Способ получения полиолефинов - RU2078771C1
Код документа: RU2078771C1
Чертежи


Описание
Изобретение касается способа получения олефиновых полимеров с узким молярно-массовым распределением, изменяющейся молярной массой и, в случае прохиральных мономеров, изменяющейся микроструктурой цепи.
Полиолефины с высокой молярной массой имеют особое значение для изготовления пленок, плит или полых изделий, как, например, труб или формованных изделий.
Полиолефины с низкой молярной массой имеют значение для получения присадок или смазок.
Известен способ получения полиолефинов (со)полимеризацией альфа-олефинов в присутствии катализатора, состоящего из алюмоксана линейного или циклического типа и металлоцена (см. EP N 310734, кл. C 08 F 4/60, опублик.1989).
Объектом данного изобретения является.
Способ получения полиолефинов (со) полимеризацией олефинов общей формулы Ra-CH=CH-Rb
или

где Ra и Rb одинаковые или различные, выбранные из группы, включающей водород и C1-C14 углеводородный остаток, при (60)-200oC, давлении 0,5 100 бар в растворе, суспензии или газовой фазе в присутствии металлоцена в качестве катализатора и циклического или линейного алюмоксана в качестве сокатализатора, отличающийся тем, что в качестве катализатора используют металлоцен общей формулы:

где M1 это металл группы IV в Периодической системы,
R1 и R2 одинаковые или разные, выбранные из группы, включающей водород, C1 -C10 алкил, C1-C10 алкоксигруппу, C6-C10 арил, C6-C10-арилокси-группу, C2-C10 -алкенил, C7 -C40-арилалкил, C7-C40-алкилалил C8-C10 арилалкенил галоген;
R3 и R4 одинаковые или различные, выбранные из группы, включающей галоген, C1-C10 алкил, галогеналкил, C6-C10-арил,
R5 и R6 одинаковые или различные, имеющие указанные для R3 и R4 значения или водород;
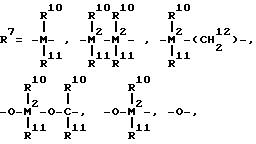
где R10 и R11 одинаковые или разные, выбранные из группы, включающей водород, галоген, C1-C10-алкил, C1-C10-фторалкил, C6 -C10-арил, C6-C10 -фторарил C1-C10-алкоксигруппу, C2-C10-алкенил, C7-C40-арилалкил,
C8-C10-арилалкенил, C7 -C40-алкиларил,
или R10 с R11 или R11 с R12 соответственно, образуют кольцо,
или R10 или R11 с R8 или R9 образуют кольцо, M2=Si;
R8 и R9 одинаковые или разные и имеющие указанные для R10 значение;
m и n одинаковые или различные 0, 1 или 2, при этом m + n 0, 1 или 2,
причем металлоцен общей формулы I, отличен от диметилсилилен-бис-(2, 4-диметил)-цирконий-дихлора и этилен-бис-(2-метил-4-метилинденил)-цирконий-дихлорида.
Предпочтительно согласно изобретению используют металлоцен общей формулы I, указанной выше, в
которой M1 означает
цирконий, R1 и R2 одинаковые или различные, означают метил или хлор, R3 и R4 одинаковые или различные, означают метил, этил,
н-пропил, изо-пропил, н-бутил,
изобутил, трет-бутил или неопентил, R5 и R6 одинаковые или различные, означает метил или этил,
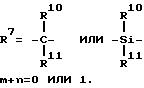
Предпочтительно также используют в составе каталитической системы металлоцен, общей формулы I, в которой заместители R1 и R2, R3 и R4, а также R5 и R6, соответственно, имеют одинаковые значения.
В частности, предпочтительно в качестве металлоцена формулы I используют рац-диметил-силилбис (1-(2-метил-4-этилендинил)-цирконийдихлорид, рац-диметилсилибис-(1-(2-метил-4-изопропилинденил)) цирконийдихлорид, рац-диметилсилилбис-(1-(2-метил-4-требутил-инденил))-цирконийдихлорид, -диметилсилилбис (1-(2-метил-4-изопропиленденил)) цирконийдихлорид, рац-диметилсилил-бис-(1-(2-этил-4-метеленденил)) цирконий дихлорид или диметилсилилбис-(1-(2-метил-4-этиленденил)) цирконийдиметил.
В каталитической системе согласно изобретению в качестве сокатализатора используют алюмоксан общей
формулы:

для линейного типа и/или общей формулы
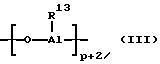
для циклического типа, где R13 одинаковые или различные, выбранные из группы, включающей (C1-C6 )-алкил, (C6-C18)-арил, водород, а "p" целое число от 2 до 50.
В частности, по способу согласно изобретению в качестве сокатализатора используют метилалюмоксан.
Предпочтительно по способу согласно изобретению металлоцен формулы (I) перед использованием в реакции полимеризации предварительно активируют алюмоксаном общей формулы (II) или (III).
Для получения изотактических поли-1-олефинов используются хиральные металлоцены в виде рецемата. Можно однако также использовать и чистую R- или S форму. С помощью этих чистых стереоизомерных форм можно получать оптически активный полимер. При этом необходимо выделить мезо-форму металлоценов, так как полимеризационно-активный центр (атом металла) в этих соединениях вследствие зеркальной симметрии на центральном металле больше не хиральный и поэтому не может производить высоко изотактическими полимерами образуется также атактический полимер. Для определения случаев применения мягкие формованные изделия, например, это может быть исключительно желательно.
Отделение стереоизомеров в принципе известно.
Вышеописанные
стереоизомеры могут быть получены по следующей схеме реакции:



Получение инденов H2Rc и/или H2Rd, 2,4-замещенных, которые служат как исходные вещества, возможно 2-мя различными путями.
а) В качестве исходного соединения служит кетоальдегид указанной в ниже приведенной схеме реакции формулы, получение которого известно.
Превращение этого кетоальдегида с циклопентадиеном осуществляется в инертном растворителе в присутствии основания. Предпочтительно применяются спирты, как метанол, этанол, трет.бутинол, в частности метанол.
Как основания можно использовать множество соединений. Как примеры cледует назвать гидроокиси щелочного металла и гидроокиси щелочноземельных металлов, щелочные алкоголяты и щелочноземельные алкоголяты, как метанолят натрия, трет.бутанолят калия, амиды, как диизопропиламид лития или амины.
При применении предпочтение отдается метаноляту натрия, трет. бутаноляту калия или гидроокиси калия. Молярные соотношения исходных
соединений, включая
применяемые
основания, могут колебаться в широких пределах. Предпочтительным является молярное отношение кетоальдегид:циклопентадиен:
основание 1:1 1,5:3, в частности
1:1, 1:2,5.
Температура реакции составляет преимущественно от -40 до 100oC, в частности 0 25oC.
Времена проведения реакции колеблются, как правило, в пределах между 10 мин и 100 ч, преимущественно между 1 и 30 ч. Заместитель во 2-положении может быть введен после превращения моно-замещенного и 2-позиции индена в 2-инданон, монозамещенного в 4-позиции, согласно общепринятой схеме в результате реакции Гриньяра. Последующее отщепление воды приводит к получению 2,4-замещенных инденов.
2,4замещенные индены образуются как изомеры с двойной связью, которые можно использовать непосредственно для синтеза соответствующих комплексов металлоценов.

