Способ отбора желательного белка и нуклеиновой кислоты, средства для его осуществления - RU2233878C2
Код документа: RU2233878C2
Чертежи
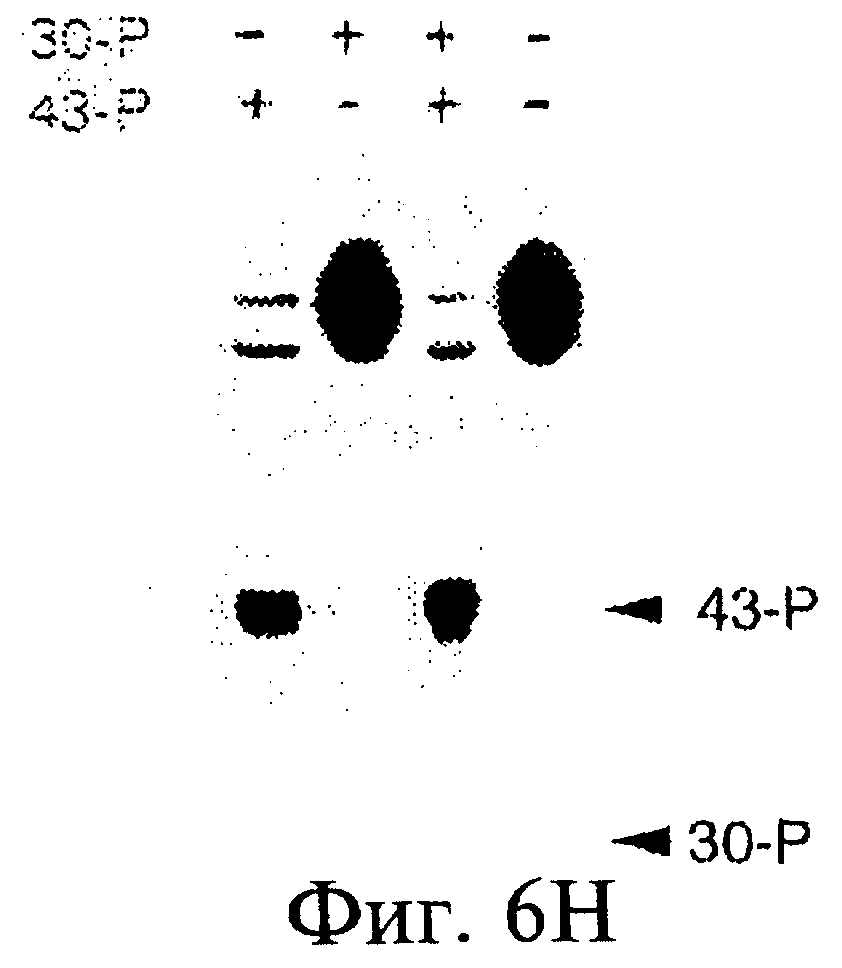



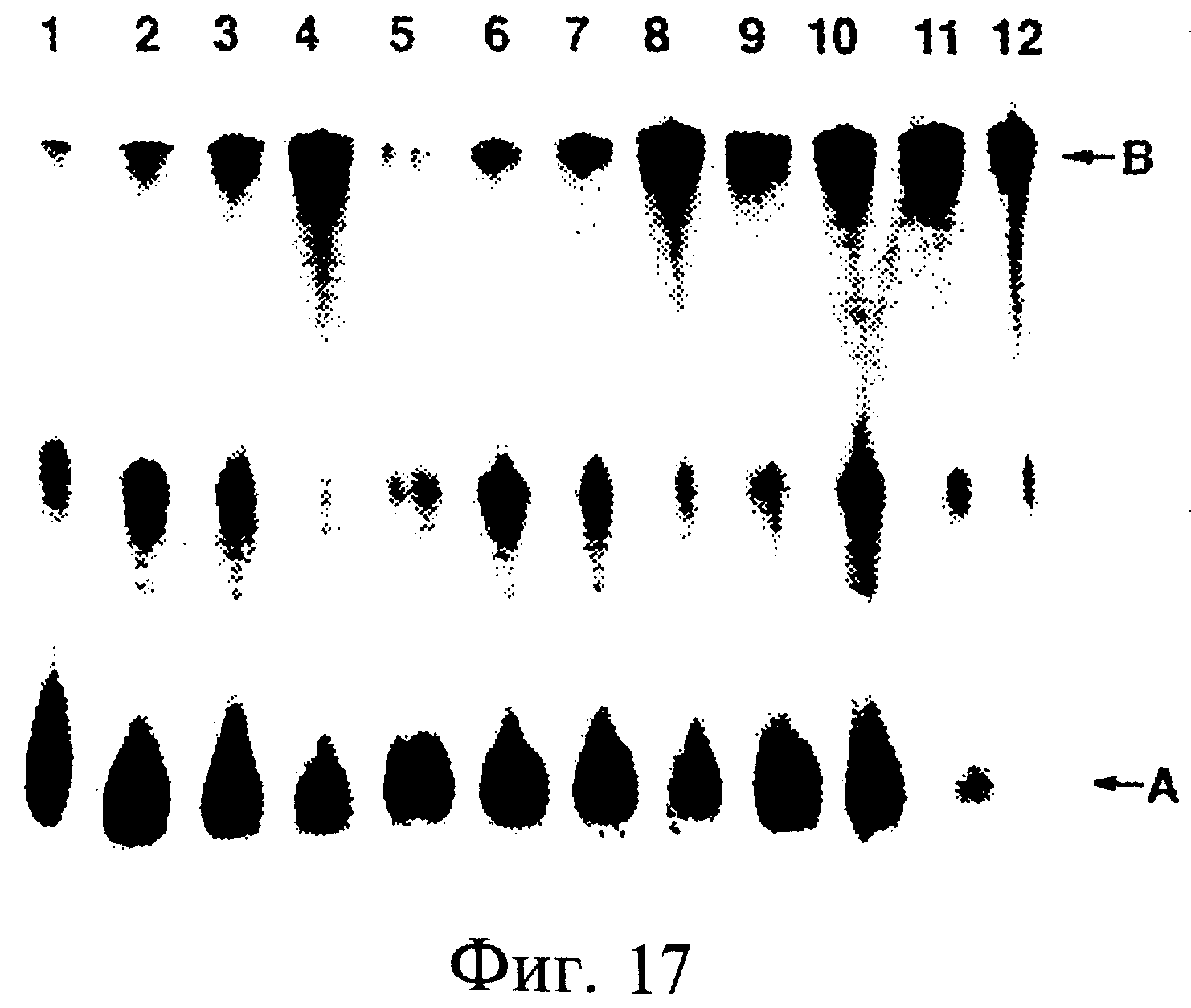

Описание
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение касается способов отбора белков.
Изобретение было осуществлено с использованием федеральной поддержки в виде грантов F32 GM 17776-01 и F32 GM 17776-02. Соответственно, федеральное правительство обладает некоторыми правами на настоящее изобретение.
Существующие в настоящее время способы выделения молекул РНК и ДНК основаны на их функциях. Например, эксперименты Ellington & Szostak (Nature, 1990, 346, 818 и Nature, 1992, 355, 850) и Tuerk & Gold (Science, 1990, 249, 505 и J. Mol. Biol., 1991, 222, 739) показали, что очень редкие (например, на уровне менее одной на 1013) молекулы нуклеиновых кислот с желательными характеристиками могут быть выделены из комплексных пулов молекул с помощью повторяющихся циклов отбора и амплификации. Эти способы обладают преимуществами по сравнению с традиционными генетическими методами отбора в том, что (1) может быть подвергнут скринингу очень большой пул молекул (более 1015), (ii) не затрагивается жизнеспособность организмов-хозяев и условия in vivo, и (iii) отбор может быть проведен даже тогда, когда не существует способа генетического скрининга в модели in vivo. Разрешающая способность отбора in vitro была продемонстрирована при определении последовательностей РНК и ДНК, обладающих очень узкоспецифичными функциями по связыванию с белками (см., например, Tuerk & Gold, 1990, Science, 249, 505; Irvine et al., 1991, J. Mol. Biol, 222, 739; Oliphant et al., 1989, Mol. Cell Biol., 9, 2944; Blackwell et al., 1990, Science, 250, 1104; Pollock & Treisman, 1990, Nucl. Acids Res., 18, 6197; Thiesen & Bach, 1990, Nucl. Acids Res., 18, 3203; Bartel et al., 1991, Cell, 57, 529; Stormo & Yoshioka, 1991, Proc. Natl. Acad. Sci. USA, 88, 5699; и Bock et al., 1992, Nature, 355, 564), функциями по связыванию с малыми молекулами (Ellington & Szostak, 1990, Nature, 346, 818); Ellington & Szostak, 1992, Nature, 355, 850) и каталитическими функциями (Green et al., 1990, Nature, 347, 406; Robertson & Joyce, 1990, Nature, 344, 467; Beaudry & Joyce, 1992, Science, 257, 635; Bartel & Szostak, 1993, Science, 261, 1411; Lorsch & Szostak, 1994, Nature, 371, 31-36; Cuenoud & Szostak, 1995, Nature, 375, 611-614; Chapman & Szostak, 1995, Chemistry and Biology, 2, 325-333; и Lohse & Szostak, 1996, Nature, 381, 442-444). Однако сходная стратегия отбора и амплификации белков пока отсутствует.
РЕЗЮМЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения явилась разработка принципов отбора in vitro и преобразования последовательностей in vitro, которые могут быть применимы в отношении белков. Изобретение представляет выделение белков, обладающих желательными характеристиками, из обширных пулов при частично или полностью случайном подборе аминокислотных последовательностей. Кроме того, изобретение решает проблему выделения и амплификации полипептидных последовательностей путем ковалентного присоединения кодирующей последовательности мРНК к молекуле белка.
В целом, представляемый изобретением способ включает транскрипционно-трансляционный протокол in vitro или in situ, с помощью которого формируется белок, ковалентно присоединенный к 3'-концу его собственной мРНК, т.е. РНК-пептидный гибрид. Это осуществляется путем синтеза и трансляции in vitro или in situ молекулы мРНК с пептидным акцептором, присоединенным к ее 3'-концу. Одним из предпочтительных пептидных акцепторов является пуромицин, являющийся аналогом нуклеозида, который присоединяется к С-концу растущей пептидной цепи и терминирующий тем самым процесс трансляции. В одном из предпочтительных вариантов последовательность ДНК включается между концом мРНК и пептидным акцептором, роль которого связана с вызыванием т.н. рибосомной паузы в конце открытой рамки считывания, что дает дополнительное время для пептидного акцептора (например, для пуромицина) присоединить новообразованную полипептидную цепь перед гидролизом пептидил-тРНК.
Если это желательно, то получаемый в результате РНК-пептидный гибрид может быть включен в циклично повторяющиеся этапы отбора и амплификации, поскольку информация о полипептидной последовательности может быть восстановлена с помощью обратной транскрипции и амплификации (например, с помощью ПЦР-амплификации или с помощью любого другого метода амплификации, включая основанные на РНК методики амплификации, такие как 3SR или TSA). Амплифицированная нуклеиновая кислота может быть затем транскрибирована, модифицирована и транслирована in vitro или in situ с целью получения мРНК-пептидных гибридов для следующего раунда отбора. Способность осуществлять многократные раунды отбора и амплификации позволяет обогащать и выделять очень редкие молекулы: например, когда одна желательная молекула содержится в пуле, состоящем из 1015 членов. С другой стороны, это позволяет выделять новые или улучшенные белки, которые специфически распознают виртуально любую мишень или которые катализируют желательные химические реакции.
Соответственно, в первом своем аспекте настоящее изобретение представляет способ отбора желательного белка, включающий следующие этапы: (а) представление популяции молекул-"кандидатов" РНК, каждая из которых включает последовательность инициации трансляции и старт-кодон, функционально связанный с последовательностью, кодирующей белок-кандидат", и каждая из которых функционально соединена с пептидным акцептором по 3'-концу последовательности, кодирующей белок-кандидат"; (b) трансляция in vitro или in situ последовательности, кодирующей белок-кандидат" с получением популяции "кандидатных" РНК-пептидных гибридов; и (с) отбор желательного РНК-пептидного гибрида, определяющий тем самым отбор желательного белка.
В близком варианте настоящее изобретение представляет способ отбора молекулы ДНК, которая кодирует желательный белок, включающий следующие этапы: (а) представление популяции молекул-кандидатов" РНК, каждая из которых включает последовательность инициации трансляции и старт-кодон, функционально связанный с последовательностью, кодирующей белок-кандидат"', и каждая из которых функционально соединена с пептидным акцептором по 3'-концу последовательности, кодирующей белок-"кандидат"; (b) трансляция in vitro или in situ последовательности, кодирующей белок-кандидат" с получением популяции "кандидатных" РНК-пептидных гибридов; (с) отбор желательного РНК-пептидного гибрида; и (а) формирование на основании РНК-компонента данного гибрида молекулы ДНК, которая кодирует желательный белок.
В другом близком варианте настоящее изобретение представляет способ отбора белка, обладающего измененными функциями по сравнению с референсным белком, включающий следующие этапы: (а) получение популяции молекул-"кандидатов" РНК из популяции ДНК-матриц, при том, что каждая из матриц-кандидатов" ДНК включает последовательность, кодирующую белок-кандидат", которая отличается от последовательности, кодирующей референсный белок, и при том, что каждая молекула РНК включает последовательность инициации трансляции и старт-кодон, функционально присоединенные к последовательности, кодирующей белок-"кандидат", и каждая из них функционально соединена с пептидным акцептором по 3'-концу; (b) трансляция in vitro или in situ последовательности, кодирующей белок-кандидат", с целью получения популяции "кандидатных" РНК-пептидных гибридов; и (с) отбор РНК-пептидных гибридов, обладающих измененными функциями, позволяющий таким образом отбирать белок, обладающий измененной функцией.
Еще в одном близком варианте настоящее изобретение представляет способ отбора молекулы ДНК, которая кодирует белок, обладающий измененной функцией по сравнению с референсным белком, включающий следующие этапы: (а) получение популяции молекул-"кандидатов" РНК из популяции ДНК-матриц, при том что каждая из матриц-"кандидатов" ДНК включает последовательность, кодирующую белок-"кандидат", которая отличается от последовательности, кодирующей референсный белок, и при том, что каждая молекула РНК включает последовательность инициации трансляции и старт-кодон, функционально присоединенные к последовательности, кодирующей белок-кандидат", и каждая из них функционально соединена с пептидным акцептором по 3'-концу; (b) трансляция in vitro или in situ последовательности, кодирующей белок-"кандидат", с целью получения популяции "кандидатных" РНК-пептидных гибридов; (с) отбор РНК-пептидных гибридов, обладающий измененной функцией; и (d) формирование на основе РНК-компонента гибрида молекулы ДНК, которая кодирует белок, обладающий измененной функцией.
Еще в одном варианте настоящее изобретение представляет способ отбора желательной РНК, включающий следующие этапы: (а) представление популяции молекул-кандидатов" РНК, каждая из которых включает последовательность инициации трансляции и старт-кодон, функционально присоединенный к последовательности, кодирующей белок-кандидат", и каждая из которых функционально соединена с пептидным акцептором по 3'-концу последовательности, кодирующей белок-кандидат"; (b) трансляция in vitro или in situ последовательностей, кодирующих белки-"кандидаты", с получением популяции "кандидатных" РНК-пептидных гибридов; и (с) отбор желательного РНК-пептидного гибрида, что позволяет таким образом отбирать желательную РНК.
В предпочтительных вариантах перечисленных выше способов пептидным акцептором является пуромицин; каждая из молекул-"кандидатов" РНК дополнительно включает последовательность-индуктор паузы или дополнительно включает последовательность ДНК или аналога ДНК, ковалентно присоединенную к 3'-концу данной молекулы РНК; популяция молекул-кандидатов" РНК включает по крайней мере 109, предпочтительно по крайней мере 1010, более предпочтительно по крайней мере 1011, 1012 или 1013 и, что наиболее предпочтительно, по крайней мере 1014 различных молекул РНК; реакция трансляции in vitro осуществляется в лизате, приготавливаемом из эукариотических клеток или их частей (например, осуществляется в лизате ретикулоцитов или в лизате зародышей пшеницы); реакция трансляции in vitro осуществляется в экстракте, приготавливаемом из прокариотических клеток (например, клеток Е.coli.) или их частей; этап отбора включает связывание желательного белка с иммобилизованным связывающим компонентом; этап отбора включает тестирование желательного белка на функциональную активность; молекула ДНК амплифицирована; способ, кроме того, включает повторяющиеся этапы перечисленных выше способов; способ также включает транскрипцию молекулы РНК с молекулы ДНК и повторяющиеся этапы от (а) до (d); после этапа трансляции in vitro данный способ также включает этап инкубации, осуществляемый в присутствии 50-100 мМ Mg2+; и РНК-пептидный гибрид также включает последовательность нуклеиновой кислоты или ее аналога, расположенную проксимально по отношению к пептидному акцептору, что обеспечивает увеличение гибкости.
В других вариантах настоящее изобретение представляет РНК-пептидный гибрид, выбираемый любым из способов по настоящему изобретению; рибонуклеиновая кислота, ковалентно соединенная по амидной связи с аминокислотной последовательностью, при том, что эта аминокислотная последовательность кодируется данной рибонуклеиновой кислотой; и рибонуклеиновая кислота, которая включает последовательность инициации трансляции и старт-кодон, функционально соединенный с последовательностью, кодирующей белок-кандидат", при том, что рибонуклеиновая кислота функционально присоединена к пептидному акцептору (например, к пуромицину) по 3'-концу последовательности, кодирующей белок-кандидат".
Во втором своем аспекте настоящее изобретение представляет способ отбора желательного белка или желательной РНК путем обогащения пула последовательностей. Способ включает следующие этапы: (а) представление популяции молекул-кандидатов" РНК, каждая из которых включает последовательность инициации трансляции и старт-кодон, функционально присоединенные к последовательности, кодирующей белок-кандидат", и каждая из которых функционально присоединена к пептидному акцептору по 3'-концу последовательности, кодирующей белок-"кандидат"; (b) трансляция in vitro или in situ последовательностей, кодирующий белки-"кандидаты", с получением популяции "кандидатных" РНК-пептидных гибридов; (с) контакт популяции РНК-пептидных гибридов со связывающим компонентом, специфичным в отношении либо РНК-компонента, либо белка-компонента РНК-пептидного гибрида в условиях, которые обеспечивают эффективное отделение комплексов РНК-пептидных гибридов со связывающим компонентом от несвязанных составляющих популяции; (d) выделение связанных РНК-пептидных гибридов из этих комплексов; и (е) контакт популяции РНК-пептидных гибридов по этапу (d) со связывающим компонентом, специфичным в отношении белкового компонента желательного РНК-пептидного гибрида в условиях, которые обеспечивают эффективное отделение комплекса РНК-пептидного гибрида и связывающего компонента от несвязанных составляющих упомянутой популяции, что таким образом обеспечивает отбор желательного белка и желательной РНК.
В предпочтительных вариантах способ также включает повторяющиеся этапы от (а) до (е). Дополнительно для этих повторяющихся этапов могут быть использованы те же самые или отличающиеся связывающие компоненты, в любом случае служащие для избирательного обогащения желательного РНК-пептидного гибрида. В другом предпочтительном варианте этап (а) включает использование связывающего компонента (например, моноклонального антитела), специфичного в отношении белковой части желательного РНК-пептидного гибрида. Этот этап предпочтительно осуществляется после обратной транскрипции РНК-компонента данного гибрида с целью формирования ДНК, которая кодирует желательный белок. Если желательно, эта ДНК может быть выделена и (или) амплифицирована с помощью ПЦР. Данная методика обогащения может быть использована для отбора желательного белка или может быть использована для отбора белка, обладающего измененной функцией по сравнению с референсным белком.
В других предпочтительных вариантах способов обогащения пептидным акцептором является пуромицин; каждая из молекул-"кандидатов" РНК также включает сайт-индуктор паузы или также включает последовательность ДНК или аналога ДНК, ковалентно присоединенную к 3'-концу данной молекулы РНК; популяция молекул-кандидатов" РНК включает по крайней мере 109, предпочтительно по крайней мере 1010, более предпочтительно по крайней мере 1011, 1012 или 1013 и, что наиболее предпочтительно, по крайней мере 1014 различных молекул РНК; реакция трансляции in vitro осуществляется в лизате, приготавливаемом из эукариотических клеток или их частей (например, осуществляется в лизате ретикулоцитов или в лизате зародышей пшеницы); реакция трансляции in vitro осуществляется в экстракте, приготавливаемом из прокариотических клеток (например, клеток Е. coli) или их частей; этап отбора включает связывание желательного белка с иммобилизованным связывающим компонентом; этап отбора включает тестирование желательного белка на функциональную активность; молекула ДНК амплифицирована; способ, кроме того, включает повторяющиеся этапы перечисленных выше способов; способ также включает транскрипцию молекулы РНК с молекулы ДНК и повторяющиеся этапы от (а) до (d); после этапа трансляции in vitro данный способ также включает этап инкубации, осуществляемый в присутствии 50-100 мМ Мg2+ и РНК-пептидный гибрид также включает последовательность нуклеиновой кислоты или ее аналога, расположенную проксимально по отношению к пептидному акцептору, что обеспечивает увеличение гибкости.
В сходном варианте настоящее изобретение представляет наборы реактивов для осуществления любого из заявлямых способов отбора.
В третьем и заключительном своем аспекте настоящее изобретение представляет микрочип, который включает набор иммобилизованных одноцепочечных нуклеиновых кислот, предназначенных для гибридизации с РНК-пептидными гибридами. Предпочтительно, чтобы белковый компонент РНК-пептидного гибрида кодировался этой РНК.
По использованию в данном тексте под термином "популяция" понимается наличие более чем одной молекулы (например, более чем одной молекулы РНК, ДНК или РНК-пептидного гибрида). Поскольку способы по настоящему изобретению представляют отбор, который начинается, если это желательно, с большого числа молекул-"кандидатов", то понятие "популяции" согласно настоящему изобретению предпочтительно соответствует более чем 109 молекулам, предпочтительнее более чем 1011, 1012 или 1013 молекулам и, что наиболее предпочтительно, более чем 1013 молекулам.
Под "отбором" понимается эффективное отделение молекулы от других молекул в популяции. По использованию здесь этап "отбора" соответствует двукратному, предпочтительно 30-кратному, более предпочтительно 100-кратному и, что наиболее предпочтительно, тысячекратному обогащению желательной молекулы по отношению к нежелательным молекулам в популяции после осуществления этапа отбора. Как определено здесь, этап отбора может быть повторен любое число раз, и при этом различные варианты этапов отбора могут быть скомбинированы в данном подходе.
Термин "белок" обозначает любые две или большее число естественно встречающихся или модифицированных аминокислот, соединенных друг с другом одной или большим числом пептидных связей. Термины "белок" ("протеин") и "пептид" используются здесь вперемешку, т.е. как синонимы.
Термин "РНК" обозначает последовательность двух или большего числа ковалентно соединенных естественно встречающихся или модифицированных рибонуклеотидов. Одним из примеров модифицированной РНК, охватываемой данным термином, является фосфоротиоат-РНК.
Термин "последовательность иниациации трансляции" обозначает любую последовательность, которая способна взаимодействовать с функциональным входным сайтом рибосомы. В бактериальных системах этот участок иногда обозначается как "последовательность Шайна-Дальгарно".
Термин "старт-кодон" обозначает триплет, являющийся сигналом начала кодирующей нуклеотидной последовательности. Чаще всего этим триплетом является триплет AUG (или ATG); однако, может быть принят и любой другой триплет, который может быть использован в данной функции.
Термин "ковалентно связанный" с пептидным акцептором обозначает, что пептидный акцептор соединен с "последовательностью, кодирующей белок" либо напрямую с помощью ковалентной связи, либо опосредованно через другую ковалентно связанную последовательность (например, ДНК, соответствующую сайту-индуктору паузы).
Под "пептидным акцептором" понимается любая молекула, которая может быть добавлена к С-концу нарастающей полипептидной цепи при наличии каталитической активности рибосомной пептидилтрансферазы. Обычно такие молекулы включают (i) нуклеотид или нуклеотидоподобную составляющую (например, аденозин или аналог аденозина (приемлемо диметилирование по N-6 амино положению)), (ii) аминокислота или подобная аминокислоте составляющая (например, любая из 20 D- или L-аминокислот или аналог любой из этих аминокислот (например, 0-метилтирозин или любой из аналогов, описанных у Е11-man et al., 1991, Methods Enzymol, 202, 301)), и (iii) сцепление между двумя этими составляющими (например, эфирное, амидное или кетонное сцепление по положению 3' или, что менее предпочтительно, по положению 2'); предпочтительно, чтобы это сцепление существенно не нарушало пространственную укладку кольца в естественной конформации рибонуклеотида. Пептидные акцепторы могут также быть нуклеофильными компонентами, т.е. могут быть, без каких-либо ограничений, аминогруппой, гидроксильной группой или сульфгидрильной группой. Кроме того, пептидные акцепторы могут быть составлены соединениями, подобными нуклеотидом, аминокислотам или комбинированными нуклеотид-аминокислотными структурами.
Под тем, что пептидный акцептор расположен "с 3'-конца" последовательности, кодирующей белок, понимается то, что молекула пептидного акцептора располагается за последним кодоном последовательности, кодирующей белок. Этот термин включает, без каких-либо ограничений, молекулу пептидного акцептора, которая расположена точно по 3'-концу последовательности, кодирующей белок, а также таковую, если она отделена от последнего кодона вставочной кодирующей или некодирующей последовательностью (например, последовательностью, соответствующей сайту - индуктору паузы). Также этот термин включает конструкции, в которых кодирующие или некодирующие последовательности следуют (т.е. являются 3"-концевыми по отношению) за молекулой пептидного акцептора. Дополнительно этот термин обозначает, без каких-либо ограничений, молекулу пептидного акцептора, которая ковалентно связана (как напрямую, так и опосредованно через нуклеотидную последовательность) с последовательностью, кодирующей белок, равно как и такую молекулу, которая соединена с последовательностью, кодирующей белок, какими-либо нековалентными способами, например, путем гибридизации с использованием второй нуклеотидной последовательности, которая связывается в 3'-конце или рядом с ним с последовательностью, кодирующей белок, таким образом оказываясь сама связанной с молекулой пептидного акцептора.
Под термином "измененная функция" понимается любое количественное или качественное изменение функции молекулы.
Под "последовательностью(сайтом)-индуктором паузы" понимается нуклеотидная последовательность, которая обусловливает замедление или остановку трансляции на рибосоме.
По использованию в данном тексте под "связывающим компонентом" понимается любая молекула, которая обладает специфичной, ковалентной или нековалентной аффинностью по отношению к одному из компонентов РНК-пептидного гибрида. Примеры связывающих компонентов включают, без каких-либо ограничений, члены пар "антиген-антитело", пар "белок-ингибитор", пар "лиганд-рецептор" (например, пар поверхностно-клеточных рецепторов и их лигандов, таких как пары, состоящие из рецепторов гормонов и самих пептидных гормонов), пар "фермент-субстрат" (например, пар киназ и субстратов), пар "лектин-углеводород", агрегаций олиго- или гетероолигомерных белков, пар ДНК-связывающихся белков и сайтов связывания в последовательности ДНК, а также нуклеотидных дуплексов, гетеродуплексов или лигированных цепей, равно как и любую молекулу, которая способна формировать одну или большее число ковалентных или иного типа связей (например, дисульфидных связей) с любым компонентом РНК-пептидного гибрида. Связывающие компоненты включают, без каких-либо ограничений, любые "селективные мотивы", представленные на фиг.2.
Под "твердой подложкой" понимается, без каких-либо ограничений, любая колонка (или материал, из которого она изготавливается), гранула, аналитическая пробирка, микротитровальная чашка, твердая субстанция (например, агароза или сефароза), микрочип (например, силикон, стекловолокно или золотой чип) или мембрана (например, мембрана липосомы или пузырька), с которым может быть соединен аффинный комплекс, как напрямую, так и опосредованно (например, с помощью других промежуточных связывающих компонентов, таких как другие антитела или белок-А), или в который аффинный комплекс может быть погружен (например, через рецептор или канал).
Представляемое изобретение обладает рядом существенных преимуществ. Начать надо с того, что впервые представляется схема для отбора и амплификации белков. Эта процедура преодолевает тот тупик, который создается необходимостью формировать нуклеотидные последовательности, соответствующие желательным выделенным белкам (поскольку только нуклеиновые кислоты могут быть реплицированы). В частности, во многих существующих методах, которые позволяют выделять белки из частично или полностью случайно подобранных пулов, это осуществлялось через этап in vivo. Методы этого типа включают технологию моноклональных антител (Milstein, 1980, Sci. Amer., 243, 66 и Schultz et al., 1990, J. Chem. Engng. News, 68, 26), конструирование фагов (Smith, 1985, Science, 228, 1315; Parmley & Smith, 1988, Gene, 73, 305; и McCafferty et al., 1990, Nature, 348, 552), соединения белков с lac-ререссором (Cull et al., 1992, Proc. Natl. Acad. Sci. USA, 89, 1865) и методы классического генетического отбора. В отличие от представляемых способов, каждый из этих методов опирается на топологическую связь между белком и нуклеиновой кислотой, так, что информация о данном белке сохраняется и может быть востребована в подлежащей считыванию форме нуклеиновой кислоты.
Кроме того, настоящее изобретение имеет преимущества по сравнению с методом "упорядоченной" трансляции (Tuerk & Gold, 1990, Science, 249, 505; Irvine et al., 1991, J. Mol. Biol, 222, 739; Korman et al., 1982; Proc. Natl. Acad. Sci. USA, 79, 1844-1848; Mattheakis et al., 1994, Proc. Natl. Acad. Sci. USA, 91, 9022-9026; Mattheakis et al., 1996, Methods Enzymol., 267, 195; и Hanes & Pluckthum, 1997, Proc. Natl. Acad. Sci. USA, 94, 4937) - методом, в котором отбор проводится с учетом свойств новообразующейся полипептидной цепи, которая сильно связана с рибосомой и мРНК. В отличие от метода "упорядоченной" трансляции, представляемый способ не опирается на поддержании целостности т.н. "тройного комплекса" (включает взаимодействующие мРНК, рибосому и новообразующуюся полипептидную цепь), т.е. комплекса, который отличается исключительной "ломкостью" и, соответственно, проявляет существенную ограниченность в типах отбора, которые могут быть технически осуществимы.
Представляемый настоящим изобретением способ обладает преимуществами над процедурой разветвленного синтеза, предложенной Бреннером-Лернером (Brenner & Lemer, 1992, Proc. Natl. Acad. Sci. USA, 89, 5381-5383), в котором синтезируются ДНК-пептидные гибриды и генетическая информация теоретически может быть воспроизведена через один раунд отбора. В отличие от метода разветвленного синтеза, представляемый способ не требует регенерации белка по ДНК-компоненту гибрида (которая в методе разветвленного синтеза в целом осуществляется в виде этапов индивидуального химического синтеза). Соответственно, представляемый способ позволяет проводить повторяющиеся раунды отбора с использованием популяций молекул-"кандидатов". Кроме того, в отличие от метода разветвленного синтеза, который в принципе ограничен отбором весьма коротких последовательностей, представляемый способ применим для отбора молекул белков значительной длины.
Еще одним преимуществом представляемого способа отбора и направленного преобразования последовательностей является возможность использования очень больших и сложных по составу библиотек последовательностей-кандидатов". С другой стороны, существующие методы отбора белков, которые опираются на этап in vivo, обычно ограничены использованием относительно небольших библиотек при достаточно ограниченной сложности их состава. Это преимущество, в частности, является важным тогда, когда проведение отбора последовательностей функциональных белков связано с представлением, например, 1013 возможных вариантов последовательности, которые может образовывать пептид только из 10-аминокислотной последовательности. В классических генетических методах - методе конструирования фага и связывания с lac-супрессором - максимальные уровни сложности обычно попадают в пределы, меньшие чем 1013 членов. Большой размер библиотеки также обеспечивает преимущество для применения в области направленного преобразования последовательностей в том, что "объем" такой последовательности может быть использован в гораздо большей степени по отношению к данной исходной последовательности.
Представляемый способ также отличается от ранее разработанных методик тем, что этап отбора является контекст-независимым. Во многих других протоколах отбора контекст, в котором, например, присутствует экспрессируемый белок, может существенно влиять на природу формируемых библиотек. Например, экспрессируемый белок может не быть правильно экспрессирован в конкретной системе или может не быть правильно размещен (например, на поверхности фаговой частицы). С другой стороны экспрессия белка может действительно служить помехой для одного или нескольких критических этапов в цикле отбора, например, таких как жизнеспособность и инфекционность фага или связывание lac-репрессора. Эти проблемы могут приводить к утрате функциональных молекул или ограничению типа процедур отбора, которые могли бы быть осуществлены.
