Покрытые частицы - RU2697455C2
Код документа: RU2697455C2
Чертежи






Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к покрытым частицам, образованным покрыванием поверхностей частиц основного материала углеродными частицами, полученными способом детонации.
УРОВЕНЬ ТЕХНИКИ
[0002] Наноразмерный алмаз (также называемый «наноалмазом») обладает большим числом замечательных свойств, таких как высокая твердость и чрезвычайно низкий коэффициент трения, и поэтому он уже используется в различных областях, и, как чрезвычайно перспективный новый материал, он продолжает исследоваться на предмет его новых применений.
[0003] Известно, что наноалмаз может быть синтезирован, например, с использованием реакции детонации высокоэнергетического взрывчатого вещества. Этот способ синтеза обычно называют детонационным способом, в котором выполняют детонацию только с исходным веществом, содержащим ароматическое соединение с тремя или более нитрогруппами, в качестве источника углерода, и атомы углерода, отщепившиеся от и высвобожденные из молекулы, составляющей исходное вещество, содержащее ароматическое соединение с тремя или более нитрогруппами, при реакции детонации образуют алмаз при высокой температуре и высоком давлении во время детонации (см., например, непатентный документ 1).
[0004] Производство наноалмазов детонационным способом до настоящего времени осуществлялось, например, в восточноевропейских странах, таких как Россия и Украина, в Соединенных Штатах Америки, Китае и т.д. Поскольку в этих странах в качестве сырья, содержащего соединение с тремя или более нитрогруппами, которое является источником углерода, доступны по низкой цене военные отходы, то использовались тринитротолуол (TNT), взрывчатая смесь TNT и гексогена (RDX: триметилентринитроамин) или октогена (HMX: циклотетраметилентетранитроамин) и т.п.
[0005] В настоящем изобретении высокоэнергетическое взрывчатое вещество означает вещество, способное осуществлять реакцию детонации, и его примеры могут включать не только сырье, содержащее соединение с тремя или более нитрогруппами, но и сырье, содержащее ароматическое соединение с двумя или менее нитрогруппами. В дополнение, взрывчатое вещество означает вещество, вызывающее внезапную реакцию сгорания, и может быть твердым или жидким при нормальной температуре и нормальном давлении.
[0006] Ожидается, что потребное количество наноалмазов будет все больше возрастать в будущем с развитием их применения. Однако, что касается производства с использованием военных отходов, содержащих соединение с тремя или более нитрогруппами, имеется некоторый предел в объеме производства. Следовательно, существует вероятность того, что поставки на мировом рынке в будущем могут сократиться. Тогда ожидается внутреннее производство. Однако на основе предыдущей оценки, сделанной авторами настоящего изобретения, было найдено, что вышеупомянутое сырье, содержащее соединение с тремя или более нитрогруппами, является достаточно дорогим, увеличивая производственные затраты и приводя тем самым к нерентабельности.
СПИСОК ЛИТЕРАТУРЫ
[0007] ПАТЕНТНАЯ ЛИТЕРАТУРА
Патентный документ 1: Японский патент № 4245310
Патентный документ 2: Японский патент № 5155975
НЕПАТЕНТНАЯ ЛИТЕРАТУРА
[0008] Непатентный документ 1: Yozo Kakudate (автор), «2-3. Dynamic High Pressure (Detonation Method)», Industrial Diamond Association of Japan (редактор), «Handbook of Diamond Technology», NGT, January 2007, pp. 28 to 33.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКИЕ ПРОБЛЕМЫ
[0009] Углеродные частицы, полученные способом детонации, содержат не только наноалмаз, но и углеродные примеси, включающие главным образом наноразмерный графитовый углерод (далее именуемый «нанографитом»), который является углеродным компонентом, не имеющим структуры алмаза. То есть, исходное вещество вызывает детонацию, вследствие чего исходное вещество разлагается до атомного уровня, и высвобождаемые из него атомы углерода без окисления агрегируются в твердом состоянии, образуя углеродные частицы. Во время детонации исходное вещество находится в состоянии высокой температуры и высокого давления из-за реакции разложения. Однако исходное вещество немедленно расширяется и охлаждается. Этот процесс перехода от состояния высокой температуры и высокого давления к состоянию пониженного давления и охлаждения происходит за очень короткий промежуток времени по сравнению с дефлаграцией, которая представляет собой явление более медленного взрыва, чем детонация или полное сгорание, и поэтому нет времени, когда агрегированный углерод мог бы расти в значительной степени. Поэтому образуется
наноразмерный алмаз. Когда высокоэнергетическое взрывчатое вещество (такое как взрывчатая смесь TNT и RDX), известное как типичное высокоэнергетическое взрывчатое вещество, вызывающее детонацию, используется в качестве исходного вещества (сырья), давление во время детонации становится достаточно высоким, чтобы позволить образующимся частицам углерода содержать много наноалмазов в соответствии с термодинамической равновесной фазовой диаграммой углерода. С другой стороны, атомы углерода, которые не формируют алмазную структуру, становятся наноразмерным графитовым углеродом (нанографитом) или т.п.
[0010] Из углеродных частиц нанографит и т.п., отличающиеся от наноалмаза, рассматриваются как нежелательные и мешающие использовать превосходные свойства наноалмаза. Следовательно, предшествующий уровень техники фокусировался на том, как в максимально возможной степени устранить углеродные примеси, такие как нанографит, чтобы тем самым очистить наноалмаз с помощью различных способов очистки или химической обработки (см., например, патентный документ 1 или 2). Однако нанографит, например, имеет более низкую твердость, чем наноалмаз, и более высокую электропроводность, чем наноалмаз. В дополнение к таким отличающимся от наноалмаза физическим свойствам, нанографит имеет ту особенность, что с ним могут быть соединены различные виды атомов или функциональных групп, отличающиеся от углерода, за счет чего могут быть приданы новые функции. Соответственно, нанографит привлек внимание как перспективный новый материал, способный обеспечивать различные свойства при его применении в отдельности или в смеси с наноалмазом.
[0011] Задача настоящего изобретения состоит в том, чтобы предложить новый материал, использующий углеродные частицы, содержащие наноразмерный графитовый углерод и алмаз и полученные детонационным способом с использованием исходного вещества, содержащего ароматическое соединение с двумя или менее нитрогруппами, которое может стабильно поставляться по низкой цене.
РЕШЕНИЕ ПРОБЛЕМ
[0012] В покрытой частице по настоящему изобретению, которая может решить вышеупомянутую(ые) проблему (проблемы), поверхность частицы основного материала покрыта углеродными частицами, содержащими графит турбостратной структуры, наноалмазы и/или углеродные нанотрубки, фуллерены и полученными с помощью следующих стадий: расположение взрывчатого вещества со скоростью детонации 6300 м/с или выше на периферии исходного вещества (сырья), содержащего ароматическое соединение с двумя или менее нитрогруппами; и детонирование этого взрывчатого вещества.
[0013] Предпочтительно, чтобы углеродные частицы были фторированы.
[0014] Настоящее изобретение охватывает функциональный материал, в котором покрытая частица поддерживается на поверхности материала подложки.
[0015] Покрытая частица в настоящем изобретении может быть получена способом, включающим в себя следующие стадии: приготовление частицы основного материала; расположение взрывчатого вещества со скоростью детонации 6300 м/с или выше на периферии исходного вещества (сырья), содержащего ароматическое соединение с двумя или менее нитрогруппами; детонирование этого взрывчатого вещества с получением углеродных частиц, содержащих графит турбостратной структуры, наноалмазы и/или углеродные нанотрубки, фуллерены; и покрывание поверхности упомянутой частицы основного материала полученными углеродными частицами способом механического объединения.
[0016] Углеродные частицы могут быть подвергнуты обработке фторированием, а затем поверхность частицы основного материала может быть покрыта углеродными частицами способом механического объединения.
[0017] Функциональный материал по настоящему изобретению может быть получен нанесением покрытой частицы, полученной вышеупомянутым детонационным способом, на поверхность материала подложки. Функциональный материал по настоящему изобретению может быть получен нанесением покрытой частицы, полученной вышеупомянутым детонационным способом, на поверхность материала подложки, с последующей обработкой фторированием. Покрытая частица может наноситься на поверхность материала подложки с помощью, например, термонапыления, прокатки или металлизации материала подложки.
ПОЛЕЗНЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0018] В настоящем изобретении устойчивая детонация может быть создана даже с помощью детонационного способа, использующего недорогое сырье, которое является исходным веществом, содержащим ароматическое соединение с двумя или менее нитрогруппами, с тем, чтобы получить углеродные частицы, содержащие наноразмерный графитовый углерод и алмаз. Когда поверхности частиц основного материала покрываются полученными таким образом углеродными частицами, может быть получен новый материал.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
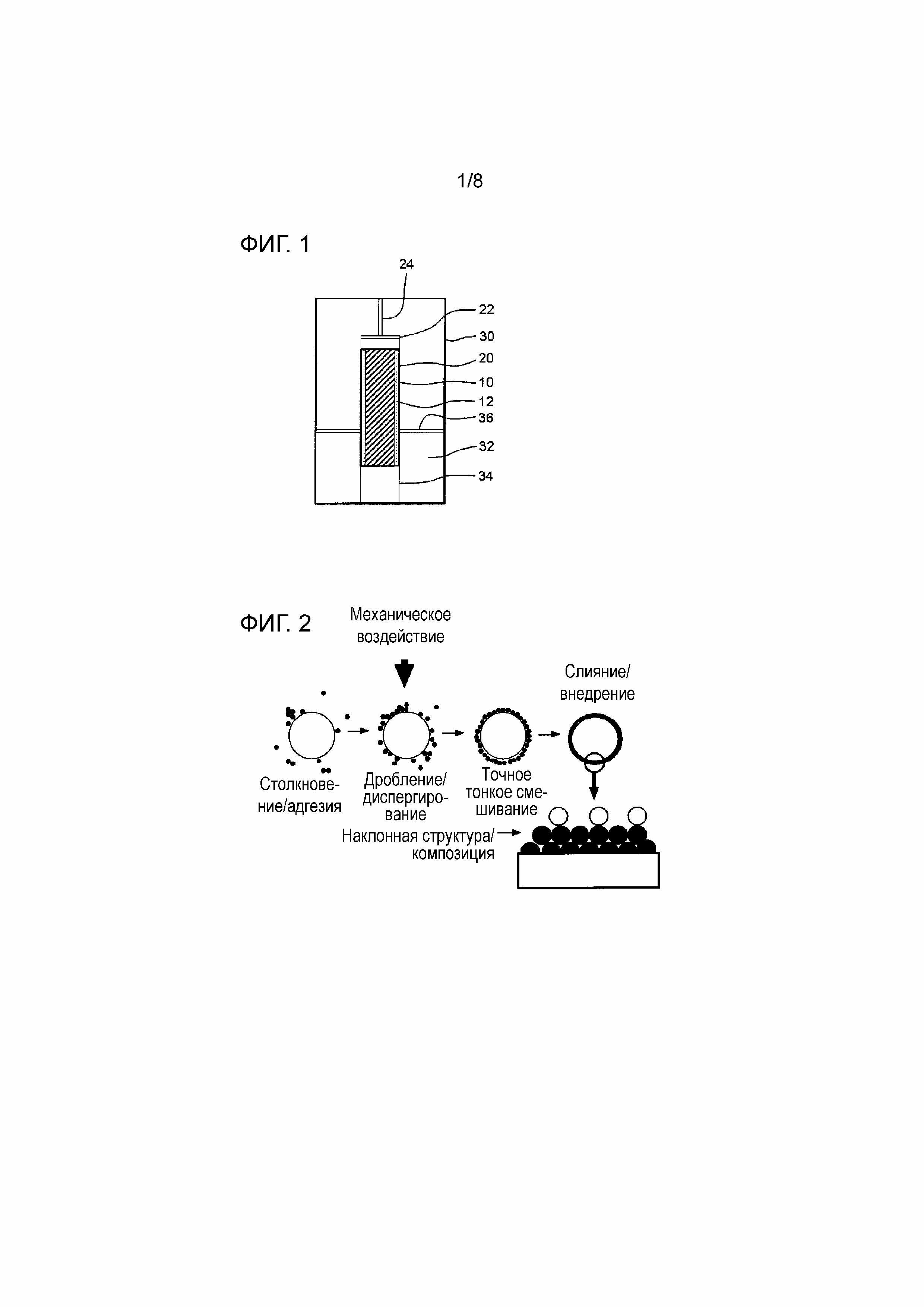
[0019] [Фиг. 1] Фиг. 1 представляет собой изображение в разрезе, схематично показывающее пример взрывного устройства, используемого в способе получения по настоящему изобретению.
[Фиг. 2] Фиг. 2 представляет собой схематическую диаграмму для пояснения процесса механического объединения.
[Фиг. 3] Фиг. 3 показывает снятые с помощью просвечивающего электронного микроскопа (ПЭМ) фотографии углеродных частиц, полученных в Экспериментальном примере 3.
[Фиг. 4] Фиг. 4 показывает снятые с помощью просвечивающего электронного микроскопа (ПЭМ) фотографии углеродных частиц, полученных в Экспериментальном примере 3.
[Фиг. 5] Фиг. 5 – рентгенодифратограмма углеродных частиц, полученных в Экспериментальном примере 3.
[Фиг. 6] Фиг. 6 – график, показывающий калибровочную кривую, используемую для определения доли содержания алмаза в углеродных частицах.
[Фиг. 7] Фиг. 7 показывает фотографии, снятые с помощью просвечивающего электронного микроскопа (ПЭМ) до и после фторирования углеродных частиц, полученных в Экспериментальном примере 4.
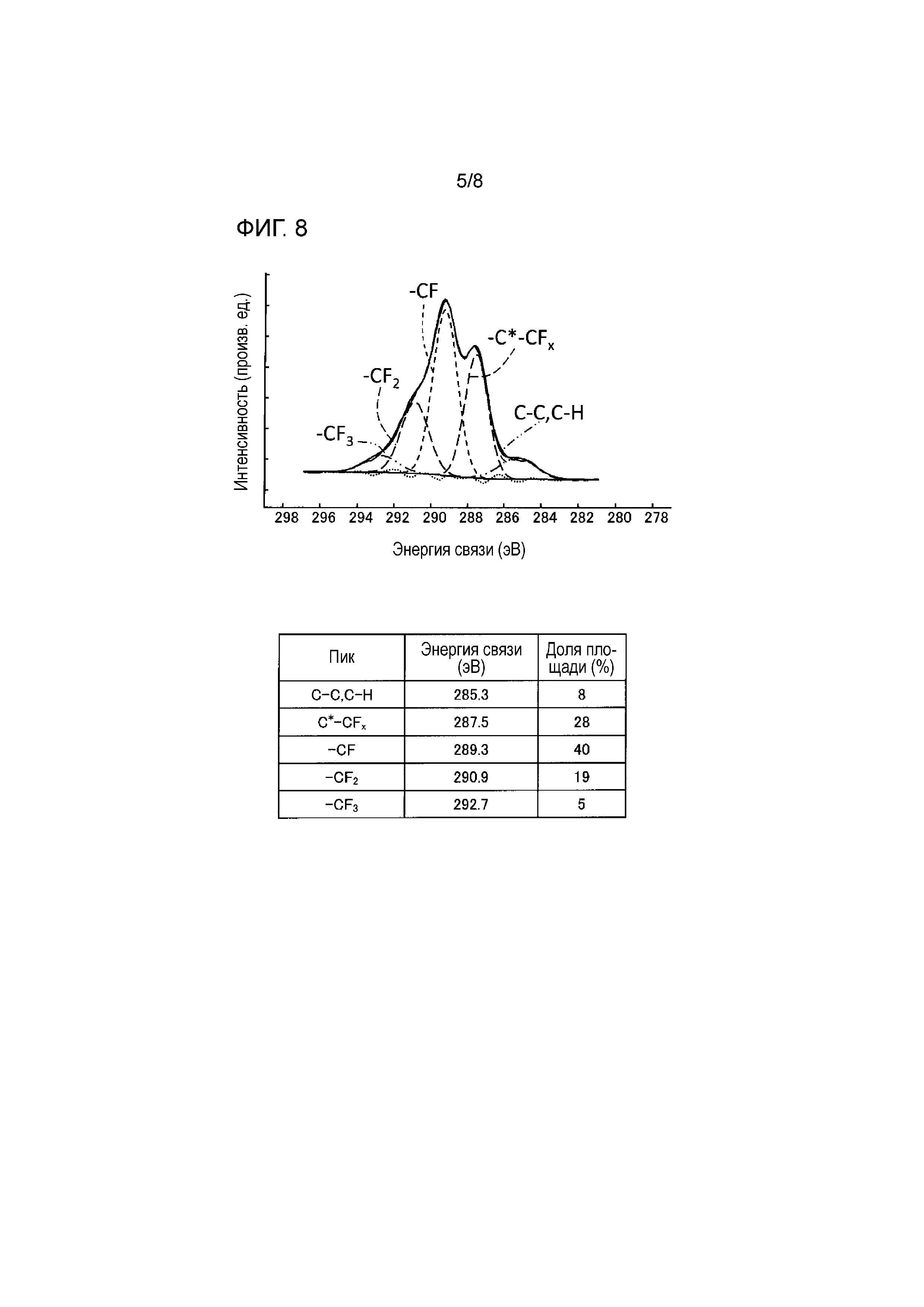
[Фиг. 8] Фиг. 8 представляет собой схематическую диаграмму, показывающую узкополосный фотоэлектронный спектр C1s, подвергнутый разделению пиков.
