Насыпные катализаторы гидропереработки из смешанного оксида металла/углерода, содержащие никель, и их применение - RU2728793C2
Код документа: RU2728793C2
Чертежи






Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Это изобретение в основном относится к насыпному катализатору для гидропереработки, содержащему никель. Катализаторы получают способом, в котором реагенты, содержащие металлы Группы VIII и Группы VIB, например, соли металлов, смешивают с по меньшей мере одной органической кислотой, полиолом или сахаром. Результирующую смесь термообрабатывают и затем сульфидируют. Катализаторы могут быть применены для гидропереработки, в особенности для гидродесульфурации и гидроденитрогенации углеводородного сырья.
УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ДАННОМУ ИЗОБРЕТЕНИЮ
[0002] Гидропереработка углеводородного сырья обычно охватывает все процессы, в которых углеводородное сырье реагирует с водородом в присутствии катализатора и при условиях гидропереработки, в типичном случае при повышенной температуре и повышенном давлении. Термин «гидропереработка» включает, однако без ограничения ими, такие процессы, как гидрирование, гидродесульфурация, гидроденитрогенация, гидродеметаллизация, гидродеароматизация, гидродеоксигенация, гидроизомеризация, гидродепарафинизация, гидрокрекинг и мягкий гидрокрекинг.
[0003] Как правило, обычные катализаторы гидропереработки состоят из носителя (или основы) с осажденным на нем компонентом металла Группы VIB и компонентом неблагородного металла Группы VIII. Такие катализаторы могут быть изготовлены посредством импрегнирования носителя водными растворами соединений желательных металлов, с последующей одной или несколькими стадиями сушки и/или обжига.
[0004] Альтернативные технологии для получения «поддерживаемых» катализаторов описаны в патенте США № 4113605, где, помимо прочего, карбонат никеля реагирует с MoO3 с образованием кристаллического молибдата никеля, который затем смешивают и экструдируют с глиноземом, и в патенте Германии № DE 3029266, где карбонат никеля смешивают с WO3, и результирующую композицию смешивают с глиноземом, импрегнированным такими соединениями, как нитрат никеля и вольфрамат аммония.
[0005] Значительное внимание в последнее время было направлено на то, чтобы предложить катализаторы, которые могут быть применены без носителя, обычно называемые насыпными катализаторами. WO 99/03578 описывает способ получения композиций насыпных катализаторов гидропереработки, содержащих насыпные частицы оксида металла, содержащие один неблагородный металл Группы VIII и два металла Группы VIB, посредством реакционного взаимодействия и совместного осаждения соединений никеля, молибдена и вольфрама при отсутствии сульфидов.
[0006] WO 00/41810 описывает способ получения катализатора гидропереработки, содержащего насыпные частицы оксида металла, где один или несколько неблагородных металлов Группы VIII и два или более металлов Группы VIB подвергают реакционному взаимодействию в протонсодержащей жидкости, где металлсодержащие соединения во время реакционного взаимодействия находятся по меньшей мере частично в твердотельном состоянии и где в конечном счете получают твердотельный материал, содержащую (нано)кристаллическую смешанную металлооксидную фазу, охарактеризованную определенной дифрактограммой, полученной дифракционным рентгеновским анализом (XRD). Также описано получение катализатора гидропереработки в подходящей форме для применения в процессе гидропереработки посредством формования, например, посредством экструзии, и посредством объединения полученных насыпных частиц оксида металла с небольшими количествами других материалов, например, связующим материалом, чтобы способствовать формованию и обеспечить механическую прочность формованному катализатору.
[0007] Патент US 7951746 описывает способ получения аморфного предшественника насыпного катализатора и конечного катализатора, содержащего (i) кобальт и молибден или вольфрам, (ii) аморфного предшественника, (iii) имеющего 20-60 масс.% углеродсодержащего соединения на основе органической комплексообразующей кислоты и (iv) имеющего площадь поверхности 16 м2/г или менее.
[0008] В US 6566296 заявляется способ получения композиции катализатора посредством объединения компонента неблагородного металла группы VIII и по меньшей мере двух компонентов из металлов группы VIB и органической добавки на любой стадии в процессе получения. Молярное отношение органической добавки к общему количеству компонентов группы VIII и группы VIB составляет по меньшей мере 0,01. Примеры описывают получение NiMoW-оксидного катализатора с применением диэтиленгликоля, добавленного во время формования катализатора или посредством последующего импрегнирования. Кроме того, получают твердотельный катализатор, содержащий (нано)кристаллическую смешанную металлооксидную фазу, как охарактеризовано присутствием определенных пиков в его дифрактограмме, полученной дифракционным рентгеновским анализом (XRD).
[0009] Хотя композиции насыпного катализатора, описанные выше, обладают превосходной активностью гидропереработки, существует постоянная потребность в данной области техники в разработке новых композиций насыпного катализатора с дополнительно улучшенной активностью гидропереработки, в частности, при гидродесульфуризации (HDS), а также при гидроденитрогенации (HDN) и гидрировании определенного целевого углеводородного сырья, такого как дизельное топливо и вакуумный газойль (VGO).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0010] Соответственно, одним аспектом данного изобретения является предшественник насыпного катализатора (т.е. материал-носитель как таковой не добавляют), содержащий Ni и Mo и/или W и органический компонент, где молярное отношение C:(Mo+W) находится в интервале от 1,5 до 10. Предшественник насыпного катализатора получают из смеси металлических предшественников с органическим агентом. Органический агент частично разлагают, чтобы образовать смешанную фазу оксид металла/C, которая является фактически предшественником насыпного катализатора. Этот предшественник насыпного катализатора (i) является практически нерастворимым в воде, (ii) не обладает каким-либо существенным объемом пор или площадью поверхности и (iii) не содержит (нано)кристаллическую металлооксидную фазу, как охарактеризовано дифракционным рентгеновским анализом (XRD). Насыпной катализатор изготавливают из данного предшественника насыпного катализатора. После обычного сульфидирования жидкой фазы, формируют активный сульфидированный насыпной катализатор, который имеет очень высокую активность в различных видах применения для гидропереработки. После сульфидирования оксидного катализатора возможно, что сульфидированный катализатор (i) показывает площадь поверхности, как измерено посредством физической адсорбции N2, и некоторые потери в адсорбции гексана (ii) для его содержания C во время сульфидирования.
[0011] В одном варианте осуществления описана композиция предшественника насыпного катализатора, содержащая никель, молибден и/или вольфрам и органический компонент, где количество оксида молибдена плюс оксид вольфрама составляет по меньшей мере 30 масс.%, где молярное отношение C:(Mo+W) находится в интервале от 1,5 до 10. Отношение Ni:(Mo+W) составляет по меньшей мере 0,05.
[0012] В другом варианте осуществления предоставлен насыпной катализатор, который получен посредством формования предшественника насыпного катализатора любым способом, известным в данной области техники, таким как экструзия, таблетирование и/или гранулирование. Насыпной катализатор характеризуется минимальной загрузкой металла, составляющей 2,0 моля молибдена плюс вольфрам на литр реактора, где молярное отношение никеля к молибдену плюс вольфрам более чем 0,05, и молярное отношение углерода к молибдену плюс вольфрам находится между 1,5 и 10. Загрузка MoO3+WO3 этого насыпного катализатора выше, чем та, что обычно применяют в поддерживаемых катализаторах гидропереработки. В другом варианте осуществления предоставляют сульфидированный катализатор, который формируют посредством сульфидирования вышеуказанной композиции насыпного катализатора.
[0013] В другом варианте осуществления описан способ получения предшественника насыпного катализатора. Данный способ включает комбинирование по меньшей мере одного соединения Ni и соединения по меньшей мере одного металла Группы VIB с по меньшей мере одним органическим агентом, формируя тем самым раствор. Раствор затем испаряют и сушат. Сушка может быть выполнена посредством применения обычно применяемых способов сушки, таких как сушка распылением, сушка сублимацией или сушка на сушильной плите, и т.п. Высушенный материал затем подвергают дополнительной термообработке при примерно 300°C до примерно 500°C, чтобы образовать предшественник насыпного катализатора, который может быть сформован любым способом, известным в данной области техники, чтобы получить насыпной катализатор. Насыпной катализатор затем сульфидируют при условиях сульфидирования, чтобы получить сульфидированный катализатор.
[0014] В другом варианте осуществления предоставлен способ гидропереработки углеводородного сырья. Данный способ включает контактирование указанного сырья с сульфидированным насыпным катализатором, данный сульфидированный насыпной катализатор сформирован посредством сульфидирования насыпного катализатора как описано выше.
