиганд и влияют на его реакционную способность.
R' и R", по-видимому, оказывают влияние на продолжительность сохранения каталитической стабильности макроциклических тетраамидных лигандов по изобретению. Несмотря на то, что каждый из них по отдельности может быть выбран из группы, состоящей из атома водорода, алкила, алкенила, арила, алкинила, атома галоида, алкоксила или феноксигруппы, предпочтительным является алкил с короткой цепью. Особо предпочтительно, когда R' и R" являются одинаковыми и выбираются из этила или метила, или когда R' и R" соединяются, образуя циклоалкильное или циклоалкенильное кольцо, особенно циклопентильное или циклогексильное. Циклоалкильное кольцо может включать, по меньшей мере, один атом, отличный от атома углерода, без ограничений такой, как атом азота, атом кислорода или атом серы.
Металл М означает переходный металл со степенью окисления I, II, III, IV, V или VI или выбирается из 6, 7, 8, 9, 10 или 11 группы Периодической таблицы элементов. Предпочтительно он выбирается из группы, состоящей из железа, марганца, хрома, меди, кобальта, никеля, молибдена, цинка или вольфрама. Возможны их смеси.
Q означает любой противоион, который сбалансировал бы заряд соединения (обычно отрицательный, предпочтительно -1) на стехиометрической основе. Таким образом, предпочтительным обычно является положительно заряженный противоион, но не ограниченный только противоионами щелочно-земельных металлов (например, ионами калия, лития, натрия), ионами NR*4 и PR*4, где каждый заместитель R*4 по отдельности выбирают из алкила, арила, алкиларила, алкенила, или они могут соединиться, образуя циклоалкильное, циклоалкенильное или арильное кольцо, которое может содержать, по меньшей мере, один атом, отличный от атома углерода.
L означает любой лабильный лиганд, который может соединяться с металлом М. Эти лиганды предпочтительно, но без ограничений, включают молекулу воды, атом хлора и группу C=N.
В связи со сложностью структуры этих соединений при описании изобретения для удобства их названия заменены на названия присутствующих в их структуре заместителей. Например, структура, представленная выше, может быть названа 5,6-(4,5-ди-Х-бензо)-3,8,11,13-тетраоксо-2,2,9,9-тетраметил-12,12-диэтил-1, 4,7,10-тетраазациклотридекан или тетраметил-диэтилди-Х-бензол (TMDE-DXB, где Х означает атом хлора, атом водорода, метил, метоксил). Таким образом, для удобства в случае вышеупомянутой структуры, где две метильные группы расположены каждая при аминогруппе лиганда и две этильные группы играют роль заместителей R' и R", соединение обозначают как TMDE. Когда оба заместителя Х и Z означают атомы хлора, соединение обозначают как DCB. Предпочтительным переходным металлом лиганда является железо, поэтому соединение может быть обозначено как FeDCB.
Поскольку макроциклические тетраамидные лиганды являются истинными катализаторами, то их количество, добавляемое к отбеливающим составам, обычно стехиометрическое. Однако, предпочтительно прибавлять без ограничений к составам по изобретению примерно 0,0001-999999 миллионных долей (м.д.), более предпочтительно 0,001-100000 м.д. лиганда.
Ниже, в экспериментальной части, приведены отдельные синтезы предпочтительных макроциклических тетраамидопроизводных. Кроме того, были проведены тесты, демонстрирующие способность к подавлению процесса переноса красителя, продолжительность сохранения каталитической активности и пятновыводящие свойства макроциклических лигандов по изобретению.
Окисляющие соединения
Окисляющие соединения могут быть органическими и
неорганическими. Предпочтительными являются перекисные соединения, содержащие перекисную связь -О-О-. Приводимые в качестве примеров соединения включают перекись водорода, аддукты перекиси водорода,
соединения, способные образовывать перекись водорода в водном растворе, органические перекиси, персульфаты, перфосфаты и персиликаты. Аддукты перекиси водорода включают пергидраты карбонатов щелочных
металлов (например, натрия, лития, калия) и перекись мочевины. Соединения, способные образовывать перекись водорода в водном растворе, включают моно- или тетрагидраты перборатов щелочных металлов
(натрия, калия, лития). Пербораты являются коммерческими соединениями и производятся такими фирмами, как Akzo N. V. и FMC Corporation. В качестве альтернативы источником перекиси водорода может
служить фермент алкоголь-оксидаза и ее субстрат - соответствующий спирт. Органические перекиси включают без ограничения гидроперекиси бензоила и кумола. Персульфаты включают моноперсульфат калия
(торговое название Oxone®, фирма E.I. du Pont de Nemours) и кислоту Каро (мононадсерную кислоту).
Эффективным количеством перекисного соединения является количество, достаточное для того, чтобы генерировать, по меньшей мере, 0,001 м.д. активного кислорода (АО). Без ограничений и в то же время предпочтительно получать от 0,001 м.д. до 1000 м.д. АО. При отбеливании тканей предпочтительным является количество от 0,01 до 50 м.д. АО. Описание и методика измерения АО приведены в публикации Sheldon N. Lewis "Окисление с помощью надкислот и перекисей" в книге "Окисление", 1969, стр. 213-258, включенной в данное изобретение в виде ссылки.
Чистящие и/или стирающие вспомогательные вещества
Макроциклические тетраамидные лиганды по
изобретению могут смешиваться с окислительной отбеливающей или моющей основой, причем упомянутая основа включает связывающие вещества, и необязательно поверхностно-активное вещество, выбранное из
группы, состоящей из анионных, неионных, катионных, амфотерных, цвиттерионных поверхностно-активных веществ и их смесей. Могут присутствовать другие вспомогательные вещества. Эти соединения могут
находиться в жидкой основе для применения на твердых поверхностях, для удаления пятен или в других процедурах очистки/отбеливания. Эти соединения могут также применяться в процессе получения
технической целлюлозы и отбеливания текстильных изделий. Каждое из этих соединений и вспомогательные вещества, пригодные для использования по изобретению, обсуждаются ниже.
А.
Связывающие вещества
Связывающими веществами обычно являются вещества со щелочными свойствами, т.е. те, которые в водном растворе имеют рН 7-14, предпочтительно 9-12. Примеры неорганических
связывающих веществ включают щелочные металлы и карбонаты аммония (включая сесквикарбонаты и бикарбонаты), фосфаты (включая ортофосфаты, политрифосфаты и тетрапирофосфаты), алюмосиликаты (как
природные, так и синтетические цеолиты) и их смеси. Для использования по данному изобретению особенно подходят карбонаты из-за их высокой щелочности и эффективности при удалении ионов, присутствующих
в жесткой воде, а также по причине их низкой стоимости. Карбонаты могут быть использованы в качестве преобладающего связывающего вещества. Могут также использоваться силикаты (Na2O: SiО2 составляет от 4:1 до 1:1, наиболее предпочтительно от 3:1 до 1: 1). Силикаты, благодаря их растворимости в воде и способности образовывать стекловидную матрицу, могут также быть
предпочтительными при использовании в качестве связывающего вещества для моющего средства.
Органические связывающие вещества также пригодны для использования и могут быть выбраны из группы, состоящей из сульфосукцинатов щелочных металлов и аммония, полиакрилатов, полималеатов, сополимеров акриловой и малеиновой кислот или малеинового ангидрида, цитратов и их смесей.
Б. Наполнители/Разбавители
Наполнители для отбеливающего или моющего состава используют для направленной доставки соответствующего количества или дозы моющего или чистящего
средства. Предпочтительными являются соли, такие, как хлорид натрия, сульфат натрия и боракс. Можно использовать органические разбавители, такие, как углеводы. В жидком исполнении в качестве
разбавителей могут использоваться растворители (без ограничения такие, как алкановые спирты, гликоли, эфиры гликолей, углеводороды, кетоны и карбоновые кислоты), жидкие поверхностно-активные вещества
и вода.
В. Поверхностно-активные вещества
Поверхностно-активные вещества обычно добавляют к отбеливающему или моющему средству для направленного удаления отдельных загрязнений,
например, неионные поверхностно-активные добавки для масляных пятен и анионные поверхностно-активные вещества для твердых загрязнений. Однако, как правило, окислительные отбеливающие составы содержат
небольшие количества или совсем не содержат поверхностно-активных веществ.
Особенно эффективными поверхностно-активными добавками являются анионные поверхностно-активные вещества. Примеры таких поверхностно-активных веществ включают соединения аммония, замещенного аммония (например, моно-, ди- и триэтаноламмоний), соли жирных кислот С6-С20 и смоляных кислот со щелочными и щелочноземельными металлами, линейные и разветвленные алкилбензолсульфонаты, алкилсульфаты, алкиловые эфиры сульфатов, алкансульфонаты, олефинсульфонаты, гидроксиалкансульфонаты, моноглицеридсульфаты жирных кислот, алкилглицериновые эфиры сульфатов, ацилсаркозинаты и ацил-N-метилтауриды. Предпочтительными являются поверхностно-активные алкиларилсульфонаты, такие, как алкилбензолсульфонаты.
Другие предпочтительные поверхностно-активные вещества включают линейные этоксилированные спирты, такие, как выпускаемые фирмой Shell Chemical Company под фирменным названием Neodol. Другие подходящие поверхностно-активные вещества могут включать другие линейные этоксилированные спирты с длиной цепи от 6 до 16 атомов углерода, обычно содержащие от 2 до 20 молей зтиленоксида на моль спирта; линейные и разветвленные, первичные и вторичные этоксилированные, пропоксилированные спирты с обычной длиной цепи от 6 до 16 атомов углерода, содержащие 0-10 молей этиленоксида и 1-10 молей пропиленоксида на моль спирта; линейные и разветвленные алкилфеноксиспирты (полиэтоксилированные), известные под названием этоксилированных алкилфенолов, с обычной длиной цепи от 8 до 16 атомов углерода, содержащие 1,5-30 молей этиленоксида на моль спирта, и их смеси.
