Ретиноиды - RU2166499C2
Код документа: RU2166499C2
Чертежи







Описание
Изобретение
относится к новым соединениям формулы
где связь C7-C8 является двойной связью и один из R1 и R2 обозначает C1-C4алкил, а другой обозначает хлор, бром или иод, или R1 и R2 вместе обозначают C3 -C13алкилен, в котором один атом углерода может быть замещен атомом серы; или связь C7-C8 является тройной связью и R1 и R2 вместе обозначают C3-C13алкилен; или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют ароматическое кольцо с 6 атомами углерода; и их фармацевтически приемлемым солям и сложным эфирам.
Когда R1 и R2 вместе обозначают C3-C13алкилен, предпочтительно чтобы они являлись C3-C6алкиленом, прежде всего С6алкиленом. R1 и R2 в качестве низшего алкила обозначают C1-C4алкил, который может быть с прямой либо с разветвленной цепью. Предпочтительной низшей алкильной группой является метил.
Предпочтительными соединениями настоящего изобретения являются соединения формулы I, где связь C7-C8 является двойной связью, R1 и R2 вместе обозначают C3-C13алкилен, в котором один атом углерода может быть замещен атомом серы, или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют ароматическое кольцо с 6 атомами углерода.
Другой предмет настоящего изобретения относится к соединениям формулы I, где связь C7-C8 является тройной связью, R1 и R2 вместе обозначают C3-C13алкилен.
Ретиноевые кислоты действуют путем связывания с семейством протеинов, известных как ядерный рецептор ретиноевой кислоты (RAR). Трех членов этого семейства обозначают как RARα, RARβ и RARγ, 9-цис-ретиноевая кислота связывается с группой рецепторов, известных как X-рецепторы ретиноевой кислоты (RXR) (Levin и др. , Nature, 355:359-361 (1992); Heyman и др., Cell, 68: 397-406 (1992)), в то время как полностью-транс- и 13-цис-ретиноевая кислота, связывающиеся с RARα, с ними не связываются. Однако 9-цис-ретиноевая кислота также действует как лиганд для RAR-группы рецепторов и, следовательно, может проявлять нежелательные побочные эффекты, характерные для полностью-транс- и 13-цис-ретиноевой кислоты. Известно, что как RAR-, так и RXR-рецепторы образуют гетеродимеры для медиирования своих биологических функций (Leid и др., Cell, 68:377-395 (1992); Yu и др., Cell, 67:1251-1266 (1991); Zhang и др., Nature, 355:441-446 (1992)).
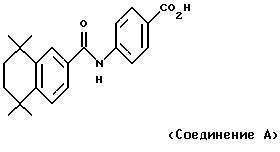
Соединения по изобретению проявляют высокую степень избирательности по отношению к семейству RXR-рецепторов и пригодны для использования в качестве антипролиферативных агентов и при дерматологических и онкологических показаниях. В частности, соединения по изобретению ингибируют пролиферацию себоцитов. Известно, что пролиферация себоцитов является причиной угрей. Поэтому ингибирование пролиферации себоцитов является известным средством лечения угрей и, таким образом, соединения по изобретению пригодны для лечения угрей.
Кроме того, избирательные по отношению к RXR соединения по изобретению в дозах, при которых они неактивны сами по себе, увеличивают активность ретиноидов, которые пригодны для лечения рака, прежде всего лейкемии, и плотных опухолей, главным образом опухолей головы, шеи и молочной железы, относящихся к плотным опухолям, прежде всего опухолей молочной железы, и они также пригодны сами по себе для лечения дерматологических нарушений, в частности угрей и солнечных ожогов кожи, с пониженными токсическими эффектами по сравнению с 9-цис- или 13-цис-ретиноевой кислотой. Ретиноиды, обладающие RARα-активностью, такие как полностью-транс-ретиноевая кислота и 13-цис-ретиноевая кислота, являются соединениями, которые обладают биологической активностью, основанной на их связывании с RARα-рецепторами и трансактивации последних. Таким образом, соединения по изобретению пригодны для использования в качестве усилителей активности ретиноидов, обладающих RARα-активностью, при лечении показаний, в отношении которых известна полезность таких ретиноидов. Назначение соединений по изобретению в сочетании с ретиноидом, обладающим RARα-активностью, позволяет использовать значительно более низкие дозы ретиноида или увеличивать эффективность ретиноида при обычной дозе при любом из таких показаний, в отношении которых известна полезность таких ретиноидов, обладающих RARα-активностью.
Кроме того, настоящее изобретение также включает способ усиления активности ретиноидов, обладающих RARα-активностью, причем количество соединения по изобретению, которое эффективно для усиления активности соединения, обладающего RARα-активностью (предпочтительно от 1- до 10-кратного количества к весу соединения, обладающего RAR α-aктивностью), назначают пациенту в сочетании с эффективным количеством соединения, обладающего RARα-активностью. "В сочетании с" означает, что избирательное в отношении RXR соединение по изобретению может назначаться в определенной последовательности с соединением, обладающим RARα-активностью, либо практически одновременно, предпочтительно, но необязательно объединенное с последним в разовую оральную стандартную дозируемую форму, либо перед или после введения соединения, обладающего RARα-активностью, так чтобы избирательное в отношении RXR соединение и соединение, обладающее RARα-активностью, одновременно присутствовали бы в кровотоке больного так, чтобы избирательное в отношении RXR соединение по изобретению присутствовало в концентрации, которая усиливает активность соединения, обладающего RARα-aктивностью. Предпочтительно избирательное в отношении RXR соединение по изобретению и соединение, обладающее RARα-активностью, назначают с временным интервалом, не превышающим 4 ч, более предпочтительно с временным интервалом, не превышающим 1 ч, и наиболее предпочтительно практически одновременно.
В соответствии с изобретением было установлено, что соединения по изобретению усиливают активность ретиноидов, обладающих RARα-активностью, обусловливающую дифференцировку клеток лейкемии человека в стандартных опытах на культуре клеток. Таким образом, соединения по изобретению обладают активностью, которая может усиливать известную активность ретиноидов, обладающих RARα-активностью, обусловливающую регресс или ремиссию гематологических опухолей, прежде всего таких опухолей, которые связаны с острой промиелоцитарной лейкемией. Такое лечение лейкемии может достигаться путем систематического назначения больному соединения по изобретению в сочетании с ретиноидом, обладающим RARα-активностью, что, как известно, является эффективным для замедления развития или для регресса гематологических опухолей. Количество лекарственного препарата зависит от количества и размера опухолей и от индивидуальных особенностей пациента.
В соответствии с изобретением было также найдено, что соединения по изобретению усиливают активность ретиноидов, обладающих RARα-активностью, обусловливающую регресс или ремиссию плотных опухолей, прежде всего указанных опухолей на голове и шее и связанных с раком молочной железы. Таким образом, соединения по изобретению обладают активностью, которая может усиливать известную активность ретиноидов, обладающих RARα-активностью, обусловливающую регресс или ремиссию плотных опухолей, прежде всего указанных опухолей, которые связаны с раком головы и шеи и с раком молочной железы. Такое лечение плотных опухолей может достигаться путем систематического назначения больному соединения по изобретению в сочетании с ретиноидом, обладающим RARα-активностью, способным вызывать регресс или ремиссию плотных опухолей, прежде всего головы, шеи и молочной железы. Количество препарата зависит от количества и размера опухолей и от индивидуальных особенностей пациента.
Для указанного выше лечения соединение по изобретению назначают систематически в виде композиции, содержащей соединение по изобретению, необязательно в комбинации с соединением, обладающим RARα-активностью, и фармацевтически приемлемый наполнитель, совместимый с указанным соединением(ями). При приготовлении такой композиции может быть использован любой обычный фармацевтически приемлемый наполнитель. Когда лекарство назначается орально, его обычно назначают через регулярные интервалы времени, обычно во время еды, или один раз в день.
Фармацевтически приемлемые соли включают любую соль, химически допустимую в данной области техники для ретиноевой кислоты и применимую для больных людей в фармацевтически приемлемых препаратах. Может быть использована любая такая обычная фармацевтически приемлемая соль соединения по изобретению. Обычные соли, которые могут быть использованы, включают основные соли, например соли щелочных металлов, таких как натрий или калий, соли щелочноземельных металлов, таких как кальций или магний, соли аммония или алкиламмония.
В соответствии с изобретением соединения по изобретению могут назначаться в форме их фармацевтически приемлемых гидролизуемых сложных эфиров. Любой из фармацевтически приемлемых гидролизуемых сложных эфиров может быть использован в композициях и способах по изобретению. Сложные эфиры включают ароматические эфиры, такие как бензиловый (OBzl) эфир или бензиловый эфир, замещенный низшим алкилом, галогеном, нитро, тиогруппой или замещенной тиогруппой, т.е. заместителем которой может быть низший алкил, трет-бутил, циклопентил, циклогексил, циклогептил и 9-флуоренилметил.
Фармацевтические композиции могут быть изготовлены в любой обычной форме, включающей: (а) твердую форму для орального применения, такую как таблетки, капсулы, пилюли, порошки, гранулы и т.д.; и (б) препараты для местного применения, такие как растворы, суспензии, мази, кремы, гели, тонкоизмельченные порошки, аэрозоли и т.п. Фармацевтические композиции могут быть стерилизованными и/или могут включать адъюванты, такие как консерванты, стабилизаторы, смачивающие агенты, эмульгаторы, соли для изменения осмотического давления и/или буферы.
В соответствии с изобретением вышеуказанные соединения по изобретению пригодны для фармацевтически приемлемых оральных способов применения. Эти фармацевтические композиции по изобретению включают указанное соединение по изобретению либо его фармацевтически приемлемые соли или его фармацевтически приемлемые гидролизуемые сложные эфиры в сочетании с совместимым фармацевтически приемлемым материалом наполнителя. Может быть использован любой обычный материал наполнителя. Материал наполнителя может быть органическим или неорганическим инертным материалом, пригодным для орального применения. Пригодные наполнители включают воду, желатин, гуммиарабик, лактозу, крахмал, стеарат магния, тальк, растительные масла, полиалкиленгликоли, вазелин и т. п. Кроме того, фармацевтические препараты могут включать другие фармацевтически активные агенты, предпочтительно ретиноид, обладающий RARα-активностью. В соответствии с принятой практикой изготовления фармацевтических композиций в них могут вводиться дополнительные добавки, такие как корригены, консерванты, стабилизаторы, эмульгаторы, буферы и т.п.
Фармацевтические препараты могут быть изготовлены в виде любой обычной дозируемой формы для орального применения, включая твердую форму для орального применения, такую как таблетки, капсулы, пилюли, порошки, гранулы и т. п. Предпочтительная форма для орального применения включает таблетки, капсулы из твердого или мягкого желатина, метилцеллюлозы или другого пригодного материала, легко растворяющегося в пищеварительном тракте. Оральные дозы в соответствии с настоящим изобретением могут варьировать в зависимости от потребностей конкретного пациента, что определяется предписанием врача.
При оральном назначении больному соединений по изобретению, предназначенных для усиления дифференцирующей активности ретиноидов, обладающих RARα-активностью, которые используют для лечения острой промиелоцитарной лейкемии или опухолей головы, шеи или молочной железы, соединение по изобретению обычно дают взрослым ежедневно в количестве, приблизительно в 1-10 раз превышающем количество ретиноида, обладающего RARα -активностью. Назначаемое количество ретиноида, обладающего RARα-активностью, составляет от приблизительно 20 до приблизительно 300 мг/м2 ежедневно, предпочтительно от приблизительно 50 до приблизительно 100 мг/м2 ежедневно, причем точная доза варьирует в зависимости от телосложения и веса пациента. Обычно комбинированное лечение проводят в течение приблизительно трехмесячного периода.
Соединения по изобретению либо их фармацевтически приемлемые соли или гидролизуемые сложные эфиры и ретиноиды, обладающие RARα-активностью, могут назначаться по отдельности в соответствии с настоящим изобретением, однако предпочтительно их назначают в виде оральной композиции, включающей соединение по изобретению, его фармацевтически приемлемые соли или сложные эфиры в комбинации с ретиноидом, обладающим RARα-активностью, в количестве, достаточном для лечения с помощью указанной комбинации острой промиелоцитарной лейкемии или опухолей головы, шеи или молочной железы.
Обычно предпочтительная стандартная оральная дозируемая форма представляет собой таблетки или капсулы, содержащие от 10 до 50 мг ретиноида, обладающего RARα-активностью, и соединение по изобретению в количестве, в 1-10 раз превышающем количество ретиноида, обладающего RARα-активностью, его фармацевтически приемлемые соли или его фармацевтически приемлемые гидролизуемые сложные эфиры. Таким образом, количество соединения по изобретению в каждой таблетке или капсуле может составлять от 10 до 500 мг. Эти таблетки или капсулы могут назначаться один или два раза в день в зависимости от веса или телосложения пациента.
В соответствии с изобретением местное или оральное применение соединений по изобретению, их фармацевтически приемлемых солей и их фармацевтически приемлемых гидролизуемых сложных эфиров эффективно при лечении всех форм угрей, как воспаленных, так и невоспаленных.
Для местного нанесения на кожу композиции, включающие соединение по изобретению в фармацевтически приемлемом наполнителе, предпочтительно готовят в виде мазей, настоек, кремов, гелей, растворов, лосьонов, распыляемых препаратов, суспензий, шампуней, мыла для волос и т.п. По сути любая обычная композиция, используемая для нанесения на волосистую часть кожи головы или на кожу, может быть использована в соответствии с изобретением. Среди предпочтительных способов применения композиции, содержащей действующее вещество по изобретению, можно указать применение действующего вещества по изобретению в форме геля, лосьона или крема. Фармацевтический препарат для местного нанесения на кожу может быть приготовлен путем смешения вышеупомянутого действующего вещества по изобретению с нетоксичными, терапевтически инертными, твердыми или жидкими наполнителями, обычно применяемыми в таких препаратах. Эти препараты должны содержать по крайней мере приблизительно 0,05 вес. % действующего вещества по изобретению по отношению к общему весу композиции. Поскольку действующее вещество по изобретению относительно нетоксично и не обладает раздражающим действием, его можно использовать в композициях для местного нанесения в количествах, превышающих 3,0%. Предпочтительно, чтобы эти препараты содержали приблизительно 1-2 вес.% действующего вещества по изобретению по отношению к общему весу композиции. Также предпочтительно наносить эти препараты на кожу один или два раза в день. Эти препараты могут наноситься в зависимости от потребности пациента. При выполнении настоящего изобретения действующее вещество по изобретению может также наноситься в виде водного раствора или спиртового раствора, например на основе этилового спирта.
При приготовлении препаратов для местного нанесения могут быть использованы описанные выше добавки, такие как консерванты, загустители, ароматизаторы и т.п., обычно используемые в области производства фармацевтических препаратов для местного нанесения. Кроме того, в препараты для местного нанесения, содержащие вышеуказанное действующее вещество по изобретению, могут быть включены обычные антиокислители. Среди обычных антиокислителей, которые могут быть использованы в этих препаратах, можно назвать N-метил-α-токофероламин, токоферолы, бутилированный гидроксианизол, бутилированный гидрокситолуол, этоксиквин и т.п.
Обычные ароматизаторы и лосьоны, обычно применяемые в препаратах, предназначенных для местного нанесения на волосы, могут быть использованы в соответствии с настоящим изобретением. Кроме того, при необходимости в препаратах по изобретению для местного нанесения могут быть использованы обычные эмульгирующие агенты.
Композиции в виде мазей, включающие действующее вещество по изобретению, могут содержать смеси из полутвердого нефтяного углеводорода с дисперсией в растворителе действующего вещества по изобретению.
Композиции в виде кремов, включающие действующее вещество по изобретению, предпочтительно содержат эмульсии, состоящие из водной фазы, включающей увлажнитель, стабилизатор вязкости и воду, и масляной фазы, включающей спирт жирной кислоты, полутвердый нефтяной углеводород и эмульгирующий агент, и фазы, включающей действующее вещество по изобретению, диспергированное в водном стабилизированном буфером растворе. К препарату для местного нанесения могут быть добавлены стабилизаторы. В соответствии с настоящим изобретением может быть использован любой обычный стабилизатор. В масляной фазе компоненты спирта жирной кислоты действуют в качестве стабилизатора. Эти компоненты спирта жирной кислоты предпочтительно образованы путем восстановления насыщенной жирной кислоты с длинной цепью по меньшей мере из приблизительно 14 атомов углерода. Фармацевтические композиции на основе крема, включающие действующее вещество по изобретению, могут состоять, например, из водных эмульсий, содержащих спирт жирной кислоты, полутвердый нефтяной углеводород, 1,2-этиленгликоль и эмульгирующий агент.
Обычно лечение угрей, как воспаленных, так и невоспаленных, может быть осуществлено путем орального введения пациенту соединения по изобретению в количестве 0,1-10 мг/кг один или два раза в день. Лечение угрей может быть осуществлено путем местного нанесения композиции для местного нанесения, содержащей соединение по изобретению в количестве приблизительно от 0,05 до приблизительно 3 вес. %, предпочтительно от приблизительно 1 до приблизительно 2 вес.% один или два раза в день.
Доза для лечения обычно зависит от пути введения, возраста, веса, телосложения и состояния болезни пациента.
Предпочтительными соединениями по настоящему изобретению являются:
(2E, 4E, 6Z,
8E)-3,6,7-триметил-9-(2,6,6-триметил-1-циклогексен-1-ил) -2,4,6,8-нонатетраеновая кислота;
(2E,4E)-3-метил-5-(2-((Е)-2-(2,6,6-триметил-1-циклогексен-1-ил)- этенил)-1-циклогексен-1-ил)-2,
4-пентадиеновая кислота;
(полностью-E)-6-бром-3,7-диметил-9-(2,6,6-триметил-1-циклогексен-1- ил)-2,4,6,8-нонатетраеновая кислота;
(полностью-E)-6-йод-3,7-диметил-9-(2,6,
6-триметил-1-циклогексен-1- ил)-2,4,6,8-нонатетраеновая кислота;
(полностью-E)-6-хлор-3,7-диметил-9-(2,6,6-триметил-1-циклогексен-1-ил) -2,4,6,8-нонатетраеновая кислота;
(2E,
4E)-3-метил-5-(2-((Е)-2-(2,6,6-триметил-1-циклогексен-1-ил) этенил)-1-циклопентен-1-ил)-2,4-пентадиеновая кислота;
(2E, 4E)-3-метил-5-(2-((Е)-2-(2,6,6-триметил-1-циклогексен-1-ил)
этенил)-1-циклогептен-1-ил)-2,4-пентадиеновая кислота;
(2E, 4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен-1-ил) этенил)-1-циклооктен-1-ил)-2,4-пентадиеновая кислота;
(2E,
4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен-1-ил) этенил)-1-фенил)-2,4-пентадиеновая кислота;
(2E,4E)-3-метил-5-(3-((E)-2-(2,6,6-триметил-1- циклогексен-1-ил)этенил)-2-тиенил)-2,
4-пентадиеновая кислота;
(2E,4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен-1- ил)этенил)-3-тиенил)-2,4-пентадиеновая кислота;
(2E, 4E)-3-метил-5-(4-((E)-2-(2,6,
6-триметил-1-циклогексен-1-ил) этенил)-3-тиенил)-2,4-пентадиеновая кислота;
(2E, 4E)-3-метил-5-[2-(2,6,6-триметилциклогекс-1-енилэтинил) циклопент-1-енил]пента-2,4-диеновая кислота;
(2E, 4E)-3-метил-5-[2-(2,6,6-триметилциклогекс-1- енилэтинил)циклогепт-1-енил]пента-2,4-диеновая кислота; и
(2E,4E)-3-метил-5- [2-(2,6,6-триметилциклогекс-1-енилэтинил)фенил]пента-2,
4-диеновая кислота.
