Способы получения топлив и растворителей - RU2458972C2
Код документа: RU2458972C2
Чертежи












































Описание
Настоящая заявка испрашивает приоритет предварительной заявки США с серийным номером 60/807358, поданной 14 июля 2006 года. Указанная заявка включена в настоящую заявку в виде ссылки во всей своей полноте для всех ее целей.
Уровень техники
В обществе и экономике возрастает острая потребность в развитии возобновляемых источников энергии, а также возобновляемых и биоразлагаемых промышленных и потребительских продуктов и материалов. Каталитическая конверсия от природных сырьевых материалов до экономически оправданных продуктов привела к новым подходам и технологиям, применение которых распространяется в традиционных секторах экономики. Актуальным является производство биотоплива, которое может быть представлено как обработка сырьевых материалов сельскохозяйственного и лесотехнического происхождения, приобретающая все возрастающее значение в плане переработки указанных материалов в многообразные продукты, включающие базисное промышленное химическое сырье, топлива и потребительские продукты. Ранее была досконально исследована конверсия твердого жира и других органических масел в биодизельное топливо. Традиционно эта конверсия включает переэтерификацию триглицерида с образованием трех метиловых сложных эфиров жирных кислот и молекулы свободного глицерина. Также были обстоятельно исследованы химические, реологические свойства и характеристики процессов сгорания полученного «биодизеля». К сожалению, было показано, что эти топлива на основе метиловых сложных эфиров гораздо более чувствительны к окислению и имеют более низкую теплотворную способность, чем традиционные дизельные топлива на основе нефти. В результате этого нужно смешивать традиционные биодизельные топлива с существующим дизельным ассортиментом, и может быть также необходимость введения добавок антиоксидантов для продления срока годности и во избежание формирования отложений в топливных баках, топливных системах и фильтрах.
Если этерификацию с образованием метиловых сложных эфиров можно рассматривать как четко контролируемую реакцию, то относительно грубой альтернативой, которая ранее применялась в промышленности, является пиролиз. Пиролиз включает применение термической обработки сырья сельскохозяйственного происхождения с образованием жидкого топливного продукта. Большинство литературных источников описывает употребление необработанных сельскохозяйственных сырьевых материалов для получения экономически оправданного топлива. Многие различные подходы к пиролизу как механизму получения жидкого топлива были описаны в литературе и относятся к различным режимам, включающим флэш-пиролиз, медленный и быстрый пиролиз. Ранее был изучен пиролиз многообразных сельскохозяйственных продуктов при этих различных режимах, включая касторовое масло, древесину хвойных пород, сорго сахарное и рапс. В зависимости от применяемых условий, включая используемую температуру, время пребывания и чистоту субстрата, качественный состав продуктов варьирует в плане соотношения между парообразными, жидкими и остаточными твердыми веществами (древесный уголь).
Одно из немногих исследований, посвященных пиролизу жирных кислот вместо триглицеридов или более сложных субстратов, было сосредоточено на пиролизе соли жирной кислоты. Условия, использованные в исследовании, были такими, что однородный продукт декарбоксилирования не образовывался. Вместо этого получалась смесь углеводородных продуктов разложения, которая не была идентифицирована авторами. В общем, в литературе нет четкого понимания процесса декарбоксилирования карбоновых кислот, которые не содержат других взаимодействующих функциональных групп, при высоких температуре и давлении. Достижение более глубокого фундаментального понимания химических основ и методологий, необходимых для стимулирования реакции декарбоксилирования жирных кислот, или реакций крекинга, до более мелких молекул алканов и алкенов, может обеспечить возможность дальнейшего развития новых технологий получения топлива и растворителей. В одном аспекте здесь описывается термическая обработка протонированных свободных жирных кислот в бескислородных условиях. Процессы этого характера являются перспективными для производства топлива более высокого качества, чем традиционные биодизельные топлива, и, кроме того, в принципе могли бы давать более высокие выходы желательных продуктов, чем пиролиз.
Сущность изобретения
Здесь описаны способы получения топлив и растворителей из источников жирных кислот. Также здесь представлены топлива и растворители, полученные описываемыми здесь способами. Преимущества материалов, способов и изделий, описанных здесь, будут отчасти изложены в нижеприведенном описании, или могут быть выявлены при осуществлении нижеописанных аспектов. Нижеописанные преимущества будут реализованы и достигнуты с помощью элементов и комбинаций, конкретно указанных в прилагаемых пунктах формулы изобретения. Должно быть понятно, что как вышеприведенное общее описание, так и последующее подробное описание являются только показательными, предназначенными для объяснения, и не являются ограничительными.
Краткое описание фигур
Сопроводительные фигуры, которые включены в настоящее описание и являются его частью, иллюстрируют некоторые аспекты, описанные ниже.
Фигура 1 показывает компоновку (а) песчаной бани и системы продувки и (b) микрореактора.
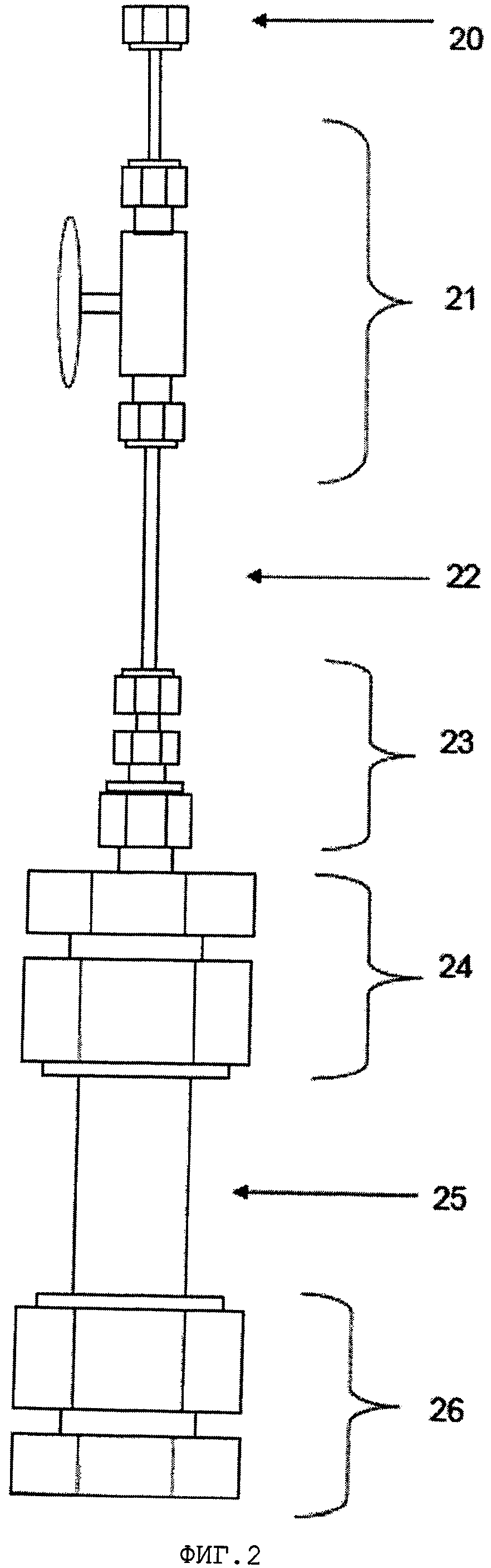
Фигура 2 показывает схему конструкции закрытого микрореактора с трубным штуцером размера 1/8′′ для присоединения микрореактора к системе продувки (20); игольчатым клапаном размера 1/8′′, соединенным с трубными штуцерами размера 1/8′′ (21); трубкой размера 1/8′′ (22); переходной муфтой с размера 1/4′′ на размер 1/8′′ (23); крышкой реактора с арматурой из гайки и втулки с отверстием размера 3/8′′ (24); трубчатым корпусом размера 3/8′′ (25) и донной частью реактора с арматурой из гайки и втулки с отверстием размера 3/8′′ (26).
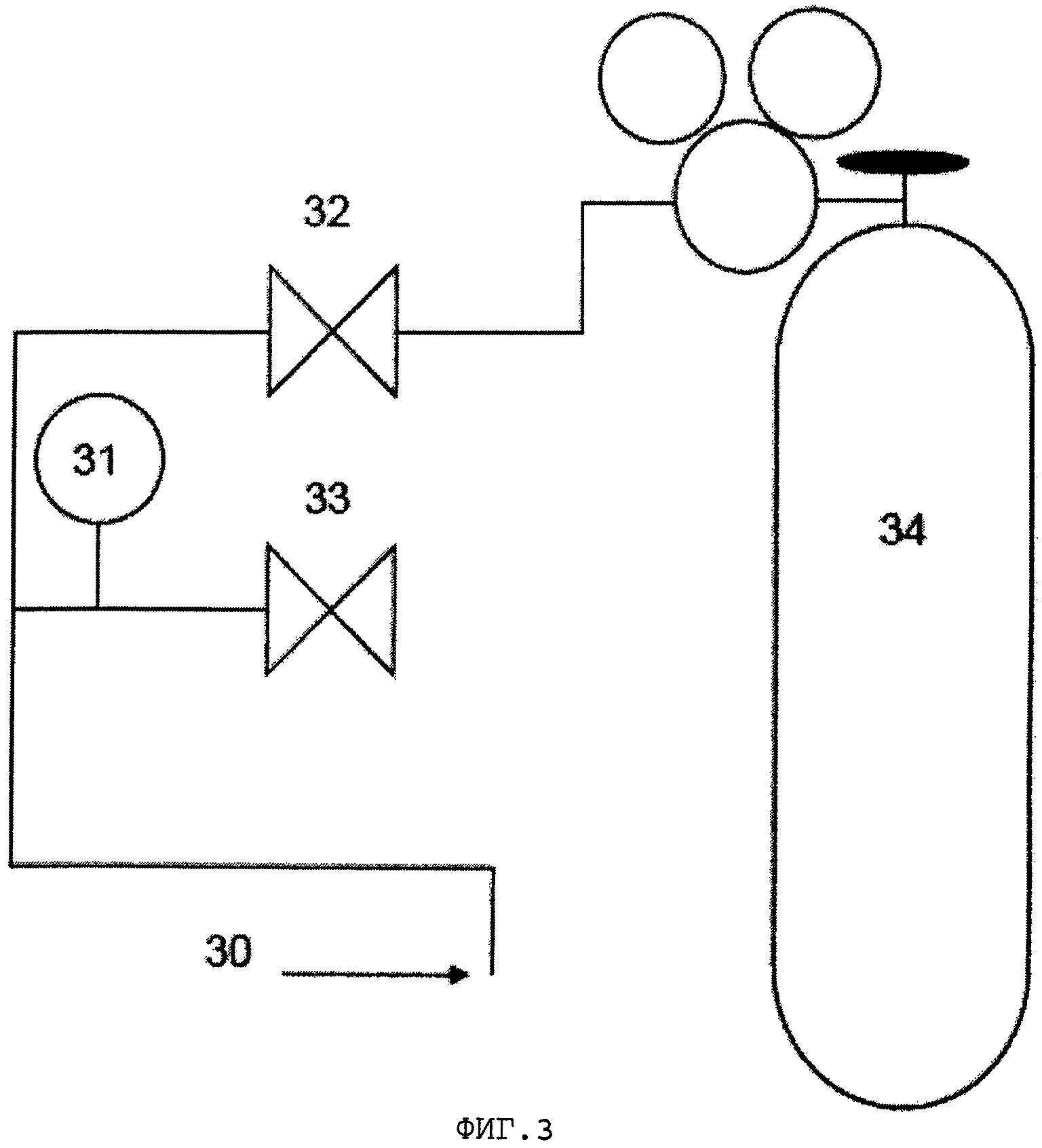
Фигура 3 показывает схему компоновки системы продувки микрореактора с микрореактором (30); запорным клапаном (31); первым клапаном (32); вторым клапаном (33) и баллоном азота (34).
Фигура 4 показывает систему песчаной бани с водяной баней (40); штангой (41); направляющей (42); мотором и колесом (43); системой продувки (44); регулятором температуры TC-8D (45) и песчаной баней SBS-4 (46).
Фигура 5 показывает схему компоновки песчаной бани Techne SBS-4 с отверстием для воздуха (50); термопарой (51); нагревательным элементом (52) и пористой пластиной (53).
Фигура 6 показывает схему компоновки модифицированного реактора для измерения внутренней температуры в реакторе.
Фигура 7 показывает схему компоновки модифицированного реактора для измерения внутреннего давления в реакторе.
Фигура 8 показывает температурный профиль GC-FID (газовая хроматография с пламенно-ионизационным детектором) для анализа жидкости.
Фигура 9 показывает температурный профиль GC-TCD (газовая хроматография с детектором по теплопроводности (катарометром)) для анализа газа.
Фигура 10 показывает внутреннюю температуру в реакторе во время пиролиза стеариновой кислоты как функцию времени для заданных температур настройки регулятора температуры при 370ºC, 410ºC и 450ºC, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 11 показывает GC-FID-хроматограмму растворимых в пентане продуктов пиролиза стеариновой кислоты после 30-минутных продолжительностей реакций при температурах между 350ºC и 500ºC, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 12 показывает GC-FID-хроматограмму растворимых в пентане продуктов пиролиза стеариновой кислоты после 5-минутных продолжительностей реакций при температурах между 400ºC и 550ºC, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 13 показывает идентификацию типичной серии сигналов, образованной после нагревания стеариновой кислоты в течение 5 минут при температуре 500ºC (хроматограмма на фигуре 12), где реакция проводилась в атмосфере N2 и первоначально была при атмосферном давлении.
Фигура 14 показывает типичные растворимые в пентане продукты пиролиза стеариновой кислоты после 5 минут при температуре 500ºC, подтвержденные экспериментом с внешними стандартами, где реакция проводилась в атмосфере N2 и первоначально была при атмосферном давлении.
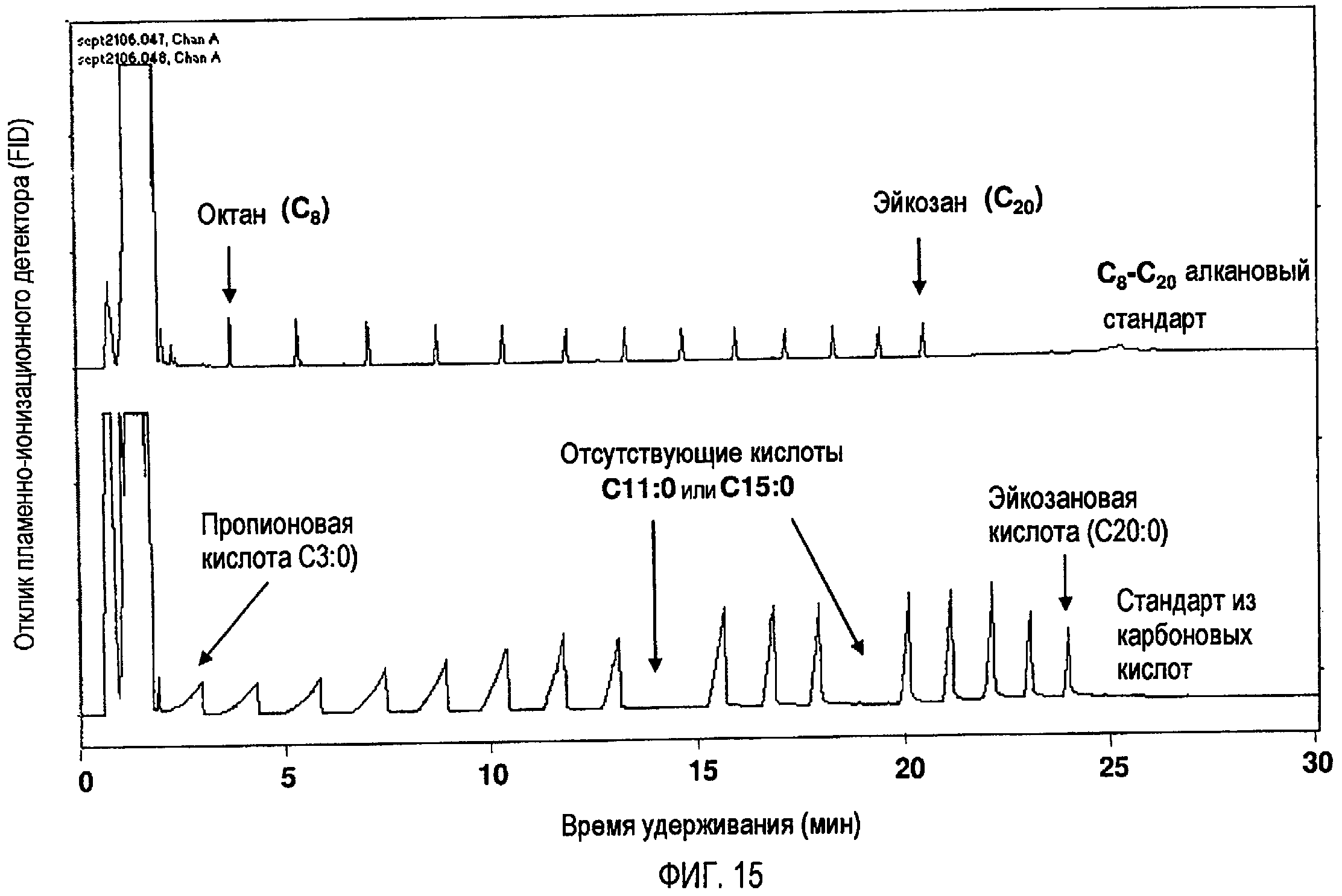
Фигура 15 представляет собой GC-FID-хроматограмму, показывающую эксперимент с внешними стандартами для подтверждения продуктов пиролиза, где стандарты представляли собой (1) приобретенную смесь С8-С20-алканов и (2) стандарт из карбоновых кислот.
