Нуклеофильное фторирование в твердой фазе - RU2315769C2
Код документа: RU2315769C2
Описание
Настоящее изобретение относится к новым твердофазным способам приготовления радиоизотопных индикаторов, в частности, для приготовления соединений, меченных18F, которые могут быть пригодными для использования в качестве радиоактивных индикаторов для позитронной эмиссионной томографии (ПЭТ). Изобретение также включает наборы реагентов для приготовления радиофармпрепарата с помощью указанного нового способа.
Предпочтительным радиоизотопом, используемым в ПЭТ, является18F, имеющий относительно короткий период полураспада - 110 минут. Таким образом, индикаторы для ПЭТ, меченные18F, должны быть синтезированы и очищены как можно быстрее, в идеале - в пределах часа до клинического использования. Стандартные синтетические методики введения фтора-18 относительно медленны и требуют послереакционной очистки (например, с помощью высокоэффективной жидкостной хроматографии, ВЭЖХ), т.е. индикатор для клинического использования, меченный18F, сложно получить с хорошим радиохимическим выходом. Возникает также необходимость в автоматизации для защиты оператора от воздействия радиоактивного излучения. Многие из способов введения радиоактивного фтора - сложные процедуры, и для их автоматизации их необходимо упростить.
В настоящем изобретении предложены твердофазные способы быстрого получения меченных18F индикаторов, имеющих высокую удельную активность, не выполняя при этом длительных по времени операций очистки, так что получаемый при этом индикатор, меченный18F, пригоден для использования в ПЭТ. Твердофазные способы также могут быть автоматизированы, что облегчает приготовление и увеличивает производительность. Настоящее изобретение также включает наборы реагентов для приготовления радиофармпрепарата с помощью таких способов и предоставляет радиофармакологам и клиницистам удобный способ приготовления индикаторов, меченных18F.
В общем аспекте, в настоящем изобретении предложен способ приготовления индикатора, меченного18F, включающий обработку закрепленного на смоле предшественника формулы (I)

ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (II)

где18F-ИНДИКАТОР выбран из 2-фтор-2-дезокси-D-глюкозы (18F-FDG), 2-(1, 1-дицианопропен-2-ил)-6-(2-фторэтил)-(метиламино)-нафталина (18F-FDDNP), 3'-дезокси-3'-фтортимидина (18F-FLT) и 6-L-18F-DOPA (18F-FDOPA). В предпочтительных аспектах настоящего изобретения получаемый индикатор выбран из FDG или FDOPA.
Поскольку меченный18F индикатор формулы (II) извлекают из твердой фазы в раствор, весь непрореагировавший предшественник остается связанным со смолой и может быть отделен простым фильтрованием, что, таким образом, исключает сложную очистку, например, с помощью ВЭЖХ. Меченный18F индикатор формулы (II) может быть очищен от избытка F18 с помощью, например, ионообменной хроматографии и/или путем полного удаления органического растворителя. Полученный меченный18F индикатор формулы (II) затем может быть введен в водную лекарственную форму для клинического использования.
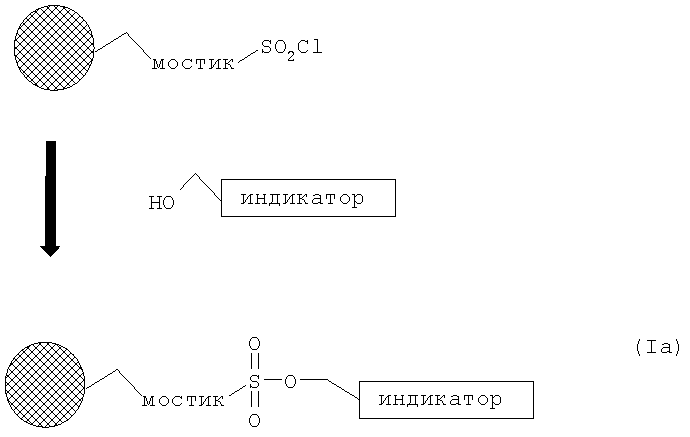
В соединениях формулы (I) X - это группа, выбранная из SO2O, как в указанной ниже формуле (Ia), и I+Y-, где Y представляет собой трифлатный анион, как в указанной ниже формуле (Id).
В следующем аспекте изобретения предложен способ приготовления меченного18F индикатора, включающий обработку закрепленного на смоле предшественника формулы (Ia)

ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (II)
Подходящим ИНДИКАТОРОМ в соединении формулы (Ia) является FDG, FLT, FDDNP или предшественник указанных соединений, в котором одна или более групп защищены, или активированный предшественник FDOPA. Наиболее предпочтительно, ИНДИКАТОР в соединении формулы (Ia) - это FDG или его предшественник.
Как показано на Схеме 1, соединение формулы (Ia) может быть удобным образом приготовлено из любой коммерчески доступной смолы, функционализированной сульфоновыми кислотами, такой как Merrifield Resin, NovaSyn® TG Bromo Resin, (бромметил)феноксиметилполистирол или Wang Resin, которые могут быть введены в реакцию с хлорирующим агентом с получением соответствующей сульфонилхлоридной смолы. Это можно осуществить, обрабатывая смолу, например, пентахлоридом фосфора, трихлоридом фосфора, оксалилхлоридом или тионилхлоридом в подходящем инертном растворителе, таком как дихлорметан, хлороформ или ацетонитрил, и нагревая при повышенной температуре в течение некоторого времени. Избыток реагента затем может быть удален из смолы последующим промыванием несколькими порциями инертного растворителя. Затем сульфонилхлоридная смола может быть введена в реакцию со спиртовым аналогом индикатора с образованием закрепленного на смоле предшественника формулы (Ia). Эта процедура может быть выполнена путем обработки смолы раствором спирта в инертном растворителе, таком как хлороформ, дихлорметан, ацетонитрил или тетрагидрофуран, содержащем растворимое ненуклеофильное основание, такое как гидрид натрия, или триалкиламин, например триэтиламин или диизопропилэтиламин. Реакция может быть проведена при температуре от 10 до 80°С, оптимально - при обычной температуре, в течение периода времени, приблизительно равного от 1 до 24 часов. Избыток спирта и основания затем может быть удален с твердого носителя последующим промыванием несколькими порциями инертного растворителя, такого как хлороформ, дихлорметан или тетрагидрофуран.
Схема 1
В соединениях, имеющих формулы (I) и (Ia), и в последующих более специфических аспектах настоящего изобретения «ТВЕРДЫЙ НОСИТЕЛЬ» может быть любым подходящим твердофазным носителем, нерастворимым в любых используемых в настоящем способе растворителях, и на котором ЛИНКЕР и/или ИНДИКАТОР может быть закреплен при помощи ковалентной связи. Примеры подходящего ТВЕРДОГО НОСИТЕЛЯ включают полимеры, такие как полистирол (который может быть привитым блоками, например, полиэтиленгликоля), полиакриламид или полипропилен, либо стекло или кремний, покрытые таким полимером. Твердый носитель может находиться в форме мелких дискретных частиц, таких как шарики или лучинки, или в форме покрытия, нанесенного на внутреннюю поверхность картриджа или на микрообработанный сосуд.
В соединениях, имеющих формулы (I) и (Ia), и в последующих более специфических аспектах настоящего изобретения «ЛИНКЕР» может быть любой подходящей органической группой, которая служит для достаточного пространственного разделения реакционного центра и структуры твердого носителя для того, чтобы обеспечить максимальную реакционную способность. Подходящим образом, ЛИНКЕР включает от нуля до четырех арильных групп (пригодны фенильные) и/или C1-6-алкильную или C1-6-галогеноалкильную группу (пригодна C1-6-фторалкильная группа) и, возможно, от одной до четырех дополнительных функциональных групп, таких как амидные или сульфонамидные группы. Примеры таких линкеров хорошо известны специалистам в твердофазной химии (химии твердого тела) и включают:





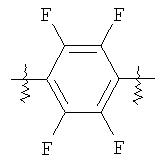

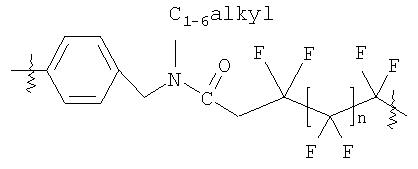

где, в каждом случае, n - это целое число от 0 до 3.
Специалистам в данной области техники понятно, что может возникнуть необходимость защиты функциональных групп ИНДИКАТОРА для предотвращения нежелательных реакций при введении радиоактивного изотопа. Такую защиту можно осуществить с помощью стандартных способов, известных в химии защитных групп. После завершения введения радиоактивного изотопа любые защитные группы могут быть удалены с помощью простых способов, также считающихся стандартными в указанной области техники. Подходящие способы введения и снятия защиты могут быть найдены, например, в книге Protecting Groups in Organic Synthesis, Teodora W. Greene and Peter G.M. Wuts, опубликованной John Wiley & Sons Inc.
Обработку соединений, имеющих формулы (I) и (Ia), изотопом18F- можно осуществить с помощью обработки любым подходящим источником18F-, таким как Na18F, K18 F, Cs18F,18F-фторид тетраалкиламмония или18F-фторид тетраалкилфосфония. Для увеличения реакционной способности фторида может быть добавлен катализатор фазового переноса, такой как 4,7,13,16,21,24-гексаокса-1,10-диазабицикло[8,8,8]гексакозан, и реакция может быть проведена в апротонном растворителе. В этих условиях получают реакционноспособные фторидные ионы. Обработку18F- удобно проводить в присутствии подходящего органического растворителя, такого как ацетонитрил, диметилформамид, диметилсульфоксид, тетрагидрофуран, диоксан, 1, 2-диметоксиэтан, сульфолан, N-метилпирролидинон, при умеренной температуре, например от 15°С до 180°С, предпочтительно при повышенной температуре. После завершения реакции меченный18F индикатор формулы (II), растворенный в растворителе, удобно отделять от твердой фазы фильтрованием. Те же самые методики фторирования могут быть использованы в следующих конкретных аспектах настоящего изобретения.
Любой избыток18F- может быть удален из раствора18F-индикатора любым подходящим способом, например, с помощью ионообменной хроматографии или твердофазных абсорбентов. Подходящие ионообменные смолы включают BIO-RAD AG 1-X8 или Waters QMA, а подходящие твердофазные абсорбенты включают оксид алюминия. Избыток18 F- может быть удален с помощью указанных твердых веществ при комнатной температуре в апротонных растворителях.
Любой органический растворитель может быть удален любым стандартным способом, таким как испарение при повышенной температуре в вакууме или пропускание над раствором потока инертного газа, такого как азот или аргон.
Перед использованием индикатора, меченного18F, возможно, следует приготовить состав на его основе, например, в виде водного раствора путем растворения индикатора, меченного18F, в стерильном изотоническом солевом растворе, который может содержать до 10% подходящего органического растворителя, такого как этанол, или подходящего буферного раствора, такого как фосфатный буфер. Для снижения радиолиза могут быть также добавлены другие добавки, такие как аскорбиновая кислота.
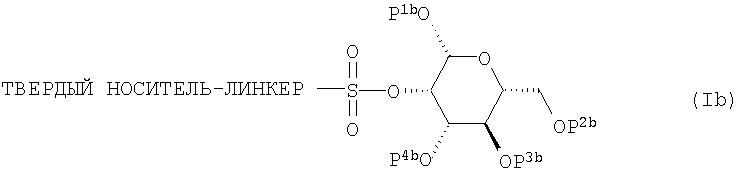
Еще одним аспектом настоящего изобретения является обеспечение способа получения 2-18 F-фтор-2-дезокси-D-глюкозы (18F-FDG), который включает обработку закрепленного на твердой подложке предшественника формулы (Ib):

