Каталитические системы для тетрамеризации этилена и способ получения 1-октена с их применением - RU2461423C2
Код документа: RU2461423C2
Чертежи
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к каталитической системе для тетрамеризации этилена и способу получения 1-октена тетрамеризацией этилена с применением каталитической системы.
УРОВЕНЬ ТЕХНИКИ
1-Октен, который является мономером или сомономером для получения линейного полиэтилена с низкой плотностью, является коммерчески важным исходным веществом, которое широко применяют в способе полимеризации и применяют в качестве специфического лекарственного средства.
Высшие α-олефины, необходимые для получения линейного полиэтилена с низкой плотностью, получают посредством реакции олигомеризации этилена. Однако реакция олигомеризации этилена является неэффективной в том отношении, что вместе с полиэтиленом образуется большое количество бутена, октена, производных октана и определенных высших олигомеров.
В общепринятых технологиях олигомеризации этилена обычно образуются различные α-олефины, зависящие от распределения продукта по Schulz-Flory или Poisson, и поэтому выход требуемых продуктов ограничивается. В связи с этим в патенте США №6184428 описан катализатор на основе никеля, содержащий 2-дифенилфосфинобензойную кислоту в качестве хелатного лиганда, NiCl2 6H2O в качестве предшественника никеля и тетрафенилборат натрия в качестве активатора катализатора. В этом патентном документе описано также, что при олигомеризации этилена с применением катализатора на основе никеля селективность получения 1-октена составляет 19%.
Кроме того, в патенте Германии №1443927 и патенте США №3906053 описаны катализаторы Циглера, полученные на основе катализатора триалкилалюминия. В этих патентных документах описано также, что с применением катализатора Циглера можно получить 13-15 мас.% 1-октена в расчете на общее количество смеси олефинов.
За последнее время проведены исследования по способам получения 1-октена селективной тетрамеризацией этилена с помощью катализа переходными металлами. В них наиболее обычно известными катализаторами переходных металлов являются катализаторы на основе хрома.
Недавно в WO 04/056479 описано, что 1-октен получают тетрамеризацией этилена с применением катализатора на основе хрома, включающего в себя гетероатомный лиганд, имеющий в качестве гетероатомов атомы фосфора и азота. В этой заявке примеры гетероатомного лиганда, который применяют для катализатора тетрамеризации этилена, могут включать в себя (фенил)2РN(изопропил)Р(фенил)2 и тому подобное.
В вышеуказанной общепринятой технологии описано также, что 1-октен можно получить при селективности больше 70 мас.% тетрамеризацией этилена с применением катализатора на основе хрома, включающего в себя гетероатомный лиганд, имеющий атомы фосфора и азота в качестве гетероатомов, без заместителей, которые являются полярными по сравнению с углеводородными группами или гетероуглеводородными группами, которые связаны с атомом фосфора.
Однако общепринятые технологии являются проблематичными в том, что относится к структуре лигандов, включающих в себя гетероатомы, в частности, они явно не могут демонстрировать очень селективное образование 1-октена при тетрамеризации этилена, когда катализаторы включают в себя такой тип лиганда, в котором они могут представлять только лиганд со структурой главной цепи PNP, например, (R1)(R2)P-(R5)N-P(R3)(R4), в качестве лиганда, имеющего селективность в отношении 1-октена приблизительно 70 мас.%, и в том, что структура заместителей лигандов, включающих в себя гетероатомы, также описана ограниченно. Кроме того, общепринятый лиганд со структурой главной цепи PNP, имеющий гетероатомы, также является проблематичным в том, что при получении 1-октена активность реакции нельзя поддерживать постоянной и скорость реакции также быстро снижается с прохождением времени реакции.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ПРОБЛЕМА
Авторы настоящего изобретения разрешили указанные выше проблемы обнаружением того факта, что каталитическая система на основе хрома, имеющая лиганд со структурой главной цепи Р-С-С-Р, не содержащий атом азота, можно применять для получения 1-октена тетрамеризацией этилена при селективности более 70 мас.%, и того, что активность каталитической системе на основе хрома поддерживается стабильной и таким образом можно предотвратить снижение скорости реакции с прохождением времени реакции. На основании полученных данных настоящее изобретение было завершено
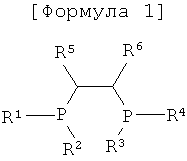
Соответственно этому настоящее изобретение было осуществлено с учетом указанных выше проблем, существующих в известном уровне техники, и задачей настоящего изобретения является обеспечение способа получения 1-октена при высокой активности и высокой селективности при стабильном поддержании реакционной способности тетрамеризацией этилена с применением каталитической системы на основе хрома, содержащей переходный металл или предшественник переходного металла, сокатализатор и лиганд со структурой главной цепи Р-С-С-Р, представленный следующей формулой 1
ТЕХНИЧЕСКОЕ РЕШЕНИЕ
Для выполнения указанной выше задачи в настоящем изобретении предложена каталитическая система для тетрамеризации этилена, содержащая переходный металл или предшественник переходного металла, сокатализатор и лиганд со структурой главной цепи Р-С-С-Р, представленный следующей формулой 1
где R1, R2, R3 и R4 представляют собой, каждый независимо, углеводородную группу, замещенную углеводородную группу, гетероуглеводородную группу и замещенную гетероуглеводородную группу, каждый из R1, R2, R3 и R4 не имеет заместитель на атоме, соседнем с атомом, связанным с Р, и R5 и R6 не являются водородом, но каждый независимо представляет собой углеводородную группу и замещенную углеводородную группу.
