Способ синтеза химического промышленного сырья и высокооктанового топлива, и состава высокооктанового топлива - RU2191769C2
Код документа: RU2191769C2
Чертежи



Описание
Область техники
Изобретение относится к способу производства химического промышленного сырья, высокооктанового топлива и их смеси из этанола
посредством использования в качестве катализатора фосфата кальция или фосфата кальция, несущего на себе металл.
Предпосылки создания изобретения
Как широко известно, в
последнее время предпринималось множество попыток получения больших партий химического промышленного сырья при использовании спиртов, в особенности этанола, в качестве сырьевого материала вместо
химического промышленного сырья, получаемого из нефти.
Способ получения этилена из этанола в качестве сырьевого материала, в котором в качестве катализатора используется фосфат кальция, известен, но этот способ характеризуется низкой активностью и экономически невыгоден. Также в качестве способа дегидратации спирта известно использование кислотного катализатора на твердом носителе, такого как цеолит и т.д., но в этом способе алюминий в цеолитовой структуре высвобождается под действием воды, образуемой в ходе реакции, что приводит к понижению активности катализатора, и этот способ не может использоваться в промышленности в течение длительного периода времени.
Известен способ получения ацетальдегида из этанола при использовании фосфата кальция или фосфата кальция, несущего металлы, такие как Cu, Ni и т.д. , но его активность и селективность низки, и этот способ экономически невыгоден. Также в качестве способа дегидратации этанола известно использование основного катализатора на твердом носителе, такого как катализатор платиновой группы на носителе, MgO и т.д., но существуют проблемы в разбросе характеристик по примесям и стабильности.
В качестве способа получения диэтилового простого эфира из этанола известно использование кислотного катализатора на твердом носителе, такого как цеолит и т.д., но алюминий в цеолитовой структуре высвобождается под действием воды, образуемой в ходе реакции, что приводит к понижению каталитической активности, и этот катализатор не может использоваться в течение длительного периода времени.
В качестве способа получения 1,3-бутадиена из этанола предлагаются способ с использованием в качестве катализатора Al2O3•ZnO (6:4) [S.K. Bhattcharyya и N.O. Ganguly; J. Appl. Chem.; 12, 105 (1962)] и способ, использующий сепиолит, адсорбированный металлом (Mn, V, Мо, W и т.д.) (японская заявка на патент 178281/1980 и 157814/1981). Однако в первом способе имеются сложности, связанные со стабильным получением катализатора и термической стабильностью самого катализатора, и второй способ приводит к возникновению на уровне запуска серийного производства, и таким образом, в самом способе проблем с точки зрения массового получения. Соответственно 1,3-бутадиен главным образом получают из ископаемого топлива, содержащего бутены. В качестве промышленного способа известны синтез 1-бутанола, ацетальдегидный способ, способ Реппе и т.д., но эти способы сложны и малоэффективны. Японская патентная публикация 305238/1993 раскрывает способ получения углеводородов базового бензина из низших спиртов при использовании катализатора, образованного на основе фосфата кальция, несущего металл.
Раскрытие изобретения
Целью
настоящего изобретения является разработка способа производства для эффективного получения химического промышленного сырья, такого как этилен, ацетальдегид, диэтиловый простой эфир, 1-бутанол, 1,
3-бутадиен и т.д., высокооктанового топлива и его смеси при использовании этанола в качестве исходного материала.
В результате детальных исследований способа получения химического промышленного сырья, такого как этилен, ацетальдегид, диэтиловый простой эфир, 1-бутанол, 1,3-бутадиен и т.д., и высокооктанового топлива и т.д., при использовании этанола в качестве исходного материала на основе выгодного с промышленной точки зрения способа в настоящем изобретении было найдено, что описанная выше цель может быть достигнута при использовании катализатора на основе фосфата кальция.
Краткое описание изобретения
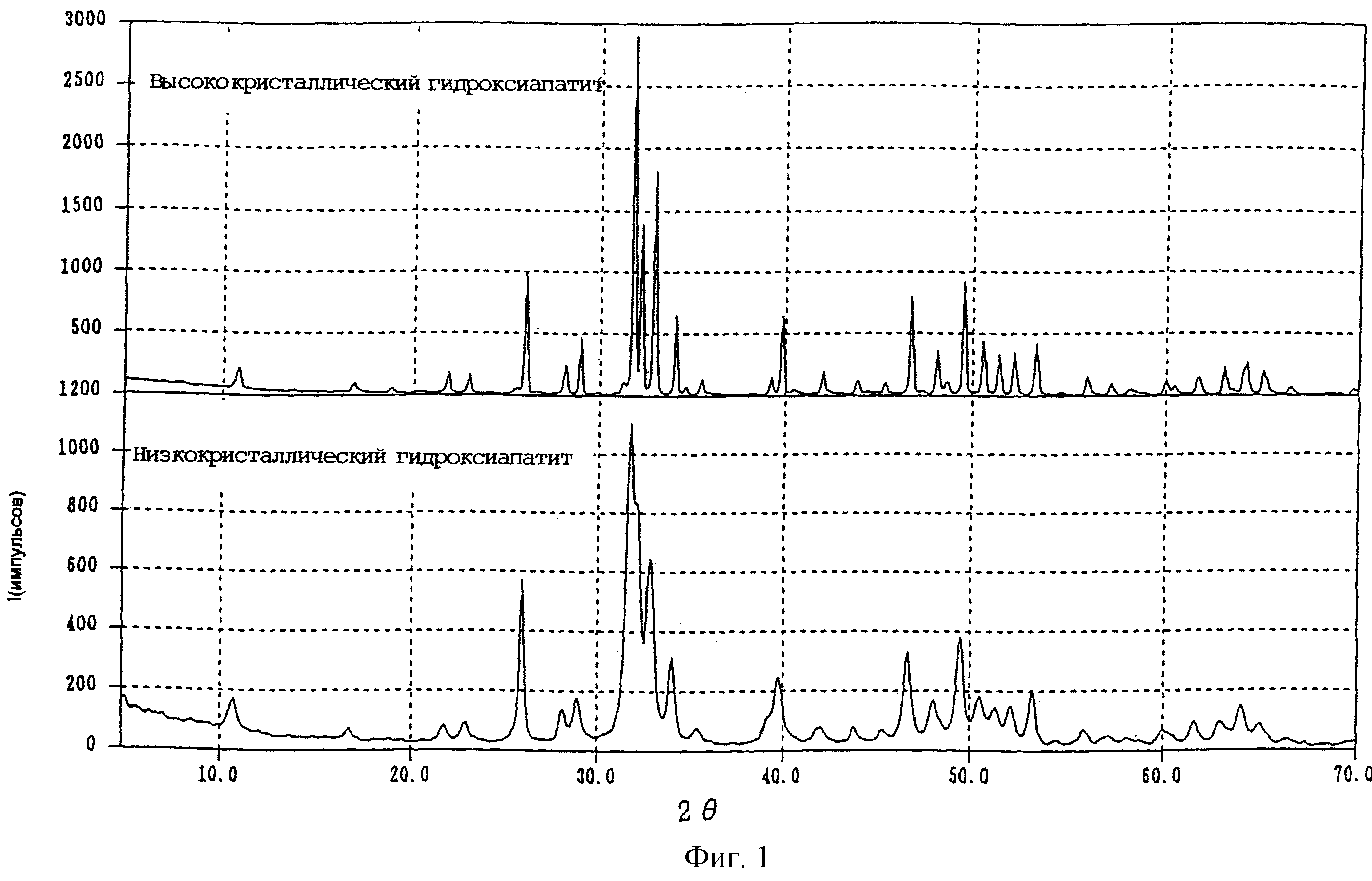
Фиг.1 представляет собой графическое представление рентгенодифракционной картины высококристаллического порошка и низкокристаллического
порошка,
фиг. 2 представляет собой изображение, показывающее реакционный аппарат, используемый в примере по изобретению, и
фиг. 3 представляет собой вид, показывающий зависимость
времени реакции катализатора, подвергающегося повторяющейся регенерации, и конверсии этанола.
Лучший способ выполнения изобретения
Далее изобретение описывается подробно.
Известно, что фосфат кальция существует в виде гидроксиапатита [Са10(РO4)6(ОН)2] , трифосфата кальция [Са3(РO4)2],
гидрофосфата кальция [СаН(РO4)•(от 0 до 2) Н2O], дифосфата кальция (Ca2P2O7), дигидрофосфата октокальция [Са8Н2
(РO4)6• 5Н2О], тетракальция фосфата [Са4(РO4)2O], аморфного фосфата кальция [Са3(РO4)2•
nН2O] и т.д.
Гидроксиапатит обычно представляют описанным выше стехиометрическим составом, но его свойства таковы, что даже если состав не удовлетворяет этому
стехиометрическому составу, гидроксиапатит может обладать структурой апатита. Гидроксиапатит с таким стехиометрическим составом может быть представлен на основе [Са10-z(НРO4)z(PO4)6-z(OH)2-z•nHO2 {0 Также аморфным фосфатом кальция является фосфат кальция, который
дает гало при рентгеновской дифракции. Низкокристаллическим порошком в изобретении является низкокристаллический порошок, рентгенодифракционный пик которого уширен по сравнению с
высококристаллическим порошком. Например, при использовании гидроксиапатита на фиг.1 представлена рентгенодифракционная картина высококристаллического и низкокристаллического порошка.