Наконец, представляемый способ является преимущественным потому, что он обеспечивает контроль за репертуаром белков, которые могут быть протестированы. В некоторых методах (например, в методе отбора с помощью антител) имеет место слабый контроль за природой исходного пула, или такой контроль отсутствует вовсе. В ряде других методов (например, в методе lac-связывания и конструирования фагов) пул молекул-"кандидатов" может быть экспрессирован в контексте участвующего в гибриде белка. С другой стороны, конструкции РНК-пептидных гибридов обеспечивают контроль за природой пула молекул-"кандидатов", пригодный для скрининга. Кроме того, размер пула молекул-"кандидатов" потенциально может быть таким же большим, как пулы РНК или ДНК (примерно 1015членов), ограничиваясь только размером трансляционной реакции, осуществляемой in vitro. Также состав пула молекул-кандидатов" полностью зависит от схемы эксперимента; случайные участки могут быть скринированы изолированно или в пределах контекста желательного белка, входящего в состав гибрида, и большинство, а то и все возможные последовательности могут быть экспрессированы в составе "кандидатных" пулов РНК-пептидных гибридов.
Другие характеристики и преимущества настоящего изобретения будут ясны из нижеследующего подробного описания и из формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ
Вначале кратко описаны чертежи.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
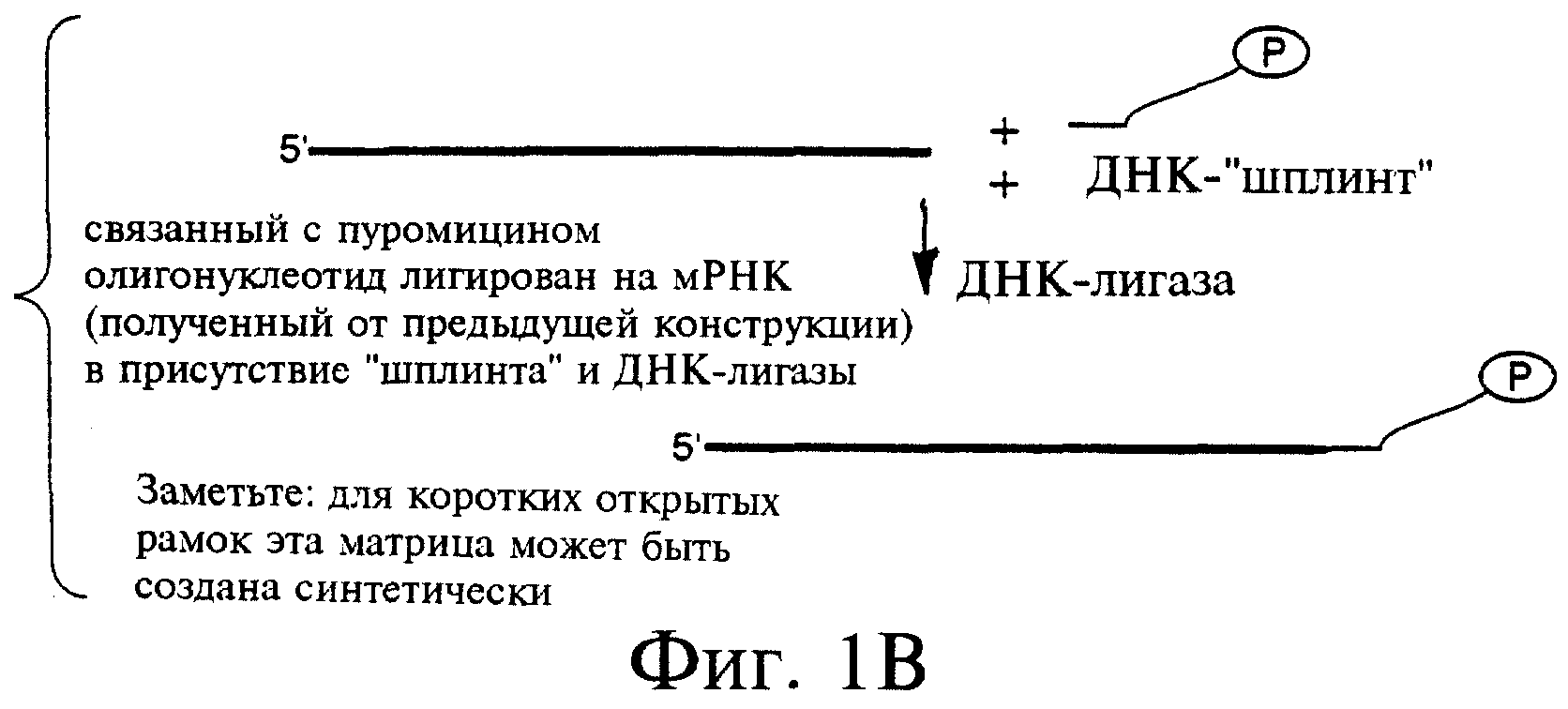
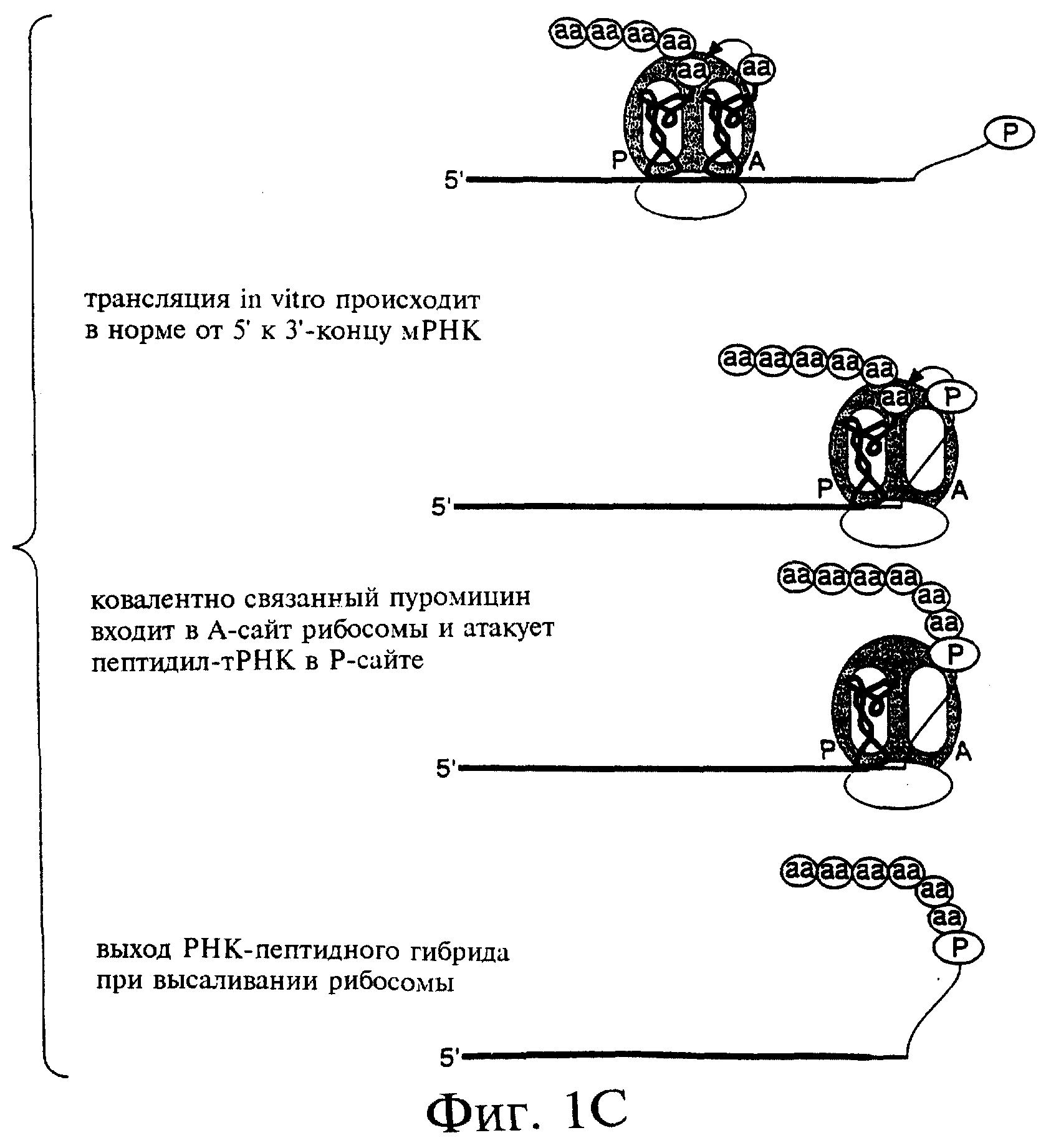
Фиг.1А-1С являются схемами этапов получения РНК-пептидных гибридов. Фиг.1А показывает образец конструкции ДНК для получения РНК-компонента гибрида. Фиг.1В показывает получение РНК-пуромицинового конъюгата. И фиг.1С показывает получение РНК-пептидного гибрида.
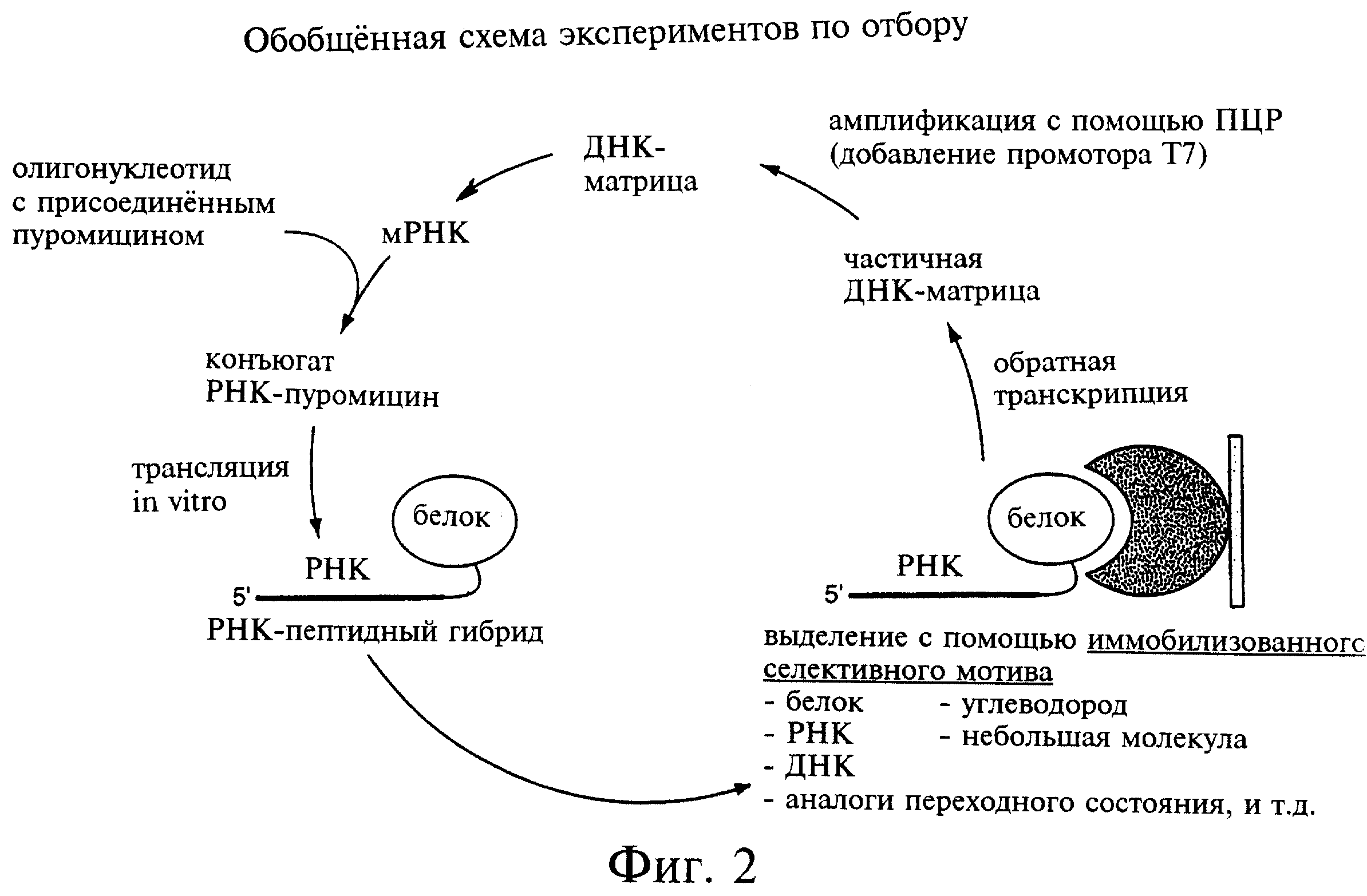
Фиг.2 является схемой обобщенного способа отбора в соответствии с настоящим изобретением.
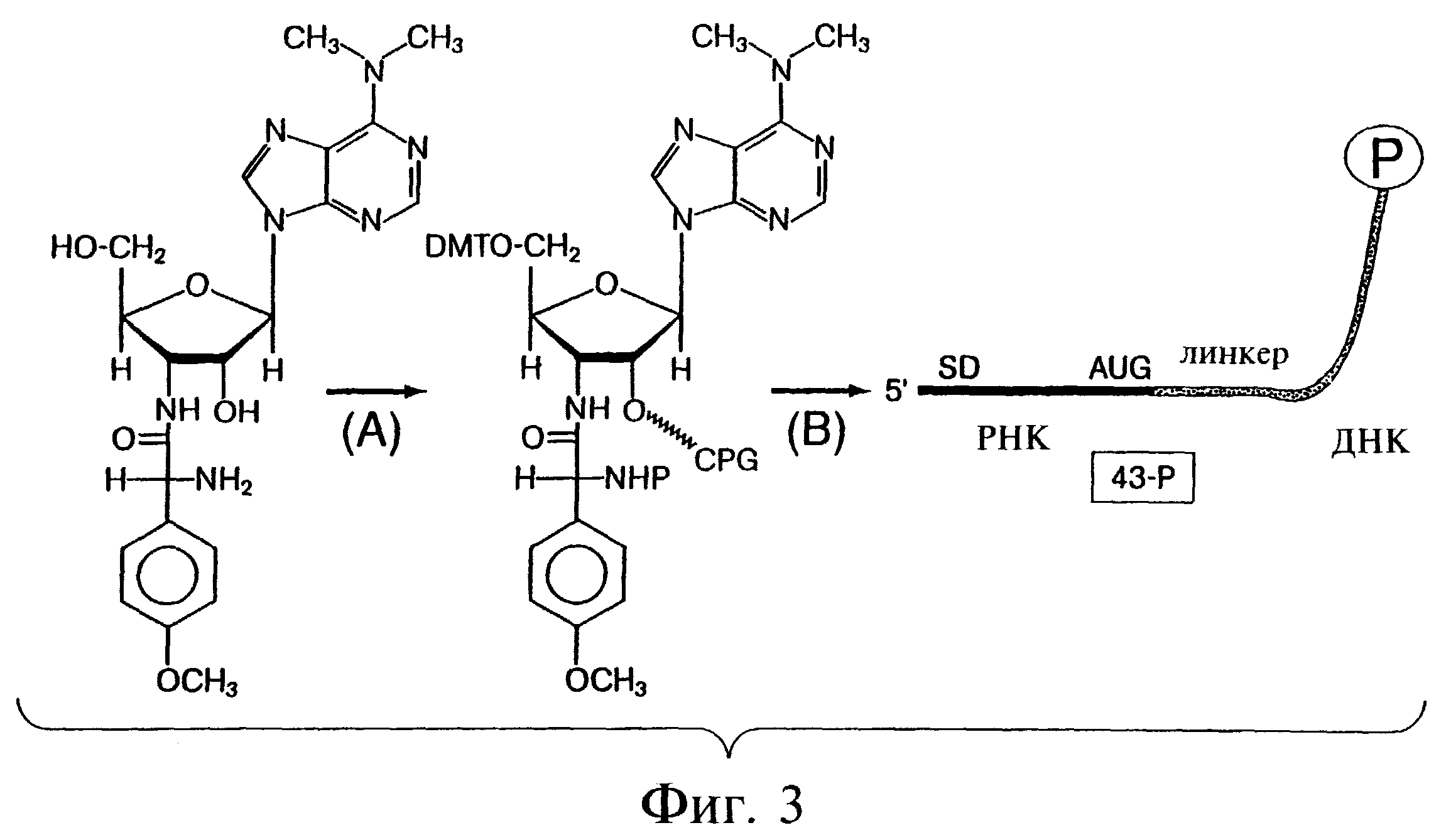
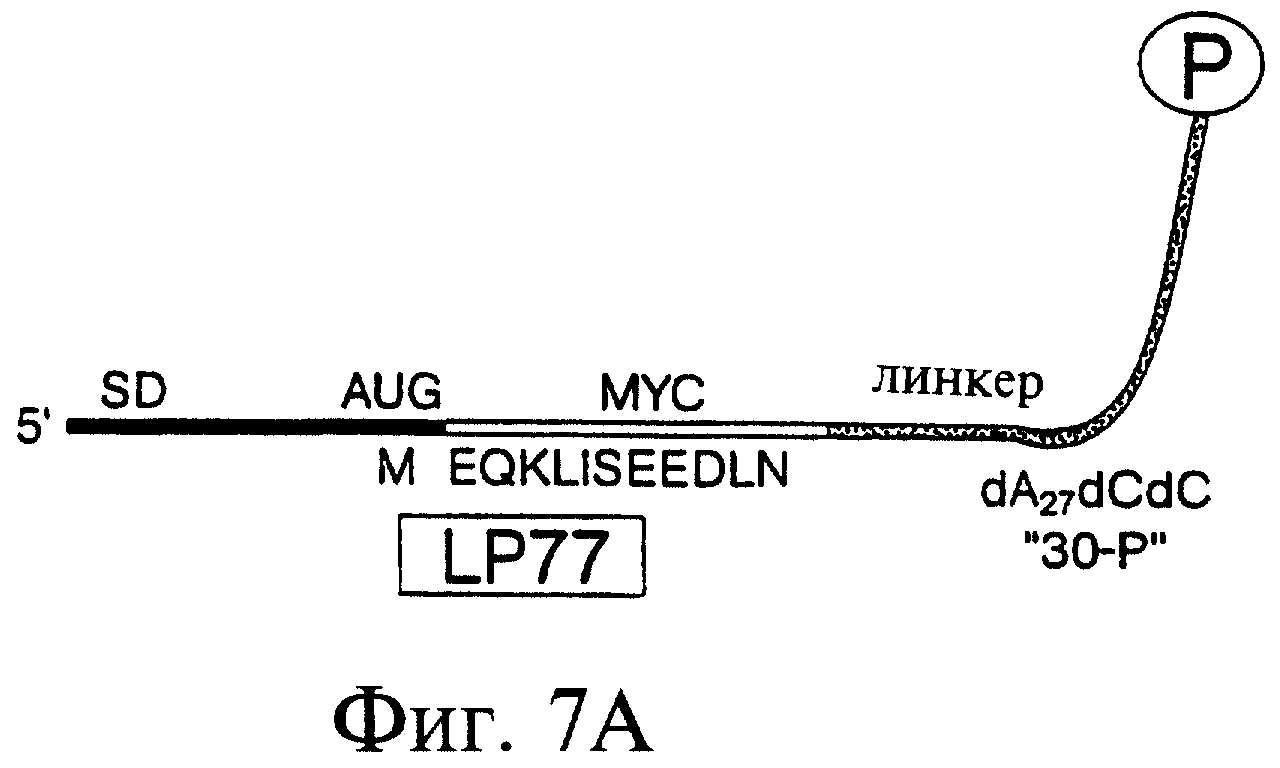
Фиг.3 является схемой протокола синтеза минимальной трансляционной матрицы, включающей пуромицин с 3'-конца. Этап (А) соответствует добавлению защитных групп к реактивным функциональным группам пуромицина (5'-ОН и NH2); в случае модификации эти группы оказываются эффективно защищенными для использования в фосфорамидитном синтезе олигонуклеотидов. Выполняющий защитную роль пуромицин был присоединен к обработанному аминогексилом пористому стеклу (CPG) по группам 2'ОН с использованием стандартного метода присоединения ДНК по ее группе 3'ОН (Gait, Oligonucleotide Synthesis, A Practical Approach, The Practical Approach Series (IRL Press, Oxfgord, 1984)). На этапе (В) минимальная трансляционная матрица (обозначенная как "43-Р"), состоящая из 43 нуклеотидов, была синтезирована с использованием стандартных методов химии РНК и ДНК (Millipore, Bedford, МА), с освобождением от защитной составляющей с помощью NH4OH и TBAF, с последующей очисткой в геле. Матрица включала 13 оснований РНК со своего 5'-конца и далее 29 оснований ДНК, присоединенных 3'-концом к пуромицину по его группе 5'ОН. Последовательность РНК включала (i) консенсусную последовательность Шайна-Дальгарно, комплементарную пяти основаниям 163-рРНК (Stormo et al., 1982, Nucleic Acids Research, 10, 2971-2996; Shine & Dalgarno, 1974, Proc. Natl. Acad. Sci. USA, 71, 1342-1346; и Steitz & Jakes, 1975, Proc. Natl. Acad. Sci. USA, 72, 4734-4738), (ii) 5-нуклеотидный спейсер, и (iii) единственный старт-кодон AUG. Последовательность ДНК была следующей – dA27dCdCP, где Р - это пуромицин.
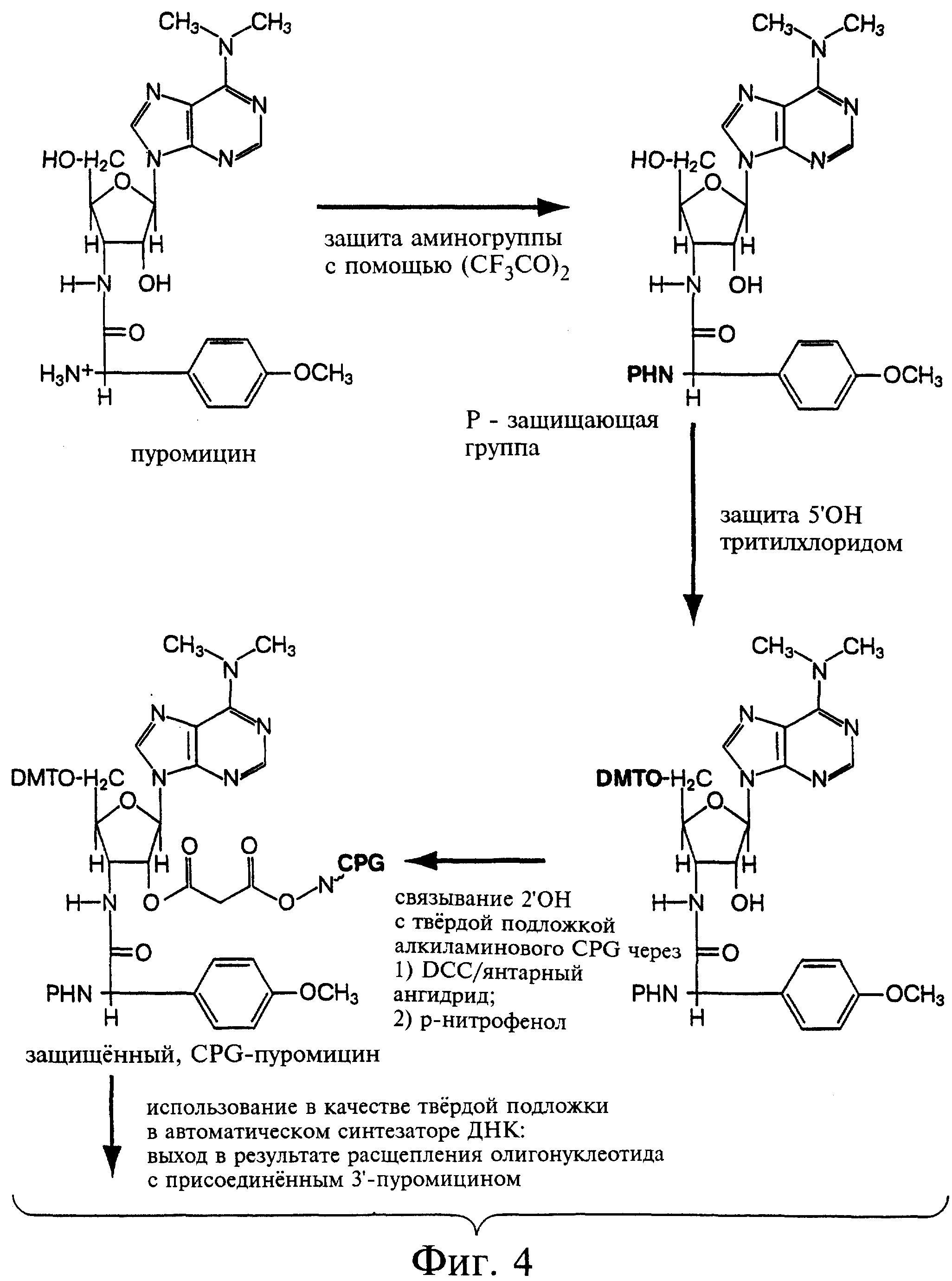
Фиг.4 является схемой предпочтительного варианта способа приготовления защищенного, связанного с CPG пуромицина.
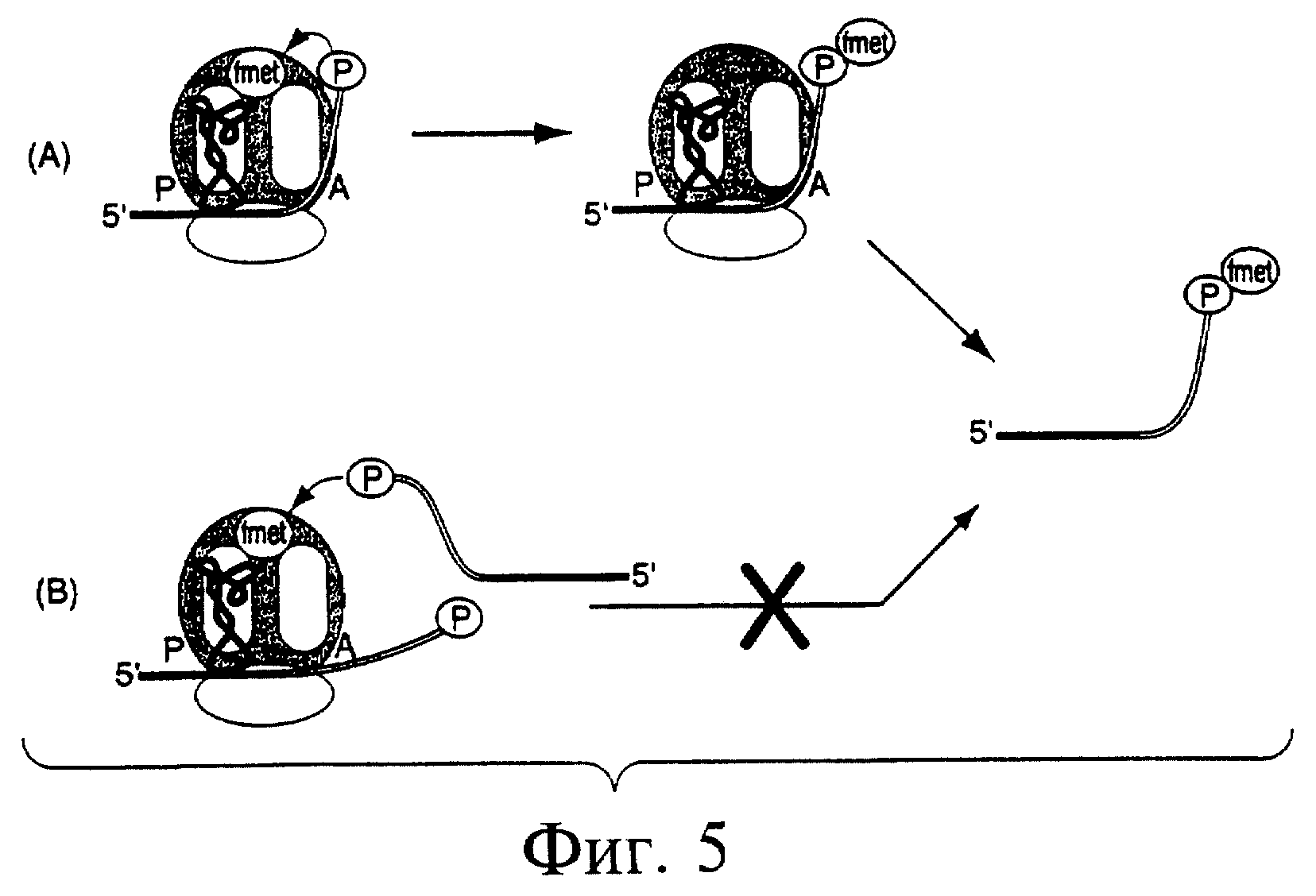
Фиг.5 является схемой, показывающей возможные пути включения метионина в матрицу по настоящему изобретению. Как показано в реакции (А), матрица связывается с рибосомой с образованием комплекса инициации 70S. тРНК формилметионина связывается с Р-сайтом и взаимодействует с матрицей согласно комплементарности оснований. Пуромицин в 3'-конце матрицы попадает в А-сайт, будучи внутримолекулярной структурой, и формирует амидную связь с N-формилметионином в пептидилтрансферазном центре, осуществляя таким образом деацилирование тРНК. Экстракия фенолом-хлороформом реакционной смеси позволяет выделить данную матрицу с ковалентно присоединенным метионином. В реакции (В) показана нежелательная межмолекулярная реакция матрицы с олигонуклеотидами, включающими пуромицин. Как и в предыдущем случае, минимальная матрица стимулирует образование субчастицы рибосомы 70S, включающей тРНК формилметионина, соединенную с Р-сайтом. После этого происходит внедрение второй матрицы (т.е. транс-матрицы по отношению к первой матрице) с появлением ковалентно присоединенного метионина.