[Фиг. 9] Фиг. 9 показывает снятую с помощью сканирующего электронного микроскопа с автоэлектронной эмиссией (FE-SEM) фотографию поверхностей покрытых частиц, полученных в примере.
[Фиг. 10] Фиг. 10 показывает фотографию, на которой часть поверхностного слоя покрытой частицы, полученной в примере, была вырезана устройством с фокусированным ионным пучком (ФИП) так, чтобы внутренняя частица уретановой смолы могла быть сравнена с углеродной частицей, покрывающей эту частицу уретановой смолы.
[Фиг. 11] Фиг. 11 показывает снятые с помощью сканирующего электронного микроскопа с автоэлектронной эмиссией (FE-SEM) фотографии, на которых изображено сечение покрытой частицы, полученной в примере.
[Фиг. 12] Фиг. 12 показывает фотографии, на которых сняты лист нержавеющей стали SUS304, использовавшийся в качестве материала подложки, и полученный функциональный материал.
[Фиг. 13] Фиг. 13 показывает снятые с помощью сканирующего электронного микроскопа с автоэлектронной эмиссией (FE-SEM) фотографии сечения функционального материала, показанного на Фиг. 12, который был разрезан тонким резаком.
[Фиг. 14] Фиг. 14 представляет собой график, показывающий коэффициенты трения образцов, полученных в примерах.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0020] Авторы настоящего изобретения исследовали способ, по которому углеродные частицы, содержащие наноразмерный графитовый углерод и алмаз, могут быть получены с малыми затратами детонационным способом. В результате авторы настоящего изобретения нашли, что недорогое производство может быть достигнуто с помощью способа получения, включающего в себя стадию расположения взрывчатого вещества со скоростью детонации 6300 м/с или выше на периферии исходного вещества (сырья), содержащего ароматическое соединение с двумя или менее нитрогруппами, и стадию детонирования взрывчатого вещества. Поэтому авторы настоящего изобретения подали заявку на японский патент № 2013-273468. После этого, в результате дальнейших обширных исследований авторы изобретения нашли, что покрытые частицы, в которых поверхности частиц основного материала покрыты углеродными частицами, полученными вышеупомянутым способом получения, полезны в качестве нового материала. Таким образом авторы изобретения создали настоящее изобретение.
[0021] Сначала будет приведено описание способа получения вышеупомянутых углеродных частиц. Способ получения углеродных частиц, который будет описан ниже, является тем же самым, что и в вышеупомянутой заявке на японский патент № 2013-273468.
<<Способ получения углеродных частиц>>
[0022] В качестве покрытых частиц, предназначенных для использования в настоящем изобретении, детонационным способом получают углеродные частицы, содержащие наноразмерный графитовый углерод и алмаз. Более конкретно, углеродные частицы могут быть произведены способом получения, включающим в себя стадию расположения взрывчатого вещества со скоростью детонации 6300 м/с или выше на периферии исходного вещества, содержащего ароматическое соединение с двумя или менее нитрогруппами, а также стадию детонирования взрывчатого вещества.
[0023] В вышеупомянутом способе получения сначала располагают взрывчатое вещество со скоростью детонации 6300 м/с или выше на периферии исходного вещества, содержащего ароматическое соединение с двумя или менее нитрогруппами. Ароматическое соединение с двумя или менее нитрогруппами является содержится в исходном веществе в качестве источника углерода для детонационного способа. Взрывчатое вещество со скоростью детонации 6300 м/с или выше является веществом, вызывающим устойчивую детонацию с получением углеродных частиц из исходного вещества. Когда молекула, образующая взрывчатое вещество, содержит атомы углерода, взрывчатое вещество может служить источником углерода вместе с исходным веществом.
[0024] Примеры ароматического соединения с двумя или менее нитрогруппами могут включать соединение, имеющее структуру, в которой 0, 1 или 2 атом(а) водорода ароматического кольца, такого как бензол, толуол, ксилол, нафталин или антрацен, замещены нитрогруппой(ами).
[0025] Вышеупомянутое ароматическое соединение может иметь иной заместитель, отличающийся от нитрогрупп. Примеры таких заместителей могут включать алкильную группу, гидроксильную группу, гидроксиалкильную группу, аминогруппу, галогеновую группу и т.п.
[0026] Может иметь место случай, когда присутствуют позиционные изомеры в зависимости от взаимного расположения нитрогрупп или заместителей. Однако все позиционные изомеры могут использоваться в вышеупомянутом способе получения. Например, когда ароматическое соединение является нитротолуолом, возможны 3 вида изомеров, т.е. 2-, 3- и 4-нитротолуолы.
[0027] Примеры такого ароматического соединения с двумя или менее нитрогруппами могут включать бензол, толуол, ксилол, нафталин, антрацен, нитробензол, нитротолуол, нитроксилол, нитронафталин, нитроантрацен, динитробензол, динитротолуол, динитроксилол, динитронафталин, динитроантрацен и т.д. Каждое из этих ароматических соединений с двумя или менее нитрогруппами может использоваться по отдельности, или же два или более вида этих ароматических соединений с двумя или менее нитрогруппами могут использоваться вместе.
[0028] Предпочтительно, чтобы ароматическое соединение с двумя или менее нитрогруппами являлось соединением, имеющим структуру, в которой 1 или 2 атом(а) водорода ароматического кольца замещены нитрогруппой(ами). Из таких ароматических соединений с двумя или менее нитрогруппами, например, динитротолуол (DNT), динитробензол (DNB), динитроксилол (DNX) и т.п. являются предпочтительными с точки зрения их низких температур плавления и легкости формования.
[0029] Исходное вещество может содержать сырье, содержащее соединение с тремя или более нитрогруппами, в дополнение к ароматическому соединению с двумя или менее нитрогруппами. Сырье, содержащее соединение с тремя или более нитрогруппами, обычно является, например, нитросоединением, используемым для проведения взрыва. Примеры таких нитросоединений могут включать тринитротолуол (TNT), гексоген (RDX; циклотриметилентринитроамин), октоген (HMX; циклотетраиметилентетранитроамин), тетранитрат пентаэритрита (PETN), тетрил (тетранитрометиланилин) и т.п. Каждое из этих нитросоединений может использоваться по отдельности, или же два или более вида этих нитросоединений могут использоваться вместе.
[0030] Доля содержания ароматического соединения с двумя или менее нитрогруппами в вышеупомянутом исходном веществе обычно составляет 50 мас.% или более, предпочтительно 80 мас.% или более, предпочтительнее 90 мас.% или более, а еще предпочтительнее 95 мас.% или более, по отношению к общей массе исходного вещества. Когда ароматическое соединение с двумя или менее нитрогруппами, которое является недорогим сырьем, являющимся исходным веществом, содержащим ароматическое соединение с двумя или менее нитрогруппами, содержится с большой долей, доля содержания соединения с тремя или более нитрогруппами, которое является дорогим сырьем, может быть уменьшена. Следовательно, наиболее предпочтительно, доля содержания ароматического соединения с двумя или менее нитрогруппами составляет 100 мас.% в качестве ее верхнего предела. Однако ее верхний предел может предпочтительно составлять примерно 99 мас.% или примерно 98 мас.%.
[0031] Скорость детонации взрывчатого вещества, расположенного на периферии вышеупомянутого исходного вещества, должна быть более высокой, чем скорость детонации исходного вещества. То есть, из содержащихся в исходном веществе ароматических соединений с двумя или менее нитрогруппами, например, динитротолуол (DNT, истинная плотность: 1,52 г/см3, точка плавления: 67-70°C), который является недорогим и легким в применении, является настолько устойчивым, что его невозможно легко детонировать. Однако его скорость детонации может быть оценена как примерно 6000 м/с, если его удается детонировать. Следовательно, необходимо сделать скорость детонации взрывчатого вещества не ниже этой скорости. Скорость детонации типичного взрывчатого вещества обычно составляет 10000 м/с или ниже. Поэтому в настоящем изобретении скорость детонации взрывчатого вещества составляет 6300 м/с или выше, а качестве ее верхнего предела, она предпочтительно составляет 10000 м/с или ниже. Что касается скорости детонации DNT, то можно сослаться на публикацию Combustion and Flames, Vol. 14 (1970), pp. 145.
[0032] Скорость детонации означает скорость распространения детонации, когда детонация вызывается взрывчатым веществом.
[0033] Из таких взрывчатых веществ примеры твердых могут включать TNT, RDX, HMX, PETN, тетрил, взрывчатую смесь (например, Состав B), содержащую RDX и TNT в качестве своих главных компонентов, взрывчатую смесь (например, октол), содержащую HMX и TNT в качестве своих главных компонентов, и т.д.
[0034] В качестве взрывчатого вещества может быть также использовано жидкое высокоэнергетическое взрывчатое вещество. Когда в качестве взрывчатого вещества используется жидкое высокоэнергетическое взрывчатое вещество, высока степень
свободы в выборе формы, увеличение размера является легким, а технологичность или безопасность может быть повышена по сравнению со случаем использования твердого высокоэнергетического взрывчатого вещества. Примеры таких жидких высокоэнергетических взрывчатых веществ могут включать смесь гидразина (включая гидрат гидразина) и нитрата гидразина, смесь гидразина и нитрата аммония, смесь гидразина, нитрата гидразина и нитрата аммония, нитрометан, смесь гидразина и нитрометана и т.п.
[0035] Из вышеупомянутых взрывчатых веществ легко формуемый TNT, Состав B, содержащий TNT в качестве своего главного компонента, или т.п. являются предпочтительными в качестве твердых взрывчатых веществ благодаря их низкой точке плавления. Каждое из вышеупомянутых взрывчатых веществ может использоваться по отдельности, или же два или более вида этих взрывчатых веществ могут использоваться вместе. Свойства и скорости детонации типичных взрывчатых веществ показаны в следующей Таблице 1. Взрывчатые вещества, способные вызывать устойчивую детонацию, показаны в следующей Таблице 1.
[0036] Таблица 1
1) Плотность во время измерения скорости детонации
2) Взрывчатая смесь из 59,5 мас.% RDX, 39,5 мас.% TNT и 1,0 мас.% воска
3) Взрывчатая смесь из 75 мас.% HMX и 25 мас.% TNT
4) Жидкое высокоэнергетическое взрывчатое вещество на основе гидразина, в котором нитрат гидразина (H2N-NH2⋅HNO3) и гидрат гидразина (H2N-NH2⋅H2O) смешаны в массовом соотношении 3:1
[0037] Скорость детонации нитрометана взята из публикации Kusakabe and Fujiwara «Studies regarding Detonation of Liquid High Explosives (First Report)», Journal of the Industrial Explosives Society, Japan, Vol. 40, No. 2 (1979), p. 109. Скорость детонации NH+HH (нитрата гидразина (H2N-NH2⋅HNO3) и гидрата гидразина (H2N-NH2⋅H2O)) взята из публикации Kusakabe et al. «Studies regarding Detonation of Liquid High Explosives (Third
Report)», Journal of the Industrial Explosives Society, Japan, Vol. 41, No. 1 (1980), p. 23. Скорости детонации других веществ, отличающихся от нитрометана и NH+HH, взяты из публикации LASL Explosive Properties Date, ed. Gibbs, T. R. and Propolato, A., University of California Press, Berkeley, Los Angels, London, 1980.
[0038] Используемые количества вышеупомянутого исходного вещества и вышеупомянутого взрывчатого вещества могут регулироваться индивидуально и подходящим образом в зависимости от желаемого количества углеродных частиц. Хотя оно конкретно не ограничено, их массовое соотношение (взрывчатое вещество/исходное вещество) предпочтительно составляет 0,1 или более, а предпочтительнее 0,2 или более. В дополнение к этому, это массовое соотношение предпочтительно составляет 1 или менее, предпочтительнее 0,9 или менее, а еще предпочтительнее 0,8 или менее. Когда соотношение используемых количеств составляет менее 0,1, невозможно вызвать реакцию детонации, достаточную для создания углеродных частиц. Поэтому выход может уменьшиться. С другой стороны, когда соотношение используемых количеств превышает 1, взрывчатого вещества используется больше, чем необходимо. Поэтому производственные затраты могут увеличиться.
[0039] Выше был описан способ получения углеродных частиц.
[0040] Далее со ссылкой на чертежи будут подробно описаны варианты осуществления этого способа получения углеродных частиц. Фиг. 1 представляет собой изображение в разрезе, схематично показывающее пример взрывного устройства для использования в вышеупомянутом способе получения. Взрывное устройство, показанное на Фиг. 1, является всего лишь примерным. Оно не предназначено для какого-либо ограничения настоящего изобретения.
[0041] Сначала на периферии исходного вещества 10 (сырья) размещают взрывчатое вещество 12. При размещении взрывчатого вещества 12 на периферии исходного вещества 10 предпочтительно симметрично располагать исходное вещество 10 и взрывчатое вещество 12 таким образом, чтобы высокая температура и высокое давление, связанные с ударной волной, создаваемой детонацией взрывчатого вещества 12, прикладывались к исходному веществу 10 как можно более равномерно, а именно таким образом, чтобы обеспечивалась симметрия формы взрыва. Например, в случае (a), в котором исходное вещество 10 и взрывчатое вещество 12 являются твердыми, исходное вещество 10 и взрывчатое вещество 12 могут быть залиты или запрессованы в цилиндрические разъемные пресс-формы так, чтобы приготовить концентрические столбчатые формованные тела. В случае (b), в котором исходное вещество 10 является твердым, а взрывчатое вещество 12 является жидким высокоэнергетическим взрывчатым
веществом, исходное вещество 10 может быть залито или запрессовано так, чтобы приготовить столбчатое формованное тело, и это формованное тело может быть размещено в центральной части внутри цилиндрического контейнера строго по его оси, после чего жидкое высокоэнергетическое взрывчатое вещество может быть залито в периферийное пространство. В случае (c), в котором исходное вещество 10 является жидким, а взрывчатое вещество 12 является твердым, взрывчатое вещество 12 может быть залито или запрессовано в концентрическое полое столбчатое формованное тело, и жидкое исходное вещество 10 может быть залито внутрь него. Контейнер 20, вмещающий исходное вещество 10 и взрывчатое вещество 12, будет именоваться в дальнейшем «взрывной контейнер». В качестве взрывного контейнера 20 предпочтительно использовать контейнер из синтетической смолы, такой как акриловая смола, поскольку это позволяет предотвратить загрязнение примесями, такими как металлы.