БЛАГОПРИЯТНЫЕ ЭФФЕКТЫ
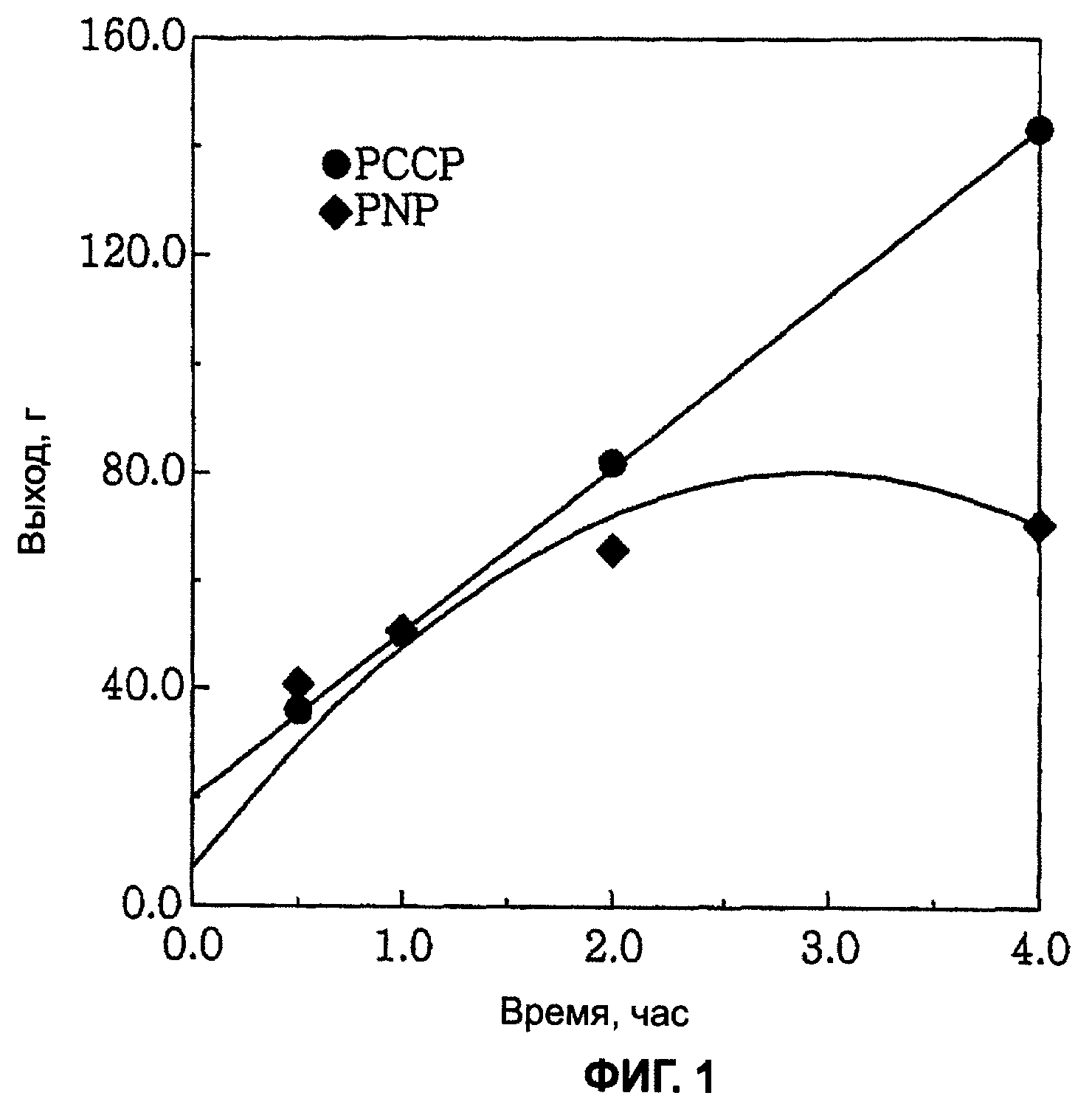
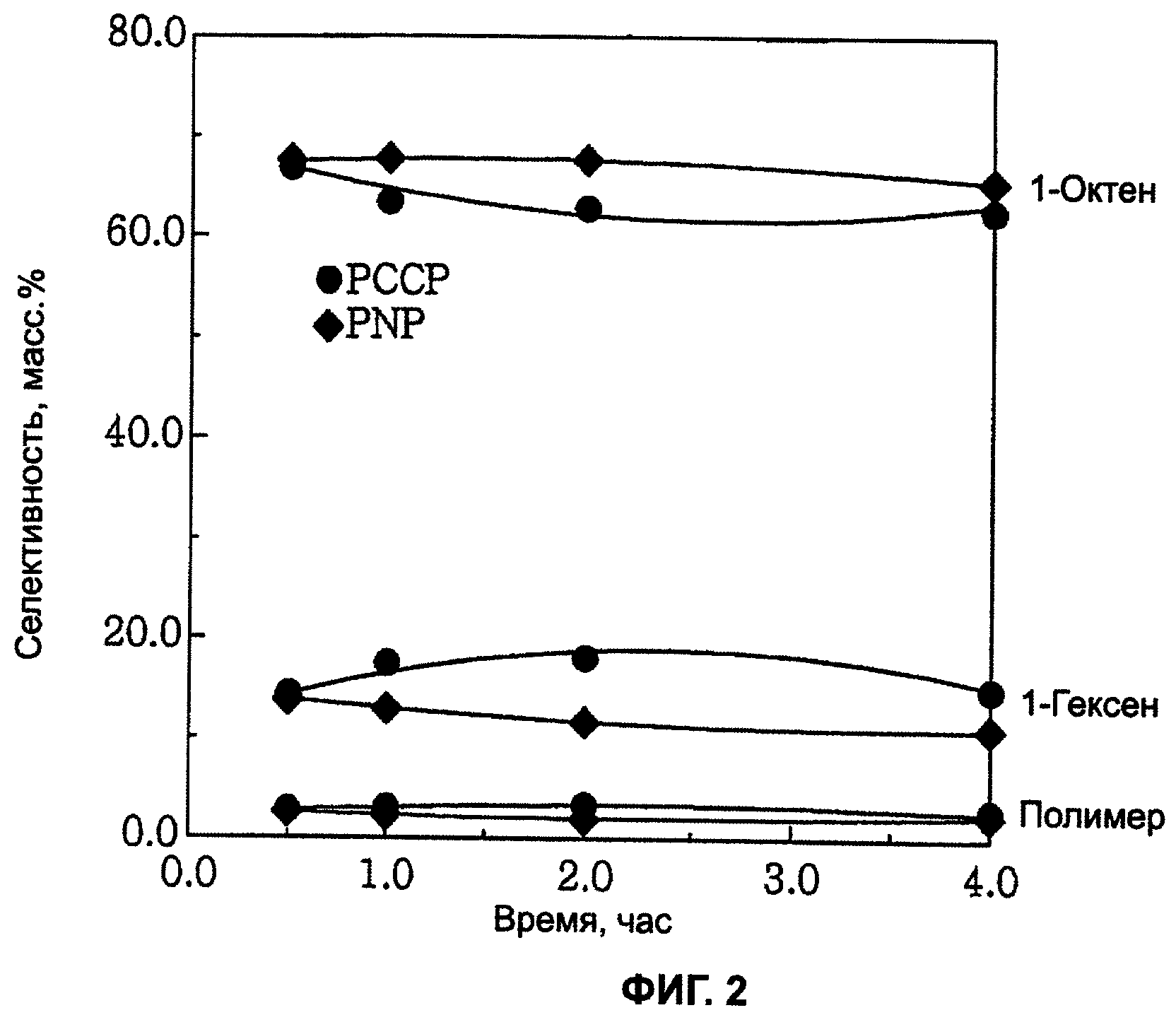
При тетрамеризации этилена с применением каталитической системы на основе хрома, включающей в себя лиганд со структурой главной цепи Р-С-С-Р, настоящего изобретения благоприятным является то, что можно получить очень чистый 1-октен, поскольку каталитическая система на основе хрома имеет высокую каталитическую активность и высокую селективность в отношении образования 1-октена, и то, что активность каталитической системы на основе хрома поддерживается стабильной и таким образом можно предотвратить снижение скорости реакции с прохождением времени реакции.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг.1 является графиком, показывающим изменение каталитической активности при реакции тетрамеризации этилена с применением каталитической системы согласно настоящему изобретению и
фиг.2 является графиком, показывающим изменение селективности при реакции тетрамеризации этилена с применением каталитической системы согласно настоящему изобретению.
ЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Далее настоящее изобретение будет описано подробно.
Настоящее изобретение обеспечивает каталитическую систему для тетрамеризации этилена, включающую в себя переходный металл или предшественник переходного металла, сокатализатор и лиганд со структурой главной цепи Р-С-С-Р, представленный указанной ниже формулой
где R1, R2, R3 и R4 которые являются произвольно выбранными заместителями и представляют собой, каждый независимо, углеводородную группу, замещенную углеводородную группу, гетероуглеводородную группу и замещенную гетероуглеводородную группу, соседние с атомами Р, не являются донорами электронов и могут быть неполярными группами; предпочтительно R1, R2, R3 и R4представляют собой, каждый независимо, замещенную ароматическую группу и замещенную гетероароматическую группу, которая не включает в себя доноры электронов на атомах, соседних с атомами, связанными с атомами Р.
В лиганде со структурой главной цепи Р-С-С-Р, представленном показанной выше формулой 1, R1, R2, R3 и R4 могут быть выбраны, каждый независимо, из группы, состоящей из фенила, бензила, нафтила, антраценила, мезитила, ксилила, метила, этила, этиленила, пропила, пропенила, пропинила, бутила, циклогексила, 4-метилциклогексила, 4-этилциклогексила, 4-изопропилциклогексила, толила, 4-метилфенила, 4-этилфенила, 4-изопропилфенила, 4-трет-бутилфенила, 4-метоксифенила, 4-изопропоксифенила, кумила, метокси, этокси, фенокси, толилокси, диметиламино, тиометила, триметилсилила и диметилгидразила. R1, R2, R3 и R4 могут быть, каждый независимо, предпочтительно выбраны из группы, состоящей из фенила, бензила, нафтила, 4-метилфенила, 4-этилфенила, 4-изопропилфенила, 4-трет-бутилфенила, 4-метоксифенила и 4-изопропоксифенила.