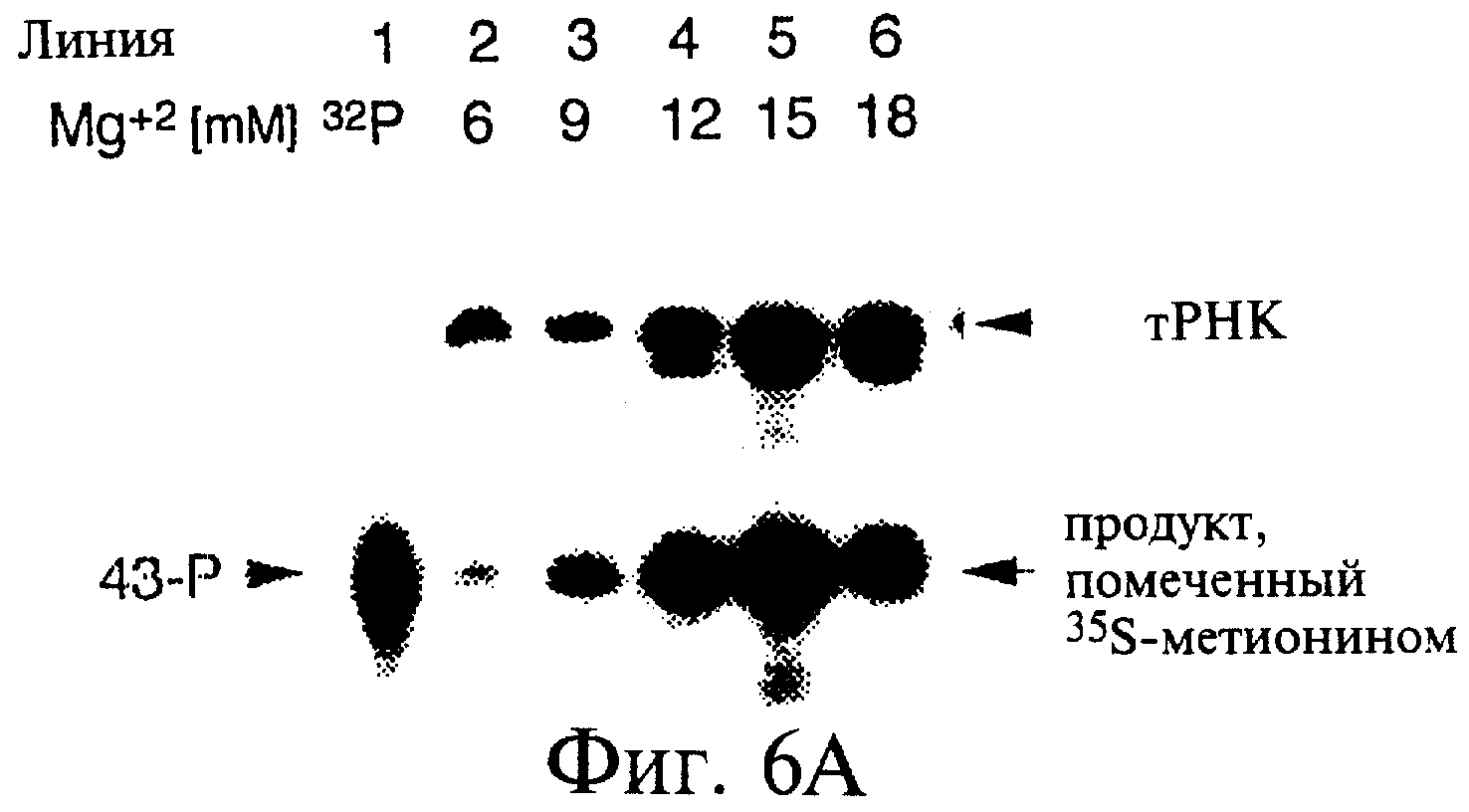
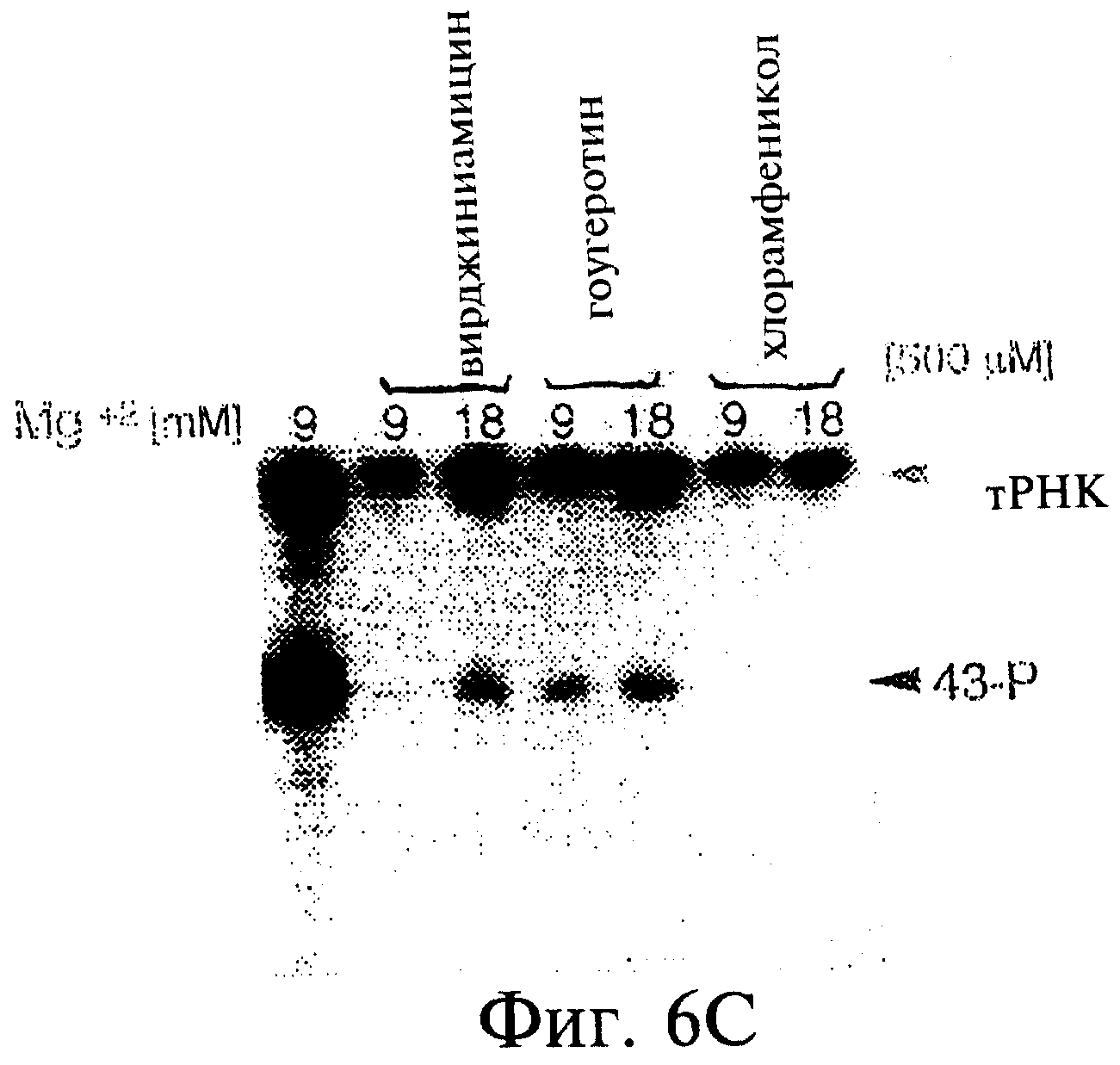
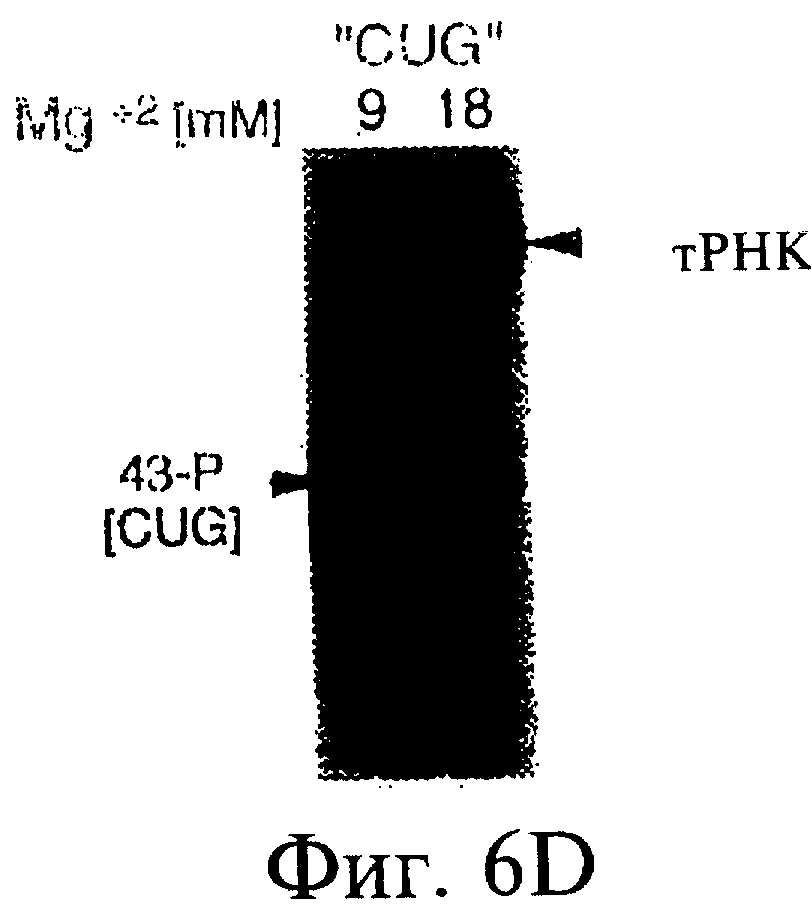
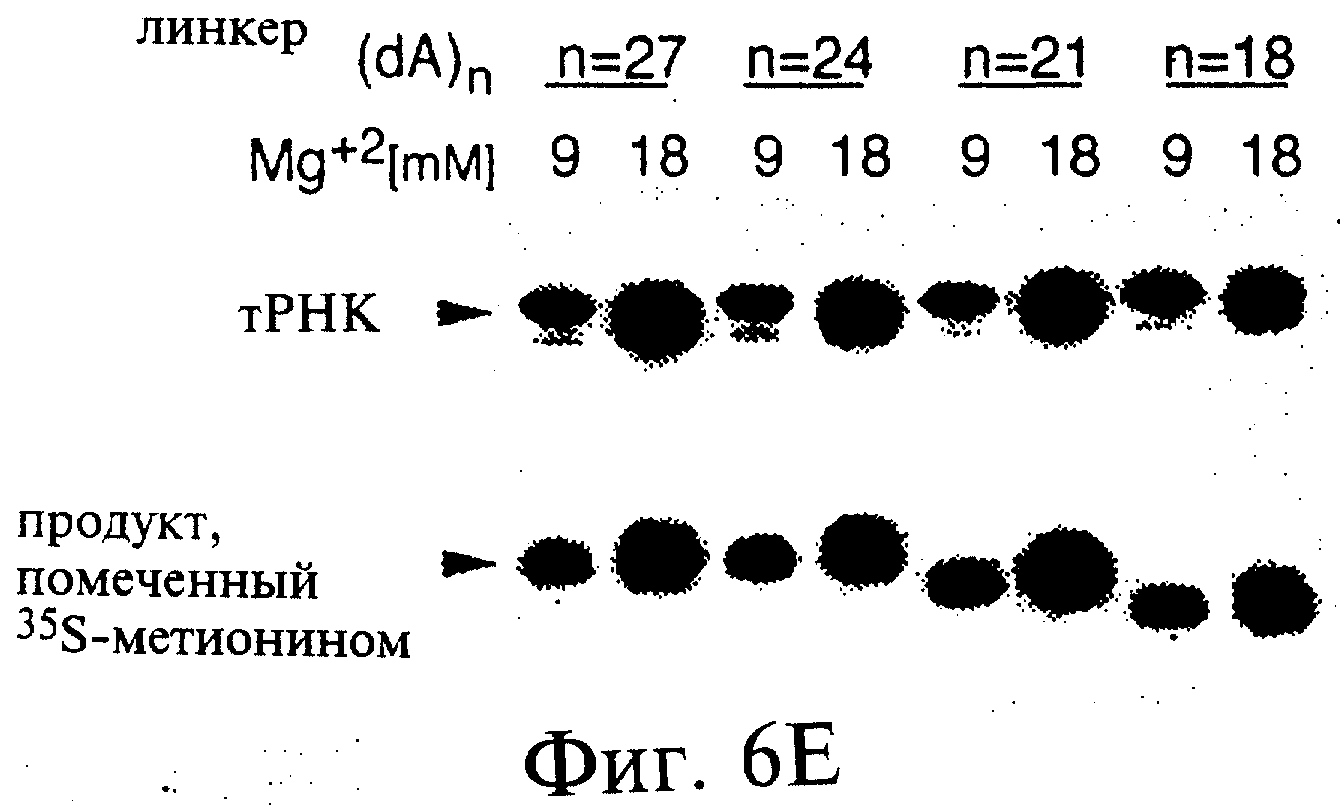
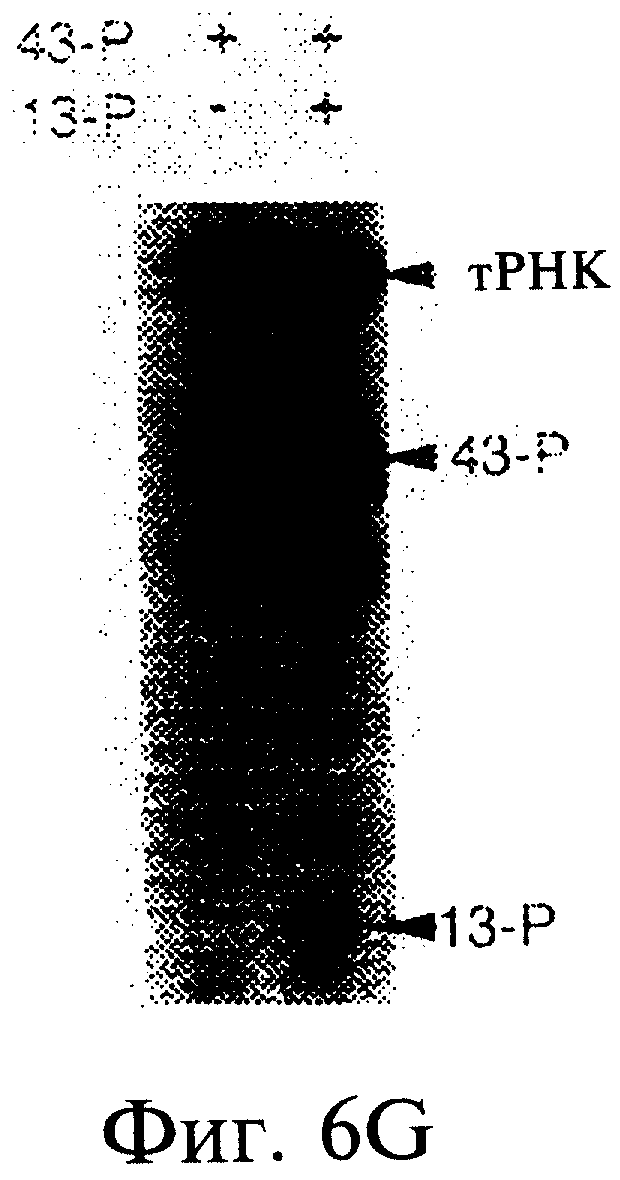
Фиг.6А-6Н являются фотографиями, показывающими включение меченого35 S-метионина (35S-met) в состав трансляционной матрицы. Фиг.6А показывает зависимость этой реакции от ионов магния (Mg2+). Фиг.6В показывает стабильность оснований получаемого продукта; изменение подвижности соответствует утрате 5'-последовательности РНК 43-Р (также обозначаемой как "Met-матрица") с получением ДНК-пуромицинового компонента, обозначаемого 30-Р. Сохранение метки после обработки оснований соответствовало образованию пептидной связи между35S-метионином и 3'-пуромицином матрицы. Фиг.6С показывает ингибирование образования продукта реакции в присутствии ингибиторов пептидилтрансферазы. Фиг.6D показывает зависимость включения35S-метионина от кодирующей последовательности в составе матрицы. Фиг.6Е показывает зависимость включения35S-метионина от длины ДНК-матрицы. Фиг.6F показывает образование продуктов реакции в цис- и транс-положении с использованием матриц 43-Р и 25-Р. Фиг.6G показывает образование продуктов реакции в цис- и транс-положении с использованием матриц 43-Р и 13-Р. Фиг.6Н показывает образование продукта реакции в цис- и транс-положении при использовании матриц 43-Р и 30-Р в системе лизата ретикулоцитов.
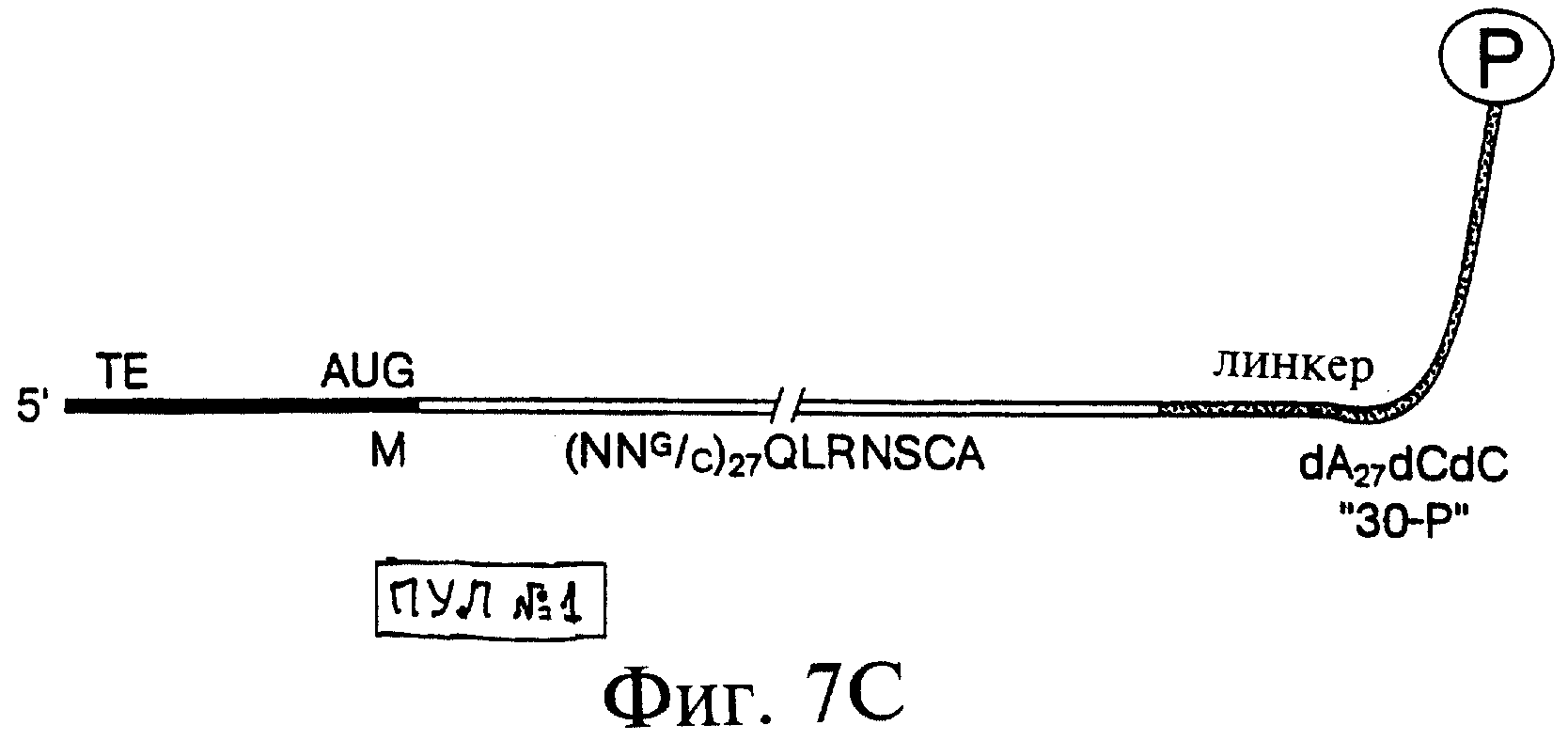
Фиг.7А-7С являются схематическим изображением конструкций, предназначенных для тестирования образования и отбора пептидных гибридов. Фиг.7А показывает LP77 ("лигированный продукт" длиной "77" нуклеотидов) (также обозначаемый как "короткая myс-матрица") (SEQ ID N0:l). Эта последовательность включает эпитоп-метку для моноклонального антитела к с-myс - EQKLISEEDL (SEQ ID NO: 2) (Evan et al., 1985, Mol. Cell. Biol., 5, 3610-3616), - фланкированную старт-кодоном с 5'-конца и линкером с 3'-конца. Участок с 5'-конца включает бактериальную последовательность Шайна-Дальгарно, идентичную той, что имеется в составе 43-Р. Кодирующая последовательность была оптимизирована для трансляции в бактериальных системах. В частности, 5'-UTRs в составе 43-Р и LP77 включали последовательность Шайна-Дальгарно, комплементарную пяти основаниям в 163-рРНК (Steitz & Jakes, 1975, Proc. Natl. Acad. Sci. USA, 72, 4734-4738) и организованную так же, как в последовательностях рибосомных белков (Stormo et al., 1982, Nucleic Acids Research, 10, 2971-2996). Фиг.7В показывает LP154 (лигированный продукт длиной 154 нуклеотида) (также обозначаемый как "длинная myс-матрица") (SEQ ID N0:3). Эта последовательность включает код пептида, используемого для получения антитела к с-myс. В 5'-концевой части находится укороченый вариант последовательности TMV (обозначенный "ТЕ"). Этот участок 5'-UTR включает 22-нуклеотидную последовательность, производную от TMV 5'-UTR, включающую два прямых повтора ACAAAUUAC (Gallie et al., 1988, Nucl. Acids Res., 16, 883). Фиг.7С показывает пул №1 (SEQ ID NO 4), являющийся примером последовательности, которая может быть использована для отбора белка. Заключительные 7 аминокислот из нативного полипептида myc были включены в матрицу в качестве 3'-константного сегмента, необходимого для ПЦР-амплификации этой матрицы. Известно, что эта последовательность не входит в состав эпитопа, с которым связывается антитело.
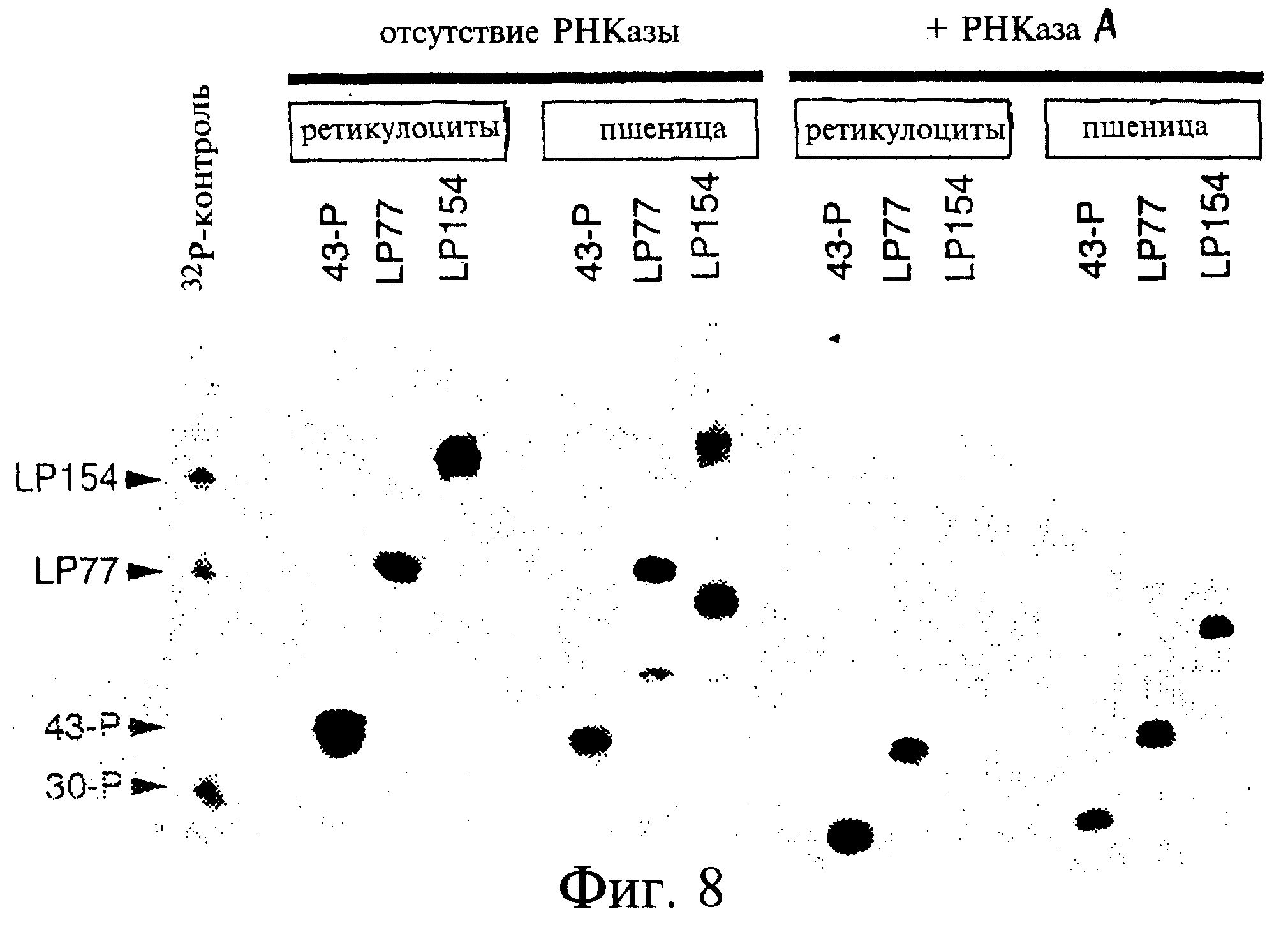
Фиг.8 является фотографией, показывающей синтез РНК-пептидных гибридов с использованием матриц 43-Р, LP77 и LP154 в трансляционных системах ретикулоцитов ("Retic") и зародышей пшеницы ("Wheat"). Левая половина фигуры показывает включение35S-метионина в состав каждой из трех матриц. Правая половина фигуры показывает полученные продукты после обработки РНКазой-А, каждой из трех матриц с целью удаления РНК-кодирующего сегмента; показаны помеченные35S-метионином ДНК-пептидные гибриды. ДНК-компонент каждого из них был идентичным олигомерной последовательности 30-Р. Таким образом, различия в подвижности были пропорциональны длине кодирующих сегментов, что соответствует существованию белков разной длины в каждом из этих случаев.
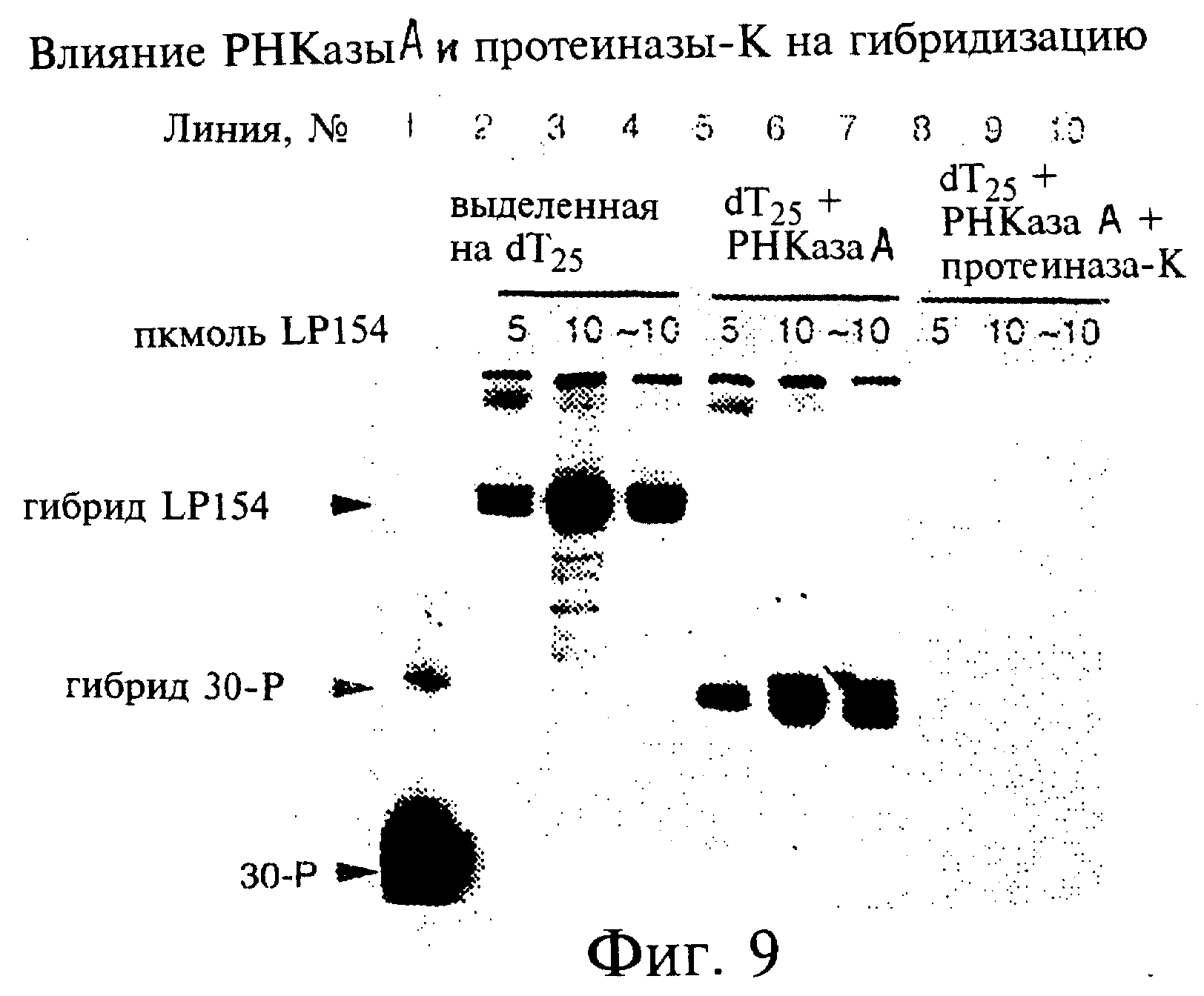
Фиг.9 является фотографией, показывающей чувствительность к действию протеазы РНК-пептидного гибрида, синтезированного на матрице LP154 и проанализированного с применением электрофореза в полиакриламидном геле в денатурирующих условиях. Линия 1 включает35Р-помеченную 30-Р. Линии 2-4, 5-7 и 8-10 включают35S-помеченные трансляционные матрицы, выделенные из лизатов ретикулоцитов либо без обработки, либо с обработкой РНКазой-А, либо с обработкой РНКазой и протеиназой-К, соответственно.
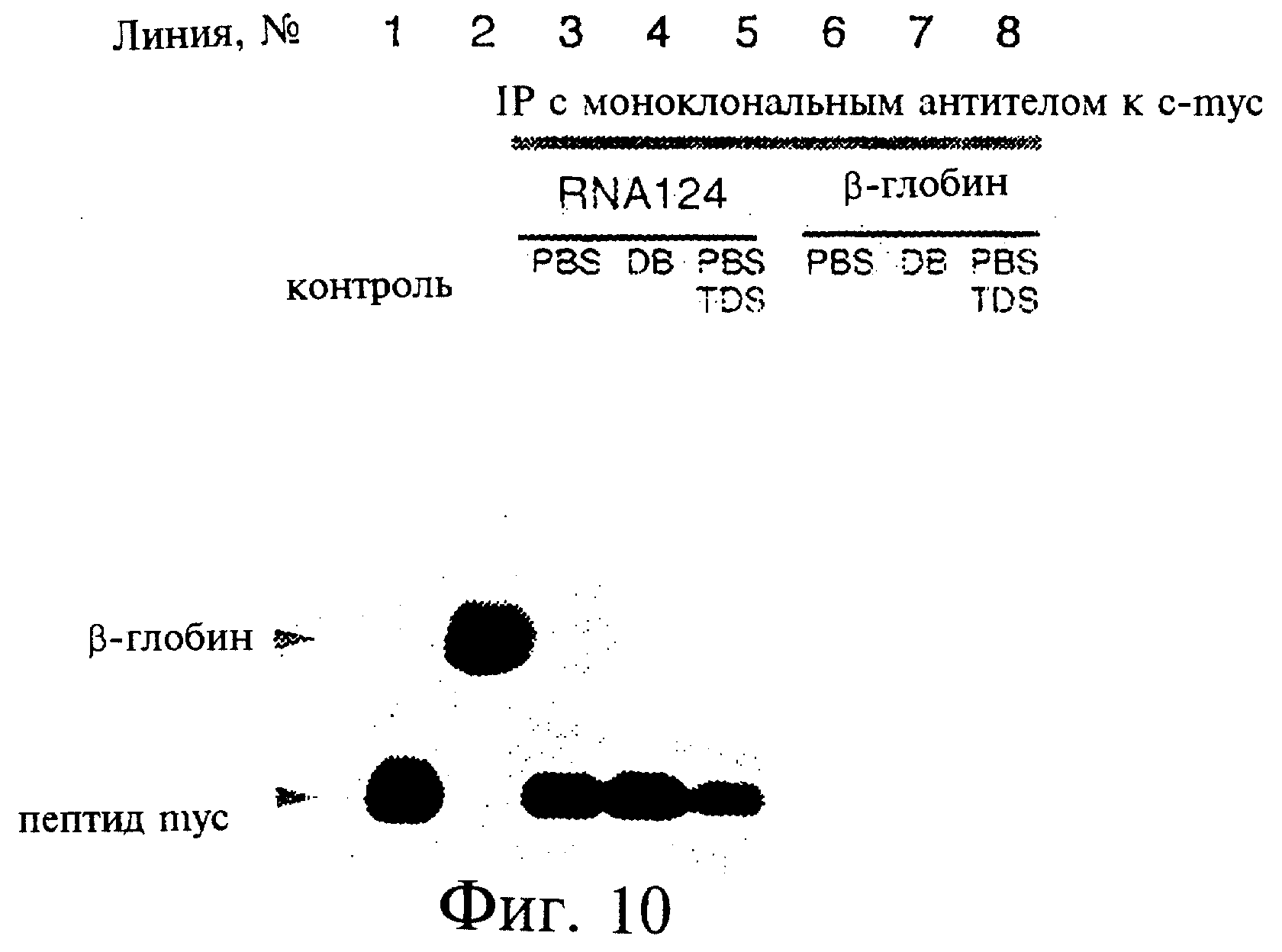
Фиг.10 является фотографией, показывающей результаты иммунопреципитации с использованием транслированного in vitro белка, включающего 33-аминокислотный эпитоп myc. Линии 1 и 2 показывают продукты трансляции с матриц белка myс-эпитопа и Р-глобина, соответственно. Линии 3-5 показывают результаты иммунопреципитации полипептида myс-эпитопа с использованием моноклонального антитела к с-myс и промывочных буферов PBS, DB и PBSTDS, соответственно. Линии 6-8 показывают аналогичные иммунопреципитационные реакции, но с использованием продукта трансляции Р-глобина.