Кроме того, подходящие неионные поверхностно-активные вещества могут включать эфиры полиоксиэтиленкарбоновых кислот, глицериновые эфиры жирных кислот, алканоламиды жирных кислот и этоксилированных жирных кислот, некоторые блочные сополимеры пропиленоксида и этиленоксида, блочные полимеры пропиленоксида и этиленоксида с пропоксилированным этилендиамином. Кроме того, они могут включать такие семиполярные неионные поверхностно-активные вещества, как аминооксиды, фосфинооксиды, сульфоксиды и их этоксилдированные производные.
Подходящие катионные поверхностно-активные вещества могут включать аммониевые производные, в которых обычно одной из групп, связанных с атомом азота, является алкильная группа C12-C18, а другие группы представляют собой алкильные группы с короткой цепью, которые могут содержать такие заместители, как фенил.
Кроме того, подходящие амфотерные и цвиттерионные поверхностно-активные вещества, содержащие анионную группу, способствующую растворимости соединения в воде, катионную группу и гидрофобную органическую группу, могут включать карбоновые аминокислоты и их соли, дикарбоновые аминокислоты и их соли, алкилбетаины, алкиламинопропилбетаины, сульфобетаины, производные алкилимидазолия, некоторые четвертичные аммониевые соединения, некоторые четвертичные фосфониевые соединения и некоторые третичные сульфониевые соединения. Другие примеры потенциально пригодных цвиттерионных поверхностно-активных веществ можно найти в составленном Jones патенте США 4,005,029, столбцы 11-15, включенном в данное изобретение в виде ссылки.
Кроме того, примеры анионных, неионных, катионных и амфотерных поверхностно-активных веществ, которые можно использовать по данному изобретению, описаны в монографиях Kirk-Othmer "Энциклопедия химической технологии", третье издание, том 22, стр. 347-387 и "Детергенты и эмульгаторы МакКатчеона", Североамериканское издание, 1983, которые включены в данное изобретение в виде ссылок.
Как
уже упоминалось выше, другие известные детергентные вспомогательные вещества могут быть добавлены при необходимости к отбеливающему или моющему отбеливающему составу. Если, например, нужен моющий
состав, то применимы следующие пределы, вес.%:
Источник перекиси водорода - 0,5-50,0%
Активатор - 0,0001-10000 м.д.
Поверхностно-активное вещество - 1,0-50,0%
Связывающее вещество - 1,0-50,0%
Наполнитель, стабилизаторы, красители, отдушки, оптические отбеливатели и т.д. - 5,0-99,9%
Г. Хелатируюшие агенты
В некоторые составы по
изобретению особо предпочтительным является включение хелатирующего агента, наиболее предпочтительно - аминополифосфоната. Эти хелатирующие агенты способствуют поддержанию стабильности оксиданта в
растворе для достижения оптимального эффекта. Их действие сводится к образованию комплексов (хелатов) со свободными ионами тяжелых металлов. Хелатирующий агент выбирают из большого числа известных
агентов, которые являются эффективными при образовании комплексов со свободными ионами тяжелых металлов. Хелатирующий агент должен быть устойчив к гидролизу и быстрому окислению под воздействием
оксиданта. Предпочтительно он должен иметь константу кислотной диссоциации (рКа) в интервале 1-9, что свидетельствует о его диссоциации при низких значениях рН, что усиливает его связывание
с катионами металлов. Наиболее предпочтительным хелатирующим агентом является аминополифосфонат, который коммерчески доступен под торговым названием Dequest от фирмы Monsanto Company. Примерами
хелатирующих агентов являются Dequest 2000, 2041, 2060 и 2066 (см. также Bossu, патент США, 4,473,594, столбец 12, строка 63 до столбца 13, строки 22, включенного в данное изобретение в виде ссылки).
Можно также использовать такой полифосфонат, как Dequest 2010. Другие хелатирующие агенты, такие, как этилендиаминтетра-уксусная кислота (EDTA) и нитрилотриуксусная кислота (NTA) также могут быть
использованы. Кроме того, новыми предпочтительными хелатирующими агентами являются пропилендиаминтетраацетаты, такие, как Hampshire 1,3 PDTA от W.R. Geace и Chel DTPA 100# F от фирм Ciba-Geigy A.G.
Применимы смеси упомянутых хелатирующих агентов. Эффективные количества хелатирующего агента находятся в пределах 1-1000 м. д. , более предпочтительно 5-500 м.д., наиболее предпочтительно 10-100 м.д.
хелатирующего агента в моющей жидкости.
Д. Другие вспомогательные вещества
Стандартные моющие вспомогательные или окислительные отбеливающие вспомогательные вещества могут
быть включены в настоящее изобретение.
В них входят ферменты, присутствие которых в моющих и окислительных отбеливающих составах наиболее желательно. Однако может быть предпочтительным включение и ферментного стабилизатора.
Протеазы составляют особо предпочтительный класс ферментов. Их выбирают из группы, состоящей из кислых, нейтральных и щелочных протеаз. Термины "кислый", "нейтральный" и "щелочной" в данном случае относятся к значению рН, при котором активность ферментов является оптимальной. Примеры нейтральных протеаз включают милезим (от фирмы Miles Laboratory) и трипсин, являющийся природной протеазой. Щелочные протеазы доступны от разнообразных источников, обычно их получают от различных микроорганизмов (например, Bacillis subtilisis). Типичные примеры щелочных протеаз включают максатазу и максакэл от фирмы International BioSynthetics, алкалазу, савиназу и эсперазу, все от фирмы Novo Industri A/S. См. также Stanislowski и др., патент США 4,511,490, включенный в данное изобретение в виде ссылки.
Кроме того, подходящими ферментами являются амилазы, представляющие собой углевод-гидролизующие ферменты. Предпочтительным также является включение смесей амилаз и протеаз. Приемлемые амилазы включают рапидазу от фирмы Societe Rapidase, милезим от фирмы Miles Laboratory и максамил от фирмы International BioSynthetics.
Другими приемлемыми ферментами являются целлюлазы, такие, как описанные Tai, патент США 4,479,881, Murata и др., патент США 4,443,355, Barbesgaard и др. , патент США 4, 435,307 и Ohya и др., патент США 3,983,082, включенные в данное изобретение в виде ссылок.
Еще одной группой приемлемых ферментов являются липазы, такие, как описанные Silver, патент США 3,950,277 и Thom, патент США 4,707,291, включенные в данное изобретение в виде ссылок.
Кроме того, представляющими интерес ферментами по данному изобретению являются пероксидазы, такие, как пероксидаза хрена и ферменты, раскрытые в международной заявке WO 93/24628, которая включена в данное изобретение в виде ссылки.
Фермент может присутствовать в количестве 0-5%, более предпочтительно 0,01-3%, наиболее предпочтительно 0,1-2% по весу от моющей/отбеливающей/очищающей основы. Желательно использовать смеси любых упомянутых выше гидролаз, особенно смесь протеаза/амилаза.
Кроме того, необязательные вспомогательные вещества включают красители, такие, как краситель Monastral blue и антрахиноновые красители (как описанные Zielske, патент США 4,661,293 и патент США 4,746,461).
Пигменты, которые также являются приемлемыми красящими веществами, могут быть выбраны без ограничений из диоксида титана, ультрамарина голубого (см., например, Chang и др., патент США 4,708,816) и цветных алюмосиликатов.
Флуоресцентные отбеливающие агенты представляют еще одну группу приемлемых вспомогательных веществ. Они включают производные стильбена, стирена и нафталина, которые при облучении ультрафиолетовым светом испускают или поглощают свет в видимой области спектра. Для улучшения вида текстильных изделий, которые потускнели от повторных загрязнений или стирок, используют FWA или оптические отбеливатели. Предпочтительными TWА являются Tinopal 5BMX-C и Tinopal RBS, оба от фирмы Ciba Geigy A.G., а также Phorwite RKH от фирмы Mobay Chemicals. Примеры приемлемых FWA можно найти в патентах США 1,298,577, 2,076,011, 2,026,054, 2.026.566. 1,393.042, а также в патентах США 3,951,960, 4,298,290, 3,993,659, 3,980.713 и 3,627.758, включенных в данное изобретение в виде ссылок.
Агенты, препятствующие повторному отложению загрязнения, такие, как карбоксиметилцеллюлоза, являются потенциально пригодными для использования по изобретению. Кроме того, в него могут быть включены пенные усилители, такие, как соответствующие поверхностно-активные вещества. Кроме того, в случае избыточного пенообразования, возникающего при использовании некоторых поверхностно-активных веществ, желательно использовать противопенные агенты, такие, как алкилированные полисилоксаны, например, диметилполисилоксан. Отдушки также являются желательными вспомогательными веществами в этих составах.
Дополнительные органические активаторы отбеливания могут быть введены в состав, включая, но не только, эфиры (см. Fong и др., патент США 4,778,618 и Rowland и др., патент США 5,182,045), кетоны, имиды (см. Kaaret, патент США 5,478,569) и нитрилы.
Добавки могут присутствовать в количестве 0-50%, более предпочтительно 0-30%, наиболее предпочтительно 0-10%. В некоторых случаях отдельные вспомогательные вещества могут дублироваться в других категориях. Однако настоящее изобретение охватывает каждое из вспомогательных веществ как обеспечивающее конкретное преимущество в различных категориях.
Экспериментальная часть
Синтез устойчивых
к окислению тетраамидных лигандов
Материалы. Все растворители и реагенты были химически чистыми (фирм Aldrich, Aldrich Sure-Seal, Fisher) и использовались без очистки. Микроанализы проводила
фирма Midwest Microlabs, Индианаполис, штат Индиана, США.