Фигура 16 представляет собой GC-FID-хроматограмму, показывающую продукты пиролиза при температуре 400ºC в течение 5 минут в пентане (первая экстракция) и толуоле (вторая экстракция), где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 17 представляет собой GC-FID-хроматограмму, показывающую продукты пиролиза при температуре 450ºC в течение 5 минут в пентане (первая экстракция) и толуоле (вторая экстракция), где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 18 представляет собой GC-FID-хроматограмму, показывающую различие в распределении продуктов перед (а) и после высушивания и повторного суспендирования (b) продуктов пиролиза стеариновой кислоты для 1-часовых реакций, проведенных при температуре 450ºC, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
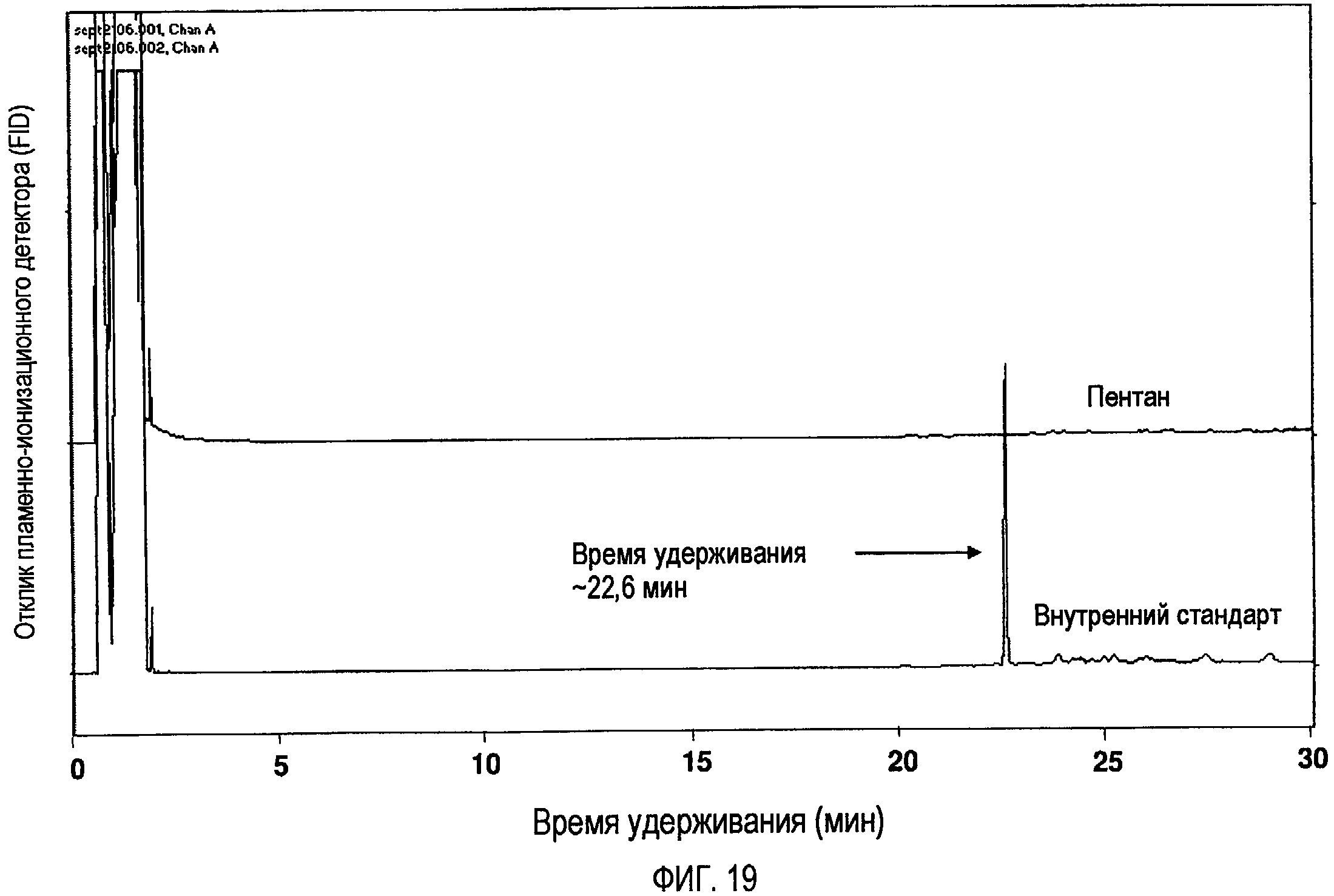
Фигура 19 представляет собой GC-FID-хроматограмму, показывающую растворитель для экстракции, пентан и раствор внутреннего стандарта (метилового эфира нонадекановой кислоты в пентане).
Фигура 20 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=350ºС и времени t=4 и 8 часов, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 21 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=370ºС и времени t=1-8 часов, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
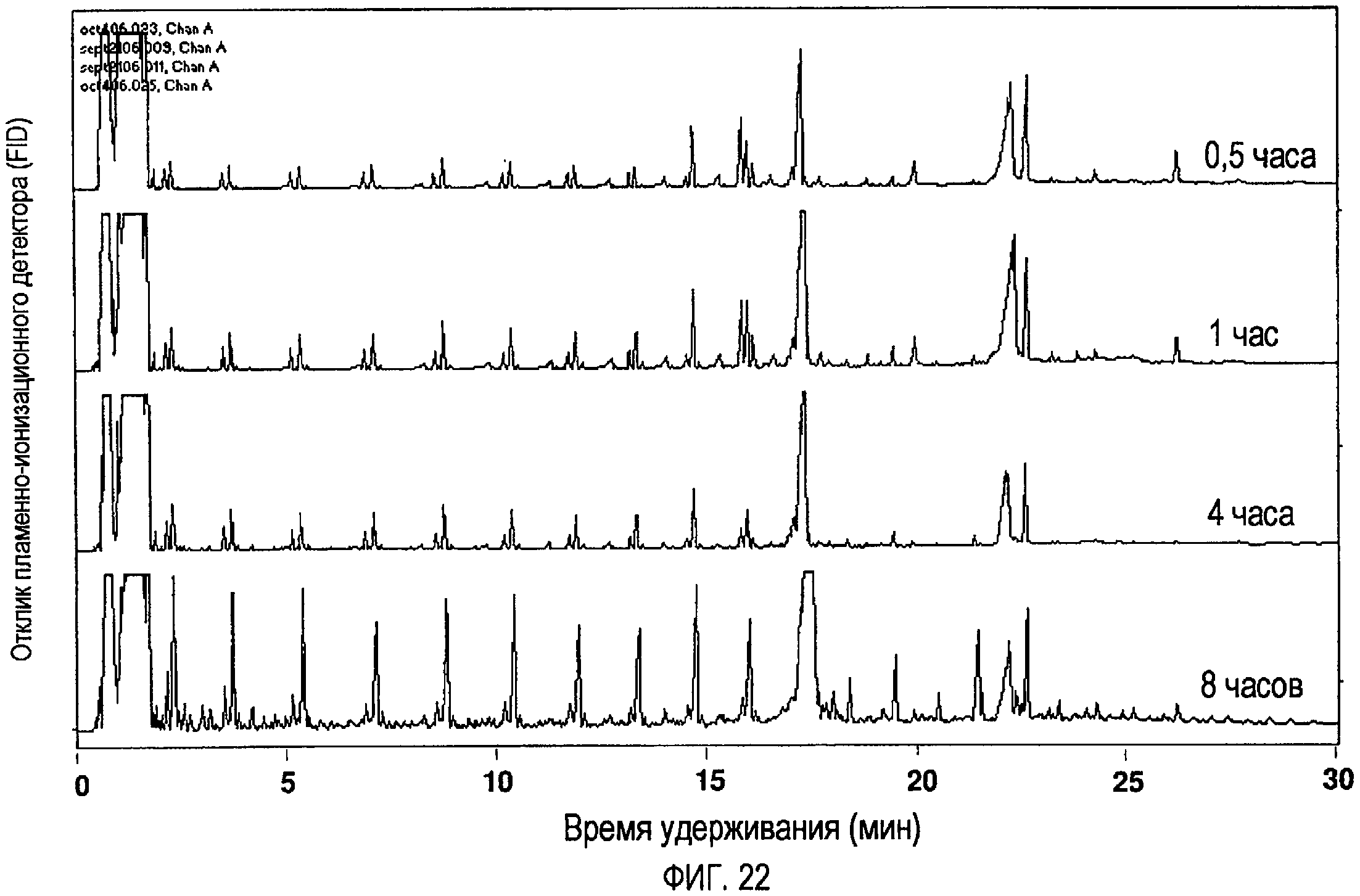
Фигура 22 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=390ºС и времени t=0,5-8 часов, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 23 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=410ºС и времени t=0,5-8 часов, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 24 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=430ºС и времени t=0,5-8 часов, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 25 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=450ºС и времени t=0,5-8 часов, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 26 представляет собой GC-FID-хроматограмму, показывающую растворимые в пентане продукты пиролиза стеариновой кислоты из реакционной смеси при температуре Т=500ºС и времени t=0,5-4 часа, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 27 показывает процентные доли образовавшихся С8-С20-алканов в зависимости от температуры и времени.
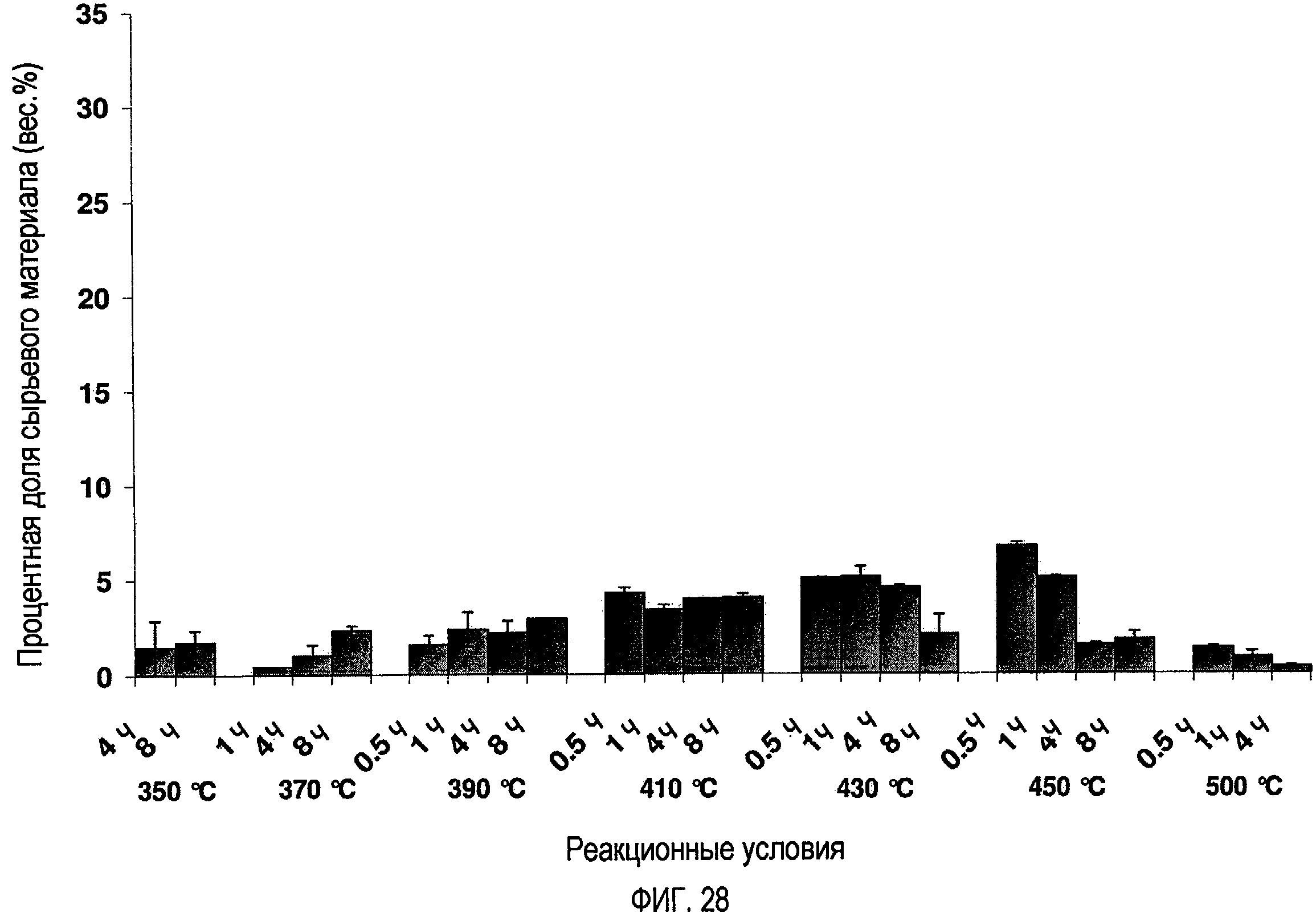
Фигура 28 показывает процентные доли образовавшихся С8-С20-алкенов в зависимости от температуры и времени.
Фигура 29 показывает молярные выходы С8-С20-алканов в зависимости от температуры для реакций продолжительностью 0,5 часа.
Фигура 30 показывает молярные выходы С8-С20-алканов в зависимости от температуры для реакций продолжительностью 1 час.
Фигура 31 показывает молярные выходы С8-С20-алканов в зависимости от температуры для реакций продолжительностью 4 часа.
Фигура 32 показывает молярные выходы С8-С20-алканов в зависимости от температуры для реакций продолжительностью 8 часов.
Фигура 33 показывает молярное отношение алканов к алкенам в зависимости от числа атомов углерода и продолжительности реакции при температуре Т=390ºС.
Фигура 34 показывает молярное отношение алканов к алкенам в зависимости от числа атомов углерода и продолжительности реакции при температуре Т=410ºС.
Фигура 35 показывает молярное отношение алканов к алкенам в зависимости от числа атомов углерода и продолжительности реакции при температуре Т=430ºС.