где Р1b, Р2b, Р3b и P4b независимо друг от друга представляют собой водород или защитную группу;
ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (IIb)

где Р1b, Р2b, Р3b и Р4b независимо друг от друга представляют собой водород или защитную группу,
с последующим удалением защитных групп в случае, если Р1b-Р4bпредставляют собой защитные группы.
Возможно также последующее удаление избытка18F-, например, с помощью ионообменной хроматографии; и/или удаление органического растворителя; и/или приготовление состава на основе полученного соединения формулы (IIb) в виде водного раствора.
В соединении формулы (Ib) ЛИНКЕР, предпочтительно, представляет собой

или
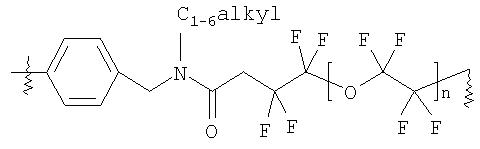
где n равно от 0 до 3, и, более предпочтительно,
или

и подходящий ТВЕРДЫЙ НОСИТЕЛЬ является полистирольной смолой.
Удаление любых защитных групп из соединения формулы (IIb) может быть осуществлено с помощью стандартных способов, как было указано выше. В предпочтительном варианте осуществления этого аспекта настоящего изобретения для защиты гидроксильных групп сахаров их сохраняют в виде сложных эфиров, подходящими являются эфиры C1-6-алкановых кислот, предпочтительно эфиры уксусной кислоты; или в виде простых эфиров, предпочтительно C1-6-алкоксиметиловых эфиров, или ацеталей. Эфирные, сложноэфирные и ацетальные защитные группировки могут быть подходящим образом удалены с помощью гидролиза, например, в присутствии кислоты или основания. Такое снятие защиты может быть осуществлено при помощи кислотного или основного катализатора, закрепленного на твердом носителе, что позволяет избежать операции нейтрализации после снятия защиты.
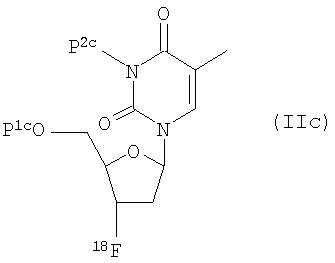
Еще в одном аспекте настоящего изобретения предложен способ получения 3'-дезокси-3'-18F-фтортимидина (18F-FLT), который включает обработку закрепленного на твердой подложке предшественника формулы (Ic):

где P1c и Р2c независимо друг от друга представляют собой водород или защитную группу;
ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (IIc)

где Р1c и Р2c независимо друг от друга представляют собой водород или защитную группу;
с последующим удалением защитных групп в случае, если Р1c и Р2c представляют собой защитные группы.
Возможно также удаление избытка18F-, например, с помощью ионообменной хроматографии; и/или удаление органического растворителя; и/или приготовление состава на основе полученного соединения формулы (IIc) в виде водного раствора.
В данном аспекте настоящего изобретения функциональные аминогруппы и гидроксильные группы в тимидиновом предшественнике подходящим образом защищают при помощи стандартных способов, как было указано выше. Аминогруппы и гидроксильные группы удобно защищать, сохраняя их в виде сложноэфирных; удобно в виде C1-6-алкильных эфиров, предпочтительно ацильных эфиров. Сложноэфирные группировки могут быть легко удалены с помощью гидролиза, например, в присутствии кислоты или основания. Такое снятие защиты может быть осуществлено при помощи кислотного или основного катализатора, закрепленного на твердом носителе, что позволяет избежать операции нейтрализации после снятия защиты.
В соединении формулы (Ic) ЛИНКЕР, предпочтительно, представляет собой

где n равно от 0 до 3.
Еще в одном аспекте настоящего изобретения предложен способ получения 6-L-18F-фтордопа (18F-FDOPA), включающий обработку закрепленного на твердой подложке предшественника формулы (Ig):

где Р1g, Р3g и Р4g независимо друг от друга представляют собой водород или защитную группу, такую как трет-бутоксикарбонил;
ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (IIg)

где Р19, Р39 и Р49 независимо друг от друга представляют собой водород или защитную группу, такую как трет-бутоксикарбонил;
с последующим удалением защитных групп в случае, если Р1g, Р3g и Р4gпредставляют собой защитные группы.
Возможно также последующее удаление избытка18F-, например, с помощью ионообменной хроматографии; и/или превращение группы -С(O)CF3 в гидроксильную группу; и/или удаление органического растворителя; и/или приготовление состава на основе полученного FDOPA в виде водного раствора.
В данном аспекте настоящего изобретения функциональную гидроксильную группу в исходном материале на основе «допа» (DOPA) удобно защищать, сохраняя в виде сложноэфирной, приемлемо в виде эфиров C1-6-алкановых кислот, предпочтительно ацетатных эфиров, или карбонатных эфиров, таких как трет-бутоксикарбонильные эфиры. Кислотная группировка может быть защищена превращением в C1-6-алкиловый сложный эфир, предпочтительно в этиловый сложный эфир, а аминогруппа может быть защищена превращением в амид, предпочтительно формиламид, или уретан, предпочтительно трет-бутоксикарбонилуретан. Сложноэфирные, формильные и уретановые защитные группы могут быть легко удалены с помощью гидролиза, например, в присутствии кислоты или основания. Такое снятие защиты может быть осуществлено при помощи кислотного или основного катализатора, закрепленного на твердом носителе, что позволяет избежать операции нейтрализации после снятия защиты. Превращение группы -С(O)CF3 в гидроксильную группу может быть осуществлено обработкой окислителем, таким как мета-хлорнадбензойная кислота, с последующим мягким кислотным гидролизом. В данном аспекте настоящего изобретения особенно подходящим ЛИНКЕРОМ является