[0015] В соответствии с другим аспектом данного изобретения предоставлен способ гидропереработки углеводородного сырья, где сырье приводят в контактирование при условиях гидропереработки с вышеуказанной композицией насыпного катализатора. Композиция насыпного катализатора в соответствии с данным изобретением может быть применена в практически всех процессах гидропереработки, чтобы обрабатывать несколько видов сырья при широком диапазоне условий реакционного взаимодействия, включая, однако не ограничиваясь ими, предварительную обработку сырья перед его гидрокрекингом, предварительную обработку сырья перед его каталитическим крекингом или обработку сырья, чтобы создавать транспортное топливо с определенной максимальной концентрацией серы. Обычно, эти условия реакционного взаимодействия включают температуру в интервале от примерно 200° до примерно 450°C, величины давления водорода в интервале от примерно 5 до примерно 300 бар, часовые объемные скорости жидкости (LHSV) в интервале от примерно 0,1 до примерно 10 ч-1 и отношения H2/нефтяной продукт в интервале от примерно 50 до примерно 2000 норм. л/л. Однако предпочтительным для улучшения катализатора данного изобретения для гидропереработки и, более конкретно, гидродесульфуризации (HDS), гидроденитрогенации (HDN) и гидродеароматизация (HDA) видов сырья, содержащих дизельное топливо или вакуумный газойль, при условиях, по меньшей мере включающих часовые объемные скорости жидкости (LHSV) в интервале от примерно 0,1 до примерно 10 ч-1 и отношения H2/нефтяной продукт в интервале от примерно 50 до примерно 2000 норм. л/л. Было найдено, что композиция предшественника насыпного катализатора проявляет улучшенную активность гидродесульфуризации при применении в интервале от 30 до 80 бар при обработке различных исходных потоков дистиллята. Вполне можно полагать, что предшественник насыпного катализатора данного изобретения будет обладать преимуществами в других видах применения гидропереработки, таких как обработка фракции вакуумного газойля (VGO) и также в более широком интервале давлений.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
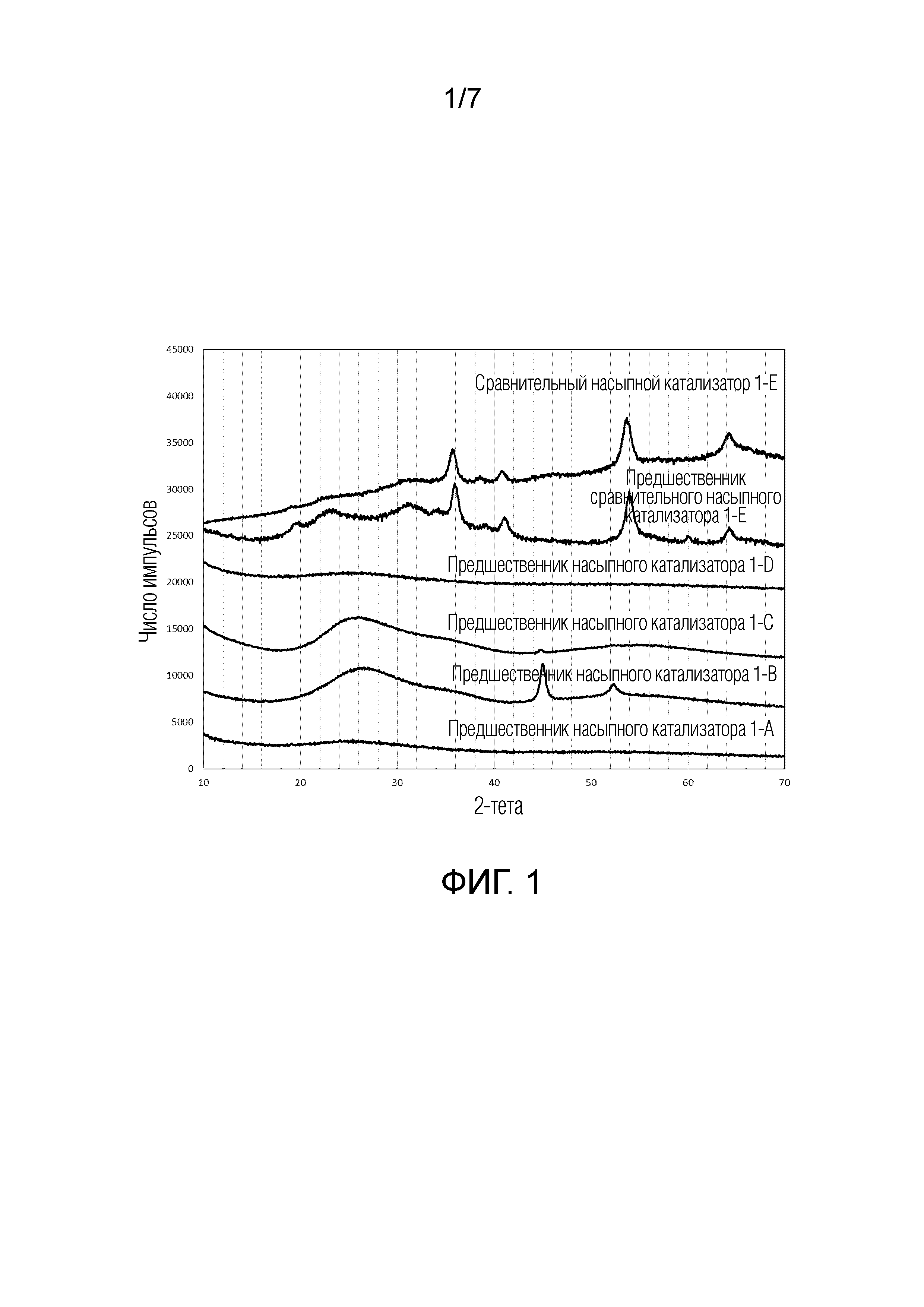
[0016] Фиг. 1. Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD), предшественников насыпного катализатора 1-A по 1-D в соответствии с данным изобретением, предшественника сравнительного насыпного катализатора 1-E и сравнительного насыпного катализатора 1-E.
[0017] Фиг. 2. Изображение, полученное просвечивающим электронным микроскопом (TEM), предшественника насыпного катализатора 1-A при высоком увеличении.
[0018] Фиг. 3. Изображение, полученное просвечивающим электронным микроскопом (TEM), предшественника насыпного катализатора 1-B при высоком увеличении.
[0019] Фиг. 4. Изображение, полученное просвечивающим электронным микроскопом (TEM), предшественника насыпного катализатора 1-C при высоком увеличении.
[0020] Фиг. 5. Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD), предшественника насыпного катализатора 2-A в соответствии с данным изобретением и предшественника сравнительного насыпного катализатора 2-B.
[0021] Фиг. 6. Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD), предшественника насыпного катализатора 3-A в соответствии с данным изобретением и предшественника сравнительного насыпного катализатора 3-B.
[0022] Фиг. 7. Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD), насыпных катализаторов 4-A и 4-B в соответствии с данным изобретением.
ПОДРОБНОЕ ОПИСАНИЕ ДАННОГО ИЗОБРЕТЕНИЯ
[0023] Было найдено, что предшественник насыпного катализатора (т.е. материал-носитель как таковой не добавляется), содержащий Ni и Mo и/или W и органическую фазу, где молярное отношение C:(Mo+W) находится между 1,5 и 10, который (i) является практически нерастворимым в воде, (ii) не обладает каким-либо существенным объемом пор или площадью поверхности и (iii) не показывает присутствие (нано)кристаллической металлооксидной фазы, как подтверждено дифракционным рентгеновским анализом (XRD), обладает множеством преимуществ над соответствующими насыпными катализаторами, полученными другим образом.
[0024] Способ изготовления, описанный в данном патенте, отличается от способа, применяемого для насыпных катализаторов в предшествующем уровне техники. Предшественник насыпного катализатора получают посредством сушки раствора NiW, NiMo или NiMoW, содержащего органический агент, с последующим разложением при высокой температуре (T), приводящим к фазе NiMo/W-C, в основном аморфной, которая составляет предшественник насыпного катализатора. Предшественники насыпного катализатора по данному изобретению отличаются отсутствием кристаллической металлооксидной фазы. Как можно сделать вывод из предшествующего уровня техники, (нано)кристаллическая металлооксидная фаза обычно обнаруживается в предшественниках насыпного катализатора, как это следует из присутствия определенных пиков на дифрактограммах этих материалов, полученных дифракционным рентгеновским анализом (XRD).
[0025] Отсутствие поддерживающего материала в насыпных катализаторах делает крайне затрудненным поддерживание металлооксидной фазы в высокодиспергированном состоянии в системе этого типа. Во время процессов осаждения или термообработки поэтому обычно формируются (нано)кристаллические металлооксидные фазы. Несмотря на высокую концентрацию металлических оксидов в предшественниках насыпного катализатора по данному изобретению, такая кристаллическая фаза неожиданным образом отсутствует. Может быть предусмотрено, что в предшественниках насыпных катализаторов по данному изобретению углеродсодержащая фаза, которая остается после термической обработки, действует в качестве диспергирующего агента для металлооксидной фазы, приводя к предотвращению формирования кристаллической металлооксидной фазы.
[0026] Без намерения быть связанными какой-либо теорией, можно предполагать, что отсутствие каких-либо кристаллических металлооксидных фаз в предшественнике оксидного катализатора указывает на высокую дисперсию металлооксидной фазы, приводящую к катализатору с высоким количеством активных центров, когда оксидная фаза преобразована в активные сульфиды металлов. Более высокая активность наблюдается для новоизобретенного катализатора в отличие от катализатора, полученного посредством способов предшествующего уровня техники, рассмотренных в этом случае.
[0027] Твердотельный предшественник катализатора получают посредством испарения до сухого состояния раствора, содержащего металлические предшественники. Это обеспечивает полную вариабельность в композиции катализатора: большинство, если не все, предшественников металлов, которые присутствуют в растворе, оказываются в предшественнике насыпного катализатора. При осаждении определенной металлооксидной фазы, которая обычно создана при получении других насыпных катализаторов, известных в предшествующем уровне техники, с другой стороны, композиция определена стехиометрией ее нерастворимой фазы. Например, отношение Ni:(Mo+W) катализатора может быть легко отрегулировано в катализаторах по данному изобретению. Как правило, отношение Ni:(Mo+W) между 0,20 и 0,75 применяют в видах применения для гидропереработки, поскольку количество Ni является достаточным для образования кристаллитов MoS2 и/или WS2, которые полностью декорированы атомами Ni, которые действуют в качестве промотора активной фазы. Однако в некоторых случаях более низкое отношение может являться предпочтительным, поскольку это приводит к более низким затратам. Отношение Ni:(Mo+W), более высокое, чем 0,75, приводит обычно к формированию отдельной Ni-сульфидной фазы в конечном катализаторе, который применяют в определенных случаях, когда функциональность Ni-сульфидных фаз является желательной.
[0028] Избегание процесса осаждения устраняет необходимость заниматься растворителем, загрязненным металлом, после фильтрования. Для коммерческого производства катализаторов, это не является несущественным преимуществом.
[0029] Было найдено, что для насыпных катализаторов, полученных способом, описанным ниже, формирование металлических Ni-кристаллов может быть обнаружено в смешанной фазе оксид металла/C, которая образует предшественник насыпного катализатора в результате термообработки, при применении дифракционного рентгеновского анализа (XRD) или просвечивающей электронной микроскопии (TEM). Характеристические пики Ni(0) могут наблюдаться в дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора, при 45° и 52° 2-тета, которые являются показателем присутствия металлических кристаллов Ni(0). Не может быть исключено присутствие C, растворенного в решетчатой структуре Ni, поскольку формирование такой фазы NiCx не приводит к заметно отличающейся дифрактограмме, полученной дифракционным рентгеновским анализом (XRD). Для простоты, в последующем, на кристаллы Ni(0) или NiCx будет делаться ссылка как на Ni-кристаллы. В результате формирования Ni-кристаллов в предшественнике насыпного катализатора, Ni-сульфидные кристаллы будут присутствовать в сульфидированном катализаторе. Эти Ni-кристаллы сформированы при условиях, которые имеют место во время термообработки при температуре >350°C, в качестве стадии при получении предшественника катализатора. Разложение органических веществ во время термообработки приводит к восстановительной окружающей среде, которая совместно с температурой приводит к восстановлению Ni-оксидной фазы и формированию Ni-кристаллов. Хотя результирующий предшественник насыпного катализатора не содержит какой-либо кристаллической металлооксидной фазы, она может не являться поэтому полностью аморфной. В дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) для предшественников насыпного катализатора по данному изобретению, обожженных при температуре >350°C, может наблюдаться наличие пика при 45° 2-тета, которое может быть обусловлено присутствием Ni-кристаллов. Отличительным признаком этого типа катализаторов является то, что, когда Ni-кристаллы сформированы, распределение их частиц по размеру является очень четко определенным, и кристаллы гомогенным образом распределены на протяжении фазы предшественника катализатора, как это может наблюдаться посредством электронной микроскопии. Характерная высокая степень дисперсности Ni-кристаллов указывает на то, что углеродная матрица, которая сформирована, является эффективным диспергирующим агентом для активной фазы. Таким же образом, как и Ni- кристаллы поддерживаются отдельно во время получения катализатора, для смешанных Ni(Mo/W)-сульфидных кристаллитов в активном катализаторе также предусматривается их поддержание в высокодиспергированном состоянии.