R1, R2, R3 и R4 могут быть, каждый независимо, ароматической группой и замещенной ароматической группой, каждый из R1, R2, R3 и R4 может быть замещен группой, не являющейся донором электронов, по меньшей мере на одном его атоме, который не является соседним с атомом, связанным с атомом Р, и каждый из R1, R2, R3 и R4 может быть замещен неполярной группой по меньшей мере на одном его атоме, который не является соседним с атомом, связанным с атомом Р.
Кроме того, в лиганде со структурой главной цепи Р-С-С-Р, представленном указанной выше формулой I, R5 и R6 не являются атомами водорода, но каждый независимо представляет собой углеводородную группу и замещенную углеводородную группу В частности, R5 и R6 могут быть, каждый независимо, выбраны из группы, состоящей из алкила, замещенного алкила, арила, замещенного арила, арилокси, замещенного арилокси, алкоксикарбонила, карбонилокси, алкокси, аминокарбонила, карбониламино, диалкиламино, силила и его производных и арила, замещенного этими произвольными заместителями.
Лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению может быть лигандом со структурой главной цепи мульти-Р-С-С-Р, представленной (R1)(R2P-(R5)CHCH(R6)-P(R3(R4), в котором в два или более лигандов со структурой главной цепи Р-С-С-Р связаны друг с другом. Этот лиганд со структурой главной цепи мульти-Р-С-С-Р включает в себя, но не ограничивается указанным, лиганды, у которых дендримерные лиганды связаны со соответствующим лигандом со структурой главной цепи Р-С-С-Р через одну или несколько групп R. Типичные примеры такого лиганда со структурой главной цепи мульти-Р-С-С-Р могут включать в себя 1,2,4,5-тетра-(Р-(4-этилфенил)2)циклогексан, 1,2,4,5-тетра-(Р-(4-этилфенил)2)бензол, 1,2,3,4-тетра-(Р-(4-этилфенил)2)циклопентан и тому подобное.
Примеры лиганда со структурой главной цепи Р-С-С-Р согласно настоящему изобретению могут включать в себя, но не ограничиваются перечисленным, (фенил)2Р-СН(метил)СН(метил)-Р(фенил)2, (4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенил)2, (4-метилфенил)2Р-СН(метил)СН(метил)-Р(4-метилфенил)2, (4-этилфенил)2Р-СН(метил)СН(метил)-Р(фенил)2, (4-этилфенил)2Р-СН(этил)СН(метил)-Р(4-этилфенил)2, (4-метоксифенил)2Р-СН(этил)СН(метил)-Р(фенил)2, (4-этилфенил)2Р-СН(этил)СН(этил)-Р(4-этилфенил)2, (фенил)2Р-СН(этил)СН(этил)-Р(фенил)2, (фенил)2Р-СН(изопропил)СН(метил)-Р(фенил)2, (4-метоксифенил)2Р-СН(изопропил)СН(метил)-Р(4-метоксифенил)2, (4-этилфенил)2Р-СН(изопропил)СН(метил)-Р(4-этилфенил)2, (фенил)2P-СН(н-пропил)СН(метил)-Р(фенил)2, (4-метоксифенил)2Р-СН(н-пропил)СН(метил)-Р(4-метоксифенил)2, (4-этилфенил)2Р-СН(н-пропил)СН(метил)-Р(4-этилфенил)2, (фенил)2P-СН(изопропил)СН(этил)-Р(фенил)2, (4-метоксифенил)2Р-СН(изопропил)СН(этил)-Р(4-метоксифенил)2, (4-этилфенил)2Р-СН(изопропил)СН(этил)-Р(4-этилфенил)2, 1,2-ди-(Р(фенил)2)циклогексан, 1,2-ди-(Р(4-метоксифенил)2)циклогексан, 1,2-ди-(Р(4-этилфенил)2)циклогексан, 1,2-ди-(Р(фенил)2)циклопентан, 1,2-ди-(Р(4-метоксифенил)2)циклопентан, 1,2-ди-(Р(4-этилфенил)2)циклопентан, 3,4-ди-(Р(фенил)2)пиррол, 3,4-ди-(Р(4-метоксифенил)2)пиррол, 3,4-ди-(Р(фенил)2)пиррол, 3,4-ди-(Р(4-метоксифенил)2)пиррол, 3,4-ди-(Р(4-этилфенил)2)пиррол, 3,4-ди-(Р(4-этилфенил)2)имидазол, (4-этилфенил)2P-СН(диметиламин)СН(диметиламин)-Р(4-этилфенил)2, (3-метоксифенил)2Р-СН(метил)СН(метил)-Р(3-метоксифенил)2, (4-этоксифенил)2Р-СН(метил)СН(метил)-Р(о-этоксифенил)2, (4-диметиламинфенил)2Р-СН(метил)СН(метил)Р(4-диметиламинофенил)2 и (4-этилциклогексил)2РСН(метил)СН(метил)Р(4-этилциклогексил)2. Лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению можно получить с применением различных способов, обычно известных специалистам в данной области.
Лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению, который является лигандом, имеющим независимую структуру, в отличие от обычного гетеролиганда (R)nPN(R')P(R)m имеет только один гетероатом, атом фосфора (Р), в его структуре главной цепи. То есть лиганд, который применяют в каталитической системе настоящего изобретения, имеет две углерод-углеродные главные цепи между двумя атомами фосфора, без атомов азота, так что проявляется превосходная каталитическая активность, и высокую селективность образования 1-октена 70 мас.% или выше можно достичь регулированием подходящим образом пространственной структуры лиганда варьированием заместителей, соседних с атомами углерода.
Для получения 1-октена при высокой селективности каталитическую систему, содержащую лиганд настоящего изобретения, можно изготовить посредством способа смешивания соединений переходных металлов и сокатализаторов добавлением в произвольном порядке.
Каталитическую систему согласно настоящему изобретению можно изготовить посредством способа получения координационного комплекса лиганда с применением соединений переходных металлов и лиганда со структурой главной цепи Р-С-С-Р. Здесь координационный комплекс лиганда, имеющего структуру главной цепи Р-С-С-Р, можно также получить in situ добавлением предварительно полученного координационного комплекса лиганда, который получают с применением соединений переходных металлов, и лиганда со структурой главной цепи Р-С-С-Р к реакционной смеси или дополнительно добавлением соединений переходных металлов и лиганда со структурой главной цепи Р-С-С-Р в реактор. Тот факт, что координационный комплекс лиганда, имеющего структуру главной цепи Р-С-С-Р, получают in situ, означает, что комплекс образуется в среде, в которой проводят каталитическую реакцию. Для получения in situ координационного комплекса лиганда соединение переходного металла и лиганд со структурой главной цепи Р-С-С-Р смешивают так, чтобы отношение металла к лиганду в смеси было приблизительно 0,01:1-100:1, предпочтительно приблизительно 0,1:1-10:1 и более предпочтительно 0,5:1-2.1. Переходный металл, который применяют для каталитической системы настоящего изобретения, можно выбрать из хрома, молибдена, вольфрама, титана, тантала, ванадия и циркония, предпочтительно им может быть хром. Соединением переходного металла для катализа тетрамеризации этилена согласно настоящему изобретению может быть неорганическая соль, органическая соль, координационный комплекс металла или металлоорганический комплекс, предпочтительно им может быть соединение хрома или предшественник соединения хрома. Здесь соединение хрома или предшественник соединения хрома может быть выбран из группы, состоящей из ацетилацетоната хрома(III), комплекса трихлорид хрома-тристетрагидрофуран и 2-этилгексаноата хрома(III).