Способы определения связывания и трансактивационных способностей ретиноидов в отношении RAR-рецепторов и RXR-рецепторов известны в данной области техники, например, см. Levin и др., Nature, 355:359-361 (January 23, 1992); Allenby и др., Proc. Natl. Acad. Sci. USA, 90:30-34 (January, 1993); Allenby и др. , J.Biol.Chem., 269:16689-16695 (June 17, 1994). Для определения связывания и трансактивационных способностей ретиноидов использовали способы, в основном такие же, как описанные в указанных публикациях, они изложены в приведенных в настоящем описании примерах.
Активность соединений по изобретению при использовании для лечения угрей можно определить любым из обычных способов. Например, способность соединений по изобретению ингибировать пролиферацию себоцитов является стандартной моделью лечения угрей. Антипролиферационная активность соединений по изобретению в отношении себоцитов может быть определена в соответствии с процедурой примера 20(1).
Кроме того, способность соединений по изобретению уменьшать размер кожных мешочков у мышей линии Rhino является другой стандартной моделью лечения угрей. Эта активность может быть определена в соответствии с процедурой примера 20(2).
Далее способность соединений по изобретению уменьшать размер сальных желез золотистого хомячка является еще одной стандартной моделью лечения угрей. Эта активность может быть определена в соответствии с процедурой примера 20(3).
Способность соединений по изобретению усиливать RARα -активность ретиноидов может быть определена любым из обычных способов. Так, способность соединений по изобретению усиливать RARα-активность ретиноидов может быть определена путем измерения активности комбинации соединения по изобретению с ретиноидом, обладающим RARα-активностью, путем стандартного метода скрининга, для которого известно, что он релевантен при таком состоянии заболевания, для которого известно, что ретиноид пригоден для лечения. Уменьшение концентрации ретиноида, обладающего RARα-активностью, необходимой для получения конкретного уровня активности, или увеличение активности ретиноида при постоянной концентрации будет свидетельствовать об усиливающем действии соединений по изобретению, если концентрация соединения по изобретению является такой, при которой соединение по изобретению само по себе обладает низкой активностью или не обладает ею совсем.
Например, приведенные ниже результаты исследования способности соединений по изобретению усиливать активность ретиноидов при дифференцировке клеток HL-60 (стандартная модель лейкемии человека) подтверждают усиливающее действие соединений по изобретению. Способность соединений по изобретению усиливать активность ретиноидов, обладающих RARα-активностью, при дифференцировке клеток HL-60 может быть определена в соответствии с примером 18.
Кроме того, представленные ниже результаты исследования способности соединений по изобретению усиливать активность ретиноидов, обладающих RARα-активностью, при ингибировании активации мышиных B-клеток (стандартная модель иммуносупрессии) подтверждают усиливающее действие соединений по изобретению. Способность соединений по изобретению усиливать активность ретиноидов при ингибировании активации мышиных B-клеток может быть определена в соответствии с примером 19.
Далее приведенные ниже результаты исследования способности соединений по изобретению усиливать активность ретиноидов, обладающих RARα-активностью, при ингибировании роста линии клеток карциномы молочной железы (модель плотной опухоли) подтверждают усиливающее действие соединений по изобретению. Способность соединений по изобретению усиливать активность ретиноидов при ингибировании роста линии клеток карциномы молочной железы может быть определена в соответствии с примером 19.
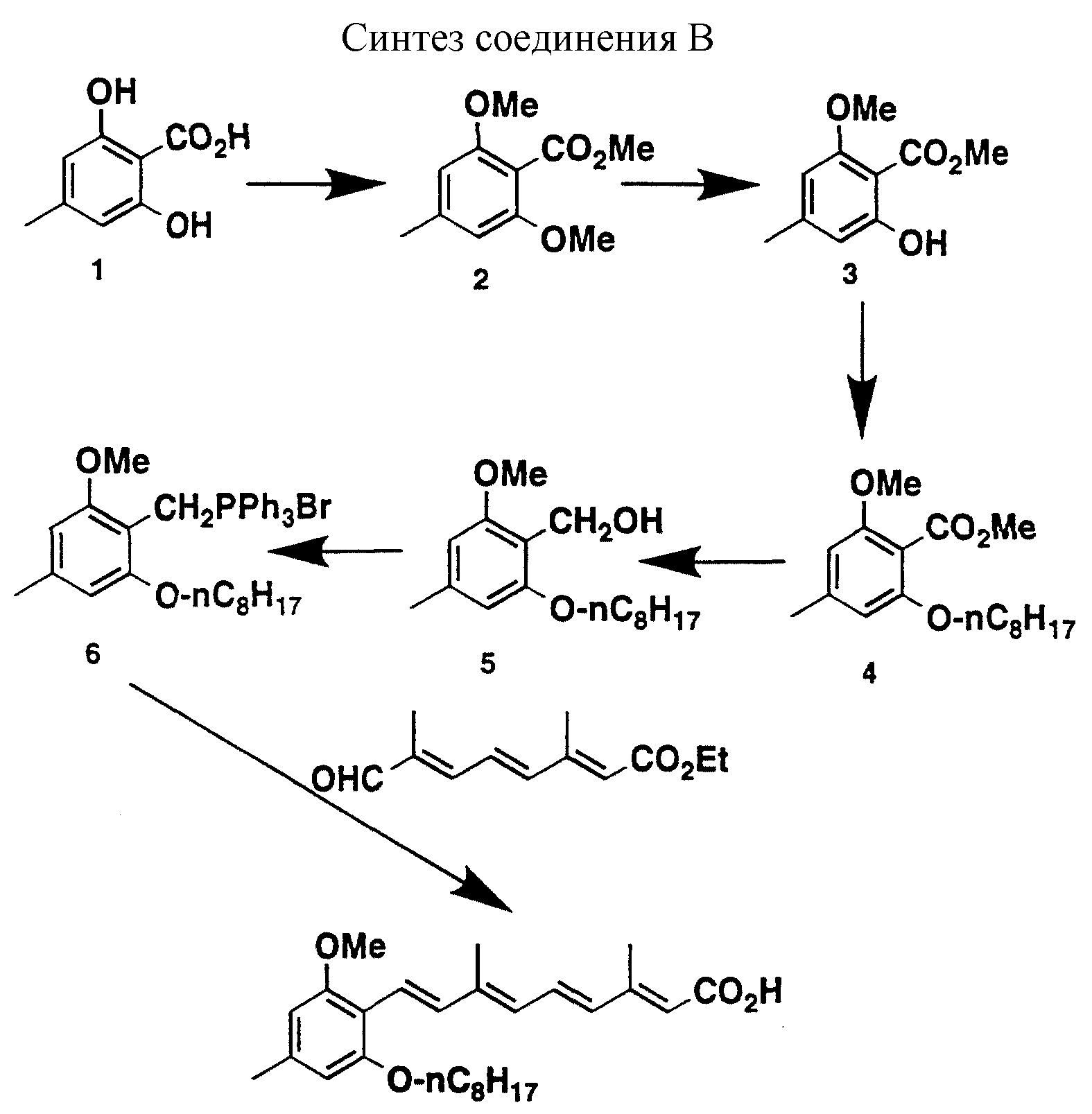
Соединения по изобретению могут быть получены любым из известных способов. Предпочтительные способы приведены на схемах 1b-1d, 2-9 (см. в конце описания).
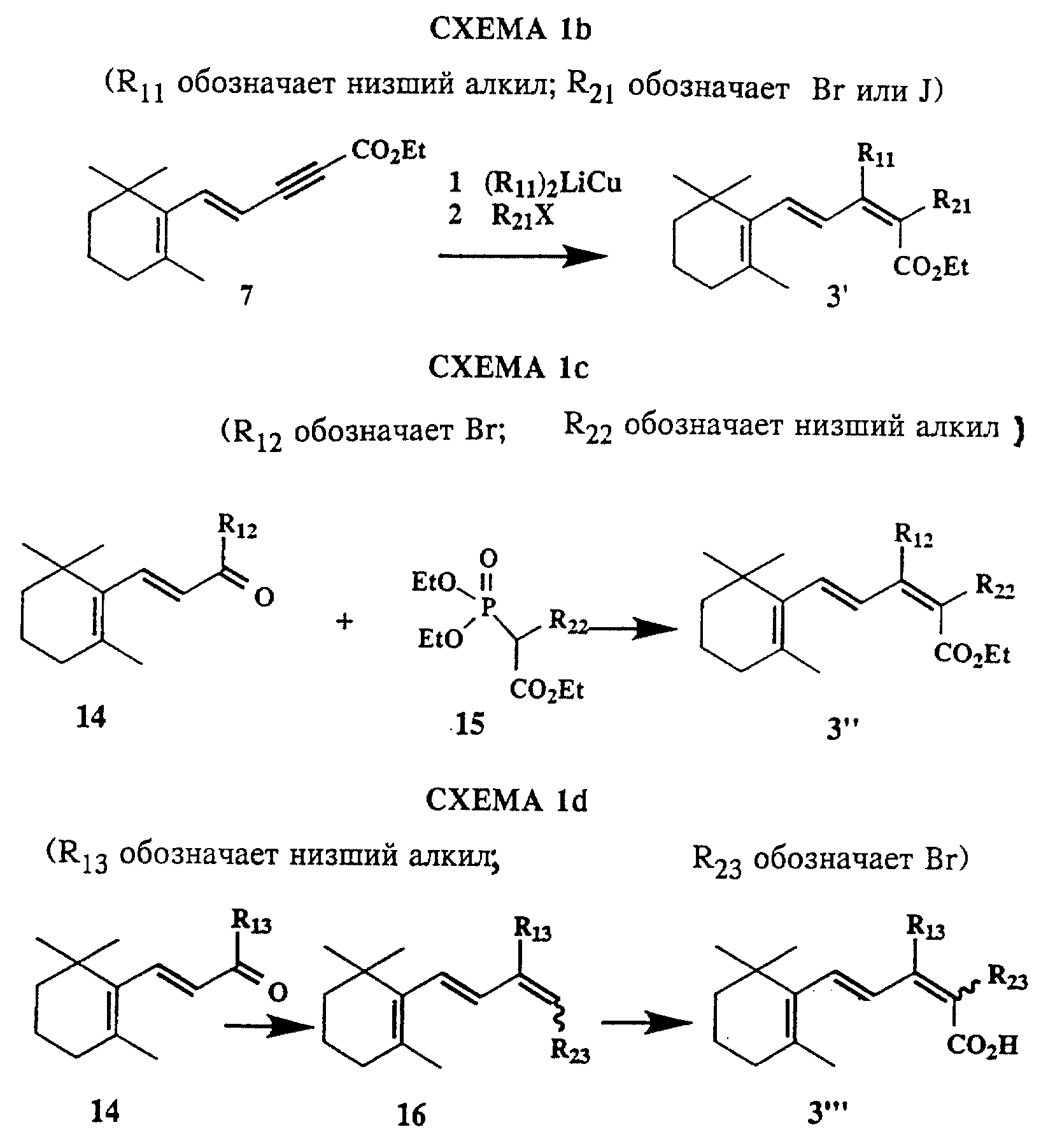
На схемах 1b-1d приведены предпочтительные способы получения промежуточных сложных эфиров для использования при получении соединений формулы I, в которых R1 и R2 вместе не образуют кольцо. На схеме 1b представлен путь получения соединений формулы I, в которых R1 обозначает низший алкил и R2 обозначает бром или иод. На схеме 1c представлен путь получения соединений формулы 1, в которых R1 обозначает бром или иод и R2 обозначает низший алкил. На схеме 1d представлен другой путь получения соединений формулы I, в которых R1 обозначает низший алкил и R2 обозначает бром. Однако, когда R1 обозначает алкил, соединения формулы I, где R2 обозначает бром, предпочтительно получают путем, представленным на схеме 1b.
Схему 1b предпочтительно используют для получения соединений формулы I, где R2 обозначает галоген. Однако схему 1b также можно использовать для получения соединений формулы I, где R2 обозначает низший алкил. Путем присоединения купрата (R1)2LiCu к ацетилену 7 (Carotenoids, Otto Isled, изд-во (Birkhauser Verlage, Basel, 1971)) получают новый купрат в результате син-присоединения, после взаимодействия которого с соответствующим галогенидом (например, йодидом в диоксане) получают соединение 3'; R2 обозначает I. Взаимодействие со смесью купрата с галоидалкилом приводит к получению сложного эфира 3', где R2 обозначает алкил. Преимущество применения купрата состоит в образовании в большинстве случаев отдельного изомера.
Схему 1c предпочтительно используют для получения соединений формулы I, где R2 обозначает низший алкил. Коммерчески доступные исходные материалы, например β-ионон 14 (R12 обозначает CH3) подвергают сочетанию с пригодным фосфонатом 15 (Carotenoids, см. выше) для получения требуемого сложного эфира 3''.
Схему 1d можно использовать для получения соединений формулы I, где R2 обозначает бром (G. Kobrich и др., Liebigs Ann. Chem., 51:704 (1967)). Кетон 14 превращают в алкен 16 соответствующим способом. Известную смесь бромзамещенных кислот 16 превращают в смесь сложных эфиров 3''' (например, R1 обозначает Me; R2 обозначает Br), которые затем разделяют с помощью ЖХВР и далее трансформируют в конечные продукты, как указано ранее.
Имея в наличии сложные эфиры 3, 3', 3'' или 3''', конечные продукты получают с помощью стандартных способов (Carotenoids, см. выше). Например, реакция может быть выполнена в соответствии со схемой 2. На схеме 2 восстановление любого из сложных эфиров 3, 3', 3'' или 3''' до спирта с последующим повторным окислением двуокисью марганца приводит к получению альдегида 4. Затем альдегид 4 подвергают сочетанию с фосфонатом 5, получая конечный продукт в виде метилового эфира 6. В результате щелочного гидролиза соединения 6 получают свободную кислоту.
Соединения формулы I, в которых R1 и R2 вместе обозначают C3-C13алкилен, легко получают, как показано на схеме 3 (R.M. Coates и др., J. Org. Chem., 47: 3597 (1982)). Так, циклический кетон 8 (например, циклопентанон или циклогексанон и т. д. ) подвергают воздействию трехбромистого фосфора в диметилформамиде для получения бромальдегида 9 (R.M. Coates и др., см. выше), который затем подвергают сочетанию с солью фосфония 10 для получения винилбромида 11. Этот винилбромид затем превращают в альдегид 12 путем переметаллирования и взаимодействия с диметилформамидом. Имея в наличии новый альдегид, конечный продукт 13 легко получают путем сочетания соединения 12 с фосфонатом 5. После этого в результате гидролиза получают требуемую кислоту.
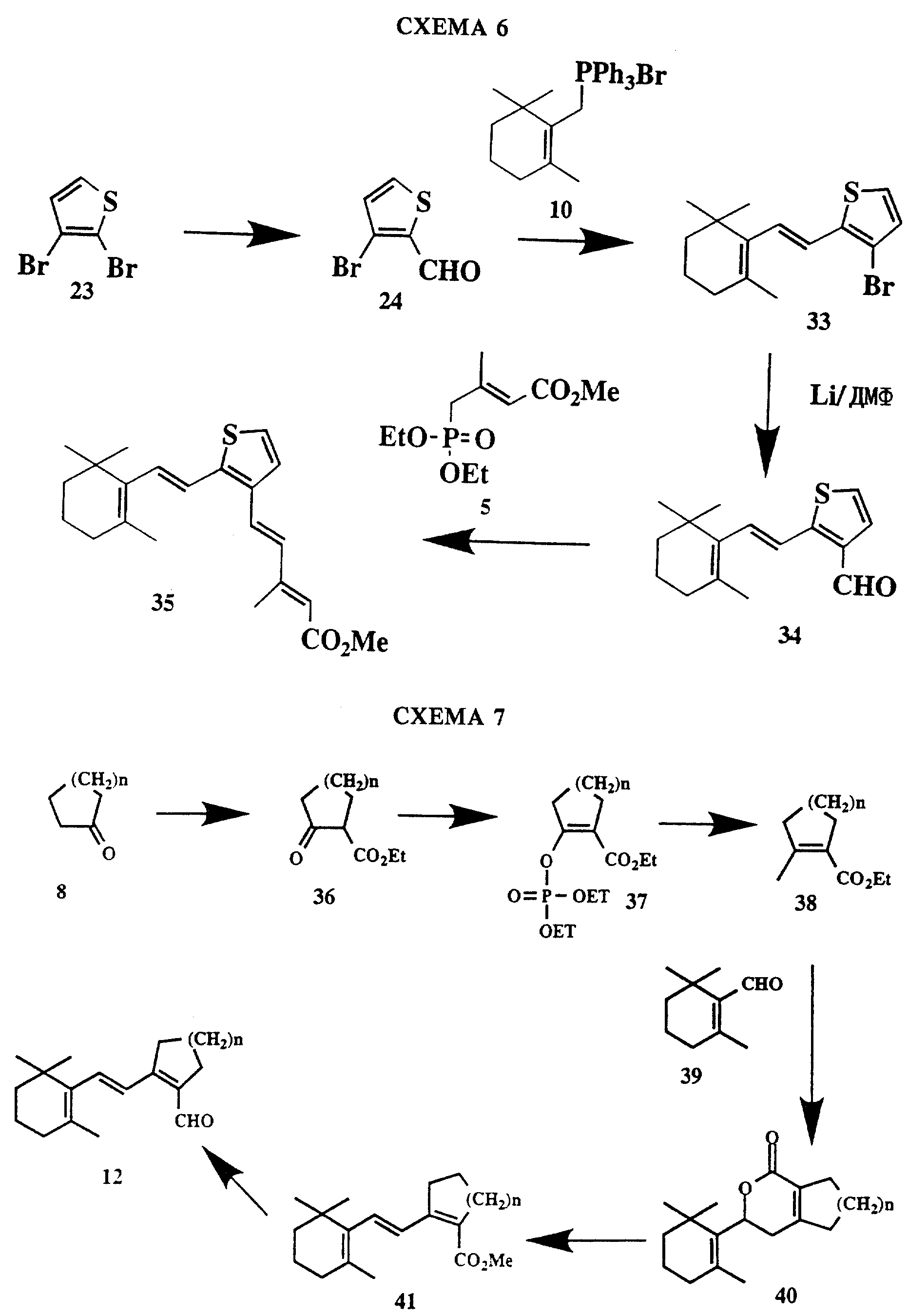
На схеме 7 показан предпочтительный способ получения промежуточного продукта 12 для использования в схеме 3, где R1 и R2 вместе обозначают C3-C13алкилен. Так, кетон 8 превращают в сложный эфир 36 известными способами (см., например, Org. Synthesis, Coll, vol. 5:198 (1973)) и затем трансформируют в энолфосфат 37. В результате последующего замещения фосфатной группы диметилкупратом (J. Org. Chem., 53:2984 (1988)) получают сложный эфир 38. Соединение 38 подвергают воздействию диизопропиламида лития с последующим присоединением циклоцитраля 39, получая лактон 40 (см. Tetr. Letters, 21: 2509 (1980)). Обработкой лактона 40 трет-бутоксидом калия и последующим взаимодействием продукта реакции с метилйодидом получают сложный эфир 41. Путем восстановления с помощью гидрида диизобутилалюминия и последующего окисления двуокисью марганца затем получают альдегид 12.