[0042] В вышеупомянутом способе получения взрывчатое вещество 12 затем детонируют с образованием углеродных частиц из исходного вещества 10. Ударная волна, создаваемая в результате реакции детонации взрывчатого вещества 12, распространяется к исходному веществу 10, исходное вещество 10 сжимается этой ударной волной, вызывая детонацию, и атомы углерода, отщепившиеся от и высвободившиеся из органических молекул, составляющих исходное вещество 10, превращаются в углеродные частицы, содержащие графитовый углерод и наноалмаз.
[0043] Детонация может быть осуществлена в открытой системе или в закрытой системе. Для того, чтобы осуществить детонацию в открытой системе, детонация может выполняться, например, в подземных полостях или галереях, образующихся при подземных работах. Детонация в закрытой системе предпочтительно выполняется в таком состоянии, когда сделанная из металла камера заполнена исходным веществом и взрывчатым веществом. Состояние, в котором сделанная из металла камера заполнена исходным веществом и взрывчатым веществом, является, например, таким состоянием, при котором формованное тело исходного вещества и взрывчатого вещества или взрывной контейнер, содержащий исходное вещество и взрывчатое вещество, подвешивают в этой камере. Предпочтительно выполнять детонацию в закрытой системе, потому что это помогает предотвратить разброс остатка по большой площади. Камера, используемая для выполнения детонации, в дальнейшем будет упоминаться как «взрывная камера». Когда атмосфера во взрывной камере практически не содержит кислорода при появлении детонации, реакция окисления углеродной фракции может быть ингибирована. Поэтому выход может быть увеличен. Для того, чтобы получить такую атмосферу, например, газ во взрывной камере может быть замещен инертным газом, таким как
газообразный азот, газообразный аргон или газообразный диоксид углерода; взрывная камера может быть вакуумирована до давления примерно от -0,1 до -0,01 МПа маном. (здесь «маном.», стоящее после единицы давления, означает, что давление является манометрическим, причем то же применимо ниже); или после удаления воздуха (кислорода) из взрывной камеры путем вакуумирования такой инертный газ может быть введен во взрывную камеру до слабого положительного давления примерно от +0,000 до +0,001 МПа маном. Камера не ограничена сделанной из металла камерой при условии, что эта камера имеет достаточно высокую прочность для того, чтобы выдержать детонацию. Например, камера может быть выполнена из бетона.
[0044] Кроме того, предпочтительно располагать охладитель вокруг исходного вещества и взрывчатого вещества во взрывной камере. Когда используется охладитель, образовавшийся алмаз может быть быстро охлажден для предотвращения его фазового перехода в графитовый углерод. Для того чтобы расположить охладитель, например, вышеупомянутое формованное тело или взрывной контейнер 20 могут быть размещены в охлаждающем контейнере 30, и охладитель 32 может быть загружен в зазор между охлаждающим контейнером 30 и формованным телом или взрывным контейнером 20. При этом, когда охладитель 32 является веществом, которое может по существу предотвратить образование окислительного вещества, такого как кислород или озон, реакция окисления углеродной фракции может быть ингибирована. Поэтому выход может быть увеличен. Для того чтобы получить подобный охладитель 32, например, может быть удален газообразный кислород, растворенный в охладителе 32, или же может использоваться охладитель 32, который не содержит составляющих его элементов, образующих какое-либо окисляющее вещество, такое как кислород или озон. Примеры таких охладителей 32 могут включать воду, галогенированные алкилы (такие как хлорфторуглероды и тетрахлорид углерода) и т.п. Вода является особенно предпочтительной, потому что она по существу не оказывает неблагоприятного влияния на окружающую среду.
[0045] Хотя взрывчатое вещество 12 обычно взрывается при использовании детонатора или детонирующего шнура, для того чтобы более надежно вызвать детонацию, может использоваться усилитель 22 детонатора между взрывчатым веществом 12 и детонатором или детонирующим шнуром. В этом случае после того, как усилитель 22 детонатора и детонатор или детонирующий шнур 24 присоединены к формованному телу или взрывному контейнеру 20, они, например, загружаются во взрывную камеру. Примеры таких усилителей 22 детонатора могут включать в себя Состав C-4, SEP производства компании Asahi Kasei Chemicals Corporation и т.п.
[0046] При использовании охладителя 32 предпочтительно, чтобы
вышеупомянутое формованное тело или взрывной контейнер 20 размещались во непроницаемом контейнере (например, мешке из олефиновой синтетической смолы, такой как полиэтилен или полипропилен, в качестве сырья), чтобы охладитель 32 не мог проникнуть во взрывной контейнер 20. После установки таким образом, когда взрывчатое вещество 12 взрывается с осуществлением детонации, в остатке получаются углеродные частицы, содержащие графитовый углерод и алмаз.
[0047] В вышеупомянутом способе получения остаток, полученный на стадии детонации, может необязательно содержать в качестве примесей взорванные обломки, такие как обломки контейнера, проводов или проволоки. В таком случае предпочтительно предусмотреть стадию удаления обломков из остатка, полученного после стадии детонации, чтобы извлечь углеродные частицы. На этой стадии извлечения углеродных частиц, например, когда выполняется обработка классификацией/очисткой, углеродные частицы могут быть получены в виде сухого порошка, имеющего желаемый размер частиц. Как правило, сначала, после удаления грубых обломков из остатка, полученного на стадии детонации, получившийся остаток просеивают на сите или т.п. и разделяют на прошедший через сито материал (просев) и остаток на сите, и просев извлекают. Остаток на сите может быть измельчен, а затем снова классифицирован. Воду отделяют от полученного в итоге просева, чтобы приготовить сухой порошок. При этом размер отверстий сита подбирают должным образом, и обработку классификацией/очисткой повторяют. Тогда просев сита с отверстиями, соответствующими желаемому размеру частиц, может быть получен в качестве продукта. Более подробно, например, когда детонация выполняется во взрывной камере с использованием воды в качестве охладителя 32, содержащую остаток воду извлекают, с последующим разделением отстаиванием. После удаления грубых обломков надосадочную жидкость извлекают в качестве жидких отходов, а осадок классифицируют на сите или т.п., чтобы получить просев. Возможно, часть образовавшихся углеродных компонентов может прилипать к обломкам. Поэтому остаток на сите измельчают (дробят) и разделяют посредством ультразвуковой вибрации или т.п. и снова классифицируют на сите и т.п. Например, остаток на сите с размером отверстий примерно 100 мкм является главным образом обломками после взрыва, такими как обломки взрывного контейнера 20, проводов или проволоки. Следовательно, такой остаток на сите утилизируют как промышленные отходы после извлечения. Из частиц, прошедших через сито с отверстием примерно 100 мкм, остаток на сите с отверстием примерно 32 мкм может быть измельчен и отделен посредством ультразвуковой вибрации или т.п. и снова классифицирован на сите и т.п. Предпочтительно в качестве конечного продукта извлекать посредством этих операций просев, прошедший через сито с размером
отверстий примерно 32 мкм. Что касается извлеченного продукта, воду отделяют от него посредством центрифугирования или т.п., а затем его сушат, получая порошок углеродных частиц с желаемым их размером.
[0048] Например, когда в качестве взрывного контейнера 20 используется контейнер из акриловой смолы, возможно, получаемый на стадии детонации остаток может быть загрязнен частицами или порошком акриловой смолы. В этом случае акриловая смола может быть удалена, например, вымывания акриловой смолы ацетоном.
[0049] Кроме того, в некоторых приложениях иногда нежелательно загрязнение металлом, таким как железо. В таком случае, например, металл, такой как железо, может быть удален с помощью обработки горячей концентрированной азотной кислотой.
[0050] Полученный порошок представляет собой наноразмерные углеродные частицы, содержащие много графитового углерода, а также наноалмаз. Однако в некоторых применениях требуется использовать превосходные свойства алмаза.
[0051] В углеродных частицах, полученных вышеупомянутым способом получения, массовое соотношение G/D составляет 2,5 или более, где G обозначает массу графитового углерода, а D обозначает массу алмаза. Далее будут подробно описаны состав и физические свойства углеродных частиц для использования в настоящем изобретении.
[0052] Углеродные частицы для использования в настоящем изобретении могут быть охарактеризованы долей содержания углеродного компонента, выражаемой массовым соотношением. Как было описано выше, исходное вещество вызывает детонацию, вследствие чего исходное вещество разлагается до атомного уровня, и высвобождаемые из него атомы углерода без окисления агрегируются в твердом состоянии, образуя углеродные частицы. Во время детонации исходное вещество находится в состоянии высокой температуры и высокого давления из-за реакции разложения. Однако исходное вещество немедленно расширяется и охлаждается. Этот процесс перехода от состояния высокой температуры и высокого давления к состоянию пониженного давления и охлаждения происходит за очень короткий промежуток времени по сравнению с обычным горением или дефлаграцией, которая представляет собой явление более медленного взрыва, чем детонация, и поэтому нет времени, когда агрегированный углерод мог бы расти в значительной степени. Поэтому формируются наноразмерные углеродные частицы.
[0053] Когда в качестве исходного вещества используется высокоэнергетическое взрывчатое вещество, такое как взрывчатая смесь TNT и RDX, известная как типичное высокоэнергетическое взрывчатое вещество, вызывающее детонацию, давление во время
детонации становится достаточно высоким, чтобы позволить образующимся частицам углерода содержать много наноалмазов, чего легко ожидать исходя из фазовой диаграммы состояния термодинамического равновесия углерода. С другой стороны, когда в качестве исходного вещества используется невысокоэнергетическое взрывчатое вещество, давление во время детонации является недостаточно высоким для того, чтобы синтезировать алмаз. Поэтому получаются наноразмерные углеродные частицы, отличающиеся от алмаза. Углеродные частицы содержат много наноразмерного графитового углерода.
[0054] Таким образом, соотношением между наноалмазом и нанографитом можно управлять давлением во время детонации исходного вещества. То есть, при использовании исходного вещества, которое не является высокоэнергетическим взрывчатым веществом, доля содержания нанографита может быть увеличена. Однако когда давление во время детонации исходного вещества является более низким, чем у высокоэнергетического взрывчатого вещества, становится трудно детонировать исходное вещество, или даже если исходное вещество удается детонировать, вероятно возникновение такого явления, что детонация может прерываться. Это означает, что трудно устойчиво детонировать одно только исходное вещество. Следовательно, когда давление во время детонации исходного вещества является низким, вызывающее детонацию взрывчатое вещество должно быть расположено на периферии исходного вещества, чтобы тем самым гарантированно детонировать исходное вещество. В любом случае, в дополнение важно выбирать исходное вещество, имеющее такой состав, который не окисляет высвобождающийся углерод.
[0055] Кроме того, предпочтительно, чтобы окисляющее вещество, такое как кислород или озон, которое может окислить высвобожденный углерод с образованием газа, такого как CO или CO2, было удалено из системы детонации в максимально возможной степени.
[0056] В дополнение к этому, когда детонируют исходное вещество, содержащее соединение с тремя или более нитрогруппами, или ароматическое соединение с двумя или менее нитрогруппами, предполагается, что производятся любые разновидности наноразмерных углеродных частиц, такие как наноалмаз, нанографит, тонкодисперсные углеродные нанотрубки, фуллерены и т.д.
[0057] Из литературы (Satoshi Tomita et al., «Diamond nanoparticles to carbon onions transformation: X-ray diffraction studies», Carbon 40, pp.1469-1474 (2002), Dilip K. Singh et al, «Diameter dependence of interwall separation and strain in multiwalled carbon nanotubes probed by X-ray diffraction and Raman scattering studies», Diamond & Related Materials 19,
pp.1281-1288 (2010), и т.д.) и полученных результатов рентгенодифракционных данных по детонационным наноалмазам, описанных ниже, можно предположить, что пик, в котором дифракционный угол 2θ рентгенодифракционных данных, измеренных с помощью трубки Cu(Kα), находится вблизи 24-26° (именуемый в дальнейшем «пиком около 26°»), происходит от наноуглеродного вещества, состоящего главным образом из слоистой структуры sp2 углерода. В дополнение к этому, появляется пик в этой области и в отношении (многослойных) углеродных нанотрубок из двух слоев, трех слоев или т.п.
[0058] Результаты наблюдения изображения решетки на снятых в просвечивающем электронном микроскопе (ПЭМ) фотографиях углеродных частиц, полученных в описываемом ниже Экспериментальном примере 3, показаны на Фиг. 4. На Фиг. 4 наблюдались два вида форм изображений решетки. А именно, наблюдались круглая сферическая форма и слоистая форма (графит), показанные соответственно символами D и G. Обе они являются наноразмерными, и принимая во внимание их количества, обе они предполагаются частицами, содержащими углерод в качестве главного компонента. Поскольку наблюдаемые на них частицы углерода предполагаются наноалмазом и графитовым углеродом, измеряли и сравнивали их период решетки и межплоскостное расстояние. Что касается шкалы (5 нм и 10 нм) и увеличения ПЭМ, то в качестве стандартного образца (эталона) используется образец, в котором многослойная пленка SiGe прикреплена к монокристаллу Si, и при высоком увеличении производится калибровка на основе межплоскостного расстояния в 3,1355 Å между плоскостями 111 Si. Эта операция калибровки выполнялась каждый месяц с подтверждением того, что погрешность измерения составляет не более 5%.
[0059] В алмазе (символ D), снятом в той же самой области на Фиг. 4, наблюдали плоскость 111 D, и результат измерения периода решетки составил 2,11 Å. Обычно говорят, что период решетки по плоскостям 111 D в кубическом алмазе составляет 2,06 Å при порошковой дифрактометрии, что дает отклонение в 2,4%. С другой стороны, результат измерения межплоскостного расстояния слоев, наблюдаемого в части, показанной символом G на Фиг. 4, составил 3,46 Å. Говорят, что расстояние между плоскостями 002 G в слоях гексагонального графита составляет 3,37 Å при порошковой дифрактометрии, что дает отклонение в 2,4%. Таким образом, наблюдаемое межплоскостное расстояние слоев по существу согласуется с межплоскостным расстоянием слоев графита. Поэтому считается, что слоистые наноразмерные углеродные частицы состоят из графита, составляя основную долю углеродных частиц.
[0060] По рентгенодифракционным данным можно подтвердить наличие наноалмаза. Однако, что касается наноразмерных углеродных частиц, неясно, какая
именно разновидность вещества содержится в них помимо нанографита и тонкодисперсных многослойных углеродных нанотрубок, дающих пик около 26°. Тонкодисперсные монослойные (однослойные) углеродные нанотрубки или различные фуллерены не сносят своего вклада в пик около 26°. Следовательно, их полученное количество не включается в количественный результат, основанный на пике около 26°. Кроме того, можно предположить, например, что наноразмерные углеродные частицы, слоистая (графитовая) структура которых была изменена на турбостратную структуру, также включаются в пик около 26°. Нельзя исключить и того, что смесь пиков этих деформированных наноразмерных углеродных частиц может служить растягиванию ширины пика около 26°. Однако из ПЭМ-фотографий найдено, что полученное количество тонкодисперсных монослойных (однослойных) углеродных нанотрубок, различных фуллеренов и т.д. мало. То есть, когда углеродные частицы получают способом детонации, предполагается, что производимое количество наноразмерных углеродных частиц, которые не проявляются пиком около 26°, может попадать в пределы определенного диапазона низких массовых долей. Поэтому предполагается, что большой погрешности не возникает даже в том случае, когда весь отличающийся от алмаза углерод рассматривают как графитовый углерод. Кроме того, предполагается, что углерод с другой структурой является редким.