Масс-спектрометрия. Масс-спектры (MS) при ионизации электроспреем получены на приборе Finnigan-MAT SSQ700 (Сан-Хосе, штат Калифорния, США), снабженном устройством для электроспрея фирмы Analytica of Brandford. Электроспрей проводили при напряжении 2400-3400 В. Образцы растворяли в ацетонитриле или в дихлорметане при концентрации приблизительно 10 пмоль/л и вводили в устройство ESI перед накоплением данных путем прямого ввода при скорости потока 1 л/мин и вводили до накопления данных. Масс-спектры при ионизации электронным ударом (770 эВ) получали на квадрупольном масс-спектрометре Finnigan-MAT 4615, соединенном с системой обработки данных INCOS. Температура ионного источника составила 150oС, температура накопительной камеры 100oС. Образцы вводили с помощью газового хроматографа или зонда прямого ввода. Масс-спектры в режиме бомбардировки быстрыми атомами (FAB) получали на приборе Finnigan-MAT 212 с магнитным сектором, соединенном с системой обработки данных INCOS. Ускоряющее напряжение 3 кВ, температура ионного источника приблизительно 70oС. Использовали седлопольный инжектор быстрых атомов фирмы Ion Tech с ксеноном при 8 кэВ. Тиоглицерин использовали в качестве матрицы FAB. Эксперименты MS/MS при ионизации электронным ударом (70 кВ) проводили на тандемном квадрупольном масс-спектрометре Finnigan-MAT TSQ/700. Образец вводили с помощью зонда прямого ввода. Поддерживали температуру ионного источника при 150oС, в накопительной камере при 70oС. Индуцированная соударением диссоциация (СID) достигалась путем введения аргона в центр коллюзионного октаполя до тех пор, пока давление в накопителе не достигало 0,9-2,5х10-6 торр (120-130 мкПа). Номинальная ионная кинетическая энергия для ионов, полученных СID, составляла менее 35 эВ (лабораторный эталон). Данные высокого разрешения получали на масс-спектрометре Jeol JMS AX-505H с двойным фокусированном в конфигурации ЕВ, используя разрешение 7500. Образец вводили с помощью газового хроматографа или зонда прямого ввода. При накоплении в ионный источник вводили перфторкеросин с помощью нагретого ввода. Точные отнесения масс получали компьютерным интерполированием масс перфторкеросина. Условия хромато-масс-спектрометрии (GC/MS): колонка DB-1701 20 м х 0,25 мм (фирмы J& W Scientific), носитель - гелий с линейной скоростью 40 см/сек, температура инжектора 125oС, температура колонки 35oС в течение 3 минут, затем повышение со скоростью 10oС/мин до 100oС, разделение примерно 50:1.
Спектроскопические методы. Спектры1Н-ЯМР с рабочей частотой 300 МГц и13С-ЯМР с рабочей частотой 75 МГц получали на приборе IBM AF300, снабженном суперпроводящей магнитной системой Oxford, данные накопления контролировали с помощью программного обеспечения фирмы Bruker. Инфракрасные спектры получали на спектрометре Mattson Galaxy 5000 FTIR под контролем компьютера Macintosh II. Спектры Уф/видимая область снимали на спектрофотометре Hewlett Packard 8452A, снабженном компьютером Zenith Z-425/SX. Спектры ЭПР записывали на спектрометре Bruker ER300, снабженном гелиевым криостатом Oxford ESR-900. Мессбауэровские спектры получали на приборе постоянного ускорения, изомерные сдвиги выражали относительно железного стандарта при 298К. Для того чтобы избежать ориентации поликристаллических образцов с помощью наложенного магнитного поля, образцы суспендировали в замороженном нуйоле.
Синтезы макроциклических тетраамидо-N-донорных лигандов
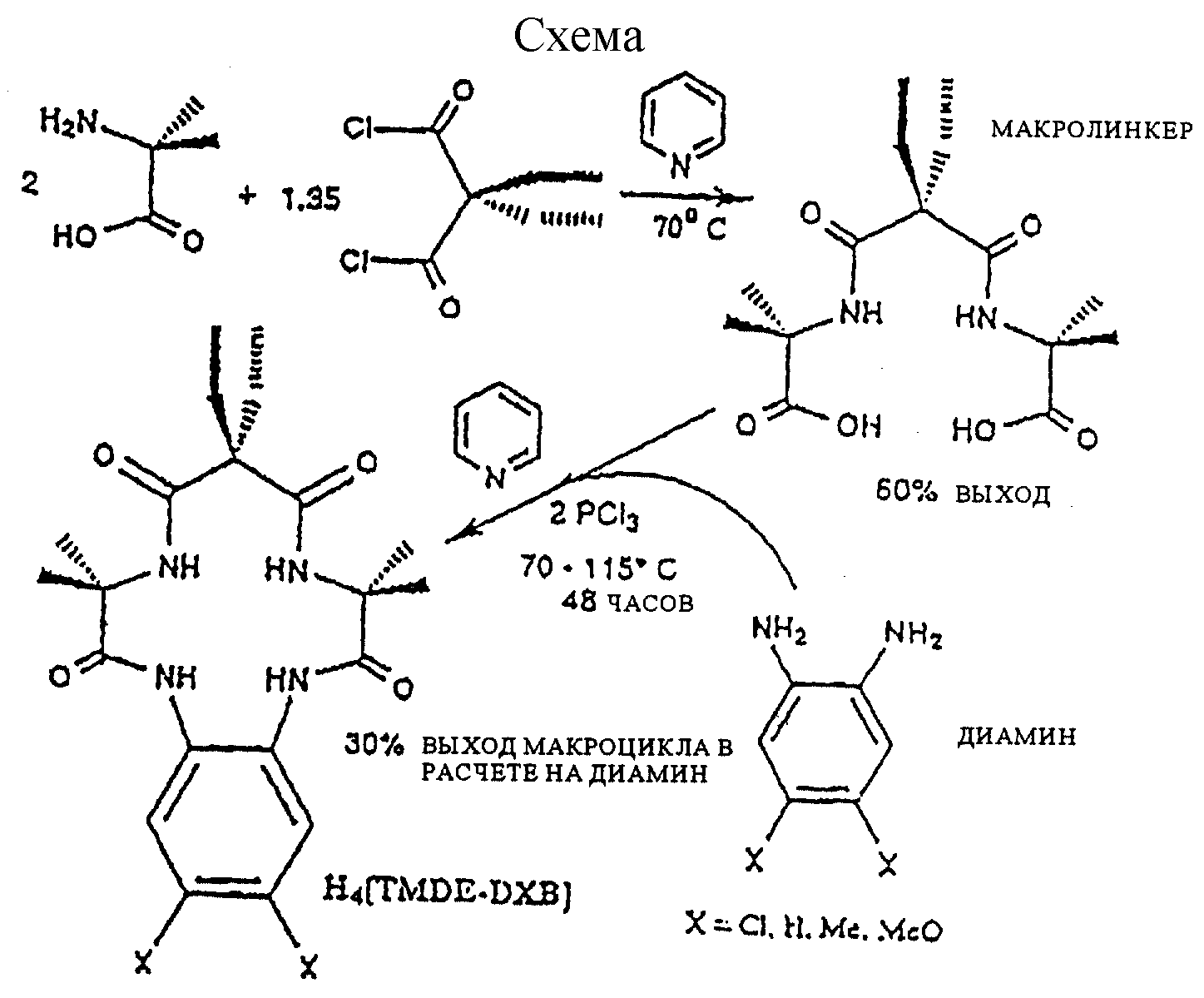
Общая схема
Приведенная в конце описания схема представляет предпочтительную
последовательность реакций для получения макроциклических тетраамидных лигандов по изобретению.
Карбоновую α-аминокислоту смешивали с активированным малонатом в пиридине при температуре ниже 70oС. После двойной, проходящей селективно конденсации (72-144 часа), выделяли макролинкер (A-L-A). На второй стадии диамин, предпочтительно о-фенилендиамин, прибавляли к раствору макролинкера в пиридине в присутствии конденсирующего агента, предпочтительно треххлористого фосфора или пивалоилхлорида. Реакция двойного замыкания (двойной конденсации) протекала при кипении в течение 48-110 часов, после чего целевой макроциклический тетраамид выделяли с хорошим выходом.
В последующих примерах 1-25 описываются различные этапы схемы. Примеры 26-32 демонстрируют характерные признаки осуществления изобретения и его преимущества.
Пример 1
Синтез макролинкерного промежуточного соединения (A-L-A) из α-метилаланина и
диэтилмалонилдихлорида (тетраметилдиэтил замещенное промежуточное соединение)
В двугорлую колбу на 1 л, снабженную капельной воронкой (250 мл) с перетекателем для выравнивания давления и
перегородкой, помещали в токе азота α-аминоизомасляную кислоту (т.е. α-метилаланин) (20,62 г, 0,2 моль) и безводный пиридин (250 мл, высушенный над молекулярными ситами 0,4 нм/4 А),
смесь перемешивали при нагревании до 60-70oС, затем с помощью капельной воронки добавляли раствор диэтилмалонилдихлорида (23,23 мл, 0,135 моль) в безводном пиридине (100 мл, высушенный над
молекулярными ситами 0,4 нм/4 А) в течение 1 часа, ацилирование продолжалось в течение 30-36 часов при 60-70oС в токе азота либо с защитой от влаги воздуха с помощью трубки с осушителем.
Реакцию останавливали прибавлением воды (30 мл) и перемешивали 24 часа при 60-70oС. Растворитель упаривали на роторном испарителе, получая масло, затем прибавляли концентрированную соляную
кислоту (примерно 25 мл) до рН 2-3. Раствор охлаждали в холодильнике (4oС, 15 часов), полученный желтовато-коричневый продукт собирали путем фильтрации на стеклянном пористом фильтре,
тщательно промывали ацетонитрилом (2х100 мл). Высушенный на воздухе продукт (16,5-19,8 г, выход 50-60%) следует хранить в эксикаторе. Этот продукт является достаточно чистым для того, чтобы быть
использованным в реакции замыкания цикла, однако в некоторых случаях может потребоваться кристаллизация. Характеристика: спектр1Н-ЯМР (дейтеропиридин), δ, м.д.: 8.9 (s, 2H, NH
амида), 2.2 (q. 4Н), 1.8 (s. 12H), 1.2 (t, 6H). ИК-спектр (нуйол), см-1: 3310 (NH амида), 1721 (СО карбоксила), 1623 (СО амида). Анализ. Вычислено для C19H21N2О9: С 54,53; Н 7.93; N 8.48. Найдено: С 54.48; Н 7.88; N 8.47.