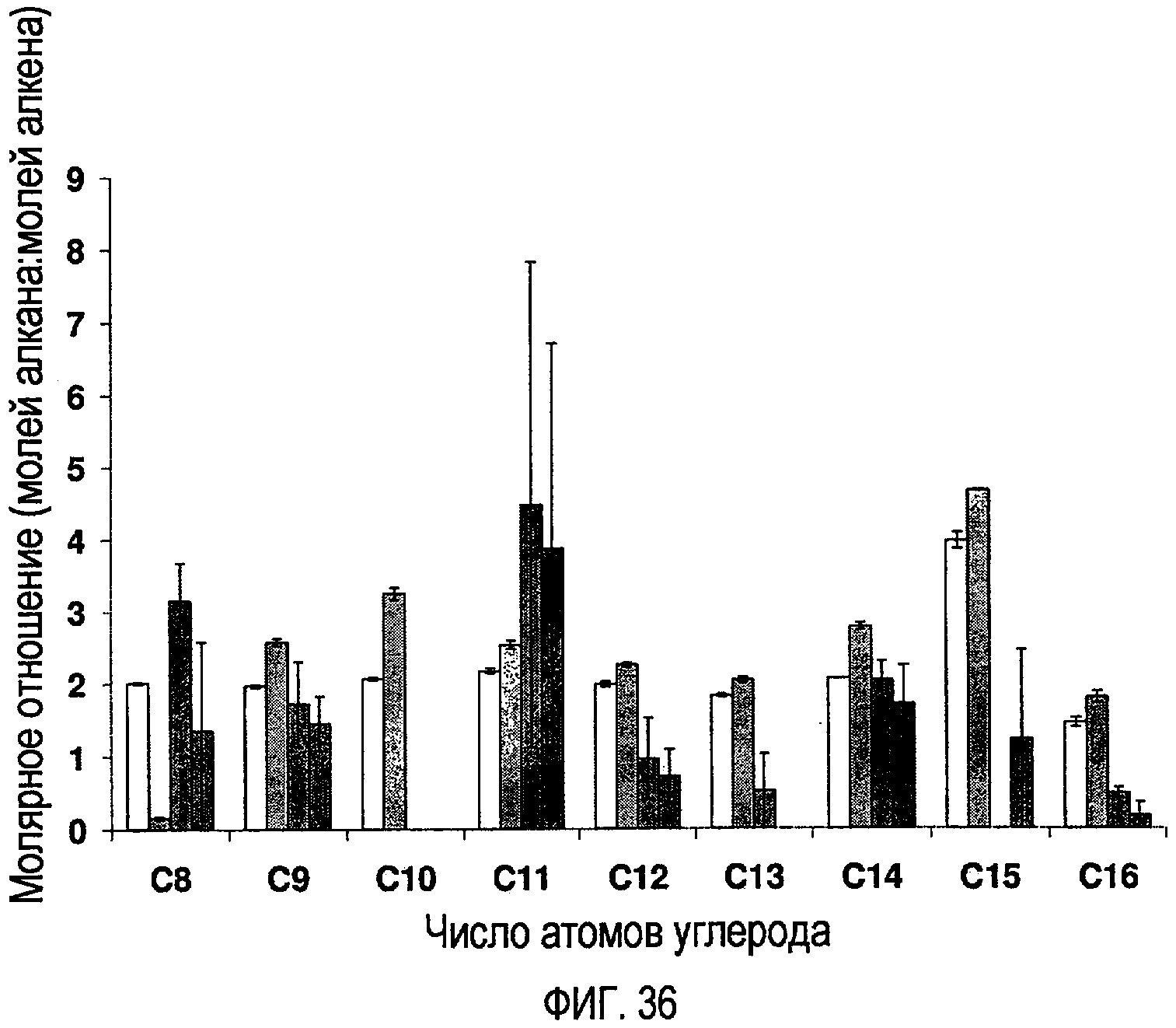
Фигура 36 показывает молярное отношение алканов к алкенам в зависимости от числа атомов углерода и продолжительности реакции при температуре Т=450ºС.
Фигура 37 показывает молярное отношение алканов к алкенам для С17 как функцию температуры и времени.
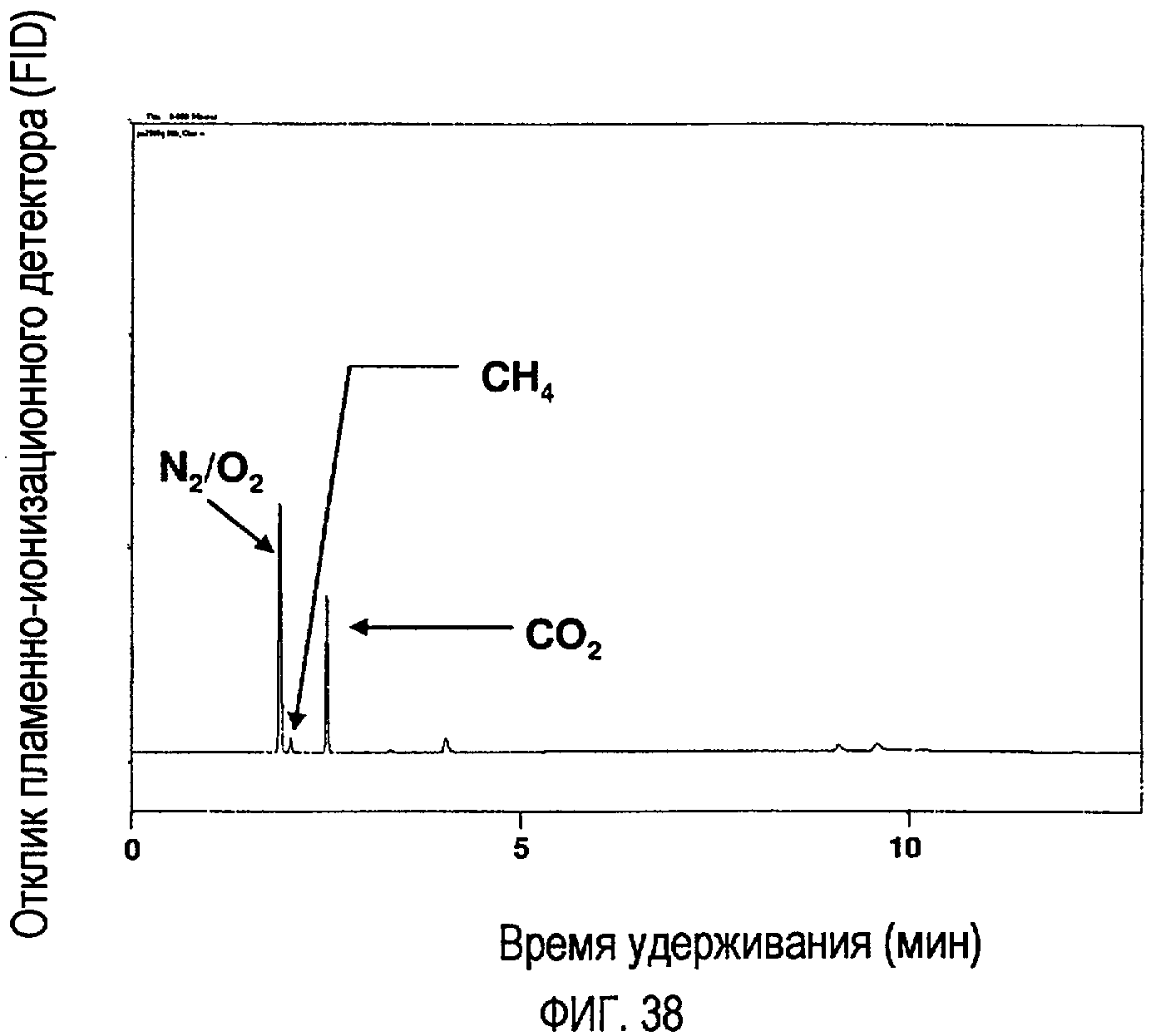
Фигура 38 показывает состав типичной газовой смеси из пиролиза стеариновой кислоты из реакции в течение 1 часа при температуре 410ºС по данным GC-TCD-анализа.
Фигура 39 показывает метан (СН4), диоксид углерода (СО2) и воздух в качестве стандартов, по данным GC-TCD-анализа.
Фигура 40 показывает процентные доли газообразных продуктов, образованных во время реакций пиролиза стеариновой кислоты в течение 1 часа, в зависимости от температуры, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
Фигура 41 показывает процентные доли жидких продуктов, образованных во время пиролиза стеариновой кислоты, в зависимости от температуры и времени, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
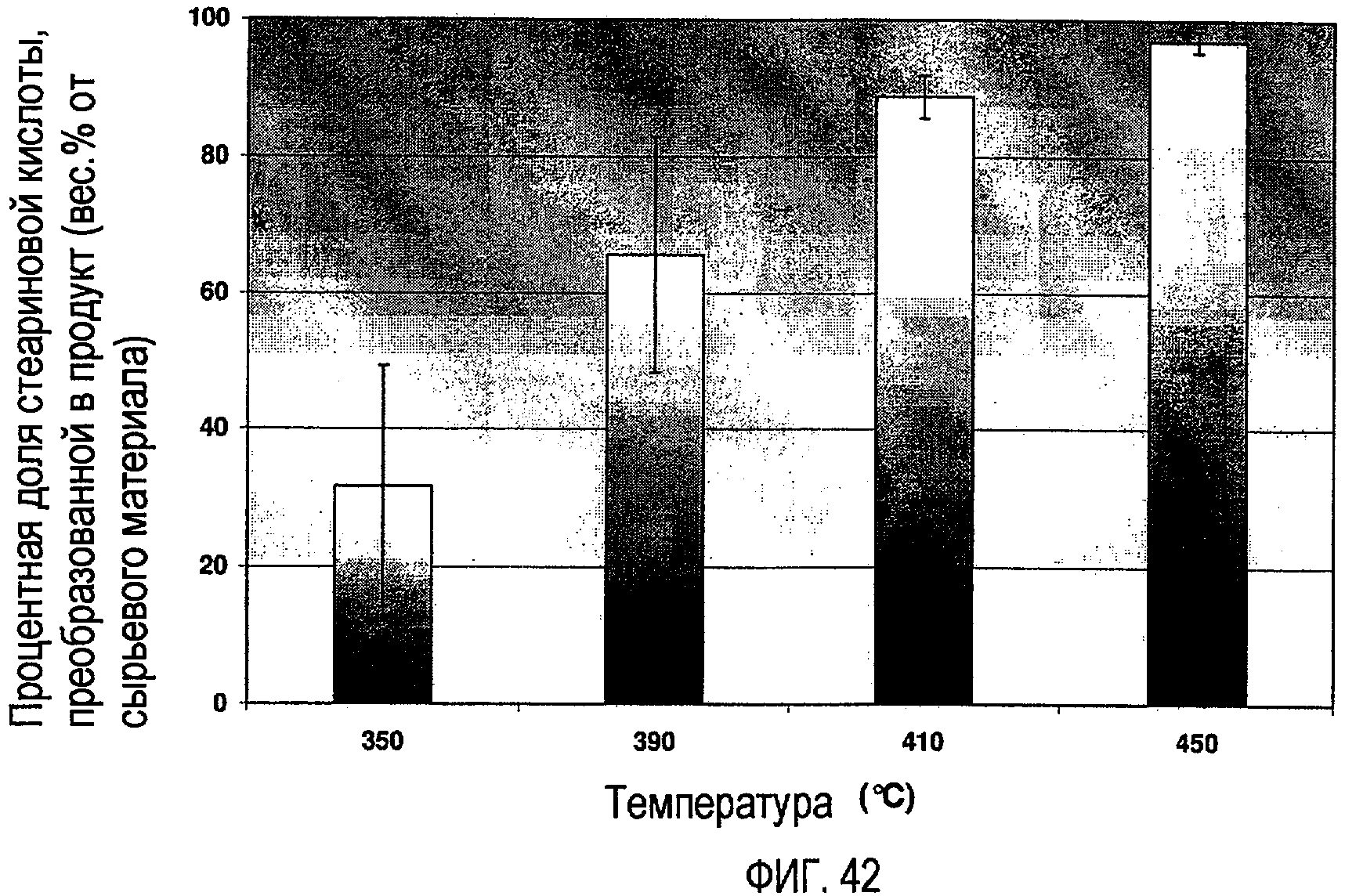
Фигура 42 показывает процентные доли начальной сырьевой стеариновой кислоты, которая была преобразована во время реакций пиролиза в течение 1 часа, в зависимости от температуры, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
Фигура 43 представляет собой хроматограмму (GC-FID), показывающую продукты пиролиза стеариновой кислоты после 4-часовой реакции при температуре 255ºС, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
Фигура 44 представляет собой хроматограмму (GC-TCD), показывающую газообразные продукты реакции после 4-часовой реакции при температуре 255ºС, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
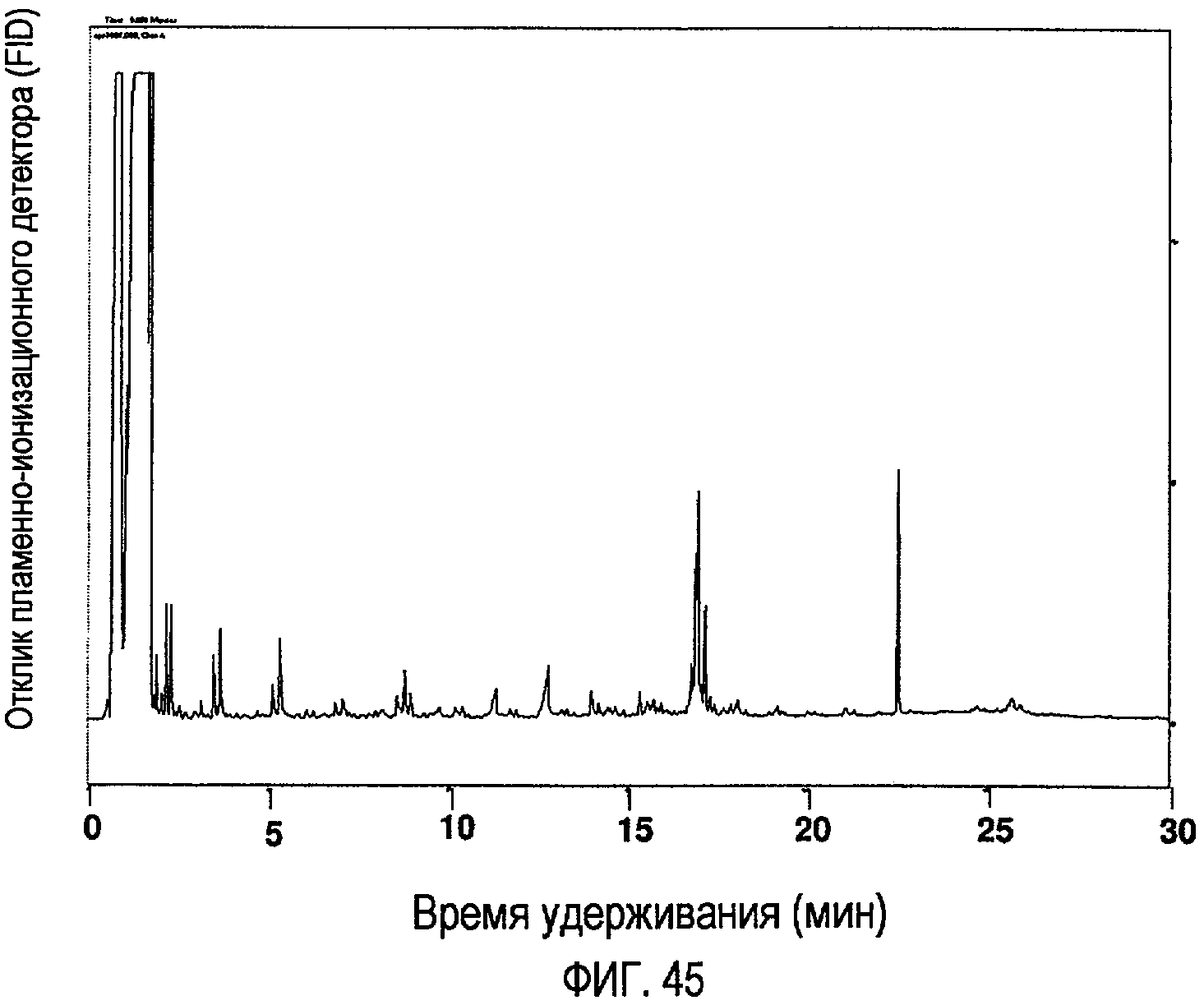
Фигура 45 представляет собой хроматограмму (GC-FID), показывающую продукты пиролиза олеиновой кислоты после 1-часовой реакции при температуре 410ºС, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
Фигура 46 показывает главные продукты пиролиза олеиновой кислоты после 1 часа при температуре 410ºС.
Фигура 47 представляет собой дублирующие хроматограммы (GC-TCD), показывающие газообразные продукты из пиролиза олеиновой кислоты после 1 часа при температуре 410ºС, где начальное давление было атмосферным, и реакции проводились в атмосфере N2.