[0061] Исходя из вышеупомянутых предпосылок предполагается, что если виды, количества и строения исходного вещества и взрывчатого вещества определяются в описанном способе получения, наноалмаз и нанографит, производимые этим способом получения, могут попадать в пределы определенного диапазона значений массового соотношения. Поэтому предполагается, что большой погрешности не возникает даже тогда, когда весь отличающийся от алмаза углерод рассматривают как графитовый углерод. Соответственно предполагается, что углерод с другой структурой, чем у алмаза и графитового углерода, является редким. Таким образом, соотношение между ними получают при допущении, что отличающийся от алмаза углерод представляет собой графитовый углерод.
[0062] Исходя из вышеупомянутых предпосылок, в настоящем изобретении используются углеродные частицы, содержащие наноразмерные графитовый углерод и алмаз и имеющие такую особенность, что доля содержания графитового углерода является более высокой, чем в обычном продукте, получаемом при использовании сырья, содержащего соединение с тремя или более нитрогруппами. Более конкретно, когда G обозначает массу графитового углерода, а D обозначает массу алмаза, массовое соотношение G/D составляет 2,5 или более, предпочтительно 3 или более,
предпочтительнее 3,5 или более, а еще предпочтительнее 4 или более. Верхний предел массового соотношения G/D конкретно не ограничен. Когда учитывают, что алмаз содержится, массовое соотношение G/D предпочтительно составляет 100 или менее, предпочтительнее 50 или менее, а еще предпочтительнее 20 или менее. Массовое соотношение G/D получают способом, который будет описан в следующих примерах.
[0063] Покрытые частицы в настоящем изобретении имеют строение, при котором поверхности частиц основного материала покрыты углеродными частицами, полученными вышеупомянутым способом получения. Когда поверхности частиц основного материала покрыты вышеупомянутыми углеродными частицами, эти покрытые частицы могут использоваться в качестве нового сырья в различных приложениях.
[0064] В покрытых частицах поверхности частиц основного материала могут быть покрыты углеродными частицами так, чтобы толщина пленки достигала 0,004 мкм. То есть, когда углеродные частицы наносятся самым тонким образом, толщина пленки может составлять 0,004 мкм. Толщина пленки может составлять 1 мкм или более. Верхний предел толщины пленки конкретно не ограничен. Например, толщина пленки может составлять 10 мкм или менее.
[0065] Вид частиц основного материала конкретно не ограничен. Примеры частиц основного материала могут включать углерод, полимер, стекло, керамику, металл, природные исходные материалы и т.д. Примеры углерода могут включать искусственный графит. Примеры полимеров могут включать акриловую смолу, уретановую смолу, нейлон, полиэтилен, высокомолекулярный полиэтилен, политетрафторэтилен и т.д. Примеры стекол могут включать различные аморфные стекла, кристаллизованные стекла и т.д. Примеры керамики могут включать SiC, неактивный глинозем, кремнезем, диоксид титана, диоксид циркония и т.д. Примеры металлов могут включать алюминий, чистую медь, бронзу, латунь, углеродистую сталь, нержавеющую сталь, мартенситностареющую сталь, сплавы на основе никеля и т.д. В дополнение, может использоваться синтетический цеолит, природные исходные материалы, такие как древесная щепа или стружка, минералы, угли и каменистые породы, и т.д.
[0066] Размер частиц основного материала конкретно не ограничен. Например, частицы основного материала могут иметь размер примерно от 2 до 550 мкм.
[0067] Предпочтительно, чтобы поверхности частиц основного материала были полностью закрыты углеродными частицами. Однако настоящее изобретение не ограничивается этим. Углеродные частицы могут прилипать только к части поверхностей частиц основного материала.
[0068] Покрытые частицы могут быть получены способом, включающим в себя стадию расположения взрывчатого вещества со скоростью детонации 6300 м/с или выше на периферии исходного вещества, содержащего ароматическое соединение с двумя или менее нитрогруппами, стадию детонирования взрывчатого вещества и стадию покрытия поверхностей частиц основного материала полученными углеродными частицами способом механического объединения.
[0069] Способ механического объединения означает смешивание или дробление. То есть, из-за связи между функциональным выражением частиц и энергией, приложенной к ним во время механической обработки порошка, при увеличении приложенной энергии со стороны частиц достигается не только простая позиционная замена (смешивание), но и придание дополнительных функциональных возможностей, таких как равномерное диспергирование, измельчение (дробление) и поверхностное покрытие (объединение).
[0070] Когда к смеси порошков, размеры частиц которых в значительной степени различаются, применяется механическое сдвиговое/ударное воздействие, поверхности частиц становятся аморфными, а активность на этих поверхностях повышается. Когда в одной системе существуют разные порошки, из-за взаимодействия порошков образуются композитные частицы, поверхности которых покрыты мелкодисперсными частицами, имеющими меньшие размеры частиц. Поверхность каждой из частиц, служащих в качестве ядер объединения (называемых частицами-ядрами или материнскими частицами), имеет регулярную структуру аналогичным образом. Следовательно, это объединение частиц также называется «регулярным смешиванием» или «точным тонким смешиванием».
[0071] В состоянии, когда механическая энергия еще выше, возникает твердофазная реакция или механохимическая реакция. То есть взаимодействие происходит в масштабе, соответствующем приложенной энергии, и при более высокой энергии появляется взаимодействие на молекулярном/атомном уровне.
[0072] Обычно композитные частицы принимают следующие две формы. Одна форма представляет собой покрытую композитную частицу, в которой поверхность частицы-ядра покрыта очень мелкодисперсными частицами (дочерними частицами), а другая форма представляет собой распределенную композитную частицу, в которой дочерняя частица входит внутрь частицы-ядра или дочерняя частица и частица-ядро образуют структуру, в которой они спутаны друг с другом. Композитная частица-капсула относится к типу ядро-оболочка. То, какие именно композитные частицы получаются, зависит от физических и химических свойств частиц-ядер и дочерних частиц, а также зависит от величины механического воздействия, оказываемого на комбинацию или
атмосферу.
[0073] Как показано на Фиг. 2, процесс механического объединения включает в себя (1) столкновение/адгезию частиц, (2) дробление/диспергирование частиц, (3) точное тонкое смешивание частиц и (4) слияние/внедрение частиц. Этим этапам процесса способствуют мощные ударные, сжимающие и сдвиговые воздействия, прикладываемые к частицам между ротором или шаром, вращающимся с высокой скоростью, и контейнером или внутренней деталью. Таким образом можно управлять объединением или поверхностными свойствами.
[0074] Примеры устройства объединения могут включать в себя систему гибридизации (ударный метод высокоскоростного воздушного потока), систему механосинтеза и т.п. Эти устройства объединения обладают соответственно разными механическими действиями. Например, в системе гибридизации из-за столкновения с лопастью или корпусом очень к частицам прикладывается мощное ударное воздействие, так что различные вещества могут внедряться друг в друга или сливаться друг с другом. В дополнение к этому, можно также ожидать, что применима система механосинтеза для механического сплавления из-за действия мощного сжимающего усилия и мощного усилия сдвига. С другой стороны, в некоторых случаях такие сильные механические воздействия могут ингибировать функциональное проявление в композитных частицах. Например, ухудшение функции или изменение кристаллической структуры могут быть вызваны внезапным повышением температуры или ударом. Против таких случаев также был разработан относительно мягкий подход. Этот подход достигается с помощью перемешивающего смесителя, такого как смеситель Хеншеля, в котором мелкодисперсные частицы могут быть хорошо диспергированы за счет вращения перемешивающей лопасти. Это удобно с целью точного тонкого смешивания на поверхностях частиц. Кроме того, существует другой подход с использованием θ-синтезатора, который может надежно фиксировать вещество без изменения его структуры, то есть можно ожидать, что он будет иметь так называемое промежуточное механическое действие. В примерах, которые будут описаны ниже, используется аппарат смеситель-синтезатор типа MP5 (от англ. «MP5 type mixer (compositor)») производства компании NIPPON COKE & ENGINEERING Co., Ltd., который является усовершенствованием вышеупомянутого смесителя Хеншеля с функцией, аналогичной системе гибридизации. Однако покрытые частицы в настоящем изобретении не обязательно получают только с помощью такого аппарата, но они могут быть получены вышеуказанными различными способами механического объединения тем же самым образом.
[0075] В настоящем изобретении углеродные частицы могут быть фторированы, а затем поверхности частиц основного материала могут быть покрыты углеродными частицами. Когда углеродные частицы фторируют заранее, покрытым частицам могут быть приданы функции самого фтора, такие как водоотталкивающие свойства, маслоотталкивающие свойства, разделяемость, неадгезивность, противообрастающее свойство, химическая стойкость, смазывающая способность, противобактериальное свойство, окисляемость и т.д. В дополнение к этому, покрытые частицы могут быть легко диспергированы как в воде, так и в органическом растворителе.
[0076] Что касается вышеупомянутой обработки фторированием, например, может использоваться способ прямого фторирования для того, чтобы заставить углеродные частицы реагировать с газообразным фтором или фторирующим агентом, производным от газообразного фтора. В дополнение, может использоваться способ фторирования частиц основного материала вследствие реакции с плазмой фтора. Кроме того, может использоваться способ фторирования частиц основного материала в растворе фторирующим агентом, таким как содержащий фторалкильную группу олигомер. Кроме того, может использоваться фторирование фторирующим агентом в ионной жидкости.
[0077] В связи с его химическими и физическими свойствами внимание было обращено на фторид графита в качестве нового промышленного материала. Фторид графита представляет собой белый порошкообразный неорганический полимер паркетной (пластинчатой) структуры, получаемый путем прямой реакции углерода с фтором. Хотя фторид графита можно отождествить с фторуглеродом, таким как CF4,C2F6 или {CF2-CF2}n, фторид графита обладает такими признаками, как: фторид графита, полученный из графита, становится кристаллическим; фторид графита – это твердый полимер, который не может быть синтезирован посредством поликонденсации или тому подобного; и т.д. Именно поэтому его называют фторидом графита в отличие от обычных соединений углерода и фтора. Такие углеродные материалы образуют систему, относимую к категории веществ, находящихся в пограничной области между органической химией и неорганической химией из-за предысторий их получения и т.д.
[0078] До сих пор считалось, что фторид графита, полученный из различных углеродных исходных материалов, таких как аморфный углерод, углеродная сажа, нефтяной кокс, графит и т.д., может быть выражен формулой (CF)n. Однако во фториде графита по настоящему изобретению наиболее распространена связь C-F, но также выявлены связи C-F2 и C-F3, как будет описано ниже.
[0079] Покрытые частицы содержат графитовый углерод и алмаз на поверхностях частиц основного материала. Следовательно, используя превосходные свойства алмаза, такие как шлифуемость, долговечность, износостойкость и т.д., покрытые частицы полезны для применения в качестве износостойкого агента, смазки и т.д. В дополнение к этому, используя превосходные свойства графитового углерода, такие как электропроводность, водоотталкивание, биосовместимость и т.д., покрытые частицы полезны для применения в качестве волокнистого материала, полимерного покрытия для обеспечения функциональности, системы доставки лекарственного средства, покрытия электронного устройства, электродного материала для батареи, проводящей пленки, армированной резины/водоотталкивающей резины, катализатора, адсорбционного материала и т.д.
[0080] Настоящее изобретение также включает в себя функциональный материал, в котором вышеупомянутые покрытые частицы поддерживаются на поверхности материала подложки. Когда покрытые частицы поддерживаются на нем, могут быть получены следующие эффекты. В зависимости от вида материала подложки, поддерживающего покрытые частицы, может быть увеличена твердость поверхности материала подложки; может быть понижен коэффициент трения для улучшения смазывающей способности; может быть улучшена износостойкость; может быть улучшено каталитическое свойство (активность в реакции); может быть улучшена электропроводность; может быть улучшена теплопроводность; может быть улучшено антиобрастающее свойство; или, если покрытые частицы фторированы, могут быть улучшены водоотталкивающие свойства, маслоотталкивающие свойства, разделяемость, неадгезивность, антиобрастающее свойство, химическая стойкость, смазывающая способность, противобактериальное свойство или окисляемость.
[0081] Материал подложки, поддерживающий покрытые частицы, конкретно не ограничен. Примеры таких материалов подложки могут включать углерод, древесину, стекло, полимер, керамику, металл, бетон, наружных стеновые материалы и т.д.
[0082] Примеры таких углеродов могут включать графит, стеклоуглерод, искусственный графит, изотропный графит, углеродную сажу, тонкодисперсный углерод, композит C/C (армированный углеродным волокном углеродный композит), углеродное волокно и т.д.
[0083] Примеры таких стекол могут включать стекло «Пирекс» (Pyrex - зарегистрированный товарный знак), аморфное стекло, такое как кварцевое стекло, кристаллизованное стекло, такое как литиево-алюмосиликатное стекло или магниево-алюмосиликатное стекло, специальное стекло, такое как проводящее стекло, и т.д.
[0084] Примеры таких полимеров могут включить термопластичную смолу, термореактивную смолу, нейлон, конструкционный пластик и т.д. Примеры таких
термопластичных смол могут включать полиэтилен, полипропилен, поливинилхлорид, полистирол, поливинилацетат, полиуретан, политетрафторэтилен, смолу АБС, акриловую смолу, поликарбонат и т.д. Примеры таких термореактивных смол могут включать фенольную смолу, эпоксидную смолу, полиэфирную смолу и т.д. Примеры таких конструкционных пластиков могут включать в себя полиацеталь, бакелит, эпоксидное стекло, ультравысокомолекулярный полиэтилен, полиамид, модифицированный полифениленовый эфир, полиэтилентерефталат, полибутилентерефталат, полифениленсульфид, полиарилат, полиамидоимид, полиэфиримид, полиэфиркетон, полиэфирэфиркетон, полисульфон, полиэфирсульфон, фторопласт и т.д.
[0085] Примеры такой керамики могут включать оксидную керамику, такую как глинозем, кремнезем и кварц, карбидную керамику, такую как карбид кремния, нитридную керамику, такую как нитрид кремния и нитрид алюминия, диоксид титана, диоксид циркония и т.д.
[0086] Примеры таких металлов могут включать металлы на основе железа, такие как обыкновенная сталь, инструментальная сталь, подшипниковая сталь, нержавеющая сталь, железо и чугун, цветные металлы, такие как медь, медные сплавы, алюминий, алюминиевые сплавы, никель, сплавы на основе никеля, олово, свинец, кобальт, титан, хром, золото, серебро, платина, палладий, магний, марганец и цинк, и т.д. Кроме того, также могут использоваться сплавы этих металлов или оксиды этих металлов.
[0087] Форма материала подложки конкретно не ограничена. Примеры таких форм могут включать листовую форму, столбчатую форму, цилиндрическую форму и т.д.
[0088] Функциональный материал может быть получен путем нанесения покрытых частиц на поверхность материала подложки.
[0089] В качестве способа нанесения покрытых частиц на поверхность материала подложки может использоваться, например, (1) термонапыление, (2) прокатка, (3) металлизация и т.д. (1) Термонапыление и (2) прокатка могут вместе упоминаться как способ термомеханической обработки. Кроме того, может использоваться метод столкновения частиц с использованием механизма, при котором тепло генерируется при высокоскоростном столкновении мелких частиц (также иногда называемый WPC (широкая дробеструйная обработка).
(1) Термонапыление
[0090] Функциональный материал, поддерживающий покрытые частицы на поверхности материала подложки, может быть получен путем термонапыления покрытых частиц на материал подложки.