В настоящем изобретении при использовании катализатора, изготовленного из этих фосфатов кальция, в особенности низкокристаллических фосфатов кальция самих по себе или в виде смеси, в которых мольное
отношение Са/Р регулируют от 1,4 до 1,8, как такового, или катализатора, несущего активирующий металл или его оксид на катализаторе, такой, что (Са + металл)/P мольное отношение составляет от 1 до 2,
эффективно получают описанное выше химическое промышленное сырье и высокооктановое топливо. В этом изобретении не существует особых ограничений на способ получения фосфата(ов) кальция,
используемых в качестве катализатора, и фосфат(ы) кальция могут быть синтезированы при помощи известного способа синтеза, такого как способ сухой твердофазной реакции, реакционный способ влажного
осаждения, способ влажной твердофазной реакции, синтетический гидротермальный способ и т.д. Также низкокристаллический(е) фосфат(ы) кальция может быть получен обжиганием фосфата(ов) кальция,
синтезированного при помощи описанного выше способа, в области низкой температуры или при помощи механохимического размалывания сожженного порошка, также при использовании подходящего темплата, поры
которого можно контролировать. Более того, мольное отношение Са/Р может быть должным образом изменено при получении фосфата(ов) кальция. Например, в случае синтеза гидроксиапатита,
добавляют по каплям к водному раствору раствор соли кальция или раствор фосфата определенной концентрации при перемешивании при комнатной температуре, при контроле рН, выпадающий в осадок продукт
отбирают, промывают, сушат, измельчают и, если необходимо, обжигают для получения каталитического исходного материала. При использовании соли кальция предпочтительны Са(ОН)2 или Са(NО3)2 и при использовании фосфата предпочтителен фосфат аммония. Контроль мольного отношения Са/Р гидроксиапатита проводят при помощи контроля отношения составов солей сырьевых
материалов и условий синтеза. Например, при синтезе, если водный раствор регулируют до основного при помощи водного раствора аммиака и т.д., мольное отношение Са/Р увеличивается, и если водный раствор
регулируют до нейтрального или слабокислотного при помощи разбавленной кислоты, мольное отношение Са/Р уменьшается. Также гидроксиапатит может быть получен при смешивании фосфатов кальция, имеющих
известное мольное отношение Са/Р, при помощи обжига смеси во влажной атмосфере. Если гидроксиапатит используется в качестве катализатора, мольное отношение Са/Р регулируют от 1,4 до 1,
8 и предпочтительно от 1,5 до 1,7 и, если желательно, может быть выбрана температура обжига и атмосфера обжига. В этом случае желательно, чтобы удельная площадь катализатора составляла по крайней мере
2 м2/г. Контроль мольного отношения Са/Р в фосфате кальция означает контроль качества и распределение плотностей кислотных центров на твердом носителе и основных центров на
твердом носителе, которые представляют собой каталитически активные центры поверхности катализатора. В этом случае сила и количество кислотных центров и основных центров могут быть определены на
основе NН3-TPD и СО3-TPD или способа адсорбции пиридина, способа индикатора и т.д. Также в качестве способа контроля кислотности и основности поверхности
катализатора известно нанесение металла как общеизвестного способа. Например в случае, если гидроксиапатит несет металл, ускоряющий реакцию дегидрогенизации, такой как обычно Ni, Zn,
Cu, Pd или Pt, достигается тот же эффект, что и при увеличении мольного отношения Са/Р, то есть увеличение основности твердого носителя. Также в случае, если гидроксиапатит несет
металл, ускоряющий реакцию дегидрогенизации, такой как обычно Al на гидроксиапатите, достигается тот же эффект, что и при уменьшении мольного отношения Са/Р, то есть увеличение кислотных характеристик
твердого носителя. Таким образом, вместо изменения мольного отношения Са/Р нанесение на гидроксиапатит такого металла также позволит изменить кислотность/основность поверхности
твердого гидроксиапатитного катализатора. Также согласно заданному продукту предпочтительно, чтобы наносилось одновременно множество металлов для получения синергетического эффекта или для увеличения
долговечности. Примеры множества металлов, которые могут наноситься одновременно, включают переходные металлы, такие как, Zn, Co, Cr, Mo, W, Fe, Ni, Cu, Mn, Ti, V, Ga, Zr, Nb, Cd, In, Sn, Sb, Pb, La,
Се, Eu, Y и т.д., благородные металлы, такие как Pt, Pd, Rh, Au, Ir, Ru, Ag и т.д., щелочные металлы или щелочноземельные металлы, такие как Ва, Na, К, Li, Sr, Ca, Mg, Cs, Rb и т.д. Также в некоторых
случаях могут использоваться оксиды или сульфиды этих металлов. Это множество металлов, которые могут наноситься одновременно, могут использоваться в диапазоне от 0,05 до 70 мольных % по отношению к
кальцию в кальций-фосфатном катализаторе, и их тип выбирается, исходя из целей. Эти металлы, оксиды металлов или сульфиды металлов наносят на фосфат кальция при использовании
традиционного способа. Например, определенные количества солей металлов, которые наносят на фосфат кальция, добавляют к жидкости, содержащей фосфат(ы) кальция, получаемый описанной выше обработкой, и
эту смесь отверждают путем выпаривания воды. Или раствор этих солей металлов, которые наносят на фосфат кальция, распыляют на полученные фосфат(ы) кальция и после высушивания эту смесь обжигают на
воздухе или в восстановительной атмосфере. Также катализатор, одновременно имеющий характеристики кислотных центров на твердом носителе и основных центров на твердом носителе, может
быть синтезирован подходящим смешиванием кислотного катализатора на твердом носителе и основного катализатора на твердом носителе. В настоящем изобретении синтез химического
промышленного сырья и высокооктанового топлива из этанола в качестве исходного материала проводится путем подходящего выбора используемого фосфата(тов) кальция, мольного отношения Са/Р, активирующих
металлов и условий реакции (температура, объемная скорость, давление и т.д.). Например, гидроксиапатит, Са/Р, мольное отношение которого устанавливается 1,6 или ниже, или трифосфат
кальция, имеющий увеличенное значение удельной площади, имеют характеристики кислотного твердого носителя. В этом случае нанесение металла, ускоряющего реакцию дегидратации, такого как Al и т. д. , в
описанный выше фосфат кальция усиливает свойства катализатора как кислотного твердого носителя. Количество добавляемого металла одного типа или его оксида находится в диапазоне от 0,05
до 50 мольных % по отношению к кальцию в фосфате кальция. Если количество добавляемого металла меньше чем 0,05 мольных %, то эффект от добавления металла не достигается. Если добавляется по крайней
мере 50 мольных % компонентов одного металла, то основным составляющим становится фосфат металла, который занимает позиции кальция в катализаторе, содержащем фосфат кальция. Если в
качестве катализатора реакции конверсии этанола используется такой фосфат кальция или металлнесущий фосфат кальция, имеющий усиленные свойства кислотного твердого катализатора, то может быть увеличена
селективность этилена и диэтилового простого эфира в реакционном продукте. Например, в случае этилена использование описанного выше фосфата кальция, замещенного и/или несущего 3
мольных % Al, увеличивает каталитическую активность по сравнению с использованием описанного выше фосфата кальция, не несущего металл, и температура, при которой конверсия или селективность этанола
100%, понижается на 50oС или более. Также в случае диэтилового простого эфира использование описанного выше фосфата кальция, несущего Al, сильно увеличивает каталитическую
активность по сравнению с использованием описанного выше фосфата кальция, не несущего металл. Также свойства катализатора как основного твердого катализатора увеличиваются при
нанесении комбинации по крайней мере двух металлов, ускоряющих реакцию дегидрогенизации, таких как Ni, Zn, Cu, Fe, Al, In, Pd и т.д. на гидроксиапатит, в котором мольное отношение Са/Р устанавливают
до по крайней мере 1,55, или фосфат кальция, имеющий мольное отношение (Са + металл)/P от 1 до 2. В этом случае количество добавляемых металлов изменяется в диапазоне от 1 до 70 мольных % в сумме
(верхний предел количества одного добавляемого металла составляет 30 мольных %) по отношению к кальцию в фосфате кальция. Если добавляемые количества меньше чем 1 мольный %, то описанный выше
катализатор не является улучшенным по сравнению с существующими известными катализаторами с добавлением металлов. С другой стороны, в случае добавления суммарно по крайней мере 70 мольных % нескольких
металлов возникает проблема стабильности фосфата кальция. В том случае, если в реакции конверсии этанола в качестве катализатора используется такой гидроксиапатит, имеющий улучшенные
свойства как основного твердого катализатора, то может быть увеличена селективность ацетальдегида в реакционном продукте. Например, при использовании фосфата кальция, несущего Cu, Fe и
Al и имеющего мольное отношение (Са + металл)/Р от 1 до 2, конверсия и селективность ацетальдегида значительно увеличиваются по сравнению с использованием известных существующих катализаторов. Если в реакции конверсии этанола используется такой катализатор, что кислота и основание могут сосуществовать, изготовленный из гидроксиапатита, мольное отношение Са/Р которого
устанавливается от 1,60 до 1,80, или катализатор, изготовленный из фосфата кальция, имеющий мольное отношение (Са + металл)/P от 1 до 2 и несущий по крайней мере один металл, выбираемый из описанных
выше металлов, ускоряющих реакцию дегидрогенизации, и металлов, ускоряющих реакцию дегидратации, таких как Ва, Na, К, Li, Cs, Sr, Y, Ce, Sb, Eu, Ti, W и Zr, или оксиды и сульфиды этих металлов в
пределах, не превышающих 50 мольных % по отношению к кальцию в фосфате кальция, то селективность 1-бутанола может быть увеличена в диапазоне от 300oС до 450oС.
Например, гидроксиапатит, в котором мольное отношение Са/Р устанавливается до 1,65, или описанный выше фосфат кальция, несущий Се, могут увеличивать селективность 1-бутанола от 60 до 65% при примерно
400oС. Также если в реакции конверсии этанола используется такой катализатор, что кислота и основание могут сосуществовать, изготовленный из гидроксиапатита, мольное
отношение Са/Р которого устанавливается от 1,55 до 1,80, или катализатор, изготовленный из фосфата кальция, имеющий мольное отношение (Са + металл)/P от 1 до 2 и несущий по крайней мере один металл,
выбираемый из описанных выше металлов, ускоряющих реакцию дегидрогенизации, и металлов, ускоряющих реакцию дегидратации, таких как W, Zr, Al, Zn, Ti, Sb, Y, La, Au и Na, или оксиды и сульфиды этих
металлов в пределах, не превышающих 50 мольных % по отношению к кальцию в фосфате кальция, то селективность 1,3-бутадиена может быть увеличена в диапазоне от 450oС до 700oС. Например, гидроксиапатит, в котором мольное отношение Са/Р устанавливается до 1,62, или описанный выше фосфат кальция, несущий Al или Zr, могут увеличивать селективность 1,3-бутадиена от
35 до 50% при примерно 500oС. Более того, если в реакции конверсии этанола используется такой катализатор, что кислота и основание могут сосуществовать, изготовленный из
гидроксиапатита, мольное отношение Са/Р которого устанавливается от 1,55 до 1,80, или катализатор, изготовленный из фосфата кальция, имеющий мольное отношение (Са + металл)/P от 1 до 2 и несущий по
крайней мере один металл, выбираемый из описанных выше металлов, ускоряющих реакцию дегидрогенизации, и металлов, ускоряющих реакцию дегидратации, таких как Ni, Ва, Li, Cs, Zn, Ag, Mn, Се, Sr, Y, Co,
Fe, Sb, Eu, Ti и W, или оксиды и сульфиды этих металлов в пределах, не превышающих 50 мольных % по отношению к кальцию в фосфате кальция, то селективность высокооктанового топлива может быть увеличена
в диапазоне от 300oС до 700oС. Например, гидроксиапатит, в котором мольное отношение Са/Р устанавливается до 1,65, или фосфат кальция, несущий Li или Zn и имеющий
мольное отношение (Са + металл)/P от 1 до 2, могут увеличивать селективность высокооктанового топлива до по крайней мере от 80% при примерно от 300oС до 700oС.
Высокооктановое топливо в данном изобретении представляет собой автомобильное топливо, имеющее октановое число по крайней мере 96,0, согласно экспериментальному способу теста на октановое число,
регламентированному JIS К 2280, содержащее в компонентах средние жидкие углеводородные соединения, содержащие кислород, такие как спирты, простые эфиры и т.д., которые известны как присадки,
повышающие октановое число. Поскольку в настоящее время бензиновый состав представляет собой неполярные углеводороды, когда небольшой % воды смешан с бензином, наблюдаются проблемы при
запуске мотора и, следовательно, необходим осушающий агент, но высокооктановое топливо по данному изобретению содержит большое количество полярных кислородсодержащих углеводородов, таких как
непрореагировавший этанол, бутанол и т.д., и представляет собой топливо, для которого нет необходимости добавления осушающего агента. В настоящий момент количество присутствующих
кислородсодержащих соединений в топливе по изобретению может контролироваться на основе Са/Р мольного отношения, добавления металла и температуры реакции. То есть присутствующее количество
кислородсодержащих соединений в топливе может быть увеличено при помощи увеличения мольного отношения Са/Р на основе добавления металла, ускоряющего реакцию дегидрогенизации, или при помощи уменьшения
температуры реакции. Дополнительно, если необходимо, в случае уменьшения содержания кислородсодержащих соединений, мольное отношение Са/Р может быть уменьшено добавлением металла,
ускоряющего реакцию дегидратации, или может быть повышена температура реакции. Фосфат кальция, полученный, как описано выше, может использоваться в любой форме, такой как, например,
гранулы, порошки и т.д., и если необходимо, после формовки фосфата кальция до оптимальной формы, такой как сферы, гранулы, сотовой формы и т.д., формованный продукт сушат и обжигают при использовании.
Фосфат кальция может наноситься на носитель, такой как глинозем, кремнезем, глинозем-кремнезем, цеолит, глина и т.д., хорошо известный специалистам в данной области. Обжиг проводится при температуре
от 200oС до 1200oС и предпочтительно от 500oС до 700oС. В общем, если используется твердый катализатор, считается, что наличие воды в
исходных материалах нежелательно, поскольку активность катализаторов понижается. Однако в катализаторе по данному изобретению при наличии воды понижение активности катализатора не наблюдается и,
следовательно, катализатор может использоваться в течение длительного периода времени. Поэтому в случае этанола, содержащего воду, реакция протекает в достаточной мере, и, следовательно, катализатор
по данному изобретению имеет преимущество, состоящее в том, что неочищенный этанол, который представляет собой простой дистиллят этанола, полученного при ферментном брожении при помощи биотехнологии,
может использоваться в промышленной установке. Температура реакции по изобретению, при которой этанол вводят в контакт с фосфат кальциевым катализатором, обычно изменяется в диапазоне
от 200oС до 700oС, и оптимальная температура реакции может быть соответствующим образом выбрана согласно типу катализатора и типу синтезируемого продукта. В реакции с
катализатором по данному изобретению ацетальдегид, диэтиловый простой эфир и 1-бутанол получают с высокой селективностью в относительно низкотемпературной области и этилен и 1,3-бутадиен получают с
высокой селективностью в относительно высокотемпературной области. Среднечасовая скорость подачи газа (GHSV) составляет от 100 до 100000 (л/ч) и предпочтительно от 5000 до 50000 (л/ч). Также
пространственная скорость ниже чем 100 экономически невыгодна. Также, если пространственная скорость выше чем 100000 становится трудным проводить температурный эндотермический или экзотермический
контроль внутри реакционной колонны, что вызывает распределение температуры, которое приводит к уменьшению селективности заданного синтетического продукта. При помощи введения этанола в контакт с
катализатором в паровой фазе непосредственно или при наличии инертного газа носителя, такого как азот или гелий, этанол может реагировать с высокой эффективностью. В этом случае, для
сохранения каталитической активности реакционно активный газ, такой как водород, углеводород и т.д., могут добавлять в газ-носитель. В качестве реакционной системы реакционной колонны может
использоваться любой способ, такой как периодическая система, непрерывная система, система неподвижного слоя, система подвижного слоя или система жидкого слоя, и реакция может проводиться при
нормальном давлении или при повышенном давлении. В случае реакции осаждения большое количество углерода осаждается на катализатор, желательно производство на основе описанной выше системы, включающей
аппарат регенерации катализатора. Если катализатор, который используется для реакции синтеза описанного выше химического промышленного сырья, отличающейся от реакции синтеза этилена и диэтилового
простого эфира, используется в течение длительного периода времени, углерод осаждается на поверхность этого катализатора, понижая конверсию этанола. Поэтому катализатор периодически подвергают
обработке для регенерации путем нагревания в атмосфере кислорода, посредством чего активность катализатора может быть регенерирована. Фиг. 2 представляет собой схематическое представление
конструктивного исполнения аппарата для осуществления настоящего изобретения. Как показано на фиг.2, этанол в качестве исходного материала подают в спиртовой карбюратор 2 через
микрофидер (жидкостной подающий насос среднего давления) 1. В спиртовой карбюратор 2 подается газ-носитель на основе 1% Ar/Не из газового баллона 3 и газ из модели газового смесителя 4, они входят в
реакционную трубу 5 совместно с этанолом и продукты реакции отводятся согласно температуре реакции. Продукты реакции подтверждаются химически при помощи анализатора 6. Полученные таким
образом продукты реакции могут быть выделены и очищены при использовании традиционных способов разделения и очистки, таких как фракционирование, экстракция и адсорбционный способ.