Координационный комплекс соединения переходного металла и лиганда со структурой главной цепи Р-С-С-Р можно растворить при комнатной или более высокой температуре, но можно трансформировать для прикрепления к полимерным цепям так, чтобы он был нерастворимым. Кроме того, лиганд со структурой главной цепи Р-С-С-Р или соединение переходного металла можно фиксировать связыванием его с диоксидом кремния, силикагелем, полисилоксаном, оксидом алюминия или тому подобное.
Сокатализатором, который применяют в настоящем изобретении, может быть соединение, применяемое для активации катализатора, когда его смешивают с лигандом со структурой главной цепи Р-С-С-Р и соединением переходного металла. Активатором катализатора может быть отдельное соединение или смесь соединений. Предпочтительные примеры активатора катализатора могут включать в себя органические соединения алюминия, органические соединения бора и органические соли.
Органическое соединение алюминия может включать в себя соединение, представленное AlR3 (где каждый R представляет собой независимо алкил с 1-12 атомами углерода, кислородсодержащая алкильная группа или галогенид), и соединение, представленное LiAlH4. Примеры органического соединения алюминия могут включать в себя триметилалюминий (ТMА), триэтилалюминий (TEA), триизобутилалюминий (TIBA), три-н-октилалюминий, метилалюминийдихлорид, этилалюминийдихлорид, диметилалюминийхлорид, диэтилалюминийхлорид, изопропоксид алюминия, этилалюминийсесквихлорид, метилалюминийсесквихлорид и алюминоксан. Алюминоксан является хорошо известным в данной области как олигомерное соединение, которое можно получить смешиванием алкилалюминийсоединения, такого как триметилалюминий, с водой. Алюминоксановое олигомерное соединение может быть линейным алюминоксаном, циклическим алюминоксаном, каркасным алюминоксаном или смесью двух или более различных алюминоксанов.
Примеры органического соединения бора могут включать в себя бороксин, NaBH4, триэтилборан, трифенилборан, комплекс трифенилборана и аммиака, трибутилборат, триизопропилборат, трис(пентафторфенил)боран, тритил(тетрапентафторфенил)борат, диметилфениламмоний(тетрапентафторфенил)борат, диэтилфениламмоний(тетрапентафторфенил)борат, метилдифениламмоний(тетрапентафторфенил)борат, этилдифениламмоний(тетрапентафторфенил)борат и тому подобное Это органическое соединение бора можно применять в форме, в которой он смешан с органическим соединением алюминий.
Кроме того, алюминоксан, применяемый в качестве сокатализатора каталитической системы согласно настоящему изобретению, можно выбрать из алкилалюминоксана, такого как метилалюминоксан (MАО) и этилалюминоксан (ЕАО), и модифицированного алкилалюминоксана, такого как модифицированный метилалюминоксан (MMАО). Модифицированный метилалюминоксан (MMАО), изготовленный Akzo Nobel Corp., включает в себя гибридную алкильную группу, такую как изобутильная группа или н-октильная группа, а также метильная группа.
Алюминоксан может быть в комбинации с соединением переходного металла, особенно соединением хрома, так чтобы отношение алюминия к металлу в комбинации было приблизительно 1:1-10000:1 и предпочтительно приблизительно 1:1-1000:1.
Каталитические компоненты, образовывающие каталитическую систему настоящего изобретения, можно комбинировать друг с другом одновременным или последовательным добавлением в присутствии растворителя или в отсутствие растворителя. Смешивание каталитических компонентов можно проводить при температуре от -20 до 250°С и предпочтительно 20-100°С. В то время как каталитические компоненты смешивают, олефин проявляет защитное действие, таким образом улучшая рабочую характеристику катализатора.
Продукт реакции согласно настоящему изобретению, то есть 1-октен, можно получить посредством реакции в гомогенной жидкости, которую проводят в присутствии неактивного растворителя или в отсутствие неактивного растворителя с применением каталитической системы, обычной аппаратуры и общепринятой технологии контактирования, реакции в суспензии, при которой часть каталитической системы или вся каталитическая система не растворена, реакции в двухфазной жидкостно-жидкостной системе или реакции в объемной фазе или газовой фазе, в которой олефин действует в качестве основной среды.
Когда 1-октен получают с применением каталитической системы настоящего изобретения в присутствии неактивного растворителя, в качестве неактивного растворителя можно применять произвольные неактивные растворители, которые не реагируют с каждым каталитическим компонентом и сокатализатором. Такие неактивные растворители могут включать в себя насыщенные алифатические углеводороды, ненасыщенные алифатические углеводороды, ароматические углеводороды и галогенированные углеводороды и предпочтительно могут включать в себя, но не ограничиваются перечисленным, бензол, толуол, ксилол, кумол, гептан, циклогексан, метилциклогексан, метилциклопентан, н-гексан, 1-гексен и тому подобное.
Способ получения 1-октена с применением каталитической системы настоящего изобретения можно проводить при температуре от -20 до 250°С, предпочтительно от 15 до 130°С и более предпочтительно от 30 до 70°С и давлении реакции от атмосферного давления до 5×107 Па (500 бар), предпочтительно 106 Па (10 бар) - 7×106 Па (70 бар) и более предпочтительно 3×106 Па (30 бар) - 5×106 Па (50 бар).
В предпочтительном варианте осуществления настоящего изобретения координационный комплекс лиганда со структурой главной цепи Р-С-С-Р и условия реакции выбирают так, чтобы выход 1-октена из этилена был 30 мас.% или больше, и предпочтительно 50 мас.% или больше. В этом случае выход 1-октена означает массовое отношение 1-октена к общим продуктам реакции. Кроме того, в способе настоящего изобретения помимо 1-октена можно получить большое или небольшое количество 1-бутена, 1-гексена, метилциклопентана, метиленциклопентана и пропилциклопентана и большое количество высших олигомеров и полиэтилена в зависимости от лиганда со структурой главной цепи Р-С-С-Р и условия для реакции.
Способ тетрамеризации этилена согласно настоящему изобретению можно проводить с применением установки, снабженной различными типами реакторов. Примеры реакторов могут включать в себя, но не ограничиваются перечисленным, реактор периодического действия, реактор полупериодического действия и реактор непрерывного действия. Установка может включать в себя реактор, реактор для олефина и впускное отверстие для подачи каталитической системы в реакторе, линия для разгрузки олигомеризованных продуктов из реактора и по меньшей мере один сепаратор для разделения олигомеризованных продуктов.
1-Октен можно получить при высокой активности и высокой селективности при стабильно поддерживаемой реакционной способности тетрамеризацией этилена с применением каталитической системы настоящего изобретения.
СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В дальнейшем настоящее изобретение будет описано более подробно посредством нижеследующих примеров, которые приводятся для иллюстрации, но не должны истолковываться как ограничение настоящего изобретения.
ПРИМЕРЫ
Пример 1 получения катализатора: получение лиганда S.S-(фенил)2РСН(метил)СН(метил)Р(фенил)2
Лиганд S,S-(фенил)2РСН(метил)СН(метил)Р(фенил)2 получали, как описано в публикации "В. Bosnich et al, J. Am. Chem. Soc. 99(19) (1977) 6262".
