Способ синтеза олефинов - RU2356876C2
Код документа: RU2356876C2
Чертежи




Описание
Перекрестная ссылка на родственные заявки
Данная заявка представляет собой частичное продолжение более ранней заявки с регистрационным номером 10/365346, зарегистрированной 12 февраля 2003 года и находящейся в настоящее время на рассмотрении, которая представляет собой продолжение более ранней заявки с регистрационным номером 10/298440, зарегистрированной 20 ноября 2002 года, абандонированной, которая представляет собой частичное продолжение более ранней заявки с регистрационным номером 10/208068, зарегистрированной 29 июля 2002 года, абандонированной, которая представляет собой частичное продолжение более ранней заявки с регистрационным номером 10/054004, зарегистрированной 24 января 2002 года, теперь патент США No.6486368, которая представляет собой частичное продолжение более ранней заявки с регистрационным номером 09/951739, зарегистрированной 11 сентября 2001 года, теперь патент США No.64 65696, которая представляет собой частичное продолжение более ранней заявки с регистрационным номером 09/886078, зарегистрированной 20 июня 2001 года, теперь патент США No.6472572.
Область техники, к которой относится изобретение
Данное изобретение относится в основном к способам и аппаратуре для синтеза олефинов, спиртов, простых эфиров и альдегидов из алканов, алкенов и ароматических соединений и, в частности, к повышению избирательности реакций, описанных в патентах и патентных заявках, идентифицированных в данной работе, и к их конкретным применениям.
Уровень техники и сущность изобретения
В заявке с регистрационным номером 10/208068, зарегистрированная 29 июня 2002 года, раскрывается способ превращения этана в диэтиловый эфир, этанол и этилацетат, в котором этанол реагирует с галогеном, выбранным из группы, включающей хлор, бром и иод. Например, этан подвергается взаимодействию с бромом с образованием бромэтана и HBr. Бромэтан затем реагирует с оксидом металла с образованием диэтилового эфира, этанола, этилацетата и бромида металла. Бромид металла реагирует с кислородом или воздухом с регенерацией исходного оксида металла. По данному способу бром и оксид металла рециклируются.
В заявке с регистрационным номером 10/365346, зарегистрированная 12 февраля 2003 года, раскрывается способ, в котором реагент, включающий алкан, алкен или ароматическое соединение, подвергается взаимодействию с галогенидом металла для получения галогенида реагента и восстановленного металла. Восстановленный металл окисляется воздухом или кислородом с образованием соответствующего оксида металла. Оксид металла подвергается взаимодействию с галогенидом реагента с получением спирта и/или простого эфира, соответствующего исходному алкану, алкену или ароматическому соединению, и исходного галогенида металла, который рециклируется.
Данная заявка охватывает способы для синтеза олефинов, спиртов, простых эфиров и альдегидов, которые включают применение твердой фазы катализаторы/реагенты в дополнение к оксидам металла и галогенидам металла, раскрытым в выше представленных заявках. Данная заявка дополнительно включает методики для повышения избирательности реакций, раскрытых в ранее зарегистрированных заявках, которые включают, например, температурный контроль. Данная заявка, кроме того, охватывает конкретные применения раскрытых способов.
Краткое описание чертежей
Более полное понимание данного изобретения может быть достигнуто при обращении к последующему подробному описанию в сочетании с сопровождающими чертежами, в которых:
фигура 1 представляет собой диаграмму-иллюстрацию первого варианта осуществления изобретения;
фигура 2 представляет собой диаграмму-иллюстрацию второго варианта осуществления изобретения;
фигура 3 представляет собой диаграмму-иллюстрацию третьего варианта осуществления изобретения;
фигура 4 представляет собой диаграмму-иллюстрацию четвертого варианта осуществления изобретения;
фигура 5 представляет собой диаграмму-иллюстрацию пятого варианта осуществления изобретения;
фигура 6 представляет собой диаграмму-иллюстрацию шестого варианта осуществления изобретения.
Подробное описание
На графиках, в частности на фигуре 1, показан способ и аппаратура 10 для синтеза спиртов и/или простых эфиров, составляющих первый вариант осуществления изобретения. Способ и аппаратура 10, а также другие варианты осуществления изобретения, описанные здесь далее, могут быть применены для синтеза олефинов, спиртов, простых эфиров и/или альдегидов. Следующее описание, где спирты и/или простые эфиры синтезированы из алкенов, является типичным.
Выбранный алкан, который может включать метан, этан, пропан, бутан, изобутан, пентан, гексан, циклогексан и т.д., получают в первом реакторе 12 из подходящего источника 14 по линии 16. Реактор 12 также получает галогенид металла по линии 18. Галогенид, включая галогенид металла, который получен в реакторе 12, выбран из группы, включающей хлор, бром и иод.
Взаимодействие алкана с галогенидом металла приводит к образованию соответствующего алкилгалогенида, который отводится по линии 20. По реакции также образуется металл в восстановленной форме, такой как гидрид металла, который отводится по линии 22 и направляется во второй реактор 24. Второй реактор также получает кислород и/или воздух из источника 26 по линии 28.
Функции второго реактора 24 состоят в превращении восстановленного металла, полученного по линии 22, до оксида металла, который отводят по линии 30 и направляют в третий реактор 32. Температура реакции внутри второго реактора 24 является достаточно низкой, такой, что любой бром, оставшийся на металле после реакции в первом реакторе 12, остается на металле, и только водород на металле замещается кислородом. Водород, выделившийся с металла, превращается в воду.
Внутри третьего реактора 32 алкилгалогенид, полученный в первом реакторе 12, реагирует с оксидом металла, полученным во втором реакторе 24, с образованием соответствующего спирта и/или простого эфира, который отделяется через выпуск 34. Реакция в третьем реакторе 32 также приводит к образованию галогенида металла, который рециклируется в первый реактор 12 по линии 18.
В соответствии с конкретным применением изобретения первый реактор 12 получает этан из источника 14 по линии 16, и галогенид металла, полученный в реакторе 12 по линии 18, включает бромид металла. Реакция внутри реактора 12 приводит к этилбромиду, который отводится по линии 20 и направляется в третий реактор 32. Реакция внутри третьего реактора 32 может быть охарактеризована следующим образом:
4CH3CH2Br + Metal Oxide+XH2O → 2CH3CH2OH + CH3CH2OCH2CH3 + Metal Bromide2.