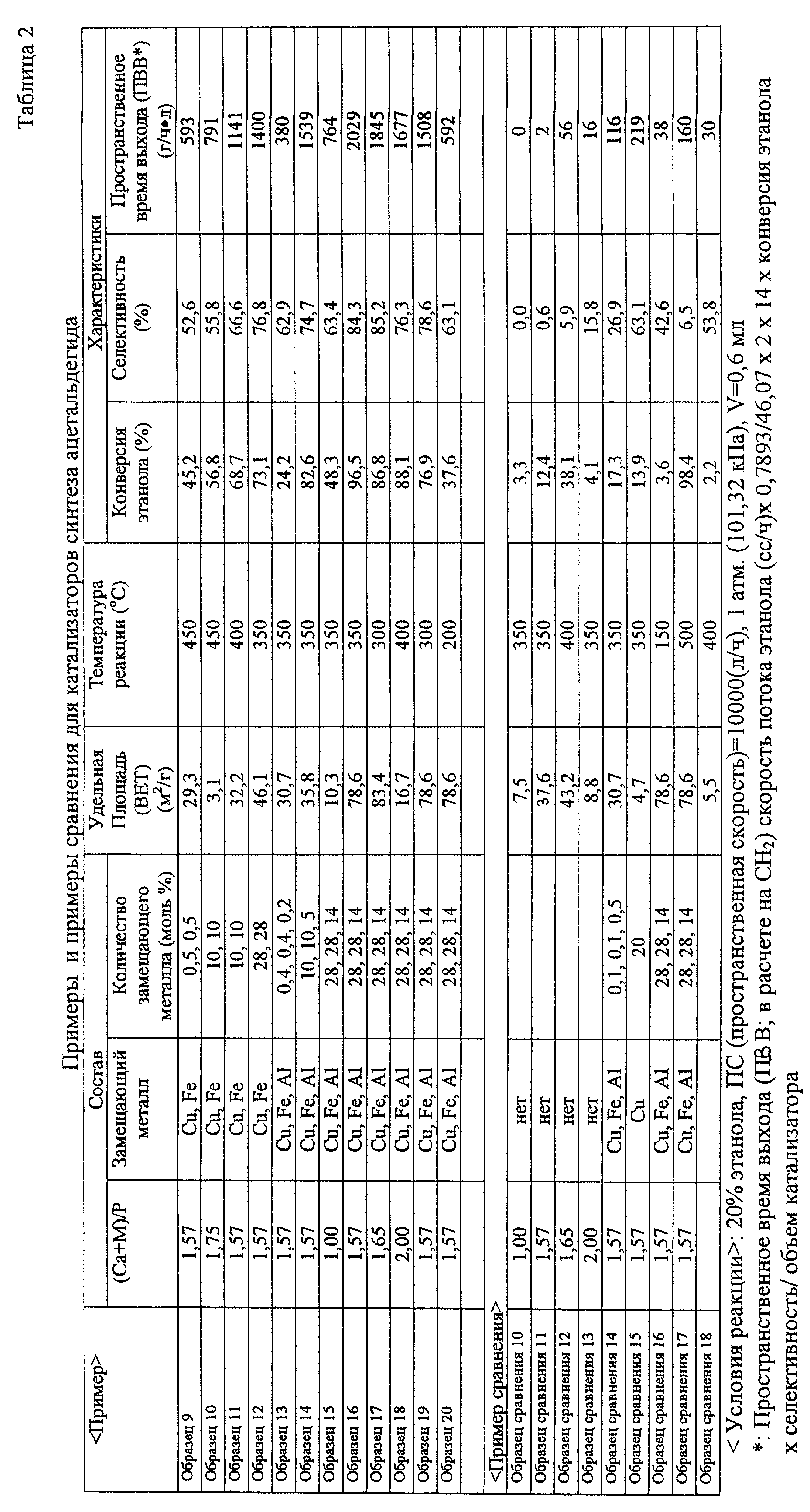
[Пример] 1) В случае катализатора синтеза
этилена: (Образец 2) (Образец 3) (Образец
4) (Образец 5) (Образец 6) (Образец 7) (Образец 8) (Образец сравнения 1) (Образец сравнения 2) (Образец сравнения 3) (Образец сравнения 4) (Образец сравнения 5) (Образец сравнения 6) (Образец сравнения 7) (Образец сравнения 8) (Образец сравнения 9) [Оценка каталитических характеристик] После завершения
этой предварительной обработки эту реакцию проводили при условиях концентрации этанола 20%, скорости потока газа носителя 80 сс/мин (общая скорость потока 100 сс/мин) и пространственной скорости
(GHSV) 10000 (л/ч) при нормальном давлении. В случае синтеза этилена температура реакции была в диапазоне от 400 до 700oС. Идентификацию компонентов
реакционного газа проводили при использовании масс-спектрометра с газовым хроматографом (GC-MS) и измерения конверсии этанола и селективности синтез-газа проводили при использовании газового
хроматографа (GC) (детектор:FID), и они определялись при помощи следующих формул из значения площади пика каждой компоненты. Конверсия этанола (%)= (1-(значение площади пика этанола
после реакции)/(значение площади пика этанола после реакции)х100 Пространственное время выхода (г/(ч•л))=введенный этанол (моль)(2•14 • конверсия этанола •
селективность/объем катализатора. В качестве реакционного аппарата использовали каталитический реакционный аппарат газового потока, показанный на фиг.2. Результаты оценки показаны в
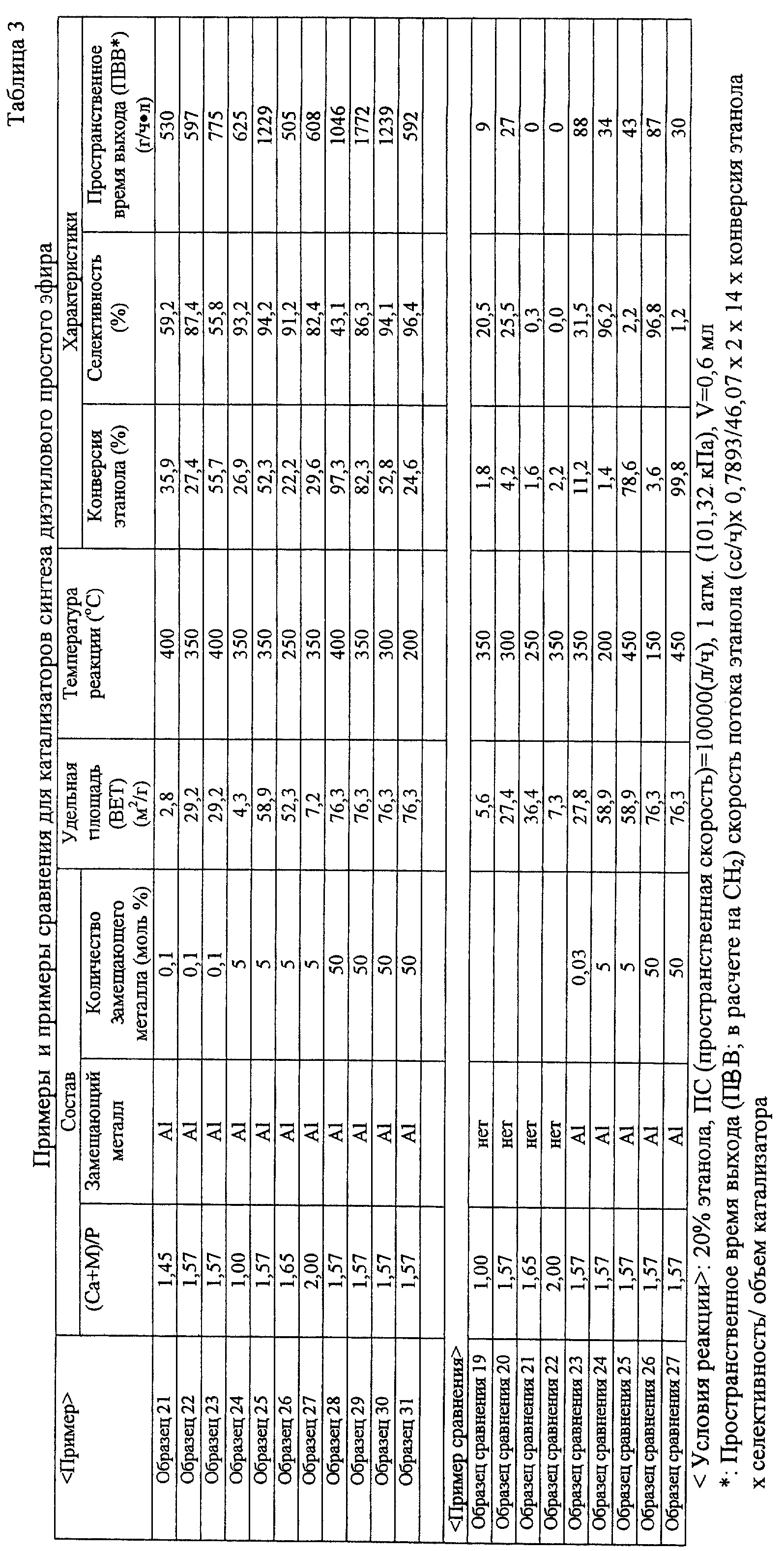
табл.1. 2) В случае катализатора синтеза ацетальдегида: (Образец 10) (Образец 11) (Образец 12) (Образец 13)
(Образец 14) (Образец 15) (Образец 16) (Образец 17) (Образец 18) (Образец 19) (Образец 20) [Образец
сравнения] (Образец сравнения 11) (Образец сравнения 12) (Образец сравнения 13) (Образец
сравнения 14) (Образец сравнения 15) (Образец сравнения 16) (Образец сравнения 17) (Образец сравнения 18) [Оценка каталитических характеристик] В случае
синтеза ацетальдегида температура реакции была в диапазоне от 200 до 450oС. Идентификацию и определение реакционного газа проводили как и в случае этилена. В качестве реакционного аппарата
использовали каталитический реакционный аппарат газового потока, показанный на фиг.2. Результаты оценки показаны в табл.2. 3) В случае катализатора синтеза диэтилового
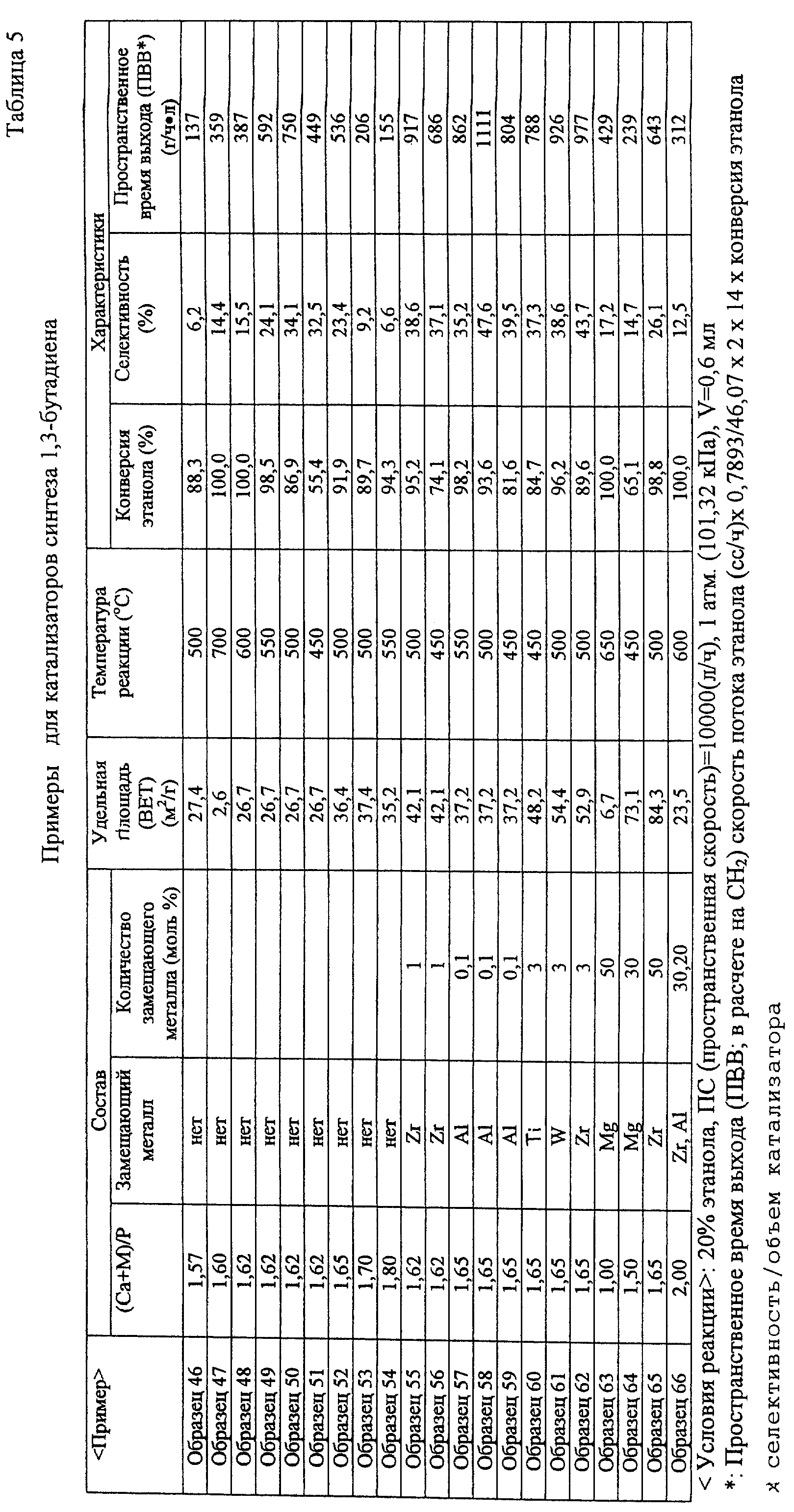
простого эфира: (Образец 22) (Образец 23) (Образец 24) (Образец 25) (Образец 26) (Образец 27) (Образец
28) (Образец 29) (Образец 30) (Образец 31) (Образец сравнения 19) (Образец сравнения 20) (Образец сравнения 21) (Образец сравнения 22) (Образец сравнения 23) (Образец сравнения 24) (Образец сравнения 25) (Образец сравнения
26) (Образец сравнения 27) [Оценка каталитических характеристик] После завершения этой предварительной обработки эту реакцию проводили при условиях концентрации этанола 20%, скорости
потока газа носителя 80 сс/мин (общая скорость потока 100 сс/мин) и пространственной скорости (GHSV) 10000 (л/ч) при нормальном давлении. В случае синтеза диэтилового эфира температура реакции была в
диапазоне от 200 до 400oС. Идентификацию и определение реакционного газа проводили при использовании тех же способов, как и в случае этилена. В качестве реакционного аппарата использовали
каталитический реакционный аппарат газового потока, показанный на фиг.2. Результаты оценки показаны в табл.3. 4) В случае катализатора синтеза 1-бутанола: (Образец 33) (Образец 34) (Образец 35) (Образец 36) (Образец 37) (Образец 38) (Образец 39) (Образец 40) (Образец 41) (Образец 42) (Образец 43) (Образец 44) (Образец 45) (Образец сравнения 28) (Образец сравнения 29) (Образец сравнения 30) (Образец сравнения
31) (Образец сравнения 32) (Образец
сравнения 33) (Образец сравнения 34) (Образец сравнения 35) [Оценка каталитических характеристик] После завершения этой предварительной обработки эту реакцию проводили при условиях концентрации этанола 20%, скорости потока газа-носителя 80 сс/мин (общая скорость потока 100 сс/мин) и
пространственной скорости (GHSV) 10000 (л/ч) при нормальном давлении. В случае синтеза 1-бутанола температура реакции была в диапазоне от 300 до 450oС. Идентификацию и определение
реакционного газа проводили при использовании тех же способов, как и в случае этилена. В качестве реакционного аппарата использовали каталитический реакционный аппарат газового потока, показанный на
фиг.2. Результаты оценки показаны в табл.4. 5) В случае катализатора синтеза 1,3-бутадиена (Образец 47) (Образец 48) (Образец сравнения 49) (Образец 50) (Образец 51) (Образец 52)