Пример 2
Крупномасштабный синтез макролинкерного промежуточного соединения (A-L-A) из α
-метилаланина и диэтилмалонилдихлорида (ТМDЕ-замещенное промежуточное соединение)
В двугорлую колбу (2 л), снабженную капельной воронкой (250 мл) с перетекателем для выравнивания давления и
перегородкой, помещали в токе азота, α-аминоизомасляную кислоту (т. е. α-метилаланин) (90,3 г, 0,9 моль), затем канюлировали в колбу безводный пиридин (1,4 л, надежно герметизирован),
реакционную смесь нагревали при 45-55oС при перемешивании. Последовательно канюлировали в капельную воронку пиридин (100 мл, надежно герметизирован) и диэтилмалонилдихлорид (104,4 мл, 0,61
моль), содержимое воронки по каплям прибавляли к реакционной смеси (3-4 часа), после чего воронку удаляли и продолжали реакцию при нагревании до 55-65oС (120-130 часов) в токе азота. По
завершении ацилирования останавливали реакцию добавлением воды (100 мл) и перемешивали в течение 24-36 часов при 60-70oС. Растворитель упаривали на роторном испарителе, получая масло, затем
прибавляли концентрированную соляную кислоту (примерно 110 мл) до конечного рН 2-3. Горячий раствор помещали в холодильник (4oС, 15 часов), полученный желтовато-коричневый продукт собирали
путем фильтрации на стеклянном пористом фильтре, тщательно промывали ацетонитрилом (700 мл, 150 мл), перемешивая в колбе Эрленмейера. Высушенный на воздухе бесцветный продукт (87,9 г, выход 60%)
измельчали с помощью пестика в ступе и хранили в эксикаторе. Амидное промежуточное соединение, полученное крупномасштабным синтезом, возможно, нуждается в перекристаллизации перед использованием в
реакциях замыкания цикла.
Пример 3
Перекристаллизация вышеупомянутого TMDE-замещенного промежуточного соединения
Неочищенное TMDE-промежуточное соединение из примера
2 (50,4 г, 0,153 моль) растворяли в воде (500 мл, деионизированная), добавляя карбонат натрия (16,2 г, 0,153 моль) тремя порциями, чтобы избежать сильного вспенивания, при перемешивании и слабом
нагревании. Раствор доводили до кипения, фильтровали и подкисляли концентрированной соляной кислотой (30 мл, 0,36 моль). Раствор охлаждали (в течение ночи, 4oС), белый осадок отделяли
фильтрованием и промывали ацетонитрилом (250 мл). Высушенный на воздухе продукт (38,8-45,4 г, перекристаллизованный, выход 77-90%) должен храниться в эксикаторе.
Пример 4
Гексаметильное (HМ) промежуточное соединение (A-L-A)
Синтез НМ-промежуточного соединения идентичен синтезу TMDE-промежуточного соединения из примера 2 за некоторыми исключениями:
диэтилмалонилдихлорид заменяли на диметилмалонилдихлорид (17,8 мл, 0,135 моль), температуру реакции снижали до 55-65oС из-за более низкой температуры кипения ацилирующего агента. Выход
гексаметильного производного 45-60%. Характеристика: спектр1Н-ЯМР (дейтеропиридин), δ, м.д.: 9.2-9.8 (br, s, 2H, ОН карбоксила), 8,23 (s, 2H, амида), 1,87 (s, 12H, (СН3),
1,74 (s, 6H, (СН3). ИК-спектр (нуйол-NaCl), см-1: 3317.0 (NH амида), 1717.9 (СО карбоксила), 1625.7 (СО амида). Анализ (образец высушен при 100oС). Вычислено для C13H22N2О6: С 51.63; Н 7.34; N 9.27. Найдено: С 51.64; Н 7.35; N 9.33.
Пример 5
Перекристаллизация НМ-промежуточного соединения
Неочищенное гексаметильное (НМ) промежуточное соединение перекристаллизовывали так же, как TMDE-амидное промежуточное соединение. Благодаря лучшей растворимости в воде НМ-амидного промежуточного
соединения, следует использовать меньшее количество воды.
Пример 6
Дициклогексилдиэтильное промежуточное соединение (DiCyHexDE-промежуточное соединение)
В
круглодонную колбу (500 мл) помещали 1-амино-1-циклогексанкарбоновую кислоту (15 г, 0,1 моль), затем вставляли капельную воронку (40 мл) с перетекателем для выравнивания давления, закрывали
перегородку и продували азотом. Безводный пиридин (300 мл) канюлировали в реакционную колбу через капельную воронку и 20 мл внутрь капельной воронки. Начинали нагрев системы до установления
температуры 60oС. Как только температура достигала этого значения, в воронку прибавляли с помощью шприца одну треть от общего количества диэтилмалонилдихлорида, которое должно было быть
использовано в этой реакции (т. е. 6 мл, 0,033 моль). Смесь пиридин/диэтилмалонилдихлорид прибавляли по каплям к реакционной массе, ацилирование продолжалось в течение 12 часов. Вторую порцию (6 мл, 0,
033 моль) и третью порцию (6 мл, 0,033 моль) прибавляли с интервалами в 12 часов. По окончании прибавления ацилирующего агента и продолжения реакции (общее время реакции 48-56 часов) добавляли по
каплям 20 мл воды. Реакционную смесь нагревали дополнительно в течение 24 часов для раскрытия кольца моно- и бисоксазолоновых промежуточных соединений с образованием диамида дикарбоновой кислоты.
Пиридин удаляли на роторном испарителе, получая светлое желтовато-коричневое масло, которое подкисляли до рН 2 концентрированной соляной кислотой. Неочищенный продукт собирали путем фильтрации,
промывали ацетонитрилом и сушили на воздухе, получая прозрачное DiCyHexDE-амидное производное (16 г, 74%). Характеристика: спектр1Н-ЯМР (дейтеропиридин), δ, м.д.: 8,30 (s, 2H, NH
амида), 2,60 (m, 4H, циклогексан), 2,25 (q, 4H, CH2 этила), 2,15 (m, 4H, циклогексан), 1,8-1,5 (m, 10Н, циклогексан), 1,25 (m, 2H, циклогексан), 1,20 (t, 6H, СН3 этила).13С-ЯМР (широкополосное декаплирование, дейтеропиридин), δ, м.д.: 178,0 (СО карбоксила), 174,3 (СО амида), 60,5 (циклогексан четв.), 59,4 (малонил четв. ), 33.0 (СН2
циклогексана), 30,3 (СН2 этила), 26,0 (СН2 циклогексана), 22,3 (СН2 циклогексана), 9,9 (СН3 этила). ИК-спектр (нуйол-NaCl), см-1: 3307 (NH амида),
3150 (sh, br, m, NH амида/ОН карбоксила), 3057 (s, str, связанный амид NH/ОН карбоксила), 1717 (s, str, CO карбоксила), 1621 (s, str, CO амида). Анализ. Вычислено для C21H24
N2О6: С 61,44; Н 8,35; N 6,82. Найдено: С 61,41; Н 8,38; N 6,90.
Пример 7
DiCyHex-диэтилмонооксазолон
Неудачной оказалась попытка остановить
реакцию DiCyHexDE-этильного промежуточного соединения (с помощью нагрева и воды, см. выше) при использовании 1,35 экв. диэтилмалонилдихлорида; 2 экв. СуНех-аминокислоты приводят к смеси
DiCyHexDE-амидного промежуточного соединения и монооксазолонового продукта. Поскольку DiCyHexDE-монооксазолоновый продукт умеренно растворим в кипящем циклогексане, а циклогексиламидное промежуточное
соединение нерастворимо, появляется возможность простого разделения смеси этих продуктов, а именно: примерно 10 г смеси в 400-500 мл циклогексана, содержащей остаточные количества хлористого метилена,
нагревали при кипении и энергичном перемешивании. Нерастворимое DiCyHexDE-амидный промежуточный продукт собирали путем горячей гравитационной фильтрации, при охлаждении фильтрата и упаривании медленно
кристаллизовался монооксазолон. Выход амидного промежуточного
соединения примерно 6 г, выход монооксазолона примерно 4 г. Характеристика монооксазолона. Спектр1H-ЯМР
(дейтеропиридин), δ, м.д.: 9,7 (s, 1H, NH амида), 2,7-1,6 (неразрешенный сигнал СуНех-групп), 1,05 (t, 6H, СН3 этила). ИК-спектр (нуйол/NaCl), см-1: 3309 (sh, w, NH амида),
3229 (s, str, связанный амидный NH/OH карбоксила), 3166 (s, str, связанный амидный NH/OH карбоксила), 3083 (s, str, связанный амидный NH/OH карбоксила), 1834 (s, str, С= О оксазалона), 1809 (s, m,
связанный С=О оксазолона), 1743 (s, str, CO карбоксила), 1663 (s, str, C=N оказолона), 1639 (s, br, str, CO амида). Анализ. Вычислено для C122H22N2О5x0,25
С6Н12: С 65,35; Н 8,53; N 6,77. Найдено: С 65,07; Н 8,67; N 6,68. Присутствие сольватного циклогексана подтверждено данными13С-ЯМР.
Реакции
макроциклизации
Примеры некоторых синтетических способов получения макроциклических тетраамидных лигандов приведены далее.
Конденсация в присутствии треххлористого фосфора
Метод конденсации амидного промежуточного соединения (A-L-A) с ароматическими 1,2-диаминами в присутствии треххлористого фосфора является безопасным, доступным и позволяет получать
макроциклические амиды с высоким выходом. Используют два различных варианта конденсации в присутствии треххлористого фосфора, различия связаны с порядком прибавления и выбором реагентов. Метод
применим для получения большого числа разнообразных макроциклов с различными по своим электронным свойствам заместителями в мостиковом диамине или с отличающимися по стерическим свойствам
заместителями в амидном промежуточном соединении, прежде всего, благодаря возможности параллельного использования амидных промежуточных соединений макролинкерного типа во всех синтезах.