Как должно быть очевидно, реакция внутри реактора 32 может быть направлена по пути получения спирта или по пути получения эфира в зависимости от количества воды, присутствующей внутри реактора. Фигура 1 иллюстрирует дополнительно операционный режим первого варианта осуществления изобретения, по которому водяной пар направляется в линию 20 по линии 36, для поступления в реактор 32 с этилбромидом и HBr, которые были получены в реакторе 12. Как очевидно из формулы, приведенной выше, введение водяного пара в реактор 32 вызывает в нем реакцию, являющуюся избирательной в направлении получения этанола в реакторе 32 в противоположность получению диэтилового эфира.
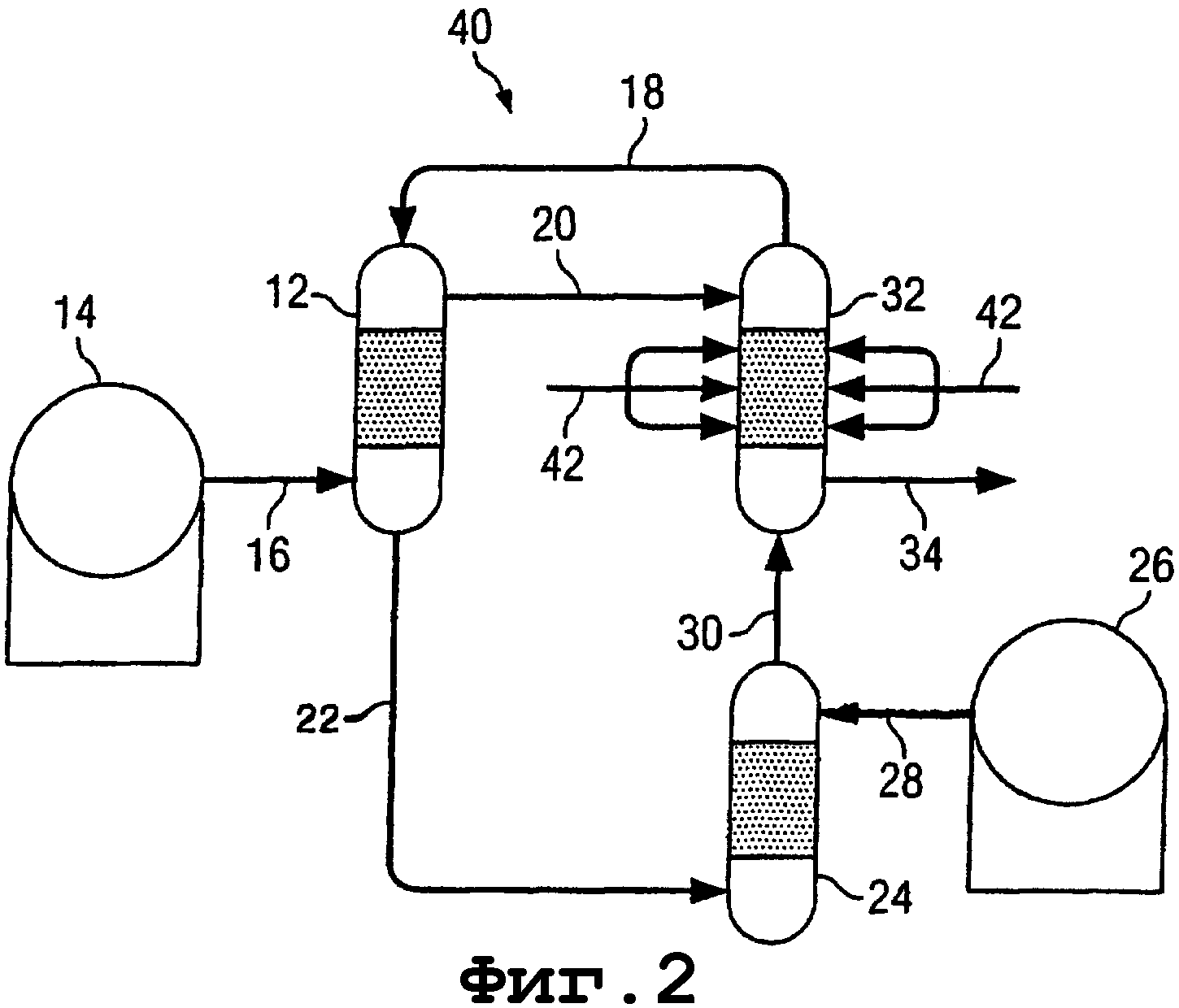
На фигуре 2 показан способ и аппаратура для синтеза олефинов, спиртов, простых эфиров и/или альдегидов, составляющих второй вариант осуществления изобретения. Многие из компонентных частей второго варианта осуществления изобретения идентичны по конструкции и функции компонентным частям первого варианта осуществления изобретения, показанным на фигуре 1 и описанным здесь выше в связи с данным вариантом. Такие идентичные компонентные части обозначены на фигуре 2 теми же самыми ссылочными номерами, примененными в описании первого варианта осуществления изобретения.
Второй вариант осуществления изобретения отличается от первого варианта осуществления изобретения тем, что вместо введения водяного пара в реактор 32 по линии 20, его вводят непосредственно в реактор 32 по линиям 42. Это позволяет добавить водяной пар в реактор 32 в определенных точках в реакторе, тем самым увеличить избирательность реакции, которая там происходит, в направлении получения спирта и избавиться от образования эфира.
Способ и аппаратура 50 для получения олефинов, спиртов, простых эфиров и/или альдегидов, составляющих третий вариант осуществления изобретения, проиллюстрирована на фигуре 3. Многие из компонентных частей третьего варианта осуществления изобретения идентичны по конструкции и функции компонентным частям первого варианта осуществления изобретения, которые показаны на фигуре 1 и описаны здесь выше в связи с данным вариантом. Такие идентичные компонентные части обозначены на фигуре 3 теми же самыми ссылочными номерами, примененными в описании первого варианта осуществления изобретения.
Третий вариант осуществления изобретения отличается от первого и второго вариантов осуществления тем, что в процессе его реализации вода из реактора 32 удаляется по линиям 52. Удаление воды выполняется либо перегонкой, либо осмосом, или и тем и другим. Как очевидно из реакции, приведенной выше, удаление воды из реактора 32 вызывает в нем реакцию, являющуюся избирательной в направлении получения эфира в противоположность получению спирта.
На фигуре 4 показан способ и аппаратура 60 для синтеза олефинов, спиртов, простых эфиров и/или альдегидов, составляющих четвертый вариант осуществления изобретения. Многие из компонентных частей четвертого варианта осуществления изобретения идентичны по конструкции и функции компонентным частям первого варианта осуществления изобретения, показанным на фигуре 1 и описанным здесь выше в связи с данным вариантом. Такие идентичные компонентные части обозначены на фигуре 4 теми же самыми ссылочными номерами, примененными в описании первого варианта осуществления изобретения.