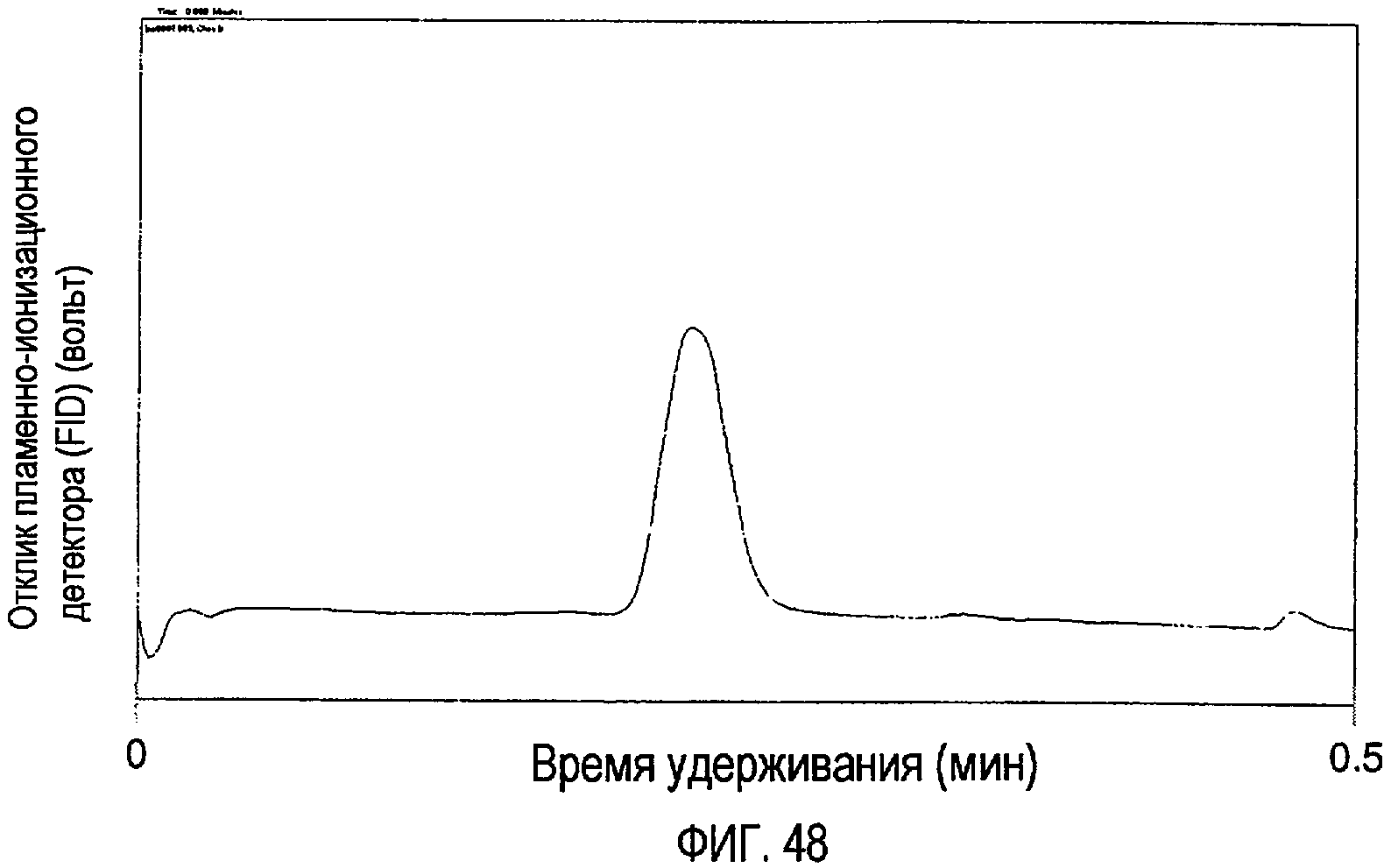
Фигура 48 представляет собой хроматограмму, показывающую гидролизаты масла канола.
Фигура 49 представляет собой TLC-FID-хроматограмму (тонкослойная хроматография с пламенно-ионизационным детектором), показывающую гидролизаты говяжьего жира “Bleached Fancy”.
Фигура 50 представляет собой TLC-FID-хроматограмму, показывающую стандартную смесь производных олеиновой кислоты.
Фигура 51 представляет собой TLC-FID-хроматограмму, показывающую стандартную смесь производных олеиновой кислоты с добавленными гидролизатами говяжьего жира “Bleached Fancy” (смесь «стандарт:образец» в объемном отношении 1:1).
Фигура 52 представляет собой TLC-FID-хроматограмму, показывающую стандартную смесь производных олеиновой кислоты с добавленными гидролизатами говяжьего жира “Bleached Fancy” (смесь «стандарт:образец» в объемном отношении 2:1).
Фигура 53 представляет собой GC-FID-хроматограмму, показывающую продукты пиролиза жира домашней птицы из 4-часовой реакции при температуре 410ºС, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 54 представляет собой GC-FID-хроматограмму, показывающую продукты пиролиза жира домашней птицы из 4-часовой реакции при температуре 410ºС после стадии водной экстракции, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 55 представляет собой GC-FID-хроматограмму, показывающую продукты пиролиза масла канола из 1-часовой реакции при температуре 410ºС, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 56 представляет собой GC-FID-хроматограмму, показывающую продукты пиролиза говяжьего жира “Bleached Fancy” из 1-часовой реакции при температуре 390ºС, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 57 представляет собой GC-FID-хроматограмму, показывающую гидролизаты говяжьего жира “Bleached Fancy” из 1-часовой реакции при температуре 410ºС, растворенные в пентане, где реакции проводились в атмосфере N2 и первоначально были при атмосферном давлении.
Фигура 58 представляет собой GC/MS-хроматограмму (газовая хроматография/масс-спектрометрия), показывающую дериватизированные и недериватизированные образцы продуктов пиролиза говяжьего жира “Bleached Fancy” после 1-часовой реакции при температуре 410ºС.
Фигура 59 представляет собой GC/MS-хроматограмму, показывающую дериватизированные образцы продуктов пиролиза говяжьего жира “Bleached Fancy” после 1-часовой реакции при температуре 410ºС.
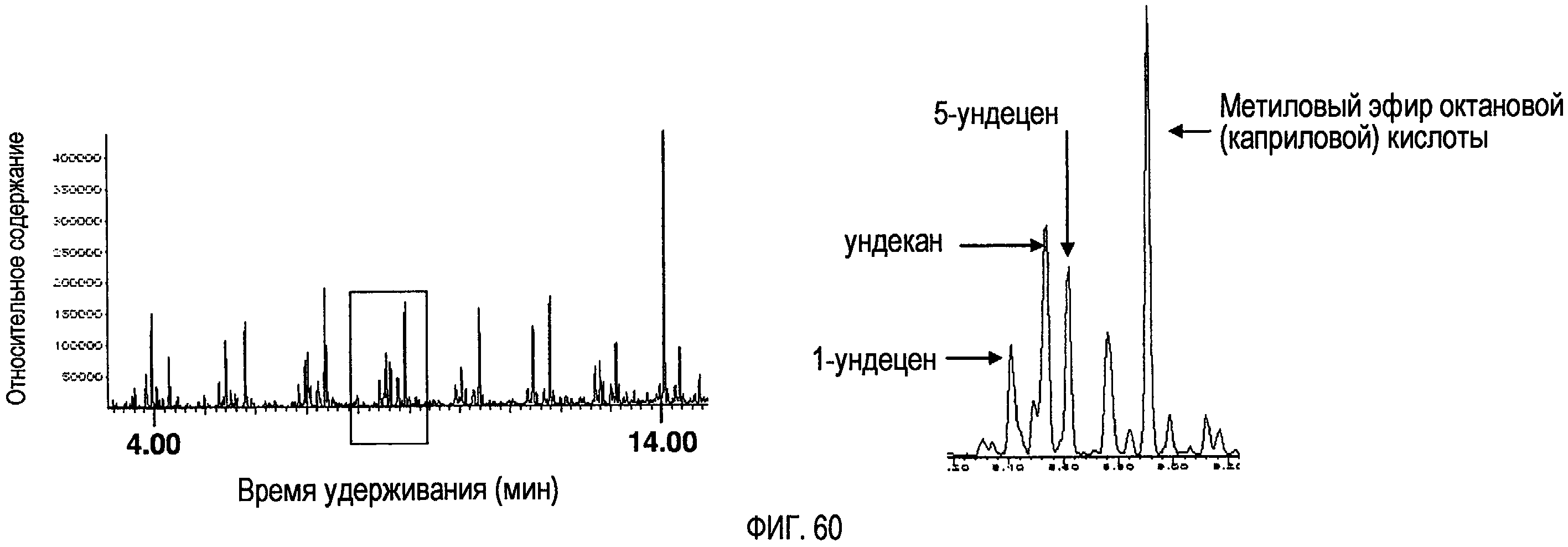
Фигура 60 представляет собой увеличенную область GC/MS-хроматограммы, показывающей дериватизированные образцы продуктов пиролиза говяжьего жира “Bleached Fancy” после 1-часовой реакции при температуре 410ºС.
Подробное описание изобретения
Прежде чем будут представлены и описаны настоящие материалы, изделия и/или способы, должно быть понятно, что нижеописанные аспекты не ограничиваются конкретными соединениями, синтетическими методами или их применением, поскольку они, конечно, могут варьироваться. Также должно быть понятно, что используемая здесь терминология предназначена только для цели описания конкретных аспектов и не предполагает ограничения.
В настоящем описании и нижеследующих пунктах формулы изобретения будет сделана ссылка на ряд терминов, которые должны быть определены как имеющие следующие значения.
На всем протяжении описания, если контекст не предусматривает иного, слово «включать», или его варианты, такие как «включает» или «включающий», будут пониматься как подразумевающие включение указанного целого числа, или стадии, или группы целых чисел или стадий, но не исключение любого другого целого числа, или стадии, или группы целых чисел или стадий.
Следует отметить, что применяемые в описании и прилагаемых пунктах формулы изобретения формы единственного числа включают множественные объекты обсуждения, если контекст ясно не оговаривает иного. Так, например, ссылка на «масло» включает масло в единственном числе или смеси двух или более масел.
«Необязательный» или «необязательно» означает, что описываемое потом событие или обстоятельство может происходить или может не происходить, и что описание включает примеры, где событие или обстоятельство имеет место, и примеры, где оно не имеет места.
Здесь описываются способы получения топлив и растворителей из источников жирных кислот. В одном аспекте способ включает:
а) выделение одной или более жирных кислот из источника жирных кислот; и
b) преобразование жирной кислоты в один или более алканов, алкенов или их смесь.
Термин «источник жирных кислот», определенный здесь, представляет собой любой источник свободной жирной кислоты или прекурсор свободной жирной кислоты с последующей обработкой. Например, триглицерид является прекурсором свободной жирной кислоты, где гидролиз функциональных групп глицерина дает свободную жирную кислоту. Примеры источников жирных кислот включают, но не ограничиваются ими, растительное масло, животные жиры, использованное масло для жарки, липиды, фосфолипиды, сырье для производства мыла или прочие источники триглицеридов, диглицеридов или моноглицеридов. В одном аспекте растительное масло включает кукурузное масло, хлопковое масло, масло канола, рапсовое масло, оливковое масло, пальмовое масло, арахисовое масло, масло земляного ореха, сафлоровое масло, кунжутное масло, соевое масло, подсолнечное масло, масло из водорослей, миндальное масло, абрикосовое масло, аргановое масло, масло авокадо, масло бенового дерева, масло кешью, касторовое масло, масло из виноградных косточек, масло лесного ореха, конопляное масло, льняное масло, горчичное масло, масло из нима, косточковое пальмовое масло, масло из тыквенных семян, масло из рисовых отрубей, масло грецкого ореха, их комбинацию. В еще одном аспекте животный жир включает ворвань, рыбий жир, топленое масло, свиное сало, твердый жир, их производные (например, солидол, использованное масло для жарки и т.д.), или их комбинацию.
Предполагается, что источник жирных кислот может быть дополнительно очищен перед стадией выделения (а). Например, источник жирных кислот может быть подвергнут перегонке или экстрагированию для удаления любых нежелательных примесей. Альтернативно, источник жирных кислот может быть использован как есть и введен в стадию выделения (а). Происхождение источника жирных кислот будет определяющим фактором в том, потребуются ли какие-нибудь стадии предварительной очистки.
Стадия выделения (а) включает удаление или выделение одной или более жирных кислот из источника жирных кислот. В технологии известен ряд различных способов выделения и очистки жирных кислот. Например, патент США № 5917501 представляет способ выделения жирных кислот. Способ включает гидролиз смеси липидов природного происхождения, содержащей фосфолипиды, триглицериды и стерины, с образованием двухфазного продукта, содержащего фазу жирных кислот, состоящую из свободных жирных кислот и стеринов, и водную фазу, включающую воду, глицерин и сложные эфиры глицерина и фосфорной кислоты. Водную фазу отделяют от фазы жирных кислот, и сырую фазу жирных кислот нагревают для преобразования свободных стеринов в сложные эфиры стеринов и жирных кислот. Свободные жирные кислоты отгоняют из сложных эфиров стеринов и жирных кислот с образованием очищенных жирных кислот, которые не содержат холестерина и других стеринов, и фосфорсодержащих соединений. В других аспектах источник жирных кислот подвергают действию кислоты, чтобы гидролизовать прекурсор жирных кислот, присутствующий в источнике жирных кислот, для получения соответствующей жирной кислоты. Например, растительные масла богаты триглицеридами, которые при кислотном гидролизе образуют свободную жирную кислоту и глицерин.
После стадии выделения желательно получить жирную кислоту в чистой или по существу чистой форме. Фраза «по существу чистый», применяемая здесь, определяется как содержание жирной кислоты больше чем 90% по весу. Присутствие примесей может оказывать вредное влияние на конечный состав топлива или растворителя. Например, если в жирной кислоте перед стадией (b) присутствуют серосодержащие, кислородсодержащие или азотсодержащие соединения, проявляются нежелательные характеристики продукта, в том числе высокие уровни выбросов серы или азота во время сгорания, или могут происходить побочные реакции во время стадии (b), такие как образование нежелательных ароматических соединений.
Природа жирной кислоты будет варьировать в зависимости от источника жирной кислоты. Жирная кислота может представлять собой насыщенную жирную кислоту, ненасыщенную жирную кислоту или их комбинацию. Примеры жирных кислот включают, но не ограничиваются ими, масляную кислоту, лауриновую кислоту, миристиновую кислоту, пальмитиновую кислоту, стеариновую кислоту, арахиновую кислоту, альфа-линоленовую кислоту, докозагексаеновую кислоту, эйкозапентаеновую кислоту, линолевую кислоту, арахидоновую кислоту, олеиновую кислоту, эруковую кислоту, жирную кислоту природного происхождения из растительного или животного источника или их комбинацию. Предполагается, что жирная кислота может быть свободной кислотой или солью/сложным эфиром таковой. Жирная кислота также может быть смесью жирных кислот.
Вторая стадия включает преобразование жирной(ых) кислоты(кислот) в один или более алканов, алкенов или смеси таковых. В общем, во время стадии конверсии жирные кислоты подвергаются декарбоксилированию и крекингу с образованием СО2 и алканов и алкенов. Длина цепи алкана или алкена будет варьировать в зависимости от природы жирной кислоты и параметров реакции, которые будут подробно обсуждены ниже. В общем алканы и алкены представляют собой углеводороды с длиной цепи от С1 до С20. Например, декарбоксилирование стеариновой кислоты, которая имеет формулу СН3(СН2)16СООН, дает СН3(СН2)15СН3, более короткие алканы и алкены и СО2.
В одном аспекте конверсия жирной кислоты в алкан и/или алкен включает нагревание жирной кислоты для преобразования всей или по существу всей жирной кислоты в алкан, алкен или смесь таковых. Температура стадии нагревания может варьировать в пределах различных параметров. В одном аспекте температура стадии нагревания составляет от 220ºС до 650ºС, от 300ºС до 650ºС, от 350ºС до 650ºС, от 350ºС до 600ºС или от 250ºС до 500ºС. Другими рассматриваемыми параметрами являются продолжительность стадии нагревания и давление, при котором проводится стадия нагревания. Давление может варьировать от атмосферного давления до 2000 фунт/кв.дюйм (13,78 МПа), и продолжительность стадии нагревания может составлять от нескольких секунд до 12 часов. В одном аспекте стадия нагревания протекает от двух секунд до 8 часов. В еще одном аспекте стадия нагревания выполняется в инертной атмосфере, например, такой как азот или аргон.
Путем варьирования реакционных условий во время преобразования жирной кислоты в алкан/алкен специалист обычной квалификации в этой области технологии может получать коротко- или длинноцепочечные алканы/алкены для топлив и растворителей. Например, при более продолжительном нагревании при повышенных температурах могут образовываться короткоцепочечные алканы/алкены, которые могут быть полезными в качестве топлив. Альтернативно, длинноцепочечные алканы/алкены могут быть получены специалистом обычной квалификации в этой области технологии при сокращении времени нагревания и снижении температуры. Если получаются короткоцепочечные алканы или алкены, условия реакции могут регулироваться так, что эти продукты представляют собой газы (например, метан, пропан, бутан и т.д.), которые могут быть легко выведены из реактора.