b) Другая преимущественная возможность соответствует следующей схеме:
Замещенный в 2-позиции бензилгалогенид подвергают реакции превращения по известному способу в двухзамещенный сложный диэфир малоновой кислоты реакцией соответственно замещенным сложным диэфиром малоновой кислоты.
Омыление сложного диэфира и декарбоксилирование по обычному способу приводит к двухзамещенному производному пропионовой кислоты.
Циклизация до 2,4-двухзамещенного 1-инданона осуществляется после перевода карбоновой кислоты в хлорид карбоновой кислоты по обычному способу.
Восстановление кетона по известным методикам и последующее отщепление воды позволяет получить 2,4-двухзамещенные индены.

Применяемый по изобретению в качестве сокатализатора алюминоксан может быть получен различными способами по различным процессам. Одна из методик, например, состоит в том, что углеводородное соединение алюминия и/или углеводородное соединение гидридоалюминия подвергают превращению с водой (газообразная, твердая, жидкая или связанная например, как кристаллизационная вода) в инертном растворителе (как, например, толуол). Для получения алюминоксана с различными алкильными группами R13 в соответствии с желаемым составом для различных триалкила алюминия AlR3 + AlR'3 превращают с водой
Точная структура алюминоксанов II и III не известна.
Независимо от типа получения всем растворам алюминоксана присуще изменяющееся содержание не превращенного исходного соединения алюминия, которое находится в свободной форме или как аддукт (продукт присоединения).
Возможно осуществление предварительной активации металлоцена перед использованием в реакции полимеризации с алюминоксаном формулы (II) и/или (III). Благодаря этому заметно повышается активность полимеризации и улучшается структура гранул.
Предварительно активируют соединения переходного металла в растворе. Предпочтительно при этом растворение металлоцена в растворе алюминоксана в инертном углеводороде. Как инертный углеводород пригоден алифатический или ароматический углеводород. Предпочтительно применяю толуол.
Концентрация алюминоксана в растворе находится в диапазоне около 1 мас. до предела насыщения, в основном от 5 до 30 мас. соответственно в расчете на общий раствор. Металлоцен можно использовать и в одинаковой концентрации, главным образом однако он используется в количестве 10-4-1 моль на каждый моль алюминоксана. Время предварительной активации составляет от 5 мин до 60 ч, преимущественно от 5 до 60 мин. Работают при температуре от -78 до 100oC, в основном от 0 до 70oC.
Металлоцен можно также форполимеризовать или наносить на носитель. Для форполимеризации применяют предпочтительно олефин (или один из олефинов), используемых в полимеризации.
Соответствующие носители это, например, силикагели, оксиды алюминия, твердый алюминоксан или другие неорганические материалы носителя. Соответствующий материал носителя это также полимерный порошок в высокодисперсной форме.
В соответствии с изобретением вместо или наряду с алюминоксаном можно применять соединения формул RxNH4-xBR'4, Rx PH4-xBR'4 или BR'3 как соответствующие сокатализаторы. В этих формулах "x" означает число 1-4, предпочтительно 3, остатки R одинаковы или различны, предпочтительно одинаковы, и означают C1 -C10-алкил, C6-C18 арил или 2 остатка R образуют вместе с соединяющими их атомами кольцо, а остатки R' одинаковы или различны, предпочтительно одинаковы, и означают C6-C18 арил, который может быть замещен алкилом, галоалкилом или фтором.
В частности R означает этил, пропил, бутил, или фенил, а R' это фенил, пентафторфенил, 3, 5-бис-трифторметилфенил, мезитил, ксилил или толил.
При применении вышеназванных сокатализаторов собственно катализатор полимеризации состоит из продукта реакции металлоцена и одного из названных соединений. Поэтому этот продукт реакции получают предпочтительно не в реакторе полимеризации на отдельном этапе при применении соответствующего растворителя (сравни пример осуществления VIII).
В принципе, в качестве сокатализатора согласно изобретению пригодно любое соединение, которое на основе своей кислотности по Льюису может перевести нейтральный металлоцен в катион и стабилизировать его ("лабильная координация"). Помимо этого, этот сокатализатор и/или образованный из него анион не может вступать ни в какие другие реакции с полученным катионом металлоцена.
Для удаления имеющихся в олефине катализаторных ядов предпочтительной является очистка с помощью алюминийалкила, например, AlMe3 или AlEt3. Такая очистка может быть осуществлена как в системе полимеризации непосредственно, так и в результате того, что олефин перед добавлением в систему полимеризации приводят в контакт с Al-соединением, а затем снова отделяют.
Полимеризация или сополимеризация осуществляются известным образом в растворе, в суспензии или в газовой фазе непрерывно или периодически, одноступенчато или многоступенчато, при температуре от -60 до 200oC, преимущественно от 30 до 80oC. Полимеризуют или сополимеризуют олефины формулы Ra -CH= CH-Rb. В этой формуле Ra и Rb - одинаковы или различны и означают атом водорода или алкильный остаток с 1 до 14 C-атомами. Ra и Rb могут однако образовывать кольцо с соединяющими их атомами. Примеры для таких олефинов это этилен, пропилен, I-бутен, 1-гексен, 4-метил-1-пентен, 1-октен, норборнен или норборнадиен. В частности подвергают полимеризации пропилен и этилен.
Как регулятор молекулярной массы, в случае необходимости, добавляют водород. Общее давление в системе полимеризации составляет 0,5 до 100 бар. Предпочтительной является полимеризация в диапазоне давления, представляющем особенно интерес в техническом плане, от 5 до 64 бар.
При этом применяют металлоцен в концентрации, в расчете на переходный металл, от 10-3 до 10-8, преимущественно 10-4 до 10-7 моль переходного металла на дм3 растворителя или на дм3 объема реактора. Алюминоксан применяют в концентрации от 10-5 до 10-1 моль, в основном 10-4 до 10-2 на каждый дм3 растворителя или на каждый дм3 объема реактора. Другие названные сокатализаторы применяют примерно в эквимолярных количествах к металлоцену. В принципе однако возможны также и более высокие концентрации.
Если полимеризацию осуществляют как суспензионную полимеризацию или как полимеризацию в растворе, то используют инертный растворитель, употребительный для способа низкого давления Циглера. Например, работают в алифатическом или циклоалифатическом углеводороде; следует назвать, например, пропан, бутан, пентан, гексан, гептан, изоктан, циклогексан, метилциклогексан.
Далее можно использовать бензиловую или гидрированную фракцию дизельного топлива. Пригоден также и толуол. Предпочтение отдают полимеризации в жидком мономере.
Если применяют инертные растворители, то в определенных дозах добавляют мономеры в газообразном или жидком виде.
Продолжительность полимеризации любая, так как применяемая согласно изобретению катализаторная система показывает лишь незначительный, зависимый от времени, спад активности полимеризации.
Предложенный способ отличается тем, что описанные металлоценкатализаторные системы, в температурном диапазоне, представляющем интерес в техническом плане, между 30 и 80oC, но особенно в диапазоне между 60 и 80oC, позволяют получать полимеры с узким молекулярно-массовым распределением и крупнозернистой структурой гранул, а также с переменной молекулярной массой и стереотактичностью. Соответственно желаемая молекулярная масса полимера и стереотактивность регулируется в результате выбора подходящих заместителей в 2- и 4-положениях системы лиганд металлоцена. Если полимеризация осуществляется без водорода как регулятора молекулярной массы, то полимеры показывают ненасыщенные конечные группы.
Следующие примеры поясняют данное изобретение более подробно.
Обозначения:
KB коэффициент
вязкости в см3/г,