Пример 8
А. Синтез макроцикла путем конденсации в присутствии треххлористого фосфора
В длинногорлую колбу (250 мл) загружали амидное промежуточное соединение (10 ммоль), описанное в
примерах 2-7, прекращали перемешивание и нагревали колбу при 80-100oС в течение 30-45 минут. В горячую колбу пускали азот, прибавляли арилдиамин (10 ммоль) и канюлировали безводный пиридин
(50 мл, с защитой от влаги воздуха). Колбу нагревали (50-60oС), прибавляли с помощью шприца треххлористый фосфор (d=l,574 г/мл, 1,72 мл, 20 ммоль) с такой скоростью, чтобы не допустить
бурного вскипания. Реакция экзотермична, поэтому следует соблюдать осторожность. Затем поднимали температуру до кипения или немного ниже (100-115oС), реакцию проводили в атмосфере азота в
течение 48 часов. По завершении ацилирования содержимое колбы подкисляли соляной кислотой (1 экв. , примерно 60 мл) до конечного рН 2. Смесь переносили в колбу Эрленмейера (использовали воду для смыва
колбы) и перемешивали с хлористым метиленом (300 мл, 2-3 часа), затем экстрагировали дополнительным количеством хлористого метилена (2х150 мл). Объединенные органические слои промывали разбавленной
соляной кислотой (0,1 М, 2х100 мл), затем разбавленным водным раствором карбоната натрия (2х5 г/100 мл). Органические растворители удаляли на роторном испарителе, получая неочищенный продукт (30%).
Обычно вес неочищенного продукта равен весу исходного диамина.
Б. Синтез макроцикла путем конденсации в присутствии треххлористого фосфора
В длинногорлую колбу (250 мл)
загружали сульфат магния (5 г), прекращали перемешивание, прибавляли арилдиамин (10 ммоль) и пиридин (50 мл, высушенный над молекулярными ситами 0.4 нм/4А), затем пускали азот и с помощью шприца
прибавляли треххлористый фосфор (d= l,574 г/мл, 1,72 мл, 20 ммоль), смесь нагревали при кипении в течение 30 минут, при этом образуется оранжево-желтый осадок. Смесь охлаждали, прибавляли амидное
промежуточное соединение (10 ммоль), после чего смесь нагревали при кипении в атмосфере азота (115oС, 48 часов). По завершении ацилирования содержимое колбы подкисляли соляной кислотой (1
экв. , примерно 60 мл) до конечного рН 2. Смесь переносили в колбу Эрленмейера и перемешивали с хлористым метиленом (2х150 мл). Объединенные органические слои промывали разбавленной соляной кислотой
(0,1 М, 2х100 мл), затем разбавленным водным раствором карбоната натрия (2х5 г/100 мл). Органические растворители удаляли на роторном испарителе, получая неочищенный продукт (30%). Обычно вес
неочищенного продукта равен весу исходного диамина.
Примечание: при крупномасштабной макроциклизации время замыкания цикла увеличивают до 4-5 суток при кипении, в конце реакции большую часть пиридина перед подкислением удаляют на роторном испарителе.
Пример 9
Получение TMDE-DCB из ТМDЕ-промежуточного соединения и DCB-диамина
Использовали 1,
2-диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) в качестве арилдиамина в реакции макроциклизации с TMDE-амидным промежуточным соединением (3,3 г, 10 ммоль) в присутствии треххлористого фосфора по методу
А или Б. Неочищенный макроциклический продукт (2,7 г) перекристаллизовывали из минимального количества горячего 95% этанола путем упаривания, получали чистый TMDE-DCB (1,5 г, 32%). Характеристика:
спектр1Н-ЯМР (CD2C12), δ, м.д.: 7,65 (s, 1H, АrН), 7,35 (s, 2H, NH амида), 6,45 (s, 2H, NH амида), 1,90 (q, 4H, СН2 этила), 1,57 (s, 12H, RСН3), 0,85 (t, 6H, СН3 этила). ИК-спектр (нуйол-NaCl), см-1: 3454 (следы ROH), 3346 (br, NH амида), 1706, 1688, 1645 (СО амида). Анализ. Вычислено для C22H28Cl2N4O4: С 53.51; Н 5.99; N 11.89. Найдено: С 53.58; Н 6.09; N 11.89.
Пример 10
Получение TMDE-B из TMDE-промежуточного соединения и
В-диамина
Использовали 1,2-диаминобензол (т.е. o-фенилендиамин) (1,08 г, 10 ммоль) в качестве арилдиамина в реакции макроциклизации с TMDE-амидным промежуточным соединением (3,3 г, 10 ммоль)
в присутствии треххлористого фосфора по методу А или Б. Неочищенный макроциклический продукт (1,5 г) перекристаллизовывали из минимального количества горячего 95% этанола путем упаривания, получали
чистый TMDE-B (25% на диамин). Характеристика: спектр1Н-ЯМР (CDCl3), δ, м. д.: 7,55 (m, 2H, АrН), 7,48 (s, br, 2H, NH ариламида), 7,17 (m, 2H, АrН), 6,46 (s, br, 2H, NH
алкиламида), 2.07 (m, br, 4Н, СН2 этила), 1,60 (s, 12H, RСН3), 0,89 (t, 6H, СН3 этил). ИК-спектр (нуйол-NaCl), см-1: 3395, 3363 (NH амида), 1702, 1680, 1652,
1635 (СО амида). Анализ. Вычислено для C21H10N4О4xH2O: С 59,98; Н 7,67; N 13,32. Найдено: С 60,18; Н 7,20; N 13,18.
Пример 11
Получение TMDE-DMB из TMDE-промежуточного соединения и DMB-диамина
Использовали 1,2-диамино-4,5-диметилбензол (1,36 г, 10 ммоль) в качестве арилдиамина в реакции макроциклизации с
TMDE-амидным промежуточным соединением (3,3 г, 10 ммоль) в присутствии треххлористого фосфора по методу А или Б. Неочищенный макроциклический продукт (1,6 г) перекристаллизовывали из минимального
количества горячего 95% этанола путем упаривания, получали чистый TMDE-DMB (25% на диамин). Характеристика: спектр1Н-ЯМР (d6-DMSO), δ, м. д. : 8,00 (s, 2H, NH амида), 7,
67 (s, 2H, NH амида), 7,28 (s, 2H, АrН), 2,17 (s, 6H, СН3 арила), 1,99 (q, 4H, СН2 этила), 1,46 (s, 12H, RCH3), 0,75 (t, 6H, СН3 этила). ИК-спектр
(нуйол-NaCl), см-1: 3446 (s, m, следы ROH), 3362 (s, str, NH амида), 3348 (sh, m, NH амида), 3332 (s, str, связанный NH), 1696 (СО амида), 1679 (СО амида), 1641 (СО амида), 1584 (s, m/w,
перекрывающиеся кольца арила и амида). Анализ. Вычислено для С23Н14N4О4: С 74,16; Н 7,96; N 13,01. Найдено: С 64,09, 64,28; Н 8,04, 7,92; N 12.86, 13,04.
Пример 12
Получение TMDE-DMOB из ТМDЕ-амидного промежуточного соединения и DМОВ-диамина
Использовали дигидробромид 1,2-диамино-4,5-диметоксибензола (5,0 г, 15 ммоль),
полученный, как описано выше, в качестве арилдиамина в масштабированной в 1,5 раза реакции макроциклизации с TMDE-амидным промежуточным соединением (5,0 г, 15 ммоль) в присутствии треххлористого
фосфора по методу А или Б. Неочищенный макроциклический продукт (3,57 г) перекристаллизовывали из минимального количества горячего 80-85% этанола (1 г/40 мл) путем упаривания, получали чистый
TMDE-DMOB (30% на диамин). Характеристика: спектр1H-ЯМР (CD2Cl2), δ, м.д.: 7,26 (s, 2H, NH амида), 7,01 (s, 2H, АrН), 6.41 (s, 2H, NH амида), 3,80 (s, 6H,
ОСН3 арила), 2.07 (q, br, 4H, СН3 этила), 1,54 (s, 12H, RСН3), 0,90 (t, 6H, СН3 этила). ИК-спектр (нуйол-NaCl), см-1: 3451 (s, m, связанный Н2О), 3391, 3347 (NH амида), 1695, 1670, 1655 (СО амида). Анализ. Вычислено для C23H34N4О6x0,22 H2O: С 58,96; Н 7,46; N 11,96. Найдено: С 58,
90; Н 7,26; N 11,76. Присутствие сольватной воды подтверждено данными1H-ЯМР и ИК-спектра.
Пример 13
Получение TMDE-Nap из ТМDЕ-промежуточного соединения и
Nap-диамина
Использовали 4,5-диамино нафталин (1,68 г, 10 ммоль) в качестве арилдиамина в реакции макроциклизации с TMDE-амидным промежуточным соединением (3,3 г, 10 ммоль) в присутствии
треххлористого фосфора по методу А или Б. Неоптимизированный выход составил 15-20% в расчете на диамин. Спектр1Н-ЯМР (CDCl3), δ, м.д.: 8,05 (s, 2H, кольцо АrН), 7,75 (m,
2H, кольцо АrН), 7,55 (s, 2H, NH ариламида), 7,35 (m, 2H, кольцо АrН), 6,45 (s, 2H, NH алкиламида), 2,15 (m, br, 4H, CH2 этила), 1,65 (s, 12H, RСН3), 0,90 (t, 6H, СН3
этила).
Пример 14
Получение HM-DCB из НМ-промежуточного соединения и DCB-диамина
Использовали 1,2-диамино-4,5-дихлорбензол (1,77 г, 10 ммоль) в качестве диамина в
реакции макроциклизации с гексаметиламидо-промежуточным соединением (3,02 г, 10 ммоль) в присутствии треххлористого фосфора по методу А или Б. Неочищенный макроциклический продукт (1,33 г, 30%)
перекристаллизовывали из минимального количества горячего пропанола путем упаривания, получали первую порцию кристаллов с выходом 60%. Характеристика: спектр1Н-ЯМР, δ, м. д. : 7,69
(s, 2H, АrН), 7.39 (s, 2H, NH амида), 6.44 (s, 2H, NH амида), 1,58 (s, 12H, СН3 плеча), 1,53 (s, 6H, СН3 малоната), отмечены небольшие пики пропанола. ИК-спектр (нуйол-NaCl),
см-1: 3503 (s, br, m-w, ОН пропанола), 3381 (sh, m, NH амида), 3338 (s, str, NH амида), 1689 (s, str, CO амида), 1643 (s, str, CO амида). Анализ. Вычислено для C17H24
N4О4Cl2x0.2C3H8О: С 51,70; Н 5,57; N 12,30. Найдено: С 51,69; Н 5.63; N 12,33.