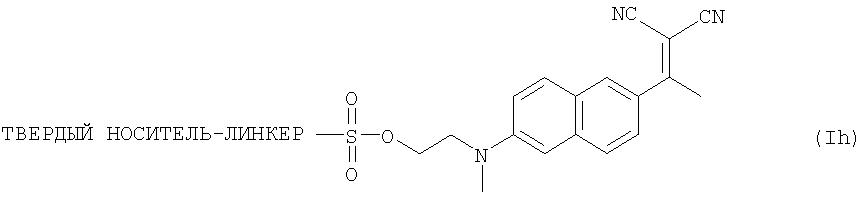
и подходящий ТВЕРДЫЙ НОСИТЕЛЬ является полистирольной смолой. Еще в одном аспекте настоящего изобретения предложен способ получения 2-(1,1-дицианпропен-2-ил)-6-((2-фторэтил)-метиламино)нафталина (FDDNP), включающий обработку закрепленного на твердой подложке предшественника формулы (Ih):

ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (IIh)

Возможно также последующее удаление непрореагировавшего18F-, например, с помощью ионообменной хроматографии; и/или удаление органического растворителя; и/или приготовление состава на основе полученного соединения формулы (IIh) в виде водного раствора.
Еще в одном аспекте настоящего изобретения предложен способ получения меченного18F индикатора, включающий обработку закрепленного на твердом носителе предшественника формулы (Id)

где Y- - трифлатный анион,
ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (IId)

Возможно также последующее удаление избытка18F-, например, с помощью ионообменной хроматографии; и/или удаление любых защитных групп; и/или удаление органического растворителя; и/или приготовление состава на основе полученного соединения формулы (IId) в виде водного раствора.
Подходящим индикатором в соединении формулы (Id) является арилсодержащее соединение, такое как фенилсодержащее соединение, предпочтительно замещенное фенильное кольцо. В одном из таких предпочтительных аспектов получаемым индикатором является FDOPA.
Соединение формулы (Id) удобно приготовить из функционализированной коммерчески доступной смолы, такой как Merrifield Resin или Wang Resin. Соответствующим образом, гидроксииодарил (такой как иодфенол), содержащий группу ЛИНКЕР, обрабатывают неорганическим основанием, таким как карбонат цезия, а затем добавляют к смоле, предварительно подвергнутой набуханию в инертном растворителе, таком как N,N-диметилформамид, и оставляют реагировать при повышенной температуре, например от 30 до 80°С. Избыток реагентов может быть удален последующим промыванием смолы несколькими порциями инертного растворителя. Полученная функционализованная иодфенолом смола может быть обработана источником ацетат-анионов (таким как уксусная кислота, уксусный ангидрид или ацетилхлорид) в присутствии окислителя, такого как пероксид водорода, с образованием соответствующей функционализованной диацетоксииодфенолом смолы. Функционализованную диацетоксииодфенолом смолу затем можно перемешать в инертном растворителе, таком как дихлорметан, в присутствии кислоты, такой как хлороводородная (соляная) кислота, трифторметансульфоновая кислота или уксусная кислота, при пониженной температуре, приемлемо при температуре от -40°С до 10°С, с последующим добавлением индикатора, подходящим образом индикатора, функционализованного в виде борной кислоты или производного триалкилолова, который может быть подвергнут сочетанию со смолой при умеренной температуре. Как и в предыдущих операциях, целевое соединение формулы (Id) может быть отделено фильтрованием и промыванием инертным растворителем.
В соединении формулы (Id) ЛИНКЕР является таким, как определено выше, но включает арильную группу (пригодной является фенильная группа), примыкающую к I+. Предпочтительные примеры включают:


В соединении формулы (Id) Y- - это трифторметилсульфонатный (трифлатный) анион.
Еще в одном аспекте настоящего изобретения предложен способ получения 6-L-18F-фтордопа (18 F-FDOPA), который включает обработку закрепленного на твердой подложке предшественника формулы (Ie):

где Р1e, Р2e, Р3e и Р4e независимо друг от друга представляют собой водород или защитную группу, а Y- - это трифлатный анион,
ионом18F- соединения, являющегося источником иона18F-, для получения меченого индикатора формулы (IIe)