Ди-п-толуолсульфонат (2R,3R)-дибутандиола получали из (2R,3R)-дибутандиола. Этот способ получения ди-п-толуолсульфоната (2R,3R)-дибутандиола проводили, как описано в публикации "R. В. Mitra et al, J. Am. Chem. Soc 84 (1962)". 100 мл (1,24 моль) высушенного пиридина помещали в колбу на 1 л, которую охлаждали на бане вода-лед и смешивали с 100 г (0,525 моль) п-толуолсульфонилхлорида и затем медленно по каплям добавляли 22 мл (0,245 моль) (2R,3R)-дибутандиола с образованием смеси. Смесь нагревали в течение 20 минут до достижения комнатной температуры и затем выдерживали в полутвердом состоянии при комнатной температуре на протяжении ночи. К смеси добавляли избыточные кусочки льда и затем ее энергично встряхивали для предотвращения агломерации. После того, как наблюдали, что из смеси медленно выделяются кристаллы порошка, смесь перемешивали в течение 2 часов вместе с кусочками льда и затем к этой смеси при перемешивании добавляли 70 мл раствора концентрированной хлористоводородной кислоты и измельченные кусочки льда, так чтобы образовывалась суспензия. Образованную таким образом суспензию фильтровали, промывали водой и затем сушили, получая при этом 85 г (86,3%) ди-п-толуолсульфоната (2R,3R)-дибутандиола, имеющего точку плавления 62-64°С.
Тем временем 95 г перекристаллизованного трифенилфосфора и 300 мл высушенного тетрагидрофурана (ТГФ) помещали в трехгорлую колбу на 1 л, снабженную 250 мл воронкой для добавления по каплям, холодильником для кипячения с обратным холодильником и вводом для азота, получая при этом раствор. К раствору в атмосфере азота при температуре 25°С и при перемешивании добавляли 5,0 г кусочков лития. После этого одновременно в растворе образовывался LiPPh2, из раствора выделялось тепло и раствор становился темного красновато-желтого цвета. Раствор медленно нагревали в течение 1 час до температуры 55°С и затем охлаждали в течение 2 час до температуры 25°С при перемешивании. Образованный фениллитий разлагали добавлением в него по каплям 33 г перегнанного и очищенного трет-бутилхлорида в течение 45 минут. Красновато-желтый раствор нагревали в течение 5 минут и затем охлаждали до температуры -4°С.
После этого 35 г полученного ди-п-толуолсульфоната (2R,3R)-дибутандиола растворяли в 100 мл высушенного тетрагидрофурана (ТГФ) и затем раствор добавляли по каплям в красновато-желтый раствор в течение 1 час с образованием смешанного раствора. Смешанный раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 минут. 300 мл азотсодержащей воды добавляли к смешанному раствору и затем тетрагидрофуран (ТГФ) удаляли из него посредством вакуумной дистилляции, тем самым экстрагируя из него бесцветный маслянистый продукт. Маслянистый продукт экстрагировали дважды с применением 150 мл простого эфира и затем сушили с применением Na2SO4 с образованием экстракта в эфире. Экстракт в эфире фильтровали в раствор 50 мл этанола и 15 г гексагидрата перхлората никеля в атмосфере азота. Na2SO4, оставшийся в отфильтрованном экстракте, в эфире полностью промывали, получая при этом раствор в эфире и затем раствор в эфире добавляли к раствору соединения никеля. В результате образовывался красновато-коричневый маслянистый продукт, содержащий желтые кристаллы, [Ni((S,S)-хирафос)2](ClO4)2. Эту маслянистую кристаллическую смесь добавляли к раствору, в котором 15 г тиоцианата натрия растворено в 50 мл этанола, с образованием раствора смеси и затем раствор смеси энергично перемешивали в течение нескольких часов, получая при этом желтовато-коричневый твердый продукт, [Ni((SS)-хирафос)2NCS]NCS. Этот твердый продукт полностью промывали этанолом и затем, наконец, промывали простым эфиром, получая при этом комплекс никеля.
К 15 г этого комплекса никеля добавляли 150 мл этанола в атмосфере азота с всплыванием комплекса на поверхность этанола и затем смесь перемешивали и нагревали. К комплексу никеля добавляли 20 г воды и 4 г цианата натрия (NaCN). В результате этого комплекс никеля медленно растворяли и таким образом превращали в красный раствор, [Ni((S,S)-xиpaфoc)2CN3]-, и затем красный раствор превращался в мутный бежевый раствор. Мутный раствор перемешивали с образованием желтой суспензии. Суспензию охлаждали с образованием твердого вещества и затем твердое вещество дважды промывали 25 мл воды и затем быстро охлаждали с применением этанола, охлажденного льдом, получая при этом бежевое твердое вещество, содержащее примеси. Бежевое твердое вещество, содержащее примеси, сушили при температуре 25°С, добавляли к 125 мл безводного этанола и затем фильтровали с применением фильтра Фритца при комнатной температуре в течение 12 час. В результате этого оставалось только бесцветное блестящее твердое вещество. Наконец, бесцветное блестящее твердое вещество перекристаллизовывали с применением 60 мл безводного этанола, получая при этом 5,5 г бесцветного чистого S,S-(фенил)2РСН(метил)СН(метил)Р(фенил)2.
Пример 1: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3 (фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
100 мл циклогексана и 4,0 ммоль Al метилалюминоксана (MAO) помещали в реактор из нержавеющей стали на 300 мл, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 3,5 мг (0,010 ммоль) Сr(III)(ацетилацетоната)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 4,3 мг (0,010 ммоль) (фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора, с получением смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем взвешивали, получая при этом 3,6 г полиэтилена. Кроме того, было найдено, что общая масса реакционной смеси, определенная при помощи ГХ, была 101,6 г.Распределение продуктов данного примера указывается в таблице 1.
Пример 2: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3 (фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
100 мл циклогексана и 2,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 0,7 мг (0,002 ммоль) Сr(III)(ацетилацетоната)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 0,86 мг (0,002 ммоль) (фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора, с получением смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 18,0 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 3: тетрамеризация этилена с применением соединений СrCl3(тетрагидрофуран)3, (фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
100 мл циклогексана и 2,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 3,75 мг (0,01 ммоль) комплекса СrСl3(тетрагидрофурана)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 4,3 мг (0,01 ммоль) (фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 36,2 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 4: тетрамеризация этилена с применением соединений Сr-(2-этилгексаноат)3, (фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
100 мл циклогексана и 4,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азоте или в вакууме, и затем нагревали до температуры 45°С 4,0 мг (0,01 ммоль) Сr-(2-этилгексаноат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 4,3 мг (0,01 ммоль) (фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 76,0 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 5: тетрамеризация этилена с применением соединений Сr-(2-этилгексаноат)3, (фенил)2РСН(метил)СН(метил)Р(фенил)2 и МАО
100 мл циклогексана и 2,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 0,8 мг (0,002 ммоль) Сr-(2-этилгексаноат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 0,86 мг (0,002 ммоль) (фенил)2PСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ.
Было найдено, что масса продукта, определенная при помощи ГХ, была 11,2 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 2 получения катализатора: получение лиганда R,R-(4-метоксифенил)2РСН(метил)СН(метил)-Р(4-метоксифенил)2
Лиганд R,R-(4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенил)2получали, как описано в публикации "В. Bosnich et al, J. Am. Chem. Soc. 99(19) (1977) 6262".
Получение ди-п-толуолсульфоната (2R,3R)-дибутандиола из (2R,3R)-дибутандиола проводили с применением такого же способа, как в примере 1 получения катализатора.