(Образец 53) (Образец 54) (Образец 55) (Образец 56) (Образец 57) (Образец 58) (Образец 59) (Образец 60) (Образец
61) (Образец 62) (Образец 63) (Образец 64) (Образец 65)
(Образец 66) Кроме того, растворы, полученные растворением каждого из 7,23 г хлорида циркония (IV) (ZrCl4) и 9,18 г нитрата
алюминия [Al(NО3)3)•9Н2O] в 500 мл дистиллированной воды соответственно, добавляли к вышеописанной смеси с последующим дополнительным перемешиванием в течение
одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 30 мольных % Zr и 20 мольных % Al по отношению к Са и имеющего (Ca+Zr+Al/P мольное отношение 2,00. (Образец сравнения 36).
Использовали образец сравнения 1. (Образец сравнения 37) (Образец сравнения 38) (Образец сравнения 39) (Образец сравнения 40) (Образец сравнения 41) (Образец сравнения 42) (Образец сравнения 43) (Образец сравнения 44) (Образец сравнения 45) (Образец сравнения 46) [Оценка каталитических характеристик] После завершения этой предварительной обработки эту реакцию
проводили при условиях концентрации этанола 20%, скорости потока газа-носителя 80 сс/мин (общая скорость потока 100 сс/мин) и пространственной скорости (GHSV) 10000 (л/ч) при нормальном давлении. В
случае синтеза 1,3-бутадиена синтез проводили при температуре в диапазоне от 450oС до 700oС. Идентификацию и определение реакционного газа проводили при использовании тех же
способов, как и в случае этилена. В качестве реакционного аппарата использовали каталитический реакционный аппарат газового потока, показанный на фиг.2. Результаты
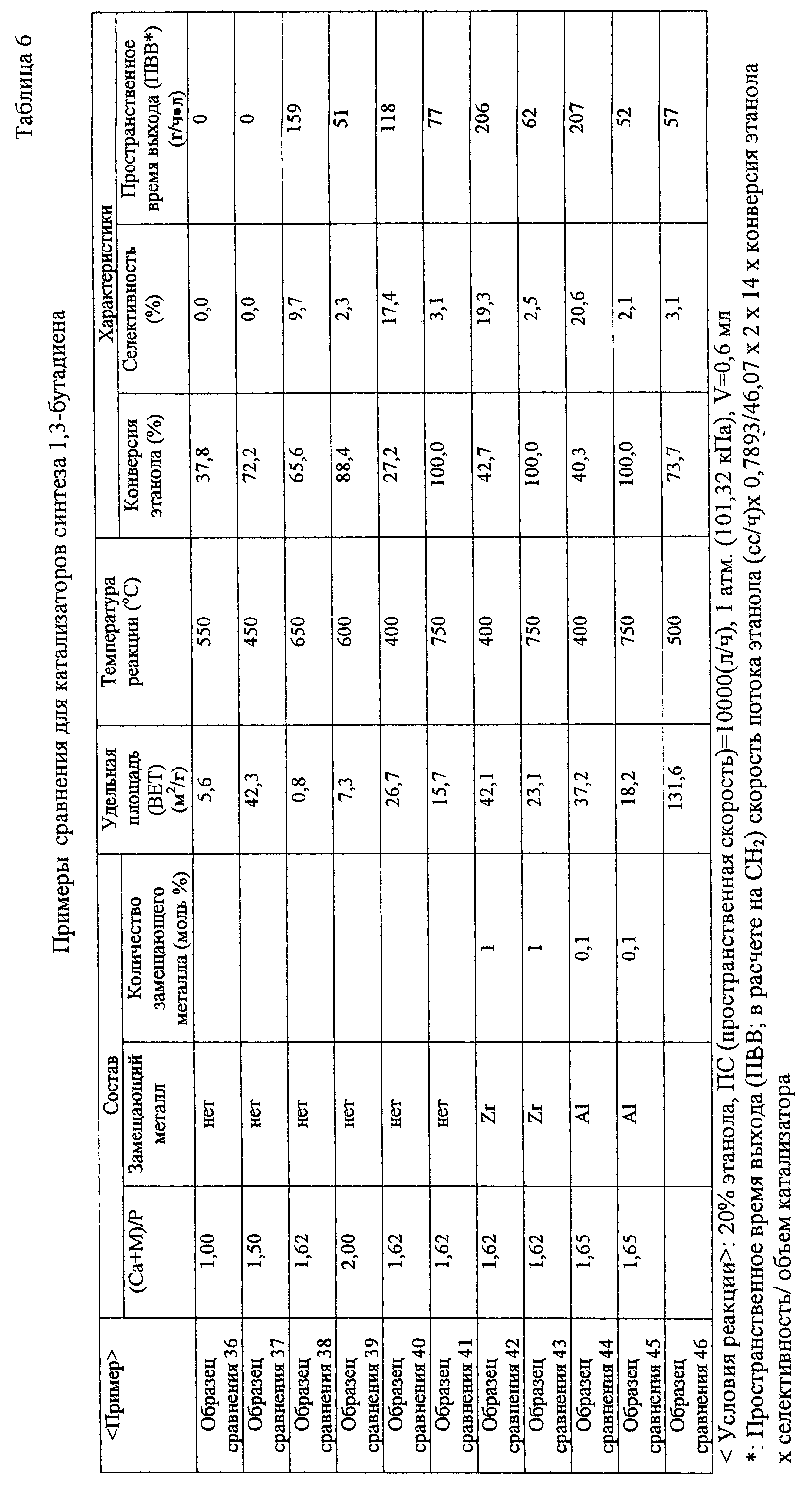
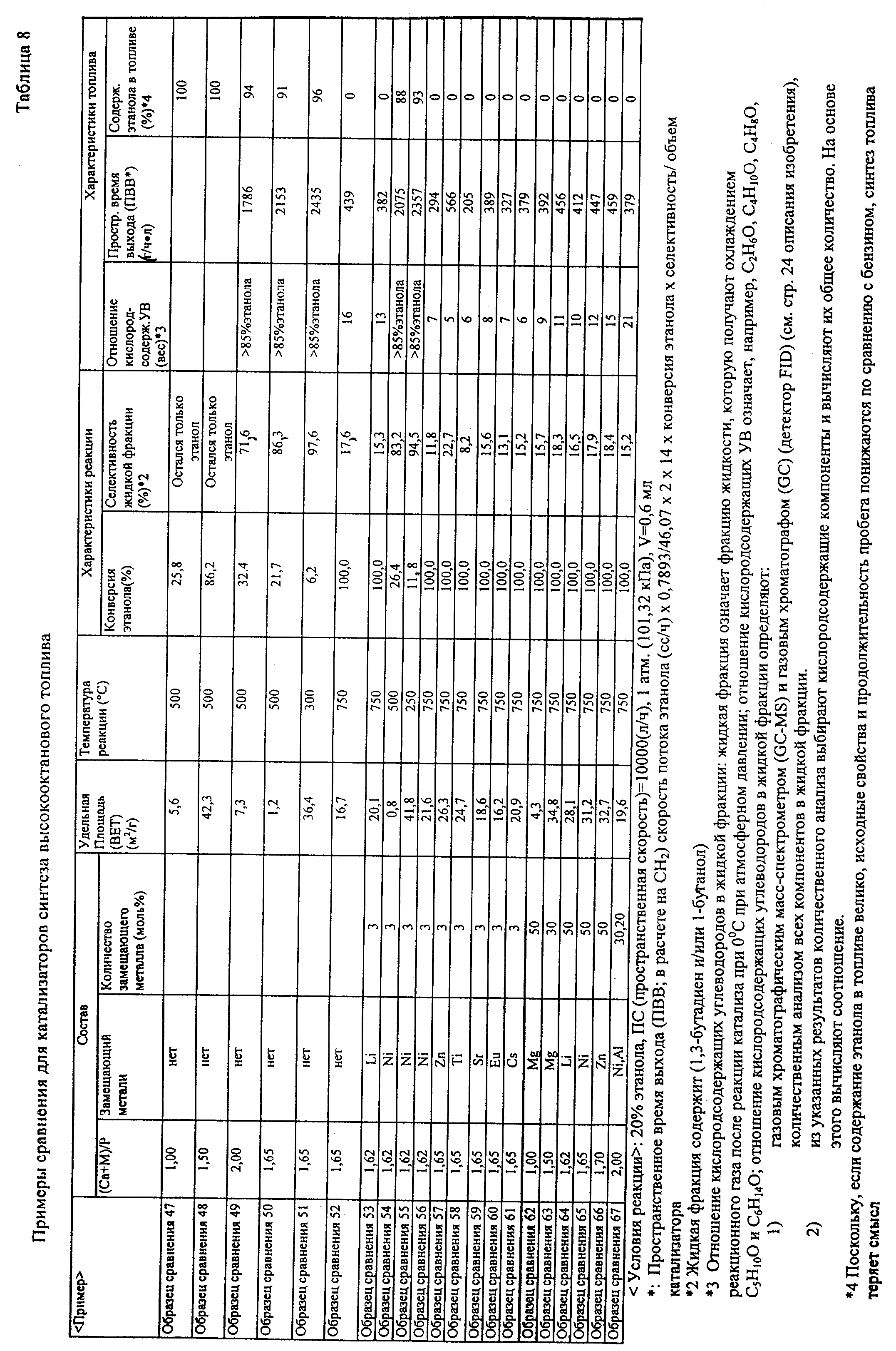
оценки показаны в табл.5 и табл.6. 6) В случае катализатора синтеза высокооктанового топлива: (Образец сравнения 68) (Образец 69) (Образец 70)
(Образец 71) (Образец 72) (Образец 73) (Образец 74) (Образец 75) (Образец 76) (Образец 77) (Образец 78) (Образец 79) (Образец 80) (Образец 81) (Образец 82) (Образец 83) (Образец 84) (Образец 85) (Образец 86) (Образец 87) (Образец 88) (Образец 89)
(Образец 90) (Образец 91) (Образец 92) (Образец 93) (Образец 94) (Образец 95) (Образец 96) (Образец сравнения 47) (Образец сравнения 48) (Образец
сравнения 49) (Образец сравнения 50) (Образец сравнения 51) (Образец сравнения 52) (Образец сравнения 53) (Образец сравнения 54) (Образец сравнения 55) (Образец сравнения 56)
(Образец сравнения 57) (Образец сравнения 58) (Образец сравнения 59) (Образец сравнения 60) (Образец сравнения 61) (Образец сравнения 62) (Образец сравнения 63) (Образец сравнения 64) (Образец сравнения 65) (Образец сравнения 66) (Образец сравнения 67) [Оценка
каталитических характеристик] После завершения этой предварительной обработки эту реакцию проводили при условиях концентрации этанола 20%, концентрации метанола
20%, скорости потока газа носителя 80 сс/мин (общая скорость потока 100 сс/мин) и пространственной скорости (GHSV) 10000 (л/ч) при нормальном давлении. В случае синтеза высокооктанового топлива синтез
проводили при температуре реакции в диапазоне от 300 до 700oС. Идентификацию и определение реакционного газа проводили при использовании тех же способов, как и в случае этилена. В качестве
реакционного аппарата использовали каталитический реакционный аппарат газового потока, показанный на фиг.2. Результаты оценки показаны в табл.7 и табл.8 ниже. В таблице жидкая фракция
представляет собой реакционный газ, сжиженный в трубе охлаждения при 0oС. Оценку жидкой фракции проводили способом оценки жидкой фракции JIS. В качестве примера анализа
состава жидкой фракции ниже показаны аналитические данные для образца 74. Основными компонентами были кислородсодержащие углеводородные соединения и их содержание было следующим.
Этанол= 10% (непрореагировавший этанол), С4Н10=6%, С4Н8О=4%, C5H10O=3%, С6Н14О=2%, С6Н10
=5%, ароматика=8% остальные углеводороды. Компоненты других жидких фракций, образованных в ходе реакций с использованием катализаторов примеров, были практически теми же, что и
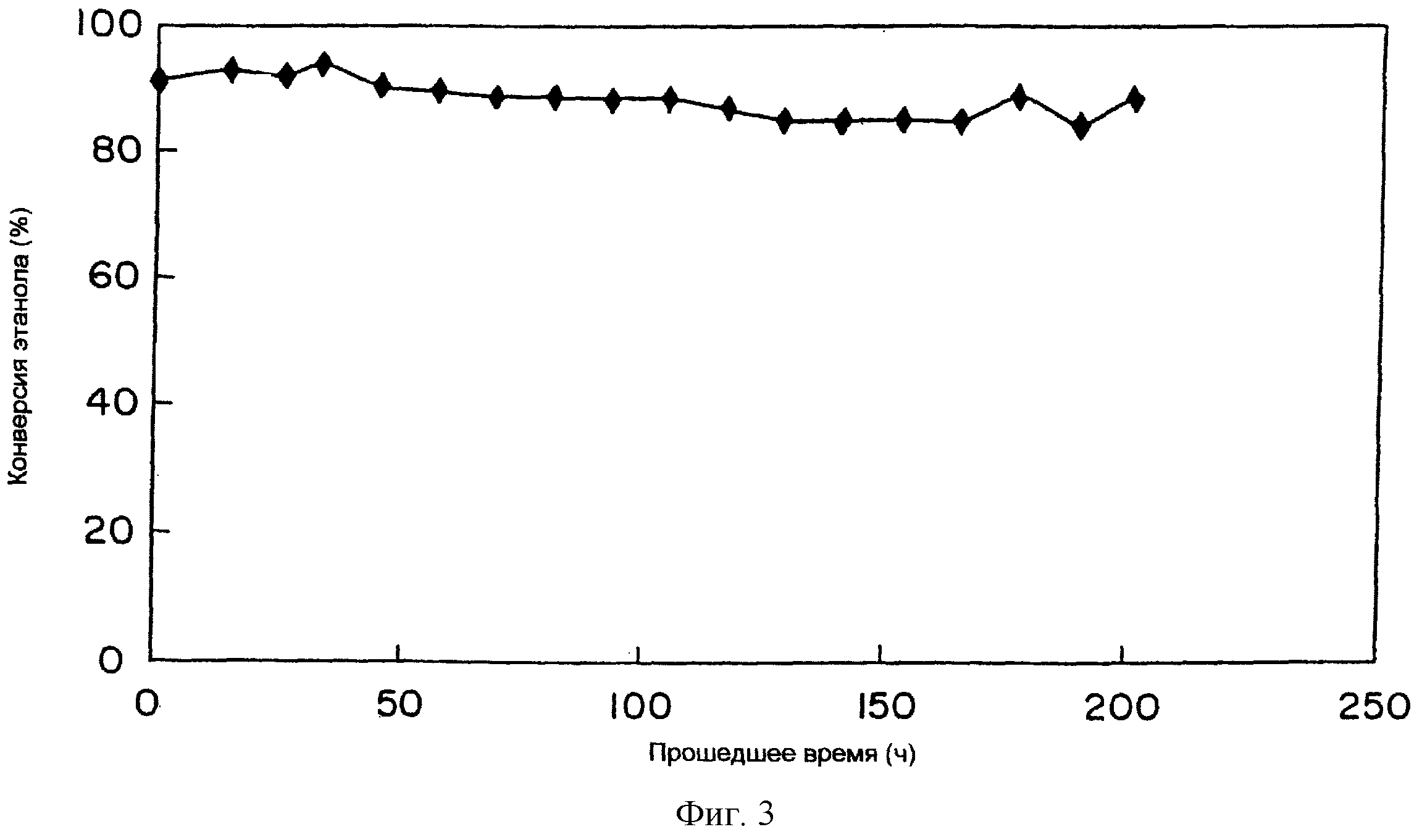
компоненты в образце 73, но содержание этих компонентов в этих топливах было различным в зависимости от типа катализатора и температуры реакции. После работы в течение 12 часов при
использовании 0,6 мл таблеток #14 до # 26, сформированных из образца 73, при условиях температуры реакции 500oС, концентрации этанола 20%, пространственной скорости (GHSV) 10000 (л/ч),
нормальном давлении, в качестве обработки для регенерации катализатора катализатор обрабатывали в атмосфере 2% кислорода при 480o в течение 15 минут, катализатор успешно использовали при