Т. пл. температура плавления, определяемая посредством ДСК/дифференциальная сканирующая калометрия (20oC/мин. скорость нагревания/скорость охлаждения),
uu изотактический индекс (uu мм + 1/2mr), установленный в результате 13C-NMR-спектроскопии (ядерно-магнитный резонанс) (ЯМР),
mmmm доля изотактических пентаденов в 13C-NMR-спектре в
НПл насыпная плотность полимера в г/см3,
d50 средний диаметр гранулы полимера в мкм.
Синтез применяемых в примерах металлоценов:
Металлоцен A:
rac-диметилсилилбис(1-(2-метил-4-этилинденил)циркондихлорид
1.1 4-этилинден (а2).
20,7 г (181, 7 ммоль) 4-оксокапрональдегид (а1, полученный из хлорида пропионовой кислоты и алкилхлорида; сравни Synthesis, (1985) 1058) растворяли в 10 мл абс. метанола и при охлаждении раствором циклопентадиена в количестве 13,2 г (199 ммоль) добавляли в 5 мл абс. метанола. Эту смесь при температуре 0oC в течение 35 мин закапывали в раствор 51 г (454 ммоль) третичного бутилата калия в 100 мл абс. метанола, при этом наступало темно-коричневое окрашивание. Через 2 4 ч помешивания при 0oC и дальнейшего помешивания в течение 2 ч при комнатной температуре эту смесь наливали на лед, устанавливали на pH 6 и экстрагировали посредством хлористого метилена. Органическую фазу промывали с помощью насыщенного NaCl-раствора, высушивали над сульфатом натрия и выпаривали. Сырой продукт хроматографировали на 750 г силикагель 60. Посредством гексан/хлористого метилена (20 1 до 10 1) можно было отделить 11,1 г (43%) индена а2 (2 изомера с двойной связью 3 2).
1.2.4-этил-2-инданон (а3).
33,9 г (235 ммоль) 4-этилинден (а2) при ледяном охлаждении медленно закапывали в смесь 141 мл муравьиной кислоты (98 100%) и 33 мл (340 ммоль) H2 O2 (35%) (сильно экзотермическая реакция). После этого осуществляли перемешивание еще в течение 2,5 ч при комнатной температуре. Из образующейся суспензии желто-оранжевого цвета в вакууме, получаемом с помощью водоструйного насоса, удаляли избыточную муравьиную кислоту. Оставшееся желтое масло разбавляли 900 мл 2N серной кислоты. При дополнительном дозировании воды перегоняли всего 3 г воды, при этом продукт осаждался в приемнике как желтоватое масло. Дистиллят нейтрализовали с помощью насыщенного раствора карбоната натрия и извлекали эфиром. Эфирную фазу высушивали над сульфатом натри и выпаривали. Получали 22,4 г (59%) соединения а3 как твердое вещество белого цвета.
1.3. 2-метил-4-этилинден (а4).
Раствор в количестве 22,4 г (140 ммоль) соединения а3 в 500 мл диэтилэфира разбавляли при комнатной температуре в атмосфере Ar защиты в течение 1 ч 140 мл (420 ммоль) 3 М эфирного раствора метилбромид магния. После этого осуществляли перемешивание еще в течение 2 ч при комнатной температуре при обратном потоке, а следующие 15 ч перемешивали при комнатной температуре. Эту смесь выливали на HCl-кислый лет и извлекали эфиром. После высушивания над сульфатом натрия растворитель удаляли. Оставшееся масло желтого цвета (20,3 г) поглощалось в 800 мл толуола (ч.д.а), затем его разбавляли 2,2 г (11,5 ммоль) пара-толуолсульфокислотой и повторно обезжиривали. После охлаждения этот раствор многократно промывали водой, высушивали над сульфатом натрия и выпаривали. Остаток подвергали хроматографическому анализу на 620 г силикагеля 60. С помощью смеси гексан/хлористый метилен (20 1) можно было элюировать 5,5 г (25%) индена а4 (масло желтоватого цвета). С помощью смеси гексан /уксусный эфир (9 1) можно было регенерировать еще неиспользованный эдукт а3.
1.4. Диметилсилилбис(2-метил-4-этилинден) (а5).
Раствор 5,5 г (34,8 ммоль) соединения а4 в 30 мл THF в атмосфере Ar - защиты при 0oC медленно разбавляли 14 мл (34,7 ммоль) 2,5 М раствора н.-бутиллития в гексане, а затем в течение 2 ч нагревали при обратном потоке. Раствор темно-коричневого цвета закапывали затем в раствор 2,2 г (17,4 мл) диметилдихлорсилана в 15 мл ТГФ. Эту смесь в общем в течение 10 ч нагревали при обратном потоке и в течение ночи перемешивали при комнатной температуре, затем выливали на лед и экстрагировали с помощью диэтилэфира. Оставшийся после удаления растворителя остаток подвергали хроматографическому анализу на 200 г силикагеля. Посредством смеси гексан/хлористый метилен (от 20 1 до 10 1) вымывали сначала 2,0 г неизрасходованного эдукта. После этого следовали 3,1 г продукта а5 (48% выхода относительно St, 75% относительно непревращенного эдукта). Это соединение образуется как масло желтоватого цвета (2 изомера 3:1).
1.5. рац-диметилсилилбис{ 1-(2-метил-4-этилинденил)} -цирконийдихлорид (A).
Раствор 3,1 г (8,3 ммоль) системы лиганд a5 в 30
мл диэтилэфира разбавляли при комнатной температуре в
условиях Ar защиты
посредством 10 мл (25 ммоль) 2,5 М раствора бутиллития в гексане. Сначала появлялось оранжевое окрашивание, а через 45 мин
помутнение раствора. После помешивания в течение ночи
окрашенную теперь
суспензию разбавляли 10 мл гексана и фильтровали через УЗ-фритту. Осадок промывали 20 мл гексана и длительное время высушивали
в вакууме, созданном масляным насосом. Почти бесцветный
порошок при
температуре 78oC быстро добавляли к суспензии 1,8 г (7,72 ммоль) цирконтетрахлорида в 30 мл хлористого метилена. Эту
смесь нагревали 1-2 ч до комнатной температуры и полностью
выпаривали
через 30 мин помешивания при комнатной температуре. Высушенный в вакууме, созданном масляным насосом, остаток промывали сначала
гексаном в количестве 60 мл. Этот продукт получали затем в
результате
многократного экстрагирования посредством 180 мл толуола. Соединенные экстракты концентрировали и оставляли для кристаллизации
при -35oC. Первая фракция давала 0,76 г цирконоцена
A в
чистом рацемическом виде (кристаллы оранжевого цвета). Другие фракции содержали увеличивающуюся долю мезо-формы. В общем были
получены 1,78 г (43%) соединения A.1H-NMR (CDCl3
рацемата: 6,85-7,55 (м, 6, ароматические углеводороды -H), 6,80 (c,2, β -H), 2,72 (кв. 4,CH2), 2,20(c,6,
CH3), 1,30(т,6,CH3), 1,27 (с,6,Si-CH3),
1H-NMR (CDCl3) мезо-форма: 6,6-7,6 (м,6,ароматические углеводороды -H), 6,68(c, 2, b -H), 2,7