где Р1e, Р2e, Р3e и Р4e независимо друг от друга представляют собой водород или защитную группу;
с последующим удалением защитных групп в случае, если Р1e-Р4e представляют собой защитные группы.
Возможно также последующее удаление избытка18F-, например, с помощью ионообменной хроматографии; и/или удаление органического растворителя; и/или приготовление состава на основе полученного соединения формулы (IIe) в виде водного раствора.
В данном аспекте настоящего изобретения функциональную гидроксильную группу, аминогруппу и кислотную группу в исходном материале DOPA удобно защищать в виде сложных эфиров, приемлемо C1-6-алкиловых сложных эфиров, предпочтительно ацильных эфиров, такие как трет-бутоксикарбонильные эфиры, или простых эфиров, предпочтительно C1-6-алкиловые простые эфиры, или амиды. Эти защитные группы могут быть легко удалены с помощью гидролиза, например, в присутствии кислоты или основания. Такое снятие защиты может быть осуществлено при помощи кислотного или основного катализатора, закрепленного на твердом носителе, что позволяет избежать операции нейтрализации после снятия защиты.
В соединениях формулы (Ie) предпочтительными ЛИНКЕРАМИ являются группы, описанные выше для соединений формулы (Id), а подходящим ТВЕРДЫМ НОСИТЕЛЕМ является полистирольная смола.
Некоторые из соединений формулы (I) являются новыми и поэтому составляют еще один аспект настоящего изобретения. Так, например, соединения формулы (Ia), в частности соединения формулы (Ib), (Ic), (Ig) и (Ih), и соединения формулы (Id), в частности соединения формулы (Ie), все в виде, определенном выше, образуют отдельные аспекты настоящего изобретения.
Как описано выше, преимущества такого твердофазного способа приготовления меченных18F индикаторов включают относительную быстроту способа, упрощенные способы очистки и легкость автоматизации - все это означает, что эти способы пригодны для приготовления меченных18F индикаторов для позитронной эмиссионной томографии (ПЭТ). Соответственно, настоящее изобретение дает возможность применения способа изготовления меченных18F индикаторов формулы (II) или (от IIа до IIh) для применения в ПЭТ.
Соответствующим образом, закрепленный на твердом носителе предшественник формулы (I) может быть предложен в виде части набора реагентов для приготовления радиофармпрепарата. Набор может содержать картридж, который может быть вставлен в автоматическую установку для синтеза, адаптированную соответствующим образом. Картридж, кроме закрепленного на твердом носителе предшественника, может содержать колонку для удаления нежелательных фторид-ионов и соответствующий сосуд, соединенный таким образом, чтобы реакционная смесь могла испаряться, а продукт мог быть введен в требуемую рецептуру. Туда также могут быть включены реактивы, растворители и иные расходные материалы, требуемые для синтеза, а также компакт-диск с записанным программным обеспечением, которое дает возможность управлять установкой для синтеза так, чтобы наилучшим образом удовлетворять требованиям потребителей с точки зрения концентрации радиоактивного компонента, объемов, производительности и т.д.
Удобно, если все компоненты набора являются одноразовыми, чтобы свести к минимуму возможность загрязнения в промежутках между циклами, и также могут быть стерильными и иметь сертификаты качества.
Далее, в изобретении также предложен радиофармацевтический набор реагентов для приготовления меченного18F индикатора для ПЭТ, который включает:
(i) сосуд, содержащий соединение формулы (I) или (от Ia до Ih); и
(ii) средства для элюирования сосуда источником18F-;
(iii) ионообменный картридж для удаления избытка18F-; и, возможно,
(iv) картридж для твердофазного снятия защиты конечного продукта формулы
(II) или (от IIa до IIh).
Далее, в изобретении также предложен картридж для радиофармацевтического набора реагентов для приготовления меченного18F индикатора для ПЭТ, включающий:
(i) сосуд, содержащий соединение формулы (I) или (от Ia до Ih); и
(ii) средства для элюирования сосуда источником18F-.
Средства элюирования сосуда источником18F- могут включать средства подачи в указанный сосуд раствора, содержащего18F-, и, возможно, растворителя, а также необходимые для этой подачи соединительные линии. Такие средства могут быть аналогичными, например, средствам элюирования, показанным на фиг.3 в статье S.A.Toorongian et al., Nucl. Med. Biol., 1990, v.17, No.3, р.275 или на фиг.1.1 в книге F.Z.Dorwald, "Organic Synthesis on Solid Phase", p.2.
Еще в одном аспекте настоящего изобретения предложено применение набора реагентов для приготовления радиофармпрепарата или картриджа для радиофармацевтического набора реагентов, описанных выше, для получения диагностического ПЭТ-изображения.
Далее изобретение будет проиллюстрировано с помощью следующих примеров.
Ниже даны расшифровки аббревиатур, используемых во всех примерах: ДМФА: N,N-диметилформамид; М/о: масса/объем; Ч.: час(ы); ТСХ: тонкослойная хроматография; ТГФ: тетрагидрофуран; Экв.: эквиваленты.
Примеры
Пример 1. Синтез 2[18F]-Фтор-2-дезокси-D-глюкозы (FDG)
Интермедиат 1
Приготовление метил-4,6-O-бензилидин-3-этоксиметил-α-D-маннопиранозида
Операция 1: Синтез метил-4,6-O-бензилидин-α-D-глюкопиранозида

Следуя указаниям Evans, M. E. Carbohydrate Research (1972), 21(3), 473-5, метил-α-D-глюкопиранозид (Aldrich, 257 ммоль) в ДМФА (200 мл) обрабатывали α,α-диметокситолуолом (39,0 г, 257 ммоль) и моногидратом толуолсульфокислоты (100 мг) в 1-литровой круглодонной колбе. Колба была соединена с установкой Buchi, ее откачивали и вращали. Колбу погружали в водяную баню с температурой 65°С и доводили ДМФА до слабого кипения с орошением паропровода, но так, чтобы отгонка ДМФА не происходила. Затем температуру водяной бани повысили до 100°С и отогнали ДМФА из реакции. После прекращения отгонки реакционную смесь охладили и обработали раствором гидрокарбоната натрия (5 г) в воде (750 мл) и этиловом спирте (250 мл). Реакционную смесь нагрели до 95°С на паровой бане и перемешивали до перехода продукта в мелкодисперсное состояние. Затем реакционную смесь охладили до 4°С, продукт отфильтровали, хорошо промыли водой и высушили в вакууме. Т.пл. 207-208,5.
Операция 2: Приготовление метил-4,6-O-бензилидин-3-этоксиметил-α-D-глюкопиранозида