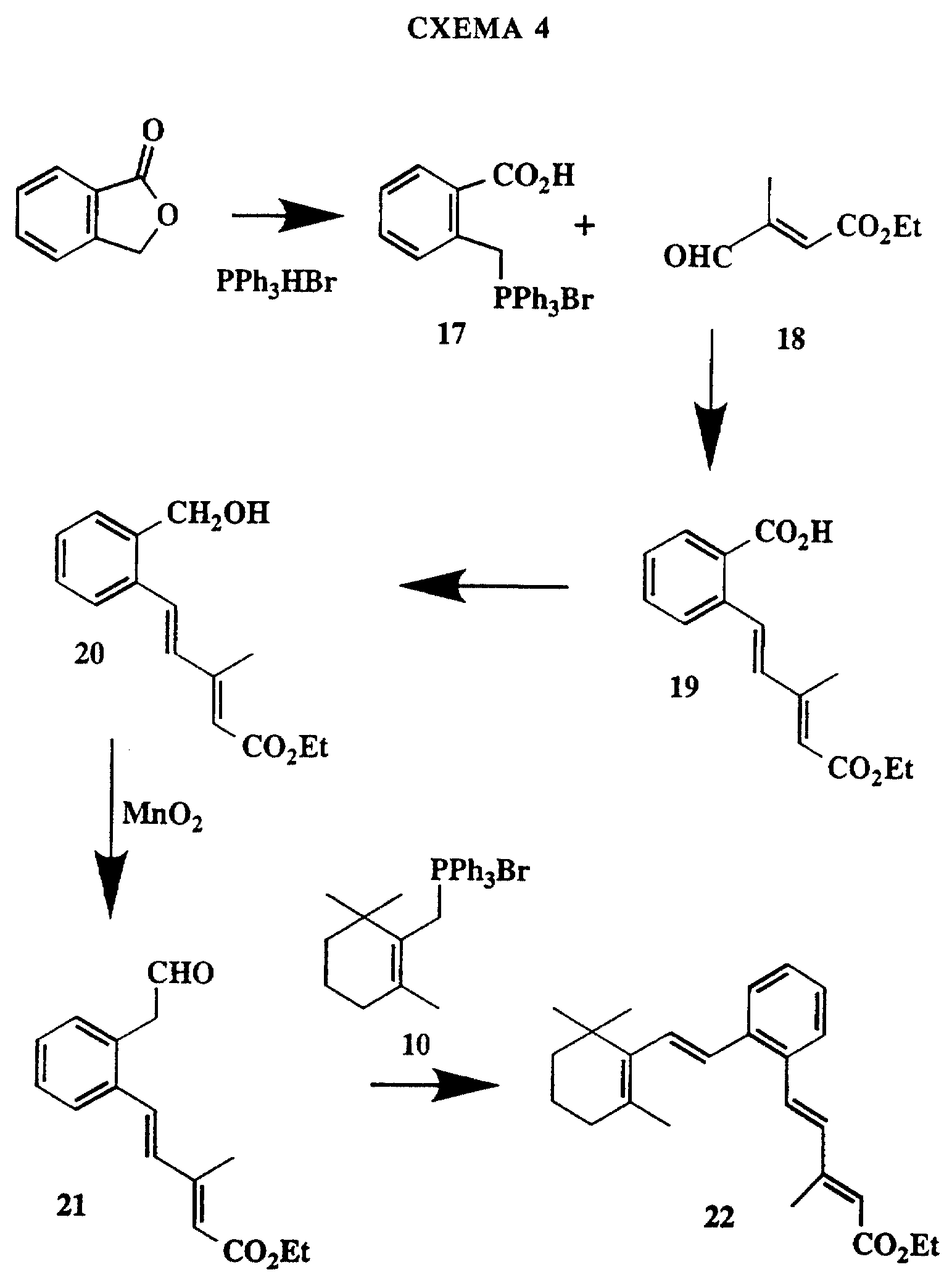
Пути, показанные на схемах 4, 5 и 6, могут быть применены для получения соединений формулы I, в которых R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют 5-6-членные ароматические кольца (например, тиофен, бензол, пиридин и т.д.). Так, на схеме 4 показан путь получения бензолсодержащей структуры 22. Фталид обрабатывают гидробромидом трифенилфосфина, и образовавшуюся соль 17 затем подвергают сочетанию с альдегидом 18 для получения, как показано, эфира кислоты 19. Восстановлением эфира кислоты. 19 с помощью хлорангидрида (не показано) получают бензиловый спирт 20, который затем превращают в альдегид 21 и далее подвергают сочетанию с циклогеранилфосфонийилидом, получая сложный эфир 22, из которого после гидролиза получают кислоту.
Различные производные тиофена получают из пригодного бромтиофена, как показано на схемах 5 и 6. Например, на схеме 5 2,3-дибромтиофен 23 избирательно металлируют и подвергают воздействию диметилформамида, получая альдегид 24, который затем превращают в сложный эфир 25 с помощью триэтилфосфонацетата. Восстановлением эфирной группы получают спирт 26, который защищают путем образования ацеталя 27, который теперь можно вновь металлировать и обрабатывать диметилформамидом, получая альдегид 28. Конденсацией соединения 28 с циклогеранилфосфонийилидом и последующей обработкой кислотой получают спирт 29. Затем спирт 29 окисляют до альдегида 30 с помощью двуокиси марганца, подвергают воздействию метиллития и вновь окисляют двуокисью марганца, получая метилкетон 31. Путем последующего удлинения цепи с помощью триэтилфосфонацетата получают требуемый материал в виде сложного эфира 32, из которого в результате гидролиза получают кислоту. Другие аналоги могут быть получены подобным же образом (см. примеры).
На схеме 6 альдегид 24 подвергают сочетанию с циклогеранилфосфонийилидом 10, получая соединение 33, которое избирательно металлируют и подвергают воздействию диметилформамида, получая альдегид 34. Путем последующего удлинения цепи с помощью триэтилфосфонацетата получают требуемый материал в виде сложного эфира 35, из которого в результате гидролиза получают кислоту.
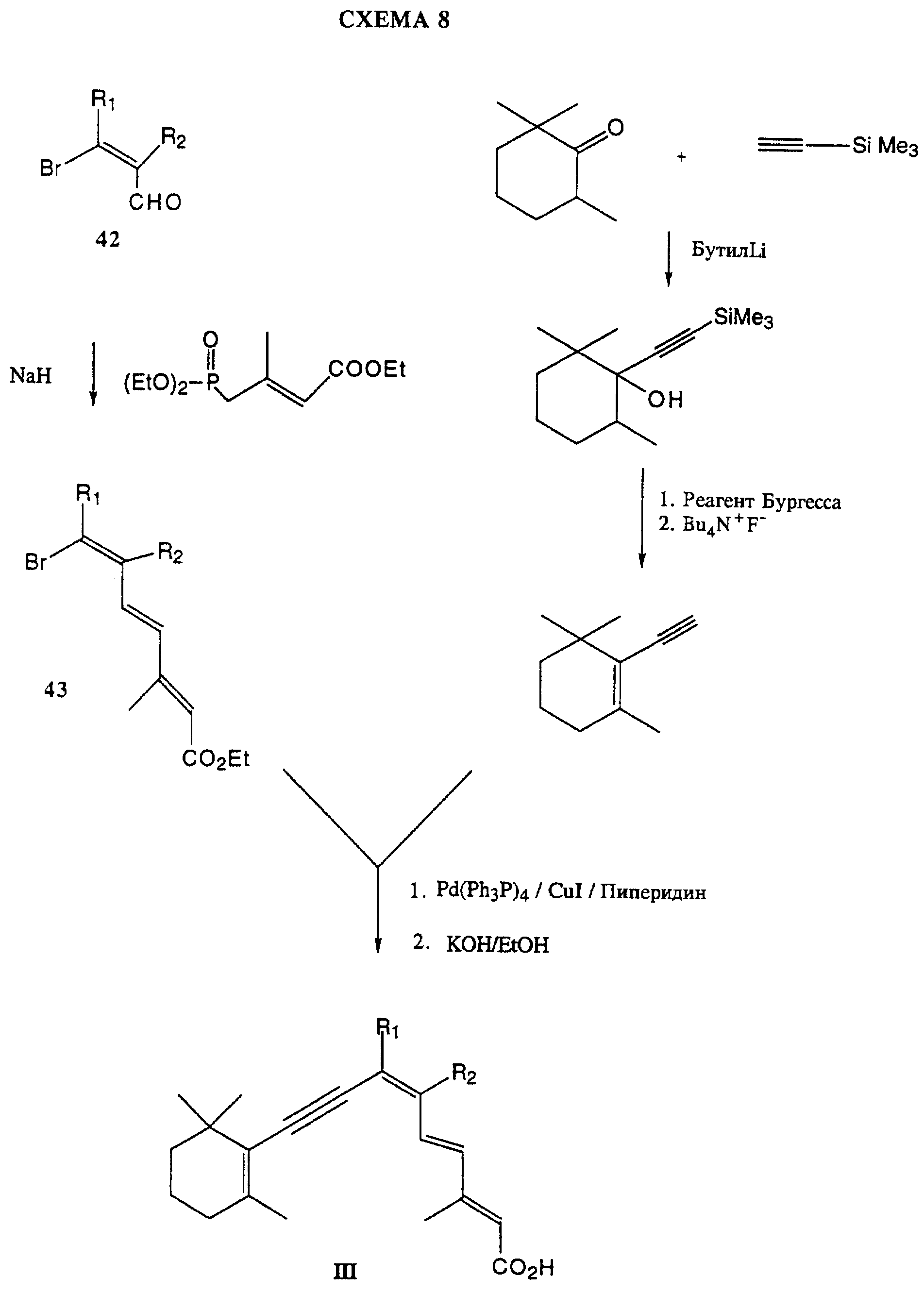
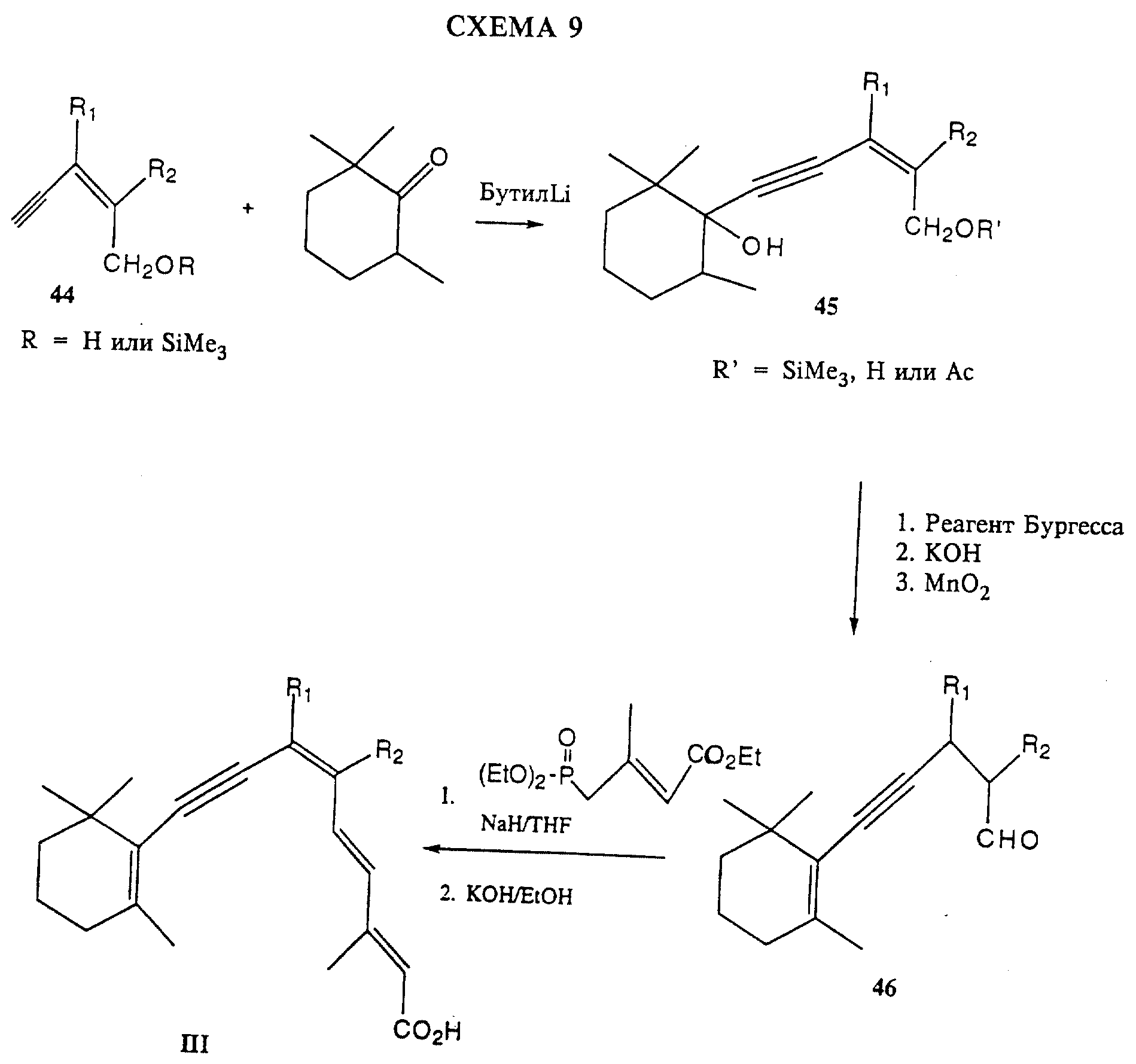
На схемах 8 и 9 показаны предпочтительные способы получения соединений формулы I, где связь C7-C8 является тройной связью.
На схеме 8 альдегид 42 подвергают сочетанию путем реакции Виттига-Хорнера с этиловым эфиром 4-(диэтоксифосфонил)-3-метилбут- 2-еновой кислоты в присутствии основания, например NaH, для получения сложного эфира 43.
2,2,6-триметилциклогексанон подвергают взаимодействию с триметилсилилацетиленом (ТМС-ацетилен) с использованием сильного основания, например бутиллития. В результате удаления воды с помощью реагента Бургесса (гидроксид метоксикарбонилсульфамоилтриэтиламмония, внутренняя соль) и удаления силильной защитной группы с помощью фторида тетрабутиламмония получают 2-этинил-1,3,3-триметил-1-циклогексен.
Это производное циклогексена подвергают сочетанию со сложным эфиром 43, используя в качестве катализатора комплекс Pd(Ph3 P)4/CuI и пиперидин в качестве растворителя. Получившийся сложный эфир затем подвергают гидролизу для получения кислоты III.
На схеме 9 2-этинилбензиловый спирт (соединение 44, где R обозначает H) подвергают взаимодействию с триметилсилилхлоридом. Защищенный спирт (44, R обозначает SiMe3) подвергают взаимодействию с 2,2,6-триметилциклогексаноном после депротонирования с помощью бутиллития, получая продукт присоединения 45, где R' обозначает SiMe3. Путем удаления силильной защитной группы c помощью основания, например водного гидроксида калия, с последующим ацетилированием ацетилхлоридом и триэтиламином получают ацетат 45 (R' обозначает ацетил).
Воду удаляют из производного циклогексанола 45 путем обработки реагентом Бургесса, получая соответствующее производное циклогексена. Далее проводят гидролиз ацетильной группы и окисление с помощью MnO2 первичного спирта, получая альдегид 46. Альдегид 46 подвергают взаимодействию с этиловым эфиром 4- (диэтоксифосфонил)-3-метилбут-2-еновой кислоты в реакции Виттига-Хорнера, получая кислоту III после гидролиза ее соответствующего эфира.
Следующие примеры иллюстрируют настоящее изобретение. В данных примерах концентрирование включает выпаривание растворителя с помощью роторного испарителя при 40oC и в вакууме при 20 мм рт.ст. Аббревиатура ЖХВР относится к жидкостной хроматографии высокого разрешения с использованием хроматографа Waters Prep. 500 с кремниевыми картриджами. Все промежуточные и конечные продукты характеризовали с помощью H-ЯМР-спектроскопии.
Пример 1
Получение (2E,4E,6Z,8E)-3,6,7-триметил-9-(2,6,6-триметил-1- циклогексен -1-ил)-2,4,6,8-нонатетраеновой кислоты
(Полностью-Е)-этил-3-метил-5-(2,6,6-триметил-1-циклогексен-1-ил)- 2,4-пентадиеноат (соединение 1; R1 обозначает Me; 13,1 г) растворяли в тетрагидрофуране (25 мл) и затем добавляли к
свежеприготовленному раствору диизопропиламида лития (1,2 экв. по отношению к исходному сложному эфиру диизопропиламина и н-бутиллития; 1,6M в гексанах), растворенному в тетрагидрофуране (100 мл) при
-70oC. Образовавшийся раствор дополнительно перемешивали в течение 1 ч при этой температуре и затем обрабатывали метилйодидом (10 мл) и давали нагреться до комнатной температуры. Добавляли
насыщенный водный хлорид амммония, и органические материалы отделяли путем экстракции смесью гексан/этилацетат (4:1). При удалении растворителей в вакууме получали алкилированный продукт (14 г) в виде
масла. Этот материал растворяли в метаноле (100 мл), содержащем метоксид натрия (10 мл; 4,6М в метаноле), и нагревали с обратным холодильником в течение 2 ч. После указанного промежутка времени смесь
охлаждали до комнатной температуры, обрабатывали водой и экстрагировали, как указано выше, смесью гексан/этилацетат. При удалении растворителей в вакууме получали неочищенный метил- (2Z,4E)-2,
3-диметил-5-(2,6,6-триметил-1-циклогексен-1-ил)-2,4- пентадиеноат (13,2 г) в виде смеси изомеров 9: 1 относительно двойной связи 2,3. Этот материал растворяли в гексане (150 мл), охлажденном до
-70oC, и затем обрабатывали избытком гидрида диизобутилалюминия (1M в гексане) и нагревали до 0oC. Затем добавляли диэтиловый эфир (200 мл), а затем водный раствор сегнетовой
соли (20%; 20 мл). Затем смесь осторожно нагревали до комнатной температуры, при которой начиналась слабая экзотермическая реакция. Смеси давали нагреться до 35oC и затем охлаждали до
комнатной температуры. Затем добавляли твердый сульфат магния (50 г) и отфильтровывали твердые частицы.