(кв.4,CH2), 2,48(c,6,CH3), 1,13-1,43 (м,12,Et-CH3 Si -CH3).
II. Металлоцен В: рац-диметилсилилбис1-(2-метил-4-изопропилинденил)}-цирконийдихлорид.
II.1 4-Изопропилинден b2).
В результате превращения хлорангидрида изомасляной кислоты и аллилхлорида был получен 5-метил-4-оксокапрональдегид (b1) аналогично a1 (см.1.1), 45,6 г (356 ммоль) b1 превращали аналогично описанному в 1.1. с использованием циклопентадиена и трет.бутилата калия. Хроматография на колонне позволила получить 19,6 г (35%) индена b2 как масло желтого цвета (2 изомера с двойной связью).
II.24-изопропил-2-инданон (b3).
33,8 г (213 ммоль) соединения b2 окисляли аналогично описанному 1.2 и дистиллировали с помощью воды. Получали 22,6 г (61%) инданона b3 как твердое вещество желтоватого цвета.
II.3 2-метил-4-изопропилинден (b4).
11,1 г (63,8 ммоль) инданона b3 подвергали реакции с 2,5 экв.метилового бромида магния аналогично описанному в 1.3. Время реакции составляло 17 ч при комнатной температуре. После этого повторно обезжиривали с помощью гидрата p-толуолсульфоновой кислоты в течение 25 мин. Хроматография позволила получить 3,9 г (36%) индена b4 бесцветное масло.
II.4 диметилсилилбис/2-метил-4-изопропилинден/(b5).
3,9 г (22,7 ммоль) индена b4 подвергали реакции аналогично описанному в 1.4. с использованием диметилдихлорсилана и продукт перерабатывают. Хроматография на колонне дала наряду с 0,44 г неизрасходованного индена 3,0 г продукта b5 как масло желтого цвета (изомеры). Выход составлял 65% относительно Si и 73% относительно непревращенного эдукта.
II. 5. рац-диметилсилилбис{1-/2-метил-4-изопропилинденил/}- цирконийдихлорид (B).
3,0 г системы лиганд b5 были депротонированы аналогично описанному в 1,5 и превращены с использованием 1 экв.цирконтетрахлорида в 20 мл хлористого метилена. После промывки сырого продукта 40 мл гексана продукт экстрагировали посредством 120 мл толуола. Экстракт толуола выпаривали в вакууме масляного насоса. Получали 1.7 г (46%) цирконоцена как органический порошок. Рацемат и мезо-форма имелись в отношении 1:1. В результате перекристаллизации из небольшого количества толуола или смесей толуол /гексана можно было отделить в чистом воде рацемическую форму.
1 H-NMR рацемата (CDCl3): 6,7 7,5 (м,6, ароматические углеводороды H), 6,85 (с, 2, b -H), 3,0(м,2,изо-Pr-CH) 2,23 (с,6,CH3), 1,17 1,37 (д,12, i-Pr-CH3) 1, 27 (с,6, Si-CH3).
1H-NMR мезо-формы (CDCl3):6,5-7,5 (м,6,аромат, углеводород.H) 6,75 (с,2, b -H) 3,0 (м,2, изо-Pr-CH) 2,48 (с,6,CH3), 1,10-1, 45 (м,18, изо Pr-CH3, Si-CH3 ).
III. Металлоцен C: рац-диметилсилилбис1-(2-метил-трет.бутилинденил)} цирконийдихлорид.
III.1. 4-трет.бутилинден (c2).
В результате реакции хлорида пивалиновой кислоты и аллилхлорида был получен 5,5-диметил-4-оксокапрональдегид c1 аналогично a1 (см.1.1.) 41 г (195 ммоль)- с 1 подвергали реакции аналогично описанному в 1.1 посредством циклопентадиена и трет.бутилата калия и перерабатывали. Время реакции составляло 19 часов при комнатной температуре. Хроматография на колонне дала 3,2 г (10%) индена как масло желтого цвета (2 изомера с двойной связью).
III.2. 4-трет.бутил-2-инданон (c3).
8,5 г (49; 4 ммоля) соединения c2 окисляли аналогично описанному 1.2 и дистиллировали с помощью воды. Время реакции составляло 4 ч при комнатной температуре. Получали 2,8 г (30%) инданона c3 в виде желтых кристаллов.
III.3. 2-метил-4-трет.бутилинден (c4).
3,6 г (19 ммоля) инданона c3 подвергали реакции с помощью 30 эк. метилового бромида магния аналогично 1.3. и продукт перерабатывали. Время реакции составляло 17 ч при комнатной температуре и следующие 4 ч при обратном потоке. После этого производили повторное обезжиривание посредством гидрата пара-толуолсульфокислоты в течение 25 мин. Хроматографический анализ позволил получить 1,2 г (33%) индана c4 как масло желтого цвета. С помощью смеси гексан/уксусный эфир (9:1) можно было регенерировать неиспользованный эдукт.
III. 4 Диметилсилилбис-(2-метил-4-трет.бутилинден) (c5).
1,2 г (6,4 ммоля) индена c4 превращали аналогично 1.4 с диметилдихлорсиланом и продукт перерабатывали. Хроматография на колонне дала наряду с 0,48 г неиспольздованного индена c4 0,40 г продукта c5 как желтое масло (изомеры). Выход составлял 29% относительно Si и 49% относит. превращенного эдукта c4.
III.5. рац-диметилсилил1-(2-метил-4-трет.бутилинденил)} цирконийдихлорид (C).
0,40 г (0,93 ммоля) системы лиганд c5 в мл диэтилового эфира при Ar-защите разбавляли с помощью 0,74 мл (1,86 ммоля) 2,5 М раствора н.-бутиллития в гексане. После перемешивания в течение ночи был полностью выпарен оранжевый раствор. Остаток высушивали продолжительное время в вакууме, полученном масляным насосом и быстро, при температуре 78oC добавляли в суспензию 225 мг (0,96 ммоля) цирконтетрахлорида в 5 мл хлористого метилена. Смесь перемешивали в течение 2 ч при температуре 0o C и в течение 30 мин при комнатной температуре и полностью выпаривали. Продукт экстрагировали с помощью 8 мл толуола. После удаления толуола получали 210 мг (37%) цирконоцена в виде порошка оранжевого цвета. Отношение рацемата к мезо-форме составляло 1:1. В результате перекристаллизации из толуол/гексана можно было получить чистую рацемическую форму.
1H-NMR рацемата (CDCl3 6,8 7,5 (м, 6, аромат. углеводороды-Н), 6,92 (с, 2, b -H), 2,27 (с, 6, CH3), 1,22-1,41 (м, 24, трет.Bu, Si-CH3).
1H-NMR мезо-формы (CDCl3), 6,7 7,6 (м, 6, аромат, углеводороды-H), 6,7 (с, 2, b -H), 2,50 (с, 6, CH3), 1,1 1,5 (м, 24, трет.Bu, Si-CH3).
IV. Металлоцен D:
rac-метилфенилсилилбис1-(2-метил-4-изопропилинденил)}
цирконийдихлорид
IV. 1. Метилфенилсилилбис-(2-метил-4-изопропилинден) (d5).