Фиг.11 является фотографией, показывающей иммунопреципитацию РНК-пептидного гибрида из реакции трансляции, проводимой in vitro. Количество матрицы, используемой в данной реакции, определено в пикомолях. Линии 1-4 показывают RNA124 (РНК-компонент гибрида LP154), а линии 5-7 показывают РНК-пептидный гибрид LP154. После иммунопреципитации с использованием моноклонального антитела к с-myс и сефарозы с белком-G образцы были обработаны РНКазой-А и полинук-леотидкиназой-Т4, затем загружены в полиакриламидный гель с денатурирующим агентом (мочевина) с целью визуализации гибрида. В линиях 1-4, где образцы либо не содержали матрицу, либо содержали только РНК-компонент "длинной myс-матрицы" (RNA124), гибриды не видны. В линиях 5-7 отчетливо визуализуются бэнды, соответствующие гибридам. Определено положение32Р-помеченной 30-Р, а количество внесенной матрицы показано вверху фигуры.
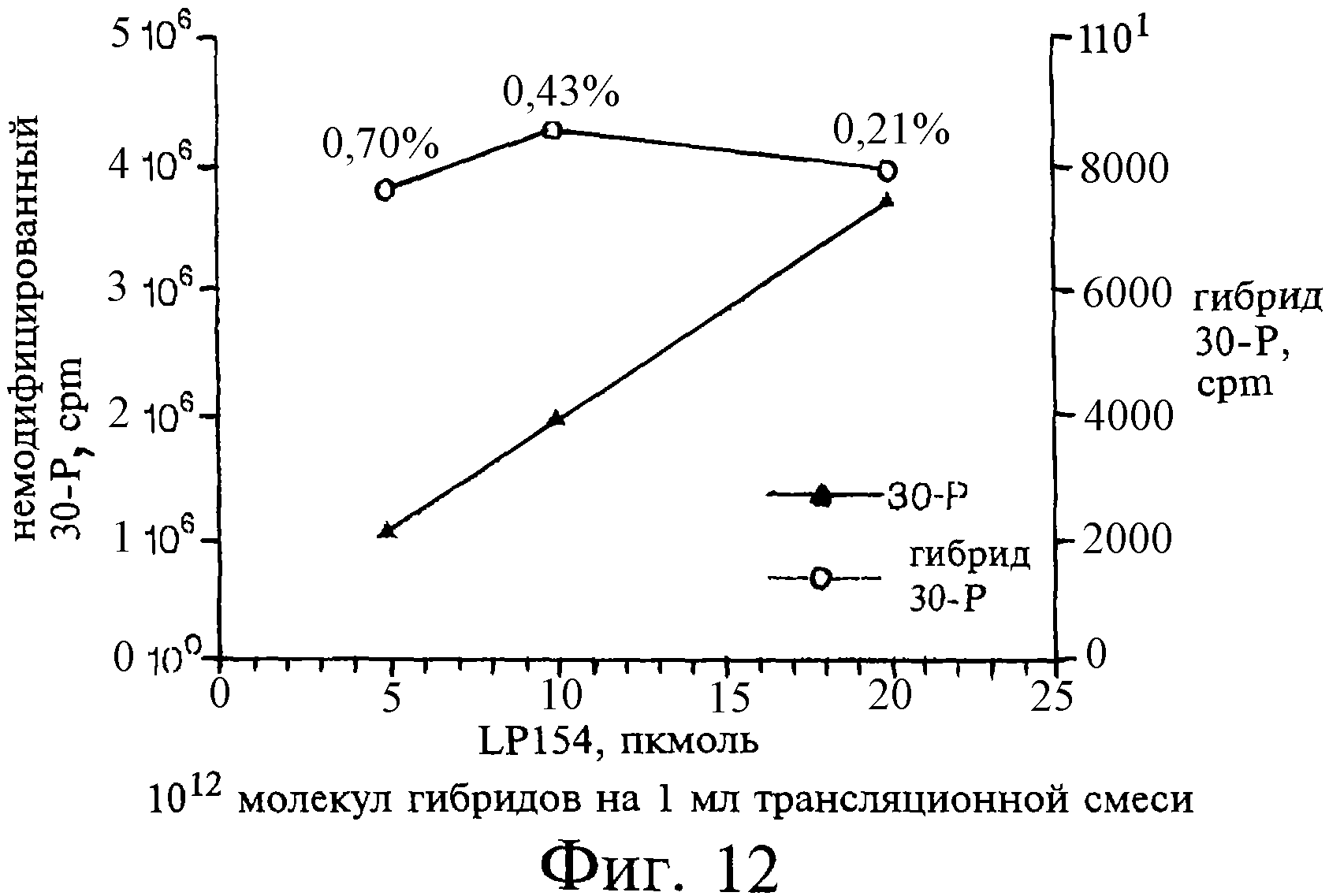
Фиг.12 является графиком, показывающим данные количественной оценки гибридов, полученных с помощью трансляции in vitro. Интенсивность бэндов гибридов, показанных в линиях 5-7 фиг.11, и бэнда 30-Р (выделенного в параллельном варианте на dT25: данные не приведены) были оценены количественно на фосфорных пластинах и показаны на графике в виде функции концентраций вносимой матрицы LP154. Количество выделенной модифицированной 30-Р (левая ордината) было прямо пропорционально количеству вносимой матрицы (абцисса), в то время как количество комплекса пептид-линкер (правая ордината) было постоянным. Исходя из данных этого анализа, было подсчитано, что примерно 1012 гибридов было образовано в расчете на 1 мл образца трансляционной реакции.
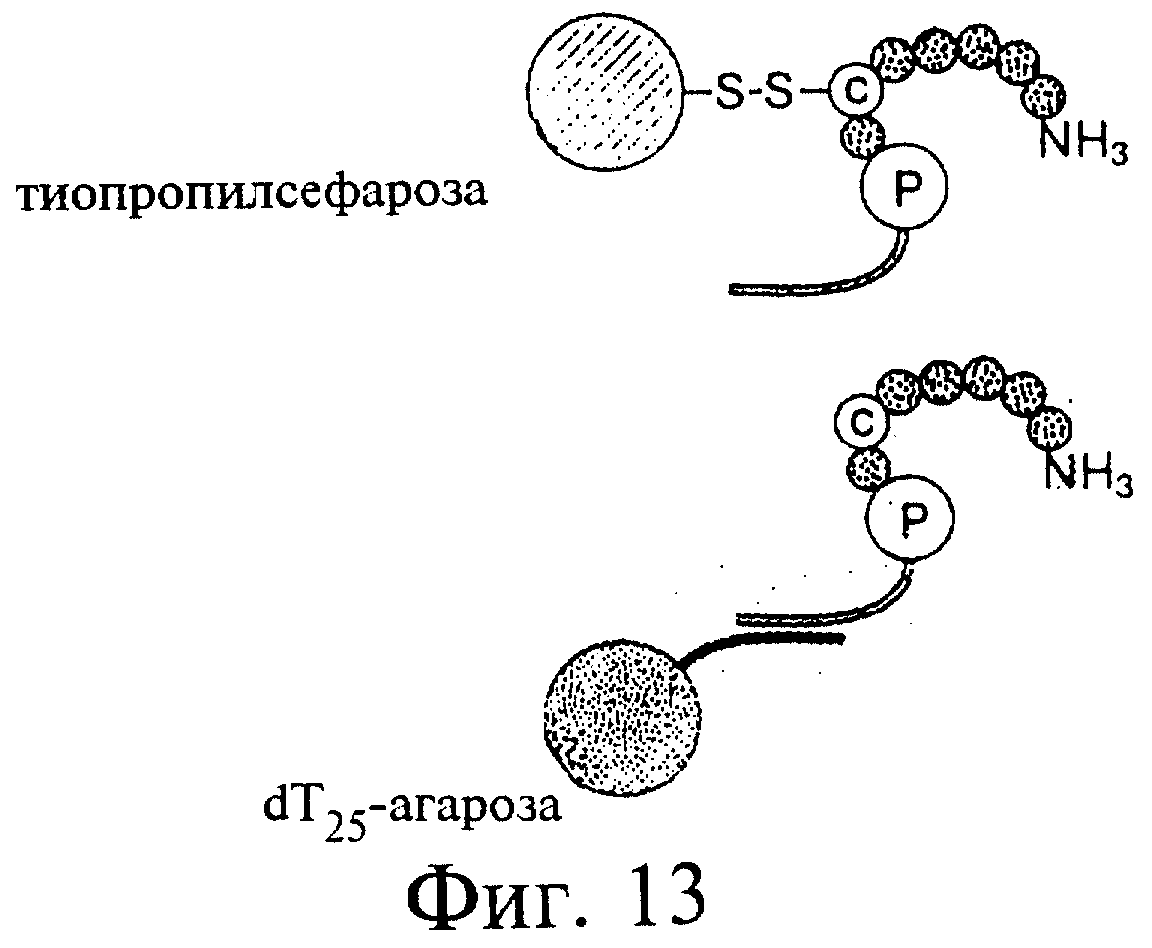
Фиг.13 является схемой представляющей тиопропиловую сефарозу и агарозу-dТ25, и способности этих субстратов взаимодействовать с РНК-пептидными гибридами по настоящему изобретению.
Фиг.14 является фотографией, показывающей результаты последовательного выделения гибридов по настоящему изобретению. Линия 1 включает32Р-помеченную 30-Р. Линии 2 и 3 показывают LP154, выделенную из трансляционной реакционной смеси и обработанную РНКазой-А. В линии 2 LP154 была выделена последовательность с использованием тиопропиловой сефарозы и затем агарозы-dТ25. Линия 3 показывает выделение с использованием только агарозы-dТ25. Данные определили, что полученный в результате продукт содержит свободный тиол, по-видимому, представляющий предпоследний остаток цистеина в последовательности, кодирующей эпитоп myc.
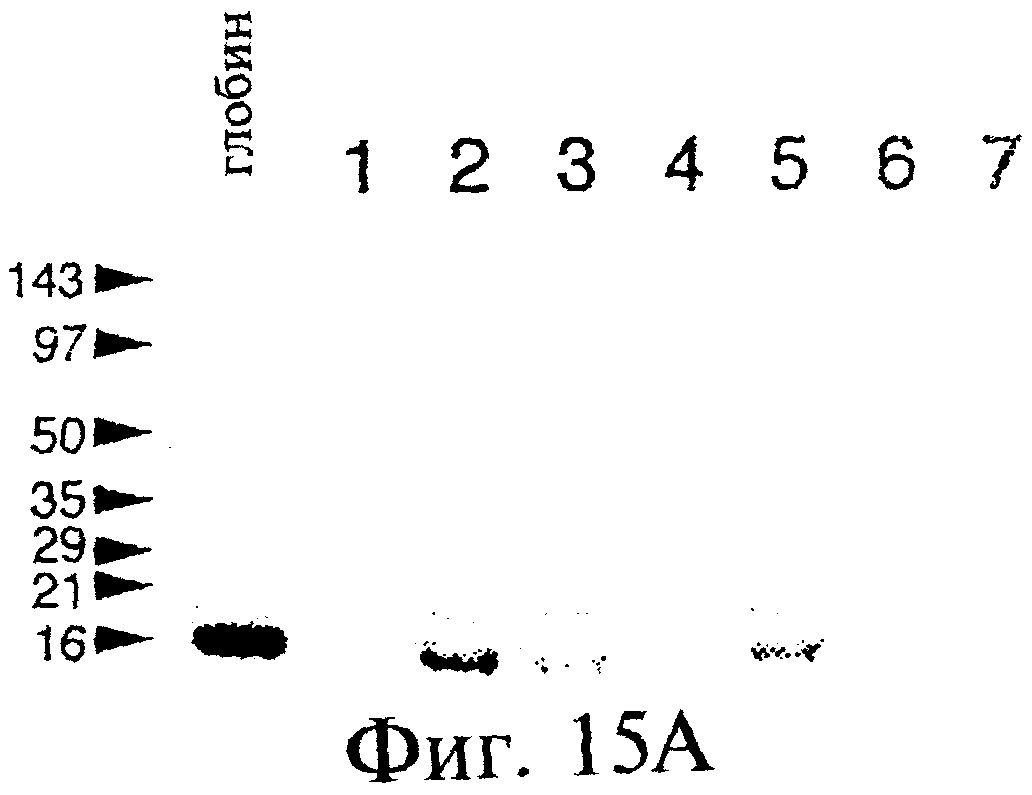
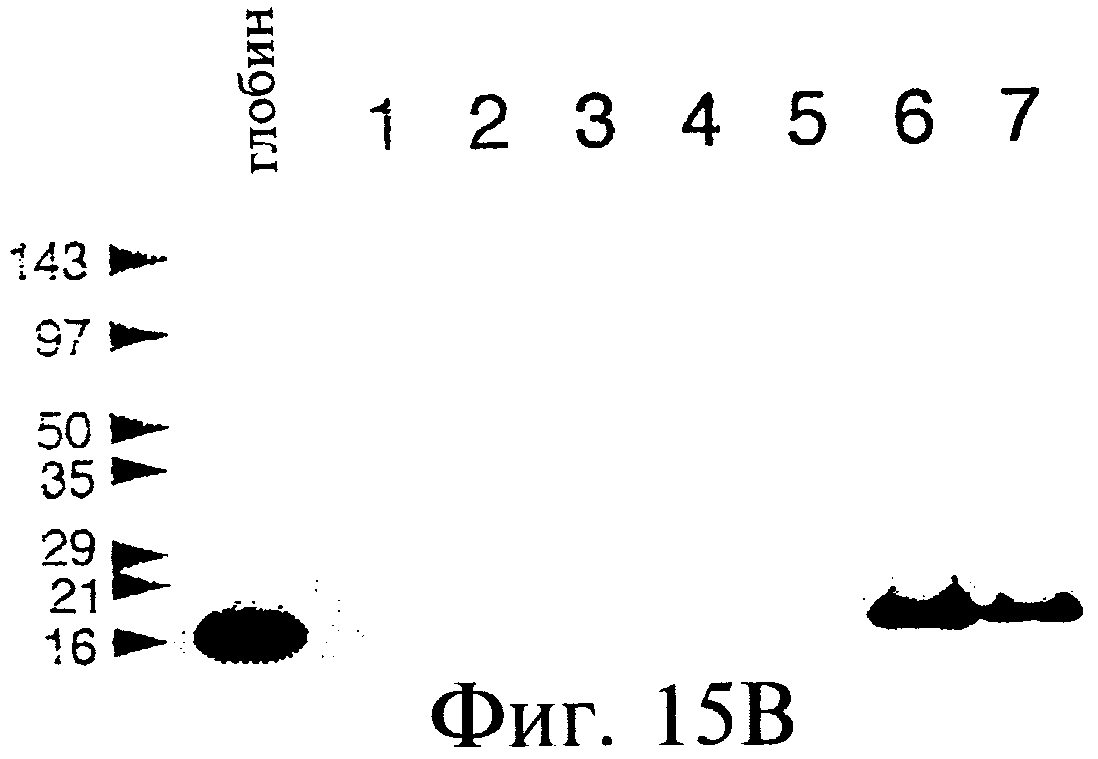
Фиг.15А и 15В являются фотографиями, показывающими образование продуктов гибрида с использованием матриц β-глобина с использованием SDS-fricene-PAGE (электрофорез в полиакриламидном геле). Фиг.15А показывает включение35S при отсутствии матрицы (линия 1), с использованием матрицы syn-β-глобина (линии 2-4) или матрицу LP-β-глобина (линии 5-7). Фиг.15В (линии, помеченные так же, как на фиг.15А) показывает35 S-помеченный материал, выделенный с использованием аффинной к олигонуклеотидам хроматографии. При отсутствии "хвоста" 30-Р (линии 2-4) такой материал выделен не был.
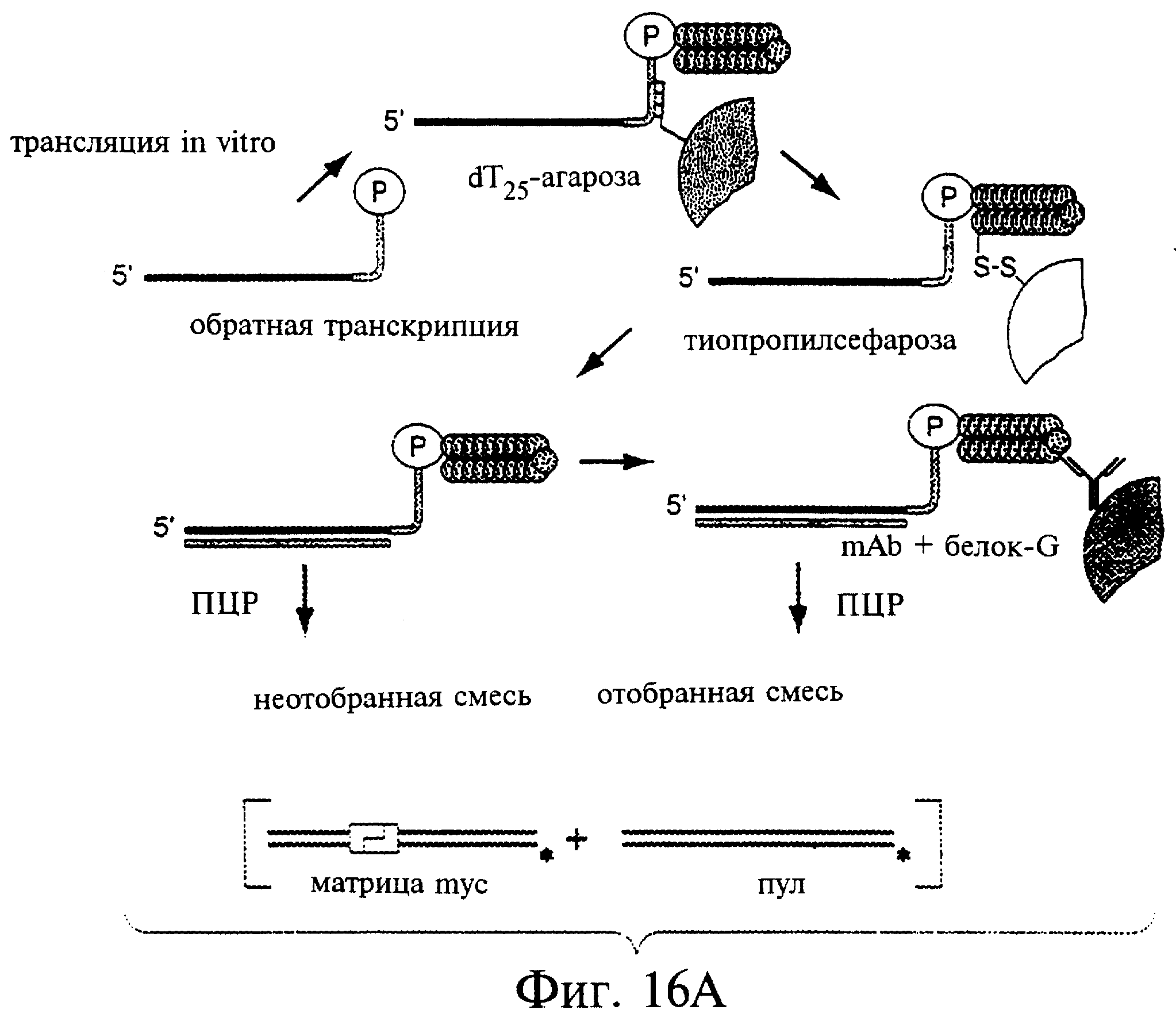
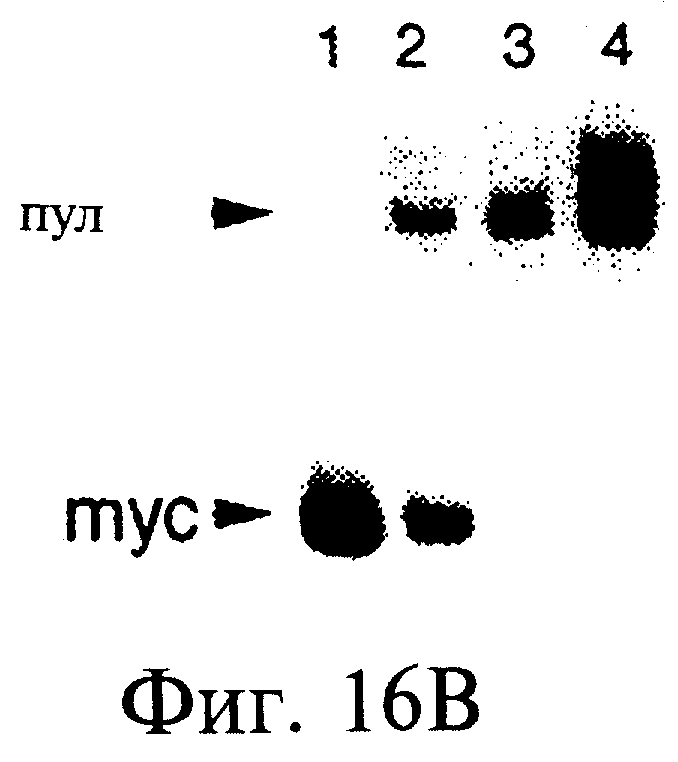
Фиг.16А-16С являются диаграммами и фотографиями, показывающими обогащение дцДНК myc в противопоставлении пулу дцДНК при отборе in vitro. Фиг.16А - схема протокола отбора. Четыре смеси матриц myc смешанного пула были транслированы in vitro и выделены с помощью агарозы-dТ25 и затем сефарозы-ТР с целью очистки прореагировавших матриц от немодифицированных матриц. РНК-пептидные гибриды были затем подвергнуты обратной транскрипции с целью подавления возникновения любой вторичной или третичной структуры, характерной для матриц. Аликвоты каждой смеси были выделены как до (фиг.16В), так и после (фиг.16С) отбора по аффинности, амплифицированы с помощью ПЦР в присутствии меченой затравки и расщеплены рестриктазой, которая расщепляет исключительно ДНК myc. Вносившиеся смеси матриц были такими: чистая матрица myc (линия 1) или myc:пул в соотношении 1:20, 1:200 или 1:2000 (линии 2-4). Неотобранный материал отклонялся от вносимых соотношений из-за предпочтительной трансляции и обратной транскрипции матрицы myc. Обогащение матрицы myc в процессе этапа отбора было подсчитано, исходя из изменения соотношения myc:пул до и после проведения отбора.
Фиг.17 является фотографией, показывающей трансляцию матриц myc-PHK. Были использованы следующие линкеры: линии 1-4 – dA27dCdCP; линии 5-8 – dA27CrCP; и линии 9-12 –dA21C9 C9C9dAdCdCP. В каждой линии концентрация матрицы РНК составляла 600 нМ, а для метки использовали35S-метионин. Условия реакции были следующими: линии 1, 5 и 9 - 1 час при 30°С; линии 2, 6 и 10 - 2 часа при 30°С; линии 3, 7 и 11 - 1 час при 30°С и 16 часов при -20°С; и линии 4, 8 и 12 - 1 час при 30°С и 16 часов при -20°С в присутствии 50 мМ Мg2+. Здесь "А" обозначает свободный пептид и "В" соответствует РНК-пептидному гибриду.
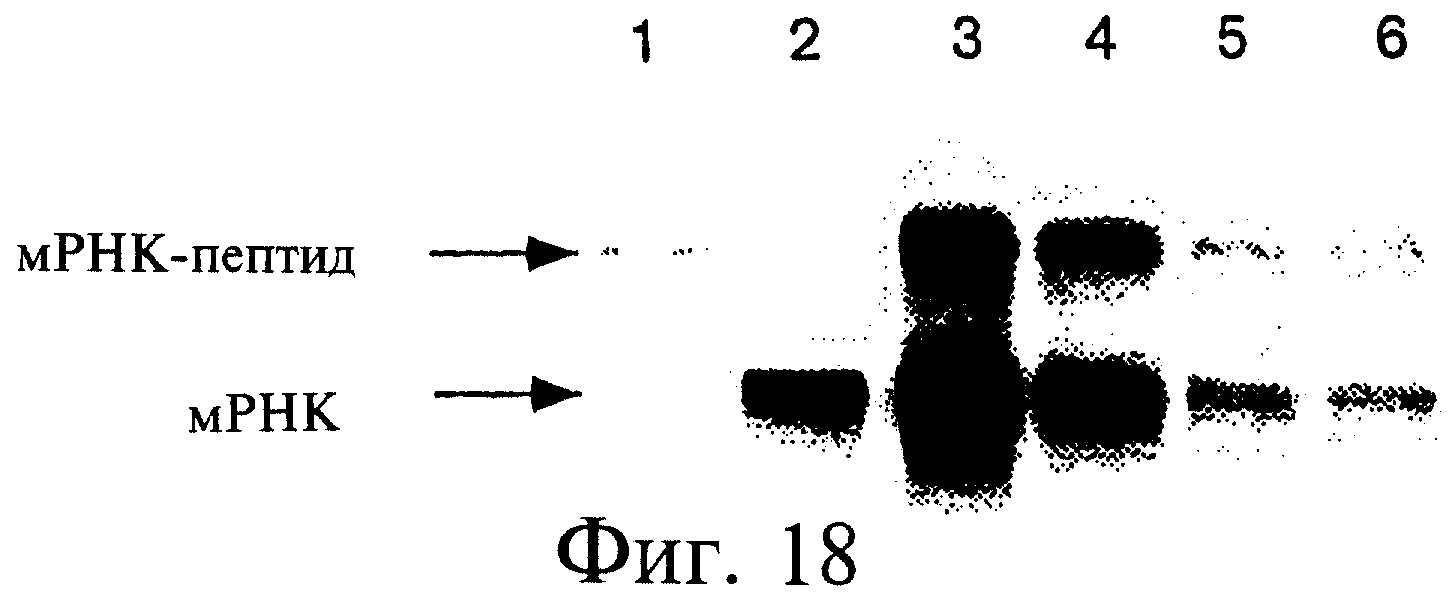
Фиг.18 является фотографией, показывающей трансляцию матриц myc-PHK, помеченных32Р. Использовали линкер dA21C9C9C9dAdCdCP. Трансляция была осуществлена в течение 90 минут при 30°С, а инкубацию проводили при -20°С в течение 2 дней без добавления Мg2+. Концентрации матриц мРНК составили 400 нМ (линия 3), 200 нМ (линия 4), 100 нМ (линия 5) и 100 нМ (линия 6). Линия 1 показывает мРНК-пептидный гибрид, помеченный35S-метионином. Линия 2 показывает мРНК, помеченную32Р. В линии 6 реакция была проведена в присутствии 0,5 мМ кэп-аналога.
Фиг.19 является фотографией, показывающей трансляцию матрицы myc-РНК при использовании лизатов, полученных от производителей Ambion (линия 1), Novagen (линия 2) и Amersham (линия 3). Использовавшийся линкер – dA27dCdCP. Концентрация матрицы составляла 600 нМ, а для метки использовали35S-метионин. Трансляция была проведена в течение 1 часа при 30°С, а инкубация - при -20°С в течение ночи в присутствии 50 мМ Mg2+.
Описанным здесь является базовый способ отбора белков, обладающих желательными функциями, с использованием гибридов, в составе которых эти белки ковалентно соединены с их собственными информационными РНК. Эти РНК-пептидные гибриды синтезируются в ходе трансляции in vitro или in situ пулов мРНК, включающих пептидный акцептор, присоединенный к их 3'-концам (фиг.1В). В одном из предпочтительных вариантов после прочтения открытой рамки рибосома переходит в стадию паузы по достижении желательного сайта-индуктора паузы, когда акцепторная составляющая занимает А-сайт рибосомы и присоединяет новообразованную полипептидную цепь из пептидил-тРНК в Р-сайте с образованием РНК-пептидного гибрида (фиг.1С). Ковалентная связь между белком и РНК (в форме амидной связи между 3'-концом этой мРНК и С-концом белка, который она кодирует) обеспечивает получение и амплификацию (например, с помощью ПЦР) генетической информации о белке с последующим отбором с применением обратной транскрипции с этой РНК. Когда гибрид сформирован, отбор и обогащение проводятся на основе свойств РНК-пептидного гибрида или, с другой стороны, обратная транскрипция может быть осуществлена с использованием матрицы мРНК тогда, когда она соединена с белком с целью предотвращения какого-либо влияния одноцепочечной РНК на проводимый отбор. Когда используется мРНК-пептидная конструкция, отбираемые гибриды могут быть тестированы с целью определения того, какая из составляющих (белок, РНК или они обе) обеспечивают желательные функции.
В одном из предпочтительных вариантов пуромицин (который напоминает тирозиладенозин) действует как акцептор присоединения нарастающей цепи полипептида к его мРНК. Пуромицин является антибиотиком, который действует как терминатор элонгации полипептида. В качестве соединения, миметирующего аминоацил-тРНК, он действует как универсальный ингибитор белкового синтеза благодаря связыванию с А-сайтом, акцептируя нарастающую полипептидную цепь и отделяясь от рибосомы (при Кd=10-4 М) (Traut & Monro, 1964, J. Mol. Biol, 10, 63; Smith et al., 1965, J. Mol. Biol, 13, 617). Одним из наиболее привлекательных свойств пуромицина является то, что он способен образовывать стабильную амидную связь с нарастающей полипептидной цепью, обеспечивая тем самым более стабильные гибриды в сравнении с потенциальными акцепторами, которые образовывают нестабильные сложноэфирные связи. В частности, пептидилпуромициновая молекула включает стабильную амидную связь между пептидом и 0-метилтирозиновой составляющей пуромицина. В свою очередь 0-метилтирозин соединяется с помощью стабильной амидной связи с 3'-аминогруппой модифицированной аденозиновой составляющей пуромицина.
Другие возможные выборы акцепторов включают тРНК-подобные структуры по 3'-концу данной мРНК, а также другие соединения, которые обладают типом активности, сходным с таковой у пуромицина. Такие соединения включают, без каких-либо ограничений, любое соединение, которое содержит в своем составе аминокислоту, соединенную с аденином или адениноподобным соединением, такие как аминокислотные нуклеотиды фенилаланинаденозин (A-Phe), тирозиладенозин (А-Тyr) и аланиладенозин (А-А1а), так же как и связанные с амидами структуры, такие как фенилаланил-3'-дезокси-3'-аминоаденозин, аланил-3'-дезокси-3'-аминоаденозин и тирозил-3'-дезокси-3'-аминоаденозин; в составе любого из этих соединений может быть использована любая из встречающихся в природе L-аминокислот. Кроме того, согласно настоящему изобретению также может быть использована комбинированная тРНК-подобная структура, соединенная по 3'-концу с пуромицином.
На фиг.2 показана предпочтительная схема отбора в соответствии с настоящим изобретением. Этапы, составляющие данную процедуру отбора, в целом выполняются следующим образом.
Этап 1. Приготовление матрицы ДНК. Осуществляется синтез РНК-компонента гибрида, что является этапом создания РНК-пептидных гибридов по настоящему изобретению. Этап может осуществляться путем прямого химического синтеза РНК или, что более обычно, проводиться путем транскрипции подходящей матрицы, являющейся двухцепочечной ДНК.
Такие ДНК-матрицы могут быть созданы с помощью любого стандартного метода (включая любую методику получения рекомбинантных ДНК, химический синтез или оба эти пути). В принципе, для данной цели может быть применен любой метод, позволяющий получать одну или большее число матриц, включающих известную, случайную, рандомизированную или мутантную последовательность. В одном конкретном случае олигонуклеотид (например, состоящий из случайного набора оснований) синтезируют и амплифицируют (например, с помощью ПЦР) перед транскрипцией. Также может быть использован химический синтез при получении случайной кассеты, которую затем вносят в среднюю часть последовательности, кодирующей известный белок (см., например, главу 8.2 в руководстве Ausubel et al., 1994, Current Protocols in Molecular Biology, John Wiley & Sons and Greene Publishing Company, NY). Этот подход позволяет получать значительное число мутаций вблизи специфичного сайта в составе белка, представляющего интерес.
Альтернативой пути полной рандомизации последовательности ДНК-матрицы является путь частичной рандомизации: пул последовательностей, синтезированных в этом случае, обычно обозначают как "легированный" пул. Пример такого подхода в приложении к последовательности РНК был описан, например, у Экланда с соавт. (Ekiand et al., 1995, Nucl. Acids Research, 23, 3231). Частичная рандомизация может быть осуществлена химическим путем с помощью смещения реакций синтеза таким образом, что смесь реакции прибавления каждого из оснований содержит избыток одного основания при очень небольшом количестве каждого из других нуклеотидов; путем тщательного контроля концентрации основания таким образом может быть достигнута желательная частота мутаций. Частично рандомизованные пулы также могут быть сформированы с использованием методик ПЦР с ошибками включения, так, как это, например, было ранее описано (Beaudry & Joyce, 1992, Science, 257, 635; и Barfcel & Szostak, 1993, Science, 261, 1411).
Также пригодны и многочисленные другие методы получения конструкций ДНК, начинающихся известной последовательностью и затем образующих пул мутантных ДНК. Примеры таких методик описаны (Ausubel et el., цитировано выше, глава 8; и Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual, глава 15, Cold Spring Harbor Press, New York, 2d ed.). Также случайные последовательности могут быть сформированы путем методик "перемешивания", в общих чертах описанных Стеммером (Stemmer, 1994, Nature, 370, 389).