тех же условиях (500oС, концентрация EtOH= 20%, GHSV= 10000 (л/ч) и V=0,6 мл), измеряли конверсию этанола, посредством чего измеряли стабильность каталитической активности с течением
времени. Результат показан на фиг.3. Результат показывает, что путем периодической регенерации каталитической активности активность катализатора полностью сохраняется.
Катализатор синтезировали следующим образом. Также для измерения удельной площади полученного порошка использовали SA3100, производимый COLTER CO. , LTD. , и для измерения мольного
отношения Са/Р и мольного отношения (Са + металл)/Р использовали флуоресцентный рентгеновский анализатор, RIX1000, производимый Rigaku Denki Kogyo К.К.
[Получение катализатора]
(Образец 1)
После легкого измельчения в ступке фосфата кальция, имеющего мольное отношение Са/Р 1,50, размолотый продукт обжигали при
700oС в течение 2 часов и размалывали в ступке с получением порошка. После растворения 0,037 г нитрата алюминия [Al(NО3)3•9Н2O] в 50 мл
дистиллированной воды 10 г описанного выше порошка добавляли к раствору и после перемешивания смеси в течение одного дня смесь сушили при 140oС. Высушенный продукт размалывали и обжигали на
воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,1 мольного % алюминия.
Раствор, полученный
растворением 232,3 г нитрата кальция [Са(NО3)2•4Н2О] в 5,0 литрах дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 литрах дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота, и полученную смесь
перемешивали в течение одного дня. После этого продукт отбирали при помощи фильтрации, промывали водой и сушили при 140oС для получения порошка, имеющего мольное отношение Са/Р 1,65. После
растворения 0,037 г нитрата алюминия [Al(NО3)3• 9Н2О] в 50 мл дистиллированной воды, 10,0 г описанного выше порошка фосфата кальция добавляли к раствору с
последующим перемешиванием в течение одного дня, смесь сушили при 140oС, размалывали и обжигали в воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 0,1 мольного % Al по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,65.
Раствор, полученный растворением 13,65 г
нитрата кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота, с последующим перемешиванием в течение
одного дня.
Кроме того, раствор, полученный растворением 0,67 г нитрата алюминия [Al(NO3)3•9Н2О] в 50 мл дистиллированной воды, добавляли к описанной выше смеси и
полученную смесь перемешивали в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения высушенного продукта размолотый продукт обжигали на воздухе при 700o
С в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,0.
После легкого измельчения в ступке фосфата кальция, имеющего мольное отношение Са/Р 1,50, раствор, полученный растворением 0,67 г нитрата алюминия [Al(NО3)3•
9Н2O] в 50 мл дистиллированной воды, добавляли к порошку и далее смесь перемешивали в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый
продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % алюминия по отношению к Са и имеющего мольное
отношение (Са+Аl)/Р 1,50.
Раствор, полученный растворением 22,53 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл
дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору
аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота с последующим перемешиванием в течение одного дня. Кроме того, раствор, полученный растворением 0,67 г нитрата алюминия [Al(NО3)3•9Н2О] в 50 мл дистиллированной воды, добавляли к описанной выше смеси и полученную смесь дополнительно перемешивали в течение одного дня. Затем смесь упаривали
досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3
мольных % алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,65.
Раствор, полученный растворением 27,31 г нитрата кальция [Са(NО3)2•4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды,
добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота, с последующим перемешиванием в течение одного дня. Кроме того, раствор, полученный растворением
0,67 г нитрата алюминия [Al(NО3)3•9Н2O] в 50 мл дистиллированной воды, добавляли к описанной выше смеси и полученную смесь дополнительно перемешивали в течение
одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 3 мольных % алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 2,00.
Обжигом порошка, синтезированного при
помощи той же процедуры, что и в случае получения образца 5, на воздухе при 750oС в течение 2 часов был получен порошкообразный каталитический состав, содержащий 3 мольных % алюминия по
отношению к Са и имеющий мольное отношение (Са+Al)/Р 1,65.