В соответствии с четвертым вариантом осуществления изобретения кислород и/или воздух направляется во второй реактор 24 с такой скоростью, что в дополнение к окислению восстановленный металл обращается в оксид металла, дополнительный кислород добавляется к оксиду металла и освобождается молекулярный галогенид. Продукты реакции из второго реактора 24 направляются в сепаратор 62. Сепаратор 62 направляет оксид металла в реактор 32 по линии 64, возвращает кислород к источнику 26 по линии 66 и направляет галогенид в бак-хранилище галогенида 68 по линии 70. Из бака-хранилища 68 галогенид направляется в реактор 12 по линии 72 и линии 18, при этом обеспечивается оптимальный уровень галогенида внутри реактора 12 на протяжении всего времени.
Обращение к фигуре 5 показывает способ и аппаратуру 80 для получения олефинов, спиртов, простых эфиров и/или альдегидов, составляющих пятый вариант осуществления изобретения. Пятый вариант осуществления изобретения особенно применим в производстве олефинов.
Выбранный алкан, который может включать метан, этан, пропан, бутан, изобутан, пентан, гексан, циклогексан и т.д. получается в первом реакторе 82 из подходящего источника 84 по линии 86. Реактор 82 также получает галогенид из контейнера для хранения 88 по линии 90 и линии 86. Галогенид, который получен в реакторе 82, выбран из группы, включающей хлор, бром и иод.
Полученные продукты реакции от операции в реакторе 82 направляются в дистилляционный сепаратор 92 по линии 94. Непрореагировавший алкан возвращается из сепаратора 92 в реактор 82 по линии 96 и линии 86. Галогенводород направляется из сепаратора 92 во второй реактор 98 по линии 100. Моноалкилгалогенид направляется из сепаратора 92 в третий реактор 102 по линии 104. Высшие галогениды возвращаются из сепаратора 92 в реактор 82 по линии 106.
Внутри второго реактора 98 галогенводород, полученный по линии 100, подвергается взаимодействию с оксидом металла для получения галогенида металла и воды. Воду отводят из второго реактора 98 в контейнер-хранилище сточной воды 108 по линии 110. Галогенид металла, включающий галогеновый компонент галогенводорода, полученный в реактор 98 по линии 100, направляется по линии 112 в четвертый реактор 114. Реактор 114 получает также воздух или кислород из подходящего источника 116 по линии 118. Реактор 114 производит генерированный оксид, который возвращается во второй реактор 98 по линии 120.
Четвертый реактор 114 также производит галогенид, диоксид углерода и воду, и все это направляется в сепаратор 122 по линии 124. Сепаратор 122 отделяет галогенид от воды и диоксида углерода и направляет отделенный галогенид к контейнер для хранения галогенида 88 по линии 126. Воду и диоксид углерода отделяют из сепаратора 122 по линии 128.
В дополнение к полученному моноалкилгалогениду по линии 104 третий реактор 102 получает генерированный оксид металла по линии 120 и линии 130. Реакция внутри третьего реактора 120 приводит к образованию галогенида металла, который направляется в четвертый реактор 114 по линии 132 и линии 112. Оставшиеся продукты реакции, полученные при операции в третьем реакторе 102, направляются в дистилляционный сепаратор 134 по линии 136.
Процесс в дистилляционном сепараторе 134 приводит к целевому олефину, который отводится по линии 138. Летучие побочные продукты отводятся по линии 140, и высококипящие побочные продукты отводятся по линии 142. Непрореагировавший моноалкилгалогенид направляется из сепаратора 134 в третий реактор 142 по линии 144. Воду, образующуюся при операции в сепараторе 134, направляют в контейнер для сточной воды 108 по линии 146.
На фигуре 6 показан способ и аппаратура для синтеза олефинов, спиртов, простых эфиров и/или альдегидов, составляющих шестой вариант осуществления изобретения. Многие из компонентных частей шестого варианта осуществления изобретения идентичны по конструкции и функции компонентным частям пятого варианта осуществления изобретения, показанным на фигуре 5 и описанным здесь выше в связи с данным вариантом. Такие идентичные компонентные части обозначены на фигуре 6 теми же самыми ссылочными номерами, примененными в описании пятого варианта осуществления изобретения.
Шестой вариант осуществления изобретения отличается от пятого варианта осуществления изобретения тем, что продукты реакции, полученные при операции в третьем реакторе 102, кроме галогенида металла, направляются по линии 136 в пятый реактор 152, функции которого состоят в превращении продуктов реакции, полученных при операции во втором реакторе 102, в соответствующие спирты. Продукты реакции, полученные при операции в пятом реакторе 152, направляются по линии 154 в дистилляционный сепаратор 156. Целевой спирт отводится из сепаратора 156 по линии 158. Летучие побочные продукты отводятся по линии 160, и высококипящие побочные продукты отводятся по линии 162. Воду, образующуюся при операции в сепараторе 156, направляют в контейнер для сбора сточной воды 108 по линии 163.
Шестой вариант осуществления изобретения также отличается от пятого варианта осуществления изобретения тем, что сточная вода из контейнера для сбора сточной воды 108 направляется в систему для обработки сточной воды 164 по линии 166. Обработанная сточная вода, полученная при операции в системе 164, направляется в сепаратор 168 по линии 170. Обработанная сточная вода отводится из сепаратора 168 по линии 171 и вода, питающая бойлер, отводится из сепаратора 168 по линии 172. Вода, питающая бойлер, направляется в бойлер 174, который производит водяной пар. Пар, полученный при операции в бойлере 174, направляется во второй реактор 102 по линии 176 и линии 130.
В соответствии с седьмым вариантом осуществления изобретения было установлено, что в дополнение к оксидам металлов и галогенидам металлов, которые были раскрыты как реагенты, применимые для превращения алкилгалогенида в продукт, а также для активной нейтрализации галогенводорода, для данной цели применимы также гидроксиды металлов. Таким образом, хотя галогенид металла, как предполагают, должен быть продуктом, получающимся по реакции оксида металла, также возможно, что продукт реакции оксида металла представляет собой оксигалогенид металла.
В соответствии с восьмым вариантом осуществления изобретения доказана применимость других композиций в качестве катализаторов/реагентов. Например, композиция твердого реагента может принимать форму твердого гидрата, такого как оксигидрат металла или гидраты галогенидов, сульфидов, карбонатов, фосфатов, фосфидов, нитридов и нитратов.
В соответствии с девятым вариантом осуществления изобретения было определено, что углеводороды, выше метана, этана и пропана, и выбранные из группы, включающей парафиновые и нафтеновые углеводороды, также могут быть использованы в качестве исходных продуктов для данного способа. Данное определение расширяет общую применимость способа к углеводородам с числом атомов углерода вплоть до 16 и применимость к нормальным, разветвленным и циклическим алканам.