Пример 15
Получение НМ-DМОВ и НМ-В из
НМ-промежуточного соединения и DMOB или В-диамина
Использовали НМ-промежуточное соединение для синтеза НМ-В и НМ-DMOB тем же методом и с результатами, приведенными в примере 14 для
хлорпроизводного. Спектр1Н-ЯМР для HM-DMOB (СDС13), δ, м.д.: 7,65 (s, 2H, NH амида), 7,21 (s, 2H, арил), 6,72 (s, 2H, NH амида), 4,00 (s, 6H, СН3 метокси), 1,
76 (s, 12H, СН3 плеча), 1,58 (s, 6H, СН3 малоната).1Н-ЯМР для НМ-В (дейтеропиридин), δ, м. д. : 8,55 (s, 2H, NH амида), 8,40 (s, 2H, NH амида), 7,81 (m, 2H,
aa'bb' АrН), 7,10 (m, 2H, aa'bb' АrН), 1,77 (s, 12H, СН3), 1,73 (s, 6H, СН3 малоната). Сигнал амидного протона сдвигается на несколько десятых м.д. в присутствии таких примесей,
как вода, кислоты и т.д.
Пример 16
Получение DiCyHexDE из DiCyHexDE-промежуточного соединения и DCВ-диамина
Использовали 1,2-диамино-4,5-дихлорбензол (1,77 г, 10
ммоль) в качестве арилдиамина в реакции макроциклизации с DiСуНехDЕамидным промежуточным соединением (3,3 г, 10 ммоль) в присутствии треххлористого фосфора по методу А или Б. В связи со стерической
затрудненностью рекомендуется увеличить время реакции замыкания цикла (3-4 суток по сравнению с обычными 48 часами). СуНех-оксазолоны, образующиеся в качестве побочного продукта реакции, не удаляются
при кислотной обработке реакционной смеси, поэтому для их удаления необходимо выделенный первоначально продукт, растворимый в хлористом метилене, обрабатывать/промывать пентаном. Упаривание пентана
позволяет регенерировать оксазолоны. Неочищенный не растворимый в пентане продукт перекристаллизовывали путем растворения в хлористом метилене или хлороформе с добавлением циклогексана до легкого
помутнения, после испарения растворителя на воздухе (1-2 дня) получали DiCyHexDE-DCB в виде бесцветного микрокристаллического продукта, который собирали путем фильтрации (1,38 г, 25% в расчете на
диамин). Перекристаллизация из горячего толуола с упариванием представляется также перспективной. Характеристика: спектр1Н-ЯМР (CDCl3), δ, м.д.: 7,70 (s, 2H, АrН), 7,45
(s, 2H, NH амида). 6,45 (s, 2H, NH амида), 2,35 (m, br, 4H, циклогексан), 2,00 (m, br, 8H, СН3 циклогексана/этила), 1,70 (m, br. 8H, циклогексан), 1,30 (m, br, 4H, циклогексан), 0,90 (t, 6H,
СН3 этила). Анализ (вещество
высушено при 100oС). Вычислено для С37Н34Сl2N4О4x0.2C6H12: С 59,60;
Н 6,81; N 9,86. Найдено: С 59,60; Н 6,77; N 9,77. Присутствие циклогексана подтверждено данными1Н- и13С-ЯМР.
Пример 17
Получение DiCyHexDE-B из
DiCyHexDE-промежуточного соединения и В-диамина
Использовали 1,2-диаминобензол (о-фенилендиамин) (1,08 г, 10 ммоль) в качестве арилдиамина в синтезе, описанном для DiCyHexDE-DCB, получали
DiCyHexDE-B (1,25 г, 26% в расчете на диамин). Характеристика: спектр1Н-ЯМР (CD3CN), δ, м. д.: 7,62 (s, 2H, NH ариламида), 7.51 (m, 2H, АrН), 7,18 (m, 2H, АrН), 6.71 (s,
2H, NH алкиламида), 2,12 (m, 6H, циклогексан), 1,85 (q&m, СН2 циклогексана/этила), 1,62 (m, циклогексан), 1,37 (m, циклогексан), 0,90 (t, 6Н, СН3 этила), 0,85 (m,
циклогексан). ИК-спектр (нуйол-NaCl), см-1: 3750 (s, m, H2О), 3385 (s, str, NH амида), 3314 (s, str, NH амида), 3258 (s, m, br, связанный NH амида), 1694 (s, str, CO амида), 1651
(s, str, CO амида), 1594 (s, m, арильное кольцо/ амид).
Пример 18
Получение DiCyHexDE-бисоксазолона
Этот продукт получали в качестве побочного соединения в реакции
макроциклизации DiCyHexDE-амидного промежуточного соединения с o-фенилендиамином в присутствии треххлористого фосфора. Бисоксазолон не удалялся путем кислотной и щелочной обработки реакционной смеси
(это нейтральная молекула, хорошо растворимая в органических растворителях). При промывании неочищенной смеси макроциклического продукта и оксазолона большая часть оксазолона переходит в пентан. После
испарения пентана на воздухе получают чистый бисоксазолон в виде больших (1х1х0,5 см) прозрачных призм. Из-за наличия объемистых гидрофобных СуНех-групп этот оксазолон намного более устойчив к
гидролизу, чем соответствующие метильные производные. Характеристика бисоксазолона: спектр1Н-ЯМР (СD3СN), δ, м.д.: 2,05 (q, 4H, СН3 этила), 1,8-1,4
(неразрешенный сигнал циклогексановых групп), 0,88 (t, t Н, СН3 этила). Спектр13С-ЯМР (широкополосное декаплирование, СD3СN), δ, м.д.: 181,0 (С=О оксазолона),
162,7 (C=N оксазолона), 69,0 (кв, циклогексан оксазолона), 49,0 (кв, малонат), 34,3 (СН3 циклогексана), 25,5 (СН2 циклогексана), 24,9 (СН2 малоната), 21,8 (СН2 циклогексана), 8,3 (СН3 этила). ИК-спектр (нуйол-NaCl), см-1: 1822 (s, str, br, C=О оксазолона), 1662 (s, str, C=N оксазолона). Анализ. Вычислено для C22H10N2О4: С 67,36; Н 8,07; N 7,48. Найдено: С 67,26; Н 8,15; N 7,64.
Синтезы хелатных комплексов
Пример 19
Синтез [Et4N]2 и
[Еt4N]3 [тетраэтиламмониевых солей железо(III)хлорТМDЕ-DСВ-моноаниона и железо(III)аквоТМDЕ-DСВ-моноаниона, соответственно].
Исходный макроциклический тетраамид (525 мг, 1,1 ммоль), описанный в любом из приведенных выше примеров 10-18, растворяли в тетрагидрофуране (фирмы Aldrich, 40 мл) в токе азота. К раствору прибавляли трет.-бутиллитий (2,6 мл, 4,4 ммоль, 1,7 М в 2, 4-диметилпентане, фирма Aldrich) в токе азота при -108oС, используя прибор Шленка. Затем прибавляли хлорид железа (безводный, 155 мг, 1,2 ммоль, фирма Alfa), раствору при перемешивании давали нагреться до комнатной температуры (16 часов), получали чувствительный к влаге воздуха комплекс FeII. Воздух впускали через трубку с осушителем (2 часа), твердое оранжевое вещество собирали и промывали хлористым метиленом (2х10 мл). Полученный оранжевый порошок сушили при пониженном давлении. Выход 595 мг (93%). В связи с изменяющейся сольватацией и ограниченной растворимостью и для удобства использования в дальнейшем литиевую соль превращали в тетраэтиламмониевую соль. Литиевую соль (595 мг) в метаноле (50 мл) наносили на ионообменную колонку (Дауэкс 50х2-100, 25 г, 2х12,5 см), которая была предварительно насыщена катионом [Et4N]+, оранжевую полосу элюировали метанолом (100 мл). Растворитель удаляли при пониженном давлении. Остаток суспендировали в хлористом метилене (20 мл), смесь фильтровали. Растворитель удаляли из фильтрата при пониженном давлении, получая оранжевый гигроскопичный стекловидный остаток [Et4N]22, который использовали без окончательной очистки. ИК-спектр (нуйол-NaCl), см-1: 1619 (СО амида), 1575 (СО амида), 1534 (СО амида). Тщательная очистка исходного вещества (железо(III)) была более легко достижима в случае аксиального аквомоноанионного комплекса, а не этого аксиального хлордианионного комплекса. Растворяли [Et4N]22 (550 мг, примерно 0.7 ммоль) в ацетонитриле (50 мл). Тетрафторборат серебра (140 мг, 0.7 ммоль) растворяли в ацетонитриле (2 мл) и прибавляли к раствору при перемешивании (1 час). Осадок хлорида серебра отделяли, растворитель удаляли при пониженном давлении. Полученный [Et4N]23 очищали колоночной хроматографией на силикагеле (8% метанол в хлористом метилене). Растворитель удаляли при пониженном давлении, продукт перекристаллизовывали из воды. Выход 360 мг (77% неустойчивых сольватов с водой найдено в различных микрокристаллических образцах). ИК-спектр (нуйол-NaCl), см-1: 1590 (СО амида), 1565 (СО амида), 1535 (СО амида). Анализ. Вычислено для С29Н46С12FеN5О5(H2O): С 50,52; Н 7,02; С1 10,28; N 10,16. Найдено: С 50,24; Н 6,84; С1 10,32; N 9,82. Масс-спектр (электроспрей, отрицательный ион), m/z: 522,2 [3-Н2O]1-, (100%); 269,7 [3-Н-]2-, (18%).