Раствор, полученный растворением 11,61 г нитрата кальция [Са(NO3)2•4H2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к
водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота с последующим перемешиванием в течение одного дня. Кроме того, раствор, полученный растворением 19,73 г нитрата
алюминия [Al(NО3)3•9Н2O] в 500 мл дистиллированной воды, добавляли к описанной выше смеси и полученную смесь дополнительно перемешивали в течение одного дня.
Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического
состава, содержащего 50 мольных % алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,65.
После легкого измельчения в ступке фосфата
кальция, имеющего мольное отношение Са/Р 1,00, размолотый продукт обжигали на воздухе при 700oС в течение 2 часов и размалывали в ступке для получения порошка для примера сравнения.
После легкого измельчения в ступке фосфата кальция, имеющего мольное отношение Са/Р 1,50, размолотый продукт обжигали на воздухе при 700oС в течение
2 часов и размалывали в ступке для получения порошка для примера сравнения.
Раствор, полученный растворением 232,3 г нитрата кальция [Са(NO3
)2•4Н2О] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NН4)2НРO4] в 3,0 л дистиллированной
воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота с последующим перемешиванием в течение одного дня. Затем продукт отделяли фильтрацией,
промывали водой, сушили при 140oС и обжигали на воздухе при 700oС в течение 2 часов для получения порошка для примера сравнения, имеющего мольное отношение Са/Р 1,65.
После легкого измельчения в ступке фосфата кальция, имеющего мольное отношение Са/Р 2,00, размолотый продукт обжигали на воздухе при 700oС в течение
2 часов и размалывали в ступке для получения порошка для примера сравнения.
После растворения 0,037 г нитрата алюминия [Al(NO3)3
• 9Н2О] в 50 мл дистиллированной воды, 10 г порошка образца сравнения 3 добавляли к раствору с последующим перемешиванием в течение одного дня, смесь сушили при 140oС и
после сушки обжигали на воздухе при 700oС в течение 2 часов для получения порошка для примера сравнения, содержащего 0,03 мольного % алюминия.
Образец 5 оценивали при температуре реакции 350oС.
Порошок, полученный при использовании способа синтеза образца 5, обжигали на воздухе при
800oС в течение 2 часов для получения порошка образца сравнения. Образец оценивали при температуре реакции 750oС.
Образец 8
оценивали при температуре реакции 350oС.
Порошок, полученный при использовании способа синтеза образца 8, обжигали на воздухе при 800oС в течение 2 часов для получения порошка образца сравнения. Образец оценивали при температуре реакции 750oС.
Каждый
приготовленный образец из образцов с 1 по 8 и образцов сравнения с 1 по 9 формировали в таблетки от #14 до #26. Затем 0,6 мл таблеток паковали в кварцевую трубку и проводили предварительную обработку
в случае образца, не несущего металл, обработку нагреванием (дегидратация) в газе-носителе (1% Ar/Не основа: скорость потока 80 сс/мин) при 500oС в течение 30 минут и в случае образца,
несущего металл, обработку восстановлением содержащегося металла в 20% H2 (He основа: скорость потока 100 сс/мин) при 500oС в течение 30 минут.
Селективность синтез-газа (%)= (значение площади пика)/(значение общей площади пика - значение площади пика остаточного
этанола)х100
Пространственное время выхода (ПВВ) определяли как выход (г) углеводородов на 1 литр катализатора и 1 час и, рассматривая углеводороды как CH2xn, пространственное
время выхода рассчитывали по следующей формуле.
[Получение катализатора]
(Образец 9)
Раствор, полученный растворением 21,88 г нитрата кальция
[Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл
дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, раствор,
полученный растворением 0,120 г нитрата меди [Cu(NO3)2 •3Н2O] и 0,201 г нитрата железа (III) [Fe(NО3)3 • 9Н2О] в 50 мл
дистиллированной воды, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт
обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,5 мольных % каждого из Cu и Fe по отношению к Са и имеющего
(Ca+Cu+Fe)/P мольное отношение 1,57.
Раствор, полученный растворением 19,71 г нитрата кальция [Са(NО3)2•4Н2О] в 500 мл
дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору
аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 2,46 г нитрата меди
[Cu(NО3)2•3Н2O] и 4,15 г нитрата железа (III) [Fe(NО3)3•9Н2O] в 200 мл дистиллированной воды, соответственно, добавляли
к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 10 мольных % каждого из Cu и Fe по отношению к Са и имеющего (Ca+Cu+Fe)/P мольное отношение 1,75.
Раствор, полученный растворением 17,68 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный
растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11 в
атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 2,21 г нитрата меди [Cu(NО3)2•3Н2
O] и 3,72 г нитрата железа (III) [Fe(NО3)3•9Н2О] в 200 мл дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием
в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 10 мольных % каждого из Cu и Fe по отношению к Са и имеющего (Ca+Cu+Fe)/P мольное отношение 1,57.
Раствор, полученный
растворением 9,72 г нитрата кальция [Са(NO3)2•4H2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4
)2НРO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в
течение одного дня. Кроме того, растворы, полученные растворением каждого из 5,94 г нитрата меди [Cu(NО3)2•3Н2О] и 10,14 г нитрата железа (III) [Fe(NО3)3•9Н2О] в 500 мл дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали
досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 28
мольных % каждого из Cu и Fe по отношению к Са и имеющего (Ca+Cu+Fe)/P мольное отношение 1,57.
Раствор, полученный растворением 21,88 г нитрата кальция [Са(NО3)2•4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл
дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того,
растворы, полученные растворением каждого из 0,096 г нитрата меди [Cu(NО3)2•3Н2O] , 0,161 г нитрата железа (III) [Fe(NО3)3•9Н2O] и 0,075 г нитрата алюминия [Al(NО3)3•9Н2O] в 50 мл дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным
перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения
порошкообразного каталитического состава, содержащего 0,4 мольных % Cu, 0,4 мольных % Fe и 0,2 мольных % Al по отношению к Са и имеющего (Ca+Cu+Fe+Al)/P мольное отношение 1,57.
Раствор, полученный растворением 16,57 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный
растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11,
в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 2,21 г нитрата меди [Cu(NО3)2•3Н2О] , 3,72 г нитрата железа (III) [Fe(NО3)3•9Н2О] и 1,77 г нитрата алюминия [Al(NО3)2•9Н2О] в 200 мл
дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения
размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 10 мольных % Cu, 10 мольных % Fe и 5 мольных % Al по
отношению к Са и имеющего (Ca+Cu+Fe+Al)/P мольное отношение 1,57.
Раствор, полученный растворением 4,22 г нитрата кальция [Са(NО3)2•
4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого
из 3,78 г нитрата меди [Cu(NO3)2•3Н2О] , 6,46 г нитрата железа (III) [Fe(NO3)3•9Н2О] в 3,19 г нитрата алюминия
[Al(NО3)3•9Н2O] в 500 мл дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем
смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава,
содержащего 28 мольных % Cu, 28 мольных % Fe и 14 мольных % Al по отношению к Са и имеющего (Ca+Cu+Fe+Al)/P мольное отношение 1,00.
Раствор, полученный
растворением 6,63 г нитрата кальция [Са(NO3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4
)2НРО4] в 300 мл дистиллированной воды, добавляли по каплям к водному
раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь
перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 5,94 г нитрата меди [Cu(NО3)2•3Н2O], 10,14 г нитрата железа (III)
[Fe(NО3)3•9Н2О] и 5,01 г нитрата алюминия [Al(NО3)3•9H2O] в 500 мл дистиллированной воды соответственно, добавляли к
раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 28 мольных % Cu, 28 мольных % Fe и 14 мольных % Al по отношению к Са и имеющего (Ca+Cu+Fe+Al)/Р мольное
отношение 1,57.
Раствор, полученный растворением 6,97 г нитрата кальция [Са(NО3)2•4Н2О] в 500 мл дистиллированной воды, и
раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему
установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 6,24 г нитрата меди [Cu(NO3
)2•3H2O], 10,66 г нитрата железа (III) [Fe(NО3)3•9Н2O] и 5,26 г нитрата алюминия [Al(NО3)3•9Н2O] в 500 мл дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и
после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 28 мольных % Cu, 28 мольных % Fe и
14 мольных % Al по отношению к Са и имеющего (Ca+Cu+Fe+Al)/P мольное отношение 1,65.
Раствор, полученный растворением 8,45 г нитрата кальция [Са(NО3
)2•4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной
воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные
растворением каждого из 7,57 г нитрата меди [Cu(NО3)2•3Н2О], 12,92 г нитрата железа (III) [Fe(NО3)3•9Н2О] и 6,38 г
нитрата алюминия [Al(NО3)3•9Н2О] в 500 мл дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием в течение
одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 28 мольных % Cu, 28 мольных % Fe и 14 мольных % Al по отношению к Са и имеющего (Ca+Cu+Fe+Al)/P мольное отношение 2,00.
Образец 16 оценивали при температуре реакции 300oС.
Образец 16 оценивали при температуре реакции 200oС.
(Образец сравнения 10)
Фосфат кальция образца сравнения обрабатывали, как и в случае образца сравнения 1, и обжигали на воздухе при 500oС в течение 2 часов для
получения порошка образца сравнения. Образец оценивали при температуре реакции 350oС.
Раствор, полученный растворением 22,10 г нитрата
кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4]
в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Затем
смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава,
имеющего Са/Р мольное отношение 1,57.
Порошок, полученный при использовании способа синтеза образца 3, обрабатывали также как образец сравнения 3 и
обжигали на воздухе при 500oС в течение 2 часов для получения порошка образца сравнения.
Порошок, полученный при использовании способа
синтеза образца сравнения 4, обрабатывали также, как образец сравнения 4 и обжигали на воздухе при 500oС в течение 2 часов для получения порошка образца сравнения.
Раствор, полученный растворением 22,04 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный
растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11,
в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 0,023 г нитрата меди [Cu(NО3)2•3Н2О] , 0,038 г нитрата железа (III) [Fe(NО3)3•9Н2О] и 0,018 г нитрата алюминия [Al(NО3)3•9Н2O] в 50 мл
дистиллированной воды соответственно, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения
размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,1 мольного % Cu, 0,1 мольного % Fe и 0,05 мольного %
Al по отношению к Са и имеющего (Ca+Cu+Fe+Al)/Р мольное отношение 1,57.
Раствор, полученный растворением 17,68 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл дистиллированной воды,
добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и полученную смесь перемешивали в течение одного дня. Кроме того, раствор, полученный
растворением 4,320 г нитрата меди [Cu(NО3)2•3Н2O] в 200 мл дистиллированной воды, добавляли к описанному выше раствору и смесь перемешивали в течение одного
дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 500oС в течение 2 часов для получения порошкообразного каталитического
состава, содержащего 20 мольных % Cu по отношению к Са и имеющего (Са+Cu)/Р мольное отношение 1,57.
Образец 16 оценивали при температуре реакции 150oС.
Образец 16 оценивали при температуре реакции 500oС.
Использовали MgO (реагент),
который являлся обычным основным твердым носителем.
Каждый приготовленный образец из образцов с 9 по 20 и образцов сравнения с 10 по 18
формировали в таблетки от #14 до #26. Затем для образца проводили предварительную обработку как и в случае этилена. После завершения этой предварительной обработки эту реакцию проводили при условиях
концентрации этанола 20%, скорости потока газа носителя 80 сс/мин (общая скорость потока 100 сс/мин) и пространственной скорости (GHSV) 10000 (л/ч) при нормальном давлении.
[Получение катализатора]
(Образец 21)
После легкого измельчения в ступке фосфата кальция, имеющего мольное отношение Са/Р 1,45, размолотый продукт обжигали
при 700oС в течение 2 часов и размалывали в ступке с получением порошка. После растворения 0,037 г нитрата алюминия [Al(NO3)3•9Н2О] в 50 мл
дистиллированной воды, 10 г описанного выше порошка добавляли к раствору и после перемешивания смеси в течение одного дня смесь сушили при 140oС и высушенный продукт размалывали и обжигали
на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,1 мольного % алюминия.
После растворения
0,037 г нитрата алюминия [Al(NО3)3•9Н2О] в 50 мл дистиллированной воды, 10 г порошка, полученного в способе синтеза образца сравнения 11, добавляли к раствору с
последующим перемешиванием в течение одного дня, смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 0,1 мольного % алюминия.
Образец 22 оценивали при температуре реакции 400oС.
При помощи легкого измельчения в ступке фосфата кальция, имеющего мольное отношение Сa/Р 1,00, получали порошок. После растворения 1,24 г нитрата алюминия [Al(NО3)3•
9Н2О] в 200 мл дистиллированной воды, 10 г описанного выше порошка добавляли к раствору с последующим перемешиванием в течение одного дня, смесь сушили при 140oС и после
измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 5 мольных % Al по отношению к Са и имеющего мольное отношение
(Са+Al)/Р примерно 1,0.