С целью оптимизации схемы отбора по настоящему изобретению также могут быть изменены последовательности и структура 5'- и 3'-концевых участков матрицы. Предпочтительно осуществлять это в двух раундах отбора, в каждом случае включая вставку случайных доменов в матрицу в проксимальном положении по отношению к анализируемому концу с последующим отбором. Эти раунды отбора могут служить (i) для увеличения количества получаемого гибрида (и таким образом увеличения комплексности библиотеки) или (ii) для обеспечения оптимизированной трансляции последовательностей. Кроме того, данный способ может быть в принципе применим, в сочетании с мутагенезной ПЦР, для оптимизации трансляции матриц как для кодирующих, так и некодирующих последовательностей.
Этап 2. Создание РНК. Как отмечалось выше, РНК-компонент РНК-пептидного гибрида может быть синтезирован химическим путем с использованием стандартных методик синтеза олигонуклеотидов. С другой стороны, и особенно если используются более длинные последовательности РНК, РНК-компонент создается с помощью транскрипции in vitro матрицы ДНК. В одном из предпочтительных подходов полимераза Т7 используется для ферментативного формирования цепи РНК. Другими подходящими РНК-полимеразами для этой цели являются, без каких-либо ограничений, полимеразы SP6, Т3 и РНК-полимеразы Е. coli (описанные, например, в главе 3 цитировавшегося выше руководства Ausubel et al.). Дополнительно синтезированная РНК может быть полностью или частично модифицированной РНК. В одном из предпочтительных вариантов фосфоротиоат-РНК может быть получен (например, при транскрипции с участием полимеразы Т7) с использованием модифицированных рибонуклеотидов и стандартных методик. Такие модифицированные РНК обеспечивают определенные преимущества, т.к. являются устойчивыми к действию нуклеаз.
Этап 3. Лигирование пуромицина на матрицу. Следующим шагом является ковалентное связывание пуромицина (или любого другого подходящего пептидного акцептора) с последовательностью матрицы. Этот этап может осуществляться с использованием РНК-лигазы Т4 с целью присоединения пуромицина напрямую к последовательности РНК, или предпочтительно присоединение пуромицина опосредованно через ДНК-"шплинт" с использованием ДНК-лигазы Т4 или любого другого фермента, который способен объединять вместе две нуклеотидные последовательности (см. фиг.1В) (см. также, например, разделы 14 и 15 главы 3 цитировавшегося выше руководства Ausubel et а1.). тРНК-синтетазы также могут быть использованы для присоединения пуромициноподобных соединений к РНК. Например, фенилаланил-тРНК-синтетаза контролирует присоединение фенилаланина к молекулам фенилаланил-тРНК, включающим 3'-аминогруппу, образуя молекулы РНК с пуромицино-подобными 3'-концами (Fraser & Rich, 1973, Ргос. Natl. Acad. Sci. USA, 70, 2671). Другие пептидные акцепторы, которые могут быть использованы, включают, без каких-либо ограничений, любые соединения, которые включают аминокислоту, соединенную с аденином или адениноподобным соединением, такие как аминокислотные нуклеотиды фенилаланиладенозин (A-Phe), тирозиладенозин (А-Тyr) и аланиладенозин (А-А1а), так же как и связанные с амидами структуры, такие как фенилаланил-3'-дезокси-3'-аминоаденозин, аланил-3'-дезокси-3'-аминоаденозин и тирозил-3'-дезокси-3'-аминоаденозин; в составе любого из этих соединений может быть использована любая из встречающихся в природе L-аминокислот или их аналоги. Значительное число пептидных акцепторов было описано, например, у Краевской и Кухановой (Krayevsky & Kukhanova, 1979, Progress Nucleic Acids Research & Molecular Biology, 23, 1).
Этап 4. Формирование и получение РНК-пептидных гибридов. Для создания РНК-пептидных гибридов может быть использована любая трансляционная система in vitro или in situ. Как показано ниже, предпочтительными являются эукариотические системы, а две конкретные предпочтительные системы - это системы лизатов зародышей пшеницы и ретикулоцитов. В принципе, однако, в соответствии с настоящим изобретением может быть использована любая трансляционная система, которая позволяет образовывать РНК-пептидный гибрид и при которой не происходит существенной деградации РНК-компонента гибрида. Кроме того, для снижения уровня деградации РНК в любой из этих систем в трансляционную реакционную смесь могут быть внесены антисмысловые олигонуклеотиды, обладающие активностью по блокированию такой деградации; такие олигонуклеотиды специфически гибридизуют с последовательностями и покрывают их в пределах РНК-компонента молекулы, что предотвращает деградацию (см., например, Hanes & Pluckthun, 1997, Proc. Natl. Acad. Sci. USA, 94, 4937).
Как отмечалось выше, для использования в соответствии с настоящим изобретением пригодно любое число эукариотических трансляционных систем. Эти системы включают, без каких-либо ограничений, лизаты дрожжей, асцитных вытяжек, опухолевых клеток (Leibowitz et al., 1991, Methods Enzymol, 194, 536) и ооцитов икры шпорцевой лягушки. Могут быть использованы бактериальные трансляционные системы in vitro, которые включают, без каких-либо ограничений, описанные ранее (Zubay, 1973, Ann. Rev. Genet., 7, 267; Chen & Zubay, 1983, Methods Enzymol, 101, 44; и Ellman, 1991, Methods Enzymol, 202, 301).
Кроме того, реакции трансляции могут быть осуществлены in situ. В одном из предпочтительных примеров трансляция может быть осуществлена путем инъецирования мРНК в икру шпорцевой лягушки с применением стандартных методов.
После формирования РНК-пептидные гибриды могут быть выделены из реакционной трансляционной смеси путем любой стандартной методики очистки белка или РНК. Обычно применяются методики очистки белков. Как показано далее, например, очистка гибрида может быть осуществлена с использованием подходящих хроматографических реагентов, таких как агароза-dT25 или тиопропилсефароза. Однако, очистка может дополнительно или самостоятельно включать очистку РНК-компонента данного гибрида; методики такой очистки описаны, например, в главе 4 цитированного выше руководства Ausubel et al.
Этап 5. Отбор желательного РНК-пептидного гибрида. Отбор желательного РНК-пептидного гибрида может быть проведен любыми способами, пригодными для избирательного разделения или выделения желательного гибрида из популяции гибридов-"кандидатов". Примерами методик выделения, без каких-либо ограничений, являются избирательное связывание, например, со связывающим компонентом, который напрямую или опосредованно иммобилизован на колонку, гранулу, мембрану или другую твердую подложку, и иммунопреципитация с использованием антитела, специфичного для данного белкового компонента гибрида. Первый из этих методов основан на использовании иммобилизованного избирательного мотива, который может содержать молекулу любого типа, с которой возможно связывание. Список возможных селективных мотивов представлен на фиг.2. Отбор также может быть основан на использовании являющихся субстратами молекул, соединенным с меткой аффинности (например, связка субстрата с биотином), которая реагирует с молекулой-"кандидатом", или на любом другом типе взаимодействий с молекулой гибрида. Дополнительно белки могут быть отобраны на основе их каталитической активности путем, аналогичным тому, который был описан Бартелем и Шостаком при выделении РНК энзимов (см. цитированную выше ссылку); в соответствии с этой конкретной методикой желательные молекулы отбирают на основе их способности связывать на себя молекулы-мишени: затем функциональные молекулы выделяют на основе присутствия такой мишени. Схемы отбора для выделения новых или улучшенных по каталитическим свойствам белков с использованием такой схемы отбора или любого другого отбора по функциональности могут быть приспособлены для целей настоящего изобретения.
Дополнительно, как это описано в данном тексте, отбор желательного РНК-пептидного гибрида (или его ДНК-копии) может быть осуществлен путем обогащения пула молекул-"кандидатов" по данному гибриду. Для проведения такого факультативного обогащения осуществляют контакт популяции РНК-пептидных гибридов-"кандидатов" со связывающим компонентом (например, с одним из связывающих компонентов, описанных выше), который специфичен в отношении либо РНК-компонента, либо белкового компонента данного гибрида, в условиях, которые пригодны для отделения комплекса, образуемого гибридом и связывающим компонентом, от несвязанного материала в образце. Этот этап может быть повторен, и методика предпочтительно включает по крайней мере два последовательных этапа обогащения - в одном из них гибриды отбирают с использованием связывающих компонентов, специфичных в отношении РНК-компонента, а в другом гибриды отбирают с использованием связывающего компонента, специфичного в отношении белкового компонента этого гибрида. Кроме того, если повторяются этапы обогащения, нацеленные на один и тот же компонент гибрида (например, на его белковый компонент), предпочтительным является использование разных связывающих компонентов. В одном из предпочтительных вариантов, описанных здесь, популяцию молекул обогащают по желательным гибридам сначала с использованием связывающего компонента, специфичного в отношении РНК-компонента гибрида, и затем, в ходе двух последующих последовательных этапов, с использованием двух различных связывающих компонентов при том, что оба они специфичны в отношении белкового компонента данного гибрида. Опять же эти комплексы могут быть отделены от составляющих образца с помощью любых стандартных методик выделения, включая, без каких-либо ограничений, аффинную хроматографию на колонках, центрифугирование или иммунопреципитацию.
Более того, элюция РНК-пептидного гибрида из обогащенного (или отобранного) комплекса может быть осуществлена с использованием ряда подходов. Например, как описано в данном тексте, можно использовать этап денатурирующей или неспецифической химической элюции с целью выделения желательного РНК-пептидного гибрида. Такой этап обеспечивает высвобождение компонентов комплекса друг от друга или из ассоциированной твердой подложки в относительно неспецифических условиях путем разрыва нековалентных связей между компонентами и(или) между компонентом и твердой подложкой. Как описано здесь, одним из являющихся примеров денатурирующих или неспецифичных химических элюентов является 4%-ная НОАс/Н2О. Другими примерами денатурирующих или неспецифических химических элюентов являются гуанидин, мочевина, высококонцентрированная соль, детергент или любое другое средство, с помощью которого нековалентные аддукты могут быть отделены. С другой стороны, может быть использован подход, связанный со специфической химической элюцией, в котором применяется химическое вещество, обеспечивающее специфическое высвобождение входящей в состав гибрида молекулы. В одном из конкретных примеров, если линкерное плечо желательного гибридного белка включает одну или большее число дисульфидных связей, аптамеры связанных гибридов могут быть элюированы путем добавления, например, дитиотрейтола, в результате чего происходит восставновление дисульфидных связей и высвобождение связанной мишени.
С другой стороны элюция может быть осуществлена путем специфического разрушения аффинных комплексов; такие методики избирательно высвобождают компоненты комплексов при добавлении избытка одного из компонентов этого комплекса. Например, при проведении АТФ-связывающего отбора элюция осуществляется путем добавления избытка АТФ в инкубационную смесь. В конце концов, может быть проведен этап ферментной элюции. При использовании такого подхода сама связанная молекула или добавляемая извне протеаза (или другой подходящий гидролитический фермент) расщепляют и высвобождают либо мишень, либо фермент. В одном из конкретных примеров сайт-мишень для протеазы может быть включен в состав любого из компонентов комплекса, а элюция связанной молекулы происходит при добавлении протеазы. С другой стороны, при проведении каталитического отбора элюция может быть использована как этап отбора при выделении молекул, способных освобождать (например, путем расщепления) самих себя от твердой подложки.
Этап 6. Создание ДНК-копий последовательностей РНК с использованием обратной транскриптазы. Если желательно, ДНК-копия отобранной последовательности РНК, входящей в состав гибрида, легко доступна для обратной транскрипции такой последовательности РНК с применением любой стандартной методики (например, с использованием обратной транскриптазы Superscript). Этот этап может быть осуществлен до проведения отбора или до этапа обогащения (например, как это показано на фиг.16), или после этого этапа. С другой стороны, процесс обратной транскрипции может быть осуществлен до выделения конкретного гибрида из трансляционной смеси в системе in vitro или in situ.
На следующем этапе амплифицируют матрицу ДНК в виде либо полной, либо частичной двухцепочечной последовательности. На этом этапе предпочтительно формировать матрицы полноразмерной ДНК, используя подходящие олигонуклеотиды и амплификацию с помощью ПЦР.
Эти этапы, а также реагенты и методики осуществления этих этапов будут теперь описаны подробно при рассмотрении конкретных примеров. Эти примеры представлены с целью проиллюстрировать настоящее изобретение и поэтому не должны рассматриваться, как в чем-то его ограничивающие.
СОЗДАНИЕ МАТРИЦ ДЛЯ РНК-ПЕПТИДНЫХ ГИБРИДОВ
Как показано на фиг.1А и 2, схема отбора по настоящему изобретению предпочтительно использует матрицы двухцепочечных ДНК, которые включают ряд желательных элементов. Первым из этих элементов является промотор, предназначенный для использования при взаимодействии с желательной РНК-полимеразой в ходе синтеза мРНК. Как показано на фиг.1А и описано здесь, предпочтительным является промотор Т7, хотя может быть использован любой другой промотор, способный направлять транскрипцию линейной двухцепочечной ДНК.
Второй элемент матрицы, показанный на фиг.1А, определяется как 5'-нетранслируемый сегмент (или 5'UTR) и соответствует участку РНК, расположенному выше сайта начала трансляции (т.е. старт-кодона). В соответствии с показанным на фиг.1А, предпочтительным 5'UTR (обозначенным как "ТЕ") является делеционный мутантный вариант 5'-нетранслируемого сегмента вируса табачной мозаики (TMV), который, в частности, соответствует основаниям, непосредственно примыкающим с 5'-стороны к сайту начала трансляции TMV; последовательность этого UTR такова - rGrGrG rArCrA rArUrU rArCrU rArUrU rUrArC rArArU rUrArC rA (при том, что первые три остатка гуанина внесены с целью усиления транскрипции) (SEQ ID NO: 5). Могут быть использованы также любые другие подходящие 5'UTR (см., например, Kozak, 1983, Microbiol. Rev., 47,1).
Третий элемент, показанный на фиг.1А, - это сайт начала трансляции. В принципе, им является старт-кодон AUG. Однако известны примеры того, когда иные, нежели AUG, кодоны используются для этой цели нативными кодирующими последовательностями - такие кодоны также могут быть использованы для схемы отбора по настоящему изобретению.
Четвертый элемент, показанный на фиг.1А, - это открытая рамка считывания соответствующего белка (обозначается как ORF), которая кодирует аминокислотную последовательность. Эта открытая рамка может кодировать нативную, случайную, рандомизированную, мутантную или полностью синтетическую полипептидную последовательность.
Пятый элемент, показанный на фиг.1А, - это 3'-константный сегмент. Эта последовательность облегчает ПЦР-амплификацию в пуле последовательностей и лигирование пуромицинсодержащего олигонуклеотида на данную мРНК. Если желательно, этот сегмент также может включать сайт-индуктор паузы - последовательность, которая обусловливает паузу на рибосоме и таким образом позволяет дать дополнительное время акцепторной составляющей (например, пуромицину) для соединения с новообразующейся полипептидной цепью из пептидил-тРНК: этот сайт-индуктор паузы будет обсужден более подробно ниже.
При разработке представляемой методологии РНК-пептидные гибриды были исходно сформированы с использованием резко упрощенных мРНК-матриц, включающих всего по 1-2 кодона. Такой подход был выбран, исходя из двух соображений. Во-первых, матрицы такого размера могут легко быть получены путем химического синтеза. И, во-вторых, маленькая открытая рамка считывания позволяет легко контролировать критические параметры реакции - такие как эффективность связывания, концевую гетерогенность, зависимость от матрицы и точность трансляции.
Создание конструкции. Базовая конструкция была использована для создания РНК-пептидных гибридов. Молекула включала мРНК с (i) последовательностью Шайна-Дальгарно (3D), необходимой для инициации трансляции, которая несла 3-нуклеотидную делецию последовательности 3D из состава рибосомного белка L1 и которая была комплементарна 5 основаниям 163-рРНК (т.е. rGrGrA rGrGrA rCrGrA rA) (SEQ ID NO: 6) (Stormo et al., 1982, Nucleic Acids Research, 10, 2971-2996; Shine & Dalgarno, '1974, Proc. Natl. Acad. 3ci. USA, 71, 1342-1346; и Steitz & Jakes, 1975, Proc. Natl. Acad. Sci. USA, 72, 4734-4738), (ii) старт-кодоном AUG, (iii) ДНК-линкером, необходимым в качестве сайта-индуктора паузы (т.е. 5'-[dA]27), (iv) dCdC-3', и (v) 3'-пуромицином (Р). Поли-dA последовательность была выбрана потому, что она известна в качестве слабой матрицы для тРНК в А-сайте (Morgan et al., 1967, J. Mol. Biol., 26, 477-497); Ricker & Kaji, 1991, Nucleic Acids Research, 19, 6573-6578) и может быть использована в качестве эффективного сайта-индуктора паузы. Длина олигоаденилового линкера была выбрана таким образом, чтобы покрывать расстояние примерно в 60-70
Химический синтез минимальной матрицы 43-Р. Для того, чтобы синтезировать конструкцию 43-Р (показана на фиг.3), пуромицин был вначале присоединен к твердой подложке таким образом, чтобы он мог быть доступен для стандартного синтеза фосфорамидитных олигонуклеотидов. Методика синтеза таких олигонуклеотидов в общих чертах схематически показана на фиг.3 и более подробно описана ниже. Для присоединения пуромицина к твердой подложке в виде регулируемого пористого стекла (CPG) аминогруппу защищали трифторацетильной группой так, как это было описано в бюллетене №49 (1988) для пользователей оборудования Applied Biosystems для модели 380 ДНК-синтезатора. На следующем этапе защиту 5'ОН осуществляли с использованием стандартного подхода DMT-C1 (Gait, Oligonucleotide Synthesis a practical approach: The Practical Approach Series (IRL Press, Oxford, 1984)), и присоединение к аминогексил-CPG по 2'ОН было проведено точно таким образом, как группа 3'ОН может быть использована для присоединения дезоксинуклеозида (см. фиг.3В и Gait, p. 47, цитировано выше). Защищенный на 5', DMT-CPG-связанный пуромицин становился затем пригоден для удлинения цепи с использованием фосфорамидитных мономеров. Синтез олигонуклеотида осуществляется в направлении 3'→5' в следующем порядке: (i) 3'-пуромицин; (ii) pdCpdC, (iii) примерно 27 мономеров dA в качестве линкера, (iv) кодон AUG, и (v) последовательность Шайна-Дальгарно. Последовательность конструкции 43-Р показана ниже.
Синтез CPG-пуромицина. Для синтеза, защищенного CPG-пуромицина использовали базовый путь для дезоксинуклеозидов, описанный в общих чертах ранее (Gait, Oligonucleotide Synthesis, A Practical Approach, The Practical Approach Series (IRL Press, Oxford, 1984)). Основные этапы включают выбор подходящей N-блокирующей группы, присоединение к твердой подложке по группе 2'ОН и реакцию связывания с твердой подложкой. В последнем случае реакцию осуществляют при очень низких концентрациях активированного нуклеотида, поскольку этот материал существенно более ценен в сравнении с твердой подложкой. Результирующий выход (примерно 20 мкмоль/г подложки) был достаточно удовлетворительным с точки зрения выбранных условий реакции.
Синтез N-трифторацетилпуромицина. 267 мг (0,490 ммоль) пуромицин-НСl сначала конвертировали в свободное основание путем растворения в воде, добавления карбонатного буфера с рН=11 и экстрагирования (3х) в хлороформе. Органическую фазу выпаривали и взвешивали (242 мг, 0,513 ммоль). Свободное основание затем растворяли в 11 мл безводного пиридина и 11 мл безводного ацетонитрила, и 139 мкл (2,0 ммоль) триэтиламина (TEA) и 139 мкл (1,0 ммоль) трифторуксусного ангидрида (TFAA) добавляли при помешивании. Затем TFAA добавляли во взмученный раствор аликвотами по 20 мкл до полного расходования из исходного материала, что оценивали с применением тонкослойной хроматографии (хлороформ и МеОН в соотношении 93:7) (общий объем 280 мкл). Реакцию проводили в течение 1 часа. На этом этапе при тонкослойной хроматографии маркировались два бэнда, причем оба характеризовалась более высокой подвижностью в сравнении с исходным материалом. Обработка реакционной смеси NH4OH и водой обусловило сведение продукта в единственный бэнд. Силикагелевая хроматография (хлороформ и МеОН в соотношении 93:7) позволила получить 293 мг (0,515 ммоль) результирующего продукта - N-TFA-Pur. Схематически продукт данной реакции показан на фиг.4.
Синтез N-трифторацетил-5'-DМТ-пуромицина. Продукт, полученный в предыдущей реакции, был разделен на аликвоты и упарен 2х вместе с безводным пиридином досуха. Набор пробирок был приготовлен для тестирования различных условий реакции. В маломасштабной реакции 27,4 мг (48,2 мкмоль) N-TFA-Pur были растворены в 480 мкл пиридина, содержащем 0,05 эквивалентного количества DMAP и 1,4 эквивалентного количества TEA. К этой смеси добавляли 20,6 мг тритилхлорида (60 мкмоль), и реакцию проводили до завершения при перемешивании. Реакцию останавливали добавлением к данному раствору равного объема воды (приблизительно 500 мкл). Поскольку данная реакция представлялась успешной, то проводили реакцию в большом масштабе. В частности, 262 мг (0,467 ммоль) N-TFA-Pur растворяли в 2,4 мл пиридина с последующим добавлением 1,4 эквивалентного количества TEA, 0,05 эквивалентного количества DMAP и 1,2 эквивалентного количества тритилхлорида. После примерно двух часов дополнительно добавляли 50 мг (0,3 эквивалентного количества) диметокситритил*С1 (DMT*C1) и реакцию проводили еще в течение 20 минут. Реакцию останавливали добавлением 3 мл воды и проводили выпаривание вместе с 3х СН3СN. Продукт реакции очищали смесью хлороформ-МеОН (95:5) на 100-мл безводных силикагелевых колонках диаметром 2 мм. Из-за неполной очистки дополнительно использовали точно такую же колонку при соотношении хлороформ/МеОН = 97,5:2,5. Общий выход составил 325 мг или 0,373 ммоль (или выход на уровне 72%). Продукт реакции схематически изображен на фиг.4.
Синтез N-трифторацетил-5'-DМТ-2'-сукцинилпуромицина. В реакции малого масштаба 32 мг (37 мкмоль) продукта, синтезированного в предыдущей реакции, соединяли с 1,2 эквивалентного количества DMAP, растворенного в 350 мкл пиридина. К этому раствору добавляли 1,2 эквивалентного количества янтарного ангидрида в 44 мкл безводного CH3CN и оставляли на ночь при перемешивании. Тестирование с помощью тонкослойной хроматографии показало сохранение лишь небольшого количества исходного материала. В крупномасштабной реакции 292 мг (336 мкмоль) продукта предыдущей реакции соединяли с 1,2 эквивалентного количества DMAP в 3 мл пиридина. К этой смеси добавляли 403 мкл 1 М янтарного ангидрида в безводном CH3CN, и полученную смесь оставляли на ночь при перемешивании. Тестирование с помощью тонкослойной хроматографии снова показало сохранение лишь небольшого количества исходного материала. Две реакции объединяли и добавляли дополнительные 0,2 экв. DMAP и сукцинат. Полученный продукт выпаривали с 1х толуолом и высушивали до получения желтой пены в условиях высокого вакуума. Добавляли CH2Cl2 (20 мл) и полученный раствор экстрагировали дважды 15 мл 10%-ной охлажденной на льду лимонной кислотой и затем еще дважды чистой водой. Полученный продукт высушивали, снова растворяли в 2 мл CH2Cl2 и осаждали добавлением 50 мл гексана при перемешивании. Полученный продукт затем взбалтывали и центрифугировали при 600 об/мин в течение 10 минут на медицинской центрифуге. Большую часть элюента отбирали, а оставшийся продукт высушивали - сначала при низком вакууме, затем при высоком вакууме в вакуумной сушилке. Выход по данной реакции составил приблизительно 260 мкмоль, т.е. на уровне примерно 70%.
Синтез N-трифторацетил-5'-DMT-2'-сукцинил-СРС-пуромицина. Продукт предыдущей реакции снова растворяли в 1 мл диоксана с последующим добавлением 0,2 мл диоксана и 0,2 мл пиридина. К этому раствору добавляли 40 мг р-нитрофенола и 140 мг дициклогексилкарбодиимида (DCC), и реакцию проводили в течение 2 часов. Полученную в результате нерастворимую циклогексилмочевину удаляли центрифугированием и полученный раствор добавляли к 5 г модифицированного аминогексилом пористого стекла (CPG), суспендированного в 22 мл безводного DMF и оставляли на ночь при перемешивании. Затем смолу промывали DMF, метанолом и эфиром и высушивали. В полученной в результате смоле определяли содержание 22,6 мкмоль тритила на 1 г, что считается хорошим показателем для приемлемого круга подложек такого типа. Подложку затем блокировали путем инкубации с 15 мл пиридина, 1 мл уксусного ангидрида и 60 мг DMAP в течение 30 минут. Полученный в результате материал колонки проявлял отрицательный (бесцветный) нингидриновый тест в сравнении с результатами, полученными до блокирования, в которых полученный реакционный материал характеризовался темно-синей окраской. Продукт этой реакции схематически показан на фиг.4.
Синтез комплекса мРНК-пуромицин. Как обсуждалось выше, олигонуклеотид с присоединенным пуромицином может быть использован двумя способами при создании комплекса мРНК-пуромицин, который служит в качестве матрицы для трансляции. Для очень коротких открытых рамок считывания пуромицин-олигонуклеотид обычно удлиняют химическим путем, используя мономеры РНК или ДНК для создания полностью синтетической матрицы. Когда желательным является получение более длинной открытой рамки считывания, олигонуклеотидные РНК или ДНК обычно лигируют на 3'-конец мРНК с использованием ДНК-"шплинта" и ДНК-лигазы Т4 так, как это было описано (Moore & Sharp, 1992, Science, 256, 992).
ТРАНСЛЯЦИЯ IN VITRO И ТЕСТИРОВАНИЕ РНК-ПЕПТИДНЫХ ГИБРИДОВ
Матрицы, синтезированные так, как это было описано выше, транслировали in vitro с использованием бактериальных и эукариотических систем для трансляции in vitro следующим образом.
Трансляция in vitro минимальных матриц. 43-Р и сходные РНК-пуромициновые комплексы добавляли в различные системы для трансляции in vitro, включающие: (i) систему 330, производную от E.coli штамма MRE600 (Zubay, 1973, Ann. Rev. Genet., 7, 267; Collins, 1979, Gene, 6, 29; Chen & Zubay, 1983, Methods Enzymol., 101, 44; Pratt, 1984, In "Transcription and Translation: A Practical Approach" eds., B.D. Hainmes & S. J. Higgins, IRL Press, Oxford, pp. 179-209; и Ellman et al., 1991, Methods Enzymol., 202, 301), приготовленную так, как было описано (Ellman et al., 1991, Methods Enzymol., 202, 301); (ii) рибосомную фракцию, выделенную из того же штамма, приготовленную так, как это было описано (Kudlicki et al., 1992, Anal. Chem., 206, 389); и (iii) систему S30, производную от Е.coli штамма BL21, приготовленную согласно описанному ранее (Lesley et al., 1991, J. Biol. Chem., 266, 2632). В каждом случае был использован премикс в соответствии с данными Лесли с соавт. (Lesley et al., 1991, J. Biol. Chem., 266, 2632), а инкубацию проводили в течение 30 минут.
Тестирование природы гибрида. Матрицу 43-Р сначала тестировали с использованием трансляционных экстрактов 330 E.coli. Фиг.5 (реакция "А") показывает желательную внутримолекулярную (т.е. "цис-") реакцию, при том, что 43-Р связывает рибосому и в то же самое время действует и как матрица, и как акцептор формилметионина. Встраивание35S-метионина и его положение в составе матрицы были протестированы вначале: результаты показаны на фиг.6А и 6В. После экстракции продукта трансляции in vitro с использованием смеси фенола и хлороформа и анализа полученного продукта с помощью SDS-электрофореза в ПААГ.35S-помеченный бэнд проявляет ту же подвижность, что и матрица 43-Р. Количество синтезируемого материала зависело от концентрации Mg2+ (фиг.6А). Оптимальная концентрация Mg2+ предположительно находится в пределах 9-18 мМ, что сходно с таким оптимумом, известным для осуществления трансляции в этой системе (Zubay, 1973, Ann. Rev. Genet., 7, 267; Collins, 1979, Gene, 6, 29; Chen & Zubay, 1983, Methods Enzymol, 101, 44; Pratt, 1984, In "Transcription and Translation: A Practical Approach", eds. B.D.Hammes & S.J.Higgins, IRL Press, Oxford, pp. 179-209; Ellman et al., 1991, Methods Enzymol, 202, 301; Kudlicki et al., 1992, Anal. Chem., 206, 389; Lesley et al., 1991, J. Biol. Chem., 266, 2632). Кроме того, включенная метка оказалась устойчивой к обработке NH4ОН (фиг.6В), что указывает на то, что метка находится в 3'-половине молекулы (ДНК-компонент со стабильными основаниями) и присоединена стабильной связью, что и предполагается для амидной связи между пуромицином и формилметионином.
Зависимость от рибосомы и матрицы. Для демонстрации того, что охарактеризованная выше реакция происходит в рибосоме, было исследовано влияние специфических ингибиторов пептидилтрансферазной функции рибосомы (фиг.6С), а также тестировали влияние изменения последовательности, кодирующей метионин (фиг.6D). Фиг.6С отчетливо показывает, что реакция жестко подавлялась факторами, являющимися ингибиторами пептидилтрансферазы - вирджиниамицином, гоугеротином и хлорамфениколом (Моnrо & Vazquez., 1967, J. Mol. Biol, 28, 161-165; и Vazquez & Monro, 1967, Biochemica et Biophysical Acta, 142, 155-173). Фиг.6D показывает, что изменения единственного основания в матрице с А на С - полностью нарушает инкорпорацию35S-метионина при 9 мМ Мg2+ и резко снижает ее при 18 мМ (что согласуется с тем фактом, что высокое содержание ионов магния приводит к ошибочному считыванию мРНК). Эти эксперименты продемонстрировали, что реакция, проходящая в рибосоме, характеризуется зависимостью от структуры матрицы.
Длина линкера. Также была исследована зависимость реакции от длины линкера (фиг.6Е). Исходная матрица была сконструирована таким образом, что линкер покрывал расстояние от декодирующего сайта (занимаемого кодоном AUG в составе матрицы) до акцепторного сайта (занимаемого пуромицином) - т.е. расстояние, имеющее примерно ту же величину, что и расстояние между антикодонной петлей и акцепторным стеблем тРНК, или примерно 60-70
Внутримолекулярные и межмолекулярные реакции. В заключение был исследован тип реакции - происходит ли она по внутримолекулярному типу (фиг.5, реакция "А"), что является желательным, - или по межмолекулярному типу (фиг.5, реакция "В"). Для проведения такого теста добавляли олигонуклеотиды с 3'-присоединенным пуромицином, но лишенные последовательности инициации трансляции, т.е. рибосомосвязывающей последовательности (т.е. матрицы 25-Р, 13-Р и 30-Р), к трансляционным реакционным смесям, включающим матрицу 43-Р (фиг.6F, 6G и 6Н). Если реакция проходит по межмолекулярному типу, более короткие олигонуклеотиды также должны метиться. Как показано на фиг.6Н, имеет место лишь слабое включение35S-метионина в состав трех коротких олигонуклеотидов: это подтверждает, что реакция протекает в основном по внутримолекулярному типу. Последовательности 25-Р (SEQ ID NO:10), 13-P (SEQ ID NO:9) и 30-Р (SEQ ID N0:8) показаны ниже.
Лизат ретикулоцитов. Фиг.6Н показывает, что35 S-метионин может быть инкорпорирован в состав матрицы 43-Р с использованием лизата ретикулоцитов кролика (см. ниже) для целей трансляции in vitro, дополнительно к системам лизатов клеток Е.coli., использовавшимся выше. Эта реакция протекает по внутримолекулярному механизму, что является желательным.
СИНТЕЗ И ТЕСТИРОВАНИЕ ГИБРИДОВ, ВКЛЮЧАЮЩИХ ЭПИТОП-МЕТКУ C-MYC
Также были получены примеры гибридов, включающих в виде белкового компоненита эпитоп-метку для моноклонального антитела 9Е10 к протоонкогену c-myc (Evan et al., 1985, Mol. Cell Biol., 5, 3610).
Конструирование матриц. Три исходные матрицы, соответствующие эпитопам-меткам (т.е. LP77, LP154 и пул №1) были сконструированы: они показаны на фиг.7А-С. Две первые матрицы содержали эпитопный мотив из состава c-myc EQKLISEEDL (SEQ ID NO:2) и третья матрица была сконструирована для использования в синтезе случайного селективного пула. LP77 кодирует 12-аминокислотную последовательность, причем состав кодонов был оптимизирован для трансляции в бактериальных клетках. LP154 и его производные включают последовательность мРНК, кодирующую 33 аминокислоты, в которой состав кодонов оптимизирован для трансляции в эукариотических системах. Кодируемая аминокислотная последовательность - MAEEQKLISEEDLLRKRREQKLKHKLEQLRNSCA (SEQ ID N0:7): она соответствует исходному полипептиду, использовавшемуся при создании моноклонального антитела 9Е10. Пул №1 включает 27 кодонов NNG/C (для формирования случайных пептидов) со следующей последовательностью, соответствующей семи аминокислотам пептида myc (которая не является частью последовательности эпитопа myc). Эти последовательности приведены ниже.