[0030] Вместе с этим, композиция NiMo, NiMoW и NiW приводит к улучшенной активности даже при условиях, когда обычным образом применяют CoMo-катализаторы. Показано, что этот тип катализатора может также быть изготовлен при применении полиола или сахара вместо комплексообразующей кислоты.
[0031] Различные варианты осуществления, относящиеся к этим результатам исследований, описаны ниже более подробно.
Получение предшественника насыпного катализатора и насыпного катализатора
[0032] Общий процесс включает следующие стадии. Во-первых, равномерное смешивание органических агентов и предшественников металлов. В идеальном случае формируются металлоорганические комплексы, однако это не требуется. На практике этого достигают посредством приготовления раствора металлических предшественников и органических соединений. Предпочтительным растворителем является вода. Во-вторых, удаление растворителя, который применяют на стадии 1. Это может быть сделано посредством термической сушки в статической печи, посредством сушки распылением или в любом другом устройстве, а также посредством сушки сублимацией или вакуумной сушки. В-третьих, частичное разложение металлоорганической фазы, чтобы образовать смешанную фазу оксид металла/углерод, которая образует предшественник насыпного катализатора. Это осуществляют посредством термической обработки, по существу в инертной атмосфере (например, азота или пара), однако воздух может быть также применен при условии предотвращения полного сгорания органических веществ. Во время этой обработки отношение C:O и C:H органической фазы будет увеличиваться, и материал будет становиться более углеродсодержащим. Это может также быть осуществлено посредством химической реакции, т.е., например, посредством обработки серной кислотой. В-четвертых, формование предшественника катализатора, чтобы получить насыпной катализатор. Это может быть выполнено посредством экструзии, таблетирования, гранулирования, прессования или любого другого подходящего способа, известного в данной области техники. В-пятых, сульфидирование насыпного катализатора, чтобы образовать сульфидированный насыпной катализатор. Это может быть выполнено in-situ в реакторе или ex-situ любым известным способом. Наряду с тем, что вышеуказанные стадии представлены в предпочтительной последовательности, возможны другие последовательности выполнения способа. Например, вы можете формовать предшественник перед разложением, и вы можете также выполнять сульфидирование перед формованием.
[0033] Первая стадия способа предназначена для создания раствора, содержащего металл Группы VIII, металл Группы VIB и органический агент. Предпочтительно, чтобы как соединение Группы VIII, так и соединение Группы VIB добавлялись в подходящей, заранее установленной концентрации, чтобы получить на выходе желательные молярные отношения. Желательно иметь молярное отношение Ni:(Mo+W), которое может варьироваться от 0,05 до 1,05. Более предпочтительно иметь отношение Ni:(Mo+W) 0,10-1,05, в частности, наряду с тем, что отношение Ni:(Mo+W) 0,20-0,75 является наиболее предпочтительным. Реагенты металлов Группы VIII и Группы VIB и органический агент смешивают с протонсодержащей жидкостью. Смесь затем, как правило, нагревают и постоянно перемешивают в течение примерно 1 часа, пока не получают прозрачный раствор. Стадия нагревания является необходимой лишь, когда реакционное взаимодействие предшественников металлов требуется, чтобы обеспечить их растворение. Хотя желательно образовывать прозрачный раствор, в котором все компоненты полностью растворены с целью наличия оптимальной гомогенности на всем протяжении катализатора, присутствие небольшого количества непрореагировавших исходных материалов или преципитата, который образован после реакционного взаимодействия исходных материалов, может, тем не менее, быть приемлемым.
[0034] Предпочтительным металлом Группы VIII является Ni. Предпочтительным металлами Группы VIB являются Mo и W. Неограничивающие примеры подходящих соединений предшественника Ni включают карбонаты и ацетаты и их смеси, включая карбонат никеля, гидроксикарбонат никеля, ацетат никеля, цитрат никеля, гидроксиды никеля, оксид никеля, нитрат никеля, сульфат никеля и их смеси. Предпочтительные соединения предшественников молибдена и вольфрама включают оксид молибдена, молибденовую кислоту, молибдаты аммония, фосфомолибдаты, силикомолибдаты, Mo-ацетилацетонаты, Na-молибдаты, вольфрамовую кислоту, вольфраматы аммония, фосфовольфраматы, силиковольфраматы, Na-вольфраматы и их смеси.
[0035] Органическими веществами, которые могут быть применены при получении, являются углеводы (молекулы, необязательно биологического происхождения, которые по меньшей мере содержат C, H и O). Органические вещества могут быть смесью различных молекул. Масс.% C в сумме органических молекул типично ниже, чем примерно 50%. Органические молекулы включают по меньшей мере 2 атома кислорода. Органические молекулы могут быть введены в качестве отдельных соединений, однако могут также быть введены посредством противоиона металлических солей. Неограничивающие примеры органических добавок или агентов, подходящих для применения, при этом включают уксусную кислоту, аспарагиновую кислоту, лимонную кислоту, муравьиную кислоту, фумаровую кислоту, глюконовую кислоту, гютаминовую кислоту, глиоксиловую кислоту, кетоглутаровую кислоту, малеиновую кислоту, оксиянтарную кислоту, щавелевоуксусную кислоту, пропионовую кислоту, пировиноградную кислоту, янтарную кислоту, фруктозу, глюкозу, лактозу, сахарозу, сорбитол, ксилитол, серин и их смеси. Во всяком случае, органическую добавку добавляют в количестве, которое приводит к молярному отношению C:(Mo+W) между 1,5 и 10 в предшественнике насыпного катализатора.
[0036] Растворитель может быть любым растворителем, который не препятствует реакциям металлсодержащих соединений. Примеры растворителей включают протонсодержащие жидкости, такие как вода, и спирты, такие как метанол, этанол, или их смеси. Предпочтительными протонсодержащими жидкостями являются смеси воды и других протонсодержащих жидкостей, такие как смеси спирта и воды, и более предпочтительной протонсодержащей жидкостью является вода сама по себе.
[0037] Очевидно, что разные протонсодержащие жидкости могут быть применены одновременно в процессе. Например, возможно добавлять суспензию металлсодержащего соединения в этаноле к водному раствору другого металлсодержащего соединения. В некоторых случаях, может быть применено металлсодержащее соединение, которое растворяется в своей собственной кристаллизационной воде. Кристаллизационная вода служит в качестве протонсодержащей жидкости в этом случае.
[0038] Вторая стадия в способе получения катализаторов является стадией сушки. Стадию сушки применяют, чтобы удалить воду или любой другой растворитель, который применяют при приготовлении первоначального раствора, из смеси. На стадии сушки разложение органического агента обычно не происходит. В пределах объема данного изобретения находится то, что нагревание и/или сушка могут быть выполнены в несколько этапов в соответствии с профилем нагревания. Стадия нагревания или сушки может быть выполнена любым способом, известным в данной области техники. В частности, стадия сушки может быть выполнена посредством конвективной сушки при применении горячего газа, например, в лотковой сушилке, или сушкой распылением. В качестве альтернативы, сушка может быть выполнена посредством контактной сушки, например, при применении сушилки с вращающимся диском, лопастной сушилки или скребкового теплообменника. Сушка посредством микроволнового нагревания, сублимационной сушки или вакуумной сушки являются другими вариантами. Сушку распылением типично выполняют при температуре на выходе в интервале от примерно 100° до примерно 200°C и предпочтительно от примерно 120° до примерно 180°C.
[0039] Третья стадия в способе получения катализаторов является частичным разложением металлоорганической фазы. Высушенный предшественник катализатора подвергают дополнительной стадии нагревания или стадии обжига. Эта дополнительная стадия нагревания может быть выполнена при температуре от примерно 300°C до примерно 500°C в течение эффективного периода времени. Этот эффективный период времени будет находиться в интервале от примерно 1 секунды до примерно 24 часов, предпочтительно от примерно 1 минуты до примерно 5 часов. Нагревание (включающее возможное разложение) может быть выполнено в присутствии протекающего кислородсодержащего газа, такого как воздух, протекающего инертного газа, такого как азот, или комбинации кислородсодержащего и инертного газов. Время, температуру и условия для этой стадии выбирают таким образом, чтобы имело место лишь частичное разложение органической добавки. Значительное количество углерода все еще присутствует после стадии термообработки, и атомное отношение C:(Mo+W) в предшественнике насыпного катализатора составляет по меньшей мере 1,5. Отношение C:O и C:H органической фазы, образованной после стадии разложения, обычно ниже, чем для органического агента, добавленного на первой стадии. Найдено, что, как правило, более высокая температура приводит к более низкой активности катализатора. Тем не менее, может являться предпочтительным выполнение обжига при более высокой температуре, поскольку полученная углеродсодержащая фаза, сформированная при более высокой температуре является более огнеупорной, имеет более высокое отношение C:O и C:H и является более стабильной при условиях гидропереработки. Как указано выше, Ni-кристаллы могут быть сформированы во время этой стадии получения. Помимо металлических оксидов и недостаточно определенной органической фазы, кристаллы металлического Ni могут присутствовать после термической обработки. Тем не менее, материал, который сформирован после стадии частичного разложения, будет называться как смешанная фаза оксид металла/C. На практике, стадии сушки и разложения могут быть выполнены в виде одной стадии процесса.