Раствор, полученный растворением 20,99 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл
дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору
аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота с последующим перемешиванием в течение одного дня. Кроме того, раствор, полученный растворением 1,77 г нитрата алюминия [Al(NО3)3•9Н2O] в 200 мл дистиллированной воды, добавляли к описанному выше раствору с последующим перемешиванием в течение одного дня. Затем смесь упаривали досуха при
140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 5 мольных %
алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,57.
Раствор, полученный растворением 22,06 г нитрата кальция [Са(NО3)2
•4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды,
добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере
азота с последующим перемешиванием в течение одного дня. Кроме того, раствор, полученный
растворением 1,86 г нитрата алюминия [Al(NО3)3•9Н2О] в 200 мл дистиллированной воды, добавляли к описанному выше раствору с последующим перемешиванием в течение
одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 5 мольных % алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,65.
При помощи легкого измельчения в ступке
фосфата кальция, имеющего мольное отношение Са/Р 2,00, получали порошок. После растворения 2,48 г нитрата алюминия [Al(NО3)3•9Н2O] в 200 мл дистиллированной
воды, 10 г описанного выше порошка добавляли к раствору с последующим перемешиванием в течение одного дня, смесь сушили при 140oС и после измельчения обжигали на воздухе при 700o
С в течение 2 часов для получения порошкообразного каталитического состава, содержащего 5 мольных % Al по отношению к Са и имеющего мольное отношение (Са+Al)/Р примерно 2,0.
Раствор, полученный растворением 11,05 г нитрата кальция [Са(NО3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г
фосфата аммония [(NH4)2НРО4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота с
последующим перемешиванием в течение одного дня. Кроме того, раствор, полученный растворением 18,78 г нитрата алюминия [Al(NO3)3•9H2O] в 500 мл дистиллированной
воды, добавляли к описанному выше раствору с последующим перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на
воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 50 мольных % алюминия по отношению к Са и имеющего мольное отношение (Са+Al)/Р 1,
57.
Образец 28 оценивали при температуре реакции 350oС.
Образец 28 оценивали при температуре реакции 300oС.
Образец 28 оценивали при температуре реакции 200oС.
Использовали образец сравнения 1.
Фосфат кальция образца сравнения 11 обрабатывали как в образце сравнения 11 и обжигали на воздухе при 700oС в течение 2 часов для получения
порошка для образца сравнения.
Использовали образец сравнения 3.
Использовали образец сравнения 4.
После растворения 0,011 г нитрата алюминия [Al(NО3)3•9Н2О] в 50 мл дистиллированной воды, 10 г порошка,
полученного в способе синтеза образца сравнения 11, добавляли к раствору с последующим перемешиванием в течение одного дня, смесь сушили при 140oС и после измельчения обжигали на воздухе
при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,03 мольного % алюминия.
Образец 25
оценивали при температуре реакции 200oС.
Образец 25 оценивали при температуре реакции 450oС.
Образец 28 оценивали при температуре реакции 150oС.
Образец 28 оценивали при температуре реакции 450oС.
Каждый приготовленный образец из образцов с 21 по 31 и образцов сравнения с 19 по 27 формировали в таблетки от #14 до #26. Затем для образца
проводили предварительную обработку, как и в случае этилена.
[Получение катализатора]
(Образец 32)
Раствор, полученный растворением 225,2 г нитрата кальция [Са(NО3)2•4Н2O] в 5,0 л дистиллированной воды,
и раствор, полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 л дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему
установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Затем этот продукт отделяли при помощи фильтрации, промывали водой и после сушки при 140oС,
обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, имеющего Са/Р мольное отношение 1,60.
Использовали образец сравнения 3.
Образец сравнения 3 оценивали при температуре реакции 400oС.
Образец
сравнения 3 оценивали при температуре реакции 350oС.
Раствор, полученный растворением 239,3 г нитрата кальция [Са(NО3)2•
4Н2O] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 л дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Затем этот продукт отделяли при помощи фильтрации, промывали водой
и после сушки при 140oС обжигали на воздухе при 900oС в течение 2 часов для получения порошкообразного каталитического состава, имеющего Са/Р мольное отношение 1,70.
Раствор, полученный растворением 253,4 г нитрата кальция [Са(NО3)2•4Н2О] в 5,0 л дистиллированной воды, и раствор, полученный
растворением 78,87 г фосфата аммония [(NН4)2НРO4] в 3,0 л дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11,
в атмосфере азота и смесь перемешивали в течение одного дня. Затем этот продукт отделяли при помощи фильтрации, промывали водой и после сушки при 140oС обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, имеющего Са/Р мольное отношение 1,80.
После растворения 1,24 г нитрата церия
[Се(NО3)3•6Н2О] в 200 мл дистиллированной воды, 9,59 г порошка, полученного на основе способа синтеза образца сравнения 3, добавляли к раствору с последующим
перемешиванием в течение одного дня. Затем продукт сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 3 мольных % Се по отношению к Са и имеющего (Са+Се)/Р мольное отношение 1,65.
Образец 38 оценивали при температуре реакции
400oС.
Образец 38 оценивали при температуре реакции 350oС.
Образец 38 оценивали при температуре
реакции 300oС.
Раствор, полученный растворением 7,04 г нитрата кальция [Са(NО3)2•4Н2О] в 500 мл
дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака,
имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, раствор, полученный растворением 8,29 г нитрата магния [Mg(NО3)2
•6Н2О] в 500 мл дистиллированной воды, добавляли к описанному выше раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 50 мольных % Mg по
отношению к Са и имеющего (Са+Mg)/Р мольное отношение 1,00.
Раствор, полученный растворением 14,78 г нитрата кальция [Са(NО3)2•
4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, раствор полученный растворением 7,22 г нитрата магния
[Mg(NO3)2•6Н2O] в 500 мл дистиллированной воды, добавляли к описанному выше раствору с последующим дополнительным перемешиванием в течение одного дня. Затем
смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава,
содержащего 30 мольных % Mg по отношению к Са и имеющего (Са+Mg)/Р мольное отношение 1,50.
Раствор, полученный растворением 11,61 г нитрата кальция [Са(NО3)2•4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл
дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, раствор,
полученный растворением 14,26 г нитрата церия [Се(NО3)3•6Н2O] в 500 мл дистиллированной воды, добавляли к описанному выше раствору с последующим дополнительным
перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения
порошкообразного каталитического состава, содержащего 50 мольных % Се по отношению к Са и имеющего (Са+Се)/Р мольное отношение 1,65.
Раствор, полученный
растворением 14,08 г нитрата кальция [Са(NO3)2•4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4
)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение
одного дня. Кроме того, растворы полученные растворением каждого из 11,96 г нитрата церия [Се(NО3)3•6Н2O] и 9,18 г нитрата алюминия [Al(NО3)3•9Н2O] в 500 мл дистиллированной воды соответственно, добавляли к описанному выше раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь
упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава,
содержащего 30 мольных % Се и 20 мольных % Al по отношению к Са и имеющего (Са+Се+Al)/Р мольное отношение 2,00.
Использовали образец сравнения 1.
Раствор, полученный растворением 211,1 г нитрата кальция [Са(NO3)2•4Н2O] в 5,0 л дистиллированной воды, и раствор,
полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 л дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11,
в атмосфере азота и смесь перемешивали в течение одного дня. Затем этот продукт отделяли при помощи фильтрации, промывали водой и после сушки при 140oС обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, имеющего Са/Р мольное отношение 1,50.
Кальций фосфат образца
сравнения 3 обрабатывали, как и в случае примера сравнения 3, и обжигали на воздухе при 1200oС в течение 2 часов для получения порошка образца сравнения.
Использовали образец сравнения 4.
Образец сравнения 3 оценивали при температуре реакции 250oС.
Образец сравнения 3 оценивали при температуре реакции 500oС.
Образец 38 оценивали при температуре реакции 250oС.
Образец 38 оценивали при температуре реакции 500oС.
Каждый
приготовленный образец из образцов с 32 по 45 и образцов сравнения с 28 по 35 формировали в таблетки от #14 до #26. Затем для образца проводили предварительную обработку, как и в случае этилена.
[Получение катализатора]
(Образец 46)
Использовали образец сравнения 20.
Фосфат кальция образца 32 обрабатывали, как и в случае образца 32, и обжигали на воздухе при 1000oС в течение 2
часов для получения порошкообразного каталитического состава.
Раствор, полученный растворением 228,0 г нитрата кальция [Са(NО3)2•
4Н2О] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 л дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Затем этот продукт отделяли при помощи фильтрации, промывали водой
и после сушки при 140oС обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, имеющего Са/Р мольное отношение 1,62.
Образец 48 оценивали при температуре реакции 550oС.
Образец 48 оценивали при температуре реакции 500oС.
Образец 48 оценивали при температуре реакции 450oС.
Использовали образец сравнения 3.
Фосфат кальция образца 36 обрабатывали, как и в случае образца 36, и затем обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава.
Использовали образец 37.
Раствор, полученный растворением 225,7 г нитрата кальция [Са(NО3)2•4Н2O] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 л
дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Затем продукт отбирали
фильтрацией, промывали водой и сушили при 140oС для получения порошка. После растворения 0,224 г хлорида циркония (IV) (ZrCl4) в 100 мл дистиллированной воды, 9,91 г описанного
выше порошка добавляли к раствору с последующим перемешиванием в течение одного дня, смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов
для получения порошкообразного каталитического состава, содержащего 1 мольный % Zr по отношению к Са и имеющего (Ca+Zr)/P мольное отношение 1,62.
Образец 55
оценивали при температуре реакции 450oС.
Использовали образец 2.
Образец 2 оценивали при температуре реакции
500oС.
Образец 2 оценивали при температуре реакции 450oС.
Раствор, полученный растворением 225,3 г
нитрата кальция [Са(NО3)2•4Н2O] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NH4)2HPO4] в 3,0 л дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Затем продукт
отбирали фильтрацией, промывали водой и сушили при 140oС для получения порошка. После растворения 0,847 г тетраизопропоксида титана {[(СН3)2СНО]4Тi} в 100
мл этанола, 9,86 г описанного выше порошка добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % Ti по отношению к Са и имеющего (Ca+Ti)/P мольное отношение 1,65.
После растворения 0,715 г вольфрамовой кислоты (H2WO4) в 100 мл дистиллированной воды, 9,47 г фосфата кальция, синтезированного в образце 60, к раствору с
последующим перемешиванием в течение одного дня. Затем смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного
каталитического состава, содержащего 3 мольных % W по отношению к Са и имеющего (Ca+W)/P мольное отношение 1,65.
После растворения 0,685 г хлорида циркония (IV)
(ZrCl4) в 100 мл дистиллированной воды, 9,73 г фосфата кальция, синтезированного в образце 60, к раствору с последующим перемешиванием в течение одного дня. Затем смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % Zr по отношению к Са и имеющего
(Ca+Zr)/P мольное отношение 1,65.
Использовали образец 42.
Использовали образец 43.
Раствор, полученный растворением 11,61 г нитрата кальция [Ca(NO3)2•4H2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата
аммония [(NН4)2НРO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь
перемешивали в течение одного дня. Кроме того, раствор, полученный растворением 9,13 г хлорида циркония (IV) (ZrCl4) в 500 мл дистиллированной воды соответственно, добавляли к описанной
выше смеси с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 50 мольных % Zr по отношению к кальцию и имеющего (Ca+Zr)/p мольное отношение 1,65.
Раствор, полученный растворением 14,08 г нитрата кальция [Ca(NO3)2•4H2O] в 500 мл дистиллированной воды, и раствор, полученный
растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11,
в атмосфере азота и смесь перемешивали в течение одного дня.
Использовали образец сравнения 29.
Фосфат кальция образца 48
обрабатывали, как в случае образца 48, и обжигали на воздухе при 1200oС в течение 2 часов для получения порошкообразного каталитического состава.
Использовали образец сравнения 4.
Образец 48 оценивали при температуре реакции 400oС.
Фосфат кальция образца 48 обрабатывали, как в случае образца 48, и обжигали на воздухе при 800oС в течение 2 часов и размалывали в ступке для получения порошка образца сравнения.
Образец 55 оценивали при температуре реакции 400oС.
Фосфат кальция образца 55 обрабатывали, как в
случае образца 55, и обжигали на воздухе при 800oС в течение 2 часов и размалывали в ступке для получения порошка образца сравнения.
Образец
2 оценивали при температуре реакции 400oС.