Раствор в количестве 2,0 г (11,8 ммоля) 2-метил-4-изопропилиндена b4 (см. П. 3) в 40 мл ТГФ разбавляли в атмосфере Ar защиты при 0oC посредством 4,8 мл раствора 2,5 М бутиллития в гексане и нагревали 90 мин. до обратного потока. Затем раствор красного цвета добавляли к раствору 1, 12 г (5,9 ммоля) метилфенилдихлорсилана в 15 мл ТГФ и в течение 7 ч нагревали при обратном потоке. Здесь выливали на лед и извлекали эфиром. Высушенная над сульфатом натрия эфировая фаза выпаривалась в вакууме. Оставшийся остаток подвергали хроматографическому анализу на 200 г силикагеля 60. С помощью смеси растворителей из гексана/хлористого метилена (10:1) регенерировали сначала 0,57 г неиспользованного индена b4. Смесью гексан/хлористый метилен (10:2) извлекали 1,2 г продукта d5. Выход составлял 44% относительно Si и 61% относительно непреобразованного индена b4.
IV. 2. Рац-метилфенилоилилбис{1-(2-метил-4-изопроприлинденил)} -цирконийдихлорид /D/.
Раствор 1,28 г (2,76 ммоля) лигандной системой d5 в 20 мл диэтилового эфира при комнатной температуре в атмосфере Ar защиты медленно разбавляли 3,3 мл (8,3 ммоля) раствором 2,5 М бутиллития в гексане и перемешивали в течение ночи. Окрашенный в оранжевый цвет раствор полностью выпаривали, продолжительное время высушивали в вакууме, полученном масляным насосом и промывали 20 мл гексана. Остаток долго высушивали в вакууме масляного насоса при температуре 40oC и превращали в порошок. Порошок желтого цвета при -78oC добавляли в суспензию 0,62 г (2,66 ммоля) циркотетрахлорида в 15 мл хлористого метилена. Эту смесь в течение 1 ч нагревали до 0oC, и следующие 2 ч перемешивали при комнатной температуре. Суспензию красно-коричневого цвета полностью выпаривали и высушивали в вакууме масляного насоса. С помощью толуола экстрагировали 1,05 г (63%) цирконоцена (порошок оранжевого цвета). В сыром продукте имелись 2 рацемическая и 2-мезо-формы в отношении 2:1:1. В результате перекристаллизации из смеси толуол /гексан можно было выделить рацемическую форму.
1H-NMR смеси изомеров (CDCl3); 6,4 8,2 (м ароматич. углеводор. -H), 3,1 (шир. изо-Pr-CH), 2,55 (c, CH3), 2,33 (c, CH3), 2,22 (c CH3), 1,95 (c, CH3), 1,13 1,47 (м, изо-Pr-CH3, Si-CH3).
V. Металлоцен E: Рац-диметилсилилбис{1-(2-эти-4-метилинденил)} цирконийдихлорид.
V.1. 2-(2-метилбензил)-масляная кислота (e1).
14,2 г (0,62 мол) натрия растворяли в 250 мл этанола и добавляли 118,4 г (0,63 мол. ) этил-диметилмалонового эфира. Закапывали 118,5 г (0,64 мол.) 2-метил-бензилбромида таким образом, чтобы смесь слегка кипела. После этого нагревали 4 часа при обратном потоке. Суспензию помещали на воду, экстрагировали посредством эфира, в соединенные органические фазы высушивали над MgSO4. Растворитель удаляли, а полученный сырой продукт (187 г) без дальнейшей очистки подвергали дальнейшей реакции.
Для омыления в присутствии 139 г KOH в 355 мл этанола и 170 мл H2O осуществляли нагревание в обратном потоке. Растворитель отводили, а остаток при охлаждении разбавляли концентрированной соляной кислотой. Экстрагировали эфиром, при этом соединенные органические фазы промывали насыщенным водным раствором NaCl и высушивали над MgSO4. Растворитель удалили, а остаток для декарбоксилирования нагревали до 170oC, при этом подвергали отгонке продукт e1 (140 145oC/0.1 тор).
Выход: 96,0 г (81%).
V.2. Хлорангидрид 2-/2-метил-бензил/-масляной кислоты (е2).
96 г (0,5 моля) 2-(о-ксилил)-масляной кислоты (eI) медленно нагревали с 89 г (0,75 моль) SOCl2 и повторно обезжиривали до конца выделения газа (1 ч) Избыточный тионилхлорид отгоняли, а остатки удаляли в результате трехкратного отвода соответственно 50 мл толуола i.v. Сырой продукт подвергали очистке посредством дистилляции (103oC/1 тор).
Выход: 101,7 г (96% 0,48 моль).
V.3. 2-этил-4-метил-1-инденон (е3).
101,7 г (0,48 моль) хлорангидрида 2-(2-метил-бензил масляной кислоты (e2) закапывали к 191 г (1,43 моль) AlCl3 в 600 мл толуола и нагревали в течение около 3,5 ч до температуры 80oC. Эту реакционную смесь выливали на 1л смеси льда (конц. HCl, и фазы разделяли. Водную фазу 4 раза экстрагировали соответственно посредством 250 мл толуола, соединенные органические фазы промывали насыщенным водным раствором NaHCO3 и NaCl, и высушивали над MgSO4. Растворитель удаляли i.V. а остаток дистиллировали (78oC/0,2 тор).
Выход 81 г (98% 0,464 моль).
V.4 2-этил-4-метил-инден (e4).
34.1 г (196 ммоля) 2-этил-4-метил-1-инданона (e3) в 210 мл смеси ТГФ (метанол) (2:1) порционно разбавляли с помощью 11,1 г (294 ммоля/ NaBH4) и в течение 15 ч перемешивали при комнатной температуре. Эту реакционную смесь выливали на лед и разбавляли конц. HCl до pH 1. После экстракции эфиром соединенные органические фазы промывали насыщенным водным раствором NaHCO3 и NaCl и высушивали над MgSO4. Освобожденный от растворителя остаток (36,2 г) непосредственно подвергали дальнейшему превращению для последующего выделения.
Неочищенный 2-этил-2-метил-1-инданон обрабатывали в 700 мл толуола в присутствии 0,75 г моногидрата толуолсульфокислоты в течение 2 часов на паровой бане. Растворитель удаляли i.v. остаток поглощается эфиром, промывался насыщенным раствором NaHCO3 и NaCl и высушивался над MgSO4. Растворитель удаляли i.v. а остаток подвергали дистилляции (62oC 0,2 тор.).
Выход: 25,7 г (83% 162 ммоля).
V.5. Диметилсилилбис(2-этил-4-метилинден) (e5).
К 10,4 г (65,5 ммоля) 2-этил-4-метил-индена (e4) в 50 мл абс. ТГФ медленно закапывали 26,2 мл (65,6 ммоля) 2,5 М BuLi- раствора в гексане и в течение 2 ч продолжали помешивание при 50oC. В это время помещали 3,95 мл Me2SiCl2 в 50 мл абс. ТГФ, а затем закапывали Li соль в течение 8 ч. Перемешивали в течение 15 ч, удаляли растворитель, суспендировали в н.-пентане и снова фильтровали. После удаления смеси растворителей очищали посредством хроматографии на колонне над силикагелем (н.-гексан (CH2Cl2 9:1).
Выход: 15,1 г (63% 41 ммоля).
V.6.Рац-диметилсилилбис{1-/2-этил-4-метилинденил/} цирконийдихлорид (E).