Метил-4,6-О-бензилидин-α-D-глюкопиранозид (19,2 г, 68 ммоль), этоксиметилхлорид (9,7 г, 81,6 ммоль) и гидроксид тетрабутиламмония (5 мл 40%-ного масс./об. раствора) в дихлорметане (150 мл) энергично перемешивали с 10%-ным водным раствором гидроксида натрия (200 мл) при комнатной температуре. Через 5 часов водную фазу заменили свежим 10%-ным водным раствором гидроксида натрия (200 мл), к которому добавили гидроксид тетрабутиламмония (5 мл 40%-ного масс./об. раствора), и продолжали быстрое перемешивание в течение ночи. Органическую фазу затем отделяли, сушили над сульфатом натрия и испаряли в вакууме. С помощью тонкослойной хроматографии остатка (40-60 гексаны - этилацетат, 2:1) на силикагеле, проявленной распылением молибдата церия-аммония (см. выше), определили присутствие трех продуктов алкилирования. С помощью хроматографии на силикагеле (1 кг, сухая масса) при градиенте 40-60 гексанов - этилацетата от 2:1 до 1:1 получили три фракции, которые по данным ЯМР-анализа представляли собой:
Фракция 1: Метил-2,3-диэтоксиметил-4,6-O-бензилидин-α-D-глюкопиранозид
Фракция 2: Метил-2-этоксиметил-4, 6-O-бензилидин-α-D-глюкопиранозид
Фракция 3: Метил-3-этоксиметил-4,6-O-бензилидин-α-D-глюкопиранозид
Операция 3: Приготовление метил-2-кето-3-этоксиметил-4,6-O-бензилидин-α-D-глюкопиранозида
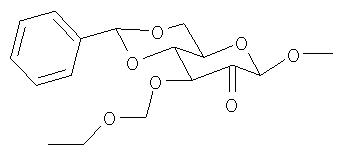
Метил-3-этоксиметил-4,6-O-бензилидин-α-D-глюкопиранозид (3 г, 8,0 ммоль) обрабатывали метилсульфоксидом (50 мл) и уксусным ангидридом (25 мл) при комнатной температуре в течение 24 ч до тех пор, пока реакцию, по данным ТСХ (петролейный эфир/этилацетат, 1:1), проявленной молибдатом церия-аммония, не посчитали законченной. Раствор затем разбавили диэтиловым эфиром (200 мл) и промыли 10%-ным водным раствором карбоната калия, чтобы гидролизовать избыток уксусного ангидрида. Эфирный слой отделили и промыли водой (100 мл). Эфирный слой отделили, сушили над сульфатом натрия и концентрировали в вакууме с получением кристаллического твердого вещества. Его перекристаллизовывали из эфира/бензина и получили 1,5 г метил-2-кето-3-этоксиметил-4,6-O-бензилидин-α-D-глюкопиранозида.
Операция 4: Приготовление метил-4,6-O-бензилидин-3-этоксиметил-α-D-маннопиранозида
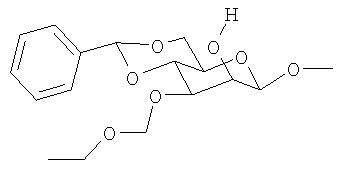
Метил-2-кето-3-этоксиметил-4,6-O-бензилидин-α-D-глюкопиранозид (0,5 г, 1,3 ммоль) в метаноле (50 мл) и ТГФ (10 мл) обрабатывали борогидридом натрия (200 мг, 5,3 ммоль) при комнатной температуре и постоянном перемешивании. Реакционную смесь затем концентрировали в вакууме до смолообразного состояния и продукт разделяли в системе этилацетат (50 мл) - 10%-ный водный раствор карбоната калия (50 мл). Раствор в этилацетате отделили, сушили над сульфатом натрия и концентрировали в вакууме с получением метил-3-этоксиметил-4,6-O-бензилидин-α -D-маннопиранозида.
Пример 1(i): Приготовление перфторбутан-1,4-бис-сульфонилхлорида
(По способу Weiming Qiu и Donald J. Burton, Journal of Fluorine Chemistry, 60(1993)93-100)

Смесь 1,4-дииодперфторбутана (I(CF2)4I) (24,14 г, 53,2 ммоль), дитионита натрия Na2S2O4 (24 г, 117,2 ммоль) и гидросульфата натрия МаНСО3 (12,8 г, 152,4 ммоль) в воде H2O (36 мл)/ацетонитриле CH3CN (36 мл) перемешивали при комнатной температуре в течение 2 часов. Смесь отфильтровали, фильтрат концентрировали под уменьшенным давлением для удаления ацетонитрила. К остатку прибавили H2O (100 мл). Полученный таким образом раствор при энергичном перемешивании обрабатывали газообразным хлором Cl2 при 0°С до исчезновения цвета I2. Затем прибавили дихлорметан CH2Cl2 (100 мл) и смесь энергично встряхивали. Органическую фазу отделили и водную фазу экстрагировали CH2Cl2. Объединенные органические фазы промыли водой Н2О, раствором соли, сушили сульфатом натрия Na2SO4 и концентрировали с получением воскообразного желтого кристаллического твердого вещества (15,4 г, 74%). После перекристаллизации из гексана получили почти белые иглы перфторбутан-1,4-бис-сульфонилхлорида.
19F ЯМР (CDCl3, эталон CFCl3) δ: -104,4; -119,1.
Пример 1(ii): Приготовление дикалиевой соли перфторбутан-1,4-бис-сульфоната

К раствору гидроксида калия КОН (9,8 г, 5 экв.) в воде Н2О (19 мл) постепенно прибавили при перемешивании перфторбутан-1, 4-бис-сульфонилхлорид (14 г, 35 ммоль) при 85-90°С. После прибавления реакцию продолжали еще 4 часа при той же температуре, затем охлаждали в течение ночи. Смесь отфильтровали, твердое вещество промыли небольшим количеством охлажденной воды и сушили в вакууме с получением дикалиевой соли перфторбутан-1,4-бис-сульфоната.
19F ЯМР (CD3OD, эталон CFCl3) δ: -114,00; -120,11.
Пример 1(iii): Приготовление перфторбутан-1,4-бис-сульфоновой кислоты (по способу, описанному в патенте США 4329478, Fred E. Behr)