Получение три-(4-метоксифенил)фосфора проводили следующим образом. Кусочки магния (91,1 г, 3,75 моль) добавляли порциями в 95 мл (0,75 моль) 4-броманизола в 2 л тетрагидрофурана (ТГФ). Смесь бурно реагировала и затем ее нагревали и кипятили с обратным холодильником в течение 2 час с получением реактива Гриньяра. Реактив Гриньяра добавляли по каплям в 17,5 мл (0,2 моль) раствора РСl3 в 2 л тетрагидрофурана (ТГФ) при температуре -78°С в течение 2 час при перемешивании. После этого баню сухого льда и ацетона для охлаждения реакционной смеси убирали и затем продукт реакции нагревали до комнатной температуры. Продукт реакции перемешивали на протяжении ночи и растворитель из него удаляли в вакууме, получая при этом фосфин. Весь продукт реакции применяли в следующих процессах без выделения из него фосфина.
Тем временем 70 г перекристаллизованного три-(4-метоксифенил)фосфора и 300 мл высушенного тетрагидрофурана (ТГФ) помещали в трехгорлую колбу на 1 л, снабженную 250 мл воронкой для добавления по каплям, холодильником для кипячения с обратным холодильником и вводом для азота, с образованием раствора. К раствору в атмосфере азота при температуре 25°С и при перемешивании добавляли 2,8 г кусочков лития. После этого одновременно в растворе образовывался LiP(4-OMe-Ph)2, в растворе выделялось тепло и раствор становился темным красновато-желтым. Раствор медленно нагревали в течение 1 час до температуры 55°С и затем охлаждали в течение 2 час до температуры 25°С при перемешивании. Таким образом образованный 4-метоксифениллитий разлагали добавлением в него по каплям 18,5 г перегнанного и очищенного трет-бутилхлорида в течение 45 минут. Красновато-желтый раствор нагревали в течение 5 минут и затем охлаждали до температуры -4°С.
После этого 19,6 г таким образом полученного ди-п-толуолсульфоната (2R,3R)-дибутандиола растворяли в 100 мл высушенного тетрагидрофурана (ТГФ) и затем раствор добавляли по каплям в красновато-желтый раствор на протяжении 1 час с образованием смешанного раствора. Смешанный раствор медленно нагревали до комнатной температуры и затем перемешивали в течение 30 минут. 300 мл азотсодержащей воды добавляли к смешанному раствору и затем тетрагидрофуран (ТГФ) удаляли из него посредством вакуумной дистилляции, тем самым экстрагируя из него бесцветный маслянистый продукт. Маслянистый продукт экстрагировали дважды с применением 150 мл простого эфира и затем сушили с применением Na2SO4 с образованием экстракта в эфире. Экстракт в эфире фильтровали в раствор 50 мл этанола и 8,4 г гексагидрата перхлората никеля в атмосфере азота. Na2SO4, оставшийся в отфильтрованном экстракте, в эфире полностью промывали, получая при этом раствор в эфире и затем раствор в эфире добавляли к раствору соединения никеля. В результате этого образовывался красновато-коричневый маслянистый продукт, содержащий желтые кристаллы, [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2](СlO4)2. Эту смесь масла с кристаллами добавляли к раствору, в котором 8,4 г тиоцианата натрия растворено в 50 мл этанола, с образованием раствора смеси и затем раствор смеси энергично перемешивали в течение нескольких часов, получая при этом желтовато-коричневый твердый продукт, [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2NCS]NCS. Этот твердый продукт полностью промывали этанолом и затем, наконец, промывали простым эфиром, получая при этом комплекс никеля.
К 17 г этого комплекса никеля добавляли 150 мл этанола в атмосфере азота с всплыванием комплекса на поверхность этанола и затем комплекс перемешивали и нагревали. К комплексу никеля добавляли 20 г воды и 4 г цианата натрия (NaCN). Тем самым комплекс никеля медленно растворяли и таким образом его превращали в красный раствор, [Ni((2S,3S)-бис(ди-п-метоксифенил)фосфорбутан)2CN3]-, и затем красный раствор превращали в мутный бежевый раствор. Мутный раствор перемешивали с образованием желтой суспензии. Суспензию охлаждали с образованием твердого вещества и затем твердое вещество дважды промывали 25 мл воды и затем быстро охлаждали с применением этанола, охлажденного льдом, получая при этом бежевое твердое вещество, содержащее примеси. Бежевое твердое вещество, содержащее примеси, сушили при температуре 25°С, добавляли к 125 мл кипящего безводного этанола и затем фильтровали с применением фильтра Фритца при комнатной температуре в течение 12 час. В результате этого оставалось только бесцветное блестящее твердое вещество. Наконец, бесцветное блестящее твердое вещество перекристаллизовывали с применением 60 мл безводного этанола, получая при этом 6,2 г бесцветного чистого соединения S,S-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2.
Пример 6: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, R,R-(4-метоксифенил)2Р-СН(метил)СН(метил)Р(4-метоксифенил)2и МАО
100 мл циклогексана и 4,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азоте или в вакууме, и затем нагревали до температуры 45°С. 3,5 мг (0,010 ммоль) Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 5,5 мг (0,010 ммоль) R,R-(4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенил)2, полученного в примере 2 получения катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем взвешивали и показали, что получили 1,9 г полиэтилена. Кроме того, было показано, что общая масса реакционной смеси, определенная с помощью ГХ, была 45,5 г.Распределение продуктов данного примера указывается в таблице 1.
Пример 7: тетрамеризация этилена с применением соединений СrСl3(тетрагидрофуран)3, R,R-(4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенил): и МАО
100 мл циклогексана и 4,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 3,75 мг (0,010 ммоль) СrСl3(тетрагидрофуран)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 5,5 мг (0,010 ммоль) R,R-(4-метоксифенил)2РСН(метил)СН(метил)Р-(4-метоксифенил)2, полученного в примере 2 получения катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было показано, что масса продукта, определенная при помощи ГХ, была 25,3 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 8: тетрамеризация этилена с применением соединений Сr(2-этилгексаноат)3. R,R-(4-метоксифенил)2Р-СН(метил)СН(метил)-Р(4-метоксифенил)2 и МАО
100 мл циклогексана и 4,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 4,0 мг (0,01 ммоль) Сr(2-этилгексаноат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 5,5 мг (0,010 ммоль) R,R-(4-метоксифенил)2РСН(метил)СН(метил)Р(4-метоксифенил)2, полученного в примере 2 получения катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было показано, что масса продукта, определенная при помощи ГХ, была 48,2 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 1 получения сравнительного катализатора: получение лиганда (фенил)2РN(изопропил)Р(фенил)2
Смешанный гетероатомный лиганд PNP получали реакцией амина с хлоридом фосфина (R2PCl), как описано в публикациях: (a) "Ewart et al, J. Chem. Soc. 1964, 1543"; (b) "Dossett, S. J. et. al, Chem.Commun., 2001, 8, 699" и (с) "Balakrishna, М. S. et al, J. Organomet. Chem. 1990, 390, 2, 203". Кроме того, реакционноспособный хлорид фосфина (R2PCl) получали, как описано в публикациях: "Casalnuovo, A. L. et al, J. Am. Chem. Soc. 1994, 116, 22, 9869" и "Rajanbabu, Т. V. et al, J. Org. Chem. 1997, 62, 17, 6012".
15 мл триэтиламина и 28 ммоль хлордифенилфосфина растворяли в 80 мл DCM и затем к раствору добавляли 1,11 мл (13 ммоль) изопропиламина. Реакционную смесь перемешивали в течение 30 минут и затем из нее удаляли примеси. Реакционную смесь далее перемешивали в течение 24 час и затем фильтровали для удаления из нее солей триэтиламмония. Продукт кристаллизовали и затем выделяли, получая при этом лиганд (фенил)2РN(изопропил)Р(фенил)2 с выходом 85%.
Сравнительный пример 1: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2РN(изопропил)Р(фенил)2 и МАО
100 мл циклогексана и 3,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 5,2 мг (0,015 ммоль) Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 6,4 мг (0,015 ммоль) (фенил)2PN(изопропил)Р(фенил)2, полученного в примере 1 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ.