[0040] После этой стадии получают предшественник насыпного катализатора, который типично имеет следующие композиционные свойства:
MoO3+WO3 масс.% между 30-85 масс.%
Молярное отношение Ni:(Mo+W) выше, чем 0,05
Молярное отношение C:(Mo+W) между 1,5 и 10
Площадь поверхности по БЭТ, измеренная посредством физической адсорбции N2, <40 м2/г
[0041] Четвертая стадия в способе получения катализаторов является стадией формования. Композиция предшественника насыпного катализатора, полученная после нагревания, может быть непосредственным образом сформована в виде форм, подходящих для желательного каталитического конечного назначения, чтобы образовать насыпной катализатор. Формование может также выполняться перед второй стадией нагревания/обжига. Формование включает экструзию, таблетирование, гранулирование и/или сушку распылением. Следует заметить, что если композиция насыпного катализатора должна быть применена в суспензионных реакторах, псевдоожиженных слоях, подвижных слоях или увеличенных в объеме слоях, то обычно применяют сушку распылением или гранулирование. Для видов применения с неподвижным слоем или кипящим слоем, как правило, композицию насыпного катализатора экструдируют, таблетируют и/или гранулируют. В случае экструдирования, таблетирования или гранулирования, на любой стадии перед стадией формования или после нее, могут быть добавлены любые добавки, которые обычно применяют, чтобы способствовать формованию. Эти добавки могут содержать стеарат алюминия, поверхностно-активные вещества, графит, крахмал, метилцеллюлозу, бентонит, аттапульгит, полиэтиленгликоли, полиэтиленоксиды или их смеси.
[0042] Для того, чтобы получить экструдаты насыпного катализатора, предшественник насыпного катализатора может быть смешан с неорганической добавкой и водой и экструдирован в присутствии органической добавки для облегчения экструзии. Применяемые связующие материалы могут быть любыми материалами, обычно применяемыми в качестве связующих в катализаторах гидропереработки. Примерами являются кремнезем, кремнезем-глинозем, например, обычный кремнезем-глинозем, глинозем, покрытый кремнеземом, и кремнезем, покрытый глиноземом, глиноземы, такие как (псевдо)бемит или гиббсит, диоксид титана, глинозем, покрытый диоксидом титана, диоксид циркония, катионоактивные глины или анионоактивные глины, такие как сапонит, бентонит, аттапульгит, каолин, сепиолит или гидротальцит, или их смеси. Предпочтительными связующими являются кремнезем, кремнезем-глинозем, глинозем, диоксид титана, глинозем, покрытый диоксидом титана, диоксид циркония, бентонит, аттапульгит или их смеси. Эти связующие могут быть применены непосредственным образом или после пептизации. В некоторых случаях предшественник насыпного катализатора измельчают, чтобы получить частицы меньшего размера, которые способствуют достижению более высокой уплотненной насыпной плотности (CBD) в реакторе с неподвижным слоем. Это может быть выгодным, чтобы получить высокие загрузки металла в расчете на объем реактора, и это может также увеличивать прочность уплотненных частиц. Результирующие экструдаты сушат при 120°C или подвергают дополнительной термообработке при температуре ниже, чем температуры, применяемые во время стадии 2 (стадии сушки) в процессе получения.
[0043] Связующие материалы могут уже быть добавлены во время или после стадии 1 (приготовления раствора) или стадии 2 (стадии сушки) в процессе получения. Это может являться предпочтительным для того, чтобы сделать возможным улучшенное распределение связующих материалов на всем протяжении экструдатов катализатора. Следует понимать, что эти связующие материалы не рассматриваются как являющиеся частью предшественника насыпных катализаторов, поскольку они добавлены исключительно, чтобы предоставлять целостность и прочность катализатору, и не содействуют активности катализатора.
[0044] Формованный материал, который получают после стадии 4, называют насыпным катализатором, который характеризуется следующим:
Молярное отношение Ni:(Mo+W) выше, чем 0,05
Молярное отношение C:(Mo+W) между 1,5 и 10
Минимальная загрузка металла 2,0 моля (Mo+W)/литр объема реактора
[0045] Способ необязательно может включать стадию сульфидирования (стадию 5). Сульфидирование обычно выполняют посредством контактирования предшественника насыпного катализатора, непосредственно после его приготовления или после любой из стадий процесса, с серосодержащим соединением, таким как элементарная сера, сероводород, диметилдисульфид (DMDS) или органические или неорганические полисульфиды. Стадия сульфидирования может быть выполнена в жидкой и газообразной фазе. Сульфидирование может быть выполнено после получения композиции насыпного катализатора. Предпочтительно, чтобы сульфидирование не выполнялось перед любой стадией процесса, посредством которой полученные сульфиды металлов снова становятся их оксидами. Такими стадиями процесса являются, например, термическая обработка или сушка распылением или любая другая высокотемпературная обработка, если она выполняется кислородсодержащей атмосфере. Поэтому, если композицию насыпного катализатора подвергают сушке распылением и/или любым альтернативным способом или термической обработке в кислородсодержащей атмосфере, сульфидирование предпочтительно выполняют после применения любого из этих способов. Конечно, если эти стадии выполняют в инертной атмосфере, сульфидирование может также быть выполнено перед этими стадиями. Если композицию насыпного катализатора применяют в процессах с неподвижным слоем, сульфидирование предпочтительно выполняют после стадии формования и, если это применимо, после последней термической обработки в окислительной атмосфере.
[0046] Сульфидирование может обычно быть выполнено in situ и/или ex situ. Предпочтительно, сульфидирование выполняют in situ, т.е. сульфидирование выполняют в реакторе гидропереработки после того, как композиция оксидного насыпного катализатора загружена в узел гидропереработки.
[0047] Композиция насыпного катализатора в соответствии с данным изобретением в особенности применима для гидропереработки углеводородного сырья. Соответственно, данное изобретение относится к способу гидропереработки углеводородного сырья, данный способ включает осуществление контакта углеводородного сырья в условиях гидропереработки с композицией катализатора, содержащей фазу оксид металла/C, которая содержит по меньшей мере один неблагородный металл Группы VIII, по меньшей мере один металл Группы VIB и необязательно Ni-кристаллы.
Охарактеризование предшественника насыпного катализатора и насыпных катализаторов
[0048] Изотермы адсорбции N2 для катализаторов были получены при применении анализатора Micromeretics Gemini-V. Образцы подвергали воздействию 120°C и вакуума в качестве предварительной обработки перед измерениями. Величины для площади поверхности были получены при применении так называемого метода Брунауэра-Эмметта-Теллера (БЭТ), на данные величины будет делаться ссылка как на SA-BET (площадь поверхности по БЭТ) в последующем тексте.
[0049] Состав предшественников насыпного катализатора или насыпных катализаторов определяли при применении рентгенофлуоресцентной спектроскопии (XRF) и отдельного измерения содержания C. Содержание C определяли на предшественнике катализатора при применении метода анализа сжиганием и определении количества образованного CO2, в расчете на количество образца. Перед измерением рентгенофлуоресцентной спектроскопией (XRF) предшественник катализатора подвергали обжигу, типично до 600°C таким образом, что любые органические вещества были удалены, и получали металлооксидную фазу. Вместе с этим измеряли потерю массы во время этой процедуры обжига. При применении потери массы во время обжига (LOI600°C) и содержания металлов в композиции оксида металла, полученного после обжига, как определено рентгенофлуоресцентной спектроскопией (XRF) [MeOx (масс.% в соответствии с рентгенофлуоресцентной спектроскопией (XRF))], фактический состав композиции предшественника насыпного катализатора или насыпного катализатора рассчитывали при применении Уравнения 1.
Уравнение 1: MeOx (масс.%)=(100% - LOI600°C) * MeOx (масс.% по XRF)
[0050] Измерения рентгеновской дифракцией выполняли по геометрии Q-Q Брэгга-Брентано при применении дифрактометра Bruker D8Advance, который был снабжен анодом из Cu (при применении рентгеновского излучения с длиной волны 1,54 Å) и детектором LYNXEYE. Образец измеряли от 4 до 70,0 °2q при размере шага 0,05 °2q при применении фиксированной щели расходимости и противорассеивающей щели 0,5°. В данной области техники известно, что присутствие любой из кристаллических металлооксидных фаз с релевантными составами (т.е. содержащих Ni и Mo и/или W), будет приводить к наличию по меньшей мере одного пика в дифрактограмме, полученной дифракционным рентгеновским анализом (XRD), в интервале 10-40° 2-тета.
[0051] Ширина пика в дифрактограммах, полученных дифракционным рентгеновским анализом (XRD), является функцией среднего размера кристаллитов наблюдаемой фазы. Уравнение Шеррера, как представлено в Уравнении 2, обычно применяют, чтобы установить размер кристаллита (τ) из ширины (β, полная ширина на половине максимума или FWHM в радианах) пика в позиции θ в дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) (A. L. Patterson, Phys. Rev. 56, 978 1939). Величину 0,9 часто применяют для безразмерной формы коэффициента K, в то время как α представляет собой длину волны применяемого рентгеновского излучения: в этом случае 1,54 Å. Можно легко определить, что для кристаллической фазы с отражением при 40° 2-тета, размер кристаллов 5 нм будет приводить к полной ширине на половине максимума (FWHM) 2° 2-тета. Для кристаллов меньше чем 5 нм, ширина пика будет еще большей.
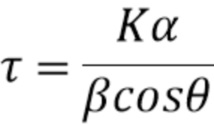
[0052] С учетом вышеизложенного, кристаллическая металлооксидная фаза присутствует, когда размер кристаллов металлооксидных кристаллических доменов больше чем 5 нм. Поэтому, когда установлено, что какая-либо из кристаллических металлооксидных фаз отсутствует в предшественниках катализатора по данному изобретению, это означает, что дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) для предшественника катализатора по данному изобретению, не показывает какого-либо пика с полной шириной на половине максимума (FWHM) менее чем 2° 2-тета в интервале 10-40° 2-тета.
[0053] Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) предшественников NiW, NiMo насыпного катализатора 1-A по 1-D и (сравнительного) предшественника NiMoW насыпного катализатора 1-E и катализатора, который сформирован из этого предшественника, представлены на Фиг. 1. Можно видеть, что для предшественников насыпного катализатора по данному изобретению дифрактограммы, полученные дифракционным рентгеновским анализом (XRD), либо не показывают каких-либо пиков, показывая, что материал является практически аморфным (предшественник насыпного катализатора 1-A и 1-D), либо показывают некоторые очень широкие пики с полной шириной на половине максимума (FWHM) более чем 2° 2-тета, которые могут быть обусловлены углеродной фазой, которая сформирована (предшественники насыпного катализатора 1-B и 1-C) и/или острые пики, расположенные при 2-тета=45° и 52°, которые могут быть обусловлены Ni-кристаллами, сформированными во время стадии частичного разложения (предшественники насыпного катализатора 1-B и 1-C). Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) других предшественников насыпного катализатора по данному изобретению (2-A и 3-A), представлены на Фиг. 5 и 6, наряду с тем, что дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) насыпных катализаторов 4-A и 4-B по данному изобретению, представлены на Фиг. 7. Ни одна из дифрактограмм, полученных дифракционным рентгеновским анализом (XRD) предшественников насыпного катализатора или насыпных катализаторов по данному изобретению не показывает каких-либо пиков с полной шириной на половине максимума (FWHM) менее чем 2° 2-тета в интервале 10-40° 2-тета. Это означает, что кристаллическая металлооксидная фаза не присутствует в этих образцах.