В соответствии с десятым вариантом осуществления изобретения было определено, что контроль над избирательностью реакции и композицией продукта специально может быть осуществлен преднамеренными изменениями в композиции и форме (в зависимости от синтетического пути) твердого оксида металла. В частности, небольшие изменения в атомном составе оксида металла (состоящего из одного или нескольких атомов металла) действуют на реакционную способность и/или избирательность и различные синтетические пути тех же самых композиций приводят к повышению различий в реакционной способности и/или избирательности.
Давление и температура проведения реакции также являются контролируемыми параметрами, применимыми в регулировании реакции в направлении образования определенных продуктов. Например, оксид металла, который дает преимущественно спирт при температуре примерно 250°С, может быть переключен на получение преимущественно олефина повышением температуры взаимодействия оксида металла от примерно 350°С до примерно 400°С. Повышение температуры создает предпочтение реакции элиминирования бета-водорода над гидроксилированием. Однако, важно отметить, что возможны другие механизмы для генерации олефина. Другая польза от повышения температуры проведения реакции состоит в полученном увеличении скорости превращения алкилбромида, приводящем к более короткому времени протекания реакции и к реактору меньшего размера.
Так как отмеченные температурные эффекты наблюдались на большом наборе продуктов без исключения, то данный эффект широко применим для ряда элементов Периодической таблицы. Образцы продуктов включают MgO, CaO, FeMoOx, La2O3, Ca(OH)2, PbO, CuO, Bi2O3, MgZrOx, ZnO. Самым важным был пример смешанного оксида кобальта и циркония (стехиометрия CoZrOx).
Пример I-CoZrOx
Образец готовят смешиванием равных количеств 0,5 М раствора нитрата кобальта и 0,5 М раствора пропоксида циркония с последующей сушкой при 120°С и прокаливанием в течение ночи при 500°С.
Этан подвергают взаимодействию с бромом в молярном соотношении приблизительно 10:1, получая ~ 94% (углерод) избирательности к этилбромиду плюс соединения с более высокой степенью бромирования. Также получается HBr. Пропускание данной смеси над CoZrOx приводит к конверсии этилбромида, которая повышается с температурой. Соответствующие конверсии бромированного этана при 175°С, 200°С, 225°С и 250°С равны приблизительно 41%, 56%, 69% и 82%. Проведение реакции при 350°С с применением менее 80% оксида обеспечивает лучшую конверсию, чем с 90%. Избирательности по этилену (моли этилена/моли всех определенных продуктов) приблизительно равны 1%, 7%, 20%, 34% и 90% при 175°С, 200°С, 225°С, 250°С и 350°С. При 350°С (исключая непрореагировавший этилбромид) смесь продуктов представляет собой приблизительно 90% этилена, 5% диоксида углерода, 4% винилбромида и 1% другого соединения (в основном, ацетон).
Пример II-CoZrOx
Когда этилбромид (98+%) пропускают над CoZrOx, описанным выше в примере I, при 350°С, продукты, определенные в потоке летучего продукта, представляют собой приблизительно 95% этилена с преобладанием диоксида углерода в оставшихся процентах до баланса.
Как вариант способа, оксид металла (или галогенид или оксигалогенид) может функционировать в качестве катализатора для дегидробромирования этилбромида до этилена и галогенводорода, при этом галогенводород затем нейтрализуют.
В соответствии с одиннадцатым вариантом осуществления изобретения гидроксид металла представляет собой рабочие соединения в реакции метатезиса. Пропускание алкилгалогенида над гидроксидом металла дает соответствующий спирт. Затем образовавшийся галогенид металла регенерируют с помощью воздух/кислород для превращения галогенида металла в оксид металла, который затем подвергают взаимодействию с водяным паром для регенерации гидроксида металла. В соответствии со второй частью одиннадцатого варианта осуществления поток воздух/кислород, используемый для регенерации оксида металла, смешивают с контролируемым количеством водяного пара, таким образом гидроксид металла получают без прохождения через отдельную стадию оксида металла.
Двенадцатый вариант осуществления изобретения относится к способности способа избирательно производить ряд продуктов окисления с помощью исходного материала, состоящего из различных углеводородов, что значительно упрощает производство продукта по сравнению с существующими способами.
В традиционных установках по производству олефинов этан или легкую нафту термически “крекируют” нагреванием этана или легкой нафты до высокой температуры и контактированием исходного сырья с водяным паром. “Крекинг” процесс происходит по свободнорадикальному механизму и приводит к различным продуктам, включающим алкановые углеводороды (такие как этан), алкеновые углеводороды (такие как этилен), алкиновые углеводороды (такие как ацетилен), диеновые углеводороды (такие как бутадиен), ароматические углеводороды (такие как бензол) и кокс. После получения данных продуктов требуется ряд стадий перегонки, многие из которых проводятся при низких температурах, чтобы отделить один продукт от другого для применения в качестве сырья для других химических продуктов из нефти. Как пример, разделение этилена от этана перегонкой является как капитальным, так и энергетически затратным, так как два соединения обладают очень близкими друг к другу температурами кипения.
В отличие от обычного способа олефины по данному способу получаются непосредственно из их алканового предшественника, либо из одного алканового сырьевого компонента, либо из сложной смеси. Один путь осуществления данного процесса состоит в следующем. Алкановый сырьевой компонент или сырьевую смесь подвергают контактированию с галогенидом для образования моногалогенированного аналога алканового сырьевого компонента или сырьевой смеси. После реакции галогенирования реакционную смесь пропускают над твердым оксидом металла при температуре примерно от 105°С до примерно 150°С, посредством чего галогенводород, полученный по реакции галогенирования, превращается в воду, которую затем удаляют из оставшихся моноалкилгалогенидов. Алкилмоногалогениды и оставшиеся алканы, имеющие в основном различные температуры кипения, затем разделяют перегонкой. Затем образуется один или несколько потоков, каждый из которых содержит алкилгалогенид с заранее известным числом атомов углерода. В данном способе упраздняется трудное отделение алканов от алкенов (например, этана от этилена). В альтернативном случае отделение избытка алканов может предшествовать превращению галогенводорода в воду. Далее каждый алкилмоногалогенид может быть затем введен во взаимодействие с оксидом металла при температуре примерно от 250°С до примерно 350°С для получения олефина с числом атомов углерода, соответствующим алкилмоногалогениду.
