Новые способы получения простагландинамидов - RU2618223C2
Код документа: RU2618223C2
Чертежи











Описание
Предмет настоящего изобретения представляет собой способ получения простагландинамидов общей формулы (I).
В соединениях общей формулы (I)

значения заместителей являются следующими:
связи, обозначенные пунктирными линиями, могут представлять собой единичные или двойные связи, в случае двойных связей в положениях 5, 6 и 13, 14 они могут иметь cis или trans ориентацию,
Q представляет собой гидроксигруппу и Z представляет собой гидрокси или оксогруппу,
R1 и R2 независимо представляют собой атом водорода или прямую или разветвленную C1-10 алкильную или аралкильную группу, необязательно замещенную -ONO2 группой, или аралкильную или арильную группу, которая содержит гетероатом,
R3 представляет собой прямую или разветвленную, насыщенную или ненасыщенную C4-6 углеводородную группу, или C4-10 алкилциклоалкильную или циклоалкильную группу, или фенильную, C7-10 алкиларильную или гетарильную группу, необязательно замещенную алкильной группой или атомом галогена,
Y представляет собой (CH2)n группу, или O атом, или S атом и
n=0-3.
Для получения простагландинамидных производных экономично, подходящим образом замещенную простаноевую кислоту необходимо активировать.
Согласно современному уровню техники карбоновые кислоты можно активировать превращением в их
смешанные ангидриды,
активированные эфиры или
активированные амиды,
и затем данные соединения можно далее превращать в требуемые простагландинамидные производные реакцией с подходящими аминами.
Из приведенных выше возможностей активация химически очень чувствительных простагландиновых кислот посредством образования эфиров описана, например, в EP 0660716.
Согласно данному способу исходный эфир образуется с помощью алкилгалогенидов, и затем эфир реагирует с подходящим амином, давая амидную функцию.
Недостаток данного способа заключается в том, что применение алкилгалогенидов в конце получения - на последней стадии - следует избегать, поскольку доказано, что алкилгалогениды представляют собой генотоксические агенты.
Кроме того, полученный в результате эфир необходимо обрабатывать подходящим амином при высокой температуре в течение длительного времени, и степень превращения редко превышает 50% (EP 0660716 стр. 42, пример 12). Учитывая известную чувствительность к температуре простагландинов, их обработка при высокой температуре неблагоприятно влияет на профиль чистоты и выход полученных таким образом простагландиновых производных.
Получение смешанного ангидрида и его реакция с подходящим замещенным амином показаны в WO 9153206.
Недостаток данного способа заключается в том, что доказано, что активные алкилирующие агенты, применяемые для получения смешанных ангидридов - эфиры галогенированной муравьиной кислоты, пивалоилхлориды и другие - являются генотоксичными соединениями.
В способе, описанном в WO №2005058812 (страница 23), исходную карбоновую кислоту непосредственно превращают в этиламид, применяя активирующий агент, гидрохлорид 1-(3-диметиламинопропил)-3-карбодиимида (EDC HCl) и этиламин. В процессе реакции амидирования гидроксильные группы в положениях 11 и 15 защищают тетрагидропиранильной (THP) защитной группой, которую затем удаляют.
Обнаружили, что посредством новых активированных эфиров и новых активированных амидов согласно настоящему изобретению соединения общей формулы (I) можно получить при мягких условиях реакции с высоким выходом и чистотой.
Соединения общей формулы (I) согласно настоящему изобретению можно получить реакцией кислоты общей формулы (II)

где в формуле
связи, обозначенные пунктирными линиями, могут представлять собой единичные или двойные связи, в случае двойных связей в положениях 5, 6 и 13, 14 они могут иметь цис- и трансориентацию,
Q представляет собой гидрокси группу и Z представляет собой гидрокси или оксогруппу,
R3 представляет собой прямую или разветвленную, насыщенную или ненасыщенную C4-6углеводородную группу, или C4-10 алкилциклоалкильную или циклоалкильную группу, или фенильную, C7-10алкиларильную или гетарильную группу, необязательно замещенную алкильной группой или атомом галогена,
Y представляет собой (CH2)n группу, или O атом, или S атом и
n=0-3,
i) с соединением, подходящим для введения группы R4, где R4 представляет собой

группу формулы a),
и реакцией таким образом полученного амида общей формулы (III)

где значения Q, Z, R3, R4, Y и n представляют собой, как определено выше,
с амином общей формулы (IV)

где значения R1 и R2 представляют собой, как определено выше, или
ii) с соединением, подходящим для введения группы R5, где R5 представляет собой

группу формулы b), c), d) или e), где X обозначает атом галогена или водорода,
и реакцией полученного таким образом активированного эфира общей формулы (V)