Дикалиевую соль перфторбутан-1,4-бис-сульфоната (15 г, 34,2 ммоль) растворили в горячей воде (100 мл). Смесь добавили в ионообменную колонку со смолой Amberlyst 15 (40×4 см), которую предварительно промыли избытком 6 н. HCl и сполоснули дистиллированной водой. Колонку затем медленно промывали дистиллированной водой и собрали первые 300 мл водного раствора. Раствор концентрировали в вакууме, а остаток сушили под уменьшенным давлением при 80°С с получением перфторбутан-1, 4-бис-сульфоновой кислоты (11,0 г, 30 ммоль, 88%).
1H ЯМР (CDCl3) δ: 8,00.
19F ЯМР (CDCl3, эталон CFCl3) δ: -114,7; -121,3.
Пример 1(iv): Приготовление ангидрида перфторбутан-1,4-бис-сульфоновой кислоты (по способу, описанному в патенте США 4329478, Fred E. Behr)

Перфторбутан-1,4-бис-сульфоновую кислоту (11,0 г, ˜30 ммоль) смешали с Р2 О5 (40 г, ˜10 экв.) и песком. Смесь нагревали до 140-180°С и перегоняли под уменьшенным давлением с приемником, охлаждаемым сухим льдом, с получением неочищенного продукта (5, 12 г). При повторной перегонке получен чистый ангидрид перфторбутан-1,4-бис-сульфоновой кислоты.
19F ЯМР (CDCl3, эталон CFCl3) δ: -105,7; -121, 8.
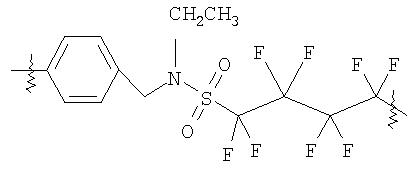
Пример 1 (v): Синтез PS-4-(бензил-этил-сульфонамид)октафторбутан-1-сульфоновой кислоты
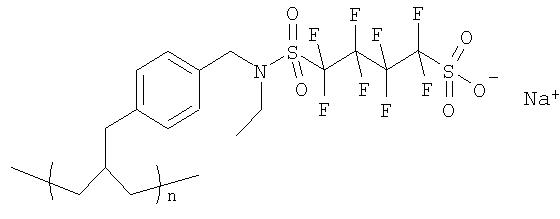
К порции полистирольной смолы (Novabiochem, смола Novasyn) (202 мг), предварительно подвергнутой набуханию в дихлорметане (2 мл) и затем суспендированной в еще одной аликвоте дихлорметана (2 мл), прибавили ангидрид перфторбутил-1,4-циклосульфоновой кислоты (116 мг, 5 экв.). Затем добавили диизопропилэтиламин (0,174 мл) и суспензию перемешивали в течение ночи при комнатной температуре. Растворитель отделили фильтрованием и смолу промыли, последовательно добавляя и отфильтровывая дихлорметан (5 мл), метанол (5 мл), ДМФА (5 мл), воду (5 мл), метанол (5 мл) и дихлорметан (5 мл). Полученную смолу затем обрабатывали NaOH (1M) в ТГФ/воде (2×2 мл), а затем последовательно промывали порциями метанола (5 мл), дихлорметана (5 мл) и вновь метанола (5 мл). Смолу затем сушили в высоком вакууме.
19F ЯМР гелевой фазы (эталон CFCl3, 300K) δ: -121,0; -114,8; -113,4.
Пример 1(vi): Синтез PS-4-(бензил-этил-сульфонамид)октафторбутан-1-сульфонилхлорида
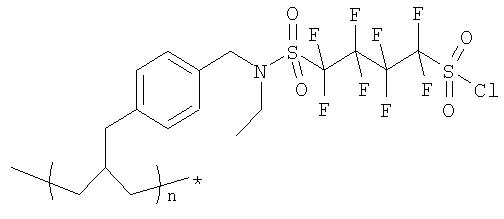
Порцию смолы, приготовленной, как указано выше в Примере 1(v), подвергали набуханию в дихлорметане (2 мл), а затем последовательно промывали HCI (1M) в ТГФ/воде (10×5 мл) с получением свободной сульфоновой кислоты. Смолу последовательно промывали дихлорметаном, метанолом и ТГФ, а затем сушили в высоком вакууме.
Смолу затем суспендировали в дихлорметане и к ней добавляли избыток обычного хлорирующего агента, такого как пентахлорид фосфора, трихлорид фосфора или тионилхлорид. Суспензию перемешивали в течение 2 часов, далее отфильтровывали, а затем промывали смолу дихлорметаном, а потом ТГФ.
Пример 1(vii): Синтез смолы с защищенной маннопиранозой
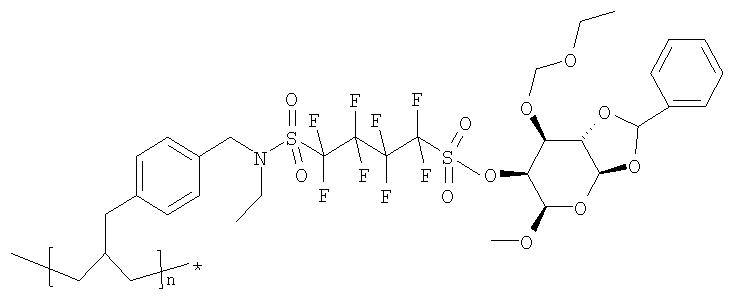
Раствор Интермедиата 1 в ТГФ прибавили к порции смолы, приготовленной, как описано выше в Примере (vi), предварительно подвергнутой набуханию в ТГФ. К полученной смеси прибавили раствор трет-бутилата (трет-бутоксида) калия в тетрагидрофуране и суспензию перемешивали в течение ночи. После отделения смолы фильтрованием ее последовательно промывали дихлорметаном и ТГФ, а затем сушили в высоком вакууме.
Пример 1(viii): Фторирование радиоактивным фтором для получения [18F]-FDG
Порцию смолы (приготовленной, как описано в Примере 1(vii)), находящуюся в картридже, добавили к раствору kryptofix, карбоната калия и [18F]-фторида в сухом ацетонитриле. Суспензию нагревали до 85°С в течение 10 минут, а затем раствор профильтровали. Раствор затем направляли на картридж для C18 твердофазной экстракции и промывали водой для удаления ацетонитрила, kryptofix и карбоната калия. Добавление дополнительного количества ацетонитрила вызывает вымывание из картриджа продукта фторирования радиоактивным фтором в раствор 0,1М HCl. Этот раствор нагревают еще 5 минут, затем нейтрализуют и анализируют.
Пример 2: Синтез 2-(1,1-дицианопропен-2-ил)-6-(2-[18F]-Фторэтил)-(метиламино)-нафталина (FDDNP)
Пример 2 (i): Синтез PS-4-(бензил-этил-сульфонамид)-бутан-1-сульфонилхлорида