В еще одном аспекте для облегчения конверсии жирной кислоты в алкан или алкен может быть предусмотрено применение катализатора декарбоксилирования. В зависимости от выбора катализатора декарбоксилирования, катализатор может снижать температуру и продолжительность нагревания. Это желательно в некоторых примерах, в особенности, если нужно избежать разложения алкана/алкена или побочных реакций (например, ароматизации). Примеры катализаторов декарбоксилирования включают, но ими не ограничиваются, катализаторы на основе активированного оксида алюминия.
Стадии (а) и/или (b) могут быть выполнены в периодическом, полупериодическом или непрерывном режимах работы. Например, в отношении стадии (b), система реактора непрерывного действия с рециркуляцией непрореагировавшей кислоты может быть использована для повышения выхода желательного алкана/алкена путем ограничения продолжительности пребывания алкана/алкена в высокотемпературном реакторе. Диоксид углерода и низкомолекулярные углеводородные продукты могут быть извлечены с газофазными углеводородами, используемыми в качестве топлива для реактора или прочих вариантов употребления. Когда применяется система реактора непрерывного действия, условия процесса могут быть оптимизированы для сведения к минимуму реакционных температур и времен пребывания, чтобы максимизировать выходы и состав продуктов. Поскольку условия реакции могут быть отрегулированы для подбора предпочтительной длины углеродной цепи (длинной, короткой или средней), технология дает возможность обогащения продуктами конкретной группы. Из этих групп индивидуальные химические соединения могут быть выделены, очищены и проданы как чистые базисные промышленные сырьевые химикаты.
Описываемые здесь способы представляют многообразные преимущества перед современными способами получения биотоплив. Как указано выше, описываемые здесь способы могут быть использованы для получения либо растворителей, либо топлив, которые подобны традиционному дизельному топливу. В способах используют возобновляемые источники для создания экологически рационального источника топлива на не нефтяной основе, не содержащего ароматических соединений. Образуемые продукты являются химически гораздо более однородными, чем продукты других высокотемпературных процессов, применяемых в настоящее время. Например, топлива или растворители, полученные здесь, по существу не содержат ароматических соединений, где термин «по существу не содержит» определяется как содержащий менее чем 5% по весу ароматических соединений. Также предполагается, что в топливах или растворителях не присутствуют ароматические соединения. Ожидается, что описываемые здесь способы будут обеспечивать более высокие выходы продуктов, чем прочие технологии пиролиза, и позволят производить топливо, гораздо более подобное дизельному, чем биодизель. Продукты не будут создавать проблем, характерных для биодизельного топлива, в том плане, что они будут более устойчивыми к окислению и будут иметь температуры застывания, подобные таковым для общеупотребительного дизельного топлива. Наконец, ожидается, что при использовании описываемых здесь способов производственные расходы будут более низкими по сравнению с конкурирующими существующими технологиями получения биодизельного топлива. В частности, процесс не требует стадии гидрирования для получения углеводородов, которая существенно увеличивает стоимость процесса.
ПРИМЕРЫ
Нижеследующие примеры представлены так, чтобы обеспечить рядовым специалистам в данной области техники полное раскрытие и описание, как получают и оценивают материалы, изделия и способы, описанные и заявленные в настоящем документе, и предназначены быть исключительно в качестве примера и не предполагают ограничения области, которая, по мнению авторов настоящего изобретения, входит в объем изобретения. Были предприняты усилия для обеспечения точности в отношении численных значений (например, количеств, температуры и т.д.), но следует учитывать некоторые ошибки и погрешности. Если не оговорено иное, части представляют собой части по весу, температура указана в ºC или является температурой окружающей среды, и давление является атмосферным или близким к таковому. Имеют место многообразные вариации и комбинации реакционных условий, например концентрации компонентов, желательные растворители, смеси растворителей, температуры, давления и прочие диапазоны и условия реакций, которые могут быть использованы для оптимизации чистоты и выхода продуктов, получаемых из описываемых процессов. Для оптимизации таких производственных условий потребуется только разумно необходимое и общепринятое экспериментирование.
I. Материалы и химические реагенты
Химические реагенты, использованные в исследовании, кроме указанных ниже сырьевых материалов для реактора, перечислены в таблице 1.
Сырьевые материалы, использованные в этих экспериментах, включают:
(1) Стеариновую кислоту (95%-ную), приобретенную в фирме Sigma (Сент-Луис, Миссури)
(2) Олеиновую кислоту, приобретенную в фирме Sigma (Сент-Луис, Миссури)
(3) Жир домашней птицы от фирмы Lomax Inc. (Монреаль, Квебек)
(4) Говяжий жир “Bleached Fancy (BF)”
(5) Говяжий жир “Yellow grease (YG)”
(6) Масло канола, приобретенное на месте в Канадском универсальном магазине.
Таблица 2 показывает жирнокислотный состав сырьевых жиров и масел. Таблица 3 показывает процентное содержание насыщенных и ненасыщенных жирных кислот в сырьевых жирах и маслах.
II. Экспериментальное оборудование
Микрореакторы и песчаная баня
Реакции пиролиза были проведены в микрореакторах периодического действия емкостью 15 мл (также называемых как реакторы), нагреваемых с помощью псевдоожиженной песчаной бани, как показано на фигуре 1. Компоновка экспериментальной установки состоит из трех главных компонентов, включающих:
(1) микрореакторы из нержавеющей стали;
(2) систему продувки микрореактора; и
(3) систему псевдоожиженной песчаной бани для нагревания.
Микрореакторы периодического действия
Микрореакторы емкостью 15 мл, использованные в этих экспериментах, были собраны из штуцеров и трубок Swagelok®, изготовленных из нержавеющей стали (S.S.). Схема компоновки закрытого микрореактора показана на фигуре 2. Микрореакторы состояли из донной крышки, центрального трубчатого корпуса и верхней крышки с отверстием диаметром 1/4 дюйма (6,4 мм). Трубка из нержавеющей стали (диаметр 1/8 дюйма (3,2 мм)), длиной приблизительно 15 см, была соединена с этим отверстием через переходную муфту, и вблизи конца этой трубки был размещен игольчатый клапан (приблизительно 13 см над верхней частью реактора) для открывания и закрывания реактора. Кронштейн (не показан в компоновочной схеме) был также прикреплен к этому трубчатому корпусу так, что микрореакторы могли быть помещены в систему песчаной бани.
Замена реакторов
Микрореакторы использовались до тех пор, пока они не могли должным образом загерметизированы или обгорали во время реакции и не могли быть открыты, в такой момент их заменяли. Обычно микрореакторы выдерживали от 10 до 20 реакций.
Система продувки микрореактора
Конструкция микрореактора позволяет обеспечить соединение с газовым баллоном для создания давления или продувки. Схематическая компоновка системы продувки микрореактора, использованной в настоящем изобретении, показана на фигуре 3. Давление настраивается путем считывания показания манометра Р1 и регулирования с помощью вентиля на баллоне. Микрореакторы были соединены с системой продувки, и вентили V1, V2 и вентиль на микрореакторе (не показан на компоновочной схеме) открывали для доступа азота в реактор.
Система песчаной бани
Микрореакторы нагревали в псевдоожиженной песчаной бане Techne Model SBS-4 (Берлингтон, Нью-Джерси). Главные компоненты системы песчаной бани подробно изображены на фигуре 4 и включают песчаную баню, двигатель и рычаг, источник подачи воздуха и регулятор температуры. Устройство песчаной бани схематически показано на фигуре 5, и ее размеры представлены в таблице 4. Песчаная баня была заполнена приблизительно до уровня 1-2 дюйма (25,4-50,8 мм) ниже верхнего края крупнозернистым оксидом алюминия в виде песка. Для псевдоожижения песка сжатый воздух вдували в баню вблизи дна и через пористую пластину для более равномерного распределения воздуха. Регулятор температуры Techne TC-8D (Берлингтон, Нью-Джерси) использовали для поддержания бани при постоянной температуре в течение всей реакции. Температуру бани измеряли с помощью термопары К-типа (хромель-алюмель), размещенной вблизи центра бани. Нагревательные элементы были расположены у дна песчаной бани, над пористой пластиной. Для перемешивания содержимого микрореактора в течение реакции использовали эксцентриковое колесо, соединенное с двигателем и рычагом.
Модифицированные реакторы для измерения внутренних реакционных условий
Микрореакторы периодического действия были модифицированы, чтобы обеспечить возможность измерения температуры и давления внутри реакторов во время протекания реакции. Термопара К-типа с размером 1/16 дюйма (1,6 мм) (фирма Aircom Industries, Эдмонтон, Альберта, Канада) была вставлена через верхнюю часть одного из реакторов так, что ее кончик располагался приблизительно на расстоянии 1 мм над дном реактора. Термопара была соединена с монтажной сборкой трубчатого корпуса реактора с использованием штуцеров Swagelok®, как показано на фигуре 6. Термопара была соединена с термоэлектрическим термометром Digi-Sense Dual JTEK (фирма Cole-Parmer Instrument Company, Вернон-Хиллз, Иллинойс) для измерения температуры. Фигура 7 показывает второй модифицированный реактор для измерения давления. Манометр Swagelok® (фирма Swagelok, Эдмонтон, Альберта, Канада) был прикреплен к монтажной сборке трубчатого корпуса реактора с помощью штуцеров Swagelok®.
III. Экспериментальная процедура
Реакции пиролиза
Все реакции пиролиза проводили в микрореакторах. Перед загрузкой реакторов включали псевдоожиженную песчаную баню и настраивали регулятор температуры на желательную температуру для данной конкретной реакции. Поток воздуха в реакторе регулировали так, чтобы псевдоожижение песка было достаточным для образования пузырьков с диаметром 1-2 дюйма (25,4-50,8 мм), или как раз вровень с верхом песчаной бани. Затем оставляли песчаную баню нагреваться, пока она не достигала установившегося температурного режима, что определялось по стабильному показанию регулятора температуры в течение по меньшей мере 15 минут. Время нагревания варьировало между 1,5 и 2,5 часами, в зависимости от предварительно установленной температуры. Когда песчаная баня нагревается, воздух также нагревается и расширяется, вызывая некоторое псевдоожижение и увеличение размера пузырьков. Чтобы поддерживать размер пузырьков на постоянном уровне, поток воздуха регулировали вручную в течение всего процесса нагревания.
Между реакциями микрореакторы тщательно очищали с помощью металлических щеток, промывали мылом и водой, ополаскивали дистиллированной водой и промывали ацетоном для обеспечения того, чтобы они были полностью очищены и не содержали остатков от предыдущей реакции. После того, как микрореакторы были полностью высушены, в реакторы отвешивали сырьевой материал. На резьбовые соединения крышки реактора наносили противозадирное смазочное средство, реактор закрывали и герметизировали. Микрореактор соединяли с системой продувки азотом, открывали все вентили, и микрореакторы испытывали на утечки с использованием жидкого течеискателя Swagelok Snoop®. Если обнаруживалась утечка, микрореактор отделяли от системы продувки и повторно герметизировали. Если достигнуть уплотнения не удавалось после нескольких попыток герметизирования, микрореактор заменяли. Когда микрореактор был полностью закрыт и не проявлял утечек, его трижды продували (заполнением и эвакуированием) перед закрыванием вентиля микрореактора и отсоединением от системы продувки.
Как только микрореактор был приготовлен для реакции, его присоединяли к штанге песчаной бани и опускали в центр песчаной бани. Положение микрореакторов на штанге поддерживали постоянным так, чтобы микрореакторы всегда располагались приблизительно в одном и том же месте бани. Микрореакторы позиционировали так, чтобы они не касались какой-либо части песчаной бани и были полностью погружены в песок. Включали двигатель и начинали отсчет времени реакции, когда рычаг начинал перемешивание. По окончании реакции микрореакторы поднимали из песчаной бани и немедленно охлаждали в ведре с водой комнатной температуры, чтобы закончить реакцию. Реакторы вентилировали в вытяжном шкафу для вывода любых газообразных продуктов, образовавшихся во время реакций, и открывали для экстракции, кроме тех случаев, когда газообразные продукты собирали для анализа, как описано ниже.
Для измерения температуры и давления внутри реактора реакторы загружали и продували, как обычно, однако использовали монтажные сборки с модифицированными реакторами, описанные ниже. Температуру регистрировали путем считывания показания цифрового термометра каждые 30 секунд в течение первых 10 минут реакции, каждую минуту в течение последующих 10-15 минут и затем опять по прошествии 30, 45 и 60 минут. Давление регистрировали в ходе всего эксперимента, а также после охлаждения для определения величины давления, развившегося вследствие образования газообразных продуктов.
Экстракция реакционных продуктов
Реакционные продукты экстрагировали из микрореактора с использованием 10 мл пентана с добавлением внутреннего стандарта, если не оговаривалось иное. В качестве внутреннего стандарта использовали метиловый эфир нонадекановой кислоты, и готовили растворы в пентане с концентрациями приблизительно 0,5 или 1 мг/мл. Смесь пентана с внутренним стандартом отмеряли в микрореактор с использованием поршневой пипетки и перемешивали так, чтобы любой твердый материал в микрореакторе был счищен со стенок микрореактора и диспергирован. Приблизительно через 15 минут жидкий экстракт переносили во флакончик для образца. Все продукты помещали во флакончики dram vial с закручивающимися крышками и прокладками Teflon® и хранили при температуре 4ºС.
Нонадекановая кислота была выбрана в качестве внутреннего стандарта потому, что она по структуре подобна исходному соединению. Когда этот стандарт анализировали в GC-FID, он давал острый четкий пик и не перекрывался ни с каким из потенциальных продуктов пиролиза.
Газовая хроматография (GC)
Жидкие экстракты
Пентановые экстракты анализировали на газовом хроматографе Varian 3400, оснащенном автоматическим пробоотборником Varian 8200 (Пало-Альто, Калифорния), соединенным с пламенно-ионизационным детектором (FID), работающим при температуре 320ºС. Для всех анализов применяли колонку RH1 фирмы Rose Scientific (Миссиссаги, Онтарио, Канада), и объем впрыска поддерживали постоянным на уровне 1 мкл. Температурный профиль показан на фигуре 8. Начальная температура колонки была установлена на 35ºС и запрограммирована на повышение до 280ºС со скоростью 10ºС/мин. Конечную температуру удерживали в течение 5,4 минуты, для общего времени прогона 29,9 минут.
Для идентификации продукта делали прогоны с двумя внешними стандартами. Таковыми были (1) смесь С8-С20-алканов (фирма Fluka) и (2) смесь карбоновых С3:0-С20:0-кислот, приготовленная в своей лаборатории с использованием карбоновых кислот, приобретенных в фирме Sigma. Эти внешние стандарты использовали во всех экспериментах с газохроматографическим анализом для учета потенциального сдвига пиков.
Газообразные образцы
Чтобы собрать газообразные образцы из микрореактора для анализа, трубный штуцер Swagelok® 1/4 с септой ввинчивали в штуцер, использованный для соединения микрореактора с системой продувки. Через септу вводили иглу стеклянного шприца, и открывали вентиль реактора. Из реактора с помощью шприца извлекали 4 мл газа и вытесняли в закрытый вакуумный контейнер емкостью 5 мл. Эту операцию повторяли для накопления в общем 8 мл газообразного продукта в каждом вакуумном контейнере емкостью 5 мл. Газообразные фракции анализировали на газовом хроматографе Hewlett Packard Series II 5890, соединенном с TCD (детектором общего состава по теплопроводности), настроенным на температуру 80ºС. 100 мкл образца вручную инъецировали в колонку Agilent HP-PLOT Q длиной 30 м с внутренним диаметром (I.D.) 0,53 мкм. Использованная программа температурного режима показана на фигуре 8. Выбранные газообразные образцы были также проанализированы с помощью GC-FID-хроматографии при условиях, описанных ниже.
Газовая хроматография - масс-спектрометрия (GC-MS)
Предварительные GC-MS-анализы были проведены на выбранных образцах с использованием прибора Waters (бывшая Micromass, Милфорд, Массачусетс) Trio 2000, оснащенного газовым хроматографом HP5890 Series II, на химическом факультете Университета Альберты. Использованный температурный профиль был таким же, как показанный на фигуре 8.