[0054] Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) NiMoW сравнительного насыпного катализатора 1-E и его предшественника, показывают пики, из которых пики с наиболее высокой интенсивностью расположены при 2-тета=36° и 54°, что соответствует формированию искаженной фазы NiWO4. Полная ширина на половине максимума (FWHM) этих пиков меньше, чем 2° 2-тета, указывая на присутствие кристаллической металлооксидной фазы в соответствии с определением, разъясненным выше. Это находится в соответствии с тем, что было в основном показано в предшествующем уровне техники для насыпных катализаторов гидропереработки с композициями NiMo/W, полученных посредством осаждения.
Применение при гидропереработки по данному изобретению
[0055] Композиция катализатора в соответствии с данным изобретением может быть применена практически во всех процессах гидропереработки, чтобы обрабатывать различные исходные материалы при условиях реакции в широких интервалах, например, при температурах от 200 до 450 °C, давлениях водорода от 5 до 300 бар, (0,5-30 МПа), часовых объемных скоростях жидкости от 0,05 до 10 ч-1 и расходах газообразного водорода для обработки от примерно 50 до примерно 2000 м3/м3 (от 280 до 11236 станд. куб футов/баррель). Термин «гидропереработка», применяемый в контексте данного изобретения, охватывает все процессы, в которых углеводородное сырье реагирует с водородом при температурах и давлениях, указанных выше, и включает гидрирование, гидродесульфурацию, гидроденитрогенацию, гидродеметаллизацию, гидродеароматизацию, гидродеоксигенацию, гидроизомеризацию, гидродепарафинизацию, гидроочистку, гидрофинишинг и гидрокрекинг.
[0056] Композиция катализатора по данному изобретению является в особенности эффективной для удаления азота и серы из углеводородного сырья. Соответственно, в предпочтительном варианте осуществления, катализатор по данному изобретению применяют для удаления серы, азота или комбинации серы и азота, из углеводородного сырья. Контактирование углеводородного сырья с композицией катализатора происходит в присутствии водородсодержащего газа для обработки, и реакцию выполняют при эффективных условиях гидропереработки. Контактирование углеводородного сырья с композицией катализатора производит углеводородный продукт, который содержит меньше азота, серы или того и другой, по сравнению с сырьем.
[0057] Углеводородное сырье является материалом, содержащим водород и углерод. Широкий ассортимент нефтепродуктов и углеводородного сырья в качестве химических реагентов может быть подвергнут гидропереработки в соответствии с данным изобретением. Углеводородное сырье включает продукты, полученные или производные от неочищенной нефти, от нефтеносных песков, от продуктов ожижения каменного угля, от сланцевой нефти и от синтеза углеводородов. Композиция катализатора по данному изобретению является в особенности эффективным для удаления серы, азота или комбинации серы и азота из углеводородного сырья. Углеводородное сырье, безусловно, часто содержит азотсодержащие и серосодержащие загрязняющие вещества, часто в форме серосодержащих и/или азотсодержащих органических соединений. Азотсодержащие загрязняющие вещества могут быть основными или неосновными.
Примеры
[0058] Приведенные ниже примеры будут служить для иллюстрирования, однако не для ограничения этого изобретения.
[0059] Пример 1 представлен для сравнения предшественников NiMo/W насыпного катализатора, полученных в соответствии с данным изобретением, с NiMoW насыпным катализатором, известным в данной области техники, и поддерживаемым ссылочным NiMo-катализатором при гидроочистке при высоком давлении (P) (80 бар) исходного тяжелого газойля (HGO).
[0060] Первый предшественник насыпного катализатора изготавливали в соответствии вариантами осуществления, описанными выше. В стеклянном сосуде, 17,01 г D-сорбитола (≥98 масс.%) растворяли в 100 мл воды без нагревания. Когда раствор становился прозрачным, добавляли 10,59 г гептамолибдата аммония (81,5 масс.% MoO3), получая в результате прозрачный раствор. Затем добавляли 9,00 г уксусной кислоты (96 масс.%-ной уксусной кислоты) и 7,47 г ацетата никеля (23,6 масс.% Ni). Получали зеленый прозрачный раствор. Этот раствор нагревали до 85°C в течение одного часа наряду с тем, что испарение воды предотвращали посредством размещения предметного стекла поверх сосуда. Раствор оставался прозрачным. Этот раствор перемещали в фарфоровую чашку и помещали в печь при 120°C на 14 часов при условиях окружающей среды. После сушки получали темно-зеленый раствор. Этот материал помещали во вращающуюся обжиговую печь и нагревали до 325°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 1. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 1. Получение изображений просвечивающим электронным микроскопом (TEM) выполняли на этом предшественнике насыпного катализатора. Характеристическое изображение при высоком увеличении представлено на Фиг. 2. Это был предшественник насыпного катализатора 1-A.
[0061] Второй предшественник насыпного катализатора изготавливали в соответствии вариантами осуществления, описанными выше. В стеклянном сосуде, 26,14 г ацетата никеля (23,58 масс.% Ni) растворяли в 30,34 г водного раствора глюконовой кислоты (50 масс.% глюконовой кислоты) без нагревания. Результирующую смесь нагревали до 60°C в течение 15 минут, получая в результате прозрачный раствор. Затем 24,64 г метавольфрамата аммония (94,10 масс.% WO3) добавляли при том. что температуру раствора поддерживали при 60°C. Снова получали прозрачный раствор. Этот раствор перемещали в фарфоровую чашку и помещали в печь при 120°C на 14 часов при условиях окружающей среды. После сушки получали темно-зеленый раствор. Этот материал помещали во вращающуюся обжиговую печь и нагревали до 400°C в потоке азота при скорости повышения температуры 5°C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 1. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 1. Получение изображений просвечивающим электронным микроскопом (TEM) выполняли на этом предшественнике насыпного катализатора. Характеристическое изображение при высоком увеличении представлено на Фиг. 3. Это был предшественник насыпного катализатора 1-B.
[0062] Третий предшественник насыпного катализатора изготавливали в соответствии вариантами осуществления, описанными выше. В стеклянном сосуде, 2,49 г ацетата никеля (23,6 масс.% Ni) растворяли в 30,34 г водного раствора глюконовой кислоты (50 масс.% глюконовой кислоты) без нагревания. Результирующую смесь нагревали до 60°C в течение 15 минут, получая в результате прозрачный раствор. Затем 24,64 г метавольфрамата аммония (94,1 масс.% WO3) добавляли при том. что температуру раствора поддерживали при 60°C. Снова получали прозрачный раствор. Этот раствор перемещали в фарфоровую чашку и помещали в печь при 120°C на 14 часов при условиях окружающей среды. После сушки получали темно-зеленый раствор. Этот материал помещали во вращающуюся обжиговую печь и нагревали до 400°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 1. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 1. Получение изображений просвечивающим электронным микроскопом (TEM) выполняли на этом предшественнике катализатора. Характеристическое изображение при высоком увеличении представлено на Фиг. 4. Это был предшественник насыпного катализатора 1-C.
[0063] Четвертый предшественник насыпного катализатора изготавливали в соответствии вариантами осуществления, описанными выше. В стеклянном сосуде, 16,38 г α-D-глюкозы (безводной, 96%) растворяли в 120 мл воды. После того, как глюкоза была растворена, добавляли 10,59 г гептамолибдата аммония (81,5 масс.% MoO3). Затем добавляли 9,00 г уксусной кислоты (96 масс.%-ной уксусной кислоты) и 7,47 г ацетата никеля (23,6 масс.% Ni). Раствор нагревали до 85°C в течение одного часа наряду с тем, что испарение воды предотвращали посредством размещения предметного стекла поверх сосуда. Результирующий раствор еще содержал небольшое количество твердотельного материала. Во втором стеклянном сосуде, 16,83 г α-D-глюкозы (безводной, 96%) растворяли в 120 мл воды. После того, как глюкоза была растворена, добавляли 10,59 г гептамолибдата аммония (81,5 масс.% MoO3). Затем добавляли 9,00 г уксусной кислоты (96 масс.%-ной уксусной кислоты) и 7,47 г ацетата никеля (23,6 масс.% Ni). Результирующий раствор содержал небольшое количество твердотельного материала неясного происхождения. Содержимое обоих стеклянных сосудов объединяли в фарфоровой чашке и помещали в печь при 120°C на 14 часов при условиях окружающей среды. После сушки получали темно-зеленый раствор. Этот материал помещали во вращающуюся обжиговую печь и нагревали до 325°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 1. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 1. Это был предшественник насыпного катализатора 1-D.
[0064] Сравнительный катализатор изготавливали в соответствии с принципами, известными в данной области техники. NiMoW насыпной катализатор получали в соответствии с принципами US 6566296. В реакторе 755 г гидроксикарбоната никеля (содержащего 70,0 масс.% Ni) суспендировали в 500 мл воды. Температуру повышали до 60°C и добавляли 90 г молибденовой кислоты (90 масс.% MoO3). Затем добавляли 137 г вольфрамовой кислоты (70,31 масс.% W). Этой смеси предоставляли возможность реакционного взаимодействия в течение достаточного времени для полного реакционного взаимодействия исходных материалов. Результирующую суспензию фильтровали, чтобы получить осадок. Он является предшественником сравнительного насыпного катализатора 1-E. Картина рентгеновской дифракции (XRD) для этого материала представлена на Фиг. 1. 597 г полученного твердотельного вещества смешивали с 241,85 г бемита и 24,37 г 65% HNO3 и перемешивали, чтобы получить гомогенную смесь. Содержание воды в экструзионной смеси регулировали (посредством нагревания или добавления воды) для того, чтобы получить экструдируемую смесь, как известно специалистам в данной области техники. Смесь экструдировали при применении отверстий диаметром 1,5 мм и экструдаты сушили в течение одного часа при 120°C. Результирующий материал помещали во вращающуюся обжиговую печь и нагревали до 385°C в потоке воздуха при скорости повышения температуры 5 °C/мин и времени выдерживания 1 час. Результирующий материал имел следующий состав, как определено рентгенофлуоресцентной спектроскопией (XRF): WO3 (31,4 масс.%), NiO (31,3 масс.%), MoO3 (20.6) и Al2O3 (15,6 масс.%). Площадь поверхности по БЭТ (SA-BET) этого материала, как измерено посредством физической адсорбции N2, составляла более чем 120 м2/г. Хотя часть этой площади поверхности (SA) создается Al2O3, низкая концентрация этого компонента не может обеспечивать эту высокую площадь поверхность (SA). Это означает, что металлооксидный предшественник насыпного катализатора 1-E также имеет значительную площадь поверхности по БЭТ (SA-BET). Затем 4,4 грамма диэтиленгликоля отвешивали и разбавляли водой в достаточном объеме, чтобы выполнить импрегнирование объема пор в экструдатах. Результирующий раствор добавляли к 50 г вышеуказанных обожженных экструдатов. Импрегнирование выполняли в течение примерно 30 минут при 120°C в закрытом контейнере при постоянном перемешивании. Затем экструдаты нагревали при вращении до тех пор, пока экструдаты не достигали температуры 90°C, как признака того, что материал являлся сухим, и вся вода была испарена. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 1. Картина рентгеновской дифракции (XRD) для этого катализатора представлена также на Фиг. 1. Он является предшественником сравнительного насыпного катализатора 1-E.