К 3.57 (9,58 ммоля) Me2Si (2-Et Me-Ind)2 в 50 мл ТГФ добавляли по каплям при комнатной температуре 7,66 мл (19,16 ммоля) 2,5 M Bu - Li раствора в н. -гексане и нагревали еще в течение 3 ч до 50oC. Выпаривали до сухого вещества, суспендировали в н. -пентане, фильтровали и высушивали. 2,23(9,58 ммоля) ZrCl4 суспендировали в 150 мл CH2Cl2 и охлаждали до температуры -78oC. Добавляли Дилитио-соль. 3 ч перемешивали при -20oC, оставляли на ночь при комнатной температуре. Производили фильтрование, а растворитель удаляли. Кристаллизация из смеси толуола /н. -гексана (25:1) дала в итоге 0,18 г кристаллов оранжевого цвета (мезо /rac 5:1). Маточный раствор концентрировали до 1/4 его объема и кристаллизовали при температуре -38oC, получали еще 0,1 г комплексной смеси. Маточный раствор выпаривали до сухого вещества, остаток суспендировали в н.-гексане, фильтровали и высушивали. Получали чистую рац-форму E как порошок оранжевого цвета.
V1.
Металлоцен F-: рац-диметилсилилбис.1-(2,4-диметилинденил)}цирконийдихлорид
VI. 1. Метиловый эфир (±
)-2-метил-3-гидрокси-3-(2-толил/пропионовой кислоты (f1).
42 г (645 ммоля) Zn в 150 мл толуола и 50 мл Et2O нагревали до 80-85oC, закапывали смесь из 51,6 г (430 ммоля) 2-толил альдегида и 62 мл (557 ммоля) 2-бромметилмалонового эфира. После 5% добавления прекращали нагревание и добавляли кристалл йода. После сильного вспенивания добавили затем по каплям остаток при температуре 80-85oC в течение 80 мин, 2 ч помешивали при 85oC и оставляли на ночь.
Брали смесь 200 г льда /30 мл H2SO4 и доливали исходную смесь. После извлечения эфиром, промывки фазы посредством NaHCO3 и NaCl раствора высушивали и дистиллировали (101oC/1 тор).
Выход: 86 г
/ 96%
VI.2. Метиловый эфир (±)-2-метил-3-(2-толил) пропионовый кислоты (f2).
К 86 г (413 ммоля) b гидроксиэфира fI в 800 мл CH2Cl2 добавляли 132 мл (826 ммоля) HSiEt3. Добавляли 102 мл (826 ммоля) BF3-эфира в 3 порции в течение 5-10 мин. Через 20 ч при комнатной температуре продукт перерабатывали. После гидролиза с помощью 220 мл NaHCO3 (pH 3) продукт извлекали эфиром, отделяли органическую фазу, промывали раствором NaCl высушивали и дистиллировали (120oC (I тор).
Выход: 58,9 г (74,1%).
VI.3. (±)-2-метил-3-(2-толил)-пропионовая кислота (f3).
38,45 г (200 ммоля) эфира f2, 850 мл 5%-ой NaOH и 850 мл MeOH повторно обезжиривали в течение 4,5 ч, отгоняли MeOH, подкисляли, экстракт эфира высушивали с помощью MgSO4 и дистиллировали (107 109oC в высоком вакууме).
Выход: 31,8 г (89%).
VI.4. Хлорангидрид (±)-2-метил-3-(2-толил)-пропионовой кислоты (f4).
16,04 г (30 ммоля) кислоты f3 медленно нагревали с 15,6 г (270 ммоля) SOCl2 до температуры 80oC и до конца выделения газа держали при такой температуре. Для удаления SOCl2 многократно выпаривали посредством толуола.
Выход: 17,7 г (сырой).
VI.5. (±)-2,4-диметилинденон (f5).
К 17,7 г (90 ммоля) хлорангидрида кислоты f4 в 50 мл толуола добавляли 36 г (270 ммоля) AlCl3 в течение 20 мин. и 4 часа помешивали при 80oC. Выливали на смесь лед/HCl, экстрагировали толуолом, промывали с помощью раствора H2O, NaHCO3 и NaCl, высушивали и дистиллировали - (109oC/1 тор) или подвергали хроматографическому анализу (н.-гексан) этилацетат 6:1, rF=0,44).
Выход: 13,75 г (95,4%).
Этапы VI. 1 до VI.5 осуществляли аналогично Synth. Comm. 20(1990) 13-87-97.
VI.6 (±)-2, 4-диметилинданол (f6).
К 10,03 г (62,6 ммоля) кетона f5 в 150 мл ТГФ (MeOH 2:1 добавляли в определенных порциях при 0oC 3,55 г (93,9 ммоля) NaBH4 в течение 1 ч. Осуществляли перемешивание в течение 2 часов при 0oC, затем оставляли на ночь при комнатной температуре. Ставили на смесь лед/HCl, pH регулировали 1, отфильтровывали осажденную на границе раздела фаз борную кислоту (?), экстрагировали посредством Et2O, промывали с помощью раствора NaHCO3 и NaCl, и высушивали на масляном насосе.
Выход: 10,24 г.
VI.7. 2,4-диметилинден (f7).
10,24 г (62 ммоля) инданола f6 растворяли в толуоле и добавляли 20 мг гидрата p-толилсульфоновой кислоты. Оставляли на паровой бане в течение 2,5 ч, добавляли небольшое количество, выпаривали органическую фазу и дистиллировали (133oC/ 10 тор).
Выход: 8,63 г (95%).
VI.8. (± )-диметилсилил-бис (2,4-диметилинден) (f8).
К 8,63 г (59,8 ммоля) лиганда f7 в 100 мл Et2O закапывали 37,4 мл 1,6 М (59,8 ммоля) раствора H.-BuLi в н.-гексане и перемешивали несколько часов при температуре 40oC. Li-соль медленно добавляли по каплям к 3,86 мл (29,9 ммоля) Me2SiCl2 в 30 мл Et2O и перемешивали в течение 2 ч. После фильтрации осуществляли выпариванием производили хроматографический анализ (н.-гексан/CH2Cl2 9:1 rF=0,29).
Фракции продукта соединяли и перекристаллизовывали из MeOH.
Выход: 1,25 г (12%).
VI.9. rac-диметилсилилбис{1-(2,4-диметилинденил)} циркондихлорид (F).
1,25 г (3,63 ммоля) хелатного лиганда f3 растворяли в 20 мл ТГФ, добавляли по каплям 2,9 М (7,26 ммоля) раствора н.-BuLi в н.-гексане и перемешивали в течение 2 ч при температуре -40oC; до окончания выделения бутана.
0,85 г (3,63 ммоля) ZrCl4 суспендировали в 30 мл CH2Cl2, после добавления дилитио-соли при -78oC медленно
нагревали до
комнатной температуры и после отстаивания фильтровали в течение ночи. Фильтрат выпаривали. Получали
комплекс смеси рацемической формы с мезо-формой в отношении 1:1 (порошок оранжевого
цвета).
Рацемическую форму можно было получить в результате перекристаллизации из толуола. Чистый выход - 15%
1H-NMR рацемата (CDCl3): 6,8 7,5 (m, 6, аромат. углевод.-6),
6,82 (S,
2, b -H), 2,3 (S, 6, OH3), 2,1 (S, 6, CH3), 1,30 (S, 6, Si-CH3).
VII. Металлоцен v rac-диметилсилилбис1-/2-метил-4-этилинденин/}циркронийдиметил.
0,26 г металлоцена А в 40 см3 Et2O при температуре -50o C по каплям разбавляли 1,3 см3 1,6 М (2,08 ммоля) эфирным MeLi раствором и в течение 2 ч при температуре -10oC производили помешивание. После замены растворителя на н-пентан перемешивали еще 1,5 ч при комнатной температуре, а отфильтрованный остаток сублимировали в вакууме. Было получено 0,15 г сублимата, соответствующего корректному элементарному анализу.
VIII. Реакция
металлоцена vc [Bu3NH][B(C6H5)4
)
0,15 г металлоцена при 0oC добавляли к 0,17 г [Bu3NH]
[B(C6H5)4)] в 25 см3 толуола. При помешивании нагревали до 50oC, и эту смесь в течение 10 мин перемешивали при такой температуре.
Глубокоокрашенную смесь выпаривали
затем до сухого вещества. Для полимеризации применили аликвотную часть реакционной
смеси(Bu=бутил)