В альтернативном случае продукты реакции галогенирования после нейтрализации галогенводорода и удаления образующейся воды могут быть направлены как смесь над оксидом металла и превращены в соответствующие олефины. После генерации олефинов олефины могут быть разделены на целевой один или несколько потоков олефинового продукта.
Если целевой продукт определенного алкана представляет собой спирт, простой эфир или альдегид, то данное изобретение особенно применимо. В производстве спиртов, простых эфиров или альдегидов способ может быть реализован в точном режиме, описанном выше для олефинов вплоть до момента, где алкилмоногалогениды отделяют один от другого. Для получения спирта, простого эфира или альдегида из определенного алкилмоногалогенида определенный алкилмоногалогенид пропускают над определенным оксидом металла, который выбирают, основываясь на его избирательности для получения требуемой функциональности (т.е. спирта, простого эфира или альдегида). Так как избирательность целевого продукта должна зависеть от количества воды, присутствующей при взаимодействии алкилмоногалогенида и оксида металла, то вода может быть добавлена к алкилмонобромидному сырьевому компоненту до соответствующего уровня. Например, вода, образующаяся при нейтрализации галогенводорода, может быть использована для данной цели.
Если требуется получить поток смешанных спиртов, простых эфиров или альдегидов, то данное изобретение можно реализовать простым удалением стадии, с помощью которой алкилмоногалогениды разделяют один от другого перед стадией взаимодействия алкилмоногалогенида с определенным оксидом металла. Предпочтительно для получения смесевого потока объединенные алкилмоногалогениды непосредственно пропускают как смесь над определенным оксидом металла и получается требуемый смесевой поток. Смесевой поток затем может быть применен сам по себе или разделен, как желательно, на один или несколько потоков продуктов.
Тринадцатый вариант осуществления данного изобретения включает способ усовершенствования существующей установки по производству олефинов для повышения ее производительности и снижения стоимости производства. Данное изобретение основывается на способах, раскрытых в патентах США под номерами: 6462243, 6472572, 6486368 и 6465696 для обеспечения избирательного неполного окисления алканов, и включает присоединение способа, раскрытого здесь, к существующей олефиновой установке таким образом, чтобы повысить производительность соответственно минимальному дополнительному вложению.
ПО общему подходу к модифицированному варианту осуществления после галогенирования алканового потока галогенводород нейтрализуют над оксидом металла, как описано ранее. Однако вместо разделения алкилмонобромидов друг от друга общий поток смешанных алкилмонобромидов пропускают над оксидом металла и превращают в смешанный поток олефинов. Образовавшийся олефиновый поток затем направляют в заводскую существующую разделительную установку вместе с продуктом, полученным из термической крекинговой камеры завода, и разделяют на определенные олефиновые потоки.
ПО более определенному примеру модифицированного варианта осуществления этан или нафту подвергают крекингу в обычной установке по производству этилена с более слабой избирательностью и превращением в этилен. По способу образуется в основном этановый рециклирующий поток. Модификация данного изобретения включает захват/поглощение всего или части этана, обычно рециклирующего в печи, и вместо этого направление его в субпроцесс, утилизирующий две стадии химии, описанные в патентах, приведенных выше. Способ избирательно превращает этан в этилен. Преимущества являются многократными:
1. За счет снижения рецикла в печи может быть введено дополнительное свежее сырье, что эффективно увеличивает производительность установки.
2. За счет обеспечения экстремально чистого сырья этилена субпроцесс снижает требование к разделительным системам, что снова эффективно увеличивает производительность установки.
3. Чистота этилена, полученного по субпроцессу, приводит к значительному снижению стоимости производства. Например, обычный этиленовый способ дает в качестве побочного продукта ацетилен, который должен быть избирательно прогидрирован до этилена, что является трудным процессом. Производство этилена, описанное в данной работе, не приводит к образованию ацетилена, тем самым уменьшается количество ацетилена, полученного в установке для производства этилена.
4. Добавление реактора позволяет произвести дополнительное расширение существующей установки без необходимости большого вложения средств, требуемых для новой установки. Данная этиленовая технология является единственной эффективной по затратам при огромных масштабах, причем она таким образом предотвращает введение небольших мощностей, когда имеется малый спрос. Способность присоединяться к существующим объектам на местах также капитализирует на существующей инфраструктуре, уменьшая стоимость расширения производства.
В дополнение к вышесказанному было установлено, что различные оксиды металлов, примененные в реакции метатезиса, могут также служить как адсорбенты/реагенты для контроля высвобождения галогенводорода или аклилгалогенида в окружающую среду. Вовлечение оксида металла в очистительную систему внутри завода не только контролирует высвобождение таких соединений, но также служит способом для возвращения брома для последующего использования по данному способу.
Пример III - Галогенирование алкана
Реакция бромирования этана
Смесь этана и брома в молярном соотношении 3,6:1 готовили пропусканием потока этана (2,0 мл/минута) через барботер с бромом, который поддерживали при 21°С. Смесь пропускали в реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), который нагревали до 400°С. Вытекающий поток идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 27,4% конверсии этана. Избирательность к этилбромиду составляла 89,9%. Другие продукты представляли собой полибромированные этаны, причем основная часть представляла собой дибромэтаны (>90% баланса).
Реакция хлорирования этана
Смесь этана и хлора в молярном соотношении 2:1 (общая скорость потока 5 мл/мин) пропускают через реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), который нагревают до 350°С. Получают 100% конверсии хлора и 45% конверсии этана с избирательностью к этилхлориду 90%.
Реакция иодирования этана
Смесь этана и иода в молярном соотношении 1:4 (общая скорость потока 5 мл/мин) пропускают через реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревают до 400°С. Получают 10% конверсии этана с избирательностью к иодэтану 85%.
Бромирование пропана
Смесь пропана и брома в молярном соотношении 3,6:1 готовили пропусканием потока пропана (2,0 мл/минута) через барботер с бромом, который поддерживали при 21,0°С. Смесь пропускали в реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), который нагревали до 350°С. Вытекающий поток идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 27% конверсии пропана. Избирательность к 2-бромпропану составляла 90%. Другие продукты представляли собой 2,2-дибромпропан (~10%) и 1-бромпропан (<1%).