Было найдено, что масса продукта, определенная при помощи ГХ, была 32,2 г. Распределение продуктов данного примера указывается в таблице 1.
Сравнительный пример 2: тетрамеризация этилена с применением соединений Сr(III)(2-этилгексаноат)3, (фенил)2РN(изопропил)Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 5,2 мг (0,015 ммоль) Сr(III)(2-этилгексаноата)2 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 6,4 мг (0,015 ммоль) (фенил)2PN(изопропил)Р(фенил)2, полученного в примере 1 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 70,0 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 2 получения сравнительного катализатора: получение лиганда (фенил)2РСН2P(фенил)2
Лиганд (фенил)2РСН2Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидрохлоридом цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к 16,6 мл безводного N.N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 А, и затем смесь перемешивали в атмосфере азота. Затем к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,09 мл (1,29 ммоль) дибромметана, после чего раствор становился белым Раствору давали возможность реагировать в течение 16 час при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMC с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в бензоле, тем самым получая чувствительные к воздуху белые кристаллы (390 мг, выход 95%).
Сравнительный пример 3: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3. (фенил)2РСН2Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 10,5 мг (0,03 ммоль) Сr(III)(ацетилацетоната)2 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 11,5 мг соединения (0,03 ммоль) (фенил)2РСН2P(фенил)2, полученного в примере 2 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×10° Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторое образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 1,47 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 3 получения сравнительного катализатора: получение лиганда (фенил)2РСН2СН2Р(фенил)2
Лиганд (фенил)2РСН2СН2Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,11 мл (1,29 ммоль) 1,2-дибромэтана, после чего раствор становился белым. Раствору давали возможность реагировать в течение 36 час при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMC с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (333 мг, выход 78%).
Сравнительный пример 4: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2РСН2СН2Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 5,2 мг (0,015 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 7,8 мг (0,02 ммоль) соединения (фенил)2РСН2СН2Р(фенил)2, полученного в примере 3 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ.
Было найдено, что масса продукта, определенная при помощи ГХ, была 10,4 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 4 получения сравнительного катализатора: получение лиганда ((фенил)2Р(СН2)3(фенил)2
Лиганд (фенил)2Р(СН2)3Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 А, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,13 мл (1,29 ммоль) 1,2-дибромпропана, после чего раствор становился белым. Раствору давали возможность реагировать в течение 45 час при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMC с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (366 мг, выход 83%).
Сравнительный пример 5: тетрамеризация этилена с применением соединений Сr(III) (ацетилацетонат)3, (фенил)2Р(СН2)3Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 9,6 мг (0,028 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 13,8 мг (0,033 ммоль) соединения (фенил)2PСН2Р(фенил)2, полученного в примере 4 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 28,8 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 5 получения сравнительного катализатора: получение лиганда (фенил)2Р(СН2)4Р(фенил)2
Лиганд (фенил)2Р(СН2)4Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 часа с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,16 мл (1,29 ммоль) 1,2-дибромбутана, после чего раствор становился белым. Раствору давали возможность реагировать в течение 48 час при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMС с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (397 мг, выход 87%).
Сравнительный пример 6: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2Р(СН2)4Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 9,7 мг (0,028 ммоль) Сr(III)(ацетилацетоната)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 15,4 мг (0,036 ммоль) (фенил)2Р(СН2)4Р(фенила)2, полученного в примере 5 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Пa (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 18,5 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 6 получения сравнительного катализатора: получение лиганда (фенил)2Р(СН=СН)Р(фенил)2
Лиганд (фенил)2Р(СН=СН)Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкена в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,11 мл (1,29 ммоль) 1,2-дибромэтилена, после чего раствор становился белым Раствору давали возможность реагировать в течение 48 часов при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMC с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (284 мг, выход 67%).
Сравнительный пример 7: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2Р(СН=СН)Р(фенил)2 и МАО
100 мл циклогексана и 9,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 10,5 мг (0,003 ммоль) Сr(III)(ацетилацетоната)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 23,8 мг (0,06 ммоль) (фенил)2Р(СН=СН)Р(фенила)2, полученного в примере 6 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 1,3 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 7 получения сравнительного катализатора: получение лиганда (фенил)2Р(1,2-фенилен)Р(фенил)2
Лиганд (фенил)2Р(1,2-фенилен)Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромбензола в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 часа с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,16 мл (1,29 ммоль) 1,2-дибромбензола, после чего раствор становился белым. Раствору давали возможность реагировать в течение 60 час при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMС с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (358 мг, выход 75%).
Сравнительный пример 8: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)2, ((фенил)2Р(1,2-фенилен)Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 10,5 мг (0,003 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 21,8 мг (0,049 ммоль) соединения (фенил)2Р(1,2-фенилен)Р(фенил)2, полученного в примере 7 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 19,6 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 8 получения сравнительного катализатора: получение лиганда (циклогексил)2РСН2Р(циклогексил)2
Лиганд (циклогексил)2РСН2Р(циклогексил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,43 мл (2,14 ммоль) дициклогексилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,09 мл (1,29 ммоль) 1,2-дибромметана, после чего раствор становился белым. Раствору давали возможность реагировать в течение 38 часов при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMC с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (372 мг, выход 85%).
Сравнительный пример 9: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)2, (циклогексил)2РСН2Р(циклогексил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 10,5 мг (0,003 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 12,2 мг (0,03 ммоль) соединения (циклогексил)2РСН2Р(циклогексил)2, полученного в примере 8 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ.
Было найдено, что масса продукта, определенная при помощи ГХ, была 9,4 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 9 получения сравнительного катализатора: получение лиганда (циклогексил)2РСН2СН2Р(циклогексил)2
Лиганд (циклогексил)2РСН2СН2Р(циклогексил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,43 мл (2,14 ммоль) дициклогексилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,11 мл (1,29 ммоль) 1,2-дибромэтана, после чего раствор становился белым Раствору давали возможность реагировать в течение 49 часов при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMС с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (366 мг, выход 81%).
Сравнительный пример 10: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (циклогексил)2РСН2СН2Р(циклогексил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 10,5 мг (0,003 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 12,7 мг (0,03 ммоль) (циклогексил)2РСН2СН2Р(циклогексил)2, полученного в примере 9 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 2,2 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 10 получения сравнительного катализатора: получение лиганда (этил)2РСН2СН2(этил)2
Лиганд (этил)2РСН2СН2Р(этил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R. N Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,25 мл (2,14 ммоль) диэтилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,11 мл (1,29 ммоль) 1,2-дибромэтана, после чего раствор становился белым. Раствору давали возможность реагировать в течение 72 часов при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMС с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (126 мг, выход 57%).
Сравнительный пример 11: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (этил)2РСН2СН2Р(этил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (MАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 5,4 мг (0,016 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 3,3 мг (0,016 ммоль) соединения (этил)2РСН2СН2Р(этил)2, полученного в примере 10 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 2,7 г. Распределение продуктов данного примера указывается в таблице 1.