[0091] Способ термонапыления представляет собой разновидность метода модификации поверхности, в котором термонапыляемый материал, такой как металл или керамику, нагревают до расплавленного или полурасплавленного состояния при помощи пламени горения, электроэнергии или т.п., и полученные таким образом частицы материала термически распыляют на поверхность материала подложки так, чтобы сформировать покрытие на поверхности материала подложки. В качестве источника тепла для плавления термонапыляемого материала, такого как порошок или проволока, используется горючий газ, плазма или т.п. Расплавленный материал преобразуется в мелкие частицы, каждая из которых имеет диаметр от нескольких мкм до сотни и нескольких десятков мкм. Эти мелкие частицы сталкиваются с поверхностью материала подложки на высокой скорости от нескольких десятков м/с до нескольких сотен м/с. Таким образом формируется покрытие путем наслаивания быстро затвердевающих (со скоростью 107°C/с или выше в случае частиц жидкого металла) плоских мелких частиц. Эта структура наслоения является отчетливым признаком термонапыленного покрытия, и она также упоминается как ламеллярная структура. Эта структура была использована для приложений следующим образом. Подлежащие термонапылению материалы термораспыляют на поверхности элементов в различных приборах или устройствах, чтобы придать им функции и качества, включая износостойкость, коррозионную стойкость, теплоизоляцию, электропроводность и т.д., отдельно от исходных материалов самих элементов. Существует большое количество способов и процессов термонапыления.
[0092] Способ термонапыления конкретно не ограничен. Примеры таких способов могут включить пламенное напыление, дуговое напыление, плазменное напыление, детонационное напыление, высокоскоростное пламенное напыление, холодное напыление и т.д. Пламенное напыление, дуговое напыление и плазменное напыление известны как основанные на температуре способы напыления, в которых удовлетворительно расплавленные частицы распыляются с низкой скоростью. С другой стороны, детонационное напыление, высокоскоростное пламенное напыление и холодное напыление известны как основанные на скорости способы напыления, в которых полурасплавленные частицы распыляются с высокой скоростью. Среди них холодное напыление представляет собой один из методов покрытия поверхности, основанных на высокоскоростном столкновении мелких частиц, и имеет ту главную особенность, что частицы ускоряются низкотемпературным и высокоскоростным рабочим газом. Поскольку температура газа при этом ниже, чем точка плавления частиц материала, эти частицы не плавятся. Поэтому в последние годы холодное напыление также использовалось для термонапыления наноуглеродных материалов, таких как углеродные
нанотрубки. Однако наноразмерные углеродные частицы, образующие часть покрытых частиц, получаются в настоящем изобретении способом детонации. При их получении углеродные частицы подвергаются воздействию высокой температуры, составляющей 800°C или выше. Поэтому предполагается, что эти углеродные частицы имеют более высокую теплостойкость, чем любой другой наноуглеродный материал, такой как углеродные нанотрубки. Так, когда для частиц основного материала выбирается металл или керамика, которые являются теплостойким материалом, с частицами основного материала можно работать даже в случае основанного на температуре способа напыления. Можно считать, что нет необходимости ограничивать способ термонапыления основанным на скорости способом напыления.
[0093] Можно считать, что, когда покрытые частицы по настоящему изобретению подвергаются термонапылению различными способами, износостойкость, способность к скольжению, электропроводность (в том случае, когда материал подложки является керамическим или полимерным) и т.д. могут быть улучшены углеродными частицами, присутствующими в термонапыленном покрытии. Кроме того, больше функций можно ожидать в том случае, когда углеродные частицы в термонапыленном покрытии рассматриваются как катализатор или носитель, или наполнитель, служащий связующим. То есть, когда углеродные частицы, поддерживаемые в покрытых частицах, превращаются во фторид графита с помощью вышеупомянутого фторирования, покрытым частицам могут быть приданы такие функции, как водоотталкивающие свойства, маслоотталкивающие свойства, разделяемость, неадгезивность, антиобрастающее свойство, химическая стойкость, смазывающая способность, противобактериальное свойство, окисляемость и т.д. Когда углеродные частицы в термонапыленных покрытых частицах фторируют тем же самым образом, функции фторида графита могут быть приданы термонапыленной поверхности материала подложки таким же образом, что и фторированным покрытым частицам. То есть, когда термонапыленная поверхность подложки фторирована, поверхности материала подложки могут быть приданы такие функции, как водоотталкивающие свойства, маслоотталкивающие свойства, разделяемость, неадгезивность, антиобрастающее свойство, химическая стойкость, смазывающая способность, противобактериальное свойство, окисляемость и т.д.
(2) Прокатка
[0094] Когда покрытые частицы прокатывают на материале подложки, может быть получен функциональный материал, поддерживающий (несущий) покрытые частицы на своей поверхности.
[0095] Способ прокатки конкретно не ограничен. Примеры таких способов могут включать способ напрессовки, такой как способ с валковым прессом или способ с ленточным прессом, способ штамповки, такой как способ плоского горячего прессования периодического типа, способ плакирования прокаткой и т.д.
[0096] Прокатка не ограничена способом, в котором подают и формуют только покрытые частицы, но может использоваться способ, в котором подают и формуют смесь покрытых частиц и связующего вещества. Когда подобрано сочетание материалов для частиц основного материала и материала подложки, покрытые частицы могут поддерживаться на поверхности материала подложки без какого-либо клейкого вещества. Например, для частиц основного материала выбирается металл, керамика или т.п. с высокой точкой плавления, а для материала подложки выбирается смола с низкой точкой плавления или т.п. Когда горячее прессование выполняют при немного более высокой температуре нагрева, чем точка плавления более легкоплавкого материала, покрытые частицы могут быть приварены к материалу подложки. С другой стороны, смола с низкой точкой плавления и т.п. может быть выбрана для частиц основного материала, а металл с высокой точкой плавления или керамика с высокой точкой плавления могут быть выбраны для материала подложки. Когда горячее прессование выполняют при немного более высокой температуре нагрева, чем точка плавления более легкоплавкого материала тем же самым образом, частицы основного материала покрытых частиц плавятся и распределяются по поверхности материала подложки. После охлаждения на поверхности материала подложки может быть сформировано слоистое покрытие из основного материала с углеродными частицами. Альтернативно, глинозем, SiC, нержавеющая сталь, мартенситностареющая сталь, инструментальная сталь и т.п., которые имеют высокую твердость, могут быть выбраны для частиц основного материала, а один из различных полимеров, алюминия, меди и т.п., которые имеют низкую твердость, могут быть выбраны для материала подложки. В этом случае покрытые частицы могут быть нанесены на поверхность материала подложки даже прокаткой при низкой температуре нагрева.
[0097] В одном примере, в котором покрытые частицы смешиваются со связующим веществом и подвергаются плакированию прокаткой, экструдированию и т.п., имеет место случай, при котором связующее вещество не может быть легко равномерно растворено в комбинации алюминия и порошка бора. В этом случае, плакированный прокаткой лист, с которым алюминий сплавляется в сильно сжатом состоянии, может быть получен способом, в котором непосредственно экструдируют порошковую смесь порошка борида и порошка алюминиевого сплава, способ, в котором экструдируют, проковывают или прокатывают заготовку из порошковой смеси, предварительно отформованную до заданной формы, и способ, в котором такие порошковую смесь и
заготовку заключают в металлический контейнер, имеющий заданную форму, и экструдируют, проковывают или прокатывают. С другой стороны, в настоящем изобретении связующее вещество не ограничено порошком алюминия или алюминиевого сплава. Может использоваться термореактивная смола или реактивный термоплавкий клей, который часто используется для производства фанеры (шпона). Таким образом, можно считать, что, когда покрытые частицы в настоящем изобретении прокатывают различными способами, за счет присутствующих в термонапыленном покрытии углеродных частиц могут быть улучшены износостойкость, способность к скольжению, электропроводность (в том случае, когда материал подложки является керамическим или полимерным) и т.д.
[0098] Кроме того, больше функций можно ожидать в том случае, когда углеродные частицы рассматриваются как катализатор или носитель, или наполнитель, служащий связующим.
[0099] В качестве покрытых частиц, как описано выше, могут использоваться покрытые частицы, в которых поверхности частиц основного материала покрыты фторированными углеродными частицами. Альтернативно, могут быть приготовлены покрытые частицы, в которых поверхности частиц основного материала покрыты углеродными частицами, которые еще не были фторированы, и может быть выполнена обработка фторированием покрытых частиц, которые были нанесены на поверхность материала подложки.
(3) Металлизация
[0100] Когда материал подложки погружают и металлизируют в ванне для нанесения покрытия, в которой были диспергированы покрытые частицы, может быть получен функциональный материал, в котором покрытые частицы нанесены на поверхность материала подложки.
[0101] Способ металлизации конкретно не ограничен. Например, может использоваться либо нанесение гальванического покрытия, либо негальваническое нанесение покрытия (химическим восстановлением). Вид наносимого металла может представлять собой единственный металл (такой как медь, никель, хром, олово, цинк, серебро, золото и т.д.) или сплав (такой как латунь, бронза, припой, сплав Zn-Ni, сплав Zn-Fe, Ni-P, Ni-B, Ni-W, Ni-Fe и т.д.), служащий в качестве способа нанесения композитного покрытия, в котором мелкие частицы, включая покрытые частицы, осаждаются на нанесенный металл.
[0102] Ванна для нанесения покрытия конкретно не ограничена при условии, что в ней были диспергированы покрытые частицы. Например, возможно использовать
коммерчески доступную ванну для нанесения покрытия, в которой были диспергированы покрытые частицы.
[0103] Вид ванны для нанесения покрытия конкретно не ограничен. Примеры таких ванн для нанесения покрытия могут включать ванну для нанесения покрытия из никеля (Ni), ванну для нанесения покрытия из Ni-P, ванну для нанесения покрытия из Ni-B, ванну для нанесения покрытия из Ni-W, ванну для нанесения покрытия из Ni-Cu-P, ванну для нанесения покрытия из Ni-S, ванну для нанесения покрытия из Cr-W, ванну для нанесения покрытия из Cr-Mo, ванну для нанесения покрытия из Cr-Fe, ванну для нанесения покрытия из Cr-C, ванну для нанесения покрытия из Cr-H, ванну для нанесения покрытия из Fe-W, ванну для нанесения покрытия из Fe-Mo, ванну для нанесения покрытия из Fe-Ni, ванну для нанесения покрытия на основе Co-W, ванну для нанесения покрытия с сульфаматом никеля, ванну для нанесения покрытия с цианидом меди, ванну для нанесения покрытия с пирофосфатом меди, ванну для нанесения покрытия с сульфатом меди, ванну для нанесения покрытия с шестивалентным хромом, ванну для нанесения покрытия с цианидом цинка, ванну для бесцианидного нанесения цинкового покрытия, щелочную ванну для нанесения оловянного покрытия, кислотную ванну для нанесения оловянного покрытия, ванну для нанесения серебряного покрытия, ванну для нанесения покрытия с цианидом золота, кислотную ванну для нанесения золотого покрытия и т.д. Кроме того, примеры таких ванн для нанесения покрытия из Ni могут включать ванну Уоттса, ванну с сульфаминовой кислотой, ударную ванну или ванну затяжки (ванну Вудса), ванну для черного никелирования и т.д.
[0104] Температура ванны во время нанесения покрытия конкретно не ограничена. Например, она может быть установлена на 50-90°C. Кроме того, раствор может перемешиваться во время нанесения покрытия.
[0105] Данная заявка на патент испрашивает право приоритета по японской патентной заявке № 2015-133153, поданной 1 июля 2015 г. Полное содержание описания вышеупомянутой японской патентной заявки № 2015-133153 включено сюда по ссылке.
Примеры
[0106] Далее настоящее изобретение будет более конкретно описано на его примерах. Однако настоящее изобретение не предназначено быть ограниченным нижеследующими примерами и может быть осуществлено с изменениями в пределах его объема, адаптируемого к сути настоящего изобретения, которое было описано выше или которое будет описано ниже. Любое из таких изменений также охватывается технической сущностью настоящего изобретения.
[0107] Поверхности частиц основного материала покрыли углеродными частицами, полученными по процедурам, описанным в следующих экспериментальных примерах 1-5, получив покрытые частицы.
<<Экспериментальный пример 1>>
[0108] В этом экспериментальном примере углеродные частицы получали детонационным способом с использованием динитротолуола (DNT) в качестве исходного вещества и с использованием жидкого высокоэнергетического взрывчатого вещества на основе гидразина в качестве взрывчатого вещества. Более конкретно, DNT (технический) плавили и загружали в качестве исходного вещества, и формовали его в столбчатую форму, имеющую диаметр 10 см и высоту 48 см. Полученное таким образом формованное тело имело массу 5,52 кг, объем 3770 см3 и плотность 1,46 г/см3. В дополнение, в качестве взрывчатого вещества приготовили 75%-й раствор нитрата гидразина в гидразин-гидрате и подразделили его на части по 2,50 кг.
[0109] Затем проводили реакцию детонации с использованием взрывного устройства, проиллюстрированного на Фиг. 1. Вышеупомянутое формованное тело в качестве исходного вещества 10 помещали в центральную часть взрывного контейнера 20, имеющего внутренний диаметр 12 см и высоту 50 см, и вышеупомянутое жидкое высокоэнергетическое взрывчатое вещество в качестве взрывчатого вещества 12 заливали в свободное место по его периферии. Усилитель 22 детонатора (SEP), детонирующий шнур и электродетонатор 24 № 6 установили на верху взрывного контейнера 20 и накрыли крышкой. После этого контейнер заключили во влагонепроницаемый полиэтиленовый мешок. В качестве охлаждающего контейнера 30 использовали контейнер емкостью 100 л. Взрывной контейнер 20 помещали в охлаждающий контейнер 30. При этом внешнюю нижнюю поверхность взрывного контейнера 20 отрегулировали так, чтобы она была размещена на высоте 15 см от внутренней нижней поверхности охлаждающего контейнера 30, используя сделанное из железа основание 34 и сделанный из железа перфорированный диск 36. Затем залили 120 л дистиллированной воды в качестве охладителя 32 в охлаждающий контейнер 30 и полиэтиленовый мешок так, чтобы зазор между охлаждающим контейнером 30 и взрывным контейнером 20 был заполнен охладителем 32. После закрывания крышкой охлаждающий контейнер 30 подвешивали во взрывной камере внутренним объемом 30 м3 к ее потолку с использованием проволочной стропы. Внутреннее пространство вышеупомянутой взрывной камеры вакуумировали от атмосферного давления, доводя количество остаточного газообразного кислорода во взрывной камере до расчетного значения примерно 279,9 г.
[0110] После проведения подготовки таким образом вышеупомянутый детонирующий шнур поджигали вышеупомянутым детонатором, тем самым детонируя взрывчатое вещество 12. Затем из внутреннего пространства вышеупомянутой взрывной камеры извлекали примерно 120 л воды, содержащей остаток, и удаляли крупные обломки с помощью разделения отстаиванием. При этом, поскольку надосадочная жидкость была сильно щелочной, к ней добавляли лимонную кислоту для того, чтобы сделать ее pH слабокислым. Полученную таким образом слабокислую надосадочную жидкость извлекали в качестве жидких отходов. Осадок классифицировали на ситах с размерами отверстий 100 мкм и 16 мкм соответственно, используя устройство вибрационного просеивания («KG-700-2W» производства компании Kowa Kogyosho Co., Ltd.). Прошедший через 16 мкм сито материал извлекали как есть. В экспериментальных примерах 2-5, которые будут описаны позже, классификацию выполняли на ситах с размерами отверстий 100 мкм и 32 мкм соответственно, и прошедший через 32 мкм сито материал извлекали как есть.
[0111] В экспериментальном примере 1 остаток от прошедшего через 100 мкм сито материала на сите, имеющем размер отверстий 16 мкм, измельчали в течение примерно 5 минут ультразвуковым вибрационным устройством («4G-250-3-TSA» производства компании Crest). В каждом из экспериментальных примеров 2-5 остаток от прошедшего через 100 мкм сито материала на сите, имеющем размер отверстий 32 мкм, измельчали тем же самым образом. Углеродную фракцию отделяли с поверхности обломков, а затем снова классифицировали на ситах, имеющих размеры отверстий 100 мкм, 32 мкм и 16 мкм соответственно, используя устройство вибрационного просеивания («KG-700-2W» производства компании Kowa Kogyosho Co., Ltd.). Прошедшие через сито материалы извлекали. Каждый из прошедших через сито материалов выдерживали в сушильной машине («OF-450S» производства компании AS ONE Corporation) при 80°C в течение 24 часов для испарения влаги, тем самым приготовив его сухой порошок.