Пример 20
Синтез [Et4N] 4 [тетраэтиламмониевой соли железо(IV)хлорТМDЕ-DСВ-моноаниона]
Растворяли [Et4N]22 (500 мг, примерно 0,6 ммоль) в хлористом метилене (30 мл). К раствору
прибавляли аммонийцерий(IV)нитрат (10,3 г, 18,3 ммоль), смесь перемешивали в течение 2 часов. Осадок солей церия удаляли путем фильтрации. После удаления растворителя при пониженном давлении и
высушивания остатка в вакууме получали пурпурный продукт. Выход 400 мг (95%). Пурпурные кристаллы получали перекристаллизацией из смеси хлористый метилен-эфир. ИК-спектр (нуйол-NaCl), см-1:
1688 (СО амида), 1611 (СО амида), 1582 (СО амида). Масс-спектр (электроспрей, отрицательный ион), m/z: 557 [4]1-, (100%); 522 [4-С1]1-. (65%).
Пример 21
Синтез [Ph4P] 5 [тетрафенилфосфониевой соли железо(IV)цианТМDЕ-DСВ-моноаниона] из [E4N]4 [тетраэтиламмониевой соли железо(IV)хлорТМDЕ-DСВ-моноаниона] и цианистого натрия
Суспендировали [Et4N] 4 [тетраэтиламмониевую соль железо(IV)хлор-TMDE-DCB-моноаниона] (225 мг, 0,33 ммоль) в воде (10 мл). Цианистый натрий (140 мг, 2,85 ммоль) растворяли в воде (10 мл),
прибавляли с суспензии, смесь облучали ультразвуком (прибор марки Branson 1200, 0,5 часа). Суспензия меняла цвет от пурпурного до темно-синего, при этом почти все твердое вещество растворялось. Смесь
фильтровали, продукт синего цвета осаждали, прибавляя PPh4Cl (хлорид тетрафенилфосфония), растворенный в воде (600 мг, 1.6 ммоль, 10 мл, фирма Aldrich). Собирали синий осадок и промывали
водой (2х10 мл). Выход 250 мг (0,28 ммоль, 85%). Это вещество (120 мг) затем очищали тонкослойной хроматографией (ТСХ) (пластина с силикагелем GF, 20х20 см, хлористый метилен-ацетонитрил, 10:1). Синее
вещество экстрагировали с силикагеля смесью ацетонитрил-хлористый метилен (1: 1, 60 мл). Растворитель удаляли при пониженном давлении, остаток растворяли в хлористом метилене (3 мл), раствор
фильтровали. При добавлении пентана (150 мл) получали синий осадок (90 мг, 0,10 ммоль). Выход после очистки 75%. ИК-спектр (нуйол-NaCl), см-1: 2129 (CN), 1659 (СО амида), 1598 (СО амида),
1571 (СО амида). Анализ. Вычислено для С46Н44Сl2FeN5ОР: С 62,18; Н 4,99; С1 7,98; N 7,88. Найдено: С 61,96; Н 5,04; С1 8,06; N 7,84. Масс-спектр
(электроспрей, отрицательный ион), m/z: 548,2[5]1-, (100%); 522,1 [5-CN]1-, (20%). Для13С-меченого цианида, m/z: 549,2[5]1-, (100%); 522,1[5-13
CN]1-. (8%).
Пример 22
Синтез [Рh4Р] 5 [тетрафенилфосфониевой соли железо(IV)цианТМDЕ-DСВ-моноаниона] из нитрилцианидного источника
[Ph4P] 5 [тетрафенилфосфониевая соль железо(IV)цианТМDЕ-DСВ-моноаниона] может получаться в присутствии и в отсутствие основания. В отсутствие основания синий цвет по мере удаления растворителя при
обработке переходит в желто-оранжевый. Следовательно, чтобы получить синий осадок, следует проводить выделение в присутствии добавленного основания при рН в интервале 9-10. В следующей ниже реакции 5
образуется при использовании любого из нитрилсодержащих растворителей (ацетонитрила, дейтероацетонитрила, пропионитрила, изобутиронитрила). В случае описанных каталитических реакций основание не
добавляется. Было показано, что синее соединение является эффективным предшественником катализатора: при добавлении выделенного [Рh4Р]5 к раствору ТВНР (трет. -бутилгидропероксида) в
ацетонитриле как растворитель, так и оксидант расходовались, свидетельствуя о том, что, хотя [Рh4Р]5 является конечным продуктом процесса каталитического окисления, он не является
дезактивированной формой катализатора.
Пример 23
Синтез [Ph4P]5 в присутствии основания
Растворяли [Et4N]4 (160 мг, 0,23 ммоля) в выбранном
нитрилсодержащем растворителе, прибавляли тетраэтиламмонийгидроксидное основание (20% весовых, 0,370 ммоль, фирмы Aldrich), после чего по каплям прибавляли трет.-бутилгидропероксид (90%, 0,605 мм, 5,4
ммоль) при перемешивании в течение 20 минут, при этом образуется синий раствор. Реакционную смесь упаривали при пониженном давлении, остающийся маслянистый остаток (15 мл) фильтровали. Синее вещество
осаждали из водного раствора PPh4Cl (800 мг, 2,1 ммоль, фирмы Aldrich), собирали и промывали водой (2х10 мл). Выход 130%. Дальнейшую очистку проводили, как описано для [Ph4
P]5.
Пример 24
Данные рентгеноструктурного анализа и очистки для [Еt4N]3•Н2О
C29H48Cl2FeN5
О6, M 689,47, триклинная пространственная группа Р-1, а= 9,899(2); b=11,771(2); с=1,4991(4) нм/14.991(4)


Пример 25
Данные рентгеноструктурного анализа и очистки для [Et4N]4
Монокристаллы [Еt4N] 4 при 20±1oС представляют собой моноклинные
пространственные P2l/c-C52h (No. 14) с а=0,9958(2) нм/(9,958(2)




Пример 26
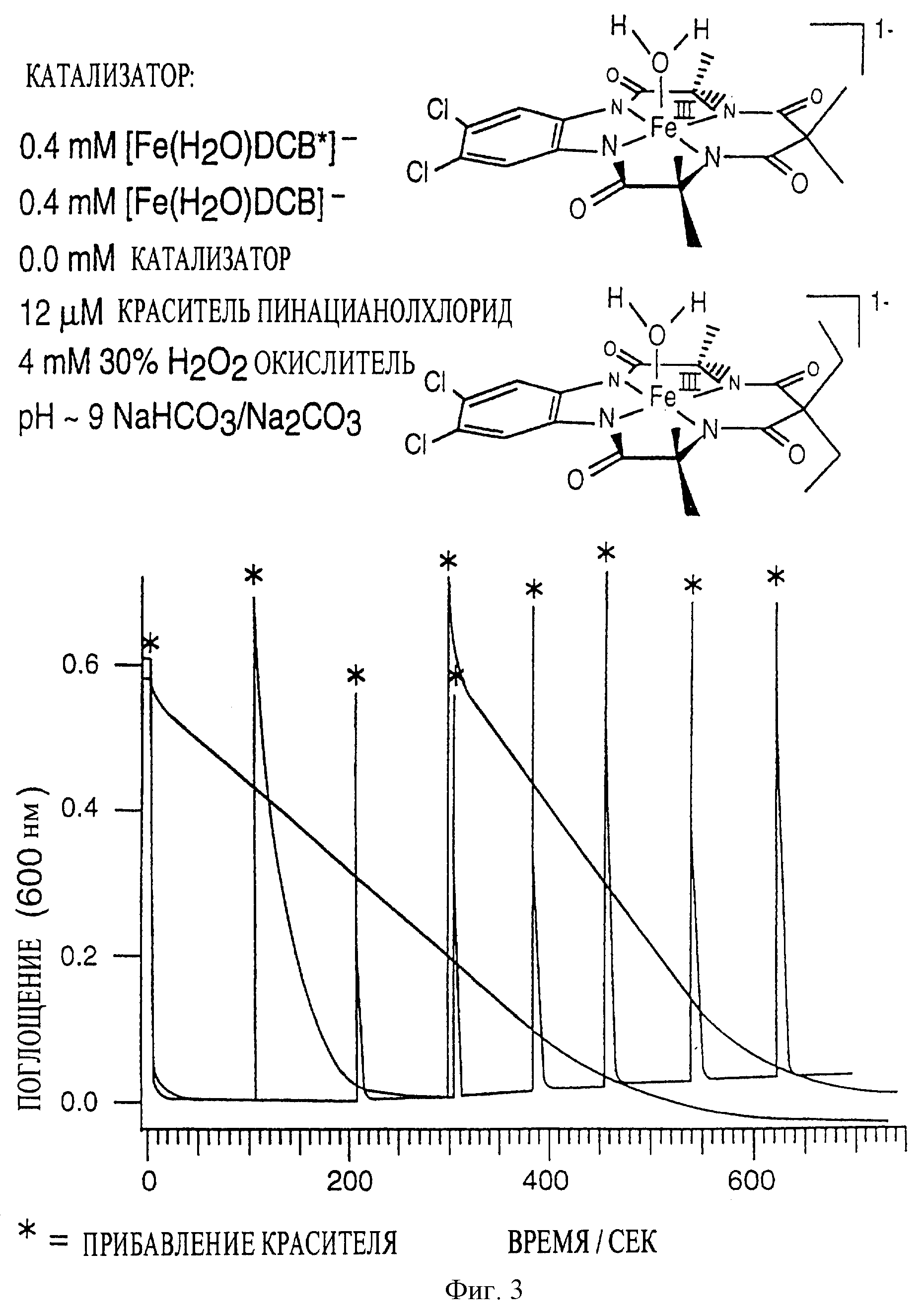
Увеличенная продолжительность каталитической стабильности
Сравнивали каталитическую долговечность в двух вариантах воплощения данного изобретения
(фиг. 3). В соединении 1 каждый из заместителей R' и R" означает метальную группу, тогда как в соединении 2 каждый из заместителей R' и R" означает этильную группу. В контроле не добавляли никакого
катализатора.
Условия: рН 9, комнатная температура (21,1oС), буфер, содержащий бикарбонат и карбонат натрия, в качестве оксиданта использовали 4 мМ (30%) перекись водорода. Каждая звездочка означает прибавление красителя - 12 мкМ пинацианолхлорида.
Как видно из графика, в случае соединения 1 каждое прибавление красителя практически сразу приводило к обесцвечиванию. В случае соединения 2 обесцвечивание наступало постепенно. В контроле обесцвечивание наступало еще медленнее.