Пример 11 получения сравнительного катализатора: получение лиганда (фенил)2РСH(метил)СН2Р(фенил)2
(Фенил)2РСН(метил)СН2Р(фенил)2 получали реакцией дифенилфосфина с 2 эквивалентами дибромалкана в диметилформамиде (ДМФА) и гидроксида цезия в атмосфере, как описано в публикации "R.N. Salvatore et al, Tetrahedron Letters 44 (2003) 8373". Сначала 360 мг (2,14 ммоль) моногидрата гидроксида цезия добавляли к суспензии 16,6 мл безводного N,N-диметилформамида, смешанного с 1,0 г порошка активированного молекулярного сита, имеющего размер пор частиц 4 Å, и затем смесь перемешивали в атмосфере азота. После этого к смеси добавляли 0,38 мл (2,14 ммоль) дифенилфосфина и затем смесь перемешивали при комнатной температуре в течение 1 час с образованием темного красновато-оранжевого раствора. В раствор по каплям добавляли 0,14 мл (1,3 ммоль) 1,2-дибромпропана, после чего раствор становился белым. Раствору давали возможность реагировать в течение 72 час при комнатной температуре и к нему добавляли 60 мл дистиллированной воды и раствор экстрагировали три раза с применением 60 мл DMС с образованием органического слоя. Органический слой промывали три раза дистиллированной водой и сушили с применением безводного сульфата натрия, растворитель удаляли из него в вакууме и затем органический слой, из которого растворитель удалили, перекристаллизовывали в растворителе бензоле, тем самым получая чувствительные к воздуху белые кристаллы (309 мг, выход 70%).
Сравнительный пример 12: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2РСН(метил)СН2Р(фенил)2 и МАО
100 мл циклогексана и 6,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 1,7 мг (0,005 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 2,1 мг соединения (0,005 ммоль) (фенил)2РСН(метил)СН2Р(фенил)2, полученного в примере 11 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об % хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 4,8 г. Распределение продуктов данного примера указывается в таблице 1.
Сравнительный пример 13: тетрамеризация этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2РСН(метил)СН2Р(фенил)2 и МАО
100 мл циклогексана и 4,0 ммоль Al метилалюминоксана (МАО) помещали в реактор из нержавеющей стали на 300 мл примера 1, который был промыт в потоке азота или в вакууме, и затем нагревали до температуры 45°С. 3,4 мг (0,010 ммоль) соединения Сr(III)(ацетилацетонат)3 в 10 мл толуола помещали в колбу Шленка на 50 мл в ящике с перчатками и затем смешивали с 4,2 мг (0,010 ммоль) соединения (фенил)2РСН(метил)СН2Р(фенил)2, полученного в примере 11 получения сравнительного катализатора, с образованием смеси и затем смесь перемешивали при комнатной температуре в течение 5 минут и затем добавляли в реактор. После этого в реактор загружали этилен при давлении 3×106 Па (30 бар) и затем перемешивали при скорости перемешивания 600 об/мин. Спустя 30 мин подачу этилена и перемешивание его прекращали и реактор охлаждали до температуры ниже 10°С.
После этого избыток этилена удаляли из реактора и затем этанол, смешанный с 10 об.% хлористоводородной кислоты, добавляли к жидкости, присутствующей в реакторе. Для анализа жидкости с применением ГХ-FID в нее добавляли нонан, служащий в качестве внутреннего стандартного вещества. Некоторые образцы органического слоя пропускали через безводный сульфат магния, сушили и затем анализировали с применением ГХ-FID. Другие образцы органического слоя фильтровали и затем из них выделяли продукт твердый парафин/полимер. Этот твердый продукт сушили в сушильном шкафу при температуре 100°С на протяжении ночи и затем анализировали с применением ГХ. Было найдено, что масса продукта, определенная при помощи ГХ, была 6,0 г. Распределение продуктов данного примера указывается в таблице 1.
Примеры 9-12: изменения в реакционной способности и селективности в зависимости от времени реакции при тетрамеризации этилена с применением соединений Сr(III)(ацетилацетонат)3, (фенил)2РСН(метил)СН(метил)Р(фенила)2 и ММАО-12
Тетрамеризацию этилена проводили с применением 1,75 мг (0,005 ммоль) соединения Сr(III)(ацетилацетонат)3, 2,2 мг(0,005 ммоль) соединения (фенил)2РСН(метил)СН(метил)Р(фенил)2, полученного в примере 1 получения катализатора, и 3 ммоль ММАО-12, изготовленного Akzo-Nobel Corp., при температуре реакции 45°С и давлении этилена 3×106 Па (30 бар) в течение времени реакции 30 минут (пример 9), 1 час (пример 10), 2 час (пример 11) и 4 час (пример 12). В других случаях реакцию и обработку продукта способа проводили, как в примере 1.
Результаты тетрамеризации этилена в примерах 9-12 указываются в таблице 2, и изменения в реакционной способности и селективности в зависимости от времени реакции показаны на фиг 1 и 2.
Сравнительные примеры 14-17: изменения в реакционной способности и селективности в зависимости от времени реакции при тетрамеризации этилена с применением соединений Сr(III)(ацетилацетонат)2, (фенил)2РN(изопропил)Р(фенил)2 и ММАО-12
Тетрамеризацию этилена проводили, как в примерах 9-12, за исключением того, что 2,1 мг (0,005 ммоль) соединения (фенил)2РN(изопропил)Р(фенил)2 примера 1 получения сравнительного катализатора применяли вместо соединения (фенил)2РСН(метил)СН(метил)Р(фенил)2.
Результаты тетрамеризации этилена в сравнительных примерах 14-17 показаны в таблице 2, и изменения в реакционной способности и селективности в зависимости от времени реакции показаны на фиг.1 и 2.
Примеры 9-12 и сравнительные примеры 14-17 проводили для сравнения выходов получения 1-октена при применении лиганда со структурой главной цепи Р-С-С-Р настоящего изобретения с выходами получения 1-октена при применении общепринятого лиганда с гетероатомной структурой PNP в зависимости от времени реакции.
На фиг.1 показано изменение каталитической активности при тетрамеризации этилена в зависимости от времени реакции. На фиг.1 можно увидеть, что при применении каталитической системы, содержащей лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению, выход 1-октена постоянно повышается с прохождением времени, но при применении общепринятой каталитической системы, содержащей лиганд PNP, выход 1-октена сначала повышается, но затем снижается, когда реакция прогрессирует. То есть можно видеть, что каталитическая система, содержащая лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению, может поддерживать стабильную каталитическую активность лучше, чем общепринятая каталитическая система, содержащая лиганд PNP.
Фиг.2 представляет собой график, показывающий селективность при тетрамеризации этилена в зависимости от времени реакции. На фиг.1 можно видеть, что как каталитическая система, содержащая лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению, так и общепринятая каталитическая система, содержащая лиганд PNP, демонстрируют высокую селективность образования 1-октена, 60% или больше.
Соответственно этому можно видеть, что при тетрамеризации этилена каталитическая система, содержащая лиганд со структурой главной цепи Р-С-С-Р согласно настоящему изобретению, обладает такой же каталитической активностью и селективностью, как общепринятая каталитическая система, и что она может стабильно поддерживать каталитическую активность лучше, чем общепринятая каталитическая система.
Реферат
Изобретение относится к способу поддержания стабильной каталитической активности в процессе тетрамеризации этилена и к способу получения 1-октена тетрамеризацией этилена. Тетрамеризацию осуществляют в присутствии каталитической системы, содержащей хром или предшественник хрома, алкилалюмоксан и лиганд со структурой главной цепи Р-С-С-Р, представленной формулой 1 ! ! где R1, R2, R3 и R4 каждый независимо представляет собой фенил или замещенный фенил и R1, R2, R3 и R4 каждый не имеет заместителей на атомах, соседних с атомами, связанными с атомами Р, и R5, R6 представляют собой метильную группу. Согласно заявленным способам стало возможно стабильно поддерживать каталитическую активность по сравнению с общепринятыми каталитическими системами. 2 н.п. ф-лы, 2 фиг., 2 табл., 17 пр.
Формула
где R1, R2, R3 и R4 каждый независимо представляет собой фенил или замещенный фенил, и R1, R2, R3 и R4 каждый не имеет заместителей на атомах, соседних с атомами, связанными с атомами Р, и R5, R6 представляют собой метильную группу, при этом сокатализатор представляет собой алкилалюмоксан.
Комментарии