При удалении растворителей в вакууме получали неочищенный спирт в виде масла. Этот материал растворяли в гексане (20 мл) и затем добавляли к суспензии диоксида марганца (102 г) в смеси гексан/эфир (1:1; 400 мл), охлажденной до 5oC, перемешивали в течение 1,5 ч при этой температуре и затем еще в течение 1 ч при комнатной температуре. Твердые частицы отфильтровывали, и раствор концентрировали в вакууме. Хроматографией остатка с помощью ЖХВР (система растворителей 5%-ный эфир/гексан) получали чистый (2Z,4E)-2,3-диметил-5-(2,6,6-триметил-1-циклогексен-1-ил)- 2,4-пентадиенал (6 г; общий выход из этилового эфира 51%).
Гидрид натрия (1,3 г; 64% в масле) промывали гексаном, сушили в вакууме и затем суспендировали в тетрагидрофуране (60 мл) при 5oC. Затем к вышеуказанной суспензии добавляли раствор метил-3-метил-4- диэтилфосфонокротоната в тетрагидрофуране (8,6 г в 30 мл), получая прозрачный раствор натриевой соли (примечание: если получается мутный раствор, то смесь фильтруют через диатомовую землю целит). Затем прозрачный раствор охлаждали до 10oC и обрабатывали вышеуказанным альдегидом (6 г), растворенным в тетрагидрофуране (20 мл), и затем перемешивали при комнатной температуре в течение 30 мин. После этого добавляли гексан и смесь промывали водой, сушили (сульфат магния), фильтровали, освобождая от твердых частиц, и концентрировали. При кристаллизации остатка из гексана получали чистый метил-(2E,4E,6Z,8E) -3,6,7-триметил-9-(2,6,6-триметил-1 -циклогексен-1-ил)-2,4,6,8-нонатетраеноат в виде желтого твердого кристаллического вещества. Этот материал растворяли в смеси этанол/вода (100 мл; 9:1), содержащей гидроксид калия (4 г), и нагревали с обратным холодильником в течение 20 мин. Эту смесь затем охлаждали до комнатной температуры, сливали в холодную водную фосфорную кислоту (2M), и твердую фракцию экстрагировали дихлорметаном. При удалении растворителей в вакууме и кристаллизации остатка из ацетонитрила получали (2E,4E,6Z,8E)- 3,6,7-триметил-9-(2,6, 6-триметил-1-циклогексен-1-ил)-2,4,6,8- нонатетраеновую кислоту (3,8 г) в виде светло-желтого твердого кристаллического вещества. Этот материал имел характерные пики в спектре протонного магнитного резонанса для E,E,Z,E-полиенной системы.
Пример 2
Получение (полностью-E)-6-бром-3,7-диметил-9-(2,6,6- триметил-1-циклогексен-1-ил)-2,4,6,8-нонатетраеновой кислоты
Смесь из (2E,4E)- и (2Z,4Е)-2-бром-3-метил-5-(2,6,6-триметил- 1-циклогексен-1-ил)-2,4-пентадиеновой кислоты (75 г) растворяли в диметилформамиде (500 мл) и медленно добавляли к суспензии гидрида
натрия (10,5 г; 65% в масле) в тетрагидрофуране (1100 мл) при 10oC и затем перемешивали до тех пор, пока полностью не прекращалось выделение водорода. Затем добавляли метилйодид (50 г), и
образовавшуюся смесь нагревали в течение 2 ч при 50-60oC, сливали в смесь льда с водой и экстрагировали гексаном. Гексановые экстракты промывали водой, сушили (сульфат магния) и
концентрировали. Смесь метиловых эфиров затем разделяли с помощью ЖХВР (2,5%-ный эфир/гексан), получая чистый метил-(2Z, 4E)-2-бром-3- метил-5-(2,6,6-триметил-1-циклогексен-1-ил)-2,4-пентадиеноат (20
г) и метил-(2E,4E)-2-бром-3-метил-5-(2,6,6-триметил-1-циклогексен- 1-ил)-2,4-пентадиеноат (37 г).
Метил-(2E, 4E)-2-бром-3-метил-5-(2,6,6-триметил-1-циклогексен- 1-ил)-2,4-пентадиеноат (37 г) восстанавливали до спирта аналогично примеру 1 и затем сразу же подвергали воздействию диоксида марганца, получая нестабильный альдегид (15 г). Затем этот материал превращали в этил-(полностью-E)-6-бром-3,7-диметил-9-(2,6,6-триметил-1-циклогексен- 1-ил)-2,4,6,8-нонатетраеноат и затем гидролизовали до (полностью-E)-6- бром-3,7-диметил-9-(2,6,6-триметил-1-циклогексен-1-ил)-2,4, 6,8- нонатетраеновой кислоты аналогично примеру 1.
Пример 3
Получение (полностью-E)-6-йод-3,7-диметил-9-(2,6,6-триметил-1-циклогексен- 1-ил)-2,4,6,8-нонатетраеновой кислоты
Суспензию из комплекса диметилсульфида с бромидом одновалентной меди (10,3 г) в тетрагидрофуране (500 мл) при 0oC обрабатывали раствором метиллития в эфире (1,4М; низший алкилбромид),
дополнительно перемешивали в течение 15 мин и затем охлаждали до -70oC. К этому холодному прозрачному раствору добавляли этил-(E)-5-(2,6,6- триметил-1 -циклогексен-1-ил)-4-пентен-2-иноат
(10 г), растворенный в тетрагидрофуране (50 мл), и эту смесь перемешивали в течение 2 ч и затем обрабатывали раствором йода в тетрагидрофуране (13 г в 100 мл) в течение 0,5 ч. По истечении этого
периода времени смесь нагревали до -60oC, сливали в раствор водного хлорида аммония, и органические материалы экстрагировали гексаном. Гексановые экстракты промывали разбавленным водным
раствором тиосульфата натрия, сушили (сульфат натрия) и концентрировали при комнатной температуре, получая нестабильный йодзамещенный сложный эфир, который очищали с помощью ЖХВР (2,5%-ный
эфир/гексан), получая чистый этил-(2E, 4E)-2-йод-3-метил-5-(2,6,6- триметил-1-циклогексен-1-ил)-2,4-пентадиеноат (5 г). Этот сложный эфир трансформировали в соответствующий альдегид и затем подвергали
сочетанию с фосфонатом, как в предыдущих примерах, и образовавшийся аналог сложного эфира ретиноевой кислоты гидролизовали, получая после кристаллизации из смеси гексан/тетрагидрофуран чистую
(полностью-Е)-6-йод-3,7-диметил-9- (2,6,6-триметил-1-циклогексен-1-ил)-2,4,6,8-нонатетраеновую кислоту в виде светло-желтого твердого вещества.
Пример 4
Получение (2E,
4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1- циклогексен-1-ил)этенил)-1-циклогексен-1-ил)-2,4-пентадиеновой кислоты
Суспензию ((2,6,6-триметил-1-циклогексен-1- ил)метил)трифенилфосфонийхлорида
(18 г) в тетрагидрофуране (300 мл) охлаждали до -60oC, обрабатывали, добавляя по каплям н-бутиллитий (1,4 М в гексане; 32 мл), и затем перемешивали еще в течение 10 мин. К этому холодному
раствору добавляли 2-бромциклогексен-1-карбоксальдегид (8,5 г; 75%-ной чистоты по H- ЯМР-анализу), растворенный в тетрагидрофуране, и этой смеси давали нагреться до комнатной температуры, после чего
ее перемешивали еще в течение 2 ч. Затем к реакционной смеси добавляли гексан, и образовавшуюся смесь промывали водой, 50%-ным водным метанолом и сушили (сульфат магния). При удалении растворителей
получали неочищенный (Е)-1-бром-2-(2-(2,6,6-триметил-1-циклогексен-1-ил) этенил) циклогексен (12,5 г) в виде смеси изомеров, которую использовали в таком виде на следующей стадии. Стандартным
переметаллированием и взаимодействием с ДМФ получали неочищенный альдегид (4,5 г), который растворяли в тетрагидрофуране (100 мл) и подвергали взаимодействию с избытком фосфоната аналогично примеру 1,
получая после очистки с помощью ЖХВР (2,5%-ный эфир в гексане) метил-(2E,4E)-3-метил-5-(2-((E)-2- (2,6,6-триметил-1-циклогексен-1-ил)этенил)-1-циклогексен-1-ил)- 2,4-пентадиеноат (3 г) в виде масла.
Путем гидролиза водным раствором гидроксида калия получали кристаллическую (2E,4E)-3- метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-1- циклогексен-1-ил)-2,4-пентадиеновую кислоту (1,6 г
из гексана/тетрагидрофурана).
Пример 5
Получение (2E, 4E)-3-метил-5-(2-((E)-2- (2,6,6-триметил-1-циклогексен-1-ил)этенил)-1- циклопентен-1-ил)-2,4-пентадиеновой кислоты
Аналогично примеру 4 2-бромциклопентен-1-карбоксальдегид превращали в этиловый эфир (2E,4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен -1-ил)этенил)-1-циклопентен-1-ил)-2,4-пентадиеновой
кислоты, из которого после гидролиза щелочью получали (2E,4E)-3-метил-5-(2- ((E)-2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-1-циклопентен-1- ил)-2,4-пентадиеновую кислоту.
Пример
6
Получение (2E, 4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1- циклогексен-1-ил)этенил)-1-циклогептен-1-ил)-2,4-пентадиеновой кислоты
Аналогично примерам 4 и 5
2-бромциклогептен-1-карбоксальдегид превращали в этиловый эфир (2E,4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен-1- ил) этенил)-1-циклогептен-1-ил)-2,4-пентадиеновой кислоты, из которого после
гидролиза щелочью получали (2E,4E)-3-метил-5- (2-((E)-2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-1-циклогептен- 1-ил)-2,4-пентадиеновую кислоту.
Пример 7
Получение (2E,
4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1- циклогексен-1-ил)этенил)-1-циклооктен-1-ил)-2,4-пентадиеновой кислоты
Аналогично вышеприведенным примерам 2-бромциклооктен-1- карбоксальдегид
превращали в этиловый эфир (2E,4E)-3-метил-5-(2- ((E)-2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-1-циклооктен-1- ил)-2,4-пентадиеновой кислоты, из которого после гидролиза щелочью получали (2E,
4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1- циклогексен-1-ил)этенил)-1-циклооктен-1-ил)-2,4-пентадиеновую кислоту.
Пример 8
Получение (2E, 4E)-3-метил-5-(2-(2-((E)-2- (2,6,
6-триметил-1-циклогексен-1-ил)этенил)-1-фенил)-2,4- пентадиеновой кислоты
Как показано на схеме 4, фталид (0,1 моль) и трифенилфосфингидробромид (0,1 моль) выдерживали при 200oC в
течение 2 ч, охлаждали до комнатной температуры и затем обрабатывали горячим ацетонитрилом, получая соль 17 (39 г) в виде белого твердого вещества. Раствор этой соли (4,6 г) в диметилсульфоксиде (ДМСО;
50 мл) при 5oС обрабатывали раствором натриевой соли ДМСО (1M; 20 мл), а затем альдегидом 18 (1,5 г), получая реакционную смесь светло-желтого цвета. Затем добавляли воду, и образовавшуюся
смесь подкисляли водной фосфорной кислотой (2M), и путем экстракции этилацетатом выделяли кислоту 19. Неочищенный продукт (4,3 г) растворяли в бензоле и подвергали воздействию оксалилхлорида (3 мл) и
диметилформамида (3 капли) и затем выдерживали при комнатной температуре в течение 30 мин. При удалении растворителей в вакууме и последующем растворении неочищенного хлорангидрида кислоты в
тетрагидрофуране (100 мл) после добавления боргидрида натрия при -20oC и нагревания до комнатной температуры получали спирт 20. Путем очистки неочищенного спирта с помощью ЖХВР и
последующей кристаллизации из смеси гексан/этилацетат получали этиловый эфир 3-метил-5-(2- гидроксиметилфенил)-2,4-пентадиеновой кислоты (соединение 20) в виде бесцветного твердого вещества (1,1 г).
Спирт 20 (1,1 г) подвергали воздействию суспензии диоксида марганца (11 г) в гексане/дихлорметане (5:1; 60 мл) при 0oC с последующим перемешиванием еще в течение 2 ч при комнатной
температуре, получая альдегид 21 (1,1 г). Затем раствор н-бутиллития и фосфонийилида, полученного из циклогераниалфосфонийбромида 10 (1,3 г), в тетрагидрофуране (20 мл) при 10oC
обрабатывали неочищенным альдегидом 21 (1,0 г) и нагревали до комнатной температуры в течение 15-30 мин. Разбавлением водой и экстракцией органических материалов смесью гексан/этилацетат (4:1)
получали неочищенный продукт присоединения 22 в виде масла. Этот материал в гексане пропускали через силикагелевый наполнитель, получая сложный эфир (0,8 г) в виде масла. Путем гидролиза этого
материала водным гидроксидом калия в кипящем этаноле получали кислоту после подкисления минеральной кислотой (2M фосфорная кислота). Кристаллизацией из смеси гексан/этилацетат получали чистую (2E,
4E)-3-метил-5-(2-(2- ((E)-2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-1-фенил)-2,4- пентадиеновую кислоту в виде белого твердого вещества.
Пример 9
Получение (2E,
4E)-3-метил-5-(3-((E)-2-(2,6,6-триметил-1- циклогексен -1-ил) этенил)-2-тиенил)-2,4-пентадиеновой кислоты
Раствор 2,3-дибромтиофена (0,1 моль) в диэтиловом эфире (приблизительно 5%-ной
концентрации) обрабатывали н-бутиллитием (1,6M в гексане; 1,1 экв.) при -70oC и перемешивали в течение 30 мин. Затем добавляли избыток диметилформамида, и смесь нагревали до комнатной
температуры, выливали в воду и затем подкисляли разбавленной водной фосфорной кислотой. Экстракцией гексаном получали неочищенный альдегид, который затем очищали с помощью ЖХВР. Все количество
альдегида из этого эксперимента затем добавляли к избытку натриевой соли триэтилфосфонацетата в тетрагидрофуране, и смесь перемешивали при комнатной температуре в течение 1 ч и затем выливали в воду и
уксусную кислоту. Неочищенный сложный эфир 25 затем добавляли к избытку гидрида диизобутилалюминия в гексане при -40oC, и эту смесь затем нагревали до 0oC, выливали в водный
раствор сегнетовой соли (20%; 10 мл) и нагревали до 32oC. Последующей экстракцией гексаном получали спирт 26, который затем превращали с помощью 2- метоксипропена в ацеталь 27. После
очистки ацеталя с помощью ЖХВР весь продукт растворяли в тетрагидрофуране (≈ 5%-ной концентрации), охлажденном до -70oC, и затем обрабатывали, как указано ранее, н-бутиллитием. После
перемешивания еще в течение 30 мин при указанной температуре добавляли избыток диметилформамида, смесь нагревали до комнатной температуры и выливали в водный раствор хлорида аммония (20%), получая
после выделения с помощью гексана и очистки ЖХВР альдегид 28. Затем этот альдегид подвергали воздействию избытка (1,2 экв.) илида, образованного из циклогераниалфосфонийбромида, и н-бутиллития в
тетрагидрофуране и гексана при -70oC и затем перемешивали еще в течение 2 ч при комнатной температуре. При последующем добавлении воды и водной разбавленной фосфорной кислоты после
экстракции гексаном получали спирт 29. Этот материал растворяли в диэтиловом эфире (минимальный объем) и затем добавляли к суспензии диоксида марганца (10-кратное количество) в избытке эфира (300 мл)
при 0oC и нагревали до комнатной температуры. После перемешивания еще в течение 1 ч твердые частицы отфильтровывали, и при удалении растворителя после очистки с помощью ЖХВР получали
альдегид 30. Затем этот материал растворяли в эфире, охлажденном до -10oC, и подвергали воздействию избытка метиллития в эфире (1,4 экв.) и после этого нагревали до комнатной температуры.
Добавлением воды и концентрированием органической фазы получали неочищенный спирт, который окисляли диоксидом марганца, как описано выше, получая кетон 31. Этот материал подвергали воздействию избытка
натриевой соли триэтилфосфонацетата в тетрагидрофуране при комнатной температуре, получая требуемый сложный эфир 32 (R обозначает этил) в виде смеси изомеров относительно вновь образованной двойной
связи (≈ 4: 1) с преобладанием требуемого изомера. Очисткой этой смеси с помощью ЖХВР и кристаллизацией основного изомера из гексана получали чистый этил-(2E, 4E)-3-метил-5-(3-((E)-2-(2,6,
6- триметил-1-циклогексен-1-ил)этенил)-2-тиенил)-2,4-пентадиеноат. Путем гидролиза водным раствором гидроксида калия в кипящем этаноле аналогично примеру 8 после кристаллизации из смеси
тетрагидрофуран/гексан получали чистую (2E,4E)-3-метил-5-(3-((E)- 2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-2-тиенил)-2,4-пентадиеновую кислоту.
Пример 10
Получение (2E,
4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен- 1-ил)этенил)-3-тиенил)-2,4-пентадиеновой кислоты
Аналогично примеру 9 2,3-дибромтиофен превращали в альдегид 24 и затем подвергали
воздействию илида, полученного из циклогераниалфосфонийбромида 10, и н-бутиллития, получая продукт присоединения 33. Обработкой бромсодержащего соединения 33 н-бутиллитием в смеси
гексан/тетрагидрофуран, как в примере 9, с последующей обработкой избытком диметилформамида получали альдегид 34 после воздействия водной фосфорной кислотой. Этот материал затем подвергали сочетанию с
фосфонатом 5, как в примере 1, получая метил-(2E, 4E)-3-метил-5-(2-((E)-2-(2,6,6- триметил-1-циклогексен-1-ил)этенил)-3-тиенил)-2,4-пентадиеноат 35 (R обозначает метил) в виде смеси изомеров
относительно концевой двойной связи. Путем очистки с помощью ЖХВР получали метил- (2E,4E)-3-метил-5-(2-((E)-2-(2,6,6-триметил-1-циклогексен-1- ил)этенил)-3-тиенил)-2,4-пентадиеноат, из которого после
гидролиза, как указано выше, после кристаллизации из смеси тетрагидрофуран/гексан получали чистую (2E, 4E)-3-метил-5-(2-((E)- 2-(2,6,6-триметил-1-циклогексен-1-ил)этенил)-3-тиенил)-2,4- пентадиеновую
кислоту.