Степень конверсии в реакции
Для определения степени конверсии в реакции было необходимо растворить всю сырьевую стеариновую кислоту, оставшуюся в реакторе. В качестве растворителя для экстракции использовали хлороформ ввиду относительно высокой растворимости стеариновой кислоты в этом растворителе по сравнению с пентаном. Реакционные продукты вымывали из реактора хлороформом в круглодонную колбу, пока внутри реактора не оставалось никаких продуктов. Затем хлороформ удаляли с использованием роторного испарителя. Во время процесса испарения и высушивания в роторном испарителе существует вероятность потери некоторой части летучих продуктов, но, поскольку требуется количественная оценка только стеариновой кислоты, это не должно повлиять на результат. 30 мл хлороформа, к которому добавлен внутренний стандарт, пипеткой вносили в колбу с остаточными продуктами и перемешивали вращательными движениями, пока все продукты не растворялись. Основываясь на растворимости стеариновой кислоты в хлороформе, 30 мл являются более чем достаточными для растворения максимально возможного количества стеариновой кислоты как продукта (1 грамм, если реакция вообще не прошла). Образцы были взяты и хранились при температуре 4ºС во флакончиках dram vial с закручивающимися крышками с прокладками из тефлона до анализа. Были проведены контрольные эксперименты с использованием процедуры экстракции без термической обработки.
Дериватизация диазометаном
Аликвоту образца объемом 250 мкл добавляли в один флакон dram vial и полностью высушивали в атмосфере азота, после чего во флакон добавляли избыточное количество диазометана собственного приготовления. После завершения реакции (то есть после прекращения образования пузырьков) образец опять высушивали азотом и затем повторно суспендировали в известном объеме хлороформа перед анализом на газовом хроматографе.
Процентное содержание жидких и газообразных фракций
Для получения приблизительной оценки выхода жидкости реактор открывали, и жидкий продукт извлекали с помощью пипетки Пастера и взвешивали. Для получения приблизительной оценки массы газообразного продукта реактор взвешивали до и после выдувания газа. Для этих реакций использовали 5,0 г стеариновой кислоты в качестве сырьевого материала вместо обычного 1,0 г, чтобы можно было легче измерить разность.
Реакции гидролиза
Перед тем, как сырые и растительные масла были подвергнуты пиролизу, они сначала были гидролизованы. Реакции гидролиза в малом масштабе были проведены в тех же микрореакторах, что и для реакций пиролиза. Приблизительно 3 грамма твердого жира или масла и 6 граммов дистиллированной воды добавляли в микрореакторы в соотношении 1:2 (по весу) масла/твердого жира к воде. Реакторы закрывали, как описано ранее, и вводили азот до давления 3,48 МПа (500 фунт/кв.дюйм). Реакцию гидролиза проводили при температуре 250ºС в течение 4 часов. Когда реакторы открывали, их помещали в стакан с горячей водой, чтобы продукты оставались в жидком состоянии, и переносили в стеклянный флакончик для образцов с помощью пипетки Пастера. Жирному слою давали возможность отделиться от водно-глицеринового слоя и с помощью пипетки переносили в отдельный стеклянный флакончик. Образцы хранили при температуре 4ºС до проведения пиролиза или дериватизации. Предполагалось, что, если в образце остается какое-то количество воды, скорость гидролиза должна быть пренебрежимо малой при такой низкой температуре. Этот слой жира или масла будет здесь называться как гидролизаты масел или жиров, чтобы не перепутать эти продукты с продуктами, образовавшимися после пиролиза (то есть пиролизатами или пиролитическим маслом).
Жирнокислотный состав сырьевого материала
Жирнокислотный состав твердого жира “Yellow Grease”, твердого жира “Bleached Fancy”, жира домашней птицы и масла канола был определен путем дериватизации образцов действием трифторида бора и анализирования их с помощью GC-FID-хроматографии. Процедура дериватизации описана ниже, и газохроматографический анализ представлял собой стандартную методику для жирных кислот, как описано выше.
Дериватизация трифторидом бора
Для дериватизации трифторидом бора приблизительно 30 мг образца отвешивали в испытательную пробирку и добавляли 5 мл смеси 14%-ного раствора трифторида бора в метаноле с метанолом и гексаном (в объемном соотношении 35:45:20). Пробирки плотно закупоривали и нагревали в кипящей воде в течение 45 минут. После охлаждения пробирок добавляли 4 мл воды и 4 мл гексана, и пробирки встряхивали в течение 1-2 минут. Давали слоям разделиться, и гексановый слой извлекали с помощью пипетки Пастера и хранили во флаконе dram vial с тефлоновой прокладкой при температуре 4ºС до анализирования.
Анализ гидролизатов с использованием TLC-FID
Состав гидролизатов определяли с использованием тонкослойной хроматографии, объединенной с пламенно-ионизационным (FID) детектором (TLC-FID). Образцы готовили для анализа путем отвешивания приблизительно 0,03 г жирных гидролизатов во флакон с закручивающейся крышкой и добавлением 5 мл гексана (качество HPLC-grade). Заданный объем образца наносили в виде пятен на кварцевые стержни Chromarods-SIII с покрытием из силикагеля, с использованием иглы и шприца порциями по 0,2 мкл. Стержни затем помещали в камеру для проявления, содержащую смесь гексан/диэтиловый эфир/уксусная кислота (в объемном соотношении 80:20:1), на 20 минут и высушивали при температуре 120ºС в течение 10 минут. Анализ липидов проводили с использованием хроматографического анализатора Iatroscan TH-10 (фирма IARON-Laboratories Inc., Токио, Япония) с давлением водорода 113 кПа, скоростью потока воздуха 2000 мл/мин и скоростью сканирования 30 с/стержень. Стандартный образец, содержащий 25% (по весу) каждого компонента из олеиновой кислоты, моноолеина, диолеина и триолеина, был получен от фирмы Nu-Chek Prep Inc. (Элизиан, Миннесота).
Анализ гидролизатов с использованием GC-FID
Для определения состава непрореагировавшего или негидролизованного сырьевого материала, если таковой имелся, проводили GC-FID-анализ с использованием дериватизированных образцов. Гидролизаты твердого жира “Bleached Fancy” были подвергнуты дериватизации четырьмя различными способами, которыми метилировались только определенные группы, как показано в таблице 5. Дериватизацию диазометаном проводили с использованием методики, описанной выше. Три других способа обсуждаются ниже.
Дериватизация метилатом натрия и метанольным раствором HCl
Такую же методику использовали для дериватизации метилатом натрия и метанольным раствором HCl. Образец масла или жира в количестве 10-30 мг отвешивали и помещали на дно испытательной пробирки с 50 мкл бензола для растворения образца. Образцу давали осесть в течение 20-30 минут, затем в испытательную пробирку добавляли 2 мл либо метилата натрия, либо метанольного раствора HCl. Затем образцы нагревали на водяной бане (30 минут для метилата натрия, 50 минут для метанольного раствора HCl) при температуре 50ºС. Образцы оставляли для охлаждения перед тем, как в испытательные пробирки добавляли 100 мкл воды и 2 мл гексана. Пробирки встряхивали и оставляли для осаждения, в течение которого сформировывались органический и водный слои. Гексановый (органический) слой отделяли и хранили во флаконе с прокладкой из Teflon® при температуре 4ºС.
IV. Внутренние температура и давление в реакторе для песчаной бани TECHNE SBS-4
Температурные профили, представляющие нагревание микрореакторов при 370, 410 и 450ºС, представлены на фигуре 10. Данные представляют среднее значение для дублирующих экспериментов, и интервалы погрешностей (не видны) представляют стандартное отклонение для этих экспериментов. Скорость нагревания материала внутри реактора проявляется как отчетливо высокая по мере того, как температура реактора, Tреактор, достигает 95% от уровня предварительно установленной температуры (отсчитывается от исходной температуры в нулевой момент времени и показана на фигуре 10 в виде пунктирной линии) в пределах 3,5, 3 и 4 минут для трех предварительно установленных температур, соответственно. Как ожидалось, имеет место снижение температуры регулятора температуры (сплошная линия) для всех трех температур, после того, как реакторы были помещены в баню. При уровне температуры 370ºС нагревание бани обратно до температуры 370ºC занимало приблизительно 6 минут, тогда как для уровня температуры 410ºC на это ушло 5,5 минут. Нагревание бани обратно до должной температуры для экспериментов, проведенных при температуре 450ºС, заняло период между 12-14 минутами.
Показания манометра регистрировали на протяжении всего течения реакции, однако компоновка затрудняла считывание вследствие перемешивания. При температуре 370ºС одна из экспериментальных реакций не привела к развитию давления во время реакции, но во втором эксперименте было достигнуто максимальное давление 1034 кПа (150 фунт/кв.дюйм). В обоих случаях манометр показал нулевое давление после охлаждения. При температуре 450ºС максимальное давление, достигнутое во время отдельных экспериментов, составило 2586 кПа (375 фунт/кв.дюйм) и 3103 кПа (450 фунт/кв.дюйм). После охлаждения в реакторах оставалось давление приблизительно 689 кПа (100 фунт/кв.дюйм). При температуре 410ºС в одном из экспериментов проявилось предельно большое повышение давления в конце опыта на уровне 4482 кПа (650 фунт/кв.дюйм). После охлаждения давление внутри реактора составляло 689 кПа (100 фунт/кв.дюйм). С учетом результатов других экспериментов это представляется необычным. Второй эксперимент при температуре 410ºC привел к результату, который можно было ожидать, основываясь на данных с другими температурами. Было достигнуто максимальное давление 1379 кПа (200 фунт/кв.дюйм), но после охлаждения манометр показал нулевое давление внутри реактора.
V. Работа с модельным соединением
Исследования предварительного пиролиза
План экспериментов для реакций предварительного пиролиза показан в таблице 6. Все реакции были проведены в азоте и первоначально были при атмосферном давлении. Немедленно после охлаждения реактор открывали и к продуктам добавляли 10 мл пентана, перемешивали вращательным движением, и растворимые в пентане продукты извлекали пипеткой в колбу. Две последующие экстракции по 10 мл также проводили для получения в целом экстракций 3×10 мл перед тем, как аликвоту переносили во флакон для образца с завинчивающейся крышкой, имеющей прокладку из Teflon®. Для этого цикла экспериментов не добавляли внутренний стандарт, но для контроля анализировали непрореагировавшую стеариновую кислоту. Жидкие экстракты анализировали с помощью GC-FID-хроматографа. Результаты показаны на фигурах 4.2 и 4.3. Дублирующие хроматограммы (не показаны) являются весьма сходными для всех температурных режимов, показывая хорошее согласование между экспериментальными реакциями.
Фигуры 11 и 12 показывают, что распределение продуктов существенно изменяется в зависимости от температуры и времени. 30-минутная реакция при температуре 350ºС (фигура 11) имеет результатом низкую конверсию, что проявляется отсутствием пиков по сравнению с другими экспериментами и относительно сильным пиком, который идентифицирован как исходный материал, то есть стеариновая кислота. Это было определено сравнением времен удерживания образца стеариновой кислоты в пентане с результатом без термической обработки. При температуре 400ºC начинает формироваться отличающаяся картина сигналов, и при температуре 450ºС эта картина сигналов продолжает развиваться. При температуре 500ºС эти сигналы начинают деградировать с проявлением множества пиков на уровне шумов с локализацией при низких временах удерживания. Такая же тенденция очевидна для 5-минутных реакций, но при слегка более высоких температурах. При температуре 400ºС (фигура 12) сигналы только лишь начинают развиваться с возрастанием интенсивности при температурах как 450ºС, так и 500ºС. Хотя сигналы все еще присутствуют при температуре 500ºС, большинство пиков начинает формироваться при временах удерживания менее чем 5 минут. При температуре 550ºС эти сигналы полностью деградировали и выродились в картину распределения, которая выглядит подобно таковой для 30-минутной реакции при температуре 500ºС.
Идентификация пиков
GC/MS-Анализ
Нижеследующие образцы были проанализированы с помощью масс-спектрометрии: (1) продукты разложения стеариновой кислоты после 5-минутной реакции при температуре 500ºС (хроматограмма, показанная на фигуре 12) и (2) продукты разложения стеариновой кислоты после пятиминутной реакции при температуре 550ºС (хроматограмма, показанная на фигуре 12). Исследование проводили с использованием библиотеки масс-спектров NIST (Национальный институт стандартов и технологий), и были выявлены наилучшие согласования спектров. Результаты показывают, что после пяти минут при температуре 500ºС формировались четыре серии сигналов, включающие алкановую серию, алкеновую серию, серию карбоновых кислот и серию ненасыщенных карбоновых кислот с одной двойной связью. Спектры показывают, что есть вероятность, что двойная связь в алкенах находится в одном положении, и в ненасыщенных карбоновых кислотах она располагается в концевом положении цепи, противоположном карбоксильной группе, однако это не было подтверждено спектрами ЯМР (ядерного магнитного резонанса). Эти сигналы иллюстрированы на фигуре 13. Результаты поиска по каталогам NIST для режима при температуре 550ºС показали, что многие из соединений были скорее всего ароматическими.
Подтверждение продуктов с использованием внешних стандартов в GC-FID
Фигура 14 показывает хроматограмму продуктов разложения стеариновой кислоты после 5-минутной реакции при температуре 500ºС. Маркированные соединения были подтверждены с использованием внешних стандартов в сочетании с результатами GC/MS-анализа. Два внешних стандарта, включающих (1) смесь С8-С20-алканов, приобретенную в фирме Fluka, и (2) смесь карбоновых кислот, приготовленную в собственной лаборатории с использованием карбоновых кислот от фирмы Sigma, были проанализированы на GC-FID с применением идентичных условий. Полученная хроматограмма показана на фигуре 15. Серия алканов от октана (С8) до гептадекана (С17), а также серия карбоновых кислот между С7:0 (гептановая кислота) и С18:0 (стеариновая кислота) были идентифицированы в пиролитической смеси.
Пиролиз со второй экстракцией с использованием толуола
После экстракции пентаном в реакторе все еще остается некоторое количество материала. Возможно, что этот материал не растворяется в пентане, или что был достигнут предел растворимости в пентане. Другими словами, последний был насыщен продуктом и неспособен растворять больше ничего. Стеариновая кислота только отчасти растворима в пентане, так что возможно, что непрореагировавший сырьевой материал также присутствовал в реакторе после экстракции пентаном. Чтобы определить, какие типы продуктов все еще находились в микрореакторе после экстракции пентаном, проводили последующую экстракцию 3×10 мл толуолом для 5-минутных экспериментов, и экстракты собирали для анализа. Избранные хроматограммы представлены на фигурах 16 и 17. Фигура 16 показывает, что толуольный экстракт содержит только стеариновую кислоту как исходное соединение. Более мелкие пики по обеим сторонам самых сильных пиков представляют собой примеси в сырьевом материале (определенные контрольными экспериментами без термической обработки), и пики с временами удерживания менее чем 10 минут представляют собой примеси в толуоле (определено газохроматографическим анализом толуола). Реактор выглядел пустым после экстракции толуолом, показывая, что пентан растворил все реакционные продукты, кроме некоторого количества непрореагировавшего кислотного сырьевого материала. Подобные результаты получены при температуре 450ºС (хроматограмма не показана). При температуре 500ºС образуется большее количество продукта и присутствует меньше непрореагировавшего сырьевого материала. При этих условиях пентан растворяет бóльшую часть продуктов в реакторе, в том числе весь непрореагировавший сырьевой материал, что проявляется в отсутствии любых соединений в толуольной фракции.
Влияние высушивания образцов на профиль продуктов
Для сведения массового баланса и количественной оценки вес растворимого в пентане продукта наиболее легко определяется высушиванием образца в атмосфере газообразного азота и затем взвешиванием. Проблема, связанная с этим методом, состоит в том, что многие реакционные продукты являются летучими и в принципе могут испариться во время процесса высушивания. Перед разработкой методологических основ экстракции представляло интерес выяснение вопроса, влияло ли высушивание под азотом на профиль продуктов. Были проведены дублирующие реакции в течение одного часа при температурах 450ºС и 500ºС. Реакторы были продуты газообразным азотом и первоначально были при атмосферном давлении. Для экстрагирования реакционных продуктов использовали 10 мл пентана, и две аликвоты по 4 мл переносили во флаконы для образцов. Один из образцов проанализировали как есть, тогда как другие образцы были высушены в азоте и затем повторно суспендированы в 4 мл пентана перед анализом. Фигура 18 показывает хроматограммы до и после высушивания продуктов, полученных при температуре 450ºС. Количества и распределение продуктов при высушивании существенно изменяются, в особенности для соединений с более низким временем удерживания. При температуре 500ºС, где продукты являются по большей части легкими фракциями и, возможно, ароматическими, процесс высушивания приводит к испарению большинства соединений.
Пиролиз при различных продолжительностях и температурах
Многочисленные реакции были проведены при разнообразных температурах и продолжительностях. Они были проведены для выявления влияния времени и температуры на продукты пиролиза в широком диапазоне условий, а также для модифицирования методики экстракции. Множество экспериментов было проведено при температурах между 350 и 500ºС и продолжительностях реакций, варьирующих от 1 до 6 часов. Результаты этих экспериментов помогли выбрать условия, использованные для эксперимента с увеличенными продолжительностью/температурой.