[0065] В качестве второго сравнительного катализатора, поддерживаемый NiMo-Al2O3 катализатор, который является коммерческим катализатором для гидроочистки исходных дистиллятов при высоком давлении (P), был включен в испытание. Состав и площадь поверхности этого катализатора при обследовании посредством физической адсорбции азота представлены в Таблице 1. Он является сравнительным катализатором 1-F.
[0066] Из данных, представленных в Таблице 1, можно заметить, что площадь поверхности (SA) предшественников насыпных катализаторов 1-A по 1-D является очень малой, во всех случаях меньше, чем та, что может быть измерена при применении метода физической адсорбции N2. Для сравнительных катализаторов 1-E и 1-F, с другой стороны, наблюдалась большая площадь поверхности (SA).
Таблица 1: Состав и площадь поверхности по БЭТ (SA-BET), как определено посредством физической адсорбции N2, предшественников насыпного катализатора (b.c.p.) 1-A - 1-D и сравнительных катализаторов 1-E и 1-F.
[0067] Предшественники насыпного катализатора 1-A - 1-D в соответствии с данным изобретением отличаются присутствием значительного количества углерода и молярным отношением C:(Mo+W) по меньшей мере 4. Кроме того, в отличие от сравнительных катализаторов 1-E и 1-F, площадь поверхности катализаторов в соответствии с данным изобретением всегда меньше, чем 5 м2/г. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD), для предшественников насыпного катализатора 1-A - 1-D в соответствии с данным изобретением, предшественника сравнительного насыпного катализатора 1-E и сравнительного насыпного катализатора 1-E, представлена на Фиг. 1. Дифрактограммы предшественника сравнительного насыпного катализатора 1-E и сравнительного насыпного катализатора 1-E показывают наиболее интенсивные пики при 2-тета=36° и 54°. Эти пики могут быть обусловлены присутствием искаженной нанокристаллической фазы NiWO4. Пики с полной шириной на половине максимума (FWHM) менее чем 2° 2-тета не присутствуют в интервале 2-тета 10-40° на дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) предшественников насыпного катализатора 1-A по 1-D в соответствии с данным изобретением. Острые пики (с полной шириной на половине максимума (FWHM) менее чем 1° 2-тета), которые наблюдаются при 45 и 52 градусах 2-тета в дифрактограмме катализаторов 1-A и 1-B, могут быть обусловлены Ni-кристаллами, сформированными и не происходящими от какой-либо кристаллической металлооксидной фазы.
[0068] В изображениях, полученных просвечивающим электронным микроскопом (TEM), предшественников насыпного катализатора 1-A, 1-B и 1-C как представлено на Фиг. 2-4, присутствие Ni-кристаллов также наблюдалось очевидным образом. Общей особенностью предшественника насыпного катализатора по данному изобретению является то, что Ni-кристаллы, которые сформированы, очень хорошо диспергированы в том смысле, что (i) пространственное распределение частиц на протяжении образца является в значительной степени гомогенным, и (ii) распределение частиц по размеру является чрезвычайно узким. Как можно видеть на Фиг. 2, в предшественнике насыпного катализатора 1-A, Ni-кристаллы являются небольшими (<5 нм в диаметре), и их концентрация низкая. По этой причине, пики не наблюдаются в соответствующей дифрактограмме, полученной дифракционным рентгеновским анализом (XRD), несмотря на присутствие кристаллической Ni-фазы. Поэтому отсутствие каких-либо пиков в дифрактограмме, полученной дифракционным рентгеновским анализом (XRD), не означает, что Ni-кристаллы не присутствуют в предшественниках насыпного катализатора. Присутствие Ni-кристаллов в микрофотографиях, полученных просвечивающей электронной микроскопией (TEM), (Фиг. 3 и 4) даже более резко выражено в предшественниках насыпного катализатора 1-B и 1-C.
[0069] Процедура испытания: Предшественники насыпного катализатора и сравнительные катализаторы калибровали до ситовой фракции 125-300 мкм и загружали в реактор при объеме 0,9 мл. Испытательная установка, применяемая для выполнения испытания, предоставляла возможность одновременного испытания различных катализаторов при идентичных условиях обработки (температуре, давлении, величине подачи и отношении H2/нефтяной продукт), наряду с тем, что часовая объемная скорость жидкости (LHSV) может быть отрегулирована для каждого катализатора, например, посредством загрузки катализатора. Катализаторы предварительно сульфидировали при применении 2,5 масс.% диметилдисульфида (DMDS) от величины подачи легкого газойля (LGO), который подавали над катализатором при часовой объемной скорости жидкости (LHSV) 3,0 при 45 бар (4,5 МПа) и при отношении H2/нефтяной продукт 300 нл/л. Температурная программа, которую применяли во время предварительного сульфидирования представлена в Таблице 2. Каталитическую активность катализаторов оценивали при давлении 80 бар (8 МПа), 341°C и отношении H2/нефтяной продукт 500 нл/л при обработке тяжелого газойля (HGO) при параметрах подачи, представленных в Таблице 3.
Таблица 2: Температурный протокол предварительного сульфидирования, применяемый для активирования предшественников насыпного катализатора 1-A - 1-D и сравнительных катализаторов 1-E и 1-F.
Таблица 3: Параметры подачи тяжелого газойля (HGO), применяемые для выполнения испытания предшественников насыпного катализатора 1-A - 1-D и сравнительных катализаторы 1-E и 1-F.
[0070] Объем и масса катализаторов в различных реакторах и содержание S и N в результирующем продукте при различных условиях реакционного взаимодействия представлены в Таблице 4. Загрузка катализатора представлена в граммах в пересчете на сухое вещество (г, в пересчете на сухое вещество). Это означает массу предшественника насыпного катализатора или катализатора после обжига при 600°C в воздушной среде. Прежде всего, можно заметить, что все предшественники насыпного катализатора являются более активными, чем сравнительный катализатор 1-F, коммерческий катализатор NiMo/Al2O3. При часовой объемной скорости жидкости (LHSV) 2,0, сравнительный катализатор 1-F был в состоянии производить продукт с 762 млн-1 S и 52 млн-1 N. Предшественники насыпного катализатора 1-A по 1-D и сравнительный насыпной катализатор 1-E могут производить продукт с более низкой концентрацией N при часовой объемной скорости жидкости (LHSV) 2,4, что указывает на то, что относительная объемная активность этих катализаторов по меньшей мере на 20% выше, чем сравнительного катализатора 1-F. Кроме того, можно видеть, что предшественники насыпного катализатора 1-A - 1-D по данному изобретению являются значительно более активными в отношении гидроденитрогенации (HDN), чем сравнительный насыпной катализатор 1-E. При часовой объемной скорости жидкости (LHSV) 2,4, сравнительные катализаторы 1-E были в состоянии производить продукт с 50 млн-1 N, в то время как катализаторы по данному изобретению производили продукт с 28 млн-1 N или менее. В ряде видов применения гидропереработки, таких как предварительная обработка гидрокрекингом и предварительная обработка каталитическим крекингом в псевдоожиженном слое (FCC), обычно вакуумного газойля в качестве сырья, удаление азота является главной целью. В этих операциях, предшественники насыпного катализатора по данному изобретению имеют все значительное преимущество по сравнению со сравнительным катализатором 1-E. Высокая активность предшественников насыпного катализатора 1-A по 1-D по данному изобретению по сравнению со сравнительными катализаторами является неожиданной, принимая во внимание низкую площадь поверхности по БЭТ (SA-BET) этих катализаторов.
Таблица 4: Загрузка катализатора и наблюдаемая конверсия для предшественников насыпного катализатора 1-A - 1-D и сравнительных катализаторов 1-E и 1-F при тестовой обработке тяжелого газойля (HGO) при 80 бар (8 МПа).
[0071] Пример 2 представлен для сравнения NiW предшественника насыпного катализатора, полученного в соответствии с данным изобретением, с CoMo предшественником насыпного катализатора, известного в данной области техники, и поддерживаемом CoMo ссылочным катализатором при гидропереработки при низком давлении (P) (30 бар (3 МПа)) исходного легкого газойля (LGO). В стеклянном сосуде, 12,44 г ацетата Ni (23,6 масс.% Ni) растворяли в 30,34 г раствора глюконовой кислоты (содержащего 50 масс.% D-глюконовой кислоты) при комнатной температуре. Добавляли 24,64 г метавольфрамата аммония (94,1 масс.% WO3) и раствор нагревали до 70°C при постоянном перемешивании, получая в результате прозрачный раствор. Этот раствор сушили в статической печи при 120°C в течение 5 часов. Результирующий коричнево-зеленоватый твердотельный материал помещали во вращающуюся обжиговую печь и нагревали до 400°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 5. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 5. Это представляет собой предшественник насыпного катализатора 2-A.
[0072] Затем приготавливали два сравнительных катализатора. Первоначально, сравнительный CoMo предшественник насыпного катализатора приготавливали посредством приведенного ниже процесса, как описано в US 7951746. В стеклянном сосуде 25,74 г ацетата кобальта (23,7 масс.% Co) растворяли в 165 мл раствора глиоксиловой кислоты раствор (50 масс.% глиоксиловой кислоты) при температуре окружающей среды. Добавляли 36,38 г гептамолибдата аммония (81,5 масс.% MoO3) и раствор нагревали до 80°C при постоянном перемешивании. Когда температура достигала примерно 60°C, реакция гептамолибдата аммония являлась более интенсивной, и наблюдалось образование пены. После перемешивания в течение часа при 80°C получают раствор, который является почти прозрачным, однако все же содержит небольшое количество твердотельного материала. Результирующую смесь сушили в течение ночи в статической печи при 120°C. Темно-окрашенное твердотельное вещество помещали во вращающуюся обжиговую печь и нагревали до 325°C в потоке сухого воздуха при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 5. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 5. Он является предшественником сравнительного насыпного катализатора 2-B.