где значения Q, Z, R3, R5, Y и n представляют собой, как определено выше,
с амином общей формулы (IV), где значения R1 и R2 представляют собой, как определено выше.
Кроме того, обнаружили, что соединения общей формулы (I) согласно настоящему изобретению можно также получить реакцией соединения общей формулы (II) с соединением общей формулы (IV), где в формулах значения заместителей представляют собой, как определено выше, в присутствии хлорида 2-хлор-1,3-диметилимидазолиния и основания (способ iii).
Промежуточные соединения общей формулы (III) и (V) представляют собой новые соединения.
Что касается соединения, подходящего для введения группы R4, предпочтительно применять 1,1'-карбонилдиимидазол (DCI) или 1,1'-тиокарбонилдиимидазол для введения группы R5, в указанном случае в присутствии активирующего агента, N-гидроксисукцинимида, N-гидроксифталимида, N-гидрокси-5-норбенэндо-2,3-дикарбоксамида, 1-гидроксибензотриазола, гексафторфосфата (бензотриазол-1-илокси)трис(диметиламино)фосфония, Ν,Ν'-дисукцинимидилкарбоната (DSC) или N,N'-дисукцинимидилоксалата, особенно Ν,Ν'-дисукцинимидилкарбоната.
Что касается активирующего агента, можно применять N,N'-диизопропилкарбодиимид, Ν,Ν'-дициклогексилкарбодиимид или хлорид 2-хлор-1,3-диметилимидазолиния, предпочтительно N,N'-диизопропилкарбодиимид.
В процессе способа i) согласно настоящему изобретению группу R4 можно вводить в растворитель эфирного типа или ароматическом растворителе, или полярном апротонном растворителе,или в их смесях, применяя, например, диизопропиловый эфир, трет-бутилметиловый эфир, 2-метилтетрагидрофуран, толуол, анизол, диметилформамид, диметилсульфоксид, N-метилпирролидон, особенно тетрагидрофуран. Полученный в результате активированный амид общей формулы (III) реагирует с амином общей формулы (IV) после выделения или без него.
Температура реакции в процессе введения группы R4 составляет 20-80°C, предпочтительно 70°C, тогда как реакцию соединений формул (III) и (IV) осуществляют при 20-80°C, предпочтительно при комнатной температуре.
В процессе способа ii) согласно настоящему изобретению введение группы R5 осуществляют в растворителе эфирного типа или в ароматическом, или полярном апротонном растворителе, или в их смесях, применяя, например, диизопропиловый эфир, трет-бутилметиловый эфир, 2-метилтетрагидрофуран, толуол, анизол, диметилформамид, диметилсульфоксид, N-метилпирролидон, особенно тетрагидрофуран. Полученный в результате активированный эфир общей формулы (V) реагирует с амином общей формулы (IV) после выделения или без него. Температура реакции в процессе введения группы R5 составляет 0-80°C, предпочтительно при комнатной температуре, тогда как реакцию соединений формул (V) и (IV) осуществляют при 20-80°C, предпочтительно при комнатной температуре.
В процессе способа iii) согласно настоящему изобретению реакцию осуществляют в растворителе эфирного типа или в ароматическом, или полярном апротонном растворителе, или в их смесях, применяя, например, диизопропиловый эфир, трет-бутилметиловый эфир, 2-метилтетрагидрофуран, толуол, анизол, диметилформамид, диметилсульфоксид, N-метилпирролидон или тетрагидрофуран. Что касается основания, можно применять обычно применяемые основания, подобные пиридину, N-метилморфолину, диизопропилэтиламину, 1,5-диазабицикло[4.3.0]нон-5-ену, 1,8-диазабицикло[5,4,0]ундец-7-ену или триэтиламину.
Реакцию осуществляют при температуре 0-70°C, таким способом, что к раствору соединения общей формулы (II) в органическом растворителе добавляют при 0-70°C, предпочтительно при 30°C, соединение общей формулы (IV), хлорид 2-хлор-1,3-диметилимидазолиния и 2 молярных эквивалентных количества основания. Смесь вначале перемешивают при данной температуре и затем постепенно нагревают до исчезновения исходных веществ. Реакцию контролируют ТСХ.
Способы i, ii или iii можно осуществлять также в условиях без выделения.
Что касается амина общей формулы (IV), можно применять амин, подходящий для конечного соединения, в случае биматопроста, этиламин.
Для получения соединений общей формулы (IA)

где в формуле значения R1, R2, R3, Y и n представляют собой, как определено выше, соединения общей формулы (IIA) согласно настоящему изобретению применяют в качестве исходных веществ.
Соединения общей формулы (IIA)

где в формуле
R3 представляет собой прямую или разветвленную, насыщенную или ненасыщенную C4-6 углеводородную группу или C4-10алкилциклоалкильную или циклоалкильную группу, или фенильную, C7-10 алкиларильную или гетарильную группу, необязательно замещенную алкильной группой или атомом галогена,
Y представляет собой (CH2)n группу, или O атом, или S атом и
n=0-3,
можно получить согласно настоящему изобретению восстановлением лактондиола общей формулы (XII)

где в формуле значения R3, Y и n определяют выше, до лактонтриола общей формулы (XIII)

где значения R3, Y и n определяют выше, затем защитную группу соединения формулы (XIII) удаляют, и таким образом полученное соединение общей формулы (XIV)

где значения R3, Y и n определяют выше, превращают реакцией Виттига в соединение общей формулы (IIA).
Восстановление соединений общей формулы (XII) можно осуществлять известными способами, например диизобутилалюмогидридом в тетрагидрофурановой среде. Защитную группу можно удалять известными способами в кислой или щелочной среде, предпочтительно в щелочной среде.
Лактолтриольные производные общей формулы (XIII)

где R3 представляет собой прямую или разветвленную, насыщенную или ненасыщенную C4-6углеводородную группу, или C4-10алкилциклоалкильную или циклоалкильную группу, или фенильную, C7-10 алкиларильную или гетарильную группу, необязательно замещенную алкильной группой или атомом галогена,
Y представляет собой (CH2)n группу или O атом или S атом, и n=0-3,
являются новыми соединениями.
Согласно следующему варианту осуществления настоящего изобретения конкретному соединению общей формулы IIA, где R3 представляет собой фенильную группу и Y представляет собой -(CH2)-группу, соединение формулы (IIB) можно получить также в кристаллической форме:

Кристаллическая форма соединения формулы (IIB) является новой.
Соединение формулы (IIB) можно получить в кристаллической форме, таким способом, как добавление к смеси, содержащей соединение формула (IIB), смесь растворителей эфирного и сложноэфирного типа.
Согласно данному способу диметиловый эфир, диэтиловый эфир, диизопропиловый эфир, предпочтительно диэтиловый эфир и диизопропиловый эфир применяют в качестве растворителей эфирного типа, и этилацетат, метилацетат, изопропилацетат, предпочтительно изопропилацетат, в качестве растворителей сложноэфирного типа.
Кристаллизацию проводят при -30°C…30°C, предпочтительно 0-25°C.
Таким образом полученную суспензию кристаллов перемешивают в течение 1-24 часов, предпочтительно 8 часов, затем фильтруют и промывают растворителем эфирного типа, предпочтительно диизопропиловым эфиром.
Отфильтрованные кристаллы сушат в вакууме при 25-50°C, предпочтительно при 35-40°C.
Соединения общих формул (II) и (XII) можно получить известными способами, например, как описано в US 5359095, WO 93/00329.
Преимущество способа согласно настоящему изобретению заключается в том, что требуемый биматопростный конечный продукт, при желании, можно получить из кристаллической биматопростной кислоты. Следующее преимущество способа согласно настоящему изобретению заключается в том, что требуемый конечный продукт получают из нового промежуточного соединения, кристаллического, активированного эфира или амида, который, при желании, можно выделить и, при желании, можно очистить кристаллизацией или хроматографией. В зависимости от применяемых агентов, активирующих карбоновые кислоты (например, DSC, DO), защита вторичных гидроксильных групп в положениях 9, 11 и 15 не является необходимой, параллельные реакции, например образование димера, не наблюдаются, или в случае активированного эфира или активированного амида, или в условиях образования конечного амида, и производные активированной карбоновых кислот согласно настоящему изобретению легко выделить с высоким выходом и чистотой.
Неожиданно было обнаружено, что кристаллические производные активированных карбоновых кислот настоящего изобретения можно легко очистить способами кристаллизации для удаления примесей и можно также превратить в требуемый амидный конечный продукт просто, в мягких условиях реакции и с высоким выходом.
Хорошо известно, что в случае активного фармацевтического ингредиента (API) количество примесей представляет собой ключевую проблему, в случае биматопроста количество каждой неизвестной примеси должно быть снижено до ниже 0,1%. Согласно данному способу настоящего изобретения - для поддержания данного очень строгого ограничения - применяют кристаллизацию биматопростной кислоты и кристаллизацию производных активных карбоновых кислот вместо дорогого препаративного ВЭЖХ разделения, описанного в WO 09153206.
Следующий вариант осуществления настоящего изобретения представляет собой способ получения высокоплавкой кристаллической формы II биматопроста формулы (IB)

Способом согласно настоящему изобретению можно получить химически и термодинамически стабильную и свободную от других кристаллических форм высокоплавкую кристаллическую форму II биматопроста.
Следующие патентные заявки рассматривают кристаллизацию биматопростного продукта: US 2005/0209337 A1, WO 2009/153206 A2, US 2009/0163596 A1.
В примере 30 US 2005/0209337 A1 высокоплавкая кристаллическая форма биматопроста (значение зпрегистрированного ДСК пика 79°C) (в дальнейшем: кристаллическая форма II) характеризуется ее рентгеновской дифракцией, ее ИК спектром в KBr кювете и ее ДСК и ТГА кривыми.
WO 2009/153206 описывает очистку биматопростного продукта препаративной ВЭЖХ, с последующей кристаллизацией. Кристаллизацию осуществляют из ацетонитрильного растворителя или из ацетонитрила в качестве растворителя и TBME (трет-бутилметилового эфира) в качестве осаждающего растворителя. Согласно данному описанию данным способом можно получить высокоплавкую кристаллическую форму биматопроста (величина зарегистрированного ДСК пика 79°C). Воспроизводя способ, не удалось получить высокоплавкую кристаллическую форму биматопроста.
US 2009/0163596 описывает кристаллическую форму I биматопроста и ее получение. Кристаллическая форма I биматопроста характеризуется ее температурой плавления (62-64°C), ДСК, рентгеновской дифракцией и ИК исследованием. Он описывает подробно способ кристаллизации, такой как способ растворения (в органическом растворителе или в смеси органического растворителя и осаждающего растворителя при температуре, близкой к температуре кипения), способ охлаждения, отделение выпавших кристаллов из маточного раствора и способ сушки (в вакууме при низкой температуре). Величину зарегистрированного ДСК пика высокоплавкой кристаллической формы 79°C биматопроста можно не получить способами, описанными в US 2009/0163596.
Способом настоящего изобретения можно получить химически и термодинамически стабильную, высокоплавкую кристаллическую форму II биматопроста (= величина зарегистрированного ДСК пика высокоплавкой кристаллической формы 79°C), которая не содержит кристаллическую форму I. Форма II характеризуют ее температурой плавления (72-78°C), ДСК исследованием и ИК исследованиями и исследованиями рентгеновской порошковой дифракцией.
Сущность данного способа заключается в том, что из содержащей биматопрост реакционной смеси после обработки и упаривания, или из любой кристаллической или некристаллической формы биматопроста или из их смесей при любом соотношении компонентов кристаллизацией из протонного растворителя или растворителя эфирного типа получают термодинамически стабильную, чистую форму II. Способ кристаллизации является следующим: к маслообразному или кристаллическому неочищенному биматопросту добавляют рассчитанное количество растворителя, затем его сушат и периодически подвергают механическому воздействию.
Согласно приведенному выше настоящее изобретение относится к способу получения формы II биматопроста формулы (IB), характеризующему тем, что к содержащей биматопрост реакционной смеси после обработки и упаривания или к любой кристаллической или некристаллической форме биматопроста или к их смесям при любом соотношении компонентов добавляют рассчитанное количество протонного растворителя или растворителя эфирного типа, при желании, полученную в результате смесь подвергают механическому воздействию, затем ее сушат и гомогенизируют.
Температура плавления кристаллической формы II, полученной в приведенном выше способе, составляет 72-78°C, эндотермический пик на основе ДСК исследования составляет 73-79°C, и теплота плавления является большей, чем 75 Дж/г.
Фигуры 6, 8 и 4 соответственно представляют собой ДСК кривую, кривую рентгеновской порошковой дифрактометрии и ИК спектр кристаллической формы II, полученной в способе согласно настоящему изобретению.
Фигуры 5, 7 и 3 соответственно представляют собой ДСК кривую, кривую рентгеновской порошковой дифрактометрии и ИК спектр кристаллической формы I.
В способе согласно настоящему изобретению применяли рассчитанное количество, желательно 20-60 мас.%, предпочтительно 35 мас.%, протонного растворителя, особенно спиртов, подобных метанолу, этанолу и/или воде. Предпочтительно в качестве протонного растворителя применяют воду.
В качестве механического воздействия применяли перемешивание или соскребание со стенок, или то и другое. Добавленный растворитель удаляют сушкой. Сушку осуществляют при температуре (-) 60°C-70°C, в частности при 35°C, в вакууме.
В качестве растворителя эфирного типа применяли рассчитанное количество, предпочтительно 2000-8000 мас.% количество диметилового эфира, диэтилового эфира, диизопропилового эфира, предпочтительно диэтилового эфира. Добавленный растворитель удаляют сушкой. Сушку осуществляют при низкой температуре, предпочтительно 0-(-)50°C пропусканием через газообразный азот.
Идентификацию продуктов осуществляют с помощью следующих аналитических приборов:
ЯМР спектры регистрировали Bruker-Avance III-500 МГц прибором, ДСК кривые Mettler-Toledo DSC 1/700 прибором, ИК спектры Perkin-Elmer Spektrum 400 FT-IR спектрофотометром, MS спектры Shimadzu LC-MS-IT-TOF прибором. Температуры плавления определяли Biichi Nelting Point B-545 прибором.
Дополнительные подробности настоящего изобретения описывают в примерах, без ограничения настоящего изобретения данными примерами.
ПРИМЕРЫ
1. Получение исходного соединения
a) Получение ((3aR,4R,5R,6aS)-гексагидро-4-[(1E,3S)-3-гидрокси-5-фенил-1-пентен-1-ил]-2-гидрокси-2H-циклопента[b]фуран-5-илового) эфира [1,1'-бифенил]-4-карбоновой кислоты (PPB-лактолтриол)
Лактоновую группу 55 г ((3aR,4R,5R,6aS)-гексагидро-4-[(1E,3S)-3-гидрокси-5-фенил-1-пентен-1-ил]-2-оксо-2H-циклопента[b]фуран-5-илового) эфира [1,1'-бифенил]-4-карбоновой кислоты (PPB-лактондиол)