Бромирование высшего алкана
Смесь алкана (CnH2n+2 3 Жидкофазное бромирование высшего алкана Додекановую жидкость под давлением смешивают с бромом в молярном соотношении 10:1. Смесь пропускают через трубчатый реактор, поддерживаемый при 250°С. Получают 100% конверсии брома с 90% избирательности к додецилбромиду. Диспропорционирование С2 соединений 100 ммоль/час этана и 100 ммоль/час брома подают в реактор, содержащий катализатор Pt/диоксид кремния, поддерживаемый при 400°С. Рециклирующий поток 46 ммоль/час этана и 50 ммоль/час полибромированных этанов также подают в реактор. В реакторе достигается равновесие со 100% конверсии брома и распределение продукта из 46 ммоль/час этана, 100 ммоль/час этилбромида и 50 ммоль/час высших бромидов. Образуется также 100 моль HBr. Продукты разделяют на 3 потока: (1) этан/HBr, (2) этилбромид и (3) высшие бромиды. Поток (1) пропускают над способным к регенерации оксидом металла, при этом нейтрализуется HBr и образуется вода. Воду отделяют перед рециклированием этана. Поток (3) также рециклируют обратно в реактор. Диспропорционирование С2 соединений без добавления галогенида 100 ммоль/час этана и 10 ммоль/час дибромэтана подают в реактор, содержащий катализатор диоксид циркония, поддерживаемый при 300°С. Получают распределение продукта из 91 ммоль/час этана, 18 ммоль/час этилбромида и 1 ммоль/час дибромэтана. Диспропорционирование С10 соединений 100 ммоль/час декана и 100 ммоль/час брома подают в реактор, содержащий катализатор Rh/диоксид кремния, поддерживаемый при 300°С. Рециклирующий поток из 100 ммоль/час декана и 80 ммоль/час дибромдекана и более высоко бромированных деканов также подают в реактор. В реакторе достигается 100% конверсии брома и распределение продукта из 100 ммоль/час декана, 100 ммоль/час децилбромида и 80 ммоль/час полибромированного декана. Образуется также 100 моль HBr. Продукты разделяют на 4 потока: (1) HBr, (2) декан, (3) децилбромид и (4) высшие бромиды. Поток (1) пропускают над способным к регенерации оксидом металла, при этом нейтрализуется HBr и образуется вода. Потоки (3) и (4) рециклируют в реактор. Диспропорционирование С10 соединений без добавления брома 100 ммоль/час декана и 10 ммоль/час дибромдеканов (смешанные изомеры) подают в реактор, содержащий катализатор Rh/диоксид кремния, поддерживаемый при 200°С. Получают распределение продукта из 91 ммоль/час декана, 18 ммоль/час децилбромида и 1 ммоль/час дибромдеканов. Диспропорционирование смешанных С1-С3 соединений Поток дибромметана (1 ммоль/час) и дибромэтана (2 ммоль/час) объединяют с потоком этана (95 ммоль/час), метана (3 ммоль/час) и пропана (2 ммоль/час) в реакторе, содержащем Ru/диоксид кремния, который поддерживают при 250°С. Продукт в реакторе представляет собой этан (96 ммоль/час), метан (4 ммоль/час), этилбромид (1 ммоль/час), дибромпропан (2 ммоль/час) и другие бромированные продукты (следы). Пример IV - Взаимодействия алкилгалогенидов с оксидами металлов и другими твердыми веществами Приготовление Zr раствора Zr(OCH2CH2CH3)4 (70 (мас.)% в изопропаноле, 112,6 мл) растворяли (при перемешивании) в растворе 56,6 г щавелевой кислоты в 200 мл воды. После перемешивания в течение 10 минут раствор разбавляли водой до получения общего объема в 500 мл. Получали раствор с Zr концентрацией, равной 0,5 М. Приготовление М1 Со(NO3)2 (0,5 М, 100,0 мл) добавляли к 100 мл перемешиваемого Zr раствора (0,5 М). После перемешивания в течение нескольких минут получали гель. Гель сушили при 120°С в течение 4 часов, затем прокаливали при 500°С в течение 4 часов. После измельчения в ступке получали М1. Приготовление М2 Fe(NO3)3 (0,5 М, 50,0 мл) и Zn(NO3)2 (0,5 М, 50,0 мл) добавляли к 100 мл перемешиваемого Zr раствора (0,5 М). После перемешивания в течение нескольких минут получали гель. Гель сушили при 120°С в течение 4 часов, затем прокаливали при 500°С в течение 4 часов. После измельчения в ступке получали М2. Приготовление М3 Со(NO3)2 (0,5 М, 80,0 мл) и борную кислоту (0,25 М, 40,0 мл) добавляли к 100 мл перемешиваемого Zr раствора (0,5 М). После перемешивания в течение нескольких минут получали гель. Гель сушили при 120°С в течение 4 часов, затем прокаливали при 500°С в течение 4 часов. После измельчения в ступке получали М3. Приготовление М4 Со(NO3)2 (0,5 М, 80,0 мл) и KNO3 (0,5 М, 20,0 мл) добавляли к 100 мл перемешиваемого Zr раствора (0,5 М). После перемешивания в течение нескольких минут получали гель. Гель сушили при 120°С в течение 4 часов, затем прокаливали при 500°С в течение 4 часов. После измельчения в ступке получали М4. Приготовление М5 Готовили взвесь хроматографического диоксида кремния и нитрата кобальта, достаточную для получения загрузки из 40 мас.% Со3О4. Взвесь прокаливали при 400°в течение 2 ч, получая М5. Бромирование этана и взаимодействие с М1 Смесь этана и брома в молярном соотношении 10:1 готовили пропусканием потока этана (5 см3/мин) через барботер с бромом, который поддерживали при 0°С. Смесь пропускали в первый реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до 350°С. Вытекающий поток предварительно идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 9,5% конверсии этана. Избирательность к этилбромиду составляла более 94%. Вытекающий поток из первого реактора пропускали во второй реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до различных температур и который содержал 5 г М1 (за исключением опыта при 350°С, в котором применен 1 г М1). Соответствующие конверсии бромированного этана при 175°С, 200°С, 225°С, 250°С и 350°С составляли приблизительно 41%, 56%, 69%, 82% и 90%, соответственно. Избирательности по этилену (моли этилена/моли всех определенных продуктов) приблизительно равны 1%, 7%, 20%, 34% и 90% при 175°С, 200°С, 225°С, 250°С и 350°С. При 350°С (исключая непрореагировавший этилбромид) смесь продуктов представляла собой приблизительно 90% этилена, 5% диоксида углерода, 4% винилбромида и 1% другого соединения (в основном, ацетон). Взаимодействие этилбромида с М1 Жидкий этилбромид (0,6 мл/час) подвергали испарению в комбинации с инертным азотом как газом-носителем. Смесь пропускали через реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), содержащий 2 г оксида металла М1, который нагревали до 350°С. Продукты анализировали c помощью ГХ (GC). Получали 100% конверсии этилбромида в течение 5 часов. Получали избирательность к этилену, равную 96%. Побочные продукты включали диоксид углерода (2%), этанол (<1%), этан (<0,5%). Взаимодействие этилхлорида с М1 Этилхлорид (1 мл/мин) пропускают через реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), содержащий 5 г оксида металла М1, который нагревают до 350°С. Получают 100% конверсии этилхлорида в течение 5 часов. Получают избирательность