Трансляционные in vitro системы зародышей пшеницы и ретикулоцитов. Матрицы 43-Р, LP77 и LP154 были протестированы в обоих трансляционных системах - ретикулоцитах кролика и экстракте зародышей пшеницы (Promega, Boehringer, Mannheim) (фиг.8). Трансляцию осуществляли при 30°С в течение 60 минут. Матрицы выделяли с использованием dТ25-агарозы при 4°С. Матрицы элюировали из агарозы с использованием 15 мМ NaOH, 1 мМ EDTA, нейтрализовали буфером NaOAc/HOAc, немедленно осаждали этанолом (2,5-3 объема), промывали 100%-ным этанолом и высушивали на скоростном вакуумном выпаривателе. Фиг.8 показывает, что35 S-метионин был инкорпорирован в состав всех трех матриц и в обеих системах - ретикулоцитах и зародышах пшеницы. Меньший уровень деградации матриц был отмечен в реакциях гибридизации в системе ретикулоцитов: соответственно, эта система является предпочтительной с точки зрения формирования РНК-пептидных гибридов. Кроме того, в целом, эукариотические системы превосходят бактериальные трансляционные системы. Поскольку эукариотические клетки имеют тенденцию содержать меньшее количество нуклеаз, время полужизни мРНК в целом на 1-2 порядка больше в этих клетках в сравнении с бактериальными клетками. В экспериментах с использованием одной конкретной трансляционной системы Е.coli. образование гибридов вообще отсутствовало при использовании матрицы, кодирующей эпитоп с-myс; мечение матрицы в различных сайтах показало, что этот отрицательный эффект может быть обусловлен деградацией и ДНК-, и РНК-компонента матрицы.
Для анализа пептидного компонента этих гибридов образцы были обработаны РНКазой с целью удаления кодирующих последовательностей. После такой обработки продукт 43-Р двигался практически с идентичной подвижностью в сравнении с32 Р-меченным олигонуклеотидом 30-Р: это соответствует тому, что к 30-Р добавлен очень небольшой пептид, возможно, состоящий из единственного метионина. В варианте с LP77 удаление кодирующей последовательности привело к получению продукта с меньшей подвижностью в сравнении с 30-Р: это соответствует тому, что к пуромицину добавлен 12-аминокислотный пептид. Наконец, в случае с LP154 удаление кодирующей последовательности позволило получить продукт со значительно меньшей подвижностью: это соответствует присоединению 33-аминокислотной последовательности к олигонуклеотиду 30-Р. Не были выявлены олигонуклеотиды в варианте с ретикулоцитной системой при обработке LP154 РНКазой, что объясняется загрузочной ошибкой. На фиг.9 показано, что подвижность данного продукта была такой же, как и у продукта, полученного в системе экстракта зародышей пшеницы. Суммируя сказанное, полученные результаты определяют, что продукты, резистентные к РНКазе, оказались добавленными к концам олигонуклеотидов 30-Р, что размер продуктов оказался пропорциональным длине кодирующих последовательностей и что полученные продукты были весьма однородны по длине. Кроме того, хотя обе трансляционные системы дали сходные гибридные продукты, система ретикулоцитов проявила некоторые преимущества благодаря большей стабильности матриц.
Чувствительность к РНКазе-А и протеиназе-К. На фиг.9 показаны данные тестирования чувствительности гибрида LP154 к РНКазе-А и протеиназе-К. Как показано на линиях 2-4, инкорпорация35S-метионина была подтверждена для матрицы LP154. Когда полученный продукт подвергали действию РНКазы-А подвижность гибрида снижалась, но при этом все-таки оставалась существенно выше, чем у32Р-меченного олигонуклеотида 30-Р: это соответствует добавлению с 3'-конца 33-пептидной последовательности. Когда этот же продукт подвергали действию протеиназы-К, сигнал35S полностью исчезал: это вновь соответствует тому, что метка присутствует в составе пептида, находящегося с 3'-конца фрагмента 30-Р. Сходные результаты были получены в аналогичных экспериментах с использованием гибридов 43-Р и LP77.
Для подтверждения того, что помеченная35S-метионином матрица является следствием трансляции и, более специфично, является результатом пептидил-трансферазной активности рибосомы, было протестировано влияние различных ингибиторов реакции мечения. Все специфические ингибиторы эукариотической пептидилтрансферазы анизомицин, гоугеротин и спарсомицин (Vazquez, 1979, "Inhibitors of Protein Biosynthesis", Springer-Verlag, New York, p. 312), равно как и ингибиторы транслокации циклогексимид и эметин (Vazquez, 1979, "Inhibitors of Protein Biosynthesis", Springer-Verlag, New York, p. 312) уменьшают образуемость РНК-пептиднх гибридов почти на 95% в вариантах длинных матриц myc и трансляционной системы лизата ретикулоцитов.
Эксперименты по иммунопреципитации. В эксперименте, разработанном для того, чтобы проиллюстрировать эффективность иммунопреципитируемости мРНК-пептидного гибрида, была предпринята попытка провести иммунопреципитацию свободного пептида с-myс, полученного в результате трансляции in vitro. Фиг.10 показывает результаты этих экспериментов по тестированию пептида с помощью SDS-электрофореза в ПААГ. Линии 1 и 2 показывают помеченный материал в трансляционных реакциях, включающих либо RNA124 (РНК-компонент гибрида LP154) или мРНК β-глобина. Линии 3-8 показывают иммунопреципитацию этих реакционных образцов с использованием моноклонального антитела 9Е10 в ряде различающихся буферных условий (описаны ниже). Линии 3-5 показывают, что пептид, производный от RNA124, эффективно иммунопреципитируется - наилучший вариант показан в линии 4, где было выделено примерно 83% общего преципитируемого материала ТСА. Линии 6-8 показывают небольшую часть β-глобина, подтверждая более чем 100-кратную степень очистки. Эти результаты определяют, что пептид, кодируемый RNA124 (и LP154), может быть выделен в иммунопреципитационной методике с учетом количественных параметров.
Иммунопреципитация гибрида. Далее была протестирована способность иммунопреципитировать гибридный РНК-пептидный продукт, используя трансляционную реакцию LP154 и моноклональное антитело 9Е10 к с-myс (фиг.11). Трансляционный продукт, полученный в системе ретикулоцитов, был выделен с помощью иммунопреципитации (как описано в данном тексте) и обработан 1 мкг РНКазы-А при комнатной температуре в течение 30 минут с целью удаления кодирующей последовательности. Это привело к образованию 5'ОН, который был помечен32P с помощью полинуклеотидкиназы Т4 и тестирован с помощью электрофореза в ПААГ в денатурирующих условиях. Фиг.11 показывает, что так был выделен продукт, характеризующийся подвижностью, сходной с таковой, которая наблюдается у гибрида эпитопа с-myс с олигонуклеотидом 30-Р, полученного в результате обработки РНКазой гибрида LP154 (см. выше); однако соответствующий продукт не был получен, когда транслировали только РНК-компонент матрицы (RNA124). На фиг.12 количество выделенного гибридного белка было определено и представлено графически по отношению к количеству немодифицированного 30-Р (не показано). Количественная оценка соотношения немодифицированного линкера к myс-линкерному гибридному пептиду показывает, что 0,2-0,7% внесенной мРНК было конвертировано в гибридный продукт. Более высокая доля внесенной РНК конвертировалась в гибридный продукт при наличии более высокого соотношения "рибосома/матрица": во всем диапазоне концентраций вносимой мРНК, которые были протестированы, приблизительно 0,8-1,0·1012гибридных молекул образовывалось в расчете на 1 мл трансляционного экстракта.
Кроме того, полученные результаты показали, что пептиды, присоединенные к различным РНК, кодировались этими мРНК: т.е. новообразующийся пептид не переносится на пуромицин каких-либо иных мРНК. Не было выявлено случаев перекрестного переноса тогда, когда линкер (30-Р) инкубировали вместе с длинной матрицей myc в трансляционных экстрактах при соотношения 20:1; также в присутствии свободного линкера не отмечено сколько-нибудь существенного уменьшения количества образующегося длинного myс-гибрида. Сходным образом котрансляция короткой и длинной матриц - 43-Р и LP154 -приводила к выработке только гибридных продуктов, которые выявлялись тогда, когда матрицы транслировались по отдельности; не было выявлено продуктов, характеризующихся промежуточной подвижностью, которые могли бы появиться в случае гибридизации короткой матрицы с длинным myс-пептидом. Все эти результаты подтвердили, что образование гибридов происходило в первую очередь между новооразующимся полипептидом и мРНК, связанной на той же самой рибосоме.
Последовательное выделение. Как дальнейшее подтверждение природы продукта, транслированного in vitro с матрицы LP154, было протестировано поведение этого продукта в двух различных типах хроматографических сред. Тио-пропилсефароза (ТР) позволяет выделять продукт, включающий свободный цистеин (например, продукт LP154, который имеет остаток цистеина рядом с С-концом) (фиг.13). Сходным образом агароза-dt25 позволяет выделять матрицы, включающие полиадениловую последовательность (например, 30-Р) (фиг.13). Фиг.14 показывает, что последовательное выделение на сефарозе ТР и затем на arapose-dt25 позволяет получать тот же продукт, что и при выделении только на агарозе-dТ25. Тот факт, что продукт трансляции in vitro включает и полиадениловый тракт, и свободный тиол, убедительно показал, что трансляционный продукт являлся желательным РНК-пептидным гибридом.
Описанные выше результаты согласуются со способностью синтезировать мРНК-пептидные гибриды и выделять их интактными из трансляционных экстрактов in vitro. Пептидные компоненты гибридов, синтезированных таким образом, представляются как имеющие выбранные последовательности, на что указывают данные иммунопреципитации и выделения с использованием подходящих хроматографических методик. В соответствии с представленными выше результатами реакции имеют внутримолокулярный тип и происходят по механизму, зависимому от параметров матрицы. В конце концов даже при модификации матрицы на уровне менее 1% представляемая система облегчает проведение отбора, основанного на комплексности пулов "кандидатов" на уровне примерно 1013 молекул.
Отбор эпитопа с-myс. Для выбора дополнительных эпитопов с-myс была сформирована большая библиотека трансляционных матриц (например, включающая 1015 членов), включающих рандомизованный участок (см. фиг.7С и ниже). Эта библиотека используется для создания примерно 1012-1013 гибридов (как описано в настоящем тексте), которые обрабатывали антителом к с-myс (например, с помощью иммунопреципитации или используя антитело, иммобилизованное на колонке или другой твердой подложке) с целью обогащения матриц, кодирующих с-myс в повторяющихся раундах отбора in vitro.
Модели формирования гибридов. Вне связи с какой-либо конкретной теорией, предложена модель, описывающая механизм образования гибрида, в котором трансляция нормально инициируется и элонгация распространяется вплоть до конца открытой рамки считывания. Когда рибосома достигает ДНК-компонента данной матрицы, трансляция приостанавливается. В этот момент комплекс может "выбирать" между двумя путями: диссоциация новообразующейся полипептидной цепи или перенос новообразующегося пептида на пуромицин, находящийся в 3'-конце данной матрицы. Эффективность реакции переноса, по-видимому, контролируется рядом факторов, которые влияют на стабильность приостановленного трансляционного комплекса и на вход остатка 3'-пуромицина в А-сайт пептидилтрансферазного центра. После реакции переноса РНК-пептидного гибрид, по-видимому, сохраняет связанность с рибосомой, при том что известные рилизинг-факторы не могут гидролизовать стабильную амидную связь между РНК и белковым компонентами.
И классическая модель элонгации Уотсона (Watson, 1964, Bull. Soc. Chim. Biol, 46, 1399), и модель промежуточных статусов (Moazed & Noller, 1989, Nature, 342, 142) предполагают обязательным, чтобы А-сайт был пустым для попадания пуромицина в пептидилтрансферазный центр. Для того чтобы пуромицин попадал в А-сайт, линкер должен либо образовывать петлю вокруг наружной части рибосомы, либо проходить напрямую от декодирующего сайта через А-сайт в пептидилтрансферазный центр. Данные, приведенные здесь, не позволяют точно разграничить эти альтернативные варианты, потому что самые короткие из тестированных линкеров (21 нуклеотид) оказываются достаточно длинными для того, чтобы проходить вокруг внешней части рибосомы. В некоторых известных моделях структуры рибосом (Frank et al., 1995, Nature, 376, 441) мРНК перемещается по каналу, который протягивается от одной из сторон декодирующего сайта: т.е. в случаях непроникновения линкера через этот канал это становится необходимым для обеспечения пуромицину достижения пептидилтрансферазного центра через А-сайт.
Перенос новообразующейся полипептидной цепи на пуромицин, по-видимому, является относительно медленным по сравнению с процессом элонгации, на что указывает однотипность и длина пептида, присоединенного к линкеру. Если пуромицин эффективно конкурирует с разными аминоацил-тРНК в ходе элонгации, комплексы пептида и линкера, имеющиеся в составе гибридного продукта, предположительно должны быть разнородными по размеру. Кроме того, рибосома, по-видимому, прочитывает линкерную последовательность, что определяется сходством величин подвижности в геле между гибридом "метионин-матрица" и немодифицированным линкером. dА3n кодирует (лизин)n, который способен снижать подвижность линкера. Низкая скорость выхода мРНК может объяснять низкий темп образования гибрида по сравнению со скоростью транслокации. Предварительные результаты подтверждают, что количество образующегося гибридного продукта существенно увеличивается вследствие удлиненной посттрансляционной инкубации при низкой температуре, по-видимому, потому, что увеличенное время нужно для переноса новообразующегося полипептида на пуромицин.
ПОДРОБНОЕ ОПИСАНИЕ МАТЕРИАЛОВ И МЕТОДОВ
Нижеследующее описание представляет подробную характеристику материалов и методов, связанных с осуществлением трансляции in vitro и тестирования РНК-пептидных гибридов, включая гибриды, в составе которых имеется эпитоп-метка myc.
Последовательности. Группа олигонуклеотидов была использована, как это отмечено выше, для формирования РНК-пептидных гибридов. Эти олигонуклеотиды характеризуются следующими нуклеотидными последовательностями:
НАЗВАНИЕ ПОСЛЕДОВАТЕЛЬНОСТЬ
30-Р : 5'-ААА ААА ААА ААА ААА ААА ААА ААА ААА ССР (SEQ ID NO:8)
13-Р : 5'-ААА ААА ААА АСС Р (SEQ ID No:9)
25-Р : 5'-CGC GGT TTT TAT TTT TTT TTT TCC P (SEQ ID NO:10)
43-Р : 5'-rGrGrA rGrGrA rCrGrA rArArU rGAA ААА ААА ААА ААА ААА ААА ААА ААА АСС Р (SEQ ID NO:11)
43-Р [CUG] : 5'-rGrGrA rGrGrA rCrGrA rArCrU rGAA ААА ААА ААА ААА ААА ААА ААА ААА АСС Р (SEQ ID NO:12)
40-Р : 5'-rGrGrA rGrGrA rCrGrA rArCrU rGAA ААА ААА ААА AAA AAA AAA AAA ACC P (SEQ ID NO:13)
37-Р : 5'-rGrGrA rGrGrA rCrGrA rArCrU rGAA AAA AAA AAA AAA AAA AAA ACC P (SEQ ID NO:14)
34-Р : 5'-rGrGrA rGrGrA rCrGrA rArCrU rGAA AAA AAA AAA AAA AAA ACC P (SEQ ID NO:15)
31-Р : 5'-rGrGrA rGrGrA rCrGrA rArCrU rGAA AAA AAA AAA AAA ACC P (SEQ ID NO:16)
LP77 : 5'-rGrGrG rArGrG rArCrG rArArA rUrGrG rArArC rArGrA rArArC rUrGrA rUrCrU rCrUrG rArArG rArArG rArCrC rUrGrA rArC AAA AAA AAA AAA AAA AAA AAA ААА ААА ССР (SEQ ID NO:l)
LP154 : 5'-rGrGrG rArCrA rArUrU rArCrU rArUrU rUrArC rArArU rUrArC rA rArUrG rGrCrU rGrArA rGrArA rCrArG rArArA rCrUrG rArUrC rUrCrU rGrArA rGrArA rGrArC rCrUrG rCrUrG rCrGrU rArArA rCrGrU rCrGrU rGrArA rCrArG rCrUrG rArArA rCrArC rArArA rCrUrG rGrArA rCrArG rCrUrG rCrGrU rArArC rUrCrU rUrGrC rGrCrU ААА ААА ААА ААА ААА ААА ААА ААА ААА ССР (SEQ ID NO:3)
LP160 : 5'-rGrGrG rArCrA rArUrU rArCrU rArUrU rUrArC rArArU rUrArC rA rArUrG rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rNrNrS rCrArG rCrUrG rCrGrU rArArC rUrCrU rUrGrC rGrCrU ААА ААА ААА ААА ААА ААА ААА ААА ААА ССР (SEQ ID NO:17)
Все перечисленные олигонуклеотиды приведены в направлении 5'→3'. Рибонуклеотидные основания определены маленькой буквой "r" перед обозначением собственно нуклеотида; Р - пуромицин; rN обозначает равновероятную встречаемость rA, rG, rC и rU; rS обозначает равновероятную встречаемость rG и rC; все остальные обозначения оснований соответствуют олигонуклеотидам ДНК.
Химические реактивы. Пуромицин/HCl, модифицированное длинноцепочечным алкиламином пористое стекло, гоугеротин, хлорамфеникол, вирджиниамицин, DMAP, диметилтритилхлорид и уксусный ангидрид были получены от Sigma Chemical (St. Louis, МО). Пиридин, диметилформамид, толуол, янтарный ангидрид и паранитрофенол были получены от Fluka Chemical (Ronkonkoma, NY). мРНК (3-глобина была получена от Novagen (Madison, WI). РНК TMV была получена от Boehringer Mannheim (Indianapolis, IN).
Ферменты. Протеиназа-К была получена от Promega (Madison, WI). Очищенная от ДНКазы РНКаза была получена согласно методике Sambrook et al. (цитировано выше) или закуплена у Boehringer Mannheim. Полимераза Т7 была получена в соответствии с опубликованной методикой Гродберга и Дунна (Grodberg & Dunn, 1988, J. Bacteriol., 170, 1245) с модификациями по Завадски и Гроссу (Zawadzki & Gross, 1991, Nucl. Acids Res., 19, 1948). ДНК-лигаза Т4 была получена от New England Biolabs (Bevely, MA).
Количественный анализ включения изотопной метки. Для анализа радиоактивных гелевых бэндов определения количества радиоактивной метки (35'S или32P), присутствующего в каждом бэнде, был применен количественный анализ с помощью либо блот-анализатора Betagen-603 (Betagen, Waltham, MA), либо с использованием фосфороимиджевых пластинок (Molecular Dynamics, Sunnyvale, CA). Для жидких и твердых образцов количество присутствующей радиоактивной метки (35S или32P) было определено с помощью сцинтиляционного счетчика (Beckman, Columbia, MD).
Имиджевый анализ гелей. Имиджевый анализ гелей проводили методом авторадиографии (с использованием фотопленки Kodak XAR) или с помощью фосфороимиджевых пластинок (Molecular Dynamics).
Синтез CPG-пуромицина. Подробное описание методики синтеза CPG-пуромицина было приведено выше.
Реакции, контролируемые ферментами. В целом, приготовление нуклеиновых кислот для воздействия киназой, транскрипции, ПЦР-амплификации и трансляции с использованием экстрактов клеток Е.coli было одинаковым. Каждая пропись приготовления начиналась с эктракции с использованием равных объемов фенола и хлороформа с последующим центрифугированием и выделением водной фазы. Добавляли ацетат натрия (рН=5,2) и спермидин до конечной концентрации 300 мМ и 1 мМ, соответственно, а образец осаждали добавлением 3 объемов 100%-ного этанола при инкубации при -70°С в течение 20 минут. Пробы центрифугировали при более чем 12000д, удаляли надосадочную фракцию и центрифугат промывали избытком 95%-ного этанола при 0°С. Полученные в результате гранулы затем высушивали в условиях вакуума и ресуспендировали.
Олигонуклеотиды. Все синтетические ДНК и РНК были синтезированы на синтезаторе Millipore Expedite с применением стандартных химических приемов для каждой из нуклеиновых кислот в соответствии с рекомендациями производителя (Milligen, Bedford, MA). Олигонуклеотиды, включающие 3'-концевой пуромицин, были синтезированы с использованием CPG-пуромициновых колонок, загруженных 30-50 мг твердой подложки (примерно 20 мкмоль пуромицина на 1 г). Олигонуклеотиды, включающие 3'-концевой биотин, были синтезированы с использованием колонок с 1 мкмоль Bioteg-CPG (Glen Research, Sterling, VA). Олигонуклеотиды, включающие 5'-концевой биотин, были синтезированы путем внесения Bioteg-фосфорамидита (Glen Research) в качестве 5'-нуклеотида. Олигонуклеотиды, предназначенные для лигирования на 3'-концы молекул РНК, были фосфорилированы либо химическими методами по 5'-концу (с использованием реагентов Glen Research для химического фосфорилирования) перед депротекцией, либо каталитически фосфорилированы с использованием АТФ и полинуклеотидкиназы Т4 (New England Biolabs) после депротекции. Депротекцию образцов, включающих только ДНК (плюс пуромицин или биотин с 3'-конца), осуществляли добавлением 25% NH4OH с последующей инкубацией при 55°С в течение 12 часов. Депротекцию образцов, включающих рибонуклеотиды (например, 43-Р), осуществляли добавлением этанола (25 об.%) в раствор NH4OH и инкубацией в течение 12 часов при 55° С. Депротекцию группы 2'ОН осуществляли с использованием 1М TBAF в THF (Sigma) в течение 48 часов при комнатной температуре. TBAF удаляли с использованием колонки NAP-25 Sephadex (Pharmacia, Piscataway, NJ).
После депротекции образцы ДНК и РНК очищали с использованием электрофореза в ПААГ в денатурирующих условиях с последующими отфильтровыванием или электроэлюцией из геля с использованием Elutrap (Schleicher & Schuell, Keene, NH) и обессоливанием с использованием либо колонки NAP-25 Sephadex, либо осаждения этанолом, как это было описано выше.
Конструирование ДНК myc. Были сконструированы две матрицы ДНК, включающие эпитоп-метку с-myс. Первая матрица была сформирована путем комбинирования олигонуклеотидов 64.27 (5'- GTT CAG GTC TTC TTG AGA GAT CAG TTT CTG TTC CAT TTC GTC CTC CCT ATA GTG AGT CGT ATT A -3') (SEQ ID NO:18) и 18.109 (5'- ТАА ТАС GAC ТСА СТА TAG -3') (SEQ ID NO:19). В результате транскрипции этой матрицы была получена РНК 47.1, которая кодирует полипептид MEQKLISEEDLN (SEQ ID NO:20). Лигирование РНК 47.1 на 30-Р привело к получению LP77, показанного на фиг.7А.
Вторая матрица была получена сначала как олигонуклеотид, состоящий из 99 оснований, обозначенный как RWR 99.6 и имевший следующую последовательность - 5'- AGC GCA AGA GTT ACG CAG CTG TTC CAG TTT GTG TTT CAG CTG TTC ACG ACG TTT ACG CAG CAG GTC TTC TTC AGA GAT CAG TTT CTG TTC TTC AGC CAT -3' (SEQ ID NO:21). Двухцепочечные транскрибируемые матрицы, включающие эту последовательность, были сконструированы с помощью ПЦР и олигонуклеотидов RWR 21.103 (5'- AGC GCA AGA GTT ACG CAG CTG -3') (SEQ ID NO: 22) и RWR 63.26 (5'- ТАА ТАС GAC ТСА СТА TAG GGA САА ТТА СТА TTT АСА ATT АСА ATG GCT GAA GAA CAG AAA CTG -3') (SEQ ID NO: 23) в соответствии с методикой, опубликованной ранее (Ausubel et al., цитировано выше, глава 15). Транкрипция с использованием этой матрицы привела к получению РНК, обозначенной RNA124, которая кодирует полипептид MAEEQKLISEEDLLRKRREQLKHKLEQLRNSCA (SEQ ID NО:24). Этот пептид включал последовательность, использовавшуюся для индукции моноклонального антитела 9Е10 при соединении с белком-носителем (Oncogene Science Technical Bulletin). RNA124 имела 124 нуклеотида в длину, а в результате лигирования RNA124 на 30-Р образовывало LP154, показанный на фиг.7В. RNA124 имеет следующую последовательность - 5'-rGrGrG rArCrA rArUrU rArCrU rArUrU rUrArC rArArU rUrArC rArArUrG rGrCrU rGrArA rGrArA rCrArG rArArA rCrUrG rArUrC rUrCrU rGrArA rGrArA rGrArC rCrUrG rCrUrG rCrGrU rArArA rCrGrU rCrGrU rGrArA rCrArG rCrUrG rArArA rCrArC rArArA rCrUrG rGrArA rCrArG rCrUrG rCrGrU rArArC rUrCrU rUrGrC rGrCrU-3' (SEQ ID NO:32).
Конструирование рандомизованного пула. Рандомизованный пул был сконструирован в виде единичного олигонуклеотида, состоящего из 130 оснований, обозначенного RWR 130.1. Начиная с 3'-конца, его последовательность была такой - 3'-CCCTGTTAATGATAAATGTTAATGTTAC (NNS)27 GTC GAC GCA TTG AGA TAG CGA -5' (SEQ ID NО:25). N обозначает случайным образом избираемое положение, а эта последовательность была сформирована на основе стандартной методики синтеза. S обозначает равновероятное наличие оснований dG и dC. ПЦР была осуществлена с использованием олигонуклеотидов 42.108 (5'- ТАА TAC GAC TCA СТА TAG GGA CAA ТТА СТА ТТТ АСА АТТ АСА) (SEQ ID NO:26) и 21.103 (5'- AGC GCA AGA GTT ACG CAG CTG) (SEQ ID NO:27). Транскрипция этой матрицы привела к получению РНК, обозначенной как пул 130.1. Лигирование пула 130.1 на 30-Р позволило получить пул №1 (также обозначаемый как LP160), показанный на фиг.7С.
Семь циклов ПЦР были проведены в соответствии с опубликованной ранее методикой (Ausubel et а1., цитировано выше) при следующих исключениях: (i) исходная концентрация RWR 130.1 составляла 30 нмоль; (ii) каждый праймер использовался в концентрации 1,5 мкМ, (iii) концентрации каждого дезоксирибонуклеотида составили по 400 мкМ, и (iv) концентрация использовавшейся полимеразы Taq (Boehringer Mannheim) составляла 5 ед. на 100 мкл. Двухцепочечный продукт был очищен с использованием электрофореза в ПААГ в неденатурирующих условиях и выделен с помощью электроэлюции. Количество ДНК было определено по уровню УФ-поглощения при длине волны 260 нм и уровню флуоресценции бромистого этидия в сравнении с известными стандартами.
Каталитический синтез РНК. Транскрипция двухцепочечной ДНК, полученной с помощью ПЦР, и синтетических олигонуклеотидов осуществляли так, как это было описано ранее (Milligan & Uhlenbeck, 1989, Methods Enzymol, 180, 51). Полноразмерные РНК очищали с помощью электрофореза в ПААГ в денатурирующих условиях, выделяли путем электроэлюции и обессоливали в соответствии с описанным выше. Концентрацию пула РНК оценивали с использованием коэффициента экстинкции света 1300 OD/мкмоль (контроль): RNA124 - 1250 OD/мкмоль, РНК 47.1 - 480 OD/мкмоль. Транскрипция пула двухцепочечной ДНК позволила получить примерно 90 нмоль пула РНК.
Каталитический синтез РНК-пуромициновых комплексов. Лигирование последовательностей myc и пула мРНК на содержащие пуромицин олигонуклеотиды было осуществлено с использованием ДНК-"шплинта", обозначенного 19.35 (5'-ТТТ ТТТ ТТТ TAG CGC AAG A) (SEQ ID NO:28) с применением процедуры, аналогичной описанной Муром и Шарпом (Moore & Sharp, 1992, Science, 250, 992). Реакционная смесь включала мРНК, "шплинт" и пуромицинолигонуклеотид (30-Р, dA27 dCdCP) в молярном соотношении 0,8:0,9:1,0 и 1-2,5 ед. ДНК-лигазы из расчета на 1 пкмоль пула мРНК. Реакцию проводили в течение одного часа при комнатной температуре. При конструировании пула РНК-гибридов использовали концентрацию мРНК около 6,6 мкмоль. После лигирования комплекс РНК-пуромицина готовили так же, как было выше описано для каталитических реакций. Преципитат ресуспендировали и полноразмерные гибриды очищали с помощью электрофореза в ПААГ в денатурирующих условиях и выделяли путем электроэлюции так, как это было описано выше. Концентрацию пула РНК оценивали с использованием коэффициента экстинкции света 1650 OD/мкмоль и матрицы myc (1600 OD/мкмоль). Таким образом было получено 2,5 нмоль данного комплекса.
Приготовление стрептавидинагарозы-dТ25. Содержащий dT25 3'-биотин (синтезированный на Bioteg-фосфорамидитных колонках (Glen Research)) инкубировали при 1-10 мкМ с суспензией стрептавидинагарозы (50% агарозы по объему: Pierce, Rockford, IL) в течение 1 часа при комнатной температуре в ТЕ (10 мМ Трис-HCl, рН 8,2, 1 мМ EDTA) и промывали. Связывающую способность агарозы оценивали затем визуально по исчезновению из раствора биотина-dТ25 и(или) путем титрования смолы с известным количеством комплементарного олигонуклеотида.
Реакция трансляции с использованием производных от E.coli экстрактов и рибосом. В целом, реакции трансляции были осуществлены с использованием закупленных реагентов (например, эктракт S30 E.coli для линейных матриц: Promega, Madison, WI). Однако, Е.coli штамма MRE600 (полученную из АТСС, Rockville, MD) также использовали для получения экстрактов S30, приготовливаемых в соответствии с опубликованными методиками (например, Ellman et al., 1991, Methods Enzymol., 202, 301), равно как и фракции рибосом готовили по Кудлицки с соавт. (Kudlicki et al., 1992, Anal Biochem., 206, 389). Стандартную реакцию проводили в объеме 50 мкл при интенсивности излучения в 20-40 мкКи использовавшегося в качестве маркера25S-метионина. Реакционная смесь включала 30 об.% эктракта, 9-18 мМ MgCl2, 40 об.% премикса без метионина (Promega) и 5 мкМ матрицы (например, 43-Р). Для экспериментов по одновременной инкубации олигонуклеотиды 13-Р и 25-Р добавляли в концентрации 5 мкМ. Для экспериментов с использованием рибосом 3 мкл рибосомного раствора добавляли в реакцию вместо лизата. Все реакции осуществляли при инкубации при 37°С в течение 30 минут. Матрицы очищали так, как это было описано выше для каталитических реакций.
Реакции трансляции в зародышах пшеницы. Реакции трансляции, изображенные на фиг.8, осуществляли с использованием закупленных реагентов, лишенных метионина (Promega), в соответствии с рекомендациями производителя. Концентрации матрицы составили 4 мкМ для 43-Р и 0,8 мкМ для LP77 и LP154. Реакции осуществляли при 25°С в общем объеме 25 мкл при интенсивности излучения35S-метионина в 30 мкКи.
Реакции трансляции в ретикулоцитах. Реакции трансляции были осуществлены либо с использованием закупленных реагентов (Novagen, Madison, WI), либо с использованием экстракта, приготовленного в соответствии с опубликованными методиками (Jackson & Hunt, 1983, Methods Enzynol, 96, 50). Богатую ретикулоцитами кровь получали от Pel-Freez Biologicals (Rogers, AK). В обоих случаях условия реакции были такими, какие рекомендуются для использования при работе с Red Nova Lysate (Novagen). Реакционные смеси содержали 100 мМ KC1, 0,5 мМ MgOAc, 2 мМ DTT, 20 мМ HEPES - pH 7,6, 8 мМ креатинфосфата, 25 мкМ каждой из аминокислот (за исключением метионина, если используется35S-метионин) и 40 об.% лизата. Инкубацию проводили в течение 1 часа при 30°С. Концентрации матриц зависели от условий эксперимента, но в целом варьировались в пределах от 50 нМ до 1 мкМ за исключением 43-Р (фиг.6Н), концентрация которой составила 4 мкМ.
Для формирования рандомизованного пула брали 10 мл реакционной трансляционной смеси при концентрации матрицы примерно 0,1 мкМ (1,25 наномоль матрицы). Дополнительно32P-меченную матрицу включали в реакцию для обеспечения возможности определения количества материала, имеющегося на каждом из этапов очистки и процедуры отбора. После трансляции при 30°С в течение 1 часа реакционную смесь охлаждали на льду в течение 30-60 минут.