Влияния продолжительности и температуры на продукты пиролиза стеариновой кислоты
На основании результатов предварительных экспериментов представляло интерес исследование продуктов пиролиза стеариновой кислоты в широком интервале температур и продолжительностей для определения, в пределах каких из этих условий формируются исследуемые продукты. В этом эксперименте реакции проводили при температурах между 350 и 500ºС и продолжительностях реакций, варьирующих от 0,5 до 8 часов. Продолжительности и температуры, выбранные для этого исследования, основывались на полученных предварительных результатах и представлены в таблице 7. Условия варьировали от мягких, где имела место очень низкая конверсия, до более жестких, где происходило значительное разложение продукта и где деградировали серии пиков, обсужденные в предыдущих разделах. В пределах этих условий формировались представляющие интерес продукты. Все реакции были проведены в атмосфере азота, и микрореакторы первоначально были при атмосферном давлении.
Распределения продуктов при различных продолжительностях и температурах
Для этих реакций метиловый эфир нонадекановой кислоты добавляли в качестве внутреннего стандарта с известными концентрациями. В условиях газовой хроматографии, выбранных в этом эксперименте, метиловый эфир нонадекановой кислоты вымывается из колонки приблизительно при 22,6 минутах, как показано на фигуре 19. Фигуры 20-26 показывают хроматограммы из реакций, проведенных в условиях, указанных в таблице 7. Эти хроматограммы дают хороший «моментальный снимок профиля» распределения продуктов при разнообразных условиях. Ввиду природы растворителя для экстракции и способа экстракции существует возможность того, что не вся стеариновая кислота, которая не очень хорошо растворяется в пентане, и гептадекан (С17-алкан), который является твердым при комнатной температуре, были растворены в пентане. Возможно, что эти пики являются преуменьшенными. В плане типов продуктов, образовавшихся при разнообразных условиях, дублирующие хроматограммы (не показаны) были в сущности идентичными. Результаты этого эксперимента подтверждают предыдущие результаты. Показано, что как продолжительность, так и температура оказывают существенное влияние на распределение продуктов. При температуре 350ºС (фигура 20) основным продуктом является гептадекан (С17-алкан). Алкановые сигналы как раз начинают формироваться при продолжительности 4 часа и слегка более развиваются при 8 часах. Также остается некоторое количество исходного сырьевого материала, однако реальное количество нельзя оценить по величине площади пика, как было разъяснено выше. Анализ количества непрореагировавшего сырьевого материала при различных условиях обсуждается в последующих разделах. По мере возрастания температуры и продолжительности очевидно развитие серий сигналов. При температуре 390ºС и продолжительности 8 часов, температуре 410ºС и продолжительности 1, 4 и 8 часов, и температуре 430ºС и продолжительности 0,5 и 1 час эти сигналы проявляются наиболее развитыми. При температуре 430ºС есть свидетельство начала формирования соединений с низким временем удерживания, возможно ароматических. При температуре 450ºС после 4 часов и при температуре 500ºС серии сигналов деградировали.
Оценка С8-С20-алканов и алкенов
Главными представляющими интерес продуктами являются алканы и алкены. Эти соединения формируют две наиболее заметные серии сигналов в продуктах пиролиза. Алканы и алкены с составом С8-С20 были идентифицированы на хроматограммах с использованием данных GC/MS и внешних стандартов. Площади пиков были использованы для полуколичественного определения количества каждого соединения в смеси продуктов относительно внутреннего стандарта с известной концентрацией. Возможно, что при более мягких реакционных условиях количество гептадекана (С17) является преуменьшенным, как описано в предыдущем разделе. Хотя это могло бы быть не вполне точным, данные все-таки должны давать для выхода, в худшем случае, оценку с запасом. Фигуры 27 и 28 показывают процентное содержание С8-С20-алканов и алкенов, соответственно, образовавшихся при различных температурах и продолжительностях. Важно отметить, что алкен, который был оценен количественно, представлял алкеновый пик, который непосредственно предшествовал алкановому пику. Как разъяснено ранее, данные от GC/MS позволяют предположить, что это скорее всего 1-алкен, однако это не было подтверждено другими методами. Данные GC/MS также свидетельствуют о том, что мелкие пики, следующие за алканом (еще один «сигнал») также представляют алкен с двойной связью в другом положении. Когда данные анализировались первый раз, эти пики были подавлены разбавлением. Флаконы для газовой хроматографии были разбавлены перед анализом, поскольку С17-пик получался чрезмерно завышенным, когда анализировали образец с более высокой концентрацией. Фигуры 20-26 представляют данные анализа для более концентрированных образцов (то есть все реакционные продукты были растворены в 10 мл пентана); однако интегрирование пиков проводили с использованием разбавленных образцов. Поскольку метод, использованный для количественной оценки соединений, соотносит их с внутренним стандартом, который был добавлен во время экстракции продуктов из реактора, это не должно сказаться на результате. Как таковой, мелкий пик алкена, который появляется после алкана на хроматограмме, не рассматривается в этом анализе, но вкратце будет обсужден ниже. Данные представляют усредненное значение дублирующих экспериментов, и интервалы погрешностей представляют стандартное отклонение между двумя экспериментами. При обращении к фигурам 27 и 28 очевидно, что образуется большее количество алканов по сравнению с алкенами. Кроме того, интервалы погрешностей при более жестких реакционных условиях являются меньшими, чем при более мягких условиях, что наблюдалось и для других результатов.
При температурах 350, 370, 390 и 410ºС количество образующихся алканов и алкенов возрастает со временем. При температуре 430ºС и выше количество алканов и алкенов в С8-С20-диапазоне начинает снижаться по мере увеличения продолжительности реакции. Например, при температуре 430ºС 4-часовая реакция имеет результатом в целом общий выход 25,2% С8-С20-алканов и алкенов, тогда как после 8-часовой реакции это значение снижается до 10,7%. При температуре 450ºС и продолжительностях реакций более чем 4 часа, и температуре 500ºС образуется относительно мало продукта в С8-С20-диапазоне. Максимальные количества С8-С20-алканов и алкенов получаются при температуре 410ºС после 4-часовой (32,7%) и 8-часовой (32,1%) реакций, и при температуре 390ºС после 8 часов (32,9%).
Картины крекинга С8-С17-углеводородов
Данные из хроматограмм представляют неплохую оценку выходов, но они также могут быть использованы для изучения параметров крекинга. Как молярная селективность, так и отношение алканов к алкенам могут дать хорошее понимание поведения при крекинге. Этот раздел будет сосредоточен на молярных выходах алканов, в то время как следующий раздел будет посвящен молярному отношению. Площади пиков при интегрировании сигналов в газовой хроматографии были преобразованы в молярные выходы С8-С20-алканов. Эти данные представлены на фигурах 29-32. Фигуры представляют усредненные значения дублирующих экспериментов, и интервалы погрешностей представляют стандартное отклонение между этими экспериментами. Для ясности, более низкие температуры (350-390ºС) иллюстрированы светлыми значками точек измерения и пунктирными линиями, тогда как более высокие температуры (430-500ºС) обозначены темными залитыми значками точек измерения и сплошными линиями. Промежуточная температура, 410ºС, иллюстрирована значками «×» и линией из более длинных штрихов (см. обозначения на фигуре). Картина крекинга алканов имеет важное значение, поскольку алканы являются главными обсуждаемыми продуктами. Параметры крекинга алкенов также являются важными и рассматриваются в следующем разделе после обсуждения молярных отношений между алканами и алкенами.
На фигурах 29-32 показано, что подобные тенденции имеют место для каждой продолжительности реакции в 0,5, 1, 4 и 8 часов, однако они проявляются при различных температурах. При самых мягких условиях (низкая температура, малая продолжительность) реакционный продукт образуется в очень малом количестве. Например, при температуре 350ºС продукты не начинают формироваться до 4-часовой продолжительности.
Отношение «алкан:алкен»
Данные этого эксперимента могут быть применены для анализа молярного отношения алканов к алкенам, важному параметру в крекинге углеводородов. Площади пиков были использованы для расчета отношения «алкан:алкен». Фигуры 33-36 показывают молярные отношения алканов к алкенам как функцию числа атомов углерода и времени при различных реакционных температурах. Цепи из 17 атомов углерода (отношение «гептадекан/гептадецен») были исключены из этих фигур, поскольку отношение было столь большим, что это делало затруднительным выявление изменений в С8-С16-отношениях. Это отношение обсуждается отдельно в следующем разделе. Как и в предыдущем разделе, фигуры представляют усреднение из дублирующих экспериментов, и интервалы погрешностей представляют стандартное отклонение между экспериментами. Отклонения для этих данных в общем были меньшими, чем для оценок выходов. Вероятно, больший процент отклонения между двумя образцами обусловливается способом экстракции и количеством экстрагированного соединения. Это, возможно, могло бы повлиять на количество соединения в экстрактах, но, скорее всего, не оказывает влияния на отношение алканов к алкенам, которое должно быть независимым от концентрации.
Важно отметить, что, поскольку эти данные представляют усредненное значение только для дублирующих экспериментов, анализы критериев значимости провести нельзя. Будут отмечены общие тенденции, основанные на графиках, но не известно, имеют ли или нет любые из упомянутых разностей истинную статистическую ценность. Для этого цикла экспериментов молярные отношения почти всегда составляют более чем 1, означая, что алканы образуются в большем количестве, чем алкены. Если возвратиться вновь к результатам первоначальных исследований (фигура 11), ясно, что во время пятиминутных реакций при температуре 500ºС алкены образовывались в бóльших количествах, чем алканы. Это также очевидно для 0,5-часовых реакций при температуре 450ºС, но в меньшей степени. Результаты настоящего эксперимента показывают, что молярное отношение является меньшим, чем единица, только для немногих условий, наиболее заметно для температуры 450ºС при 4-часовых и 8-часовых реакциях, и только для цепей с определенными числами атомов углерода, а именно С12-С14 и С16.
Изменения молярного отношения со временем
На фигурах 30-33 показано, что тенденция состоит в том, что при температурах 390ºС и 410ºС молярное отношение возрастает со временем. Более высокое молярное отношение показывает, что большее количество алканов образуется по сравнению с алкенами, или алканы формируются более предпочтительно, чем алкены. Например, при температуре 390ºС молярное отношение для С8 повышается от 1,69±0,07 после 0,5 часов до 5,55±0,09 после 8 часов. Подобным образом, молярное отношение для С16 увеличивается от 0,69±0,11 после 0,5 часов до 4,17±0,22 после 8 часов. Подобные тенденции наблюдаются для чисел атомов углерода между этими пределами. При температуре 430ºС некоторые соединения (С8, С10, С11 и С15) показывают возрастающее молярное отношение со временем, однако другие (С9, С12, С14, С16) проявляют снижение молярного отношения между 4- и 8-часовыми реакциями. При температуре 450ºС это выглядит так, будто молярное отношение начинает снижаться даже раньше, между 1 и 4 часами реакции. В целом, молярное отношение увеличивается со временем от 0,5-8 часов до определенной температуры, где более продолжительные времена реакций приводят к снижению молярного отношения.
Изменения молярного отношения с температурой
Температура не оказывает столь сильного влияния на молярное отношение, как продолжительность, при температурах между 390ºС и 430ºС. При каждой продолжительности реакции возникает максимальное отношение при определенной температуре, и по мере увеличения продолжительности реакции температура, при которой возникает максимум, снижается. Например, для 0,5-минутных реакций максимальные отношения проявляются при температурах 410ºС или 430ºС, тогда как для 8-часовых реакций максимальные отношения имеют место при гораздо более низких температурах около 370ºС или 390ºС.
Эффекты продолжительности и температуры
Хотя статистический анализ не проводился, ясно, что как температура, так и продолжительность влияют на молярное отношение. Самые мягкие условия (низкие температуры и малые продолжительности) имеют результатом относительно низкое молярное отношение, но то же происходит в наиболее жестких условиях (самые большие продолжительности и наивысшие температуры). Оптимальное отношение располагается где-то между этими двумя экстремальными ситуациями. При испытанных условиях наибольшее отношение проявляется при температуре 370ºС для 8-часовых реакций. 8-часовые реакции при температуре 350ºС проявились в слегка более низких отношениях, однако, поскольку реакции не проводились при продолжительностях, превышающих 8 часов, возможно, что реакции с продолжительностью более чем эта при температурах 350 или 370ºС могут иметь результатом более высокие молярные отношения.
Изменения молярного отношения в зависимости от числа атомов углерода
Еще одной рассматриваемой переменной величиной является число атомов углерода, которое имеют цепи алканов и алкенов. Для этого анализа были исследованы С8-С16-цепи атомов углерода. Распределения молярных отношений для каждого соединения относительно другого такового проявляются как согласующиеся при различных продолжительностях и температурах, за исключением того факта, что при более высоких температурах (430ºС и выше) молярные отношения для С8 и С9 снижаются в большей степени относительно других соединений. Для многих из температур соединения С8-С11 и С15 имеют более высокие молярные отношения, чем С12-С14 и С16. Очевидно, что С15 имеет наивысшее молярное отношение, тогда как С16 имеет наинизшее. Например, при температуре 410ºС 1-часовая реакция приводит к молярному отношению 4,75±0,06 для С15, но только 1,20±1,16 для С16. При температуре 390ºС 8-часовая реакция приводит к молярному отношению 8,86±0,07 для С15 и 4,17±0,02 для С16.
Молярные отношения для С17
Фигура 37 показывает молярное отношение для С17, или отношение гептадекана к гептадецену. Было установлено, что гептадекан является главным реакционным продуктом, и что присутствует очень малое количество гептадецена. Это отражают молярные отношения, которые являются существенно более высокими для С17, чем для С8-С16. В С8-С16-диапазоне С15 имел самые высокие молярные отношения. При температуре 390ºС молярное отношение после 8-часовой реакции составляло 8,86±0,07. Напротив, при таких же условиях молярное отношение для С17 было 39,0±0,71. Опять же, поскольку реакции дольше 8 часов не проводились, возможно, что максимальное молярное отношение находится вне испытанных условий. Наибольшее молярное отношение для С17, 43,53±3,59, проявилось при температуре 410ºС для 8-часовой реакции. Данные позволяют предположить, что для С17 существуют те же тенденции, что и для С8-С16-углеводородов. Например, при температурах 390-430ºС молярное отношение возрастает со временем, но при более высоких температурах, таких как 450ºС, увеличенные продолжительности (4 и 8 часов) приводят к пониженным отношениям. При температуре 500ºС отношения являются низкими для всех испытанных продолжительностей.
Анализ легких фракций (газообразных фракций)
Состав
Типичные хроматограммы, показывающие состав газообразной фракции по данным анализа на GC-TCD, представлены на фигуре 38. Метан (СН4), диоксид углерода (СО2) и комнатный воздух в качестве стандартов также были проанализированы и показаны на фигуре 39. Благодаря чувствительности детектора N2 и О2 проявились как одиночные пики. Это значит, что первый пик в хроматограммах может представлять собой N2, О2, воздух или любую комбинацию этих трех. Для простоты это будет называться как «N2/О2-пик». При сравнении времен удерживания для пиков из образца (фигура 37) с временами удерживания для пиков из стандартов (фигура 38) очевидно, что газообразные фракции содержат «N2/О2», СН4 и СО2. Большая часть N2/О2-пика скорее всего может быть отнесена на счет азотной атмосферы внутри реактора и малых количеств воздуха из образца вакуумного контейнера или инъекционного шприца. Небольшие количества воздуха присутствуют в стандартах СО2 и СН4 (фигура 39), показывая, что малые количества воздуха поступают в газовый хроматограф, вероятно через шприц. Есть также два набора мелких пиков при более поздних временах удерживания, которые проявляются в виде дуплетов. Эти пики скорее всего представляют собой легкие газообразные углеводороды, такие как этан и пропан, однако это не было подтверждено аналитически. Анализ газообразной фракции проводили много раз, и все композиции, полученные из фракций после одночасовых реакций при температурах 390ºС и 410ºС, а также 30-минутной реакции при температуре 500ºС, дали сходные результаты.