к этилену, равную 90%. М1 регенерируют нагреванием до 600°С в кислороде с выделением хлора. Каталитическое взаимодействие этилбромида с диоксидом титана Жидкий этилбромид (0,6 мл/мин) подвергали испарению в комбинации с инертным азотом как газом-носителем. Смесь пропускали через реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), содержащий 5 г диоксида титана (TiO2), который нагревали до 275°С. Продукты реакции пропускали через ловушку с водным гидроксидом натрия для удаления остатков HBr перед ГХ (GC) анализом. Получали 60% конверсии этилбромида с избирательностью к этилену >99% и с отсутствием этана, не наблюдаемого при пределах определения <100 ч/млн. В другом варианте очистку от HBr производят с помощью оксида меди, который можно регенерировать, вместо гидроксида натрия. Каталитическое взаимодействие алкилбромида с твердым катализатором Поток, содержащий алкилбромид (CnH2n+2 1 Бромирование пропана и последующее взаимодействие с М5 Смесь пропана и брома в молярном соотношении 3,6:1 готовили пропусканием потока пропана (5 мл/минута) через барботер с бромом, который поддерживали при 21,0°С. Смесь пропускали в первый реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), который нагревали до 350°С. Вытекающий поток предварительно идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 27% конверсии пропана. Избирательность к 2-бромпропану составляла 90%. Другие продукты представляли собой 2,2-дибромпропан (~10%) и 1-бромпропан (<1%). Вытекающий поток из первого реактора пропускали во второй реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до 350°С и который содержал 5 г М5. Продукты анализировали с помощью ГХ (GC). Избирательности (основанные на конверсии пропана) равны по пропилену (90%), ацетону (5%), другим веществам (5%). Взаимодействие 1-бромдекана с М3 Жидкий децилбромид (0,6 мл/мин) пропускали (с азотом в качестве газа-носителя) в реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), содержащий 2 г оксида металла М3, который нагревали до 200°С. Примененный образец М3 ранее был использован в более чем 5 циклах реакция/регенерация. Реакцию проводили в течение 2 часов со сбором продуктов в 6 г охлажденного CDCl3. ЯМР-анализ показал 100% конверсии и продукты преимущественно представляли собой внутренние олефины с избирательностью более чем 85%. Взаимодействие 1-бромпентана с М3 Азот (5 мл/мин) барботировали через 1-бромпентан при комнатной температуре и подавали в реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), содержащий 2 г оксида металла М3, который нагревали до 200°С. Примененный образец М3 ранее был использован в более чем 5 циклах реакция/регенерация. Реакцию проводили в течение 2 часов со сбором продуктов в 6 г охлажденного CDCl3. ЯМР-анализ показал 100% конверсии, и продукты преимущественно представляли собой внутренние олефины с избирательностью более чем 90%. Взаимодействие смешанных алкилбромидов с М1 Поток, содержащий смесь 90% алкилбромидов (CnH2n+1Br 1 Взаимодействие дибромдодекана с М1 Смешанные изомеры дибромдодекана пропускают над слоем М1 при 300°С. Получают 100% конверсии с 80% избирательности к диолефину. Взаимодействие винилбромида с оксидом меди(II) Поток, содержащий 10 мол.% винилбромида, пропускают над оксидом меди(II) при 350°С. Более чем 99,9% винилбромида удаляется из потока. Винилбромид реагирует с оксидом меди(II), образуя оксид меди(I), бромиды меди, диоксид углерода и воду. После реакции твердое вещество отжигают в кислороде при 375°С, высвобождая бром для повторного использования и регенерируя оксид меди(II). Взаимодействие смешанных бромидов с оксидом меди(II): Поток, содержащий смесь органических бромидов, пропускают над оксидом меди(II) при 350°С. Более чем 99,9% бромидов удаляется из потока. Бромиды реагируют с оксидом меди(II), образуя оксид меди(I), бромиды меди, диоксид углерода и воду. После реакции твердое вещество отжигают в кислороде при 375°С, высвобождая бром для повторного использования и регенерируя оксид меди(II). Добавление воды к взаимодействию этана Смесь этана и брома в молярном соотношении 10:1 готовили пропусканием потока этана (5 мл/минута) через барботер с бромом, который поддерживали при 0°С. Смесь пропускали в первый реактор (стеклянная трубка ID 0,038'', длина зоны нагрева 4''), который нагревали до 350°С. Вытекающий поток предварительно идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 9,5% конверсии этана. Избирательность к бромэтану составляла более 94%. Вытекающий поток из первого реактора объединяли с добавлением различных количеств избытка воды и пропускали во второй реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который содержал 5 г М1 при 200°С. Так как молярное соотношение вода: бром изменяли от 1 до 4-9, то полученный этанол повышался от 0,07 до 0,09-0,14 ммоль/час, соответственно. Соответствующие скорости образования эфира составляли 0,21, 0,22 и 0,07. Добавление воды к взаимодействию высшего алкилбромида Смесь водяного пара и додецилмонобромидов в молярном соотношении 10:1 подают на слой оксида металла М4 при 175°С. Превращается 100% додецилмонобромида с избирательностью, равной 50% к додецилспиртам и 50% к додецену (различные изомеры). Одновременная отгонка с водяным паром низкокипящих фракций высших олефинов Смесь водяного пара и додецилмонобромидов в молярном соотношении 10:1 подают на слой оксида металла М1 при 250°С. Превращается 100% додецилмонобромида с избирательностью, равной 90% к додецену (различные изомеры). Отгонка с водяным паром низкокипящих фракций высших олефинов после окончания реакции Додецилмонобромиды подают на слой оксида металла М3 при 200°С. Превращается 100% додецилмонобромида с избирательностью, равной 80% к додецену (различные изомеры). После окончания реакции 10% продукта остается адсорбированным на твердом веществе и 95% его отделяют пропусканием водяного пара через реактор со скоростью потока, превышающей скорость потока реагента в десять раз, в течение 10 миннут. Гидратация высших олефинов Додецилмонобромиды подают на слой оксида металла М1 при 200°С. Превращается 100% додецилмонобромида с избирательностью, равной 90% к додецену (различные изомеры). Смесь продуктов подают вместе с десятикратным избытком воды на слой сульфированного диоксида циркония, поддерживаемый при 175°С. Получают 50% конверсии додецена в додециловые спирты (смешанные изомеры). Нейтрализация HBr из смешанного потока оксидом кальция и последующая регенерация Поток, содержащий бромистый водород, этилбромид, этилдибромид и высшие бромиды этана, пропускают над оксидом кальция при 150°С. Бромистый водород реагирует с оксидом, образуя бромид кальция и воду. Органические бромиды проходят через реактор в основном непрореагировавшими. Бромид