Выделение гибрида с помощью стрептавидинагарозы-dТ25. После инкубации трансляционную реакционную смесь разбавляли примерно 150-кратно изоляционным буфером (1,0 М NaCl, 0,1 М Трис-HCl рН 8,2, 10 мМ EDTA, 1 мМ DTT), содержащим более чем 10-кратный молярный избыток dТ25-биотин-стрептавидинагарозы, в которой концентрация dT25 была примерно 10 мкМ (объем суспензии такой же или больший, чем объем лизата), инкубировали при перемешивании в течение одного часа при 4°С. Затем удаляли агарозу из смеси путем фильтрации (фильтры Millipore ultrafree MC) или центрифугирования и промывали холодным изоляционным буфером 2-4 раза. Затем матрицу высвобождали из стрептавидинагарозы-dТ25 путем повторного промывания аликвотами по 50-100 мкл 15 мМ NaOH, 1 мМ EDTA. Полученный элюент немедленно нейтрализовали в 3 М NaOAc pH 5,2, 10 мМ спермидина и осаждали этанолом. Для реакции пула общий уровень радиоактивности показал, что было выделено примерно 50-70% внесенной матрицы.
Выделение гибрида с использованием тиопропилсефарозы. Гибриды, включающие цистеин, могут быть очищены с использованием тиопропилсефарозы-6В, как показано на фиг.13 (Pharmacia). В описанных здесь экспериментах выделение проводили либо напрямую в трансляционной реакционной смеси или после исходного выделения гибрида (например, с использованием стрептавидинагарозы). Для образцов, очистка которых проводилась напрямую, использовали объемное соотношение лизата и сефарозы 1:10. Для пула использовали 0,5 мл сефарозной суспензии с целью выделения всего гибридного материала из 5 мл реакционной смеси. Образцы были разбавлены в объемном соотношении 50:50 суспензией тиопропилсефарозы в 1х ТЕ рН 8,2 (10 мМ Трис-HCl, 1 мМ EDTA, рН 8,2), включающей свободную от ДНКазы РНКазу (Boehringer Mannheim), и инкубировали при вращении в течение 1-2 часов при 4°С до завершения реакции. Избыток жидкости удаляли и сефарозу промывали несколько раз изоляционным буфером, включающим 20 мМ DTT, и выделяли путем центрифугирования или фильтрации. Гибриды элюировали из сефарозы с использованием раствора 25-30 мМ дитиотрейтола (DTT) в 10 мМ Трис-НСl рН 8,2, 1 мМ EDTA. Затем гибриды концентрировали путем сочетания выпаривания в высоком вакууме и осаждения этанолом в соответствии с описанным выше. Для реакции пула общая выделенная радиоактивность показала, что примерно 1% всего количества матрицы оказалось конвертированной в гибрид.
Для некоторых вариантов dT25 добавляли к данному элюату и вращали в течение 1 часа при 4°С. Агарозу промывали трижды изоляционным буфером, выделяли с помощью фильтрации и элюировали связанный материал так, как описано выше. Добавляли тРНК и гибридные продукты осаждали этанолом. Образец ресуспендировали в буфере ТЕ рН 8,2, включающем свободную от ДНКазы РНКазу-А, с целью удаления РНК-компонента матрицы.
Реакции иммунопреципитации. Иммунопреципитацию пептидов из трансляционных реакционных смесей (фиг.10) проводили путем смешения 4 мкл ретикулоцитарной трансляционной реакции, 2 мкл нормальной мышиной сыворотки и 20 мкл белка-G + А-агарозы (Calbiochem, La Jolla, CA) с 200 мкл либо фосфатного буфера PBS (58 мМ Nа2НРO4, 17 мМ NaH2PO4, 68 мМ NaCl), разбавляющего буфера (10 мМ Трис-НСl рН 8,2, 140 мМ NaCl, 1% по объему тритон Х-100) или PBSTDS (PBS + 1% Тритон Х-100, 0,5% дезоксихолата, 0,1% SDS). Затем образцы перемешивали вращением в течение одного часа при 4°С с последующим центрифугированием при 2500 об/мин в течение 15 минут. Элюент удаляли и добавляли 10 мкл моноклонального антитела 9Е10 к c-myc (Calbiochem, La Jolla, CA) и 15 мкл белка-G + А-агарозы и перемешивали вращением в течение 2 часов при 4°С. Образцы затем промывали двумя объемами по 1 мл PBS, разбавляющего буфера или PBSTDS. К полученной смеси добавляли 40 мкл гель-загрузочного буфера (Calbiochem Product Bulletin) и 20 мкл были загружены в денатурирующих условиях для электрофореза в ПААГ в соответствии с описанным у Шаггера и фон Ягова (Schagger & von Jagow, 1987, Anal. Biochem., 166, 368).
Иммунопреципитацию гибридов (как показано на фиг.11) осуществляли путем смешения 8 мкл ретикулоцитарной трансляционной реакционной смеси с 300 мкл разбавляющего буфера (10 мМ Трис-НСl рН 8,2, 140 мМ NaCl, 1 об.% Тритон X-100), 15 мкл белка-G-сефарозы (Sigma) и 10 мкл (1 мкг) антитела 9Е10 к c-myc (Calbiochem) с последующим перемешиванием вращением в течение нескольких часов при 4°С. После выделения образцы промывали, обрабатывали свободной от ДНКазы РНКазой, помечали с использованием полинуклеотидкиназы и32P-γ-ATP и разделяли с применением электрофореза в ПААГ с денатурацией мочевиной (фиг.11).
Обратная транскрипция пула гибридов. Реакции обратной транскрипции осуществляли в соответствии с рекомендациями производителя (Superscript II), за исключением того, что матрицу, воду и праймер инкубировали при 70°С в течение только двух минут (Gibco BRL, Grand Island, NY). В некоторые реакции включали 50 мкКи α-32P-dCTP с целью отслеживания наращивания цепи; в других реакциях обратную транскрипцию контролировали с использованием помеченных32P по 5'-концу праймеров, приготовленных с использованием32Р-α-АТР (New England Nuclear, Boston, MA) и полинуклеотидкиназы Т4 (New England Biolabs, Beverly, MA).
Приготовление белка-G и сефарозы с антителом. Две аликвоты по 50 мкл суспензии сефарозы с белком-G (50% твердого вещества по объему) (Sigma) промывали разбавляющим буфером (10 мМ Трис-НСl рН 8,2, 140 мМ NaCl, 0,025% NaN3, 1 объемный % Тритон Х-100) и выделяли с помощью центрифугирования. Первую аликвоту резервировали для использования в качестве предварительной колонки до формирования селективного матрикса. После ресуспендирования второй аликвоты в разбавляющем буфере добавляли 40 мкг моноклонального антитела АВ-1 к c-myc (Oncogene Science) и полученную реакционную смесь инкубировали в течение ночи при 4°С при перемешивании вращением. Затем очищали сефарозу с антителом с помощью центрифугирования в течение 15 минут при 1500-2500 об/мин на микроцентрифуге и промывали 1-2 раза разбавляющим буфером.
Отбор. После выделения гибрида и синтеза комплементарной цепи в процессе отбора напрямую использовали полную реакцию обратной транскрипции. В данном варианте охарактеризованы две методики. В первом раунде обратно-транскриптазную реакционную смесь добавляли непосредственно к сефарозе с антителом, приготовленной так, как описано выше, с последующей инкубацией в течение 2 часов. Для следующих раундов реакционную смесь инкубировали примерно 2 часа с промытой белок-С-сефарозой перед помещением на колонку с антителом с целью уменьшения количества связывающих элементов, более активно взаимодействующих с белком-G, чем с иммобилизованным антителом.
Для элюции пула из матрикса может быть использовано несколько подходов. Первый из них связан с промыванием селективного матрикса 4%-ной уксусной кислотой. Эта процедура позволяет высвободить белок из состава матрикса. С другой стороны, более жесткое промывание (например, с использованием мочевины или иного денатурирующего агента) может быть использовано вместо или в дополнение к методике, связанной с использованием уксусной кислоты.
ПЦР с выбранными гибридами. Отобранные молекулы амплифицируют с помощью ПЦР с использованием стандартных протоколов, как это было описано выше в разделе конструирования пула.
СИНТЕЗ И ТЕСТИРОВАНИЕ ГИБРИДОВ β-ГЛОБИНА
Для синтезирования гибридных конструкций с β-глобином была сформирована β-глобиновая кДНК на основе 2,5 мкг глобиновой мРНК с применением метода обратной транскрипции при 200 пкмоль затравки 18.155 (5'-GTG GTA TTT GTG AGC CAG) (SEQ ID NO:29) и с обратной транскриптазой Superscript (Gibco BRL) в соответствии с рекомендуемым производителем протоколом. Последовательность затравки была комплементарна 18 нуклеотидам участка β-глобинового гена в 5'-сторону от стоп-кодона. Для добавления промотора Т7 20 мкл обратно-транскрипционной реакционной смеси выделяли и подвергали 6 циклам ПЦР с затравками 18.155 и 40.54 (5'- ТАА ТАС GAC ТСА СТА TAG GGA CAC TTG CTT TTG АСА САА С) (SEQ ID NO:30). Полученная в результате мРНК "синтетического β-глобина" затем была сформирована путем транскрипции под контролем полимеразы Т7 в соответствии с методом Миллигана-Оленбека (Milligan & Uhlenbeck, 1989, Methods Enzymol, 180, 51): эту РНК очищали в геле, подвергали электроэлюции и обессоливали так, как это было описано выше. Затем "LP-β-глобин" был получен на базе конструкцуии "синтетического β-глобина" путем лигирования этой конструкции на 30-Р в соответствии с методом Мура-Шарпа (Moore & Sharp, 1992, Science, 256, 992) с использованием затравки 20.262 (5'-ТТТ ТТТ ТТТ Т GTG СТА ТТТ G) (SEQ ID NO:31) в качестве "шплинта". Продукт лигирования затем очищали в геле, подвергали электроэлюции и обессоливали так, как это было описано выше. Концентрацию конечного продукта определяли по поглощению при 260 нм.
Эти β-глобиновые матрицы затем транслировали in vitro в соответствии с описанным в таблице 1 при общем объеме 25 мкл в каждом случае. Mg2+ добавляли из базового 25 мМ раствора. Все реакции осуществляли путем инкубации при 30°С в течение 1 часа с последующим помещением при температуре -20°С на ночь. Затем СРМ-осаждаемость dT25 определяли дважды, используя по 6 мкл лизата и усредненный "минус-фон".
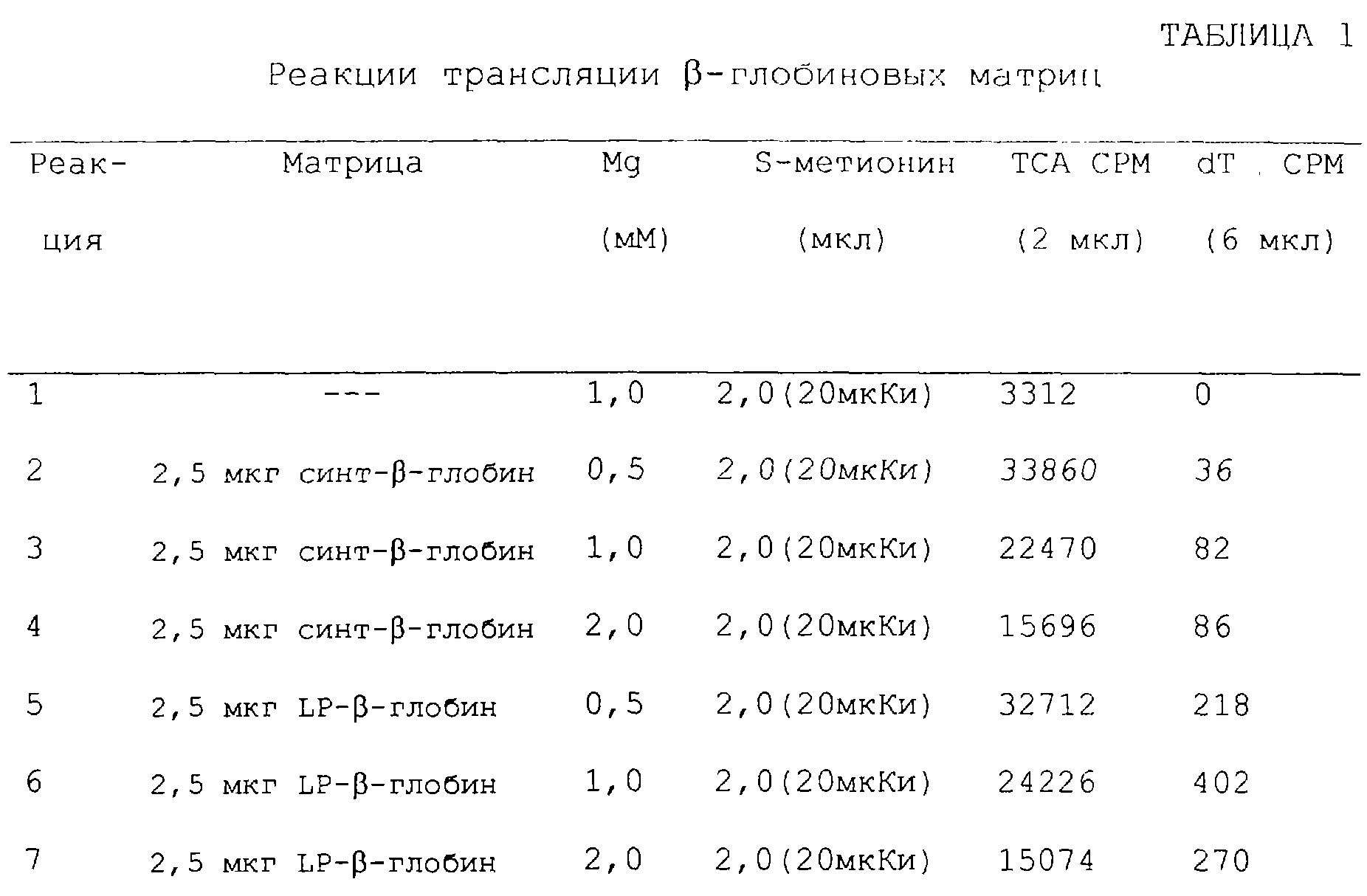
Для приготовления образцов для гель-анализа по 6 мкл каждой трансляционной реакционной смеси смешивали с 1000 мкл изоляционного буфера (1 М NaCl, 100 мМ Трис-HCl рН 8,2, 10 мМ EDTA, 0,1 мМ DTT), 1 мкл РНКазы-А (свободный от ДНКазы препарат: Boehringer Mannheim) и 20 мкл 20 мкМ dТ25-стрептавидинагарозы. Образцы инкубировали при 4°С в течение одного часа при перемешивании. Избыток изоляционного буфера удаляли, и образцы помещали на миллипоровые фильтры МС для удаления остатков изоляционного буфера. Образцы затем промывали четырежды в 50 мкл воды и дважды в 50 мкл 15 мМ NaOH, 1 мМ EDTA. Образец (300 мкл) нейтрализовали 100 мкл ТЕ рН 6,8 (10 мМ Трис-HCl, 1 мМ EDTA), добавляли 1 мкл РНКазы-А (1 мг/мл) (как описано ранее) и образцы инкубировали при 37°С. Затем добавляли дважды 10 мкл SDS-загрузочного буфера (125 мМ Трис-HCl рН 6,8, 2% SDS, 2% β-меркаптоэтанола, 20% глицерина, 0, 001% бромфенолового синего), и образец лиофилизировали до высыхания и ресуспендировали в 20 мкл воды и 1% β-меркаптоэтанола. Образцы затем загружали в пептидо-анализаторный гель по методу Шаггера-фон Ягова (Schagger & von Jagow, 1987, Anal. Biochem., 166, 368) и визуализировали с помощью авторадиографии.
Результаты этих экспериментов показаны на фиг.15А и 15В. Как представлено на фиг.15А,35S-метионин инкорпорировался в состав белкового компонента гибридных "синт-β-глобина" и "LP-β-глобина". Белок был неоднородным, однако самый мощный бэнд соответствовал по величине той подвижности, которая ожидается для мРНК β-глобина. Также, как показано на фиг.15В, после выделения в dТ25-агарозе и обработки РНКазой-А меченный35S-метионином материал в линиях "синт-β-глобина" не сохранялся (фиг.15В, линии 2-4). Напротив, в линиях LP-P-глобина был выявлен однородный по размерам продукт, помеченный35S.
Эти результаты показали, что, как и в предыдущих опытах, гибридный продукт был выделен с помощью олигонуклеотидной аффинной хроматографии только тогда, когда матрица включает пуромицин по своему 3'-концу. Это было подтверждено при использовании сцинтилляционного счетчика (см. таблицу 1). Полученный материал, как ожидается, включает линкер 30-Р, присоединенный к некоторой части β-глобина. Гибридный продукт, по-видимому, характеризуется размерной однородностью, на что указывают данные гель-анализа. Однако, поскольку полученный гибридный продукт характеризуется подвижностью, очень сходной с таковой у нативного β-глобина (контрольные линии на фиг.15А и 15В), то весьма затруднительно определить точную длину белкового компонента данного гибридного продукта.
ДАЛЬНЕЙШАЯ ОПТИМИЗАЦИЯ ОБРАЗОВАНИЯ РНК-ПЕПТИДНОГО ГИБРИДА
Были обнаружены некоторые факторы, которые способны обеспечивать повышение эффективности образования РНК-пептидных гибридов. Образование гибридов - суть перенос новообразующейся полипептидной цепи от ее тРНК на пуромициновую составляющую, находящуюся на 3'-конце данной мРНК - является достаточно медленной реакцией, которая следует за исходной относительно быстрой реакцией трансляции открытой рамки считывания, сопровождающейся образованием новой полипептидной цепи. Темп образования гибрида может быть существенно увеличен путем посттрансляционной инкубации в условиях повышенного содержания Mg2+ предпочтительно в диапазоне 50-100 мМ) и(или) путем использования более гибкого линкера, соединяющего мРНК и пуромициновый остаток. Кроме того, продолжительная инкубация (12-48 часов) при низкой температуре (предпочтительно при -20°С) также приводит к увеличению выхода гибридов при меньшей интенсивности деградации мРНК, чем это характерно для инкубации при 30°С. Путем комбинирования этих факторов вплоть до 40% внесенной мРНК может быть в конечном счете преобразовано в мРНК-пептидные гибридные продукты, как это показано далее.
Синтез комплексов "мРНК-пуромицин". В данных оптимизующих экспериментах содержащие пуромицин олигонуклеотидные линкеры лигировали на 3'-концы мРНК с использованием ДНК-лигазы фага Т4 в присутствии комплементарных ДНК-"шплинтов", в принципе так же, как это было описано выше. Поскольку ДНК-лигаза Т4 предпочтительно обеспечивает точное соответствие пар оснований вблизи точки лигирования, а продукты транскрипции при участии РНК-полимераз Т7, ТЗ или SP6 обычно оказываются неоднородными по своим 3'-концам (Nucleic Acids Research, 1987, 15, 8783), то эффективное лигирование было проведено только на тех РНК, которые включали правильные 3'-концевые нуклеотиды. Когда был использован стандартный ДНК-"шплинт", примерно 40% транскрипционного продукта оказывалось лигировано на пуромицин-олигонуклеотид. Количество лигационного продукта было увеличено с использованием избытка РНК, однако такого увеличения не происходило при использовании избытка пуромицин-олигонуклеотида. Вне связи с какой-либо теорией, предполагается, что ограничивающим фактором для реакции лигирования являлось количество РНК, которая была полностью комплементарной соответствующему участку ДНК-"шплинта".
Для проведения лигирования тех транскриптов, которые заканчиваются дополнительными не соответствующими матрице нуклеотидами по 3'-концу (обозначаемыми как "продукты N+1") использовали смесь стандартного ДНК-"шплинта" с новым ДНК-"шплинтом", включающим дополнительное, случайным образом выбираемое основание в сайте лигационного соединения. Эффективность лигирования увеличивалась до более чем 70% в варианте с РНК-матрицей myc (т.е. в варианте RNA124) в присутствии такого смешанного ДНК-"шплинта".
Дополнительно к этому подходу, связанному с использованием модифицированного ДНК-"шплинта" эффективность формирования комплекса мРНК-пуромицина могла быть далее оптимизирована путем принятия во внимание еще трех факторов. Во-первых, предпочтительно конструируют или используют такие мРНК, которые лишены 3'-концов, имеющих какие-либо существенные стабильные вторичные структуры, которые могли бы приводить к помехам из-за отжига на олигонуклеотиде "шплинта". Кроме того, поскольку высокая концентрация солей иногда обусловливала блокировку реакции лигирования, предпочтительным оказывалось включение в используемую процедуру в качестве одного из существенных этапов этапа обессоливания олигонуклеотидов с использованием колонок NAP-25. Наконец, поскольку реакция лигирования являлась относительно быстрой и обычно в целом завершалась за 40 минут при комнатной температуре, не должны использоваться существенно более длительные периоды инкубации, что часто приводило бы к необязательной деградации конкретной РНК.
С учетом определенных выше условий комплексы мРНК-пуромицина были синтезированы следующим образом. Лигирование последовательности РНК myc (RNA124) на пуромицинсодержащий олигонуклеотид осуществляли с использованием либо стандартного ДНК-"шплинта" (например, 5'-ТТТТТТТТТТА-GCGCAAGA) или "шплинта", включающего случайным образом выбираемое основание (N) в сайте лигационного соединения (например, S'-TTTTTTTTTTN-AGCGCAAGA). Реакционные смеси включали мРНК, ДНК-"шплинт" и пуромицин-олигонуклеотид в молярном соотношении 1,0:1,5-2,0:1,0. Смесь этих компонентов сначала нагревали до 94°С на 1 минуту и затем охлаждали на льду в течение 15 минут. Реакцию лигирования проводили в течение одного часа при комнатной температуре в 50 мМ Трис-НС1 (рН 7,5), 10 мМ MgCl2, 10 мМ DTT, 1 мМ АТР, 25 мкг/мл BSA, 15 мкМ пуромицин-олигонуклеотида, 15 мкМ мРНК, 22,5-30,0 мкМ ДНК-"шплинта", ингибитора РНКазы (Promega) в концентрации 1 ед./мл и 1,6 ед. ДНК-лигазы фага Т4 на 1 пкМ пуромицин-олигонуклеотида. После инкубации добавляли EDTA до достижения конечной концентрации 30 мМ и реакционную смесь экстрагировали смесью фенола и хлороформа. Полноразмерные комплексы очищали с помощью электрофореза в денатурирующем ПААГ и выделяли с помощью электроэлюции.
Основные условия трансляции в ретикулоцитах. В дополнение к улучшению синтеза комплексов мРНК-пуромицина реакции трансляции были также оптимизированы следующим образом. Реакции проводили в лизатах ретикулоцитов кролика, происходящих из разных коммерческих источников (Novagen, Madison, WI; Amersham, Arlington Heights, IL; Boehringer Mannheim, Indianapolis, IN; Ambion, Austin, TX и Promega, Madison, WI). Типовая реакционная смесь (при конечном объеме 25 мкл) включала 20 мМ HEPES (рН 7,6), 2 мМ дитиотрейтола, 8 мМ креатинфосфата, 100 мМ КС1, 0,75 мМ Мg(ОАс)2, 1 мМ АТФ, 0,2 мМ ГТФ, 25 мкМ каждой аминокислоты (0,7 мкМ метионина, если используется35S-метионин), ингибитор РНКазы в концентрации 1 ед./мл и 60 об,% лизата. Конечные концентрации матрицы были в пределах от 50 до 800 нМ. Для каждой инкубации все компоненты реакционной смеси, за исключением лизата, тщательно перемешивали на льду, а замороженный лизат растаивали непосредственно перед его использованием. После добавления лизата реакционную смесь тщательнейшим образом перемешивали, используя мягкую пипетку, и инкубировали при 30°С до начала трансляции. Оптимальные концентрации Мg2+и К+ варьировались в пределах 0,25-2 мМ и 75-200 мМ соответственно, в зависимости от различных мРНК, и предпочтительно определялись в предварительных пилотных экспериментах. В частности, для плохо транслируемых мРНК иногда также проводили оптимизацию концентраций гемина, креатин-фосфата, тРНК и аминокислот. Обычно более предпочтительным является использование хлорида калия против ацетата калия для реакций гибридизации, однако иногда лучшие результаты были получены при использовании смеси КС1 и КОАс.
После трансляции при 30°С на протяжении 30-90 минут реакционную смесь охлаждали на льду в течение 40 минут и добавляли Мg2+. Конечная концентрация Mg2+, добавляемого на этом этапе, была также оптимизирована для различных мРНК-матриц, но обычно находилась в пределах от 50 до 100 мМ (при том, что 50 мМ является предпочтительной концентрацией, используемой для пулов смешанных матриц). Полученную в результате смесь инкубировали при -20°С в течение 16-48 часов. Для визуализации помеченных гибридных продуктов 2 мкл реакционной смеси смешивали с 4 мкл загрузочного буфера и полученную смесь нагревали до 75°С на 3 минуты. Полученную в результате смесь затем загружали в SDS-ПААГ с 6% глицина (для матриц, помеченных32P) или SDS-ПААГ с 8% трицина (для матриц, помеченных35S-метионином). В качестве альтернативы этому подходу гибридные продукты также могут быть выделены с использованием dТ25-стрептавидинагарозы или тиопропилсефарозы (или их обеих), в принципе так же, как это было описано.
Для удаления РНК-компонента из состава комплекса "РНК-линкер-пуромицин-пептид" для последующего анализа с помощью электрофореза в SDS-ПААГ подходящее количество EDTA добавляли после посттрансляционной инкубации, а реакционную смесь обессоливали с использованием колонки microcon-10 (или microcon-30). 2 мкл полученной в результате смеси (при ее общем количестве примерно 25 мкл) смешивали с 18 мкл буфера РНКазы-Н (30 мМ Трис-НСl рН 7,8, 30 мМ (NH4)2SO4, 8 мМ MgCl2, 1,5 мМ β-меркаптоэтанола и подходящее количество комплементарного ДНК-"шплинта"), и полученную смесь инкубировали при 4°С в течение 45 минут. Затем добавляли РНКазу-Н и расщепление проводили при 37°С в течение 20 минут.
Качество пуромицин-олигонуклеотидов. Качество пуромицин-олигонуклеотидов также является важным фактором, влияющим на эффективность получения гибридных продуктов. Соединение 5'-DMT-2'-сукцинил-N-трифторацетилпуромицина с CPG не является столь же эффективным, как соединение обычных нуклеотидов. А раз так, то реакция соединения тщательно контролировалась с целью удаления CPG со слишком низкой концентрацией присоединенного пуромицина, а непрореагировавшие аминогруппы в CPG полностью закрывали с целью предотвращения последующего синтеза олигонуклеотидов, лишенных 3'-концевого пуромицина. Также было важно предотвратить использование CPG, содержащем очень тонкозернистые частицы, поскольку это было способно создавать проблемы ввиду засорения клапанов в процессе последующих этапов автоматического синтеза олигонуклеотидов.
Кроме того, синтезированные пуромицин-олигонуклеотиды были предпочтительно протестированы до крупномасштабного применения для того, чтобы быть уверенными в присутствии пуромицина на 3'-конце. В проведенных экспериментах отсутствие гибридизации не отмечалось, если пуромицин был замещен дезоксиаденозином, включающим первичную аминогруппу на своем 3'-конце. Для проверки присутствия 3'-гидроксильных групп (т.е. нежелательного синтеза олигонуклеотидов, лишенных на 3'-конце пуромицина) пуромицин-олигонуклеотиды могут вначале быть изотопно помечены (например, путем 5'-фосфорилирования) и затем использованы в качестве затравки для удаления с использованием терминальной дезоксинуклеотидил-трансферазы. В присутствии 3'-концевой пуромициновой составляющей удлинение контролируемого продукта наблюдаться не может.
Динамика трансляции и посттрансляционной инкубации. Реакция трансляции была относительно быстрой и обычно завершалась за 25 минут при 30°С. Реакция гибридизации, однако, медленнее. Когда был использован стандартный линкер (dA27dCdCP) при 30°С, синтез гибридов достигал своего максимального уровня при добавлении 45 минут. Посттрансляционная инкубация могла быть проведена при меньших температурах, например при комнатной температуре, 0°С или -20° С. Меньшая деградация матрицы мРНК наблюдалась при -20°С, а наилучшие результаты гибридизации были получены после инкубации при -20°С в течение 2 дней.
Влияние концентрации Мg2+. Высокая концентрация Мg2+ в ходе посттрансляционной инкубации в значительной степени стимулировала формирование гибридов. Например, для описанной выше матрицы РНК myc 3-4-кратная стимуляция образования гибридов была отмечена при использовании стандартного линкера (dA27dCdCP) в присутствии 50 мМ Mg2+ в течение 16 часов инкубации при -20°С (фиг.17, сравнение линий 3 и 4).
Сходным образом эффективное образование гибридов также наблюдалось при использовании посттрансляционной инкубации в присутствии 50-100 мМ Mg2+, если реакции проводили при комнатной температуре в течение 30-45 минут.
Длина и последовательность линкера. Также была исследована зависимость реакции гибридизации от длины линкера. В диапазоне от 21 до 30 нуклеотидов (n=18-27) были выявлены лишь небольшие изменения в уровне эффективности реакции гибридизации (как это было описано выше). Более короткие линкеры (например, 13 нуклеотидов в длину) приводят к менее эффективной гибридизации. Кроме того, хотя конкретные линкеры большей длины (такие как 45-нуклеотидные и 54-нуклеотидные) также приводят к некоторому снижению эффективности гибридизации, остается вероятным, что более длинные линкеры могут быть также использованы для оптимизации эффективности реакции гибридизации.
Что касается последовательности линкера, замещение остатков дезоксирибонуклеотидов вблизи 3'-конца остатками рибонуклеотидов существенно не изменяет эффективности гибридизации. Однако последовательность dCdCP (или rСrСР) в 3'-конце конкретного линкера оказалась важной с точки зрения образования гибрида. Замещение dCdCP на dUdUP существенно снижает эффективность образования гибридов.
Гибкость линкера. Также были исследована зависимость реакции гибридизации от гибкости линкера. В этих экспериментах было определено, что эффективность гибридизации была низкой, если жесткость конкретного линкера была увеличена путем отжига с комплементарным олигонуклеотидом вблизи 3'-конца. Сходным образом, когда был использован более гибкий линкер (например, dA21C9 C9C9dAdCdCP, где С9 представляет HO(CH2CH2O)3РO2), эффективность образования гибрида существенно повышалась. В сравнении со стандартным линкером (dA27dCdCP) использование более гибкого линкера (dА21С9С9С9dAdCdCP) приводило к повышению эффективности гибридизации матрицы RNA124 более чем в 4 раза (фиг.17, сравнение линий 1 и 9). Кроме того, в противоположность матрице со стандартным линкером, посттрансляционная гибридизация которой протекает малоэффективно в отсутствие высокой концентрации Мg2+ (фиг.17, линии 3 и 4), матрица с гибким линкером не требовала увеличения концентрации ионов магния для получения хорошего выхода гибридного продукта при удлиненной посттрансляционной инкубации при -20°С (фиг.17, сравнение линий 11 и 12). Этот линкер к тому же был особенно применим в случаях, когда повышение концентрации Mg2+ не было желательным. Кроме того, гибкий линкер также обусловливал оптимальный выход гибридных молекул и в присутствии увеличенного количества Mg2+.
Количественный анализ эффективности гибридизации. Эффективность образования гибридов может быть выражена либо как доля транслированного пептида, вошедшего в состав гибрида, либо как доля внесенной матрицы, конвертировавшейся в гибридный продукт. Для определения доли транслированного пептида, вошедшего в состав гибридного продукта, было использовано мечение транслируемого пептида35 S-метионином. В этих экспериментах, когда были использованы линкеры dA27dCdCP или dА27rСrСР, примерно 3,5% транслированного пептида были гибридизованы с его мРНК спустя 1 час трансляции при 30°С. Эта величина увеличивалась до 12% после инкубации в течение ночи при -20°С. Когда посттрансляционная инкубация была осуществлена в присутствии высокой концентрации Mg2+, более чем 50% транслированного пептида было гибридизовано с соответствующей матрицей.
Для матрицы с гибким линкером приблизительно 25% транслированного белка было гибридизовано с матрицей после 1 часа инкубации при 30°С. Это значение увеличивалось до более чем 50% после инкубации в течение ночи при -20°С и еще до более чем 75% в случае, если посттрансляционная инкубация проводилась в присутствии 50 мМ Мg2+.