Пример 11
Получение (2E, 4E)-3-метил-5-(4-((E)-2-(2,6,6-триметил-1- циклогексен-1-ил)этенил)-3-тиенил)-2,4-пентадиеновой кислоты
Путем обработки 3,
4-дибромтиофеном аналогично примеру 10 получали (2E, 4E)-3-метил-5-(4-((E)-2-(2,6,6- триметил-1-циклогексен-1-ил)этенил)-3-тиенил) -2,4-пентадиеновую кислоту.
Пример 12
Получение (полностью-E)-6-хлор-3,7-диметил-9- (2,6,6-триметил-1-циклогексен-1-ил)-2,4,6,8-нонатетраеновой кислоты
Раствор триэтил-2-хлорфосфоноацетата (J. Org. Chem., 51:5467 (1986)) (0,12M)
в тетрагидрофуране обрабатывали гидридом натрия (64%-ная суспензия в масле; 0,12M) и перемешивали при комнатной температуре, получая прозрачный раствор аниона. К этой смеси добавляли β-ионон (0,
1M), и реакционную смесь выдерживали при 45oC в течение ночи. Затем добавляли воду, и продукты экстрагировали смесью гексан/этилацетат (4:1). При удалении растворителей получали продукт
сочетания в виде смеси (1:1) изомеров относительно двойной связи, из которой с помощью ЖХВР выделяли требуемый изомер: этил-(2E,4E)-2-хлор-3-метил-5- (2,6,6-триметил-1 -циклогексен-1 -ил)-2,
4-пентадиеноат. Восстановлением этого эфира с помощью гидрида диизобутилалюминия аналогично примеру 1 с последующим окислением диоксидом марганца, как указано выше, получали (2E,
4E)-2-хлор-3-метил-5-(2,6,6-триметил- 1-циклогексен-1-ил)-2,4-пентадиенал. Затем этот материал подвергали сочетанию с метил-3-метил-4-диэтилфосфонокротонатом аналогично примеру 1, получая
метил-(полностью-Е)-6-хлор-3,7-диметил-9- (2,6,6-триметил-1-циклогексен-1-ил)-2,4,6,8-нонатетраеноат. Путем гидролиза этого сложного эфира, как указано выше, и кристаллизации кислоты из смеси
тетрагидрофурана и гексана получали чистую (полностью-Е)-6-хлор-3,7-диметил-9-(2,6,6-триметил-1-циклогексен- 1-ил)-2,4,6,8-нонатетраеновую кислоту в виде твердого вещества желтого цвета.
Пример 13
Получение (полностью-E)-3,7-диметил-9- [(2-метокси-4-метил-6-октилокси)фенил]-2,4,6,8-нонатетраеновой кислоты
Раствор 2,6-дигидрокси-4-метилбензойной кислоты в
ацетоне (50,4 г; 750 мл) обрабатывали метилйодидом (100 мл) и карбонатом калия (124,2 г) и нагревали с обратным холодильником в течение 18 ч. Затем смесь охлаждали до комнатной температуры и удаляли
твердые частицы фильтрованием. Концентрированием раствора получали неочищенный продукт, который растворяли в этилацетате и промывали водным раствором гидроксида натрия (1H; холодный). При последующем
удалении растворителей в вакууме получали метил-2,6-диметокси-4- метилбензоат (54 г). Этот материал (52,5 г) растворяли в дихлорметане (1000 мл), охлажденном до -70oC, и обрабатывали
раствором боронтрихлорида в этом же растворителе (29,3 г/100 мл; 180 мл). Затем эту смесь нагревали до комнатной температуры, перемешивали еще в течение 45 мин и затем сливали на лед. Органический
слой отделяли, промывали большим количеством воды, сушили (сульфат магния), фильтровали и концентрировали.
Затем остаток очищали с помощью препаративной жидкостной хроматографии высокого разрешения (ЖХВР), используя хроматограф Waters Prep. 500 (элюент 7%-ный этилацетат/гексан), получая чистый метил-2-гидрокси- 6-метокси-4-метилбензоат в виде твердого вещества (28,3 г). Раствор этого материала (35 г) в метилэтилкетоне (700 мл), содержащий карбонат калия (27,1 г), обрабатывали 1-октилйодидом (34,2 г) и нагревали с обратным холодильником в течение 20 ч. Реакционную смесь охлаждали, фильтровали для удаления твердых частиц, фильтрат концентрировали и затем повторно растворяли в смеси гексана и этилацетата. Затем этот раствор промывали водным раствором щелочи (1H гидроксид натрия), водой, сушили (сульфат магния) и концентрировали досуха. Путем очистки с помощью ЖХВР, как указано выше, получали чистый метил-2-метокси-6-октилокси-4- метилбензоат (51,8 г).
Раствор этого материала (51,8 г) в толуоле (500 мл) охлаждали до -60oC и обрабатывали раствором гидрида диизобутилалюминия (25% в гексане; 281 мл) и затем нагревали до комнатной температуры. Затем реакционную смесь осторожно обрабатывали водным метанолом (100 мл; 1:1), охлаждая так, чтобы температура поддерживалась на уровне 20oC, с последующим добавлением гексана (500 мл) и сульфата магния. Затем твердые частицы отфильтровывали, и растворители удаляли в вакууме, получая неочищенный спирт. Этот материал (47 г) растворяли в ацетонитриле (500 мл), содержащем трифенилфосфонийгидробромид (54,9 г), и нагревали с обратным холодильником в течение 22 ч. Затем растворители удаляли в вакууме, и неочищенную соль сушили при комнатной температуре и давлении 0,1 мм (этот материал может быть выкристаллизован из тетрагидрофурана с получением чистого [(2-метокси-6-октилокси-4-метил)фенил]метилтрифенилфосфонийбромида). Эту соль затем превращали в (полностью-Е)-3, 7- диметил-9-[(2-метокси-4-метил-6-октилокси)фенил] -2,4,6,8- нонатетраеновую кислоту, как описано в патенте США 4894480, пример 5.
Пример 14
Получение (Е)-2-(2-(2,6,
6-триметил-1-циклогексен-1- ил)этенил)-циклогептенкарбоксальдегида
Как показано на схеме 7, циклогептанон (152 г) превращали в сложный эфир (Org. Syn. Coll. , 5:198 (1973); 232 г),
трансформировали в энолфосфат 37 (274 г) и затем обрабатывали диметилкупратом лития, получая этил-2- метилциклогепт-1-еноат 38 (119 г). Раствор этил-2-метилциклогепт-1-еноата (91 г) в тетрагидрофуране
затем добавляли к раствору диизопропиламида лития (0,53 моль) и затем подвергали взаимодействию с циклоцитралем 39 (76 г), получая после обработки кислотой лактон 40 (113 г). Соединение 40 подвергали
воздействию трет-бутоксида калия в тетрагидрофуране с последующей обработкой избытком метилйодида, получая метиловый эфир 41 (111 г). Восстановлением указанного сложного эфира гидридом
диизобутилалюминия с последующим окислением с помощью диоксида марганца, как в примере 1, получали альдегид 12 (n = 3) (98 г).
Пример 15
А: Получение этилового эфира (2E,
4E)-3-метил-5-[2- (2,6,6-триметилциклогекс-1-енилэтинил) циклопент-1-енил] пента- 2,4-диеновой кислоты
Этиловый эфир (2E,4E)-5-(2-бромциклопент-1-енил)-3-метилпента- 2,4-диеновой кислоты (1,
43 г) растворяли в 5 мл бензола. При комнатной температуре последовательно добавляли 293 мг Pd(Ph3P)4, 95 мг CuI, 147 мг (Ph)3P и 8 мл пиперидина. К этой смеси через
капельную воронку в течение 1 ч добавляли 2-этинил- 1,3,3-триметил-1-циклогексен (745 мг), растворенный в 5 мл бензола. Через 4,5 ч дополнительно добавляли ацетилен (350 мг) и продолжали перемешивание
в течение 30 мин. Эту смесь затем сливали на ледяную крошку/HCl, экстрагировали EtOEt, дважды промывали водой, сушили над Na2SO4 и упаривали досуха. Путем хроматографии среднего
давления (SiO2, гексан/AcOEt = 98/2) получали 1,478 г этилового эфира (2E,4E)-3-метил-5-[2-(2,6,6-триметилциклогекс-1- енилэтинил) циклопент-1-енил] пента-2,4-диеновой кислоты в виде
желтого масла.
Необходимый этиловый эфир (2E,4E)-5-(2-бромциклопент-1-енил)-3-метилпента-2,4-диеновой кислоты синтезировали в соответствии с приведенным ниже процессом.
2,03 г NaH (50% в минеральном масле) суспендировали в 120 мл ДМФ. Добавляли при 0oC этиловый эфир 4-(диэтоксифосфинил)-3- метилбут-2-еновой кислоты (12,9 г). Смесь перемешивали в течение 15
мин при 0oC и в течение 30 мин при комнатной температуре. После повторного охлаждения до 0oC 2-бромциклопент-1-енкарбальдегид (5,72 г), растворенный в 11 мл
ДМФ,
добавляли по каплям и давали прореагировать в течение 10 мин при 0oC и в течение 2 ч при комнатной температуре. Затем смесь сливали на ледяную крошку, экстрагировали EtOEt, промывали
насыщенным раствором NaCl, сушили над Na2SO4 и упаривали досуха. Очисткой остатка с помощью быстрой хроматографии (силикагель, гексан/AcOEt= 97/3) и кристаллизацией из смеси
гексан/следовые количества AcOEt в результате получали 3,408 г чистого этилового эфира (2E,4E)-5-(2-бромциклопент-1-енил)-3-метилпента-2,4-диеновой кислоты в виде желтоватых кристаллов с температурой
плавления 85-86oC.
Необходимый 2-этинил-1,3,3-триметил-1-циклогексен синтезировали в соответствии со стандартными методами путем добавления Li-производного триметилсилилацетилена к 2,2,6- триметилциклогексанону, дегидратации путем обработки реагентом Бургесса (гидроксид метоксикарбонилсульфамоилтриэтиламмония, внутренняя соль) и десилилирования с помощью фторида тетрабутиламмония. Его необходимо немедленно использовать вследствие его нестабильности.
Б: Получение (2E,4E)-3-метил-5-[2-(2,6,
6-триметилциклогекс-1- енилэтинил)циклопент-1-енил]пента-2,4-диеновой кислоты
Этиловый эфир (2E, 4E)-3-метил-5-[2-(2,6,6- триметилциклогекс-1-енилэтинил)циклопент-1-енил] пента-2,4- диеновой
кислоты (1,47 г) растворяли в 14 мл ТГФ/EtOH = 1/1. Добавляли водный 3H NaOH (7 мл), и реакционную колбу выдерживали в темноте. После перемешивания в течение 16 ч при температуре окружающей среды
смесь сливали на ледяную крошку/HCl, дважды экстрагировали EtOEt, промывали водой, сушили над Na2SO4 и упаривали досуха. Кристаллизацией из смеси EtOEt/пентан получали 1,31 г (2E,
4E)-3-метил-5- [2-(2,6,6-триметилциклогекс-1-енилэтинил)циклопент-1-енил] пента-2,4-диеновой кислоты в виде желтых кристаллов с температурой плавления 173-174oC.
Пример
16
По аналогии с примером 15 получали (2E,4E)-3-метил-5-[2-(2,6,6-триметилциклогекс-1-енилэтинил)циклогепт-1-енил]пента-2,4-диеновую кислоту в виде желтых кристаллов с температурой плавления
166-167oC.
Пример 17
Получение (2E, 4E)-3-метил-5-[2-(2,6,6-триметилциклогекс-1- енилэтинил)фенил]пента-2,4-диеновой кислоты
229 мг этилового эфира (2E,
4E)-3-метил-5-[2-(2,6,6- триметилциклогекс-1-енилэтинил)фенил] пента-2,4-диеновой кислоты растворяли в 8 мл этанола. После добавления раствора, содержащего 411 мг гидроксида калия в 2,5 мл воды,
реакционную смесь перемешивали при 50oC в течение 2 ч. Затем смесь сливали на лед/воду, подкисляли 2H HCl и экстрагировали этилацетатом. Органическую фазу промывали водой, сушили над
сульфатом натрия и упаривали, получая бледно-желтые кристаллы, которые перекристаллизовывали из смеси этилацетат/гексан. На выходе получали 93 мг (2E,4E)-3-метил-5- [2-(2,6,
6-триметилциклогекс-1-енилэтинил)фенил] пента-2,4-диеновой кислоты с температурой плавления 194-196oC.
Используемый в этом примере исходный материал получали следующим образом.
970 мг 2-этинилбензилового спирта растворяли в 50 мл диэтилового эфира. Добавляли 1,9 мл триэтиламина, а затем по каплям добавляли 0,9 мл триметилсилилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, сливали на лед/5%-ный водный бикарбонат натрия и экстрагировали эфиром. Маслянистый остаток, полученный после высушивания и выпаривания растворителя, перегоняли, получая 1,3 г 2-этинилбензилтриметилсилилового эфира в виде бесцветного масла с температурой кипения 85-89oC при 0,8 мм.
Это бесцветное масло растворяли в 5 мл тетрагидрофурана. После добавления по каплям 3,9 мл бутиллития (1,6М в гексане) при -78oC реакционную смесь перемешивали при этой температуре в течение 30 мин. По каплям добавляли раствор, содержащий 0,59 г 2,2,6- триметилциклогексанона в 4 мл тетрагидрофурана, и реакционную смесь перемешивали при комнатной температуре в течение 5 ч, получая желтый раствор. Желтый раствор сливали на ледяную воду/10%-ный водный хлорид аммония, экстрагировали гексаном, сушили над сульфатом натрия и упаривали. Желтый маслянистый остаток очищали с помощью быстрой хроматографии (SiO2, гексан/5%-ный этилацетат), получая 1,7 г желтого масла.
Желтое масло растворяли в 90 мл тетрагидрофурана и перемешивали в течение 6 ч при комнатной температуре с 28,8 мл 0,5H водного раствора гидроксида калия. Реакционную смесь сливали на лед/воду, экстрагировали эфиром, промывали водой, сушили и упаривали. Путем очистки остатка с помощью быстрой хроматографии (SiO2, гексан/этилацетат = 7:3) и кристаллизацией из смеси этилацетат/гексан получали 1,1 г белых кристаллов с температурой плавления 120-121oC.
Белые кристаллы растворяли в 10 мл тетрагидрофурана и последовательно обрабатывали 679 мг триэтиламина и 400 мг ацетилхлорида. После 4 ч перемешивания при комнатной температуре реакционную смесь сливали на лед/1H HCl, экстрагировали эфиром, сушили над сульфатом натрия и упаривали. Путем быстрой хроматографии (SiO2, гексан/этилацетат = 4:1) получали 0,8 г бесцветного масла.
Бесцветное масло растворяли в 13 мл бензола и добавляли к раствору, содержащему 1,3 г реактива Бургесса (гидроксид метоксикарбонилсульфамоилтриэтиламмония, внутренняя соль) в 40 мл бензола. Реакционную смесь нагревали до 60oC в течение 3 ч. После отгонки большей части растворителя остаток растворяли в ледяной воде и экстрагировали эфиром. Остаток, полученный после высушивания и выпаривания растворителя, очищали с помощью быстрой хроматографии (SiO2, гексан/этилацетат = 9:1), получая 0,7 г светло-желтого масла.
Это желтое масло растворяли в 18 мл этанола и обрабатывали раствором, содержащим 0,78 г гидроксида калия в 4 мл воды, при 45oC в течение 2,5 ч. Реакционную смесь сливали на лед/насыщенный раствор хлорида аммония, экстрагировали эфиром, сушили и упаривали. Остаток очищали с помощью жидкостной хроматографии среднего давления (SiO2, гексан/этилацетат = 9:1), получая 0,46 г светло-желтого масла. Это желтое масло растворяли в 20 мл метиленхлорида и обрабатывали 1,6 г диоксида марганца в течение 15 ч с интенсивным перемешиванием при комнатной температуре. Диоксид марганца отфильтровывали, фильтрат упаривали, и остаток очищали с помощью хроматографии среднего давления ((SiO2, гексан/2%-ный этилацетат), получая 225 мг 2-(2,6,6-триметилциклогекс-1- енилэтинил)бензальдегида в виде желтого масла, которое затвердевало на холоде.
85 мг гидрида натрия (50% в минеральном масле) промывали пентаном и суспендировали в 4 мл тетрагидрофурана. 470 мг этилового эфира 4-(диэтоксифосфонил)-3- метилбут-2-еновой кислоты, растворенного в 4 мл тетрагидрофурана, добавляли по каплям при 0oC, и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После повторного охлаждения до 0oC по каплям добавляли раствор, содержащий 225 мг желтого альдегида, полученного на предыдущей стадии, в 3 мл тетрагидрофурана. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, сливали на лед/насыщенный раствор хлорида аммония, экстрагировали эфиром, промывали водой, сушили и упаривали. Остаток очищали сначала с помощью быстрой хроматографии (SiO2, гексан/5%-ный этилацетат), а затем с помощью жидкостной хроматографии среднего давления (SiO2, гексан/2%-ный этилацетат), получая 229 мг этилового эфира (2E, 4E)-3-метил-5-[2-(2,6,6- триметилциклогекс-1-енилэтинил)фенил] пента-2,4-диеновой кислоты в виде бесцветного масла.
Пример 18
Под воздействием ретиноидов клетки
HL-60 (линия клеток лейкемии спинного мозга человека) дифференцируются в гранулоциты. Эта дифференцировка клеток HL-60 в гранулоциты осуществляется посредством RARα, что обеспечивает тем самым
основу для использования ретиноидов при лечении лейкемии. Результаты этого опыта показывают, что избирательные по отношению к RXR соединения по изобретению в дозах, при которых они неактивны сами по
себе, способны усиливать практически на порядок продифференциирующие эффекты полностью-транс-ретиноевой кислоты и других избирательных по отношению к RARα ретиноидов, имеющих следующую
формулу:
Способность тестирования соединений к трансактивации ретиновых рецепторов представлена в табл. 1.