кальция затем окисляют пропусканием воздуха над продуктом при 500° с высвобождением брома и регенерацией оксида кальция. Нейтрализация HBr из смешанного потока с помощью М1 и последующая регенерация Поток, содержащий бромистый водород, этилбромид, этилдибромид и высшие бромиды этана, пропускают над М1 при 150°С. Бромистый водород реагирует с оксидом, образуя бромид кобальта и воду. Органические бромиды проходят через реактор в основном непрореагировавшими. Бромид кобальта затем окисляют пропусканием кислорода над продуктом при 350° с высвобождением брома и регенерацией М1. Нейтрализация HBr с помощью М1 и последующая регенерация Поток бромистого водорода пропускают над М1 при 150°С. Бромистый водород реагирует с оксидом, образуя бромид кобальта и воду. Бромид кобальта затем окисляют пропусканием кислорода над продуктом при 350° с высвобождением брома и регенерацией М1. Взаимодействие с оксибромидом Этилбромид пропускают над оксибромидом висмута при 350°С. Превращается 80% этилбромида с избирательностью 80% к этилену. В ходе реакции оксибромид висмута превращается в бромид висмута, который затем подвергают взаимодействию с кислородом при 500°С с высвобождением порции брома в твердом веществе и регенерацией оксибромида висмута. Взаимодействие с гидроксидом и гидратом Поток пропускают над М1 при 200°С, при этом получают гидроксид кобальта и гидрат оксида кобальта, находящиеся в диоксиде циркония. Этилбромид пропускают над этим продуктом при 200°С. Превращается 50% этилбромида с 80% избирательностью к этанолу. После данной реакции бромид кобальта окисляют до оксида кобальта пропусканием кислорода через твердое вещество, при этом высвобождается бром. Затем оксид подвергают действию водяного пара для регенерации гидроксида и гидрата. Регенерация М1 Смесь этана и брома в молярном соотношении 10:1 готовили пропусканием потока этана под давлением через барботер с бромом, который поддерживали при 7°С. Смесь пропускали в первый реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до 350°С. Вытекающий поток предварительно идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 9,5% конверсии этана. Избирательность к бромэтану составляла более 94%. Вытекающий поток из первого реактора (приблизительно 4 см3/мин) пропускали во второй реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до 200°С и который содержал 5 г М1. Продукты анализировали с помощью ГХ (GC). Через 5 часов взаимодействия реакторы продували и твердое вещество регенерировали при 350°С потоком кислорода, пока не прекращалось выделение брома. Через 26 циклов реакция/регенерация продукт оставался активным и химическое превращение (конверсия, избирательность) оставалось неизмененным в пределах экспериментальной ошибки. Регенерация М2 Смесь этана и брома в молярном соотношении 10:1 готовили пропусканием потока этана через барботер с бромом, который поддерживали при 0°С. Смесь пропускали в первый реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до 350°С. Вытекающий поток предварительно идентифицировали c помощью ГХ (GC) и ЯМР-анализа продуктов, улавливаемых в дейтерохлороформе. Получали 100% конверсии брома с 9,5% конверсии этана. Избирательность к бромэтану составляла более 94%. Вытекающий поток из первого реактора (приблизительно 4 см3/мин) пропускали во второй реактор (стеклянная трубка ID 0,38'', длина зоны нагрева 4''), который нагревали до 200°С и который содержал 5 г М2. Продукты анализировали с помощью ГХ (GC). Через 5 часов взаимодействия реакторы продували и твердое вещество регенерировали при 350°С потоком кислорода, пока не прекращалось выделение брома. Бром улавливали ловушкой с водным гидроксидом натрия и анализировали с помощью UV/VIS спектроскопии. Для четырех последующих опытов определяли выделение брома, которое составляло 94, 108, 100 и 98% от количества входящего брома с неопределенностью приблизительно 5%. Химическое превращение М2 было неизмененным в течение данных опытов. Хотя предпочтительные варианты осуществления данного изобретения были проиллюстрированы в сочетании с чертежами и описаны в вышеприведенном подробном описании, следует иметь в виду, что изобретение не ограничено только раскрытыми вариантами осуществления и в нем возможны различные перестановки, модификации и замещения частей или элементов без отклонения от сущности изобретения.
Реферат
Настоящее изобретение относится к способу синтеза олефинов, включающему следующие стадии: поступление потока алканов, представляющих собой побочные продукты, полученные при производстве олефинов; поступление второго реагента, представляющего собой галогенид, выбранный из группы, состоящей из хлора, брома и йода; взаимодействие потока алканов и второго реагента с образованием первых продуктов реакции и галогенводорода; отделение первых продуктов реакции от галогенводорода; взаимодействие первых продуктов реакции с твердым окислителем с образованием потока олефинов и отработанного окислителя; окисление отработанного окислителя, полученного при окислении первых продуктов реакции, для получения исходного твердого окислителя и исходного второго реагента; рециклирование твердого окислителя; и рециклирование второго реагента; взаимодействие галогенводорода с твердым окислителем с образованием воды и отработанного окислителя; окисление отработанного окислителя, полученного при окислении галогенводорода, для получения исходного твердого окислителя; и рециклирование твердого окислителя. Предлагаемое изобретение позволяет избирательно получать олефины с возможностью присоединения предлагаемого способа к существующей олефиновой установке для повышения производительности при минимальном дополнительном вложении. 10 з.п. ф-лы, 6 ил.
Формула
поступление потока алканов, представляющих собой побочные продукты, полученные при производстве олефинов;
поступление второго реагента, представляющего собой галогенид, выбранный из группы, состоящей из хлора, брома и йода;
взаимодействие потока алканов и второго реагента с образованием первых продуктов реакции и галогенводорода;
отделение первых продуктов реакции от галогенводорода;
взаимодействие первых продуктов реакции с твердым окислителем с образованием потока олефинов и отработанного окислителя;
окисление отработанного окислителя, полученного при окислении первых продуктов реакции, для получения исходного твердого окислителя и исходного второго реагента;
рециклирование твердого окислителя и
рециклирование второго реагента;
взаимодействие галогенводорода с твердым окислителем с образованием воды и отработанного окислителя;
окисление отработанного окислителя, полученного при окислении галогенводорода, для получения исходного твердого окислителя и
рециклирование твердого окислителя.
Комментарии