Для определения процента внесенной матрицы, конвертировавшейся в состав гибридного продукта, трансляцию проводили с использованием32Р-помеченной мРНК-линкерной матрицы. Когда был использован гибкий линкер и посттрансляционная инкубация проводилась при -20°С без добавления Mg2+, примерно 20, 40, 40, 35 и 20% внесенной матрицы было конвертировано в состав мРНК-пептидного гибрида, когда концентрация внесенной матрицы РНК составляла 800, 400, 200, 100 и 50 нМ соответственно (фиг.18). Сходные результаты были получены, когда посттрансляционная инкубация осуществлялась в присутствии 50 мМ Мg2+. Наилучшие результаты были достигнуты при использовании лизатов, полученных от фирм Novagen, Amersham или Ambion (фиг.19).
Различия в подвижности между мРНК и мРНК-пептидными гибридами по данным измерения с помощью SDS-электрофореза в ПААГ, могут быть очень незначительными, если конкретная матрица мРНК является длинной. В таких случаях матрица может быть помечена по 5'-концу линкера с помощью32P. Длинный РНК-компонент может быть затем расщеплен РНКазой-Н в присутствии комплементарного ДНК-"шплинта" после этапов "трансляции-инкубации", а эффективность образования гибрида определена путем количественного анализа соотношения немодифицированного линкера и линкер-пептидного гибрида. В сравнении с расщеплением РНКазой-А, которое образует 3'-Р и 5'-ОН, такой подход обладает преимуществом, связанным с тем, что32P на 5'-конце линкера не удаляется.
Внутримолекулярная или межмолекулярная гибридизация в процессе посттрансляционной инкубации. В дополнение к описанным выше экспериментам была исследована природа (внутри- или межмолекулярная) реакции гибрадазации, протекающей при -20°С в присутствии Mg2+. Свободный линкер (dА27dCdСР или dA21C9C9C9dAdCdCP, где С9 представляет О(CH2CH2O)3РO2-) инкубировали вместе с матрицей, включающей ДНК-линкер, но без пуромицина на 3'-конце, в условиях трансляции и посттрансляционной инкубации, которые были описаны выше. В этих экспериментах не обнаружено маркируемого количества (т.е. менее 2% от нормального уровня)35S-метионина, инкорпорированного в линкерпептидный гибридный продукт: это подтверждает, что посттрансляционная гибридизация встречалась преимущественно между новообразующимся полипептидом и мРНК, связанной на той же самой рибосоме.
Результаты оптимизации. Как было показано выше, при использовании гибкого линкера и(или) проведения посттрансляционной инкубации в присутствии высокой концентрации Mg2+ эффективность гибридизации увеличивалась до примерно 40% от количества внесенной мРНК. Эти результаты определили, что по крайней мере 1014 молекул мРНК-пептидных гибридов могут быть сформированы в 1 мл трансляционой реакционной смеси in vitro с получением пулов мРНК-пептидных гибридов, характеризующихся очень высоким уровнем комплексности, необходимых для использования в экспериментах по отбору in vitro.
ИЗБИРАТЕЛЬНОЕ ОБОГАЩЕНИЕ РНК-ПЕПТИДНЫХ ГИБРИДОВ
Была продемонстрирована пригодность использованных РНК-пептидных гибридов в экспериментах по отбору и преобразованию путем обогащения по конкретному РНК-пептидному гибриду из комплексного пула гибридов со случайными последовательностями на основе параметров кодируемого белка. В частности, была приготовлена серия смесей, в которых малое количество известной последовательности (в данном случае - длинной myс-матрицы LP154) было комбинировано с некоторым количеством пула случайных последовательностей (таких как LP160). Эти смеси были транслированы и полученные РНК-пептидные гибридные продукты отобраны с применением олигонуклеотид- и дисульфид-аффинной хроматографии так, как это было описано в данном тексте. Гибриды, включающие myс-матрицу, были избирательно иммунопреципитированы с использованием моноклонального антитела к myc (фиг.16А). Для измерения уровня обогащения, достигнутого на данном этапе отбора, аликвотные смеси кДНК/мРНК-пептидных гибридов, взятые до и после иммунопреципитации, были амплифицированы с помощью ПЦР в присутствии изотопно помеченной затравки. Амплифицированную ДНК расщепляли рестриктазой, которая разрезает последовательность myс-матрицы, но не последовательность пула (фиг.16В и 16С). Количественный анализ соотношения разрезанной и неразрезанной ДНК определил, что последовательность myc в ходе иммунопреципитации была обогащена в 20-40 раз по отношению к случайной библиотеке.
Эти эксперименты были осуществлены следующим образом. Реакции трансляции. Реакции трансляции были проведены в целом так же, как это было описано выше. Более детально, реакции проводили при 30°С в течение 1 часа в соответствии с рекомендациями производителя (Novagen) и замораживали на ночь при -20°С. Были приготовлены две версии шести образцов: одна включала35S-метионин и одна включала холодный метионин, добавляемый до конечной концентрации 52 мкМ. Реакции 1-6 включали количества матрицы, приведенные в таблице 2. Все числа в таблице 2 соответствуют пикомолям матрицы на 25 мкл реакционной смеси.

Приготовление dТ25-стрептавидинагарозы. Стрептавидинагарозу (Pierce) промывали трижды буфером ТЕ рН 8,2 (10 мМ Трис-НСl рН=8,2, 1 мМ EDTA) и ресуспендировали при объемном соотношении 1:1 в ТЕ (рН 8,2). 3'-биотинил-Т25, синтезированную с использованием Bioteg-CPG (Glen Research), добавляли затем до достижения желательной конечной концентрации (обычно 10 или 20 мкМ), а инкубацию проводили при перемешивании в течение 1 часа. Затем dТ25-стрептавидинагарозу промывали трижды буфером ТЕ (рН 8,2) и хранили при 4°С до времени использования.
Очистка матриц из трансляционных реакционных смесей. Для очистки матриц из трансляционных реакционных смесей 25 мкл каждой реакционной смеси выделяли и добавляли к 7,5 мл изоляционного буфера (1 М NaCl, 100 мМ Трис-HCl рН 8,2, 10 мМ EDTA, 0,1 мМ DTT) и 125 мкл 20 мкМ dТ25-стрептавидинагарозы. Этот раствор инкубировали при 4°С в течение 1 часа при помешивании. Пробирки центрифугировали, а элюент удаляли. Добавляли 1 мл изоляционного буфера, взвесь ресуспендировали и смеси переносили в 1,5-мл микроцентрифужные пробирки. Затем образцы промывали четыре раза 1-мл аликвотами охлажденного на льду изоляционного буфера. Затем горячие и холодные образцы одних и тех же реакционных смесей комбинировали друг с другом на миллипоровых фильтрах Millipore MC и элюировали из dТ25-стрептавидинагарозы путем промывки двумя объемами 100 мкл воды, 0,1 мМ дитиотрейтола и двумя объемами 15 мМ NaOH, 1 мМ EDTA.
К этому элюенту добавляли 40 мкл 50%-ной суспензии промытой тиопропилсефарозы (Pharmacia), а инкубацию проводили при 4°С при перемешивании в течение 1 часа. Затем образцы промывали тремя объемами по 1 мл буфера ТЕ (рН 8,2) и полученный элюент удаляли. 1 мкл 1 М DTT добавляли к твердой фракции (общий объем приблизительно 20-30 мкл), и образец инкубировали в течение нескольких часов, выделяли и промывали четырежды в 20 мкл воды (общий объем 90 мкл). Элюент содержал 2,5 мМ тиопиридона, что было определено по поглощению в ультрафиолете. 50 мкл этого образца осаждали этанолом путем добавления 6 мкл 3 М NaOAc (pH 5,2), 10 мМ спермина, 1 мкл гликогена (10 мг/мл, Boehringer Mannheim) и 170 мкл 100%-ного этилового спирта с последующей инкубацией при -70°С в течение 30 минут и центрифугированием при 13000 об/мин в течение 30 минут на микроцентрифуге.
Реакции обратной транскрипции. Реакции обратной транскрипции были осуществлены и с осажденными этанолом, и с элюированными тиопиридоном образцами следующим образом. Для образцов, осажденных этанолом, 30 мкл ресуспендированной матрицы, вода до 48 мкл и 200 пкМ затравки 21.103 (SEQ ID NO:22) прошли отжиг при 70°С в течение 5 минут и были охлаждены на льду. К этому образцу были добавлены 16 мкл "буфера первой цепи" (250 мМ Трис-НСl рН 8,3, 375 мМ КС1 и 15 мМ MgCl2: получен от Gibco BRL, Grand Island, NY), 8 мкл 100 мМ дитиотрейтола и 4 мкл 10 мМ NTP с уравновешиванием при 42°С и затем было добавлено 4 мкл обратной транскриптазы Superscript II (Gibco BRL, Grand Island, NY). 13 мкл воды было добавлено к ТР-сефарозному элюенту (35 мкл), и реакция была проведена так, как это описано выше. После инкубации в течение 1 часа одинаково пронумерованные образцы были комбинированы друг с другом (при общем объеме 160 мкл). По 10 мкл образца было запасено для проведения ПЦР каждого неотобранного образца, а 150 мкл образца было запасено для иммунопреципитации.
Иммунопреципитация. Для проведения иммунопреципитации 170 мкл обратной транскрипционной реакционной смеси добавляли к 1 мл разбавляющего буфера (10 мМ Трис-НСl рН 8,2, 140 мМ NaCl, 1 об.% Тритон Х-100) и 20 мкл конъюгата белков-G/A (Calbiochem, La Jolla, CA) и проводили предварительное осветление путем инкубации при 4°С в течение 1 часа при перемешивании. Выделяли элюент и добавляли 20 мкл конъюгата G/A и 20 мкл моноклонального антитела (2 мкг, 12 пкМ), и образцы инкубировали в течение 2 часов при 4°С при перемешивании. Конъюгат осаждали путем микроцентрифугирования при 2500 об/мин в течение 5 минут, элюент выделяли и конъюгат промывали трижды аликвотами по 1 мл охлажденного на льду разбавляющего буфера. Затем образец промывали охлажденным на льду буфером, содержащим 10 мМ Трис-НСl (рН 8,2) и 100 мМ NaCl. Связанные фрагменты удаляли с использованием трех объемов замороженной 4%-ной уксусной кислоты и образцы лиофилизировали до обезвоживания.
ПЦР отобранных и неотобранных образцов. Реакции ПЦР проводили путем добавления 20 мкл концентрированной NH4OH к 10 мкл неотобранного материала и ко всему отобранному материалу с последующей инкубацией в течение 5 минут при 55, 70 и 90°С с целью разрушения всякой РНК, которая может присутствовать в данном образце. Затем образцы выпаривали досуха с использованием вакуумной центрифуги. 200 мкл ПЦР-смеси (1 мкМ затравок 21.103 и 42.108, 200 мкМ dNTP в ПЦР-буфере плюс Мg2+ (Boehringer Mannheim) и 2 мкл полимеразы Taq (Boehringer Mannheim) добавляли к каждому образцу. Были осуществлены 16 циклов ПЦР в отношении неотобранного образца №2 и 19 циклов - в отношении всех остальных образцов.
Затем образцы были амплифицированы в присутствии32Р-помеченной 5'-затравки 21.103 в соответствии с таблицей 3 и отдельно очищали дважды с использованием набора реактивов Wizard direct PCR purification kit (Promega) с целью удаления всех затравок и коротких фрагментов.
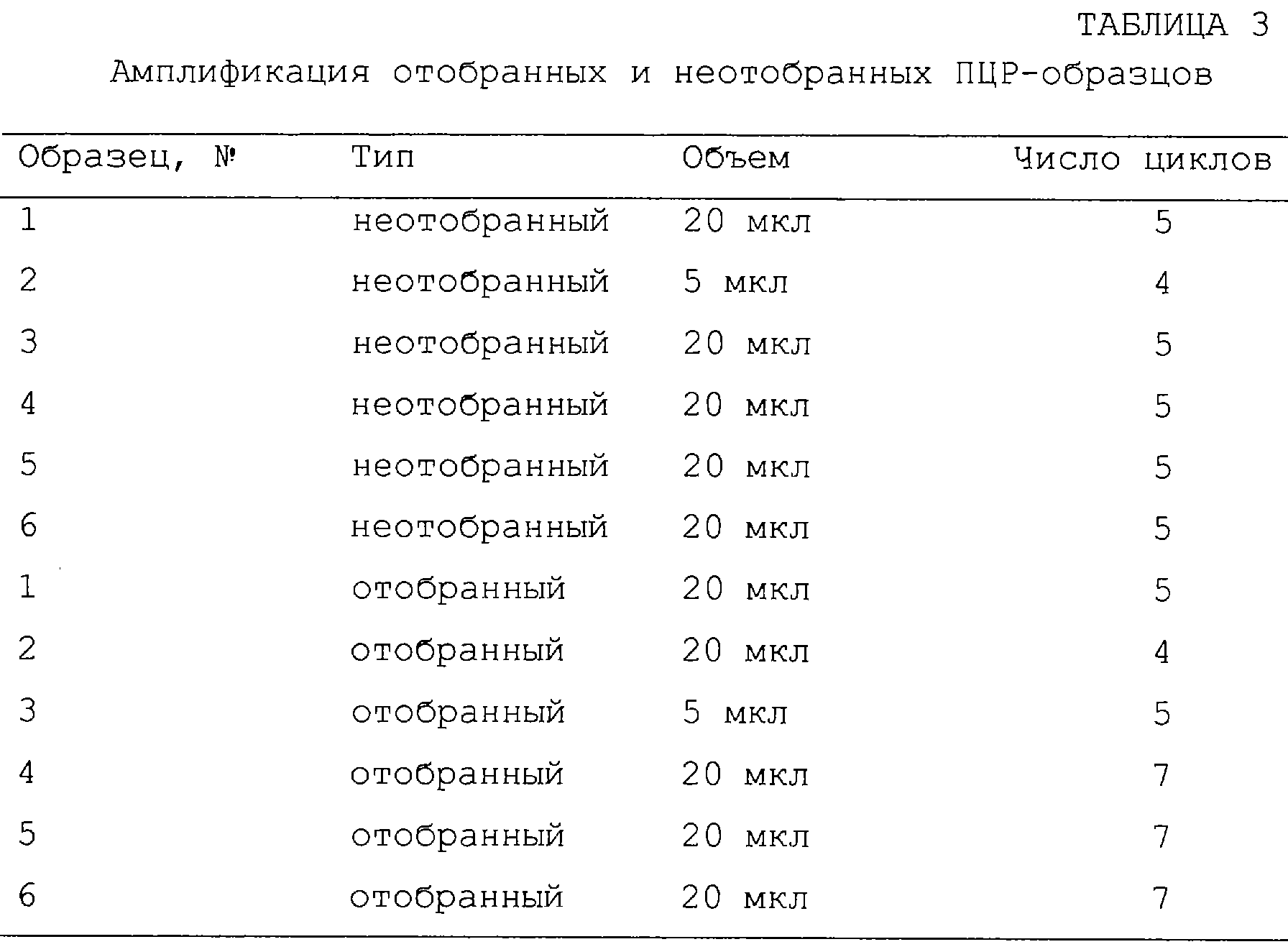
Расщепление рестриктазами.32Р-помеченную ДНК, приготовленную в каждой из вышеописанных ПЦР-реакций, добавляли в равных количествах (выраженных в срm образца) к рестрикционной реакционной смеси в соответствии с таблицей 4. Общий объем каждой реакционной смеси составлял 25 мкл. К каждой реакционной смеси добавляли 0,5 мкл рестриктазы AlwnI (5 ед.; New England Biolabs). Образцы инкубировали при 37°С в течение 1 часа и затем рестриктазу инактивировали нагреванием в течение 20-минутной инкубации при 65°С. Затем образцы перемешивали с 10 мкл денатурирующего загрузочного буфера (1 мл ультрачистого формамида (USB), 20 мкл 0,5 М EDTA и 20 мкл 1 М NaOH), нагревали до 90°С на 1 минуту, охлаждали и загружали в 12%-ный денатурирующий полиакриламидный гель, содержащий 8 М мочевины. После проведения электрофореза гель фиксировали 10%-ной уксусной кислотой (объемные проценты), 10% МеОН (объемные проценты) в воде.
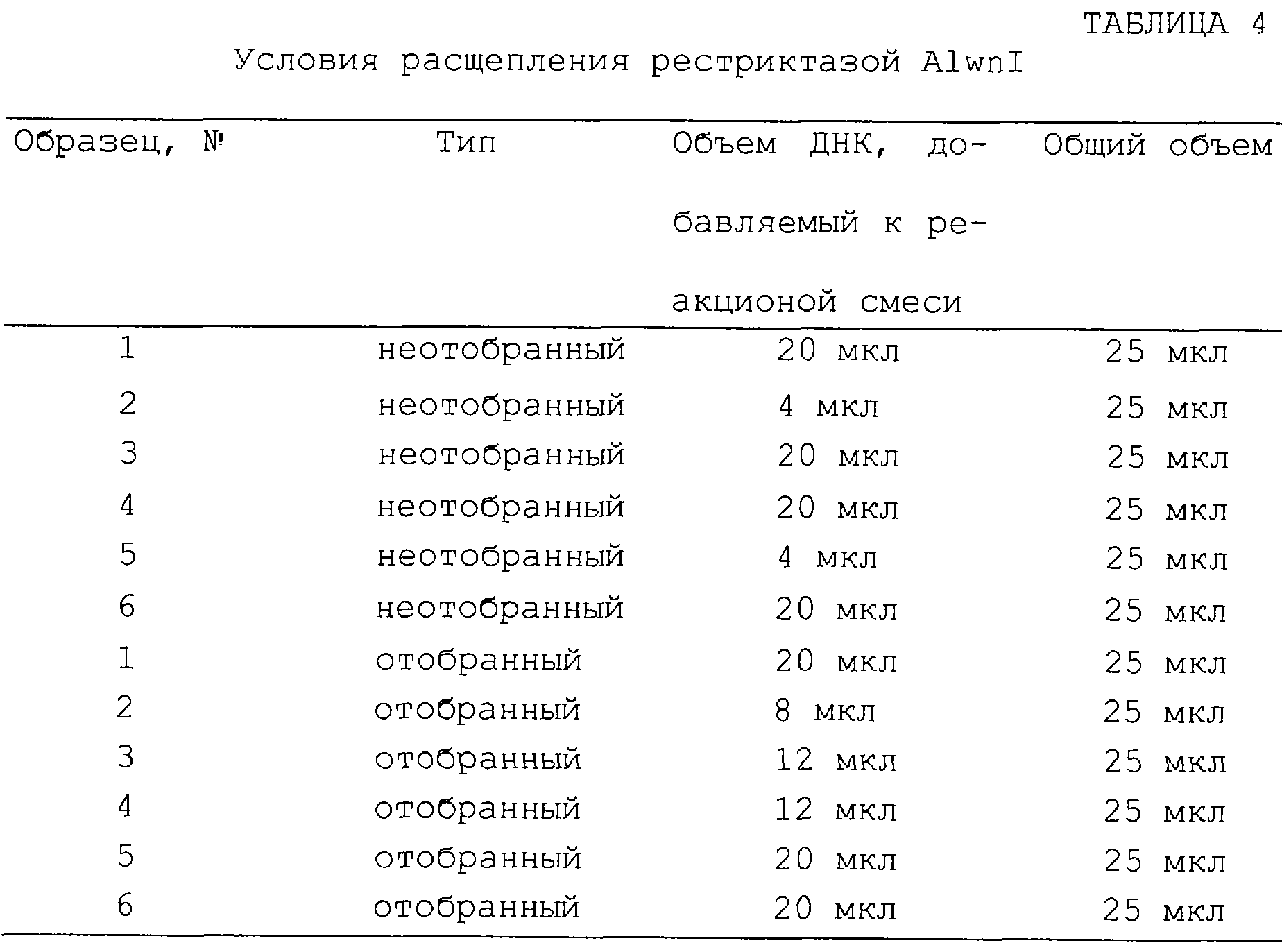
Количественный анализ расщепления. Количественный анализ содержания myc-ДНК и пула ДНК, присутствующих в образце, был проведен с использованием фосфоромиджера (Molecular Dynamics). Количество материала, содержащегося в каждом бэнде, было определено как интегрированная величина идентичных прямоугольников, которые могут быть нанесены вокруг данного бэнда на геле. Общая величина срm для каждого бэнда рассчитывалась как общая интенсивность за вычетом фона. Были использованы три показателя фона: 1) средняя величина на идентичных площадях за пределами обсчитываемой зоны геля; 2) величина в срm, характерная для линии неотобранного пула, в составе которого бэнд myc должен присутствовать (при отсутствии выраженного бэнда в данном сегменте геля); и 3) приведенное значение, которое воспроизводит наиболее близкое значение к 10-кратному приросту матрицы между линиями неотобранных образцов. Линии 2, 3 и 4 на фиг.16В и 16С показывает обогащение последовательности-мишени в противопоставление общему пулу последовательностей. Выявленное обогащение по линии 3 (неотобранный/отобранный) проявляет наибольшие значения (17-, 43- и 27-кратное обогащение в результате использования способов 1-3 соответственно) благодаря оптимизации соотношения сигнала к фону в конкретном образце. Эти данные обобщены в таблице 5.

Во второй группе экспериментов те же самые продукты
ПЦР были очищены с использованием набора реактивов Wizard direct PCR purification kit, а параметры расщепления оценивали количественно описанным выше способом (2). В этих экспериментах были получены сходные результаты; измеренные уровни обогащения - 10,7, 38 и 12 - были определены для образцов, эквивалентных тем, для которых установлены линии 2, 3 и 4, описанные выше.
ИСПОЛЬЗОВАНИЕ СИСТЕМ ОТБОРА БЕЛКОВ
Системы отбора по настоящему изобретению обладают коммерческим значением в любой области, в которой используется протеиновая технология, призванная решать терапевтические, диагностические или промышленные проблемы. Данная методология отбора применима для улучшения или изменения существующих белков, равно как и для выделения новых белков, обладающих желательными функциями. Эти белки могут быть естественно встречающимися последовательностями, могут быть измененными формами естественно встречающихся последовательностей или могут быть частично или полностью синтетическими последовательностями.
Выделение новых связывающих реагентов. В одном из конкретных приложений технология образования РНК-пептидных гибридов, описанная здесь, применима для выделения белков, обладающих специфической связывающей активностью (например, специфичностью по связыванию лигандов). Белки, проявляющие высокоспецифические связывающие свойства, могут быть использованы в качестве не являющихся антителами распознающими реагентами, обеспечивающими с помощью технологии получения РНК-пептидных гибридов возможность обойти традиционную технологию с использованием моноклональных антител. Реагенты типа антител, выделенные таким способом, могут быть использованы в любой области, где используются традиционные антитела, включая терапевтическое и диагностическое применение.
Улучшение антител человека. Также настоящее изобретение может быть применено для улучшения антител человека и гуманизованных антител, предназначенных для лечения любого из большого числа заболеваний. При таком применении формируются библиотеки антител и осуществляется их скрининг in vitro, что исключает необходимость применения таких методик, как слияние клеток или конструирование фагов. В одном из важнейших способов применения настоящее изобретение пригодно для совершенствования библиотек одноцепочечных антител (Ward et al., 1989, Nature, 341, 544; и Goulot et al., 1990, J. Mol. Biol, 213, 617). В этом случае варьирующий домен может быть сконструирован либо на основе человеческого источника (с целью минимизации возможных нежелательных иммунных реакций у реципиента) или может включать полностью рандомизованную кассету (с целью максимализации комплексности библиотеки). Для скрининга улучшенных молекул антител пул молекул-"кандидатов" тестируют по связыванию молекулы-мишени (например, антигена, иммобилизованного так, как показано на фиг.2). Затем все более высокие уровни жесткости применяют на этапе связывания в процессе перехода процесса отбора от одного раунда к следующему. Для повышения жесткости изменяли такие параметры, как число этапов промывки, концентрация избытка конкурирующего соединения, параметры буфера, продолжительность реакции связывания и выбор иммобилизующего матрикса.
Одноцепочечные антибиотики могут быть использованы либо напрямую для лечения, либо косвенно для формирования антибиотиков стандартной структуры. Такие антитела имеют целый ряд практических применений, включая выделение антител к аутоиммунным заболеваниям, обеспечение иммуносупрессии и создание вакцин к вирусным заболеваниям, таким как СПИД.
Выделение новых катализаторов. Настоящее изобретение также может быть применено для отбора новых белков, обладащих каталитическими функциями. Отбор и преобразование in vitro ранее применялись для выделения новых каталитически активных РНК и ДНК, а в соответствии с настоящим изобретением применяются для выделения новых белков-ферментов. В одном из конкретных примеров такого подхода катализатор может быть выделен опосредованно с помощью отбора по связыванию с химическим аналогом переходной формы катализатора. В другом частном варианте прямое выделение может быть осуществлено путем отбора по образованию ковалентных связей с субстратом (например, с использованием субстрата, связанного с аффинной меткой) или путем расщепления (например, путем отбора по способности разрывать специфическую связь и таким образом высвобождать обладающие каталитическими свойствами компоненты библиотеки из твердой подложки).
Такой подход к выделению новых катализаторов обладает по крайней мере двумя важными преимуществами над технологией каталитических антител (см. обзор Schultz et al., 1990, J. Chem. Engng. News, 68, 26). Во-первых, в технологии каталитических антител исходный пул обычно ограничен иммуноглобулинами; напротив, исходная библиотека РНК-пептидных гибридов может либо полностью рандомизирована, либо может содержать, без каких-либо ограничений, варианты известных ферментных структур или белковых систем. Кроме того, выделение каталитических антител обычно опирается на исходный отбор по связыванию с аналогами реакционных переходных форм с последующим достаточно сложным скринингом активных антител; опять-таки напротив, прямой отбор катализаторов возможен при использовании библиотеки РНК-пептидных гибридов в соответствии с описанным выше использованием библиотек РНК. В альтернативном способу выделения белков-ферментов подходе возможным является сочетание способов прямого отбора и тестирования по аналогам переходного состояния каталитических молекул.
Ферменты, полученные таким способом, обладают значительной ценностью. Например, в настоящее время существует насущная необходимость в новых эффективных промышленных катализаторах, которые позволили бы усовершенствовать развиваемые химические процессы. Очень существенное преимущество настоящего изобретения состоит в том, что отбор может быть осуществлен в произвольных условиях и, в частности, не ограничивается обязательными условиями in vivo. Кроме того, настоящее изобретение облегчает выделение новых ферментов или улучшенных вариантов существующих ферментов, которые обладают высокоспецифичными трансформациями (и, соответственно, минимизируют образование нежелательных побочных продуктов), обеспечивания функционирование в заранее оговоренных средах, например, в условиях повышенных температуры, давления или концентрации растворителей.
Интерактивная ловушка in vitro. Технология получения РНК-пептидных гибридов также применима для скрининга библиотек кДНК и клонирования новых генов на основе параметров взаимодействий "белок-белок". С помощью такого подхода библиотека кДНК формируется на основе желательного источника (например, с помощью метода,' описанного у Ausubel et al., цитировано выше, глава 5). На каждую молекулу-"кандидат" кДНК лигируют пептидный акцептор (например, пуромициновый "хвост") (например, с использованием методик, описанных выше при создании матриц LP77, LP154 и LP160). Затем в соответствии с настоящим изобретением формируют РНК-пептидные гибриды и в соответствии с описанным выше тестируют эти РНК-пептидные гибриды (или улучшенные варианты этих гибридов) на интерактивность по отношению к конкретным молекулам. Если желательно, в этом подходе стоп-кодоны и 3'-нетранслируемые сегменты могут быть удалены либо (i) добавлением супрессорной тРНК, обеспечивающей "проскакивание" стоп-кодона, либо (ii) удаление освобождающего фактора из состава трансляционной реакционной смеси с помощью иммунопреципитации, либо (iii) сочетанием пунктов (i) и (ii), или (iv) удалением стоп-кодонов и 3'-UTR непосредственно из последовательностей ДНК.
Тот факт, что этап взаимодействия имеет место in vitro, позволяет тщательно контролировать жесткость реакции, используя неспецифические конкурирующие соединения, температуру и концентрации ионов. Изменение нормальных небольших молекул с помощью негидролизуемых аналогов (например, АТФ против АТФ-gS) обеспечивает такой отбор, который позволяет разграничивать различные конформационные состояния одной и той же молекулы. Такой подход применим как для клонирования, так и для функциональной идентификации многих белков, поскольку последовательность РНК отобранного связывающегося компонента ковалентно присоединена и может быть, соответственно, легко выделена. Кроме того, методика применима для идентификации функций и взаимодействий примерно 50-100 тысяч генов человека, чьи последовательности в настоящее время определены международным проектом "Геном человека".
ИСПОЛЬЗОВАНИЕ РНК-ПЕПТИДНЫХ ГИБРИДОВ В ФОРМАТЕ МИКРОЧИПА
"ДНК-чипы" состоят из пространственно размеченных наборов иммобилизованных олигонуклеотидов или клонированных фрагментов кДНК или геномной ДНК и могут применяться для скоростного секвенирования и профилирования транскрипции. Путем отжига смеси РНК-пептидных гибридов (например, полученных на материале пула клеточных ДНК или РНК) на таком "ДНК-чипе" возможно формирование "белкового чипа-дисплея", в котором каждое пятно, соответствующее одной иммобилизованной последовательности, способно претерпевать отжиг на соответствующую последовательность РНК в составе пула РНК-пептидных гибридов. С помощью такого подхода соответствующий белок иммобилизуется пространственно предопределенным образом благодаря его связыванию с его собственной мРНК, а в результате чипы, содержащие наборы последовательностей ДНК, проявляют соответствующий набор белков. С другой стороны, полипептидные фрагменты таких белков также могут быть определены, если библиотека гибридов сформирована из меньших фрагментов кДНК или геномных ДНК.
Такие упорядоченные наборы белков ("белковый дисплей") и полипептидов могут быть использованы в различных целях. Например, они представляют эффективный инструмент для идентификации ранее неизвестных белок-белковых взаимодействий. В одном специфическом формате белок-зонд помечают (например, флуоресцентным красителем) и инкубируют его с "белковым чипом-дисплеем". При таком подходе идентичность белков, которые способны связываться с выбранным белковым зондом, определяется исходя из расположения пятен на чипе, которые визуально помечаются благодаря связыванию с зондом. Другим подходом является быстрое определение белков, которые химически модифицированы в результате воздействия модифицирующих ферментов (например, протеинкиназ, ацилтрансфераз и метилтрансфераз). При инкубации "белкового чипа-дисплея" с интересующим ферментом и изотопно помеченным субстратом с последующей промывкой и авторадиографическим тестированием положение и, соответственно, идентичность тех белков, которые являются субстратами для модифицирующего фермента, легко могут быть определены. Кроме того, использование такого подхода с выстроенными дисплеями коротких пептидов позволяют уточнять локализацию таких модифицируемых сайтов.
Технология "белковых дисплеев" может быть осуществлена с использованием наборов нуклеиновых кислот (включая РНК, но предпочтительно с ДНК), иммобилизованных на подходящую твердую подложку. Примерами твердых подложек могут быть такие материалы как стекло (например, предметные стекла), кварц или кварцевое стекло (например, микрочипы) или золото (например, золотые пластинки). Способы прикрепления нуклеиновых кислот к точно определенным участкам такой твердой поверхности, например, методы светолитографии, хорошо известны в соответствующей области техники и могут быть использованы для формирования твердых подложек (таких как "ДНК-чипы") для применения в соответствии с настоящим изобретением. Примерами способов, пригодных для таких целей, без каких-либо ограничений, являются: Schena et al., 1995, Science, 270, 467-470; Kozal et al., 1996, Nature Medicine, 2, 753-759; Cheng et al., 1996, Nucleic Acids Research, 24, 380-385; Lipshutz et al., 1995, BioTechniques, 19, 442-447; Pease et al., 1994, Proc. Natl. Acad. Sci. USA, 91, 5022-5026; Fodor et al., 1993, Nature, 364, 555-556; Pirrung et al., Патент США №5143854; и Fodor et el., Международная патентная публикация WO 92/10092.
Реферат
Изобретение относится к биотехнологии, в частности, молекулярной биологии. Описан способ отбора белка с использованием РНК-пептидных гибридов. РНК-пептидный гибрид включает молекулу РНК, ковалентно связанную или гибридизованную по 3’-концу последовательности, кодирующей белок, с последовательностью НК, способной приостанавливать трансляцию ДНК. Последовательность НК ковалентно присоединена к белковому акцептору, а белок кодируется ковалентно-связанной РНК. Описана также РНК, содержащая последовательность инициации трансляции и старт-кодон, функционально соединенный с последовательностью, кодирующей белок. Описан также микрочип, включающий набор иммобилизированных НК, каждая из которых гибридизуется с РНК-белковым гибридом. Способ отбора белков включает следующие этапы: представление популяции молекул РНК со свойствами, перечисленными выше; трансляция последовательности, кодирующей белок; отбор PHK-белкового гибрида путем тестирования белка на связывание или функциональную активность. Использование изобретения позволяет отбирать желательные белки из обширных пулов и преобразовывать полипептидные последовательности in vitro. 4 с. и 23 з.п. ф-лы, 19 ил., 5 табл.

Комментарии