[0112] Таким образом всего было получено 2048 г углеродных частиц, включая 584 г прошедшего через 16 мкм сито материала, 907 г прошедшего через 32 мкм сито материала и 557 г прошедшего через 100 мкм сито материала. Содержимое эксперимента, извлеченное количество и выход углеродных частиц в этом экспериментальном примере показаны в нижеследующей Таблице 2.
<<Экспериментальный пример 2>>
[0113] В этом экспериментальном примере углеродные частицы получали тем же самым образом, что и в вышеупомянутом экспериментальном примере 1, за исключением того, что используемое в качестве взрывчатого вещества количество жидкого высокоэнергетического взрывчатого вещества на основе гидразина было изменено с 2,50 кг до 2,49 кг, контейнер емкостью 100 л в качестве охлаждающего контейнера был
заменен на контейнер емкостью 200 л, а используемое в качестве охладителя количество дистиллированной воды было изменено со 120 л на 220 л. В результате было получено всего 2334 г углеродных частиц, включая 534 г прошедшего через 16 мкм сито материала, 1315 г прошедшего через 32 мкм сито материала и 485 г прошедшего через 100 мкм сито материала. Содержимое эксперимента, извлеченное количество и выход углеродных частиц в этом экспериментальном примере показаны в нижеследующей Таблице 2.
<<Экспериментальный пример 3>>
[0114] В этом экспериментальном примере углеродные частицы получали тем же самым образом, что и в вышеупомянутом экспериментальном примере 1, за исключением того, что DNT в качестве исходного вещества было взято в количестве 5,46 кг вместо 5,52 кг и 3750 см3 вместо 3770 см3 по объему, контейнер емкостью 100 л в качестве охлаждающего контейнера был заменен на контейнер емкостью 200 л, используемое в качестве охладителя количество дистиллированной воды было изменено со 120 л на 220 л, количество (расчетное значение) остаточного газообразного кислорода в камере было изменено с 279,9 г на 191,0 г, и лимонную кислоту к надосадочной жидкости не добавляли. В результате было получено всего 1645 г углеродных частиц, включая 164 г прошедшего через 16 мкм сито материала, 801 г прошедшего через 32 мкм сито материала и 680 г прошедшего через 100 мкм сито материала. Содержимое эксперимента, извлеченное количество и выход углеродных частиц в этом экспериментальном примере показаны в нижеследующей Таблице 2.
<<Экспериментальный пример 4>>
[0115] В этом экспериментальном примере углеродные частицы получали детонационным способом с использованием 2,4-динитротолуола (2,4-DNT) в качестве исходного вещества и с использованием жидкого высокоэнергетического взрывчатого вещества на основе гидразина в качестве взрывчатого вещества. Более конкретно, 2,4-DNT (технический) плавили и загружали в качестве исходного вещества, и формовали его в столбчатую форму, имеющую диаметр 10 см и высоту 48 см. Полученное таким образом формованное тело имело массу 5,48 кг, объем 3785 см3 и плотность 1,45 г/см3. В дополнение, в качестве взрывчатого вещества приготовили 75%-й раствор нитрата гидразина в гидразин-гидрате и поделили его на части по 2,49 кг.
[0116] Затем проводили реакцию детонации с использованием проиллюстрированного на Фиг. 1 взрывного устройства тем же самым образом, что и в вышеупомянутом экспериментальном примере 1. Контейнер емкостью 200 л использовали в качестве охлаждающего контейнера 30. В дополнение к этому, в качестве охладителя 32 использовали 220 л дистиллированной воды. В результате было получено всего 2059 г углеродных частиц, включая 636 г прошедшего через 16 мкм сито материала, 726 г прошедшего через 32 мкм сито материала и 697 г прошедшего через 100 мкм сито материала. Содержимое эксперимента, извлеченное количество и выход углеродных частиц в этом экспериментальном примере показаны в нижеследующей Таблице 2.
<<Экспериментальный пример 5>>
[0117] В этом экспериментальном примере углеродные частицы получали тем же самым образом, что и в вышеупомянутом экспериментальном примере 3, за исключением того, что DNT в качестве исходного вещества было взято 3800 см3 вместо 3750 см3 по объему и с плотностью 1,44 г/см3 вместо 1,46 г/см3; используемое в качестве взрывчатого вещества количество жидкого высокоэнергетического взрывчатого вещества на основе гидразина было изменено с 2,50 кг на 2,43 кг, а и количество (расчетное значение) остаточного газообразного кислорода в камере было изменено со 191,0 г на 25,52 г. В результате было получено всего 1465 г углеродных частиц, включая 177 г прошедшего через 16 мкм сито материала, 678 г прошедшего через 32 мкм сито материала и 610 г прошедшего через 100 мкм сито материала. Содержимое эксперимента, извлеченное количество и выход углеродных частиц в этом экспериментальном примере показаны в следующей Таблице 2.
[0118] Таблица 2
1) Жидкое высокоэнергетическое взрывчатое вещество на основе гидразина, полученное смешиванием нитрата гидразина (H2N-NH2⋅HNO3) и гидрата гидразина (H2N-NH2⋅H2O) в массовом соотношении 3:1
2) Выход (%) = 100 × (общее извлеченное количество (г) углеродных частиц/[масса (кг) исходного вещества × 1000])
[0119] Затем полученные углеродные частицы изучали с помощью просвечивающего электронного микроскопа (ПЭМ) по следующей процедуре.
<Наблюдение в ПЭМ>
[0120] Полученные углеродные частицы наблюдали с использованием ПЭМ, имеющего ПЗС-камеру и фотографическое увеличение и позволяющего наблюдать изображения решетки алмаза и графитового углерода со слоистой структурой. Конкретные условия измерения с помощью ПЭМ показаны ниже.
Название устройства ПЭМ: просвечивающий электронный микроскоп JEM-ARM200F производства компании JEOL Ltd.
Способ измерения: суспензионный способ, дисперсионный растворитель – метанол
Ускоряющее напряжение: 200 кВ
ПЗС-камера: UltraScan производства компании Gatan
Фотографическое увеличение: 300000-кратное и 800000-кратное
Увеличение визуализации: 2200000-кратное и 5900000-кратное при печати в формате A4
[0121] По результатам измерения с использованием ПЭМ было установлено, что углеродные частицы, полученные способом получения по настоящему изобретению, образованы из наноразмерного алмаза и графитового углерода. Подробное описание будет дано ниже.
[0122] Среди углеродных частиц, полученных в экспериментальном примере 3, полученные в ПЭМ фотографии прошедшего через 16 мкм сито материала показаны на Фиг. 3. Фиг. 4 показывает фотографию, на которой увеличено изображение части c1, изображенной на Фиг. 3с.
[0123] Среди приведенных на Фиг. 3а фотографий, на фотографии a, показанной верхней левой, углеродная частица, имеющая круглую форму и находящаяся среди углеродных частиц, была увеличена с увеличением визуализации, соответствующим 5900000-кратному. По фотографии a можно убедиться в том, что диаметр этой углеродной частицы, имеющей круглую форму, составляет примерно 7,0 нм. Также на фотографии b, показанной верхней правой на Фиг. 3, углеродная частица, имеющая круглую форму и находящаяся среди углеродных частиц, была увеличена с увеличением визуализации, соответствующим 5900000-кратному. По фотографии b можно убедиться в том, что диаметр этой углеродной частицы, имеющей круглую форму, составляет примерно 17,5 нм. Результат измерения периода решетки в углеродных частицах, имеющих круглую форму и показанных на фотографиях a и b с Фиг. 3, составил 2,11 Å. Обычно говорят, что межплоскостное расстояние между плоскостями 111 D в решетке алмаза составляет 2,06 Å, что дает отклонение в 2,4%. Следовательно, углеродные частицы, имеющие круглую форму, могут рассматриваться как алмаз.
[0124] С другой стороны, увеличение визуализации фотографии c, показанной нижней левой на Фиг. 3, соответствует 2200000-кратному. На фотографии c наблюдались углеродные частицы, имеющие круглую форму, а также углеродные частицы, имеющие нерегулярную структуру решетки. Диаметры углеродных частиц, имеющих круглую форму и наблюдаемых на фотографии c, составили примерно от 2,0 до 4,0 нм. Увеличение визуализации фотографии d, показанной внизу справа на Фиг. 3, соответствует 2200000-кратному. На фотографии d углеродные частицы, имеющие круглую форму, и углеродные частицы, имеющие нерегулярную структуру решетки, наблюдались тем же самым образом, что и на вышеупомянутой фотографии c. Диаметры углеродных частиц, имеющих круглую форму и наблюдаемых на фотографии d, составили примерно от 6,0 до 10,0 нм.
[0125] Фиг. 4 показывает фотографию, на которой увеличена часть (часть c1) фотографии c, изображенной на Фиг. 3. Как показано символом G, поле, в котором могут наблюдаться углеродные частицы, имеющие нерегулярную структуру решетки, увеличено так, чтобы было видно изображение их решетки. Результат измерения наблюдаемого межплоскостного расстояния слоев составил 3,46 Å. Говорят, что расстояние между плоскостями 002 слоев гексагонального графита (при порошковой дифрактометрии) составляет 3,37 Å, что аналогичным образом дает отклонение в 2,4%. Таким образом, наблюдаемое межплоскостное расстояние слоев по существу согласуется с межплоскостным расстоянием слоев графита. Поэтому считается, что слоистые наноразмерные углеродные частицы, показанные символом G, состоят из графитового углерода (нанографита), занимая большую часть углеродных частиц. Кроме того, на фотографии, показанной на Фиг. 4, размер в направлении, перпендикулярном направлению наслаивания, составил от 1,5 до 10 нм. Как очевидно из Фиг. 4, установлено, что направление наслаивания каждого кусочка графита является нерегулярным, и направления наслаивания смежных кусочков графита не идентичны друг другу.
[0126] Затем измеряли и оценивали полученные углеродные частицы методом рентгенофазового анализа (РФА).
<Количественный метод РФА>
[0127] Во-первых, из углеродных частиц, полученных в экспериментальном примере 3, на Фиг. 5 показана рентгенодифратограмма прошедшего через 100 мкм сито материала.
[0128] Условия измерения рентгеновской дифракции показаны ниже.
Название рентгеновского дифрактометра: горизонтальный рентгенодифракционный прибор SmartLab производства компании Rigaku Corporation
Способ измерения: θ-2θ
Рентгеновский источник: излучение Cu-Kα
Напряжение и ток возбуждения: 45 кВ-200 мА
Отклоняющая щель: 2/3°
Рассеивающая щель: 2/3°
Приемная щель: 0,6 мм
[0129] Затем у углеродных частиц, полученных в экспериментальных примерах 1-5, измеряли РФА углеродных частиц в прошедшем через 16 мкм сито материале. По результатам этих измерений была получена интегрированная интенсивность дифракционного пика около 2θ=75° на поверхности D20 алмаза, и была получена доля содержащегося в углеродных частицах алмаза с использованием каждой подготовленной заранее калибровочной кривой.
[0130] В качестве стандартного вещества для определения количества алмаза использовали алмаз, который был очищен путем удаления графитового углерода и т.п. перхлорной кислотой из алмазсодержащих углеродных частиц, отдельно произведенных в настоящем изобретении. В качестве внутреннего стандарта добавляли кремниевый порошок (Stansil-G03A производства компании Osaka Yakken Co. Ltd., D50=5,2 мкм) в количестве 10 мас.% от общего количества углерода.
[0131] Калибровочная кривая для алмаза была подготовлена с использованием 5 стандартных образцов (эталонов) путем выполнения четырехточечного измерения по отношению интегрированной интенсивности вышеупомянутого дифракционного пика и интегрированной интенсивности дифракционного пика на каждой из плоскости 220 Si и плоскости 311 Si кремниевого кристалла, добавленного к каждому из образцов. Два пика кремниевого кристалла используются для того, чтобы подавить влияние ориентации порошкового кремния.
[0132] Эти 5 стандартных образцов приготовили путем смешивания кремниевых кристаллов с алмазом так, чтобы обеспечить 0 мас.%, 25 мас.%, 50 мас.%, 75 мас.% и 100 мас.% в качестве долей содержания алмаза соответственно.
[0133] Калибровочная кривая для алмаза была получена путем построения графика с концентрацией алмаза по оси ординат и с отношением интенсивностей по площади пиков D220/(Si220+Si311) алмаза и кремния по оси абсцисс. Соотношение между концентрацией Y алмаза и отношением интенсивностей X было Y=117,12×X в соответствии с линейной аппроксимацией методом наименьших квадратов. Полученная калибровочная кривая показана на Фиг. 6.
[0134] Полученную долю содержания алмаза делили на оценочную долю содержания графита, вычисляя массовое соотношение G/D. Найдено, что алмаз и графитовый углерод были главными компонентами. Углерод с другими структурами наблюдать не удалось.
[0135] Долю содержания алмаза (D: когда углеродные частицы рассматриваются как 100 мас.%) получали у углеродных частиц, полученных в каждом из экспериментальных примеров 1-5. Из этих углеродных частиц углеродные частицы, отличающиеся от алмаза, принимали за графитовый углерод и вычисляли долю содержания (G) графитового углерода. Массовое соотношение G/D вычисляли на основе доли содержания (D) алмаза в углеродных частицах и доли содержания (G) графитового углерода в углеродных частицах. Этот результат также показан в вышеупомянутой Таблице 2.
[0136] Из Таблицы 2 видно, что графитовый углерод может быть получен детонационным способом даже тогда, когда в качестве исходного вещества используется DNT, который является недорогим сырьем, являющимся исходным веществом, содержащим ароматическое соединение с двумя или менее нитрогруппами, и даже тогда, когда в качестве взрывчатого вещества используется жидкое высокоэнергетическое взрывчатое вещество.
[0137] Для углеродных частиц, полученных в каждом из вышеупомянутых экспериментальных примеров 1-5, вычисляли значение G/(G+D) на основе доли содержания (D) алмаза в углеродных частицах и доли содержания (G) графитового углерода в углеродных частицах. Результат вычисления показан в следующей Таблице 3 вместе с вышеупомянутым массовым соотношением G/D. В дополнение к этому, следующая Таблица 3 показывает значения G/D и G/(G+D) в коммерческом продукте, доступном на рынке как наноалмаз.
[0138] Таблица 3
1) NUAC и ND62 сделаны в Китае, NanoAmand сделан в Японии, BD и UDD (УДА) сделаны в России/Украине
[0139] Из вышеупомянутой Таблицы 3 видно, что доля содержания графитового углерода в углеродных частицах, полученных в вышеупомянутых экспериментальных примерах 1-5, является более высокой, чем в коммерческих продуктах.
[0140] Затем из рентгенодифракционных данных получали размер кристаллита, используя уравнение Шеррера: D=Kλ/βcosθ. Здесь D обозначает размер кристаллита (Å), λ обозначает длину волны рентгеновской трубки (1,5418 Å для Cu-Kα излучения в примерах), β обозначает уширение дифрагированного рентгеновского излучения, вызываемое кристаллитом, θ обозначает угол дифракции (рад), и K обозначает константу Шеррера, которая была установлена равной 0,9 (см. публикацию B. D. Cullity (автор), Gentaro Matsumura (переводчик), «X-ray diffraction main theory (new edition)», Agne Shofusha; March, 1999). Уширение β получали из уравнения β=(βexp2-βi2)1/2, используя ширину βexp измеренного дифрагированного рентгеновского излучения и уширение βi дифрагированного рентгеновского излучения, вызванное прибором.