Дифференцировка клеток HL-60
Индуцируемую ретиноидами дифференцировку клеток HL-60 определяли путем измерения импульсной активности их окислительного
потенциала при восстановлении NBT (нитросинего тетразолия) [Pick и др., J.Reticuloendothelial Soc. 30:581-593 (1981)].
Клетки HL-60 выращивали в среде RPMI 1640, дополненной 10% ФТС (фетальная телячья сыворотка), 2 мМ L-глутамина, 1 мМ пирувата натрия, 1% незаменимых аминокислот, 50 ед/мл пенициллина и 50 мкг/мл стрептомицина (= RPMI/ФТС). Было обнаружено, что клетки лишены микоплазмы.
30000 клеток/100 мкл RPMI/ФТС высевали в плоскодонные лунки для микротитрования. Одновременно добавляли 10 мкл ретиноидов, растворенных в готовой среде, для получения конечных концентраций в диапазоне от 10-11 до 10-6M (маточные растворы с концентрацией 10 М в этаноле выдерживали при -20oC в защищенном от света месте). Через 3 дня среду удаляли с помощью многоканальной пипетки и замещали 100 мкл раствора NBT (1 мг/мл ЗФР (забуференный фосфатом физиологический раствор)) с 200 нМ форболмиристатацетата (ФМА)). После дополнительной инкубации в течение 1 ч при 37oC раствор NBT удаляли и добавляли 100 мкл 10%-ного ДСН (додецилсульфат натрия) в 0,01Н HCl. Количество восстановленного NBT определяли фотометрически при 540 нм с использованием автоматического планшет-ридера. Вычисляли средние значения по трем лункам. Среднеквадратичная ошибка составляла 5-10%.
Результаты
Воздействия избирательных
по отношению к RXR ретиноидов на дифференцировку клеток HL-60
В соответствии со своей слабой эффективностью в качестве RARα-активатора избирательный по отношению к RXR ретиноид из
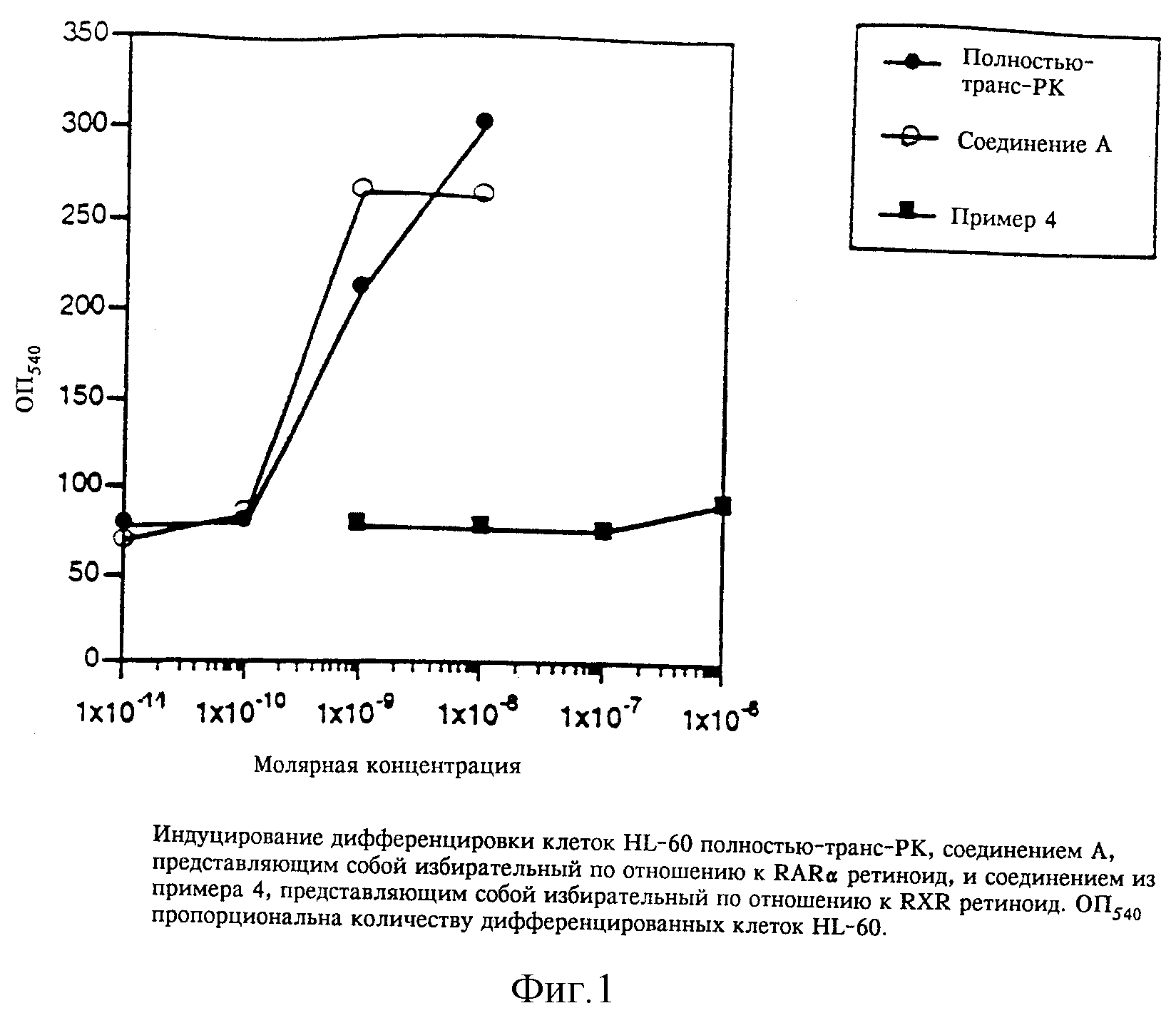
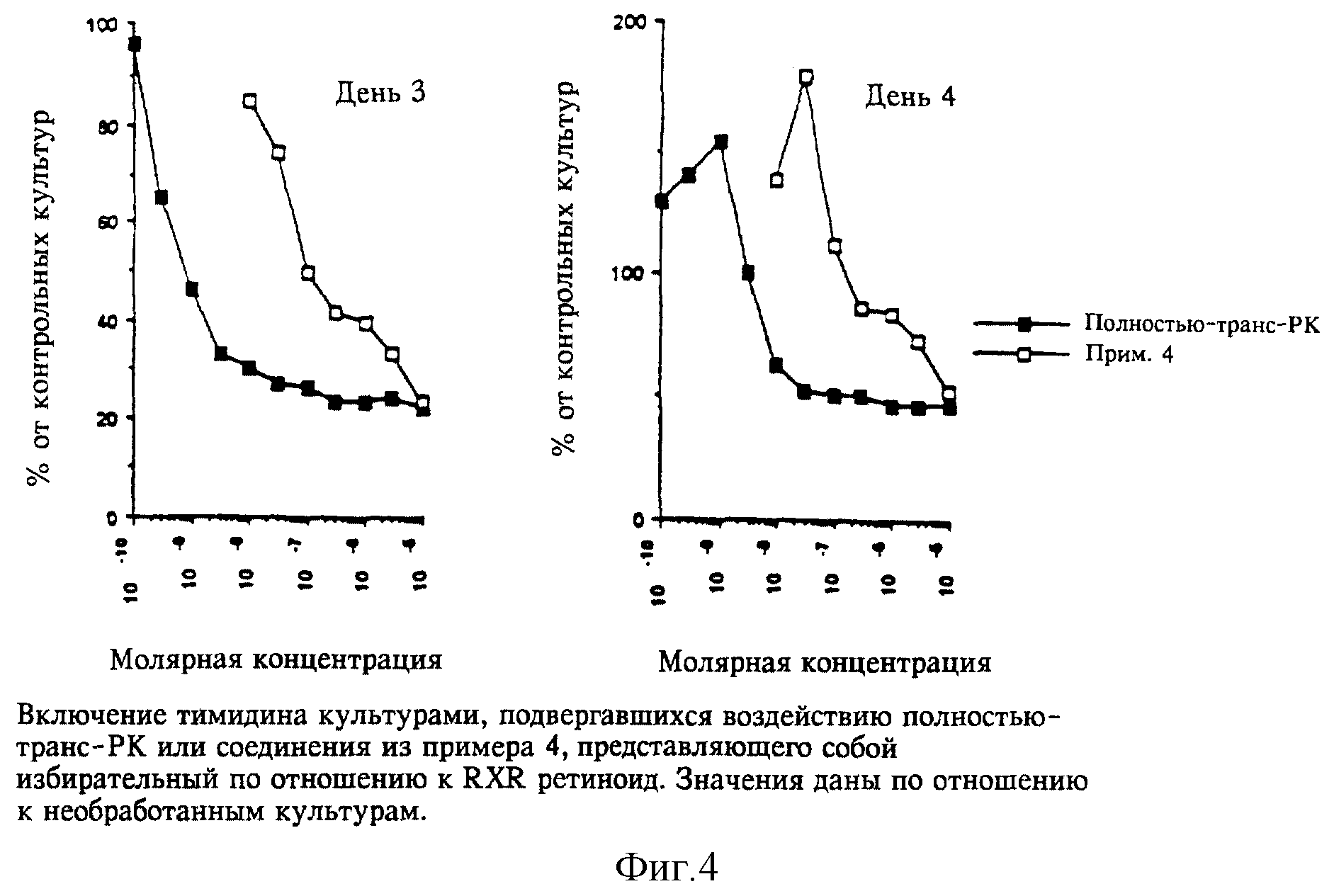
примера 4 оказался существенно менее активным, чем полностью-транс-ретиноевая кислота в качестве индуцирующего фактора дифференцировки клеток HL-60 (фиг. 1). Действительно, соединение из примера 4
оказалось фактически неактивным даже в концентрации 1 x 10-6 M, что хорошо согласуется с его трансактивирующими характеристиками. По-видимому, клетки HL-60 являются примерно в 10 раз более
чувствительными к ретиноидам, чем трансактивирующая система (ср. табл. 1, RARα и фиг. 1).
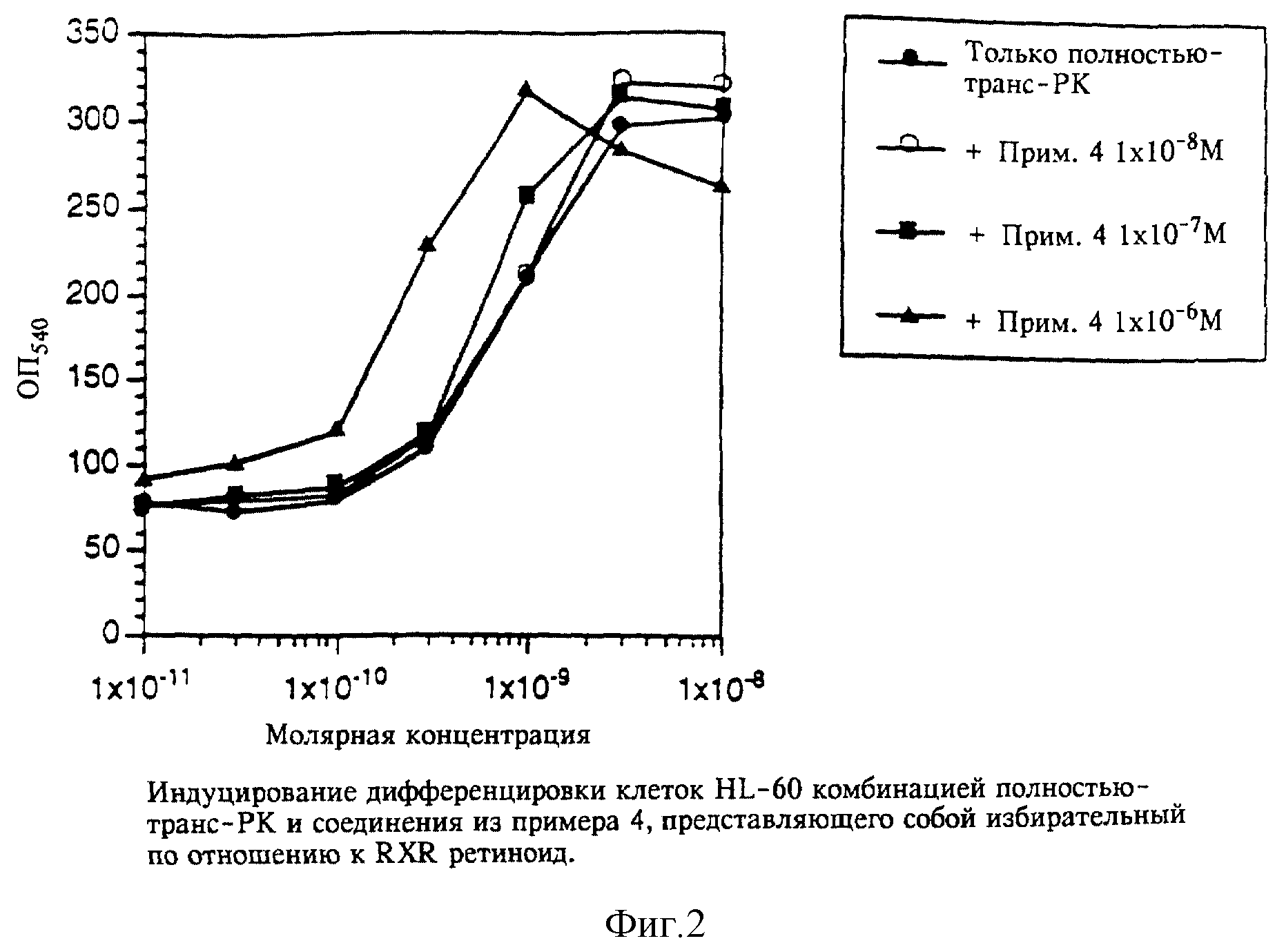
Однако, как видно на фиг. 2 и 3, избирательный по отношению к RXR ретиноид из примера 4 проявлял способность усиливать воздействия полностью-транс-ретиноевой кислоты и соединения А в концентрациях, при которых он был неактивен сам по себе. Наблюдали усиление в 3-10 раз, т.е. для того, чтобы получить сопоставимые уровни дифференцировки клеток HL-60, требовалось в 3-10 раз более высокая концентрация взятых по отдельности полностью-транс-ретиноевой кислоты или соединения А.
Из полученных результатов ясно, что воздействия избирательных по отношению к RXR лигандов на дифференцировку клеток HL-60 выше, чем просто аддитивные, и, следовательно, не могут быть объяснены остаточными RAR-активирующими воздействиями. Наблюдаемые воздействия также несколько более выражены, если используют более предпочтительный RARα-лиганд (соединение А), а не менее избирательную (по отношению к RAR по сравнению с RXR) полностью-транс-ретиноевую кислоту. Усиливающее воздействие также сопоставимо с данными, полученными in vitro, показывающими, что RXR образует гетеродимеры с RAR, что приводит к увеличению транскрипционной активности на RAR-специфичных последовательностях промотoра. До настоящего времени остается неясным, почему относительно высокие концентрации избирательных по отношению к RXR ретиноидов (10-7M) требуются для обнаружения каких-либо воздействий. Эти концентрации более чем на два порядка выше, чем значение ЕС50, полученное при изучении RXR-транскрипционной активации, однако остается неясным, изменяется ли при связывании сродство RXR к ретиноиду при образовании гетеродимера. Таким образом, значения ED50, приведенные в табл. 1, могут неполностью характеризовать воздействия, осуществляемые посредством гетеродимеров, включающих RXR.
Пример 19
Изучали способность
соединения из примера 4 в сочетании с полностью-транс-ретиноевой кислотой ингибировать пролиферацию B-клеток мышей. Избирательный по отношению к RXR лиганд по изобретению усиливал ингибирующее
действие полностью-транс-ретиноевой кислоты на пролиферацию B-клеток мышей.
Материалы и методы исследования
Ретиноиды
Характеристики связывания и трансактивирования
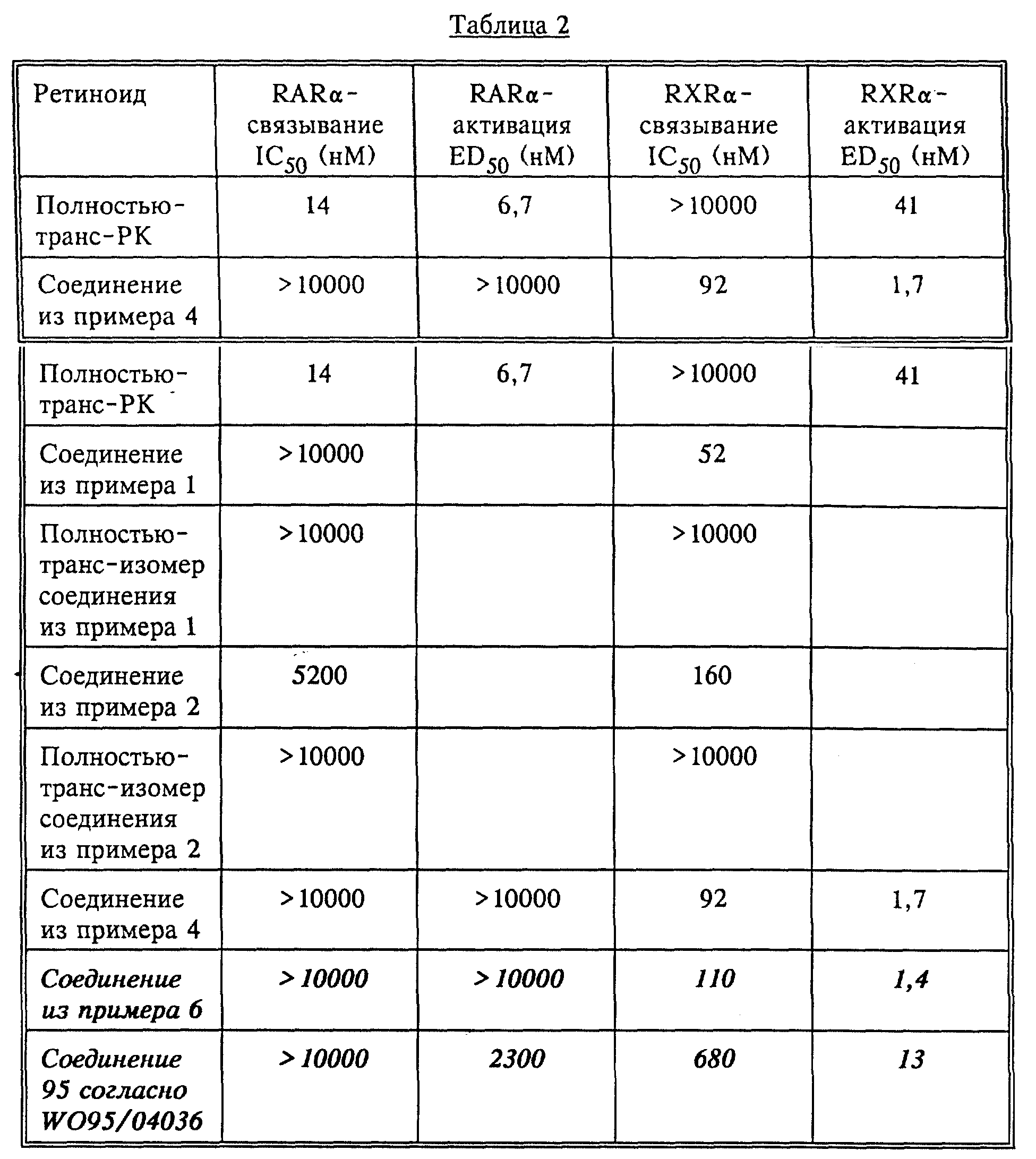
ретиноидов, использованных в данных исследованиях, приведены в табл. 2.