[0073] Поддерживаемый CoMo-Al2O3 катализатор приготавливали импрегнированием CoMo-раствором коммерческой Al2O3 основы, применяемой для получения катализаторов гидроочистки. Экструдаты γ-Al2O3 имеют площадь поверхности по БЭТ (SA-BET) 267 м2/г, средний диаметр пор, как определено десорбцией N2, 8 нм и объем пор, как определено посредством физической адсорбции N2, 0,78 мл/г. Раствор Co32+[Co2Mo10O38H4]6- приготавливали посредством загрузки металла, сравнимой с коммерческими CoMo-Al2O3 катализаторами при применении способа изготовления импрегнирующего раствора как опубликовано в статье в Langmuir 2013, 29, 207-215. Импрегнирующий раствор приготавливали смешиванием 180,0 г MoO3 (100%) с 0,80 л воды в стеклянном сосуде. Затем добавляли 612,5 г раствора H2O2 (30 масс.% H2O2) и суспензию нагревали до 40°C. После примерно 2 часов перемешивания при 40°C получают прозрачный раствор. К этому раствору добавляли 79,9 г CoCO3 (46 масс.% Co) небольшими порциями в течение периода времени 45 минут. Результирующую смесь нагревали до 90°C и предоставляли ей возможность реакционного взаимодействия в течение 2 часов. Раствор разделяли на 9 автоклавов, вмещающих каждый 50 мл раствора, которые нагревали при автогенном давлении до 150°C, где они поддерживались в течение 2 часов. Результирующий раствор сушили распылением при применении стендовой распылительной сушилки типа Buchi Mini Spraydryer B-290, снабженной инертным контуром B295. Во время сушки распылением, температура на входе составляла 180°C, и температура на выходе составляла 100-110°C. Раствор подавали в распылительную сушилку при степени пропускания материала примерно 200 мл/час. Полученный порошок повторно растворяли в воде, чтобы получить импрегнирующий раствор. Конечный катализатор получали посредством импрегнирования этим раствором объема пор глиноземного носителя, в соответствии с чем объем и концентрацию раствора регулировали, чтобы достигнуть желательного состава конечного катализатора. Конечный катализатор, содержащий 23,81% MoO3 и 6,16% CoO, при определении рентгенофлуоресцентной спектроскопией (XRF) после обжига при 600°C. Эта композиция соответствует составу коммерческих CoMo-Al2O3 катализаторов, которые обычно применяют в этом виде применения. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 5. Он является сравнительным катализатором 2-C.
[0074] Из данных, представленных в Таблице 5, можно заметить, что площадь поверхности (SA) предшественника насыпного катализатора 2-A меньше, чем та, что может быть измерена при применении метода физической адсорбции N2. Для предшественника сравнительного насыпного катализатора 2-B площадь поверхности (SA) является чрезвычайно низкой, наряду с тем, что при сравнительном катализаторе 2-C наблюдают высокую площадь поверхности (SA).
[0075] На Фиг. 5 представлены дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора 2-A и предшественника сравнительного насыпного катализатора 2-B. Отсутствуют пики в интервале 10-40° 2-тета в дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора, что показывает отсутствие какой-либо (нано)кристаллической металлооксидной фазы. Можно заметить, что на дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора 2-A, острый пик присутствует при примерно 45° 2-тета, который может быть обусловлен присутствием Ni-кристаллов. Этот пик отсутствует в предшественнике сравнительного насыпного катализатора 2-B.
Таблица 5: Состав и площадь поверхности по БЭТ (SA-BET), как определено посредством физической адсорбции N2, предшественника насыпного катализатора 2-A, предшественника сравнительного насыпного катализатора 2-B и сравнительного катализатора 2-C.
[0076] Предшественники насыпного катализатора и поддерживаемый катализатор калибровали до ситовой фракции 125-300 мкм и загружали в реактор при объеме 0,9 мл. Испытательная установка, применяемая для выполнения испытания, предоставляла возможность одновременного испытания различных катализаторов при идентичных условиях обработки. Катализаторы предварительно сульфидировали при применении 2,5 масс.% диметилдисульфида (DMDS) от величины подачи легкого газойля (LGO), который подавали над катализатором при часовой объемной скорости жидкости (LHSV) 3,0 при 30 бар (3 МПа) и при отношении H2/нефтяной продукт 300 нл/л. Температурная программа, которую применяли во время предварительного сульфидирования представлена в Таблице 6. Каталитическую активность катализаторов оценивали при давлении 30 бар (3 МПа), 350°C и отношении H2/нефтяной продукт 200 нл/л при обработке легкого газойля (LGO) при параметрах подачи, представленных в Таблице 7.
Таблица 6: Температурный протокол предварительного сульфидирования, применяемый для активирования образцов 2-A - 2-C.
Таблица 7: Параметры подачи легкого газойля (LGO), применяемые для выполнения испытания образцов 2-A - 2-C.
[0077] Объем и масса образцов в различных реакторах и содержание S в результирующем продукте при различных условиях реакционного взаимодействия представлены в Таблице 8. Можно заметить, что активность гидродесульфуризации (HDS) NiW предшественника насыпного катализатора 2-A значительно выше активностей сравнительного CoMo предшественника насыпного катализатора 2-B и сравнительного CoMo-Al2O3 катализатора 2-C. NiW предшественник насыпного катализатора 2-A обеспечивает достижение более низкой величины содержания S (12 млн-1) при часовой объемной скорости жидкости (LHSV) 1,5, чем сравнительный CoMo предшественник насыпного катализатора 2-B при часовой объемной скорости жидкости (LHSV) 1,2 (89 млн-1) и сравнительный поддерживаемых катализатор 2-C (240 млн-1) при часовой объемной скорости жидкости (LHSV) 1,5. Поскольку обычно катализаторы с CoMo составом применяют при гидропереработки при низком давлении (P) исходных дистиллятов, это является неожиданным результатом.
Таблица 8: Применяемые загрузка катализатора, часовая объемная скорость жидкости (LHSV) и наблюдаемая конверсия для предшественника насыпного катализатора 2-A, предшественника сравнительного насыпного катализатора 2-B и сравнительного катализатора 2-C при проведении тестовой обработки легкого газойля (LGO) при 30 бар (3 МПа).
[0078] Пример 3 представлен для сравнения NiMoW предшественника насыпного катализатора, полученного в соответствии с данным изобретением, с CoMo предшественником насыпного катализатора, при применении точно такого же способа получения, при среднем давлении (45 бар (4,5 МПА)) для обработки исходного легкого газойля (LGO). В стеклянном сосуде, 12,44 г ацетата никеля (23,6 масс.% Ni) растворяли в 30,34 г раствора глюконовой кислоты (50 масс.% D-глюконовой кислоты) при комнатной температуре. Добавляли 12,32 г метавольфрамата аммония (94,1 масс.% WO3) и 8,83 г гептамолибдата аммония (81,5 масс.% MoO3) и раствор нагревали до 70°C при постоянном перемешивании и поддерживали при этой температуре, наряду с предотвращением испарения воды, в течение одного часа. Результирующий раствор сушили в статической печи при 120°C в течение 5 часов. Результирующий твердотельный материал помещали во вращающуюся обжиговую печь и нагревали до 400°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 9. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 6. Это представляет собой предшественник насыпного катализатора 3-A.
[0079] Сравнительный CoMo предшественник насыпного катализатора приготавливали таким же способом. В стеклянном сосуде, 12,45 г ацетата кобальта (23,7 масс.% Co) растворяли в 30,34 г раствора глюконовой кислоты (50 масс.% D-глюконовой кислоты) при комнатной температуре. Добавляли 17,66 г гептамолибдата аммония (81,5 масс.% Mo) и раствор нагревали до 70°C при постоянном перемешивании. Результирующий раствор сушили на протяжении ночи в статической печи при 120°C в течение 5 часов. Результирующий твердотельный материал помещали во вращающуюся обжиговую печь и нагревали до 400°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Состав результирующего материала и площадь поверхности при обследовании посредством физической адсорбции азота представлены в Таблице 9. Дифрактограмма, полученная дифракционным рентгеновским анализом (XRD) этого предшественника насыпного катализатора, представлена на Фиг. 6. Результирующий материал является предшественником сравнительного насыпного катализатора 3-B.
[0080] Из данных, представленных в Таблице 9, можно заметить, что площадь поверхности (SA) обоих катализаторов меньше, чем та, что может быть измерена при применении метода физической адсорбции N2. На Фиг. 6 представлены дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора 3-A и предшественника сравнительного насыпного катализатора 3-B. Отсутствуют пики в интервале 10-40° 2-тета в дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора, что показывает отсутствие какой-либо (нано)кристаллической металлооксидной фазы. Можно заметить, что на дифрактограмме, полученной дифракционным рентгеновским анализом (XRD) предшественника насыпного катализатора 3-A, острый пик присутствует при примерно 45° 2-тета, который может быть обусловлен присутствием Ni-кристаллов. Этот пик отсутствует в предшественнике сравнительного насыпного катализатора 3-B.
Таблица 9: Состав и площадь поверхности по БЭТ (SA-BET), как определено посредством физической адсорбции N2, предшественников насыпного катализатора 3-A и 3-B.
[0081] Процедура испытания: Предшественники насыпного катализатора калибровали до ситовой фракции 125-300 мкм и загружали в реактор при объеме 0,9 мл. Испытательная установка, применяемая для выполнения испытания, предоставляла возможность одновременного испытания различных катализаторов при идентичных условиях обработки. Образцы предварительно сульфидировали при применении 2,5 масс.% диметилдисульфида (DMDS) от величины подачи легкого газойля (LGO), который подавали над катализатором при часовой объемной скорости жидкости (LHSV) 3,0 при 45 бар (4,5 МПа) и при отношении H2/нефтяной продукт 300 нл/л. Температурная программа, которую применяли во время предварительного сульфидирования представлена в Таблице 10. Каталитическую активность катализаторов оценивали при давлении 45 бар (4,5 МПа), 350°C и отношении H2/нефтяной продукт 300 нл/л при обработке легкого газойля (LGO) при параметрах подачи, представленных в Таблице 11.
Таблица 10: Температурный протокол предварительного сульфидирования, применяемый для активирования образцов 3-A и 3-B.