Сокращения: Me метил, Et=этил, Bu=бутил, Ind
инденил.
Примеры
полимеризации
Пример 1:
Сухой реактор объемом 10 дм3 промывали
азотом и заполняли 10 дм3 жидкого пропилена. Затем добавляли
30 см3 толуольного
раствора метилалюмин оксана (соответственно 45 ммоля А1, средняя степень олигомеризации n=16), и
исходную смесь перемешивали при температуре 30oC в течение
15 мин.
Параллельно этому 3,3 мг (0,006 ммоль) металлоцена B растворяли в 20 см3 толуольного раствора метилалюминоксана (30 ммоля А1) и предварительно активировали в результате 15-минутного отстаивания.
Раствор помещали в реактор, в результате подачи тепла нагревали до температуры полимеризации 70oC (4oC/мин) и полимеризационную систему удерживали в течение 1 ч в результате добавления 20 мл изопропанола. Избыточный мономер отдували газом, полимер высушивали в вакууме. Получали 1,44 кг полипропилена.
Активность катализатора составляла, таким образом, 436 кг nn/г металлоцена в час.
КВ 168 см3/г;
Темп. плавл. 149,6oC; uu 95%
mmmm
88,6%
H Пл. 0,30 г/см3; d50 2600 mm;
Mw 1,8•105 г/моль;
Mw/Mn 2,2.
Примеры 2
11
Поступали соответственно
аналогично примеру 1, причем однако варьировали следующие величины:
тип металлоцена
u
расход металлоцена (мг)
температуру
полимеризации
Варьированные параметры
полимеризации и выход полимеров указаны в табл. 1, измеренные на полимерах величины содержатся в табл. 2.
Пример 12.
Поступали так же, как в примере 1. Непосредственно после добавления металлоцена в реактор однако нагнетали еще 0,3 бар водорода на реактор. Получали 1,00 кг полимера.
Кв 76 см3/г;
t плавл. 148,
9oC;
HПл 0,20 г/см3; d50 1800 μm;
Mw 6,2•104 г/моль;
Mw
/Mn 2,8.
Пример 13.
Сухой реактор емкостью 16 дм3 промывали азотом и заполняли 10 дм3 жидкого пропилена.
Затем реакционную смесь в количестве 2,5 см3 согласно примеру VIII (соответственно 15 мг металлоцена Φ) растворяли в 20 см3 толуола и при температуре окружающей среды помещали в реактор. Раствор нагревали в результате подачи тепла до температуры полимеризации 70oC (4oC/мин), и полимеризационную систему поддерживали в течение 1 часа в результате охлаждения при 70oC. Прекращали полимеризацию в результате добавления 20 мл изопропанола. Избыточный мономер отдували, полимер высушивали в вакууме. Получали 0, 8 кг полипропилена.
КВ 120 см3/г. Температура плавления: 144,8oC.
Пример 14.
Сухой реактор емкость 70 дм3 промывали азотом и заполняли жидким пропиленом в количестве 40 дм3.
Затем добавляли 180 см3 толуольного метилалюминоксанового раствора (соответственно 270 ммоля А1, средняя степень олигомеризации n 16) и исходную смесь помешивали в течение 15 мин при температуре 30oC. После этого добавляли 35 г этилена. Параллельно этому растворяли 10,9 мг металлоцена А в 20 см3 толуольного метилалюминоксанового раствора (30 ммоля А1) и предварительно активировали в результате отстаивания в течение 15 мин.
Раствор подавали затем в реактор, реактор нагревали в течение 10 мин до температуры полимеризации 50oC, и выдерживали при перемешивании 4 ч. В течение также 4 ч непрерывно добавляли в определенных дозах 85 г этилена. Полимеризацию прекращали потом в результате добавления 20 мл изопропанола, избыточны мономер отдували, полимер высушивали в вакууме. Получали 3,5 кг статистического пропилен-этилен-мополимера с 3,0 мас. содержания этилена.
КВ 226 см3/г; Mw 2,3•105 г/мол; Mw/Mn 1,9
Пример 15.
Сухой реактор емкостью 16 дм3 промывали азотом и при температуре 20oC заполняли порцией деароматизированного бензина с диапазоном кипения 100 120oC.
Затем газовое пространство котла промывали до отсутствия азота в результате 5-кратного нагнетания 2 бар этилена и сброса давления.
После этого добавляли 30 см3 толуольного метилалюминоксанового раствора (соответственно 45 ммоля А1, молярная масса согласно криоскопическому определению 750 г/моль).
При перемешивании содержимое реактора в течение 15 мин нагревали до температуры 30oC и в результате добавления этилена, при скорости перемешивания 250 об/мин устанавливали общее давление 5 бар.
Параллельно этому 2,3 мг матллоцена растворяли в 20 см3 толуольного метилалюминоксанового раствора и предварительно активировали в результате 15-минутного отстаивания. Потом этот раствор подавали в реактор, полимеризаионную систему доводили до температуры 70oC, и в результате соответствующего охлаждения выдерживали при этой температуре 1 ч.
Общее давление в течение этого времени поддерживалось благодаря соответствующей подаче этилена до давления 5 бар.
Полимеризацию прекращали в результате добавления изопропанола, полимер отфильтровывали и высушивали в вакууме.
Получали 1,3 кг полиэтилена.
КВ 542 см3/г
Пример 16.
Повторяют пример 1, однако вводят в качестве металлоцена 3,1 мг рацемического диметилгермилбис-(2-метил-4-изопропиленденил) цирконий дихлорида.
Было получено 0,97 кг полипропилена с КВ=186 см2/г, Т. пл. 150,1oC,
изотактическим индексом
(UU) 95,2% и mmmm 89,5%
Пример 17. Повторяют пример 1, однако вводят в качестве металлоцена 1,1 мг рацемического диметилсилилбис-(2-метил-4-фенилинденил)
цирконий
дихлорида и температура
полимеризации была 50oC. Было получено 0,9 кг полипропилена с КВ 905 см3/г, т. пл. 159,4oC, изотактическим индексом 95,4% и mmmm 95,
4% Mw 11000000 г/мол;
Mw/Mn 2,5.
Пример 18. Повторяют пример 1, однако вводят в качестве металлоцена 10,5 мг рацемического тераметилдисилиленбис-(2-метил-4-фенилинденил) цирконий дихлорида и температура полимеризации была 65oC. Было получено 0,2 кг полипрорпилена с Mw 420000 г/мол. т. пл. 152oC. Активность катализатора 19 кг ПП на 1 г маталлоцена.
Пример 19. Повторяют пример 1, однако вводят в качестве металлоцена 3,7 мг рацемического этандиилбис-(2-метил-4-фенилинденил)цирконий дихлорида и температура полимеризации была 30oC. Было получено 0,35 кг полипропилена с КВ 440 см3/г, т. пл. 153oC. Активность катализатора составила 94 кг ПП/г металлоцена.
Реферат
Использование: получение полиолефинов с узким ММР. Сущность: проводят (со)полимеризацию олефинов общей формулы Ra=CH=CH=Rb или