К суспензии смолы, предварительно подвергшейся набуханию в дихлорметане (5 мл), прибавили избыток 1,4-бутан-дисульфонилхлорида в дихлорметане вместе с избытком триэтиламина. Суспензию перемешивали при комнатной температуре в течение ночи. Смолу отфильтровали, последовательно промывали дихлорметаном, метанолом, ТГФ, водой, метанолом и еще одной порцией дихлорметана. После последней промывки смолу сушили в вакууме.
Пример 2(ii): Синтез смолы с закрепленным на ней 2-(1,1-дицианпропен-2-ил)-6-(2-этил)-(метиламино)-нафталином

К суспензии вышеуказанной смолы, предварительно подвергнутой набуханию в дихлорметане (2 мл), прибавили избыток 2-(1, 1-дицианпропен-2-ил)-6-(2-гидроксиэтил)-метиламино)-нафталина в дихлорметане вместе с избытком триэтиламина. Суспензию перемешивали при комнатной температуре в течение ночи. Смолу отфильтровали, последовательно промывали дихлорметаном и ТГФ. После последней промывки смолу сушили в вакууме.
Пример 2(iii): Фторирование радиоактивным фтором для получения [18F]-FDDNP
К порции смолы, находящейся в картридже, добавили раствор kryptofix, карбоната калия и [18F]-фторида в сухом ацетонитриле. Суспензию нагревали до 85°С в течение 10 минут, а затем раствор профильтровали. Смолу затем промывали ацетонитрилом (1 мл), все содержимое собирали, далее испаряли растворитель, после чего готовили состав рецептурной формы.
Пример 3: Синтез [18F]-фторбензола
Пример 3(i): Синтез PS-иодфенилбензилового эфира

К суспензии смолы Wang Resin, предварительно подвергшейся набуханию в ДМФА (2 мл), прибавили раствор карбоната цезия и иодфенола в ДМФА. Смесь перемешивали в течение 3 часов при 60°С, а затем оставили при комнатной температуре в течение ночи. Смолу отфильтровали, последовательно промывали метанолом, дихлорметаном, ДМФА и ТГФ, а затем тщательно сушили в высоком вакууме.
Пример 3(ii): Синтез PS-диацетоксииодфенилбензилового эфира

Суспензию вышеуказанной смолы обрабатывали уксусным ангидридом и пероксидом водорода (см. способ S. Ficht, Tetrahedron, 57 (2001) 4863) в соотношении 4:1 при 40°С в течение ночи. Далее смолу отфильтровали и тщательно промывали метанолом, а затем сушили досуха в высоком вакууме.
Пример 3(iii): Синтез трифлата PS-(фенил)(4-фенилбензилового эфира)иодония

К суспензии вышеуказанной смолы в дихлорметане прибавили по каплям при температуре -30°С в течение 15 минут трифторметансульфоновую кислоту. Смесь затем нагревали до 0°С в течение еще 15 минут, а затем перемешивали при комнатной температуре в течение ночи. Суспензию затем охладили до -30°С и прибавили фенилборную кислоту, суспензию перемешивали в течение 1 часа, а затем нагрели до комнатной температуры и снова перемешивали в течение ночи. Смесь затем отфильтровали, смолу тщательно промывали дихлорметаном и диэтиловым эфиром, а затем сушили в вакууме.
Пример 3(iv): Фторирование радиоактивным фтором для получения [18F]-фторбензола
К порции смолы, находящейся в картридже, добавили раствор kryptofix, карбоната калия и [18F]-фторида в сухом ацетонитриле. Суспензию нагревали до 85°С в течение 10 минут, а затем раствор профильтровали. Смолу затем промывали ацетонитрилом (1 мл), все содержимое собирали, затем испаряли растворитель, после чего готовили состав рецептурной формы.
Реферат
Изобретение относится к усовершенствованному твердофазному способу приготовления радиоизотопных индикаторов, в частности, для приготовления соединений, меченных18F, которые могут быть применены в качестве радиоактивных индикаторов для позитронной эмиссионной томографии (ПЭТ). В частности, изобретение относится к способу приготовления меченного18Е индикатора, включающему обработку закрепленного на смоле предшественника формулы (I): ТВЕРДЫЙ НОСИТЕЛЬ-ЛИНКЕР-Х-ИНДИКАТОР, где Х - это группа, способствующая нуклеофильному замещению по определенному центру закрепленного ИНДИКАТОРА, ионом18F- для получения меченого индикатора формулы (II):18F-ИНДИКАТОР; к соединению формулы (Ib):

к соединению формулы (Ih):

к радиофармацевтическому набору реагентов для приготовления меченного18F индикатора для использования в ПЭТ и к картриджу для радиофармацевтического набора реагентов для приготовления меченного18F индикатора для использования в позитронной эмиссионной томографии. 6 н. и 7 з.п. ф-лы.
Формула









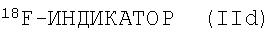



Комментарии