Таблица 11: Параметры подачи легкого газойля (LGO), применяемые для выполнения испытания образцов 3-A и 3-B.
[0082] Объем и масса предшественников насыпного катализатора в различных реакторах, объемная скорость, которая была применена, и содержание N и S в результирующем продукте при различных условиях реакционного взаимодействия представлены в Таблице 12. Можно заметить, что активность гидродесульфуризации (HDS) и гидроденитрогенации (HDN) NiMoW предшественника насыпного катализатора 3-A значительно выше активности предшественника сравнительного CoMo насыпного катализатора 3-B. Например, предшественник насыпного катализатора 3-A обеспечивает достижение значительно более низких величин содержания S (39 млн-1) при часовой объемной скорости жидкости (LHSV) 3,0, чем предшественник сравнительного насыпного катализатора 3-B (72 млн-1) при часовой объемной скорости жидкости (LHSV) 2,0. Это означает, что предшественник насыпного катализатора 3-A по данному изобретению имеет объемную активность гидродесульфуризации (HDS) более чем 150% в сравнении с предшественником сравнительного насыпного катализатора 3-B. Это является неожиданным результатом, поскольку для этого типа условий (гидроочистка при среднем давлении исходных дистиллятов) катализаторы с CoMo-композициями обычно применяют.
Таблица 12: Применяемые загрузка катализатора, часовая объемная скорость жидкости (LHSV) и наблюдаемая конверсия для предшественников насыпного катализатора 3-A и 3-B при проведении тестовой обработки легкого газойля (LGO) при 45 бар (4,5 МПа).
[0083] Пример 4 представлен для иллюстрирования формирования предшественников насыпного катализатора по данному изобретению, чтобы образовать насыпные катализаторы по данному изобретению, и их применения для гидропереработки при высоком давлении. В стеклянном сосуде, 134,66 г гидроксикарбоната никеля (48,4 масс.% Ni) суспендировали в 300 мл воды и нагревали до 75°C. Примерно через 30 минут, 217,78 г MoO3 (100 масс.% MoO3) добавляли небольшими порциями: формирование CO2 наблюдали посредством образования пузырьков. Температуру увеличивали до 90°C и смеси предоставляли возможность реакционного взаимодействия в течение 2 часов, наряду с тем, что испарение воды предотвращали посредством размещения крышки на сосуде. Затем добавляли 400 г 50 масс.%-ного раствора глюконовой кислоты. Получали прозрачный, очень темный, сине-зеленый раствор. Этот раствор сушили на протяжении ночи в статической печи при 120°C в течение 5 часов. Результирующий твердотельный материал помещали во вращающуюся обжиговую печь и нагревали до 450°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Это представляет собой предшественник насыпного катализатора 4-A.
[0084] В стеклянном сосуде, 80,79 г гидроксикарбоната никеля (48,4 масс.% Ni) суспендировали в 300 мл воды и нагревали до 75°C. Примерно через 30 минут, 130.67 г MoO3 (100 масс.% MoO3) добавляли небольшими порциями: формирование CO2 наблюдали посредством образования пузырьков. Температуру увеличивали до 90°C и смеси предоставляли возможность реакционного взаимодействия в течение 2 часов, наряду с тем, что испарение воды предотвращали посредством размещения крышки на сосуде. Затем добавляли 400 г 50 масс.%-ного раствора глюконовой кислоты. Получали прозрачный, очень темный, сине-зеленый раствор. Этот раствор сушили на протяжении ночи в статической печи при 120°C в течение 5 часов. Результирующий твердотельный материал помещали во вращающуюся обжиговую печь и нагревали до 350°C в потоке азота при скорости повышения температуры 5 °C/мин и времени выдерживания 4 часа. Это представляет собой предшественник насыпного катализатора 4-B.
[0085] Предшественники насыпного катализатора измельчали при применении шаровой мельницы и затем смешивали влажным методом с примерно 5 масс.% оксидного связующего материала (в расчете на общую массу композиции катализатора). Содержание воды в смеси регулировали для того, чтобы получить экструдируемую смесь, и смесь затем экструдировали. Результирующие твердотельные цилиндрические экструдаты сушили при 120°C в течение 16 часов (на протяжении ночи). Таким образом получали насыпные катализаторы 4-A и 4-B. Эти катализаторы показывают достаточно высокую прочность и низкое истирание, чтобы подходить для загрузки в коммерческий реактор гидроочистки с неподвижным слоем. Дифрактограммы, полученные дифракционным рентгеновским анализом (XRD) этих насыпных катализаторов представлены на Фиг. 7.
[0086] Состав насыпных катализаторов 4-A и 4-B и площадь поверхности при обследовании посредством физической адсорбции азота экструдатов представлены в Таблице 13. Можно заметить, что оба из насыпных катализаторов показывают очень низкую площадь поверхности по БЭТ (SA-BET) или ее отсутствие. На дифрактограммах, полученных дифракционным рентгеновским анализом (XRD), на Фиг. 7 можно заметить, что пики присутствуют при 45° и 52° 2-тета, указывая на присутствие Ni-кристаллов в этих насыпных катализаторах. Пики не наблюдаются в интервале 10-40° 2-тета, показывая, что нанокристаллическая металлооксидная фаза не присутствует в этих насыпных катализаторах.
Таблица 13: Состав, площадь поверхности по БЭТ (SA-BET), как определено посредством физической адсорбции N2, насыпных катализаторов 4-A и 4-B.
[0087] Экструдаты насыпного катализатора калибровали и просеивали, чтобы удалить экструдаты с длиной по отношению к диаметру более чем примерно 2,5. Выдержанные по размерам экструдаты затем загружали в реактор при объеме 10 мл. Испытательная установка, применяемая для выполнения испытания, предоставляла возможность одновременного испытания различных катализаторов при идентичных условиях обработки. Катализаторы предварительно сульфидировали при применении 2,5 масс.% диметилдисульфида (DMDS) от величины подачи легкого газойля (LGO), который подавали над катализатором при часовой объемной скорости жидкости (LHSV) 3,0 при 45 бар (4,5 МПа) и при отношении H2/нефтяной продукт 300 нл/л. Температурная программа, которую применяли во время предварительного сульфидирования представлена в Таблице 14. Каталитическую активность катализаторов оценивали при давлении 80 бар (8 МПа), 290°C и отношении H2/нефтяной продукт 500 нл/л при обработке смеси легкий газойль (LGO)/легкий рецикловый газойль (LCO) при параметрах подачи, представленных в Таблице 15. Катализатор подвергали воздействию смеси легкий газойль (LGO)/легкий рецикловый газойль (LCO) при реакционных условиях в течение примерно 8 дней.
Таблица 14: Температурный протокол предварительного сульфидирования, применяемый для активирования образцов 4-A и 4-B.
Таблица 15: Параметры подачи смеси легкий газойль (LGO)/легкий рецикловый газойль (LCO), применяемые для выполнения испытания образцов 4-A и 4-B.
[0088] Объем и масса насыпных катализаторов в различных реакторах, объемная скорость, которая была применена, и содержание N и S в результирующем продукте представлены в Таблице 16. Можно заметить, что активность гидродесульфуризации (HDS) и гидроденитрогенации (HDN) насыпного катализатора 4-B значительно выше активности насыпного катализатора 4-B, поскольку более низкие величины для S и N получены при одних и тех же условиях реакционного взаимодействия.
Таблица 16: Применяемые загрузка катализатора, часовая объемная скорость жидкости (LHSV) и наблюдаемая конверсия для насыпных катализаторов 4-A и 4-B при проведении тестовой обработки смеси легкий газойль (LGO)/легкий рецикловый газойль (LCO) при 80 бар (8 МПа).
[0089] После испытания для определения рабочих характеристик использованные катализаторы удаляли из реактора и не загружали в нефтепродукты. Затем, использованные катализаторы промывали толуолом при применении экстракционного оборудования Soxhlet, чтобы удалить любой подаваемый материал, оставшийся в порах катализатора. После этой обработки, любой оставшийся толуол удаляли испарением. Физическую адсорбцию N2 выполняли на использованных катализаторах и определяли содержание C. Результаты анализа для использованных катализаторов представлены в Таблице 17.
[0090] Анализ использованного катализатора показывает, что содержание углерода в катализаторе может быть уменьшено во время применения, как в случае насыпного катализатора 4-B, где молярное отношение C:(Mo+W) уменьшено от 4,7 до 2,1. Очевидно некоторая часть органической фазы удаляется при условиях реакционного взаимодействия. Это является неожиданным результатом, поскольку, как правило, при гидропереработки, углерод осаждается на катализаторе в форме нагара, и содержание углерода в использованном катализаторе выше, чем в свежеприготовленном катализаторе. Кроме того, для катализатора 4-B, площадь поверхности по БЭТ (SA-BET) использованного катализатора значительно больше, чем в свежеприготовленном насыпном катализаторе. Однако обычно постоянная площадь поверхности (SA) или уменьшение в площади поверхности (SA) наблюдается вследствие дезактивации катализаторов, при сравнении использованных катализаторов со свежеприготовленным катализатором.
Таблица 17: Содержание углерода и площадь поверхности по БЭТ (SA-BET) использованных насыпных катализаторов 4-A и 4-B.
Реферат
Изобретение относится к предшественнику насыпного катализатора на основе NiW, NiMo или NiMoW, предназначенного для использования в процессах гидропереработки углеводородного сырья, содержащего органические соединения серы и/или азота, где данный предшественник насыпного катализатора имеет состав, содержащий оксид никеля, а также оксид молибдена, или оксид вольфрама, или их смесь, а также органический компонент, полученный из органической добавки, где общее количество оксида молибдена и оксида вольфрама составляет от 30 мас.% до 85 мас.%, молярное отношение никеля к сумме молибдена и вольфрама составляет от 0,05 до 1,05, и молярное отношение углерода к сумме молибдена и вольфрама составляет от 1,5 до 10; и где органическая добавка выбрана из уксусной кислоты, муравьиной кислоты, глюконовой кислоты, пропионовой кислоты, фруктозы, глюкозы, лактозы, сахарозы, сорбита, ксилита и их смесей. Также изобретение относится к способу гидропереработки углеводородного сырья, содержащего органические соединения серы и азота, к способам получения предшественника катализатора на основе NiW, NiMo или NiMoW, катализатора и сульфидированного катализатора. Технический результат заключается в улучшении активности гидропереработки. 5 н. и 19 з.п. ф-лы, 17 табл., 4 пр., 7 ил.
Формула

Документы, цитированные в отчёте о поиске
Катализаторы гидроконверсии и способы их изготовления и применения
Комментарии