Формула


где M металл группы IVB Периодической системы;
R1 и R2 одинаковые или различные, выбраны из группы, включающей водород, C1 C10 -алкил, C1 - C10-алкоксигруппу, C6 C10-арил, C6 - C10 -арилоксигруппу, C2 C10-алкенил, C7 - C40-арилалкил, C7 C40-алкилалил, C8 - C10-арилалкенилгалоген, R3 и R4, одинаковые или различные, выбраны из группы, включающей галоген, C1 - C10-алкил, галогеналкил, C6 C10-арил; R5 и R6, одинаковые или различные, имеют указанные для R3 и R4 значения, или водород;


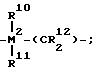
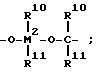

где R10 и R11 одинаковые или различные, выбраны из группы, включающей водород, галоген, C1 C10-алкил, C1 - C10-фторалкил, C6 C10 -арил, C6 - C10-фторарил, C1 C10-алкоксигруппу, C2 - C10-алкенил, C7 C40-арилалкил, C8 - C40-арилалкенил, C7 C40 - алкиларил, или R10 с R11 или R11 с R12 соответственно, образуют кольцо, или R10, или R11 с R8, или R9 образуют кольцо, R8 и R9 одинаковые или различные, имеют указанные для R10 значения;
m и n, одинаковые или различные, равные 0,1 или 2, при этом m + n 0, 1 или 2,
причем металлоцен формулы I отличен от диметилсилилен-бис-(2,4-диметил)-цирконийдихлорида и от этилен-бис-(2-метил-4-метилинденил)-цирконийдихлорида.

m + n 0 или 1.

для линейного типа и/или общей формулы III

для циклического типа,
где R13, одинаковые или различные, выбраны из группы, включающей C1 C6 -алкил, C6 C18-арил, водород;
p 2 50 целое число.
Комментарии