Процентное содержание сырьевого материала
Фигура 40 показывает процентное содержание газа, образованного при различных температурах во время 1-часовых реакций. Для этого эксперимента реакции проводили с использованием приблизительно 5,0 г сырьевой стеариновой кислоты вместо обычного 1,0 г. Большее количество исходного материала было использовано, чтобы разность весов газа, измеренная путем взвешивания реактора до и после продувки, была определяемой в масштабах, доступных для лабораторного исследования. Фигура 40 показывает, что по мере повышения температуры количество образующихся газообразных продуктов также возрастает. При температуре 370ºС образуется только 0,50 вес.% газа, но при температуре 430ºС в среднем образуются 7,89 вес.% газообразного продукта. Реакции были проведены при температуре 450ºС; однако при таком гораздо большем количестве сырьевого материала повышение давления было настолько значительным, что два из реакторов разгерметизировались, и в двух из реакторов произошли выбросы масла во время продувки, несмотря на тот факт, что образцы были оставлены для охлаждения в течение ночи, и вентиляционный клапан открывали медленно. Поскольку это привело к потере масла, точных данных получить не удалось. Форма этого графика показывает, что в испытанном температурном диапазоне образование газа не проявляет линейной зависимости от температуры.
Оценка выхода жидкости
Для получения приблизительной оценки выхода жидкости жидкий продукт извлекали из реактора с помощью пипетки Пастера (без добавления растворителя) и взвешивали. Результаты представлены на фигуре 41. При температуре 390ºС в реакторе не было никакого жидкого продукта. Продукт состоял из светло-коричневого порошка. После одночасовой реакции при температуре 410ºС в трех экспериментах (данные не показаны) также не образовалось жидкого продукта, однако в других 3 экспериментах получились жидкие продукты с выходами между 58-71%.
Степень конверсии в реакции
В общем, жирные кислоты не создают острых пиков на GC-хроматограмме. Они имеют тенденцию расползаться по колонке с образованием широких пиков в виде «акульего плавника», которые с трудом поддаются количественной оценке. По этой причине жирные кислоты перед проведением газохроматографического анализа сначала подвергают дериватизации в метиловые сложные эфиры. Во избежание изменений структуры других продуктов образца ни один из образцов перед анализом не дериватизировали. Поэтому количественное определение непрореагировавшего сырьевого материала, основанное на недериватизированных образцах, возможно, было неточным. Более того, в качестве главного растворителя для экстракции применяли пентан, который не является лучшим вариантом для растворения жирных кислот. Пик стеариновой кислоты, скорее всего, не дает в должной мере представления о реальном количестве непрореагировавшего материала. Фигура 42 показывает процентное содержание стеариновой кислоты как сырьевого материала, преобразованной в продукт во время реакций (то есть 100% непрореагировавшей стеариновой кислоты). Данные представляют усредненное значение из четырех экспериментов. Как ожидалось, по мере роста температуры будет происходить конверсия большего количества сырьевого материала. При самой низкой температуре, 350ºС, произошла конверсия только 31,70% стеариновой кислоты. При самой высокой температуре, 450ºС, конверсии подверглись почти 95% продукта. Следует также отметить, что при более высоких температурах интервалы погрешностей были гораздо меньшими, чем при более низких температурах. Вероятно, это обусловливается природой реакционного продукта, а также способом экстракции. При более низких температурах, где продукт по большей части является твердым, он прилипает к реактору, и его труднее экстрагировать. Несмотря на перемешивание этого материала во время экстракции, более вероятно, что некоторое количество соединения может не раствориться в пентане. При более высоких температурах, когда реакционные продукты являются жидкими, они растворяются в пентане легче.
Минимальная температура крекинга
Для определения минимальной температуры, при которой происходит декарбоксилирование, были проведены 4-часовые реакции, начиная с температуры 350ºС и с понижением температуры. Дублирующие эксперименты при температуре 255ºС все еще показывали наличие С17-пика (фигура 43). Анализ газообразного образца, взятого из этой реакции, показал предельно малый, но явственный пик СО2, как показано на фигуре 44.
Пиролиз олеиновой кислоты
Интересно проследить, отличаются ли параметры крекинга ненасыщенной жирной кислоты от таковых для насыщенной жирной кислоты. Олеиновую кислоту, свободную жирную кислоту с цис-двойной связью в положении 9, подвергли пиролизу в течение одного часа при температуре 410ºС с использованием стандартных методик проведения реакции и экстракции. GC-FID-хроматограмма показана на фигуре 45. Основные продукты были идентифицированы с использованием GC-FID-хроматограмм и сравнением их с такими внешними стандартами, как смеси алканов и смеси жирных кислот, а также с помощью GC/MS. Количества различных соединений были определены полуколичественно и представлены на фигуре 46. Этот график показывает усредненные данные для четырех экспериментов, и интервалы погрешностей представляют стандартное отклонение. Фигура 46 показывает главные продукты реакции, полученные при термическом крекинге олеиновой кислоты при температуре 410ºС в течение 1 часа. Наибольшее количество образовавшегося продукта составлял 8-гептадецен. 8-Гептадецен представляют два столбца, возможно вследствие конформационных различий. Это показывает, что декарбоксилирование, скорее всего, является начальной стадией термического крекинга олеиновой кислоты. Наиболее заметное различие между крекингом олеиновой кислоты и стеариновой кислоты состоит в отсутствии заметной серии сигналов алканов/алкенов при более высоких числах атомов углерода. Эти сигналы являются очевидными при более низких числах атомов углерода, С9 и ниже, и фигура 46 показывает, что гептан/гептен, октан/октен и нонан/нонен были среди образовавшихся главных продуктов реакции. Это должно согласовываться с разрывом по двойной связи цепи, полученной после декарбоксилирования. Также интересно отметить присутствие нонановой и декановой кислоты, в концентрациях 11,31±0,52 и 18,41±1,01 мг/г сырьевого материала, соответственно.
Суммарно продукты, идентифицированные на фигуре 46, представляют только 18,4% от общего количества образовавшегося продукта (включая газообразные продукты). При рассмотрении хроматограммы (фигура 45) очевидно, что присутствует ряд второстепенных пиков. Анализ этих пиков с помощью GC/MS показал, что некоторые из пиков, скорее всего, представляют циклические соединения, такие как циклопентаны и циклогексаны, с различным образом присоединенными к ним метильными и этильными группами.
Газообразные образцы также были собраны при пиролизе олеиновой кислоты. Дублирующие хроматограммы из GC-TCD-анализа показаны на фигуре 47. Результаты подобны газообразным продуктам, образованным во время пиролиза олеиновой кислоты. Результаты показывают сильный N2/O2-пик, слабый пик СН4 (метана), а также пик СО2 (диоксида углерода).
VI. Гидролиз и пиролиз неразбавленных масел и жиров
Анализ гидролизатов
TLC-FID-анализ
Фигуры 48-52 показывают избранные хроматограммы из TLC-FID-анализа гидролизатных фракций. Фигура 49 показывает хроматограмму для гидролизатов твердого жира “Bleached Fancy”, и фигура 48 показывает хроматограмму для гидролизатов масла канола. Ясно, что одиночный пик, вероятно, показывает, что присутствует только один тип или класс липида. Дублирующие хроматограммы (не показаны) продемонстрировали такие же результаты. Поскольку условия были жесткими, и очевидно наличие одиночного пика, очень вероятно, что все триглицериды (TAG) преобразованы в свободные жирные кислоты (FFA). Для подтверждения этого были определены времена удерживания для различных типов липидов путем анализирования стандартной смеси триглицеридов (TAG), диглицеридов (DAG), моноглицеридов (MAG) олеиновой кислоты и свободных жирных кислот (FFA) с использованием того же метода. Эти результаты представлены на фигуре 50. Анализ с использованием хроматографических кварцевых стержней Chromarod проводят так, что пламенно-ионизационный детектор (FID) производит сканирование вниз вдоль стержня. Это значит, что первый пик, проявляющийся на хроматограмме (наименьшее время сканирования), будет таким, который дальше всех продвигается по стержню во время выполнения тонкослойной хроматографии (TLC). В этом случае фракция триглицеридов (TAG) должна проявляться первой, за которой следуют свободные жирные кислоты (FFA), диглицериды (DAG) и затем моноглицериды (MAG). Это обозначается на стандартной кривой и было подтверждено несколько раз в других исследованиях. Пики диглицеридов (DAG) и моноглицеридов (MAG) являются четкими, однако существенного разделения пиков триглицеридов (TAG) и свободных жирных кислот (FFA) не происходит. Очевидно, что есть два отдельных пика, и что пик триглицеридов (TAG) является более сильным, чем пик свободных жирных кислот (FFA). Такая же тенденция была показана в дублирующих образцах. При сравнении одиночного пика из образцов и стандарта ясно, что одиночный пик располагается на одной линии с пиками триглицеридов (TAG) и свободных жирных кислот (FFA), однако, ввиду плохого разделения пиков, нельзя было с полной определенностью установить, что образцы содержат почти 100% свободных жирных кислот (FFA).
Поскольку результаты показывают, что диглицериды (DAG) или моноглицериды (MAG) как промежуточные соединения отсутствуют, иной возможностью является только то, что пик представляет непрореагировавшие триглицериды. Для подтверждения того, что одиночный пик действительно представляет свободные жирные кислоты (FFA), образцы были нанесены на хроматографические стержни Chromarod, и затем к ним был добавлен стандарт. Фигуры 51 и 52 показывают, что, когда к образцу добавлен стандарт, высота пика свободных жирных кислот (FFA) возрастает относительно высоты пика триглицеридов. На фигуре 51 на стержни Chromarod нанесены равные объемы образца и стандарта. Опять же нет хорошего разделения, однако это выглядит подобно двум пикам, и видно, что второй пик является настолько большим, что почти полностью перекрывает первый. Это должно показывать, что одиночный пик из образца складывается со вторым пиком свободных жирных кислот (FFA) стандарта. Фигура 52 показывает стандарт, нанесенный вместе со стандартом в соотношении 2:1 по объему «стандарт/образец». В этом случае имеет место лучшее разделение между пиками триглицеридов (TAG) и свободных жирных кислот (FFA), и пики проявляются почти равными по величине. При сравнении этого со стандартной хроматограммой, где пик триглицеридов (TAG) является явственно больше, чем пик свободных жирных кислот (FFA), очевидно, что одиночный пик складывается со вторым пиком свободных жирных кислот (FFA) из стандарта. Это показывает, что гидролизаты масла канола и твердого жира “Bleached Fancy” составлены почти полностью из свободных жирных кислот (FFA). Предполагается, что гидролизаты жира домашней птицы и твердого жира “Yellow Grease” также составлены преимущественно из свободных жирных кислот (FFA). Хотя на этих гидролизатах анализ липидов не проводили, анализ пиролитических фракций всех четырех масел и жиров кажется подобным.
GC-FID-анализ
Первоначально гидролизаты из жира домашней птицы были подвергнуты пиролизу в течение четырех часов при температуре 410ºС по нормальным методикам. Экстракты были затем проанализированы на GC-FID. Хроматограммы представлены на фигурах 53 и 54. Масло канола и два сорта говяжьего твердого жира (“Bleached Fancy (BF)” и “Yellow Grease (YG)”) были подвергнуты пиролизу при температуре 410ºС, но в течение одного часа (фигура 55). Гидролизаты говяжьего жира “Bleached Fancy” были также подвергнуты пиролизу при температуре 390ºС в течение одного часа. Хроматограммы из этих реакций представлены на фигурах 55 и 56. Следует отметить, что образцы были приготовлены с различными концентрациями. На фигурах отчетливо видна серия сигналов алканов/алкенов.
Хроматограммы показывают, что есть множество соединений в пределах этих серий сигналов. Хроматограммы неразбавленных масел и жиров не являются такими же четкими, как хроматограммы из пиролиза стеариновой кислоты. Многочисленные анализы гидролизатов показывают, что сырьевой материал содержит главным образом свободные жирные кислоты. Нет свидетельства того, что присутствовали большие количества загрязняющих примесей, которые могут проявляться в пиках, показанных на фигуре 56. Для проверки этого гидролизаты были растворены в пентане и проанализированы в GC-FID (фигура 57). Проявился массивный пик с приблизительным временем удерживания. Ввиду существенного уширения вдоль нулевой линии есть вероятность, что этот пик обусловлен смесью жирных кислот. Не видно никаких других пиков. Чтобы выяснить, не были ли какие-либо соединения в продуктах пиролиза водорастворимыми примесями, провели простую экстракцию пиролитического масла водой. После промывки водой органический образец отделили и опять проанализировали на GC-FID (фигура 57). Фигуры весьма похожи, показывая, что водная экстракция оказала малое влияние на продукты экстракции. Эти простые эксперименты показывают, что пики являются органическими по природе и, вероятно, являются результатом пиролитической реакции. Возможно, что пики представляют непрореагировавший сырьевой материал или свободные жирные кислоты с меньшим числом атомов углерода. Поскольку таковые имеют склонность к уширению вдоль нулевой линии, их трудно выделить, и они искажают нулевую линию. Во-вторых, результаты анализа олеиновой кислоты показывают, что пиролиз ненасыщенных свободных жирных кислот может приводить к образованию многочисленных циклических соединений.
GC/MS-анализ после дериватизации диазометаном
Как упомянуто ранее, недериватизированные жирные кислоты не проявляются четкими острыми пиками на хроматограммах после выхода из GC-колонки и в условиях, которые были использованы в этой работе для анализа углеводородного продукта, и поскольку они распространяются по колонке, они могут перекрываться с другими соединениями. Продукты не были подвергнуты общепринятой дериватизации ввиду возможной опасности изменения распределения продуктов во время процесса дериватизации, и также вследствие того, что стеариновая кислота как сырьевой материал дает относительно четкие хроматограммы, где жирные кислоты и углеводороды были явственно разделены. Преимущество дериватизации этих продуктов заключалось бы в получении более точной оценки содержания жирных кислот. Этот подход был использован, как описано ранее, для оценки непрореагировавшего сырьевого материала, однако в этих случаях для анализа были приняты во внимание углеводороды. Поскольку продукты пиролиза неразбавленных масел и жиров содержали многочисленные неидентифицированные соединения, было интересно проанализировать их с помощью GC/MS. В попытках очистить образец и устранить любую жирную кислоту, дающую уширенный вдоль нулевой линии пик, сначала подвергли дериватизации диазометаном пиролитическое масло из твердого жира “Bleached Fancy” (без высушивания) и проанализировали с помощью GC/MS. Недериватизированный образец также проанализировали для определения любых эффектов анализа дериватизированных образцов без стадий высушивания. Эти хроматограммы представлены на фигурах 58-60.
На протяжении всего настоящего описания упоминаются разные публикации. Содержание этих публикаций во всей их полноте включено в данное описание в качестве ссылки, чтобы более полно раскрыть соединения, составы и способы, описанные здесь.
Многообразные модификации и варианты могут быть осуществлены в отношении материалов, способов и изделий, описанных в настоящем документе. Другие аспекты описанных в настоящем документе материалов, способов и изделий будут очевидными при рассмотрении описания и реализации описанных материалов, способов и изделий. Предполагается, что описание и примеры рассматриваются как показательные.
Список химических реагентов, использованных в исследовании



Реферат
Группа изобретений относится к вариантам способа получения топлива или растворителя из источника жирных кислот. Способ включает выделение одной или более жирных кислот из источника жирных кислот, где содержание жирных кислот больше, чем 90% по весу свободных жирных кислот. Затем осуществляют нагревание свободных жирных кислот, полученных или в инертной атмосфере в отсутствии катализатора декарбоксилирования, или необязательно в инертной атмосфере для преобразования всех или по существу всех свободных жирных кислот в алкан, алкен или их смесь и диоксид углерода, в котором стадию нагревания проводят при температуре от 220°С до 650°С. Достигаемый при этом технический результат заключается в производстве топлива более высокого качества. 2 н. и 13 з.п. ф-лы, 7 табл., 60 ил.
Формула
a. выделение одной или более жирных кислот из источника жирных кислот, где содержание жирных кислот больше, чем 90% по весу свободных жирных кислот; и
b. нагревание свободных жирных кислот, полученных на стадии (а) в инертной атмосфере в отсутствии катализатора декарбоксилирования для преобразования всех или по существу всех свободных жирных кислот в алкан, алкен или их смесь и диоксид углерода, в котором стадию нагревания проводят при температуре от 220°С до 650°С.
a. выделение одной или более жирных кислот из источника жирных кислот, где содержание жирных кислот больше, чем 90% по весу свободных жирных кислот; и
b. нагревание свободных жирных кислот, полученных на стадии (а) необязательно в инертной атмосфере для преобразования всех или по существу всех свободных жирных кислот в алкан, алкен или их смесь и диоксид углерода, в котором стадию нагревания проводят при температуре от 220°С до 650°С.
Документы, цитированные в отчёте о поиске
Композиция дизельного топлива, включающая компоненты на основе биологического исходного материала, полученные гидрированием и разложением жирных кислот

Комментарии