Фосфат кальция образца 2 обрабатывали, как в случае образца 2, и обжигали на воздухе при 800oС в течение 2 часов и размалывали в ступке для получения порошка образца сравнения.
Использовали сепиолит (коммерчески доступный продукт),
упоминавшийся в примере синтеза 1,3-бутадиена из этанола.
Каждый приготовленный образец из образцов с 46 по 66 и образцов сравнения с 36
по 46 формировали в таблетки от #14 до #26. Затем для образца проводили предварительную обработку, как и в случае этилена.
[Получение катализатора]
(Образец 67)
Использовали образец
20.
Фосфат кальция образца 48 обрабатывали, как в случае образца 48, и обжигали на воздухе при 1000oС в течение 2 часов и размалывали в
ступке для получения порошка образца сравнения.
Использовали образец 48.
Использовали образец сравнения 3.
Образец сравнения 3 оценивали при температуре реакции 400oС.
Образец сравнения 3 оценивали при температуре реакции 450oС.
Образец сравнения 3 оценивали при температуре реакции 500oС.
Образец сравнения 3 оценивали при
температуре реакции 550oС.
Использовали образец 53.
Использовали образец 37.
Раствор, полученный растворением 228,0 г нитрата кальция [Са(NO3)2•4Н2О] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата
аммония [(NH4)2HPO4] в 3,0 л дистиллированной воды, добавляли по каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь
перемешивали в течение одного дня. Затем продукт отбирали фильтрацией, промывали водой и сушили при 140oС для получения порошка, имеющего мольное отношение Са/Р 1,62. После растворения 0,
007 г нитрата лития (LiNO3) в 100 мл дистиллированной воды, 10,0 г описанного выше порошка фосфата кальция добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь
сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,1 мольного % Li по
отношению к Са и имеющего (Ca+Li)/P мольное отношение 1,62.
Раствор, полученный растворением 232,3 г нитрата кальция [Са(NО3)2•
4H2O] в 5,0 л дистиллированной воды, и раствор, полученный растворением 78,87 г фосфата аммония [(NН4)2НРО4] в 3,0 л дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Затем продукт отбирали фильтрацией, промывали водой и сушили при
140oС для получения порошка, имеющего мольное отношение Са/Р 1,65. После растворения 0,029 г нитрата никеля (Ni(NО3)2•6Н2О) в 100 мл дистиллированной
воды, 10,0 г описанного выше порошка фосфата кальция добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе
при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 0,1 мольного % Ni по отношению к Са и имеющего (Ca+Ni)/P мольное отношение 1,65.
После растворения 0,202 г нитрата лития (LiNO3) в 100 мл дистиллированной воды, 9,98 г необожженного порошка фосфата кальция, синтезированного в образце 48,
добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения
порошкообразного каталитического состава, содержащего 3 мольных % Li по отношению к Са и имеющего (Ca+Li)/P мольное отношение 1,62.
Образец 79 оценивали при
температуре реакции 450oС.
Образец 79 оценивали при температуре реакции 400oС.
После растворения 0,
838 г нитрата никеля [Ni(NО3)2•6Н2O] в 100 мл дистиллированной воды, 9,83 г необожженного порошка фосфата кальция, синтезированного в образце 48, с последующим
перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического
состава, содержащего 3 мольных % Ni по отношению к Са и имеющего (Ca+Ni)/P мольное отношение 1,62.
После растворения 0,854 г нитрата никеля [Ni(NO3
)2•6H2O] в 100 мл дистиллированной воды, 9,83 г необожженного порошка фосфата кальция, синтезированного в образце сравнения 3, с последующим перемешиванием в течение
одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе при 1000oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3
мольных % Ni по отношению к Са и имеющего (Ca+Ni)/P мольное отношение 1,65.
Ni-содержащий фосфат кальция, полученный в образце 83, обрабатывали, как в случае
образца 83, обжигали на воздухе при 700oС в течение 2 часов и размалывали в ступке для получения каталитического состава.
Образец 84 оценивали при
температуре реакции 400oС.
После растворения 0,871 г нитрата цинка [Zn(NО3)2•6Н2О] в
100 мл
дистиллированной воды, 9,81 г необожженного порошка фосфата кальция, синтезированного в образце сравнения 3, добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при
140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % Zn по отношению к Са и
имеющего (Ca+Zn)/P мольное отношение 1,65.
После растворения 0,837 г тетраизопропоксида титана {[(СН3
)2СНО]4Тi} в 100 мл
этанола, 9,86 г необожженного порошка фосфата кальция, синтезированного в образце сравнения 3, добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС
и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % Ti по отношению к Са и имеющего (Ca+Ti)/P
мольное отношение 1,65.
После растворения 0,616 г нитрата стронция [Sr(NО3)2] в 100 мл дистиллированной воды, 9,74 г необожженного порошка
фосфата кальция, синтезированного в образце сравнения 3, добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на
воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % Sr по отношению к Са и имеющего (Ca+Sr)/P мольное отношение 1,65.
После растворения 1,274 г нитрата европия [Eu(NО3)3•6Н2О] в 100 мл дистиллированной воды, 9,56 г необожженного порошка фосфата
кальция, синтезированного в образце сравнения 3, добавляли к раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе при
700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 3 мольных % Eu по отношению к Са и имеющего (Са+Eu)/Р мольное отношение 1,65.
После растворения 0,483 г хлорида цезия (CsCl) в 100 мл дистиллированной воды, 9,61 г необожженного порошка фосфата кальция, синтезированного в образце сравнения 3, добавляли к
раствору с последующим перемешиванием в течение одного дня. Смесь сушили при 140oС и после измельчения обжигали на воздухе при 700oС в течение 2 часов для получения
порошкообразного каталитического состава, содержащего 3 мольных % Cs по отношению к Са и имеющего (Са+Cs)/Р мольное отношение 1,65.
Использовали образец 42.
Использовали образец 43.
Раствор, полученный растворением 11,40 г нитрата кальция [Са(NО3)2•
4Н2О] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, раствор, полученный растворением 3,99 г нитрата лития
(LiNO3) в 500 мл дистиллированной воды, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после
измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 50 мольных % Li по отношению к Са и
имеющего (Ca+Li)/P мольное отношение 1,62.
Раствор, полученный растворением 11,61 г нитрата кальция [Са(NО3)2•4Н2O] в 500
мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NН4)2НРO4] в 300 мл дистиллированной воды, добавляли по каплям к водному раствору
аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, раствор, полученный растворением 13,09 г нитрата никеля [Ni(NО3)2•6H2O] в 500 мл дистиллированной воды, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС
и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 50 мольных % Ni по отношению к Са
и имеющего (Ca+Ni)/P мольное отношение 1,65.
Раствор, полученный растворением 11,96 г нитрата кальция [Са(NО3)2•4Н2О] в
500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по каплям к водному
раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, раствор полученный растворением 13,39 г нитрата цинка [Zn(NО3
)2•6Н2О] в 500 мл дистиллированной воды, добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при 700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 50 мольных % Zn по
отношению к Са и имеющего (Ca+Zn)/P мольное отношение 1,70.
Раствор, полученный растворением 14,08 г нитрата кальция [Са(NО3)2•
4Н2O] в 500 мл дистиллированной воды, и раствор, полученный растворением 7,89 г фосфата аммония [(NH4)2HPO4] в 300 мл дистиллированной воды, добавляли по
каплям к водному раствору аммиака, имеющему установленный рН от 9 до 11, в атмосфере азота и смесь перемешивали в течение одного дня. Кроме того, растворы, полученные растворением каждого из 9,85 г
нитрата никеля [Ni(NО3)2•6Н2O] и 9,18 г нитрата алюминия [Al(NО3)3•9Н2О] в 500 мл дистиллированной воды соответственно,
добавляли к раствору с последующим дополнительным перемешиванием в течение одного дня. Затем смесь упаривали досуха при 140oС и после измельчения размолотый продукт обжигали на воздухе при
700oС в течение 2 часов для получения порошкообразного каталитического состава, содержащего 30 мольных % Ni и 20 мольных % Al по отношению к Са и имеющего (Ca+Ni+Al)/P мольное отношение 2,
00.
Использовали образец сравнения 1.
Использовали образец сравнения 29.
Использовали образец сравнения 4.
Фосфат кальция образца сравнения 3 обрабатывали, как в случае образца сравнения 3, обжигали на
воздухе при 1100oС в течение 2 часов и размалывали в ступке для получения порошка образца сравнения.
Образец сравнения 3 оценивали при
температуре реакции 300oС.
Фосфат кальция образца сравнения 3 обрабатывали, как в случае образца сравнения 3, обжигали на воздухе при
800oС в течение 2 часов и размалывали в ступке для получения порошка образца сравнения.
Фосфат кальция образца 79 обрабатывали, как в случае
образца 79, обжигали на воздухе при 800oС в течение 2 часов и размалывали в ступке для получения порошка образца сравнения.
Ni-содержащий
фосфат кальция, полученный в образце 82, обрабатывали, как в случае образца 82, и обжигали на воздухе при 1200oС в течение 2 часов и размалывали в ступке для получения каталитического
состава.
Образец 82 оценивали при температуре реакции 250oС.
Ni-содержащий фосфат кальция,
полученный в образце 82, обрабатывали, как в случае образца 82, обжигали на воздухе при 800oС в течение 2 часов и размалывали в ступке для получения каталитического состава.
Zn-содержащий фосфат кальция, полученный в образце 86, обрабатывали, как в случае образца 86, и обжигали на воздухе при 800oС в течение 2 часов, и
размалывали в ступке для получения каталитического состава.
Ti-содержащий фосфат кальция, полученный в образце 87, обрабатывали, как в случае образца
87, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения каталитического состава.
Sr-содержащий фосфат
кальция, полученный в образце 88, обрабатывали, как в случае образца 88, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения каталитического состава.
Eu-содержащий фосфат кальция, полученный в образце 89, обрабатывали, как в случае образца 89, и обжигали на воздухе при 800oС в течение 2 часов,
и размалывали в ступке для получения каталитического состава.
Cs-содержащий фосфат кальция, полученный в образце 90, обрабатывали, как в случае
образца 90, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения каталитического состава.
Mg-содержащий
фосфат кальция, полученный в образце 42, обрабатывали, как в случае образца 42, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения каталитического
состава.
Mg-содержащий фосфат кальция, полученный в образце 43, обрабатывали, как в случае образца 43, и обжигали на воздухе при 800oС в
течение 2 часов, и размалывали в ступке для получения каталитического состава.
Li-содержащий фосфат кальция, полученный в образце 93, обрабатывали, как
в случае образца 93, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения каталитического состава.
Ni-содержащий фосфат кальция, полученный в образце 94, обрабатывали, как в случае образца 94, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения
каталитического состава.
Zn-содержащий фосфат кальция, полученный в образце 95, обрабатывали, как в случае образца 95, и обжигали на воздухе при
800oС в течение 2 часов, и размалывали в ступке для получения каталитического состава.
Ni, Al-содержащий фосфат кальция, полученный в
образце 96, обрабатывали, как в случае образца 96, и обжигали на воздухе при 800oС в течение 2 часов, и размалывали в ступке для получения каталитического состава.
Каждый приготовленный образец из образцов с 67 по 96 и образцов сравнения с 47 по 67 формировали в таблетки от #14 до #26. Затем для образца проводили
предварительную обработку, как и в случае этилена.
Реферат
Изобретение относится к способам производства 1-бутанола (варианты), 1, 3-бутадиена и высокооктанового топлива из этанола. Все способы включают контактирование этанола с низкокристаллическим фосфатом кальция, имеющим удельную площадь по крайней мере 2 м2/г. Способ синтеза 1-бутанола, как правило, проводят при мольном отношении Са/Р от 1,6 до 1,8 и температуре, изменяющейся от 350 до 450oС, а в варианте синтеза 1-бутанола низкокристаллический фосфат кальция содержит в себе по крайней мере один тип металла и/или оксид металла, выбираемый из Ва, Na, К, Li, Cs, Sr, Y, Ce, Sb, Eu, Ti, W и Zr в количестве, не превышающем 50 мол.% по отношению к Са и имеющим мольное отношение (Са + металл)/Р от 1 до 2, при температуре, изменяющейся от 300 до 450oС. Способ синтеза 1,3-бутадиена обычно проводят при мольном отношении Са/Р от 1,55 до 1, 8 и температуре, изменяющейся от 450 до 700oС, а способ синтеза высокооктанового топлива - при мольном отношении Са/Р от 1,55 до 1,8 и температуре, изменяющейся от 300 до 700oС. 4 с.п.ф-лы, 3 ил., 8 табл.
Комментарии