Из вышесказанного следует, что соединения по изобретению, особенно соединение 1, являются эффективными в окислении и обесцвечивании посторонних красителей и красителей, сходящих с окрашенных текстильных изделий в процессе стирки. Сходящие красители могут абсорбироваться или адсорбироваться на адсорбент или абсорбент, такой, как другое текстильное изделие. Таким образом, макроциклические тетраамидные соединения по изобретению обеспечивают уникальное преимущество окислительной системы, а именно, придают ей свойство ловушки красителя, предотвращая тем самым перенос посторонних, и поэтому нежелательных, красителей в моющей жидкости с одного текстильного изделия на другое.
Следующие далее примеры 27-30 иллюстрируют уникальную способность активаторов отбеливания по изобретению окислять абсорбируемые или адсорбируемые вещества, такие, как красители, в растворе со скоростью, достаточной для подавления абсорбции или адсорбции их на субстрате, таком, как текстильное изделие. Способность быстро окислять в растворе придает макроциклическим тетраамидным лигандам по изобретению свойство подавлять перенос красителя. В примерах 27-28 приведенные спектры и кривые поглощения в зависимости от времени записаны на спектрофотометре Shimadzu. Образцы сканировали в интервале 350-700 нм перед добавлением перекиси или катализатора для того, чтобы определить длину волны максимума поглощения красителей. Затем спектрофотометр устанавливали на определенную длину волны и прибавляли перекись и/или катализатор. Через 2 минуты определяли изменения максимума поглощения.
В случае красителя Acid Blue 25 измерения проводили при 600 нм. Образцы измеряли при 25oС в кювете на 1 см, содержащей 2 мл раствора.
Пример 27
Отбеливание красителя Acid Blue 25 в
растворе
К раствору Acid Blue 25 (120 мг/л, содержание красителя 45%, начальное поглощение при 600 нм составляло 1,2) прибавляли: (а) 20 м.д. А.О. Н2О2; (б) 20 м. д.
А.О. Н2О2 и 1 м.д. соединения по изобретению, где Z и Y каждый означают атом водорода (в дальнейшем "FeB"); (с) 20 м.д. А.О. Н2О2 и 1 м.д. соединения по
изобретению, где Z и Y каждый означают атом хлора (в дальнейшем "FeDCB"). Как показано ниже, только с системами, содержащими катализатор, получен какой-либо отбеливающий эффект (контролируемый по
наблюдаемому изменению поглощения при 600 нм через две минуты). Для сравнения определяли падение поглощения, вызванное гипохлоритом натрия (5,25% раствор, среднее значение 20 м.д. хлора). Результаты
приведены в табл.1.
Большее падение поглощения означало более сильное обесцвечивание красителя. Приведенные выше данные показывают, что при использовании катализаторов по изобретению наблюдается эффективное подавление переноса красителя. При сопоставлении с количеством добавленного красителя (начальное поглощение 1,2) потеря красителя составила более 90% (1,15:1,2х100=95,83%).
Пример 28
Отбеливание красителя Acid Orange 8 в растворе
Эксперименты проводили, как в примере 27, за исключением того, что использовали раствор красителя Acid Orange 8
(210 мг/л, содержание красителя 65%, начальное поглощение при 490 нм составляло 1,2). Отбеливание измеряли по изменению поглощения при 490 нм (см. табл.2).
И снова при сопоставлении с количеством добавленного красителя (начальное поглощение 1.2) потеря красителя составила более 90% (1,15: 1,2х100= 95,83%). Перенос красителя (ΔЕ) рассчитывали также в соответствии с методикой, приведенной в патенте США 5,698,476 от 16 декабря 1997 года авторов Johnson и др., озаглавленном "Стиральный состав для предотвращения переноса красителя и его индикатор". Величина ΔЕ отражает среднее значение наблюдаемых изменений изделия из ткани до и после стирки в соответствии с приведенным выше уравнением. Увеличение вычисленного значения ΔЕ для испытуемого изделия, выстиранного в присутствии источника красителя, по сравнению с испытуемым изделием до стирки указывает на то, что испытуемое изделие абсорбировало краситель. Все красители получали от фирмы Aldrich Chemicals.
В приведенных ниже примерах 29 и 30 использовали следующие условия: 0,95 г детергента Ultra Tide® (Procter & Gamble) вносили в емкость для стирки Terg-О-Tometer с 1,5 л теплой воды, двумя испытуемыми изделиями из хлопка размером 20,32х20,32 см/8х8 дюймов (большой образец) и изделием, которое выделяло краситель в раствор. Назначение испытуемого изделия состояло в том, чтобы служить рецептором для любого постороннего красителя, который не был обесцвечен или окислен. Образцы перемешивали в течение 12 минут после прибавления улавливающей краску системы (перекись водорода и катализатор), используя Terg-О-Tometer, затем в течение двух минут полоскали водой с температурой окружающей среды и в течение 20 минут сушили в автоматической сушилке.
Пример 29
Перенос краски с одной ткани на другую с использованием красителя Direct Red 79
Для того чтобы продемонстрировать, что явления, наблюдаемые в описанных выше
экспериментах, проводимых в растворах, оказывают влияние на присутствующую в растворе ткань, проводили опыт, в котором чистые образцы ткани из хлопка погружали в модельную стирающую жидкость,
содержащую изделие, которое высвобождало в растворе 0,1 г красителя Direct Red 79. Количество красителя, абсорбированное испытуемым изделием, определяли путем вычисления ΔЕ. Интенсивность
подавления переноса красителя сопоставляли с действием поливинилпирролидона (PVP), стандартного ингибитора процесса переноса красителя. В приведенных данных чем меньше балл, тем лучше подавление (см.
табл.3).
Приведенные выше данные показывают, что макроциклические тетраамидные лиганды по изобретению не только проявляют высокое ингибирующее действие на процесс переноса красителя, но значительно превосходят по эффективности поливинилпирролидон, известный и эффективный ингибитор процесса переноса красителя.
Пример 30
Перенос красителя с ткани на ткань с
использованием красителя Acid Red 151
Эксперименты проводили в соответствии с примером 29, используя изделие, которое высвобождало 0,1 г красителя Acid Red 151 (см. табл.4).
В следующем примере действие FeDCB на горчицу и природный глинозем сопоставляли с таковым для системы, содержащей только перекись водорода. Соединения по изобретению проявляют превосходное специфическое в отношении красителя действие на горчицу, которая является заряженным катионным красителем.
Пример 31
Удаление пятен горчицы и грунта
Этот пример
демонстрирует удаление пятен при симулированных условиях бытовой стирки. Изделия с пятнами горчицы или природного глинозема стирали с 2 г жидкого стирального детергента All® и
оксидантной системы (либо перекись водорода, либо перекись водорода с катализатором по изобретению FeDCB). Условия стирки: средний уровень теплой воды, полоскание холодной водой с использованием
Terg-О-Tometer. Измеряли удаление пятен и вычисляли % пятноудаления (%SRE). Таким образом, предпочтительными являются более высокие баллы.
Результаты даны в табл. 5.
В следующем примере действие соединений по изобретению против повторного отложения загрязнения сопоставляли с таковым в контроле (никакого активаторного соединения) и в присутствии коммерчески доступного органического активатора отбеливания - тетраацетилэтилендиамина (TAED).
Пример 32
Сравнительное исследование действия против повторного отложения приведено
ниже.
Система - Повторное отложение
Контроль - 0
Контроль и FeDCB - 1,3
Контроль и TAED - 0,7
Повторное отложение загрязнения оценивали после
процесса стирки изделия, используя метод расчета отбеливания Stensby. Это исследование показывает, что случайные красители, разрушаясь в водной моющей жидкости, препятствуют повторному отложению
загрязнения на изделиях; и что воздействие по изобретению превосходит TAED, коммерчески доступный активатор.
Реферат
Изобретение относится к отбеливающему составу для отбеливания текстильных изделий и подавления абсорбции и адсорбции загрязнений или красителей на абсорбирующем или адсорбирующем субстрате. Описывается отбеливающий состав, содержащий эффективное количество источника окисляющего соединения и активатор отбеливателя, представляющий собой устойчивый к окислению макроциклический тетраамидолиганд формулы, где Х и Z - атом водорода, электронодонорная или электроноацепторная группа, R1 и R11 - алкил и циклоалкил, М - переходный металл со степенью окисления I, II, III, IV, V, VI, VII, VIII или выбирается из 3, 4, 5, 6, 7, 8, 9, 10 или 11 группы Периодической таблицы элементов, система IU РАС, Q - любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе, L, еcли присутствует, означает лабильный лиганд, а когда присутствует в водном растворе, L представляет собой воду. Описывается также способ отбеливания текстильных изделий и способ подавления абсорбции и адсорбции загрязнений или красителей на абсорбирующем или адсорбирующем субстрате. Изобретение позволяет повысить степень отбеливания и снизить степень переноса загрязнений или красителя на ткань за счет использования указанного активатора отбеливателя. 4 с. и 11 з.п. ф-лы, 3 ил., 5 табл.


Формула

где X и Z означают атом водорода, электронодонорную или электроноакцепторную группу;
R' и R" означают алкил и циклоалкил;
М означает переходный металл со степенью окисления I, II, III, IV, V или VI, VII или VIII или выбирается из 3, 4, 5, 6, 7, 8, 9, 10 или 11 группы Периодической таблицы элементов, система IUРАС;
Q означает любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе;
L, если присутствует, означает лабильный лиганд, а когда присутствует в водном растворе, L представляет собой воду.

где Х и Z означают атом водорода, электронодонорную или электроноакцепторную группу;
R, R' и R" означают любую комбинацию алкила и циклоалкила;
М означает переходный металл со степенью окисления I, II, III, IV, V, VI, VII или VIII или выбирается из 3, 4, 5, 6, 7, 8, 9, 10 или 11 группы Периодической таблицы элементов, система IUРАС;
Q означает любой противоион, который сбалансировал бы заряд соединения на стехиометрической основе;
L, если присутствует, означает лабильный лиганд и, когда присутствует в водном растворе, L представляет собой воду.
Комментарии