Цитoлогические исследования
Суспензии одиночных клеток селезенки мыши помещали в культуру в
плоскодонные 96-луночные планшеты для микротитрования, причем в каждую лунку помещали по 0,2 мл суспензии клеток в концентрации 2 x 105 мл в IMDM с добавлением 10%-ной фетальной телячьей
сыворотки, HEPES, антибиотиков и 50 мкМ 2-меркаптоэтанола. Добавляли специфический для митогена B-клеток Е. coli липополисахарид (ЛПС) (фирма DIFCO) в концентрации 50 мкг/мл. Культуры инкубировали при
температуре 37oC в увлажненной атмосфере с 5%-ным содержанием CO2.
Ретиноиды, предназначенные для испытания, сначала трижды титровали от 10 нМ до 10 мкМ и оставляли на весь период культивирования. Далее последовательно титровали действующие вещества до достижения величины IC50. В качестве эталона использовали циклоспорин А (фирма Sandoz AG) (от 1 нМ до 1 мкМ).
После 2, 3 и 4-го дней культивирования клетки импульсно метили [3H]-тимидином, 1 мкКи/лунку в течение 4 ч. Затем культуры собирали на фильтры из стекловолокна и измеряли включенную в ДНК радиоактивность с помощью β-жидкостного сцинтилляционного счетчика (Betaplate, Wallac Oy, Турку, Финляндия).
Результаты выражали в процентах по отношению к реакции необработанных культур. Индуцируемую митогеном пролиферацию измеряли на основании данных о включении3Н-тимидина после культивирования в течение 24, 48 и 72 ч. Ретиноиды добавляли в культуры в начале периода культивирования.
Результаты
1. Воздействие избирательных по отношению к RXR лигандов на пролиферацию B-клеток мышей
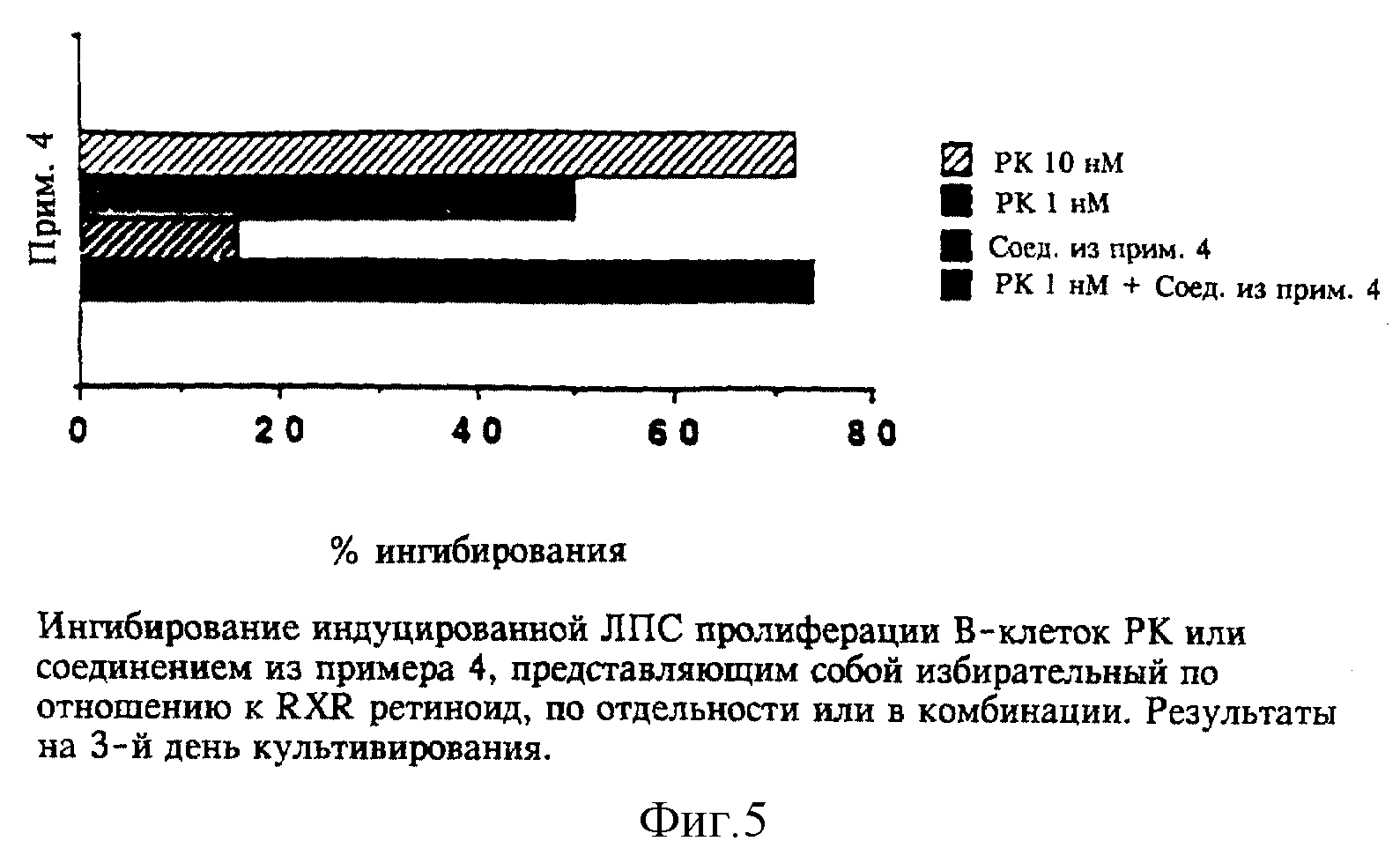
Избирательный по отношению к RXR лиганд из примера 4 сначала испытывали на непосредственное взаимодействие с индуцированной ЛПС пролиферацией B-клеток мышей. Результаты приведены на фиг. 4, они
показывают, что ретиноид из примера 4 является ингибитором, значение IC50 которого составляет 100 нМ. Эта эффективность составляет 1/100 таковой полностью-транс-ретиноевой кислоты (IC50 = 1 нМ). На четвертый день ретиноид из примера 4 в низких дозах (соответствующих ≈ IC50 на третий день) слегка увеличивает реакцию, что согласуется с результатами по
активности всех ретиноидов в этой системе.
2. Лиганды, избирательные по отношению к RXR, усиливают ингибирующее действие полностью-транс-ретиноевой кислоты на пролиферацию B-клеток
мышей
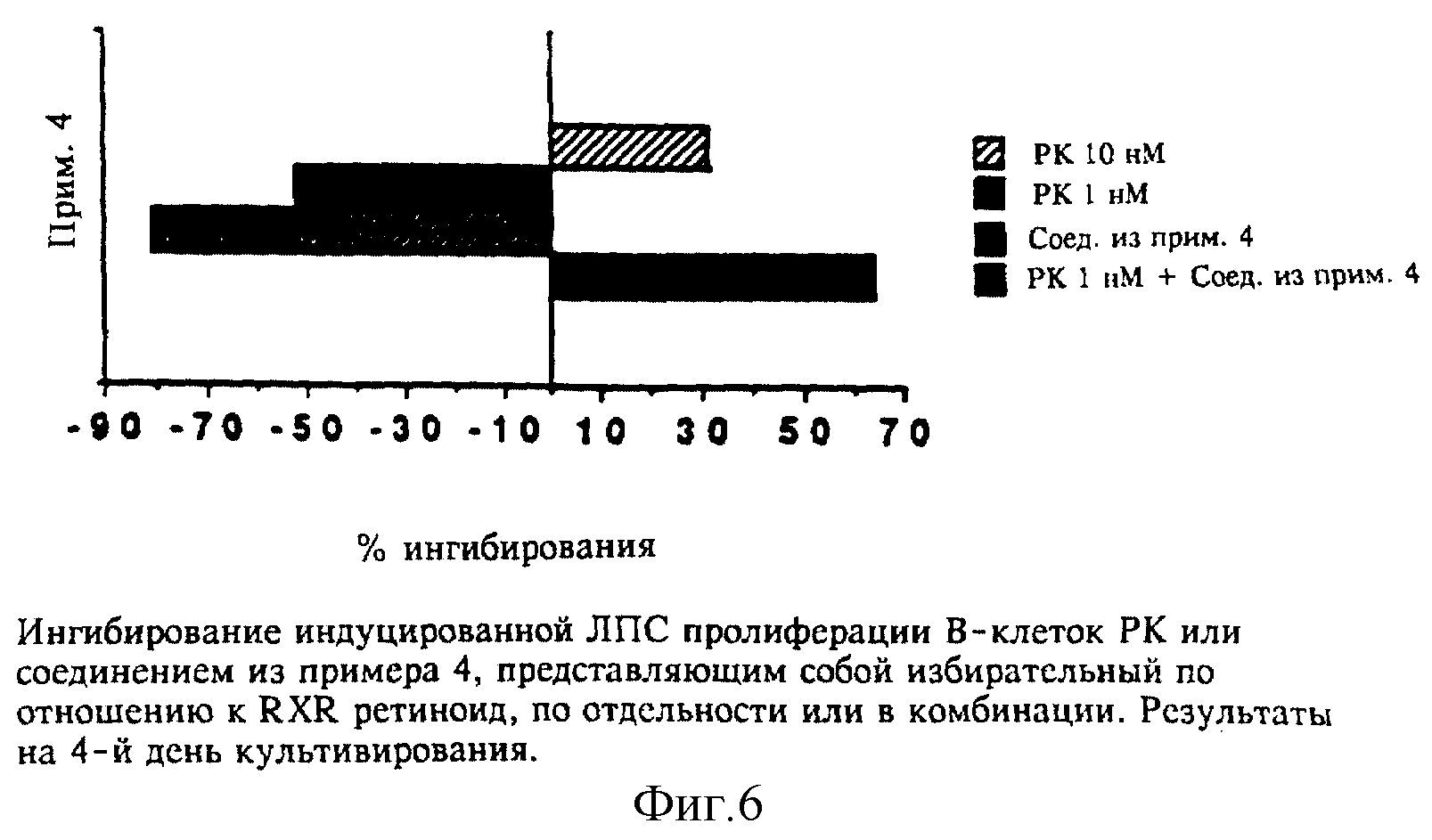
Полностью-транс-РК (РК обозначает ретиноевую кислоту) ингибирует индуцированную ЛПС пролиферацию В-клеток мышей с IC50 = 1 нМ. Максимальное ингибирование получают при 10-30
нМ, и оно никогда не превышает 75-80%. Избирательный по отношению к RXR лиганд из примера 4 по изобретению добавляли в стимулированные ЛПС культуры, подвергавшиеся воздействию 1 нМ полностью-транс-РК;
параллельно готовили культуры, подвергавшиеся воздействию 10 нМ полностью-транс-РК. Во всех случаях концентрации избирательного по отношению к RXR лиганда из примера 4 по изобретению, которые сами по
себе обладают слабой ингибирующей активностью, индуцировали усиление действия 1 нМ РК до уровня, наблюдаемого при 10 нМ РК. Аналогичные результаты получали на второй и третий день культивирования.
Данные, полученные на третий день, представлены на фиг. 5.
Поскольку соединение из примера 4 обладает активностью, то эффект может быть либо аддитивным, либо усиленным (синергистическим). Результаты, полученные на более поздних стадиях культивирования, показывают, однако, что эффект не просто аддитивный, а скорее усиленный.
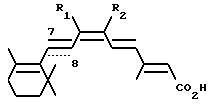
На четвертый день 1 нМ РК уже не оказывает ингибирующего действия и фактически вызывает незначительное усиление реакции, в то время как 10 нМ РК ингибирует приблизительно на 30%. Причины этого явления не установлены, но при этом может играть роль период полураспада соединения. Избирательный по отношению к RXR лиганд также обладает аналогичным действием (см. кривую на фиг. 4). Однако при этом комбинация 1 нМ РК и RXR-лиганда из примера 4 по изобретению вызывает ингибирование такого же порядка, как и индуцируемое 10 нМ РК. Результаты приведены на фиг. 6.
Результаты показывают, что избирательный по отношению к RXR ретиноид (пример 4) усиливает действие полностью-транс-РК в функциональной системе, в которой действие ретиноида осуществляется посредством RARα. Усиление получают при 10-30-кратном превышении RXR-лиганда над полностью-транс-ретиноевой кислотой. Величина усиления приблизительно десятикратная: уровень ингибирования, обычно индуцируемый 10 нМ РК, получают при использовании 1 нМ РК в комбинации с RXR-лигандом по изобретению.
Пример 20
Противоугревая активность соединений по изобретению показана на основе следующих исследований с
использованием обычных способов изучения противоугревой активности.
(1) Антипролиферативная активность в отношении себоцитов человека
Методы
Жировые клетки выделяли
из сальных желез взрослого человека с помощью комбинации ферментативных и механических методов (Doran и др., 1991). Клетки культивировали в среде Искова, содержащей 10% фетальной телячьей сыворотки и
4 мкг/мл дексаметазона на слое 3Т3-фибробластов мышей с заингибированным ростом. Клетки высевали в среду без исследуемого соединения и затем спустя 24-48 ч после первоначального посева вводили
исследуемое соединение в свежей среде. В культуры каждые 48 ч добавляли свежую среду, содержащую исследуемое соединение. В день сбора культуры промывали 0,03%-ным ЭДТК в ЗФР для удаления только
3Т3-фибробластов с последующей инкубацией в 0,05%-ном трипсине/0,03%-ном ЭДТК. Клетки суспендировали, интенсивно перемешивали для получения суспензии одиночных клеток и подсчитывали с помощью
гемоцитометра.
Маточные растворы соединений готовили в виде 10-2М растворов в 100%-ном ДМСО и хранили в темноте при -20oC. Растворы соединений доводили до комнатной температуры и разбавляли до соответствующей концентрации непосредственно в полной среде.
Способность соединений ингибировать in vitro пролиферацию роста жировых клеток изучали в концентрациях 10-6 и 10-7 M.
Изучали in vitro способность соединений из примеров 12, 2 и 4 ингибировать пролиферацию жировых клеток человека первого пассирования после 10 дней выдерживания с лекарственным препаратом. Результаты представлены в виде концентрации (в нМ), необходимой для ингибирования роста на 50% (IC50) (см. табл. 3).
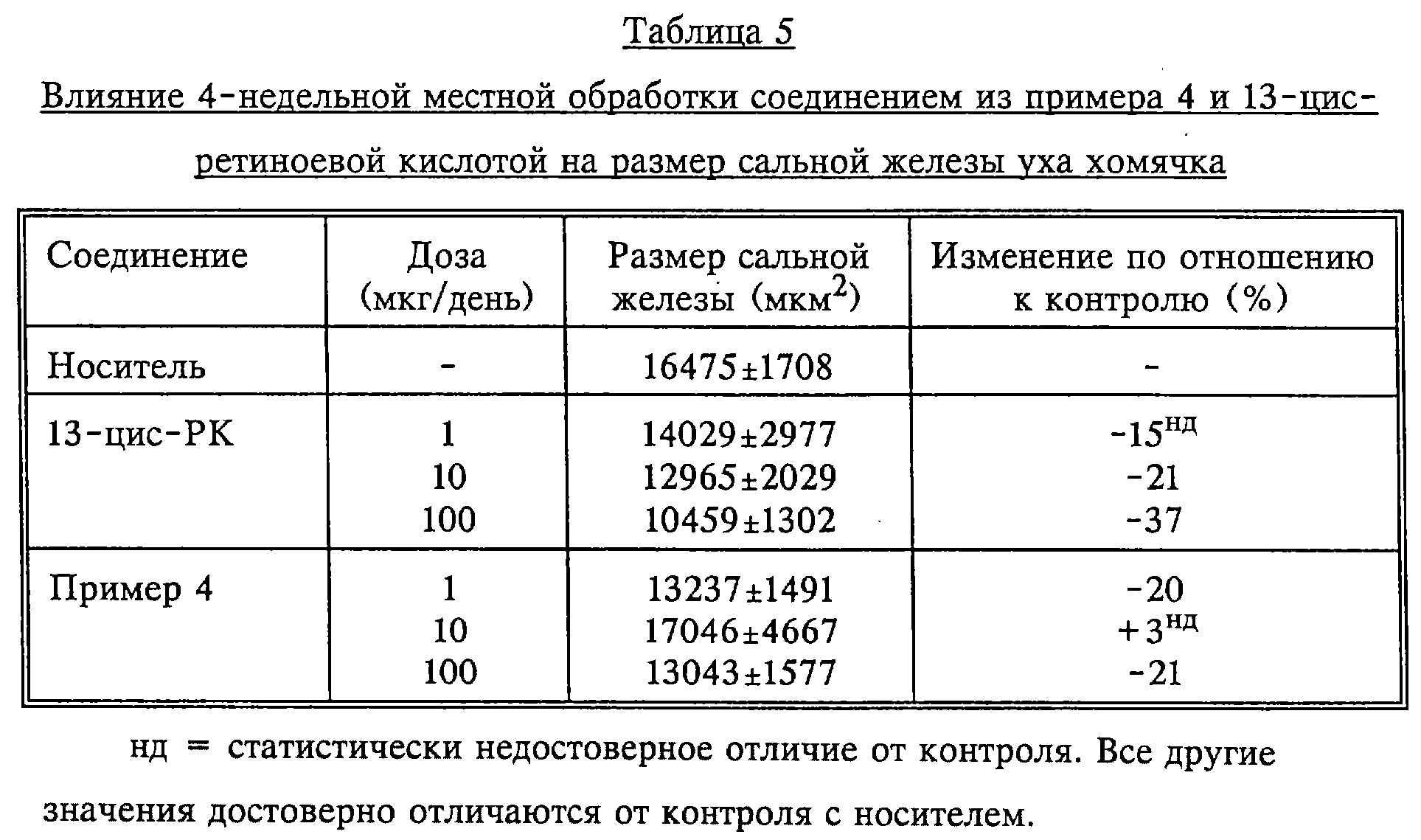
Соединение из примера 4 проявило слабую зависимость реакции от дозы, при этом подавление роста достигало 30-40%. Хотя это намного слабее, чем для 13-цис-ретиноевой кислоты, тем не менее, это свидетельствует о биологической активности in vitro. Значение IC50 соединения из примера 2 составило 100 нМ в первом эксперименте, однако доза 1000 нМ в этом эксперименте дала только 70%-ное ингибирование роста.