[0141] Измеренное дифрагированное рентгеновское излучение подвергали сглаживанию, удалению фона и удалению Kα2. После этого получали значения полуширины пика около 26° (обычно называемого G002) и пика около 43° (обычно называемого D111), и каждое из полученных значений полуширины принимали за ширину βexp по измеренному дифрагированному рентгеновскому излучению. Пик G002 является пиком, вызванным графитовым углеродом, а пик D111 – пиком, вызванным алмазом. В дополнение, добавляли 10 мас.% порошка Si (Stansil-G03A производства компании Osaka Yakken Co. Ltd., с центральным размером частиц 5,2 мкм), и значение полуширины пика около 47° (обычно называемого Si220) при рентгеновской дифракции принимали за βi.
[0142] В качестве рентгеновского дифрактометра использовали горизонтальный рентгенодифракционный прибор SmartLab производства компании Rigaku Corporation. Это тот же самый прибор, что и в вышеприведенном разделе <Количественный метод РФА>.
[0143] Размеры кристаллитов, оцененные по измеренным данным рентгеновской
дифракции углеродных частиц, полученных в вышеупомянутых экспериментальных примерах 1-5, показаны в следующей Таблице 4. В результате считается, что размер кристаллита алмаза, вычисленный на основе полуширины пика D111, составляет 2-5 нм. То есть, размер кристаллита алмаза, полученный из ширины пиков дифракции рентгеновских лучей на алмазе по уравнению Шеррера, по существу соответствует результату наблюдения в ПЭМ, которое будет описано позже.
[0144] С другой стороны, размер кристаллита графитового углерода, вычисленный на основе значения полуширины пика G002, составил 2-4 нм. Таким образом, размер кристаллита оценивается при допущении, что межплоскостное расстояние является фиксированным и отличается только размер кристаллита. Однако было найдено, что графитовый углерод имеет так называемую турбостратную структуру, в которой поверхности гексагональной сетки графита наслоены параллельно, но регулярности в ее ориентации не наблюдается. Поэтому предполагается, что размер кристаллита, полученный из смеси пиков около 26°, в которых принимают участие различные деформированные вещества, не является корректным. Поэтому оцененный таким образом размер кристаллита графитового углерода рассматривается как справочные данные.
[0145] Таблица 4
[0146] Далее измеряли размер первичных частиц алмаза, период решетки по плоскости D111 алмаза и межплоскостное расстояние в слоях графитового углерода на основе фотографий, снятых при наблюдении в ПЭМ. Результаты показаны в следующей Таблице 5. Размер кристаллита алмаза, вычисленный на основе полуширины пика D111, показанной в вышеупомянутой Таблице 4, также показан в следующей Таблице 5. Как очевидно из следующей Таблицы 5, размер первичных частиц, наблюдаемых на снятых в просвечивающем электронном микроскопе (ПЭМ) фотографиях углеродных частиц, составил от примерно нескольких до 20 нм. Однако предполагается, что частица с наименьшим размером из частиц, показанных на этих фотографиях, была выражена в размере кристаллита, оцененном по данным рентгеновской дифракции.
[0147] Таблица 5
1) Размер кристаллита по РФА: размер кристаллита алмаза (Å), полученный из ширины линии рентгеновской дифракции
2) Размер по наблюдениям в ПЭМ (Å): размер первичных частиц алмаза, период решетки по плоскости D111 алмаза и межплоскостное расстояние (Å) в слоях графитового углерода, полученные приблизительно из фотографий в просвечивающем электронном микроскопе (ПЭМ)
[0148] Далее углеродные частицы, полученные в экспериментальном примере 4, фторировали, и фтор определяли количественно путем сжигания и ионной хроматографии. В результате доля содержания фтора составила 53 мас.%.
[0149] Фиг. 7 показывает фотографии, на которых частицы, полученные фторированием углеродных частиц, полученных в экспериментальном примере 4, наблюдали в просвечивающем электронном микроскопе (ПЭМ). Устройство и условия измерения, использовавшиеся при наблюдении в ПЭМ, были теми же самыми, что и описанные выше. На Фиг. 7 фотография а показывает углеродные частицы, которые еще не были фторированы, а фотография a1 представляет собой увеличенный вид части, выделенной прямоугольником на фотографии а. На Фиг. 7 фотография b показывает углеродные частицы, которые были фторированы, а фотография b1 представляет собой увеличенный вид части, выделенной прямоугольником на фотографии b.
[0150] Из показанных на Фиг. 7 фотографий a1 и b1 найдено, что межплоскостное расстояние увеличилось с 0,35 нм до 0,60 нм при введении 53 мас.% фтора.
[0151] Кроме того, состав и состояние связывания поверхности каждой фторированной углеродной частицы исследовали с помощью рентгеновского фотоэлектронного спектроскопа (XPS). Самую внешнюю поверхность фторированной углеродной частицы проанализировали качественно путем измерения широкополосных фотоэлектронных спектров. После этого были измерены узкополосные фотоэлектронные спектры для обнаруженных при качественном анализе элементов. Элементный состав (ат.%) вычисляли из доли площади узкополосного фотоэлектронного пика и коэффициента относительной чувствительности, а состояние связывания оценивали по положению пика.
<Условия измерения XPS>
[0152] Анализатор: «Quantera SXM (полностью автоматический сканирующий рентгеновский фотоэлектронный спектроскоп)» производства компании Physical Electronics Co., Ltd.
Рентгеновский источник: монохроматический Al Kα
Мощность рентгеновского излучения: 25,1 Вт
Размер рентгеновского луча: 100 мкм ϕ
Угол экстракции фотоэлектрона: 45°
[0153] Фиг. 8 показывает результат, в котором из широкополосного фотоэлектронного спектра и узкополосного фотоэлектронного спектра, измеренных на внешней поверхности фторированных углеродных частиц, полученных в экспериментальном примере 4, узкополосный фотоэлектронный спектр C1s был подвергнут разделению пиков. Таблица, приведенная на Фиг. 8, показывает энергию связи и долю площади каждого пика, вычисленную из узкополосного фотоэлектронного спектра C1s. Найдено, что введенный фтор удерживается на углеродной частице связью C-F, связью C-F2, связью C-F3 и связью C*-Fx. Связь C-F была наиболее распространенной во фторированных углеродных частицах, полученных в экспериментальном примере 4, а связь C-F2 и связь C-F3 были второй и третьей по распространенности.
[0154] Затем поверхности частиц основного материала покрывали углеродными частицами, полученными в вышеупомянутом экспериментальном примере 3, 4 или 5. В качестве частиц основного материала использовали (a) частицы уретановой смолы, (b) частицы акриловой смолы, (c) частицы полиамидной смолы (нейлона), (d) частицы высокомолекулярной полиэтиленовой смолы, (e) частицы SiC, (f) частицы неактивного глинозема, (g) частицы нержавеющей стали SUS316L, (h) частицы меди, (i) частицы бронзы и (j) частицы мартенситностареющей стали. Конкретное описание будет дано ниже.
[0155] (a) В качестве вышеупомянутых частиц уретановой смолы использовали «C-300» (средний размер частиц ϕ22 мкм, плотность 1,16 г/см3) производства компании Negami Chemical Industrial Co., Ltd., или «JB-300T» (средний размер частиц ϕ22 мкм,
плотность 1,16 г/см3) производства компании Negami Chemical Industrial Co., Ltd.
(b) В качестве вышеупомянутых частиц акриловой смолы использовали «SE-20T» (средний размер частиц ϕ22 мкм, плотность 1,21 г/см3), производства компании Negami Chemical Industrial Co., Ltd., «GR-300T» (средний размер частиц ϕ22 мкм, плотность 1,21 г/см3) производства компании Negami Chemical Industrial Co., Ltd., «TAFTIC AR650M» (средний размер частиц ϕ30 мкм, плотность 1,35 г/см3) производства компании Toyobo Co., Ltd., «TAFTIC FH-S010» (средний размер частиц ϕ10 мкм, плотность 1,17 г/см3) производства компании Toyobo Co., Ltd., или «J-4PY» (средний размер частиц ϕ2,2 мкм, плотность 1,21 г/см3) производства компании Negami Chemical Industrial Co., Ltd.
(c) В качестве вышеупомянутых частиц полиамидной смолы (нейлона) использовали «TR-2» (средний размер частиц ϕ20 мкм, плотность 1,13 г/см3) производства компании Toray Industries, Inc.
(d) В качестве вышеупомянутых частиц высокомолекулярной полиэтиленовой смолы использовали «MIPELON XM-221U» (средний размер частиц ϕ30 мкм, плотность 0,94 г/см3) производства компании Mitsui Chemicals Co. Ltd.
(e) В качестве вышеупомянутых частиц SiC использовали «SSC-A15» (средний размер частиц ϕ18,6 мкм, плотность 1,91 г/см3) производства компании Shinano Electric Refining Co., Ltd.
(f) В качестве вышеупомянутых частиц неактивного глинозема использовали «V-250» (средний размер частиц неизвестен) производства компании Union Showa K. K. или «VERSAL-G» (средний размер частиц ϕ50 мкм, плотность 1,93 г/см3) производства компании Union Showa K. K.
(g) В качестве вышеупомянутых частиц нержавеющей стали SUS316L использовали частицы нержавеющей стали SUS316L производства компании SANYO SPECIAL STEEL Co., Ltd.. Средний размер частиц составлял ϕ20 мкм, а плотность 7,98 г/см3.
(h) В качестве вышеупомянутых частиц меди использовали частицы чистой меди производства компании HIGH PURITY CHEMICALS Co., Ltd.. Средний размер частиц составлял ϕ20 мкм, а плотность 8,82 г/см3.
(i) В качестве вышеупомянутых частиц бронзы использовали частицы бронзы производства компании Sandvik AB. Они имели состав Cu-15%Sn-0,4%P. Средний размер частиц составлял ϕ27,4 мкм, а плотность 5,2 г/см3.
(j) В качестве вышеупомянутых частиц мартенситностареющей стали использовали частицы мартенситностареющей стали производства компании Sandvik AB. Это была
сталь 18Ni300. Средний размер частиц составлял ϕ32,4 мкм, а плотность 8,0 г/см3.
[0156] Углеродные частицы, полученные в экспериментальном примере 3, 4 или 5, и вышеупомянутые частицы основного материала помещали в смеситель-синтезатор типа MP5 производства компании NIPPON COKE & ENGINEERING Co., Ltd. и механически объединяли со скоростью вращения лопастей 10000 об/мин в течение времени перемешивания 10-30 минут, получив покрытые частицы. Смеситель-синтезатор типа MP5 имел емкость резервуара 6,5 л, рабочий объем примерно 3 л и двигатель мощностью 2,2 кВт. Углеродные частицы и частицы основного материала объединяли при следующих конкретных соотношениях.
[0157] (a-1) Частицы уретановой смолы «C-300» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 2 мас.%. Толщина пленки (оценочная толщина пленки), оцененная исходя из размеров и плотностей частиц основного материала и углеродных частиц, составляла 42 нм при этих условиях. Вышеупомянутое количество углеродных частиц представляет собой значение, когда количество частиц основного материала берется за 100 мас.% (это же применимо и к следующим абзацам). Таким образом, в (a-1) 500 г частиц уретановой смолы покрывали 100 г вышеупомянутых углеродных частиц.
(a-2) Частицы уретановой смолы «JB-300T» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 42 нм.
(b-1) Частицы акриловой смолы «SE-20T» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 44 нм.
(b-2) Частицы акриловой смолы «GR-300» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 44 нм.
(b-3) Частицы акриловой смолы «TAFTIC AR650M» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 67 нм.
(b-4) Частицы акриловой смолы «TAFTIC FH-S010» брали в количестве 300 г, а углеродные частицы, полученные в экспериментальном примере 3, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 19 нм.
(b-5) Частицы акриловой смолы «J-4PY» брали в количестве 230 г, а углеродные частицы, полученные в экспериментальном примере 3, брали в количестве 5 мас.%. Оценочная толщина пленки составляла 4,4 нм.
(с) Частицы полиамидной смолы «TR-2» брали в количестве 600 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 5 мас.%. Оценочная толщина пленки составляла 93 нм.
(d) Частицы высокомолекулярной полиэтиленовой смолы «MIPELON XM-221U» брали в количестве 250 г, а углеродные частицы, полученные в экспериментальном примере 3, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 47 нм.
(e) Частицы SiC «SSC-A15» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 5 мас.%. Оценочная толщина пленки составляла 146 нм.
(f-1) Частицы неактивного глинозема «V-250» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 5 мас.%. Средний размер частиц точно неизвестен, потому что распределение частиц «V-250» по размерам не представлено. При допущении, что средний размер частиц (d50) составляет 5 мкм, оценочная толщина пленки составляет 396 нм.
(f-2) Частицы неактивного глинозема «VERSAL-G» брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 5, брали в количестве 5 мас.%. Оценочная толщина пленки составляла 396 нм.
(g) Частицы нержавеющей стали SUS316L брали в количестве 1000 г, а углеродные частицы, полученные в экспериментальном примере 3, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 259 нм.
(h) Частицы чистой меди брали в количестве 1000 г, а углеродные частицы, полученные в экспериментальном примере 4, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 286 нм.
(i) Частицы бронзы брали в количестве 500 г, а углеродные частицы, полученные в экспериментальном примере 3, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 233 нм.
(j) Частицы мартенситностареющей стали брали в количестве 1000 г, а углеродные частицы, полученные в экспериментальном примере 4, брали в количестве 2 мас.%. Оценочная толщина пленки составляла 421 нм.
[0158] Затем полученные покрытые частицы наблюдали с помощью сканирующего электронного микроскопа с автоэлектронной эмиссией (FE-SEM) и проверяли, покрыты ли поверхности частиц основного материала углеродными частицами или нет. В качестве FE-SEM использовали микроскоп «JSM-7000F» производства компании JEOL Ltd. с ускоряющим напряжением 7,5 кВ, способом визуализации во вторичных электронах и 200-3000-кратным увеличением наблюдения.
[0159] Кроме того, часть поверхностного слоя покрытой частицы вырезали устройством с фокусированным ионным пучком (ФИП) и наблюдали. В качестве устройства обработки сечения использовали «IM-4000» (установку ионного фрезерования) производства компании Hitachi High-Technologies Corporation, используя источник ионов аргона, ускоряющее напряжение 4,0 кВ и температуру обработки -10°C. Сечение наблюдали с помощью микроскопа FE-SEM марки «S-5500» производства компании Hitachi High-Technologies Corporation, используя ускоряющее напряжение 2,0 кВ, способ визуализации во вторичных электронах, изображение в отраженных электронах (LA-BSE) и 200-25000-кратное увеличение наблюдения.
[0160] Фиг. 9 показывает снятую с помощью FE-SEM фотографию, на которой изображены поверхности покрытых частиц, полученных в вышеупомянутом абзаце (a-1).
[0161] Фиг. 10 показывает фотографию, на которой часть поверхностного слоя покрытой частицы, полученной в вышеупомянутом абзаце (a-1), вырезана устройством с ФИП и снята так, чтобы покрытую углеродную частицу можно было сравнить с внутренней частицей уретановой смолы.
[0162] Кроме того, покрытая частица, полученная в вышеупомянутом абзаце (a-1), была заморожена в криоаппарате CP, а затем разрезана ионным пучком, и сечение этой покрытой частицы наблюдали и сняли с помощью FE-SEM, получив фотографию a, показанную на Фиг. 11. Фотография b, показанная на Фиг. 11, представляет собой увеличенное изображение части, ограниченной прямоугольником на фотографии а, показанной на Фиг. 11. Фотография с, показанная на Фиг. 11, представляет собой увеличенное изображение части, ограниченной прямоугольником на фотографии b, показанной на Фиг. 11.
[0163] Как очевидно из Фиг. 9, Фиг. 10 и Фиг. 11, оказалось, что поверхности частиц основного материала покрыты углеродными частицами. В дополнение к этому, как очевидно из Фиг. 11, толщина пленки наблюдаемой покрытой частицы составляла примерно 30 нм в наименьшей части и примерно 400 нм в наибольшей части. Толщина наблюдавшейся пленки составила в среднем 40-60 нм. Найдено, что наблюдаемая средняя толщина пленки по существу согласуется с оценочной толщиной пленки в 42 нм. На Фиг. 11 переосажденный слой означает, что отходы, образовавшиеся во время резания ионным пучком для получения среза, снова осели на поверхности образца.