восстанавливали в 1000 мл тетрагидрофуранового (THF) растворителя при (-)65-(-)85°C гексановым раствором 422 мл диизобутилалюмогидрида (DIBAL-H). Реакционную смесь разлагали NaHSO4 раствором, водную фазу экстрагировали этилацетатом, органическую фазу промывали NaHCO3 раствором, и растворитель удаляли при 40-50°C. Неочищенный продукт упаривали, получая 46,2 г масла.
Структурная формула полученного PPB-лактолтриола:

Из неочищенного масла после кристаллизации в смеси трет-бутилметиловый эфир (TBME):гексан получали 41,6 г белых кристаллов.
Температура плавления: 91,1-91,7°C.
ИК спектр лактолтриола примера 1a показан на фигуре 1.
13C и1H ЯМР данные:



b) Получение (3aR,4R,5R,6aS)-гексагидро-4-[(1E,3S)-3-гидрокси-5-фенил-1-пентен-1-ил]-2H-циклопента[b]фуран-2,5-диола, (лактолтриола):
46,2 г масла ((3aR,4R,5R,6aS)-гексагидро-4-[(1E,3S)-3-гидрокси-5-фенил-1-пентен-1-ил]-2-гидрокси-2H-циклопента[b]фуран-5-илового) эфира [1,1'-бифенил]-4-карбоновой кислоты (PPB-лактолтриол) растворяли в 230 мл метанола и после добавления 6,6 г K2CO3 его деацилировали при 35-45°C. pH реакционной смеси доводили до 7-8 при -5…0°C 0,5 M раствором фосфорной кислоты. Выпавшие кристаллы отфильтровывали и промывали смесью метанол:вода. Маточный раствор упаривали, экстрагировали этилацетатом, органическую фазу сушили над Na2SO4, сухой материал отфильтровывали, и продукт кристаллизовали добавлением гексана. Получали 26 г белого кристаллического материала.
Структурная формула продукта:

Температура плавления: 98-103°C.
13C и1H ЯМР данные:


c) Получение 7-[(1R,2R,3R,5S)-3,5-дигидрокси-2-[(1E,3S)-3-гидрокси-5-фенил-1-пентен-1-ил]циклопентил]-5-гептеновой кислоты, (5Z)-(биматопростной кислоты):
c1) 108 г бромида 4-карбоксибутилфосфония (KBFBr) растворяли в 800 мл THF, и раствор охлаждают до 0…-5°C. Вначале к данному раствору добавляли 91 г трет-бутилата калия (KOtBu) и затем после перемешивания и охлаждения до (-10)-(-15)°C раствор 25 г лактолтриола в THF. После достижения ожидаемой степени превращения, реакционная смесь разлагали водой, затем добавляли EtOAc. Водную фазу промывали EtOAc. Водный слой подкисляли NaHSO4 раствором до pH 2 и экстрагировали EtOAc. Объединенную органическую фазу промывали 15%-ным NaCl раствором, сушили над Na2SO4, фильтровали и упаривали. Остаток кристаллизовали из этилацетата и диизопропилового эфира. Кристаллы отфильтровывали и промывали, фильтровальный раствор упаривали. Полученное в результате желтое масло очищали хроматографией на силикагеле, применяя элюент диизопропиловый эфир - ацетон. Получали 25,5 г масла.
ИК спектр полученной биматопростной кислоты показан на фигуре 2.
c2) Продукт, полученный в примере 1/c1), растворяли в 60 мл изопропилацетата и при перемешивании добавляли к нему 40 мл диэтилового эфира. Добавляли к реакционной смеси небольшое количество затравки кристаллической биматопростной кислоты. При перемешивании и постепенном охлаждении до 0°C добавляли приблизительно 60 мл диизопропилового эфира. Суспензию перемешивали при данной температуре в течение ночи, после чего ее фильтровали и промывали диизопропиловым эфиром и сушили в вакууме. Получали 20,4 г кристаллической биматопростной кислоты.
ДСК кривая полученной биматопростной кислоты показана на фигуре 10 и кривая рентгеновской порошковой дифрактометрии на фигуре 9.
Структурная формула продукта:

Температура плавления: 63,0-65,5°C.
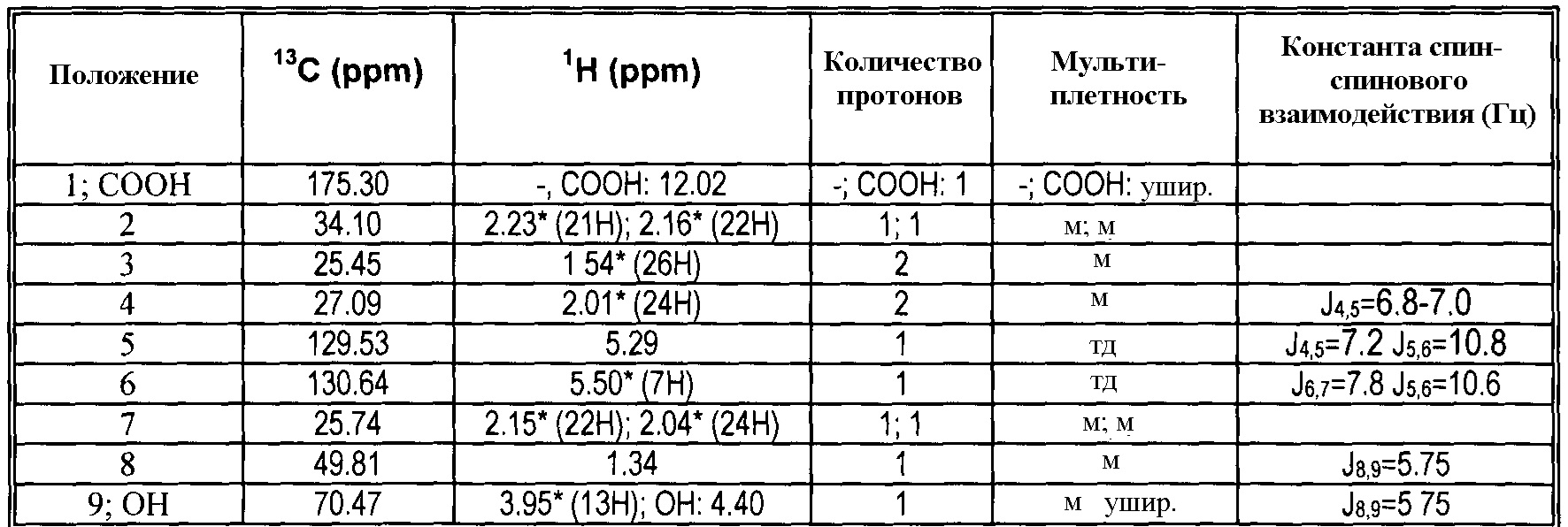
13C и1H ЯМР данные:


2. Получение (2,5-диоксопирролидин-1-илового) эфира 7-[3,5-дигидрокси-2-(3-гидрокси-5-фенилпент-1-енил)циклопентил]-5-гептеновой кислоты (активированный эфир)
27,5 г биматопростной кислоты примера 1/c2) растворяли в 270 мл THF и к ней добавляли при комнатной температуре 13,7 г Ν,Ν'-диизопропилкарбодиимида, с последующим добавлением 13,7 г N-гидроксисукцинимида. Смесь перемешивали при данной температуре, и затем выливали в смесь 1 н. NaHSO4 раствора и трет-бутилметилового эфира (TBME). Фазы разделяли. Органическую фазу промывали 1 н. NaHCO3 раствором, водно-щелочную фазу экстрагировали TBME. Объединенную органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток кристаллизовали из смеси гексан:ацетон, получая 30,04 г белого кристаллического продукта.
Продукт:

Температура плавления: 93,5-103,4°C.
3. 27,5 г биматопростной кислоты примера 1/c1) растворяли в 270 мл THF, и к данному раствору добавляли при комнатной температуре 11,5 г карбоната калия и 19,6 г Ν,Ν'-дисукцинимидилкарбоната. Реакционную смесь при перемешивании постепенно нагревали до 60°C, и затем выливали в смесь 1N NaHSO4 раствора и трет-бутилметилового эфира (TBME). Фазы разделяли, органическую фазу промывали 1N NaHCO3 раствором и водно-щелочную фазу экстрагировали TBME. Объединенную органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток кристаллизовали из смеси гексан:ацетон, получая 30,9 г белого кристаллического вещества.
Продукт:

Температура плавления: 93,5-103,4°C.
13C и1H ЯМР данные:


4. Получение 1,3-диоксо-l,3-дигидроизоиндол-2-илового эфира (активированного эфира) 7-[3,5-дигидрокси-2-(3-гидрокси-5-фенил-пент-1-енил)циклопентил]-5-гептеновой кислоты
2 г биматопростной кислоты растворяли в 20 мл THF, и к данному раствору добавляли при комнатной температуре 1 г N-гидрокси-фталимида и 1 мл Ν,Ν'-диизопропилкарбодиимида. Реакционную смесь перемешивали в течение 2 часов и затем выливали в смесь 1 н. NaHSO4 раствора и трет-бутилметилового эфира (TBME). Фазы разделяли, органическую фазу промывали 1 н. NaHCO3 раствором, и водно-щелочную фазу экстрагировали TBME. Объединенную органическую фазу сушили над Na2SO4, фильтровали и упаривали. Остаток кристаллизовали из смеси гексан:ацетон, получая 1,5 г белого кристаллического вещества.
Продукт:

Температура плавления: 83,2-84,5°C.
13C и1H ЯМР данные:



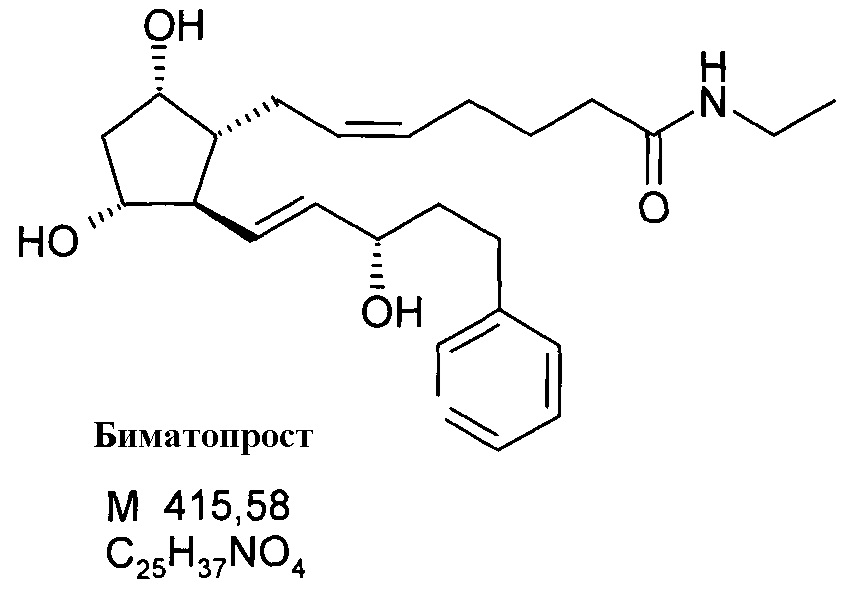
5. Получение 7-[(1R,2R,3R,5S)-3,5-дигидрокси-2-[(1E,3S)-3-гидрокси-5-фенил-1-пентенил]циклопентил]-N-этил-5-гептенамида, (5Z(-)биматопроста) через активированный эфир
27,5 г биматопростной кислоты растворяли в 270 мл THF, и к данному раствору добавляли при комнатной температуре 13,7 г Ν,Ν'-диизопропилкарбодиимида и затем 13,7 г N-гидроксисукцинимида. Смесь перемешивали при комнатной температуре. Полученный в результате активированный эфир не выделяли.

После завершения образования амида добавляли к реакционной смеси 70 мл 2M раствора этиламина в THF. Смесь перемешивали до достижения ожидаемой степени превращения, затем, ее выливали в смесь 1 н. NaHSO4 раствора и трет-бутилметилового эфира (TBME). Фазы разделяли, органическую фазу промывали 1 н. NaHCO3 раствором, и водно-щелочную фазу экстрагировали TBME. Объединенную органическую фазу сушили над Na2S04, фильтровали и упаривали, получая 25,4 г масла.
Продукт:

13C и1H ЯМР данные:



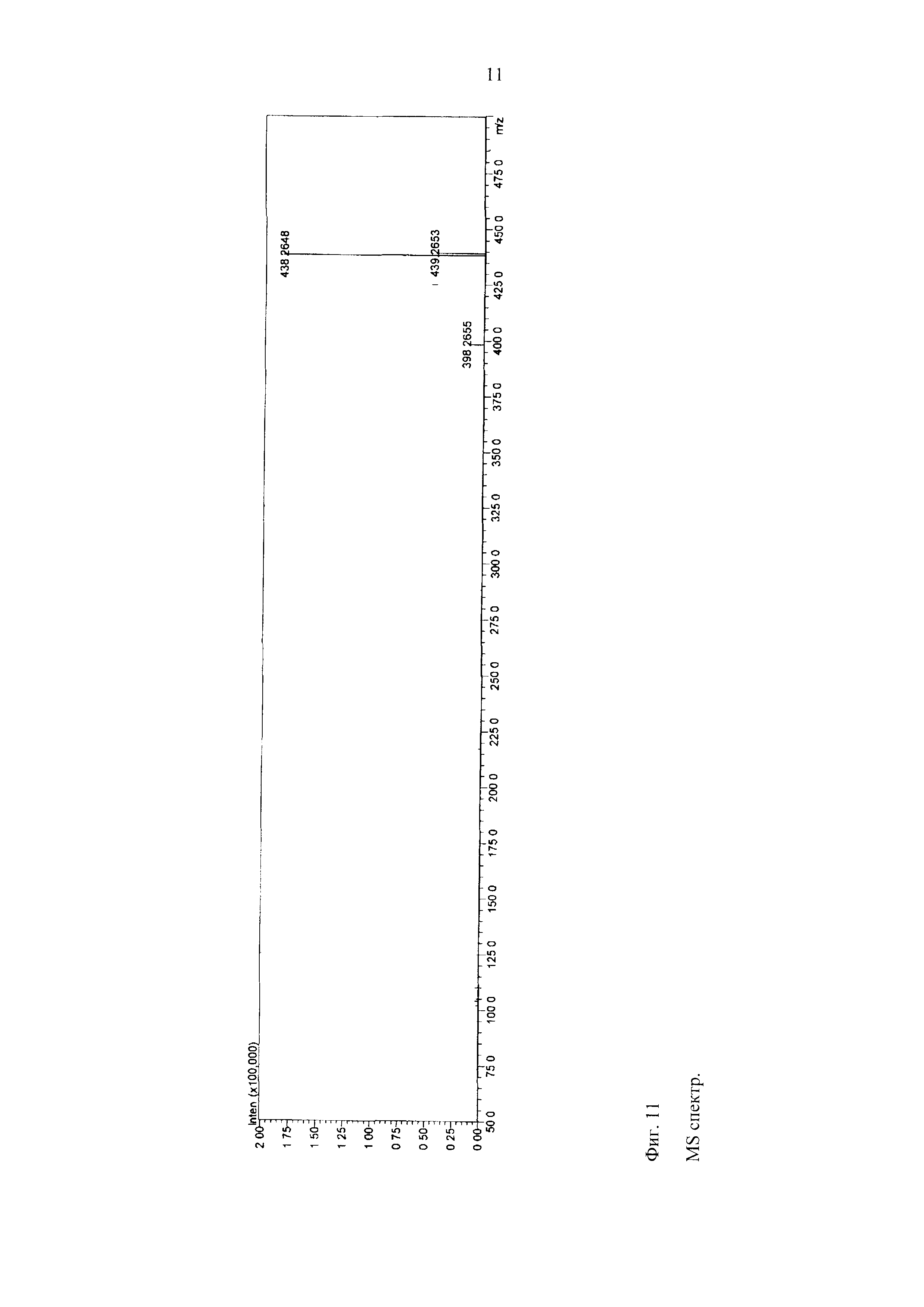
MS данные
MS спектр:
Положительная ионизация:
Фиг. 11
Ожидаемая формула:
C25H37NO4.
Измеренная точная масса: 438,2648 [M+Na]+.
Ожидаемая точная масса: 438,2615. [M+Na]+, ΔΜ=3,3 мДа и 7,53 м.д.
C25H35NO3 (M-H2O).
Измеренная точная масса: 398,2655 [M-H2O+H]+.
Ожидаемая точная масса: 398,2690 [M-H2O+H]+ ΔΜ=-3,5 мДа и 8,79 м.д.
MSMS (ион-предшественник: 438,26):
Фиг. 12
Ожидаемая формула:
C25H35NO3 (M-H2O).
Измеренная точная масса: 420,2520 [M-H2O+Na]+
Ожидаемая точная масса: 420,2509 [M-H2O+Na]+ ΔΜ=1,1 мДа и 2,62 м.д.
C25H32NO3 (M-H2O-5H).
Измеренная точная масса: 394,2366 [M-H2O-5H]+.
Ожидаемая точная масса: 394,2377 [M-H2O-5H]+ ΔΜ=-1,1 мДа и 2,79 м.д.
C25H30NO2 (M-2xH2O-5H).
Измеренная точная масса: 376,2258 [M-2xH2O-5H]+.
Ожидаемая точная масса: 376,2271 [M-2xH2O-5H]+ ΔΜ=-1,3 мДа и 3,46 м.д.
6. Получение биматопроста через активированный эфир
27,5 г биматопростной кислоты растворяли в 270 мл THF и к данному раствору добавляли при комнатной температуре 11,5 г карбоната калия и 19,6 г N,N'-дисукцинимидилкарбоната. Реакционную смесь постепенно нагревали до 60°C при перемешивании. Полученный в результате активированный эфир не выделяли.

После образования активированного эфира добавляли к реакционной смеси 70 мл 2M раствора этиламина в THF. Когда реакция завершалась, смесь выливали в смесь 1 н. NaHSO4 раствора и EtOAc. Органическую фазу промывали 1 н. NaHCO3 раствором, водно-щелочную фазу экстрагировали EtOAc. Объединенную органическую фазу промывали NaCl раствором и сушили над Na2SO4. Осушающее вещество отфильтровывали, фильтрат упаривали, получая 25,7 г масла.
Продукт:

7. Получение биматопроста через активированный амид
27,5 г биматопростной кислоты растворяли в 270 мл пиридина и добавляли к ней 13,7 г 1,1'-карбонилдиимидазола. Смесь перемешивают при 20-25°C до образования активированного амида. Полученный в результате активированный амид не выделяли.

70 мл 2M раствора этиламина в THF добавляли к реакционной смеси при комнатной температуре и смесь перемешивают до достижения ожидаемой степени превращения. Затем смесь выливали в смесь 1 н. NaHSO4 раствора и трет-бутилметилового эфира (TBME). Фазы разделяли, органическую фазу промывали 1 н. NaHCO3 раствором, и водно-щелочную фазу экстрагировали TBME. Объединенную органическую фазу сушили над Na2SO4, фильтровали, и фильтрат упаривали, получая 23,82 г масла.
Продукт:

8. Получение биматопроста из очищенного активированного эфира
30,9 г активированного эфира согласно примеру 3 растворяли в 270 мл THF и к данному раствору добавляли 70 мл 2M этиламина, растворенного в THF. После завершения реакции, смесь выливали в смесь 1N NaHSO4 раствора и EtOAc. Органическую фазу промывали 1 н. NaHCO3 раствором. Водно-щелочную фазу экстрагировали EtOAc. Объединенную органическую фазу промывали NaCl раствором и сушили над Na2SO4. Осушающее вещество отфильтровывали, и фильтрат упаривали. К полученному в результате маслу добавляли 35 мас.% воды, и продукт кристаллизовали. Получали 24,8 г белых кристаллов биматопроста с большей, чем 99,5% чистотой.
Продукт:

Температура плавления: 71,9-72,5°С.
ВЭЖХ: 99,6% биматопроста, меньше чем 0,3% транс-биматопроста, 0,1% других примесей.
9. Получение биматопроста согласно способу iii)
2,00 г биматопростной кислоты растворяли в 20 мл тетрагидрофурана (THF) и при 30°C вначале добавляли 1,29 г хлорида 2-хлор-1,3-диметилимидазолиния (DMC) и 1,44 мл триэтиламина, затем после 10 минут перемешивания 2,57 мл 2M раствора этиламина в THF. Реакционную смесь постепенно, в течение 1 часа, нагревали до 70°C, и смесь перемешивают при данной температуре до исчезновения исходных соединений (приблизительно 1 час). Реакцию контролировали ТСХ.
После завершения реакции смесь выливали в смесь 1 н. NaHSO4 раствора и изопропилацетата (iPrOAc). Органическую фазу промывали 1N NaHCO3 раствором, водно-щелочную фазу экстрагировали iPrOAc. Объединенную органическую фазу промывали NaCl раствором и сушили над Na2SO4. Осушающее вещество отфильтровывали, и фильтрат упаривали, получая 1,41 г масла.
Продукт:
10. Получение кристаллической формы II биматопроста из неочищенного масла биматопроста
К маслу биматопроста, полученному согласно примеру 6, добавляли 35 мас.% количество очищенной воды. Смесь интенсивно перемешивали и затем сушили в вакууме при максимальной температуре 35°C, между тем, каждые 2 часа ее встряхивали и соскребали со стенок. После полного высушивания смесь гомогенизировали. ИК спектр данного продукта показан на фигуре 4, и ДСК кривая данного продукта показана на фигуре 6. Кривая дифракции рентгеновских лучей полученной формы II показана на фигуре 8.
Выход: 96,9%.
Температура плавления: 78°C.
ДСК начало: 73,56°C.
11. Получение кристаллической формы II биматопроста
Неочищенный биматопрост, полученный согласно примеру 5, растворяли при нагревании в 3000-кратном количестве диэтилового эфира. Затем растворитель удаляли при (-)20-(-)30°C медленным пропусканием газообразного азота. Полученные в результате кристаллы гомогенизировали или вначале подвергали механическому воздействию и затем гомогенизировали.
Выход: 94,4%.
Температура плавления: 75,9°C.
ДСК начало: 72,92°C.
12. Получение кристаллической формы II биматопроста
К неочищенному биматопросту, полученному согласно примеру 6, добавляли 35 мас.% количество метанола. Смесь интенсивно перемешивали, и затем сушили в вакууме при максимальной температуре 35°C, между тем, каждые 2 часа ее встряхивали и соскребали со стенок. После полного высушивания смесь гомогенизировали.
Выход: 95,8%.
Температура плавления: 77,2°C.
ДСК начало: 73,07°C.
13. Получение кристаллической формы II биматопроста
К неочищенному биматопросту, полученному согласно примеру 6, добавляли 17,5 мас.% количество очищенной воды и 17,5 мас.% количество этанола. Смесь интенсивно перемешивали, и затем сушили в вакууме при максимальной температуре 35°C, между тем, каждые 2 часа ее встряхивали и соскребали со стенок. После полного высушивания смесь гомогенизировали.
Выход: 92,3%.
Температура плавления: 72,9°C.
ДСК начало: 72,96°C.
14.
a) Получение кристаллической формы I биматопроста (согласно примеру 38 патентной заявки US №20090163596)
5,2 г неочищенного биматопроста кристаллизовали из 106 г ацетонитрила: смесь нагревали до температуры, близкой к температуре кипения, горячий раствор охлаждали до комнатной температуры, и смесь перемешивали при данной температуре в течение 1 часа, затем при 0-5°C в течение 2 часов. Выпавшие кристаллы отфильтровывали, промывали 20 г холодного (0-5°C) ацетонитрила и сушили в вакууме при 0-5°C в течение 1 часа, при комнатной температуре в течение получаса и при 30-40°C в течение 2 часов.
Получали 4,3 г кристаллической формы I биматопроста. Ее ИК спектр показан на фигуре 3, ее ДСК кривая показана на фигуре 5 и ее кривая дифракции рентгеновских лучей формы I показана на фигуре 7.
Выход: 83%.
Температура плавления: 62,1°C.
ДСК начало: 63,61°C.
b) Получение кристаллической формы II биматопроста, исходя из формы I.
К кристаллической форме I биматопроста, полученной согласно примеру 14a, добавляли 35 мас.% количество очищенной воды. Смесь интенсивно перемешивали, и затем сушили в вакууме при максимальной температуре 35°C, между тем, каждые 2 часа ее встряхивали и соскребали со стенок. После полного высушивания смесь гомогенизировали.
Выход: 97,3%.
Температура плавления: 77,7°C.
ДСК начало: 73,14°C.
15. Получение кристаллической формы II смеси кристаллической формы II и I биматопроста
К 50% смеси кристаллической формы II и I биматопроста добавляли 35 мас.% количество очищенной воды. Смесь интенсивно перемешивали и затем сушили в вакууме при максимальной температуре 35°C, между тем, каждые 2 часа ее встряхивали и соскребали со стенок. После полного высушивания смесь гомогенизировали.
Выход: 97,6%.
Температура плавления: 78,2°C.
ДСК начало: 73,77°C.
Реферат
Изобретение относится к способу получения простагландинамидов общей формулы (I)соединениям общей формулы (V)способу получения соединений общей формулы (IIA)через промежуточный лактонтриол общей формулы (XIII).Технический результат: разработан новый способ получения простагландинамидов формулы (I), а также способ получения соединений формулы (IIA), получены новые соединения формулы (V) и промежуточный лактонтриол общей формулы (XIII). Производные активированных карбоновых кислот настоящего изобретения можно легко очистить способами кристаллизации для удаления примесей и можно также превратить в требуемый амидный конечный продукт просто, в мягких условиях реакции и с высоким выходом. 4 н. и 10 з.п. ф-лы, 12 ил.
Формула












Комментарии