(2) Редуцирующая активность в отношении кожных мешочков мышей линии Rhino
При использовании этой модели исследуют способность соединений
уменьшать размер зачаточных кожных мешочков, кератинизированных, связанных с сальными железами волос структур, которые напоминают комедоны человека (Mezick и др., 1985).
Метод
Использовали группы по 6 самок мышей линии Rhino (hrrhhrrh) 6-8-недельного возраста, полученных из Jackson Laboratories. Соединения растворяли в 100%-ном ацетоне и хранили при
4oС в атмосфере азота на протяжении всего эксперимента. Исследуемые соединения наносили на спинку мыши ежедневно в течение пяти дней на протяжении трех последовательных недель. Через один
день после нанесения последней дозы мышь умерщвляли путем аспирации CO2. Вырезали кусочек кожи со спинки и инкубировали в 2M бромиде натрия в течение 2-3 ч при комнатной температуре. Затем
отделяли эпидермис от собственно кожи.
Затем эпидермис обезвоживали с помощью 70%, 80%, 95%, 100%-ного этанола и ксилола. Образцы выдерживали в каждом из указанных выше растворов в течение 2 ч. Образцы кожи извлекали из ксилола и помещали на предметные микроскопические стекла. Для определения диаметра и площади кожных мешочков применяли анализ изображения с использованием компьютерной программы для анализа изображения Ultimage. Оценивали приблизительно 5 областей каждого образца, измеряя в среднем по 150 кожных мешочков у каждой мыши.
Результаты
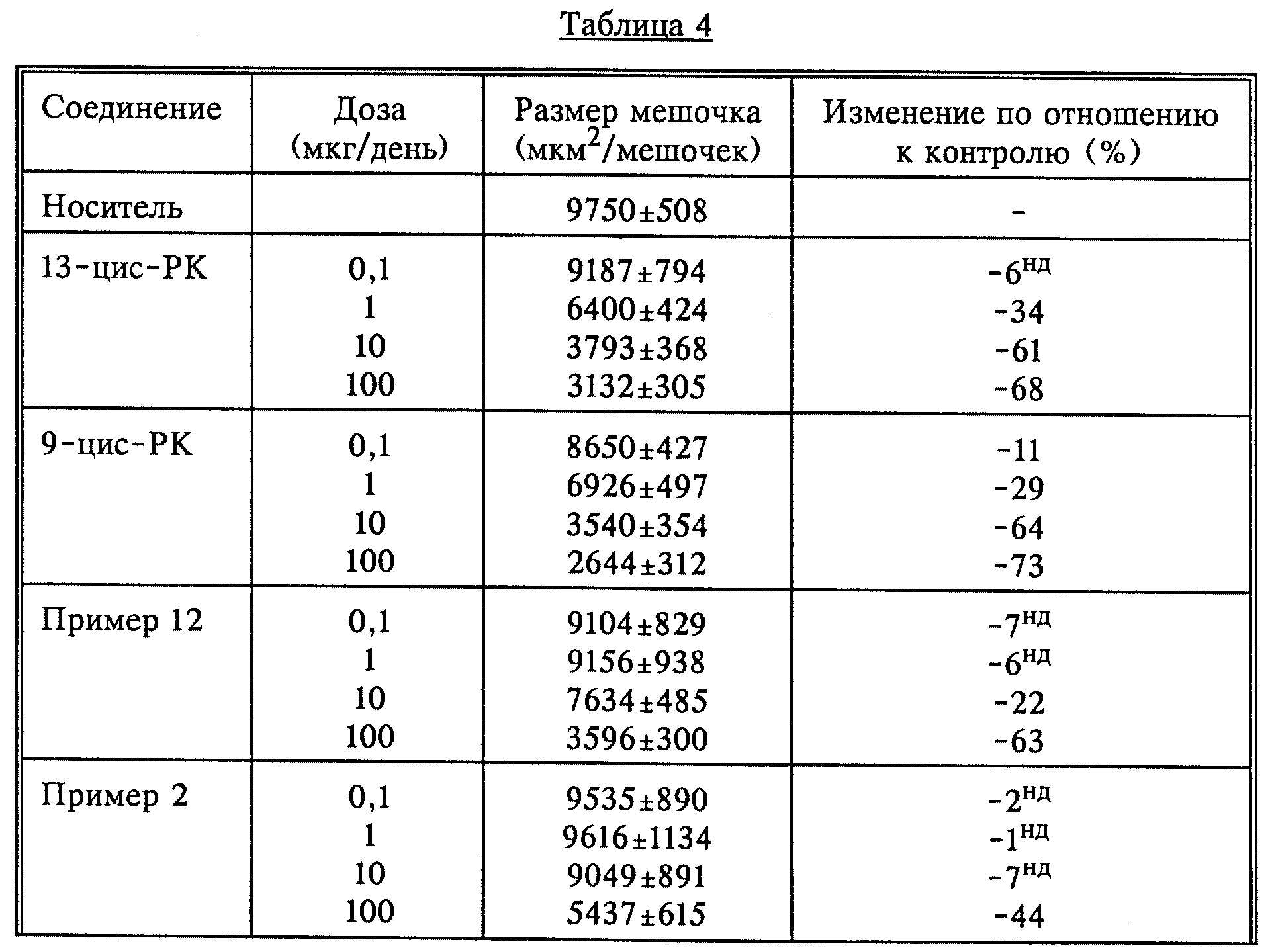
С помощью местного нанесения на мышь линии Rhino исследовали несколько соединений по изобретению (примеры 12, 2, 4). На мышей локально наносили дозы по 100 мкл 13-цис-ретиноевой кислоты,
9-цис-ретиноевой кислоты и соединений из примеров 12, 2 и 4 в ацетоне. Результаты показаны в табл. 4.
Примечания.
Во время данного эксперимента отмечали следующие побочные эффекты. У животных, обработанных дозами по 100 мкг 13-цис-ретиноевой кислоты, на пятый день наблюдали отслоение рогового слоя эпидермиса и эритему. У животных, обработанных 9-цис-ретиноевой кислотой, такие же эффекты при такой же дозе проявлялись на день раньше.
На седьмой день обработки эритему обнаруживали при использовании 100 мкг соединения из примера 12. В тот же день отмечали некоторые проявления эритемы у животных, обработанных 100 мкг соединения из примера 2. Однако в этом случае эритема была не столь интенсивной, как при использовании соединения из примера 12. За такой же интервал времени сравнимая доза соединения из примера 14 не вызвала заметной эритемы.
К четырнадцатому дню обработки отслоение рогового слоя эпидермиса наблюдали у всех животных, обработанных дозами по 100 мкг соединения из примера 12, чего не обнаруживали днем раньше. Если данные соединения, взятые в концентрации 100 мкг, выстроить в ряд в зависимости от их способности вызывать отслоение/эритему, от более серьезных проявлений к менее серьезным, то порядок будет следующим: 9-цис-РК, 13-цис-РК, соединение из примера 12, соединение из примера 2 и соединение из примера 4.
Таким образом, соединения по изобретению вызывают меньшее раздражение по сравнению с 9-цис-РК и 13-цис-РК.
(3) Воздействие in vivo соединений на размер
сальных желез золотистых хомячков
Методы
Самцам золотистого хомячка Charles River давали требуемую дозу в ацетоне. Соединения хранили при 4oC в защищенном от света месте.
Еженедельно готовили свежие растворы.
Хомячков обрабатывали ежедневно определенными дозами в течение пяти дней, делали двухдневный перерыв и обрабатывали вновь в течение пяти дней в общей сложности 20-ю дозами. Объем раствора составлял 50 мкл на ухо.
Хомячков умерщвляли с помощью аспирации CO2 и отделяли уши для гистологического изучения. Брали одно ухо и отделяли вентральную поверхность от остальной части уха. Брали образец размером 2 мм на расстоянии 5 мм от кончика уха и окрашивали в течение ночи 0,1% черного судана B, растворенного в 100%-ном пропиленгликоле. После обесцвечивания количественно оценивали размер сальных желез с помощью системы анализа изображения Donsanto.
Результаты представлены в виде средней площади 80-120 сальных желез на дозу, в процентах по отношению к таковой в контрольных срезах (обработанных только растворителем) (см. табл. 5).
Ссылки
Doran и др. "Characterisation
of human sebaceous cells in vitro", J. Invest. Dermatol., 96:341-348 (1991).
Mezick и др. "Topical and systemic effects of retinoids on horn-filled utriculus size in the rhino mouse. A model to quantify 'antikeratinizing' effects of retinoids", J. Invest. Dermatol., 83:110-113 (1985).
Пример 21
Исследовали ингибирующее действие на линию клеток карциномы
молочной железы человека соединения из примера 6 в сочетании с ретиноидом, обладающим RARα-активностью, а именно полностью-Е-3,7-диметил-9-[(2-метокси-4-метил-6-октилокси) фенил] -2,4,6,
8-нонатетраеновой кислотой (соединение В), представленного ниже:
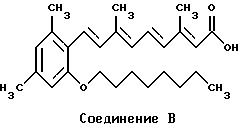
1. Синтез соединения В
Соединение получали в соответствии с процедурой, представленной в конце описания.
Результаты показали, что избирательный по отношению к RXR лиганд по изобретению усиливает ингибирующее действие соединения B на плотную опухоль.
Раствор 2,6-дигидрокси-4-метилбензойной кислоты 1 в ацетоне (50,4 г; 750 мл) обрабатывали метилйодидом (100 мл) и карбонатом калия (124,2 г) и нагревали с обратным холодильником в течение 18 ч. Затем смесь охлаждали до комнатной температуры и отфильтровывали твердые частицы. Концентрированием раствора получали неочищенный продукт, который растворяли в этилацетате и промывали водным раствором гидроксида натрия (1H, холодный). При последующем удалении растворителей в вакууме получали метил-2,6- диметокси-4-метилбензоат 2 (54 г). Этот материал (52,5 г) растворяли в дихлорметане (1000 мл), охлажденном до -70oC, и обрабатывали раствором боронтрихлорида в этом же растворителе (29,3 г/100 мл; 180 мл). Затем эту смесь нагревали до комнатной температуры, дополнительно перемешивали в течение 45 мин и затем сливали на лед. Органический слой отделяли, промывали большим количеством воды, сушили (сульфат магния), фильтровали и концентрировали. Затем остаток очищали с помощью препаративной жидкостной хроматографии высокого разрешения (ЖХВР), используя хроматограф Waters Prep. 500 (7%-ный этилацетат/гексан в качестве элюента), получая чистый метил-2-гидрокси-6-метокси-4- метилбензоат 3 в виде твердого продукта (28,3 г). Раствор этого материала (35 г) в метилэтилкетоне (700 мл), содержащем карбонат калия (27,1 г), обрабатывали 1-октилйодидом (34,2 г) и нагревали с обратным холодильником в течение 20 ч. Реакционную смесь охлаждали, отфильтровывали твердые частицы, и фильтрат концентрировали и затем повторно растворяли в смеси гексана и этилацетата. Этот раствор затем промывали водным основанием (1H гидроксид натрия), водой, сушили (сульфат магния) и концентрировали досуха. Путем очистки с помощью ЖХВР, как указано ранее, получали чистый метил-2-метокси-6-октилокси-4-метилбензоат 4 (51,8 г). Раствор этого материала (51,8 г) в толуоле (500 мл) охлаждали до -60oC и обрабатывали раствором гидрида диизобутилалюминия (25% в гексане; 281 мл) и затем нагревали до комнатной температуры. Затем реакционную смесь осторожно обрабатывали водным метанолом (100 мл; 1:1), охлаждая так, чтобы поддерживать температуру на уровне 20oC, с последующим добавлением гексана (500 мл) и сульфата магния. Затем твердые частицы отфильтровывали, и растворители удаляли в вакууме, получая неочищенный спирт 5. Этот материал (47 г) растворяли в ацетонитриле (500 мл), содержащем трифенилфосфонийгидробромид (54,9 г) и нагревали с обратным холодильником в течение 22 ч. Затем растворители удаляли в вакууме, и неочищенную соль 6 сушили при комнатной температуре и давлении 0,1 мм рт.ст. (этот материал может быть выкристаллизован из тетрагидрофурана, давая чистый [(2- метокси-6-октилокси-4-метил)фенил] метилтрифенилфосфонийбромид). Эту соль 6 затем превращали в соединение В, т.е. полностью-E-3,7- диметил-9-[(2-метокси-4-метил-6-октилокси)фенил] -2,4,6, 8- нонатетраеновую кислоту, как описано в патенте США 4894480 (пример 5).
2. Тестирование
Изучение связывания с ретиноидным рецептором, и трансактивации проводили с
использованием соединения В и соединения из примера 6, как описано выше. Получены результаты, представленные в табл. 6.
Линию клеток карциномы молочной железы, T47-D, полученную из Американской коллекции типовых культур (АТСС) [каталожный номер N НТВ133], выращивали в среде RPMI 1640 с добавлением 10% ФТС, 10 мкг/мл инсулина и 13 нг/мл гентамицина, и инкубировали при 37oС при относительной влажности воздуха 95,5%, содержащего 4,5% CO2. Клетки собирали после достижения 70-80% слияния и пеллетировали. Клетки ресуспендировали в среде и высевали (150 мкл/лунку) в 96-луночные планшеты (Corning) с плотностью, позволяющей иметь линейный рост в течение семидневного периода исследования (т.е. клетки T47D высевали из расчета 4 x 103 клеток/лунку). Планшеты помещали на ночь в инкубатор с температурой 37oC.
Лекарственные препараты (1 мМ маточного раствора в 100%-ном ДМСО) добавляли через 18-24 ч после посева. Комбинации лекарственных препаратов готовили в 96-луночных планшетах для микротитрования, и вручную добавляли в опытные планшеты. Анализы МТТ проводили в течение 3-7 дней после добавления лекарственного препарата. МТТ-анализ представляет собой анализ, основанный на определении тетразолия, который позволяет оценить жизнеспособность клеток в культуре. Маточный раствор МТТ (3-[4, 5-диметилтиазол-2- ил]-2,5-дифенилтетразолийбромид) (5 мг/мл в 1xЗФР) добавляли к опытным планшетам (50 мкл/лунку) и инкубировали в течение 2,5 ч при 37oC.
Жидкость удаляли
путем аспирации, добавляли 50 мкл 95%-ного этанола, и планшеты встряхивали (Mini-Orbital Shaker, Belico) в течение 15 мин для повышения растворимости формазанового продукта. Оптическую плотность (ОП)
каждой ячейки измеряли, используя автоматический планшетный ридер (Microplate EL320 Reader, Bio-Tek Instruments) с опытной длиной волны 570 нм и контрольной длиной волны 660 нм. Величину ингибирования
клеточного роста, вызываемую различными концентрациями соединения В в комбинации с постоянной концентрацией соединения из примера 6, определяли в соответствии со следующим уравнением:

Результаты представлены графически на фиг. 7. Эти результаты показывают, что избирательное по отношению к RXR соединение по изобретению повышает активность ретиноида, обладающего RARα-активностью, при ингибировании роста линии клеток плотной опухоли.
Пример А
Твердые
желатиновые капсулы, содержащие 20 мг активного вещества.
Композиция - одна капсула содержит, мг:
Соединение формулы I - 20,0
RARα-ретиноид - 20,0
Желатиновый блюм 30 - 70,0
Мальтодекстрин MD 05 - 108,0
dl-α-токоферол - 2,0
Аскорбат натрия - 10,0
Микрокристаллическая целлюлоза - 48,0
Стеарат
магния - 2,0
(Вес содержимого капсулы) - 280,0
Процедура
Действующие вещества измельчают во влажном состоянии в растворе желатина, мальтодекстрина, dl-α-токоферола и
аскорбата натрия. Суспензию из измельченных во влажном состоянии ингредиентов высушивают распылением.
Высушенный распылением порошок смешивают с микрокристаллической целлюлозой и стеаратом магния.
Каждую твердую желатиновую капсулу соответствующего размера и цвета заполняют 280 мг этой смеси.
Описание чертежей
Фиг. 1 - индуцирование
дифференцировки клеток HL-60 полностью-транс-ретиноевой кислотой (РК), другим избирательным по отношению к RARα ретиноидом (в данном случае соединение А) и избирательным по отношению к RXR
ретиноидом (в данном случае соединение из примера 4).
Фиг. 2 - индуцирование дифференцировки клеток HL-60 комбинацией полностью-транс-ретиноевой кислоты (РК) и соединения из примера 4.
Фиг. 3 - индуцирование дифференцировки клеток HL-60 комбинацией соединения А и соединения из примера 4.
Фиг. 4 - включение тимидина культурами B-клеток, подвергавшихся воздействию полностью-транс-РК или соединения из примера 4.
Фиг. 5 - ингибирование индуцированной ЛПС пролиферации B-клеток полностью-транс-РК и соединением из примера 4, по отдельности или в комбинации, на третий день культивирования.
Фиг. 6 - ингибирование индуцированной ЛПС пролиферации B-клеток полностью-транс-РК и соединением из примера 4, по отдельности или в комбинации, на четвертый день культивирования.
Фиг. 7 - увеличенная антипролиферативная активность соединения, обладающего RARα-активностью (соединение B), и избирательного по отношению к RXR соединения (соединение из примера 6) в отношении линии клеток рака молочной железы.
Реферат
Изобретение относится к новым ретиноидам формулы I, где связь C7-C8 является двойной связью и один из R1 и R2 обозначает C1-C4алкил, а другой обозначает хлор, бром или иод, или R1 и R2 вместе обозначают C3-C6алкилен; или связь C7-C8 является тройной связью и R1 и R2 вместе обозначают C3-C6алкилен; и их фармацевтически приемлемые соли или сложные эфиры. 7 з.п. ф-лы, 7 ил., 6 табл.
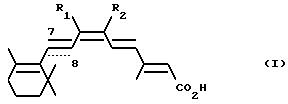
Формула
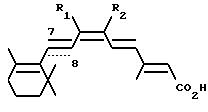
где связь C7 - C8 является двойной связью и один из R1 и R2 обозначает C1 - C4алкил, а другой обозначает хлор, бром или йод, или R1 и R2 вместе обозначают C3 - C6 алкилен; или связь C7 - C8 является тройной связью и R1 и R2 вместе обозначают C3 - C6алкилен;
и их фармацевтически приемлемые соли и сложные эфиры.
06.06.95 по пп.1 и 8;
24.02.95 по пп.2 - 7.
Комментарии