[0164] Далее, покрытые частицы, полученные путем покрывания поверхностей частиц основного материала углеродными частицами, полученными в вышеупомянутом экспериментальном примере 5 или 3, нанесли на поверхность материала подложки плазменным напылением, тем самым получив функциональный материал. Более
конкретно, в качестве вышеупомянутых покрытых частиц использовали покрытые частицы, полученные в вышеупомянутых абзацах (e), (g) и (i). То есть, в покрытых частицах (e) частицы SiC «SSC-A15» были покрыты углеродными частицами, полученными в экспериментальном примере 5. В покрытых частицах (g) частицы нержавеющей стали SUS316L были покрыты углеродными частицами, полученными в экспериментальном примере 3. В покрытых частицах (i) частицы бронзы были покрыты углеродными частицами, полученными в экспериментальном примере 3. Затем покрытые частицы были нанесены на поверхность материала подложки плазменным напылением.
[0165] В качестве материалов подложки использовали лист нержавеющей стали SUS304, лист углеродистой стали, бронзовый лист и алюминиевый лист. Плазменное напыление выполняли с использованием устройства плазменного напыления типа F4 производства компании Sulzer Metco Japan Ltd. Условия измерения плазменного напыления показаны в следующей Таблице 6.
[0166] Таблица 6
SLPM: стандартных литров в минуту (ст. л/мин)
[0167] Когда масса полученного образца металлического материала подложки была измерена после термонапыления, было обнаружено увеличение массы. Таким образом было найдено, что покрытые частицы были нанесены на поверхность материала подложки с помощью плазменного напыления.
[0168] Фиг. 12 показывает фотографии, на которых изображены использованный в качестве материала подложки лист нержавеющей стали SUS304 и полученный функциональный материал. В качестве термонапыляемого материала использовали покрытые частицы, полученные путем механического объединения 2 мас.% углеродных частиц, полученных в экспериментальном примере 3, с частицами основного материала из порошка нержавеющей стали SUS316. На фотографии а по Фиг. 12 снят материал подложки, на который покрытые частицы еще не были нанесены плазменным напылением. На фотографии b по Фиг. 12 снят функциональный материал, в котором на материал подложки были плазменным напылением нанесены покрытые частицы, полученные в вышеупомянутом абзаце (g). Когда оценили толщину пленки функционального материала по увеличению массы, оказалось, что покрытыми частицами было сформировано термонапыленное покрытие толщиной в среднем примерно 34 мкм.
[0169] На фотографии а, показанной на Фиг. 13, изображен сделанный тонким резаком и снятый с помощью FE-SEM срез функционального материала, показанного на фотографии b по Фиг. 12. На изображении b по Фиг. 13 показан увеличенный вид выделенной пунктирным прямоугольником части на фотографии а, показанной на Фиг. 13. На фотографии а, показанной на Фиг. 13, a1 обозначает термонапыленное покрытие, образованное покрытыми частицами, а a2 обозначает лист нержавеющей стали SUS304 в качестве материала подложки.
[0170] Как очевидно из Фиг. 12 и Фиг. 13, было подтверждено, что покрытые частицы могут наноситься на материал подложки плазменным напылением. В дополнение, наблюдаемая толщина пленки термонапыленного покрытия, образованного покрытыми частицами, составляла примерно 20 мкм в наименьшей части и примерно 60 мкм в наибольшей части. Наблюдаемая толщина пленки составила в среднем 40-50 мкм. Оказалось, что наблюдаемая средняя толщина пленки по существу согласуется с оценочной толщиной пленки в 34 мкм.
[0171] Толщину (мкм) термонапыленного покрытия в функциональном материале, полученном таким образом, а именно когда покрытые частицы (g) или (i), полученные путем покрывания поверхностей частиц основного материала углеродными частицами, полученными в вышеупомянутом экспериментальном примере 3, нанесли на поверхность материала подложки плазменным напылением, вычисляли по изменению массы. Результаты вычислений показаны в следующей Таблице 7.
[0172] В дополнение, измеряли твердость по Виккерсу термонапыленного покрытия в функциональном материале, полученном таким образом, а именно когда покрытые частицы (g) или (i), полученные путем покрывания поверхностей частиц
основного материала углеродными частицами, полученными в вышеупомянутом экспериментальном примере 3, нанесли на поверхность материала подложки плазменным напылением. Более конкретно, частицы покрытия (g) из частиц нержавеющей стали SUS316L, покрытых углеродными частицами, полученными в вышеупомянутом экспериментальном примере 3, или частицы покрытия (i) из частиц бронзы, покрытых углеродными частицами, полученными в вышеупомянутом экспериментальном примере 3, наносили на поверхность каждого материала подложки (листа нержавеющей стали SUS304, листа углеродистой стали, бронзового листа или алюминиевого листа) плазменным напылением при условиях, показанных в вышеупомянутой Таблице 6, тем самым получив функциональный материал.
[0173] Твердость измеряли при следующих условиях. Средние значения результатов измерения показаны в следующей Таблице 7.
<Условия измерения твердости>
Измерительный прибор: «Тестер микротвердости HM-102» производства компании Mitutoyo Corporation
Прикладываемая нагрузка: 0,1 кгс или 0,05 кгс
Продолжительность нагрузки: 10 секунд
Положение измерения: 5 точек в желаемых положениях термонапыленного покрытия
[0174] Следующая Таблица 7 показывает твердость по Виккерсу (Hv) каждого материала подложки (листа нержавеющей стали типа SUS304, листа углеродистой стали, бронзового листа и алюминиевого листа) в качестве справочных данных.
[0175] В дополнение к этому, образец, в котором частицы нержавеющей стали SUS316L производства компании SANYO SPECIAL STEEL Co., Ltd. были нанесены на поверхность каждого материала подложки, получали плазменным напылением при условиях (g), показанных в вышеупомянутой Таблице 6, и измеренная твердость по Виккерсу этого термонапыленного покрытия приведена в качестве справочных данных в следующей Таблице 7. Результаты измерений показаны в следующей Таблице 7.
[0176] Из следующей Таблицы 7 может быть сделан следующий вывод. Найдено, что при нанесении покрытых частиц по настоящему изобретению на поверхность материала подложки твердость этой поверхности по Виккерсу может быть улучшена до более высокой, чем твердость самого материала подложки. Кроме того, найдено, что при нанесении покрытых частиц по настоящему изобретению на поверхность материала подложки твердость этой поверхности по Виккерсу может быть повышена до более высокой, чем при нанесении частиц нержавеющей стали SUS316L на поверхность материала подложки. То есть оказалось, что когда используются покрытые частицы, в которых поверхности частиц нержавеющей стали SUS316L покрыты углеродными частицами, твердость по Виккерсу может быть улучшена примерно на 5-10%.
[0177] Таблица 7
[0178] Далее покрытые частицы, полученные путем покрывания поверхностей частиц основного материала углеродными частицами, полученными в вышеупомянутом экспериментальном примере 3, нанесли на поверхность материала подложки методом металлизации, тем самым получив функциональный материал. В качестве частиц основного материала использовали порошок глинозема с диаметром ϕ 4,2 мкм.
[0179] Лист из алюминиевого сплава (A5052) использовали в качестве материала подложки. Размеры этого листа составляли 80 мм × 50 мм × 0,8mm в толщину.
[0180] Углеродные частицы, полученные в экспериментальном примере 3, и вышеупомянутые частицы основного материала помещали в смеситель-синтезатор типа «MP5» производства компании NIPPON COKE & ENGINEERING Co., Ltd. и механически объединяли со скоростью вращения лопастей 10000 об/мин в течение времени перемешивания 20 минут, получив покрытые частицы. Смеситель-синтезатор типа MP5 имел емкость резервуара 6,5 л, рабочий объем примерно 3 л и двигатель мощностью 2,2 кВт. Углеродные частицы и частицы основного материала объединяли при следующих конкретных соотношениях.
(f-3) Сферический глинозем «DAW-03» производства компании Denka Company Limited брали в количестве 200 г в качестве частиц неактивного глинозема, а углеродные частицы, полученные в экспериментальном примере 3, брали в количестве 5,0 мас.%. Оценочная толщина пленки составляла 0,024 мкм.
[0181] Далее вышеупомянутые покрытые частицы нанесли на поверхность материала подложки методом металлизации. В качестве метода металлизации использовали негальваническое нанесение покрытия (химическим восстановлением). Использовали ванну для нанесения покрытия, в которой вышеупомянутые покрытые частицы диспергировались в ванне Ni-P так, чтобы концентрация покрытых частиц достигала 1,0 г/л. Температура ванны для нанесения покрытия была установлена на 80°C, а время металлизации было установлено на 60 минут. Этот раствор перемешивали во время нанесения покрытия. После металлизации выполняли термическую обработку с выдержкой при 100°C в течение 30 минут. Доля покрытой поверхности материала подложки нанесенным слоем металлического покрытия составляла 100%. При измерении массы полученного образца после металлизации было обнаружено увеличение массы. Найдено, что покрытые частицы были нанесены на поверхность материала подложки с помощью металлизации.
[0182] В качестве сравнительного образца слой металлического покрытия сформировали на поверхности материала подложки при тех же самых условиях за исключением того, что использовалась ванна Ni-P, в которой не были диспергированы вышеупомянутые покрытые частицы. Доля покрытой поверхности материала подложки нанесенным слоем металлического покрытия составляла 100%.
[0183] Толщину нанесенного слоя металлического покрытия в каждом образце после металлизации вычисляли по изменению его массы. В результате толщина нанесенного слоя металлического покрытия, образовавшегося в сочетании с покрытыми частицами, составила 38 мкм, а толщина нанесенного слоя металлического покрытия, образовавшегося без сочетания с покрытыми частицами, составила 25 мкм.
[0184] В дополнение к этому, твердость по Виккерсу каждого нанесенного слоя металлического покрытия измеряли при следующих условиях. В результате твердость нанесенного слоя металлического покрытия, образовавшегося в сочетании с покрытыми частицами, составила 586 Hv, а твердость нанесенного слоя металлического покрытия, образовавшегося без сочетания с покрытыми частицами, составила 681 Hv.
<Условия измерения твердости>
Измерительный прибор: «Тестер микротвердости HM-102» производства компании Mitutoyo Corporation
Прикладываемая нагрузка: 0,025 кгс
Продолжительность нагрузки: 10 секунд
Положение измерения: 5 точек в желаемых положениях нанесенного слоя металлического покрытия
[0185] Кроме того, измеряли шероховатость поверхности нанесенного слоя металлического покрытия в соответствии с японским промышленным стандартом JIS B0601 (2013). В качестве шероховатости поверхности измеряли среднеарифметическую шероховатость (Ra) и максимальную высоту профиля (Rt) кривой шероховатости с эталонной длиной 3 мм. В результате в нанесенном слое металлического покрытия, образовавшемся в сочетании с покрытыми частицами, среднеарифметическая шероховатость (Ra) составила 16,00 мкм, а максимальная высота профиля (Rt) кривой шероховатости составила 16,50 мкм. С другой стороны, в нанесенном слое металлического покрытия, образовавшемся без сочетания с покрытыми частицами, среднеарифметическая шероховатость (Ra) составила 0,28 мкм, а максимальная высота профиля (Rt) кривой шероховатости составила 2,20 мкм.
[0186] Как очевидно из вышеприведенных результатов, можно считать, что, особенно в применении к негальваническому нанесению покрытия, за счет сочетания покрытых частиц в дополнение к преимуществам смазывающей способности и износостойкости могут быть получены такие эффекты, как повышение производительности из-за увеличения скорости осаждения, уменьшение тумана или нагрузки на окружающую среду на фабрике из-за снижения температуры ванны, уменьшение затрат на электроэнергию, упрощение работы и т.д.
[0187] Диаметр ϕ частиц неактивного глинозема, используемых в качестве частиц основного материала, был большим, составляя 4,2 мкм. Поэтому можно считать, что примешанные покрытые частицы распределялись в поверхности нанесенного слоя металлического покрытия, делая твердость этого слоя металлического покрытия более низкой, чем в том случае, когда покрытые частицы не примешивали, так что шероховатость поверхности была более грубой.
[0188] Износостойкость функционального материала, полученного путем нанесения вышеупомянутых покрытых частиц на поверхность материала подложки методом металлизации, оценивали по следующей процедуре. А именно, измеряли коэффициент трения на поверхности функционального материала, поверхность которого была отполирована, с использованием устройства для измерения трения HEIDON, и на основе измеренного коэффициента трения оценивали износостойкость функционального материала. Испытание на истирание выполняли при следующих условиях. Число проходов туда и обратно было установлено равным 100, и коэффициенты трения, измеренные через каждые 10 проходов туда и обратно, показаны в следующей Таблице 8.
<Условия>
Измерительное устройство: тестер свойств поверхности «TYPE: 14DR» производства компании SHINTO Scientific Co., Ltd.
Индентор: шариковый индентор SUJ2, диаметр 10 мм
Скорость испытания: 3 мм/с (эквивалентно 9 проходам туда и обратно в минуту)
Нагрузка: 1 кгс
Ход: 10 мм, скольжение в продольном направлении тестового образца
Число проходов туда и обратно: 100
Среда испытания: комнатная температура, без смазки
Измерение: коэффициент трения измерялся только при движении вперед
[0189] Кроме того, в качестве сравнительного образца сформировали слой металлического покрытия на поверхности материала подложки при тех же самых условиях за исключением того, что использовалась ванна Ni-P, в которой не были диспергированы вышеупомянутые покрытые частицы. Результат измерения коэффициента трения на поверхности образца при тех же самых условиях после металлизации показан в следующей Таблице 8.
[0190] В дополнение к этому, Фиг. 14 показывает зависимость между числом проходов туда и обратно и коэффициентом трения. На Фиг. 14 сплошная линия означает результат функционального материала, полученного таким образом, что на поверхности материала подложки был сформирован слой металлического покрытия, содержащий покрытые частицы, а пунктирная линия означает результат образца, полученного таким образом, что на поверхности материала подложки был сформирован слой металлического покрытия, не содержащий покрытых частиц.
[0191] Из следующей Таблицы 8 и Фиг. 14 может быть сделан следующий вывод. Найдено, что при примешивании покрытых частиц по настоящему изобретению в нанесенный слой металлического покрытия коэффициент трения на поверхности этого слоя металлического покрытия может быть снижен, составляя примерно на 10% меньше, чем у нанесенного слоя металлического покрытия в отдельности.
[0192] Таблица 8
[0193] Описание ссылочных номеров
10 - Исходное вещество
12 - Взрывчатое вещество
20 - Взрывной контейнер
22 - Усилитель детонатора
24 - Детонатор или детонирующий шнур
30 - Охлаждающий контейнер
32 - Охладитель
34 - Основание
36 - Перфорированный диск.
Реферат
Изобретение относится к углеродсодержащим покрытым частицам для применения в качестве катализатора или адсорбционного материала и способу их получения, а также функциональному материалу, при получении которого использованы такие частицы. Покрытые частицы получают детонационным способом с использованием непорохового исходного вещества, путем расположения взрывчатого вещества со скоростью детонации 6300 м/с или более вокруг исходного вещества, включающего в себя ароматическое соединение с двумя или менее нитрогруппами, детонирования этого взрывчатого вещества с получением углеродных частиц, включающих графит турбостратной структуры, наноалмазы и/или углеродные нанотрубки, фуллерены, и нанесения полученных углеродных частиц на поверхность частиц основного материала. Для получения функционального материала покрытые частицы наносят на поверхность материала подложки. Использование покрытых частиц придает смазывающие свойства и улучшает износостойкость материала. 5 н. и 4 з.п. ф-лы, 14 ил, 8 табл.
Комментарии